

Abstract
Vertebrate cells express a family of heat shock transcription factors (HSF1 to HSF4) that coordinate the inducible regulation of heat shock genes in response to diverse signals. HSF1 is potent and activated rapidly though transiently by heat shock, whereas HSF2 is a less active transcriptional regulator but can retain its DNA binding properties for extended periods. Consequently, the differential activation of HSF1 and HSF2 by various stresses may be critical for cells to survive repeated and diverse stress challenges and to provide a mechanism for more precise regulation of heat shock gene expression. Here we show, using a novel DNA binding and detection assay, that HSF1 and HSF2 are coactivated to different levels in response to a range of conditions that cause cell stress. Above a low basal activity of both HSFs, heat shock preferentially activates HSF1, whereas the amino acid analogue azetidine or the proteasome inhibitor MG132 coactivates both HSFs to different levels and hemin preferentially induces HSF2. Unexpectedly, we also found that heat shock has dramatic adverse effects on HSF2 that lead to its reversible inactivation coincident with relocalization from the nucleus. The reversible inactivation of HSF2 is specific to heat shock and does not occur with other stressors or in cells expressing high levels of heat shock proteins. These results reveal that HSF2 activity is negatively regulated by heat and suggest a role for heat shock proteins in the positive regulation of HSF2.
The heat shock response is a cellular defense against the deleterious effects of physiological and environmental stress that is mediated by heat shock transcription factors (HSFs). In vertebrates, four members of the HSF family have been identified (27, 28, 38, 40, 46), and of these, HSF1 and HSF2 are ubiquitously expressed and conserved (27, 40). HSF1 and HSF2 are expressed as inert monomers and dimers, respectively, in unstressed cells. Upon activation, these HSFs trimerize and function as transcriptional activators that bind with similar, but not identical, specificities to the heat shock element (HSE), thus regulating heat shock gene transcription and hence the expression of diverse heat shock proteins and molecular chaperones (2, 40, 49, 62).
HSF1 and HSF2 differ in their pathways of activation and the nature of their transcriptional responses. Whereas HSF1 is activated upon exposure to a multitude of physiological and environmental stresses (41, 62), HSF2 activity appears to be more selective, being induced upon down-regulation of the ubiquitin-proteasome pathway (22), during differentiation (36, 37, 42, 49), and in early development (9, 23, 37). Comparison of the HSF1 and HSF2 transactivation domains as chimeric proteins with a minimal heterologous DNA binding domain reveals that HSF1 is a more potent transcriptional activator than HSF2 (47, 59, 60). These observations are also supported by studies comparing hsp70 and hsp90 gene transcription rates in K562 cells in which endogenous HSF1 or HSF2 was activated (49). In those studies, HSF1 transcriptional activity was found to be 10-fold greater than HSF2 activity for hsp70 and 5-fold greater for hsp90.
Despite these apparent distinctions, both HSF1 and HSF2 DNA binding activities have recently been codetected in a cell-type- and stress-specific manner (22, 34, 50, 53). The existence of multiple conditions under which coactivation occurs raises the potential for competition between these HSFs to bind the highly conserved heat shock promoter elements. Moreover, the lower transcriptional potency of HSF2 could have deleterious consequences under conditions such as heat shock that require an immediate response and rapid transcriptional activation of heat shock genes. Various observations have previously suggested that HSF2 activity was affected during heat shock. In vitro-translated HSF1 acquires DNA binding activity upon heat shock, whereas HSF2 DNA binding activity is abolished at the elevated temperature (40). It was also shown that constitutive in vivo HSF2 DNA binding activity observed in several embryonal carcinoma cells lines was labile following heat shock (26). In this study, we have examined the DNA binding properties of HSF1 and HSF2 using a novel DNA binding assay that provides a quantitative basis to measure overall HSF DNA binding activity and relative contributions by HSF1 or HSF2. Additionally, we show that HSF2 has the unique biochemical properties of temperature sensitivity in vivo. Consequently, only HSF1 is activated to function as a heat shock factor without interference from HSF2.
MATERIALS AND METHODS
Preparation of cell extracts.
The murine fibroblast cell line NIH 3T3 was grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Cells were treated for the indicated periods with the proteasome inhibitor MG132 (Peptides International) (10 μM), with heat shock (43°C), or sequentially with MG132 (4 h) followed by heat shock. Cells were alternatively treated with 20 μM cadmium chloride (Fisher Scientific) or 5 mM azetidine (Boehringer Mannheim) for 2 to 6 h. Cell extracts were prepared by freeze-thaw lysis as described previously (25). The soluble and pellet fractions were obtained by centrifugation at 50,000 rpm for 5 min in a Beckman TL100 instrument, or the extracts were used without fractionation. The protein content of cell extracts was determined by the Bradford assay (Bio-Rad).
Gel mobility shift assays.
Soluble cell extracts were analyzed for HSF DNA binding activity using labeled HSE-containing oligonucleotides in the gel mobility shift assay (25) and specific polyclonal antibodies to HSF2 (1:50 dilution in phosphate-buffered saline) or HSF1 (1:20 dilution in phosphate-buffered saline) to establish the composition of the HSE binding activities obtained with the various treatments (41).
ORIGEN assay for HSF DNA binding activity.
The assay for HSF DNA binding activity, based on IGEN International's ORIGEN technology, utilizes biotinylated oligonucleotides containing the HSE consensus sequence, which can be immobilized on streptavidin-coated magnetic beads. The identification of the proteins bound to this DNA is enabled by use of specific antibodies (in this case goat anti-rat antibodies [Rockland]) modified with electrochemiluminescent ORI-TAG labels [containing ruthenium ions as tris(bipyridine) chelates]. Ruthenium-tagged antibody molecules bound to DNA-associated proteins immobilized on the magnetic beads are brought in proximity to an electrode by a magnet. This allows discrimination of DNA-bound from unbound factors following immobilization. Application of a current results in emission of photons due to electrochemiluminescence of the ORI-TAG labels. The light emitted is thus a direct measure of the amount of antibody and hence of the protein it recognizes. The assay is performed in a 96-well format on IGEN′s M8 instrumentation, allowing measurements to be multiply replicated.
Soluble cell extracts were prepared as for the gel mobility shift assay, and 20 μg of each was incubated with 0.05 μg of monoclonal rat anti-mouse HSF1 (4B4) or HSF2 (3E2) antibodies (NeoMarkers) in a final volume of 5 to 8 μl for 20 min at room temperature. The primary antibody dilutions were made in gel mobility shift assay binding buffer (25) such that 2 μl was added per sample. To this was added 20 μl of a binding buffer mix containing biotinylated oligonucleotide (4 pmol), poly(dI)/poly(dC) (0.5 μg), and ORI-TAG tag-labeled anti-rat secondary antibodies (0.1 μg), with incubation for an additional 20 min. Streptavidin-coated Dynabeads M280 (IGEN) were added (20 μg per sample) and incubated for an additional 30 min with occasional shaking to immobilize the biotinylated oligonucleotides. The samples were normally prepared in multiple replicates and added singly to M8 plates. The volumes were made up to 250 μl per well with binding buffer, and the plate was read on an M-series M8 instrument (IGEN), using appropriate positive calibrators (IGEN) and the standard M8 assay protocol. The light emitted from the ORI-TAG-labeled antibody is a direct measure of the HSF DNA binding activity, since unbound antibody does not contribute to the signal generated. Controls for this experiment included samples lacking primary antibodies. The relative affinity of the tagged secondary antibody for the 4B4 and 3E2 antibodies was assessed in a similar fashion. Serially diluted amounts of the primary antibodies were immobilized directly on tosyl-activated Dynabeads M280 (Dynal), and the binding of the secondary antibody was assessed.
Immunological analyses.
For immunoblotting analyses, cell extracts (10- to 20-μg protein samples) were resolved by sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis and transferred to nitrocellulose, and HSF2 protein was detected using polyclonal serum raised against murine HSF2 (1:10,000 dilution of serum), as described previously (41), and visualized by ECL (Amersham). Other antibodies used were the murine HSF1-specific polyclonal serum (41) (1:2,000 dilution of serum), Hsp70-specific monoclonal antibody RPN 1197 (Amersham), and polyclonal serum raised against human Hdj-1 (S. G. Fox, B. C. Freeman, and R. I. Morimoto, unpublished data) (1:2,500 dilution of serum).
Nuclear run-on assays.
Transcriptional run-on assays were performed as described previously (3, 49) using nuclei prepared from NIH 3T3 cells treated with MG132, heat shock, or MG132 followed by heat shock, as described above, and visualized and quantitated by PhosphorImager analysis (Molecular Dynamics). Radioactively α-32P-labeled RNA was prepared and hybridized to the following plasmids immobilized on nitrocellulose filters: pH2.3 (human hsp70) (58), pUCHSP86 (mouse hsp90) (20), pHG23.1.2 (human grp78) (S. S. Watowich and R. I. Morimoto, unpublished data), pHA7.6 (human hsp72), pGEM4 (Promega), and pGAPDH (rat glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) (11).
Immunofluorescence and image acquisition.
Detection of HSF1 by immunofluorescence was performed on formaldehyde-fixed 3T3 cells as described previously (17). The monoclonal anti-HSF1 (4) and polyclonal anti-HSF2 antibodies were used at a dilution of 1:300 and detected with a goat anti-rat antibody coupled to rhodamine and a sheep anti-rabbit antibody coupled to fluorescein, respectively (Sigma). Images were acquired on a Zeiss LSM 410 confocal microscope using the 63× (1.4 NA) oil immersion objective.
RESULTS
Differential activation of HSF1 and HSF2.
To assess the relative contributions of HSF1 and HSF2 to the total amount of HSF DNA binding activity induced in response to different stresses, we employed two methods, electrophoretic gel mobility shift assays (EMSA), which, together with antibody supershift assays, have been used to assess the relative contributions of different members of the HSF family, and a novel electrochemiluminescence assay (ORIGEN) that provides a more quantitative assessment of DNA binding activities. Using EMSA and antibody supershift assays, extracts of cells exposed to heat shock were shown to contain HSF1 DNA binding activity (Fig. 1, lanes 6 to 8), whereas both HSF1 and HSF2 DNA binding activities were detected in cells treated with the proteasome inhibitor MG132 (Fig. 1, lanes 2 to 4). However, antibody supershift assays provide qualitative assessments and are incapable of revealing the fraction of total HSF DNA binding contributed by either HSF1 or HSF2. Likewise, with other stresses such as the heavy metal cadmium or the amino acid analogue azetidine, the relative contributions of both HSF1 and HSF2 to the total DNA binding activities could not be clearly established using EMSA due to the highly qualitative nature of this assay and the relatively low level of total HSF DNA binding activity induced (data not shown).
FIG. 1.

Effects of heat shock and MG132 treatments on activation of HSF1 and HSF2. Gel mobility shift analyses using extracts from untreated 3T3 control cells (C) (lane 1) or cells treated with MG132 (lanes 2 to 5), heat shock (HS) (lanes 6 to 9), or MG132 followed by heat shock (lanes 10 to 13) are shown. Cell extracts were incubated in either the presence or absence of specific antiserum to HSF1 or HSF2 or a mixture of both antisera, as indicated, prior to the gel mobility shift assay. HSF DNA binding activities and nonspecific binding (NS) are indicated by arrows. The radiolabeled complex detected at the top of the gel corresponds to retarded antibody-HSF complexes.
A quantitative measure of HSF DNA binding activities was established using the ORIGEN assay (Fig. 2A). To validate the ability of this electrochemiluminescence-based detection method to discriminate between HSF1 and HSF2 DNA binding activities, extracts from heat shock- or hemin-treated K562 cells were used, with monoclonal rat antibodies specific to each HSF and a ruthenylated anti-rat secondary antibody (Fig. 2B). The affinity of the secondary antibody was demonstrated to be equivalent for the monoclonal rat antibodies used, and hence the HSF1 and HSF2 signals may be summed, yielding a value corresponding to total HSF activity. The data in Fig. 2B are presented as the percentage of maximum HSF DNA binding activity and normalized to 100% of the signal observed following heat shock, which is comprised of 80% HSF1 and 20% HSF2. Extracts from control cells contain a low (15%) but consistently detectable HSF DNA binding activity comprised of both HSF1 (10%) and HSF2 (5%). Relative to heat shock, hemin treatment yielded approximately 80% HSF DNA binding activity, corresponding to 50% HSF2 and 30% HSF1. These results show that the ORIGEN assay, by generating a quantifiable signal, provides data that can be readily compared across experiments to assess contributions of relative DNA binding activities that recognize a common promoter element.
FIG. 2.

ORIGEN-based assay of HSF DNA binding activity. (A) Assay scheme as described in the text. Ab, antibody. (B) ORIGEN-based detection of HSF1 and HSF2 activities in K562 cells. Soluble whole-cell extracts (20 μg of protein) from control (C), heat-shock treated (HS), or hemin-induced K562 cells were incubated with 0.05 μg of rat anti-mouse HSF1 (4B4) or HSF2 (3E2) antibodies. The extracts were simultaneously incubated with biotinylated double-stranded HSE-containing oligonucleotide and an ORIGEN tag-labeled goat anti-rat antibody. The complex was allowed to bind to streptavidin-coated Dynal beads and used to measure the amount of bound HSF as described in the text. The sum of the HSF1 and HSF2 signals determined total HSF activity. The data are presented as percent maximum HSF activity, where maximum is defined by the condition which resulted in the highest total HSF activation (heat shock in this case). The data are derived from triplicate samples with coeffi- cients of variation of below 15%. Error bars indicate standard deviations. (C) HSF1 and HSF2 activities induced by heat, MG132, or azetidine treatment of 3T3 cells. 3T3 cells were treated for the lengths of time indicated with heat at 43°C (HS), 10 μM MG132 (MG), or 5 mM azetidine (Az). Extracts were prepared and used in the ORIGEN assay as described in the text. The data are derived from duplicate samples with coefficients of variation of 10% or less and are presented as percent maximum HSF activity as defined above, with maximum activity being induced in this case by the 6-h MG132 treatment. The data for both ORIGEN assays are corrected for nonspecific binding of tagged antibody determined by parallel assays containing all components except the primary antibodies.
The relative contributions of HSF1 and HSF2 using the ORIGEN assay were further assessed in 3T3 cells exposed to heat shock, MG132, or azetidine treatment (Fig. 2C). Control cells contained a low basal level (8%) of both HSF1 (3%) and HSF2 (5%) activities relative to the maximal level of HSF DNA binding activity observed in cells treated with MG132 (100%). Exposure to MG132 activated both HSF1 and HSF2 such that over a 6-h period, HSF2 corresponded to approximately 45% and HSF1 corresponded to 55% of the total amount of HSF DNA binding activity. By comparison, heat shock resulted in activation of only HSF1 to the same level as was detected following MG132 treatment, whereas HSF2 activity decreased to a level below that normally detected in control cells. Unlike the rapid attenuation of the heat shock response observed in human tissue culture cells, we observed that attenuation of HSF1 is less dramatic in murine 3T3 cells exposed to heat shock for extended periods (3 to 6 h) (25, 41).
These results show that the overall level of total HSF DNA binding activity induced by MG132 is nearly twice that induced by heat shock, with one difference being that MG132 induces both HSF1 and HSF2. These data show that HSF1 and HSF2 are activated differentially, with the consequence that the overall effect on heat shock gene expression is modulated accordingly. In further support of this, cells exposed to azetidine, a proline analogue, activated much lower overall levels of HSF DNA binding activity (30%) comprised equally of HSF1 and HSF2. The results using the ORIGEN assay provide, for the first time, a more complete description of the different degrees of stress activation of HSF1 and HSF2 as they contribute to overall HSF DNA binding activity and show that both HSFs are constitutively activated at low levels in cells maintained under control conditions.
An unexpected consequence of heat shock was the apparent reduction of HSF2 DNA binding activity in 3T3 cells. This was observed in both DNA binding assays as an inability to detect preexisting levels of HSF2 DNA binding activity primarily in MG132-treated cells exposed to heat shock (Fig. 1, compare lanes 2 and 3 to lanes 10 and 11) or the basal level of HSF2 DNA binding in control cells (Fig. 1 and 2).
Transcriptional consequences of HSF1 and HSF2 coactivation.
The stress-dependent differential activation of HSF1 and HSF2 confers a more complex regulatory control for the inducible transcription of heat shock genes. This was examined in cells where HSF1 alone was activated by heat shock and under stress conditions where both factors were activated, as occurs with MG132 and the combination of MG132 and heat shock. The levels of transcription of selected heat shock genes induced by these treatments were analyzed by nuclear run-on assays. Treatment of 3T3 cells with MG132 for up to 4 h induced hsp70 and hsp90 gene transcription 5-fold and 10-fold, respectively, relative to transcription of the GAPDH gene, a housekeeping gene that has been used as a non-heat shock response reference gene (Fig. 3, lanes 1 to 3). The rates of hsp70 and hsp90 gene transcription were enhanced 115- and 35-fold, respectively, following 30 min of heat shock at 43°C (Fig. 3, lane 4). This demonstrates that the relative rates of hsp70 and hsp90 gene transcription vary significantly following exposure to different stressors and that conditions that lead to coactivation of HSF1 and HSF2 result in lower relative levels of heat shock gene transcription. In cells treated with both MG132 and heat shock, the transcription rates for hsp70 and hsp90 were identical to the effect of heat shock alone (Fig. 3, lane 5). The observation that the level of heat shock gene transcription in cells treated initially with MG132 followed by heat shock is equivalent to that in cells treated with heat shock alone is consistent with the observed loss of HSF2 DNA binding activity following heat shock treatment.
FIG. 3.

Effects of heat shock and MG132 on rates of transcription of heat shock genes. Nuclear run-on analyses of hsp70, hsp90, grp78, hsc70, and GAPDH gene transcription in control 3T3 cells (lane 1) or cells treated with MG132 for 1 and 4 h (lanes 2 and 3, respectively), heat shock (HS) at 43°C for 0.5 h (lane 4), or MG132 for 4 h followed by heat shock at 43°C for 0.5 h (lane 5) are shown. The vector corresponds to the plasmid pGEM4, used to determine level of nonspecific hybridization. The levels of transcription were determined relative to nonspecific background and the internal control, the GAPDH gene.
HSF2 is relocalized from the nucleus and cytoplasm during heat shock.
A complementary approach to examine the properties of HSF1 and HSF2 is to visualize the subcellular distribution of these factors using monoclonal antibodies specific to each HSF and indirect immunofluorescence analyses (Fig. 4). HSF1 and HSF2 were diffusely localized in the cytoplasm and nuclei of control cells (Fig. 4a and h) and relocalized to the nucleus upon treatment with MG132 (Fig. 4b and i). Exposure of either MG132-treated cells (Fig. 4c) or control untreated cells (Fig. 4d) to heat shock resulted in a dramatic and unexpected relocalization of HSF2 to a perinuclear distribution, whereas heat shock caused the nuclear localization of HSF1 (Fig. 4j and k). The localization of HSF2 to the perinuclear region of stressed cells was demonstrated by reference to the boundaries of the nucleus established with DAPI (4′,6′-diamidino-2-phenylindole) staining of DNA (data not shown) and by comparison with the nuclear localization of HSF1. Upon recovery at 37°C (Fig. 4e and l), both HSF2 and HSF1 returned to the diffuse cytoplasmic and nuclear localization pattern. The relocalization of HSF2 to the perinuclear region is specific to heat shock. Incubation of cells with other stressors, such as cadmium or azetidine, known to coactivate both HSF2 and HSF1 did not cause perinuclear localization of HSF2 but rather resulted in increased nuclear concentration of both factors (Fig. 4f, g, m, and n).
FIG. 4.
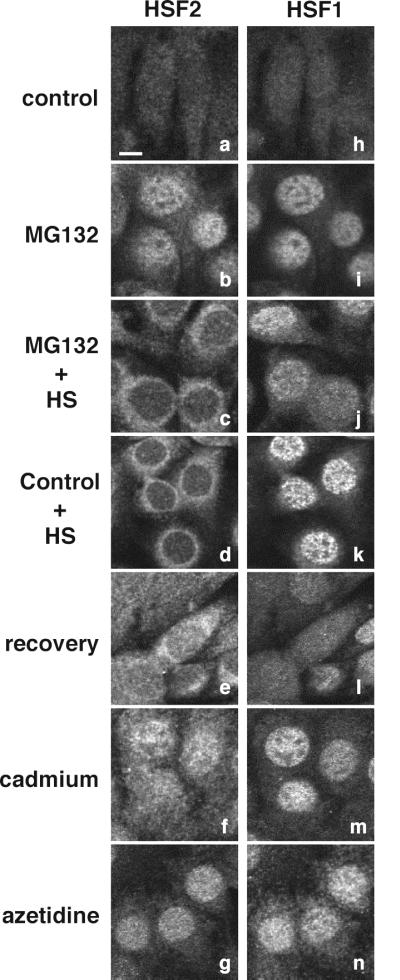
Stress-dependent changes in intracellular localization of HSF2 and HSF1. Double immunofluorescence analyses were performed using specific antibodies to HSF2 (a to g) and HSF1 (h to n) to determine their intracellular localization in control 3T3 cells (a and h) or cells treated with MG132 (4 h) (b and i), MG132 (4 h) followed by heat shock (1.5 h) (c and j), heat shock (1.5 h) (d and k), heat shock (1.5 h) followed by 6 h of recovery at 37°C (e and l), cadmium (4 h) (f and m), or azetidine (4 h) (g and n). Bar, 5 μm.
HSF2 is a temperature-sensitive protein that becomes insoluble upon heat shock.
The heat shock-induced loss of HSF2 DNA binding activity and the perinuclear relocalization of HSF2 upon heat shock of 3T3 cells led us to examine whether heat shock affected the solubility of HSF2 in whole-cell extracts. HSF2 was detected in extracts from control cells, and its levels increased after MG132 treatment (Fig. 5A, top panel, lanes 1 and 4). In contrast, soluble extracts from either control or MG132-treated 3T3 cells exposed to heat shock contained reduced levels of HSF2 (Fig. 5A, top panel, lanes 2, 3, 5, 6), although the total amount of HSF2 in unfractionated extracts remained constant (Fig. 5A, second panel, lanes 1 to 6). By comparison, HSF1 remained in the soluble fraction (Fig. 5A, third panel, lanes 1 to 6) as compared to the amount of total HSF1 (Fig. 5A, bottom panel, lanes 1 to 6) and exhibited its characteristic slower electrophoretic mobility due to inducible phosphorylation. These results show that the effects of elevated temperature on the solubility of HSF2 were specific to HSF2, as activated HSF1 trimers remained in the soluble fraction.
FIG. 5.

Heat shock affects the solubility of HSF2. (A) Western blot analyses of HSF2 and HSF1 proteins in soluble and total cell extracts from control (lanes 1 to 3) or MG132-treated (lanes 4 to 6) 3T3 cells exposed to heat shock for 0 (lanes 1 and 4), 0.5 (lanes 2 and 5), or 1 (lanes 3 and 6) h. (B) Western blot analyses of HSF2 protein present in soluble or total extracts from control 3T3 cells (C) or cells treated with cadmium (Cd) or azetidine (Az) for 2 h.
The differential solubility and temperature sensitivity of HSF2 is stress specific, as activated HSF2 remains soluble in extracts from cadmium- or azetidine-treated 3T3 cells (Fig. 5B). These results show that HSF2 is inactivated only under conditions of heat shock. Moreover, the effect of heat shock on the biochemical state of HSF2 is associated with the conversion from a soluble to an aggregated form of HSF2, coincident with its relocalization from the nucleus to the cytosol and association with the perinuclear region of the cell.
Biochemical properties of inactivated HSF2.
The heat shock-induced change in HSF2 subcellular localization coincides with a change in HSF2 biochemical properties and conversion to an inert state. This suggests that HSF2 has features in common with proteins that have acquired a biochemical phenotype of temperature sensitivity (Fig. 5). In non-heat-shocked cells, the inert and DNA binding forms of HSF2 exist as soluble dimeric and trimeric species, respectively (50). Following heat shock, these oligomeric states of HSF2 are not detected, providing additional support for elevated temperature-induced changes in the biochemical characteristics of HSF2 (data not shown). Another feature of HSF2 is its solubility in low-ionic-strength buffers and nonionic detergents, whereas the heat shock-inactivated form of HSF2 was resistant to extraction by buffers containing nondenaturing nonionic detergents, including Triton X-100 (1%), Nonidet P-40 (2%), sodium deoxycholate (1%), sarcosyl (0.5%), and Tween 20 (2%). Concentrations of NaCl (up to 2 M) sufficient for extraction of histones (16, 32) were also ineffective. Low concentrations (0.1%) of the denaturing ionic detergent sodium dodecyl sulfate could not extract inactive HSF2, while higher concentrations (0.5 to 1%), sufficient to cause disruption of noncovalent protein associations and protein denaturation (10, 39, 54), were required to extract HSF2 from heat-shocked cells (data not shown). These observations were further corroborated by using protease digestion to demonstrate that the rate of protease digestion of HSF2 decreases after heat shock treatment of cells, presumably as a consequence of its more inaccessible state (data not shown).
Isolation of nuclei from heat shock-treated cells results in copurification of HSF2. One explanation for these observations is that HSF2 becomes associated with a specific structure or structures following stress treatment. A link to components of the cytoskeleton is suggested by the observation that heat shock also causes the perinuclear collapse of the vimentin intermediate filament network (55; C. Jolly, data not shown). In these immunofluorescence studies, we observe that HSF2 and vimentin colocalize only in cells exposed to heat shock.
The thermolabile state of HSF2 is reversible and protected in thermotolerant cells.
The heat shock-induced insolubility of HSF2 is reversible, and HSF2 reappears in the soluble fraction of cells allowed to recover at 37°C for periods of 4 to 6 h (Fig. 6). This period of recovery from heat shock is also associated with the increased synthesis and accumulation of molecular chaperones such as Hsp70 and Hdj1 (Fig. 6, bottom panels). This reversibility is coincident with the relocalization of HSF2 from the perinuclear region to the diffuse distribution throughout the cell observed in the control state (Fig. 4e).
FIG. 6.
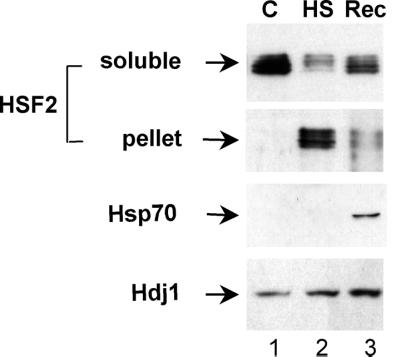
HSF2 levels recover following heat shock. Western blot analysis of HSF2, Hsp70, and Hdj1 in the soluble fractions (first, third and fourth panels) and pellet fractions from control 3T3 cells (C) (lane 1) or cells exposed to heat shock (HS) (lane 2) and allowed to recover for 6 h at 37°C (Rec) (lane 3) is shown.
We sought to determine whether the temperature-sensitive biochemical phenotype of HSF2 was associated with the levels of molecular chaperones. We established a thermotolerant state of the 3T3 cells by exposure to a transient priming heat shock treatment followed by recovery for periods of up to 24 h prior to a subsequent prolonged heat shock for 1 to 2 h. Exposure to the priming heat shock induces the synthesis and accumulation of high levels of Hsp70 and elevated levels of Hdj1 (Fig. 7, bottom two panels). Whereas HSF2 was inactivated in control cells exposed to a single heat shock (Fig. 7, top panel, lanes 1 and 2), HSF2 levels and solubility were unaffected in thermotolerant cells (Fig. 7, top panel, lanes 3 and 4). Moreover, the perinuclear relocalization of HSF2 observed in heat shock-treated cells was not observed in thermotolerant cells as examined by indirect immunofluorescence (data not shown). By comparison, the levels of HSF1 were essentially unaffected under all conditions with a difference in mobility due to stress-induced serine phosphorylation (Fig. 7, second panel).
FIG. 7.
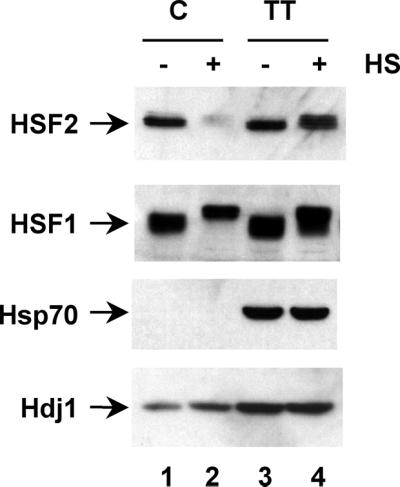
HSF2 is refractile to heat shock in thermotolerant cells. HSF2, HSF1, Hsp70, and Hdj1 were detected by Western blot analysis of soluble extracts from control (C) (lanes 1 and 2) or thermotolerant (TT) 3T3 cells treated with (lanes 2 and 4) or without (lanes 1 and 3) heat shock (HS).
These results suggest that conditions that lead to the accumulation of molecular chaperones, which are often correlated with acquisition of thermotolerance (18, 19, 21, 51), prevent the inactivation of HSF2. Since chaperone proteins inhibit protein aggregation and participate in resolubilization or renaturation of aggregated proteins (12, 15, 33, 57), they may serve as positive regulators of HSF2 by maintaining HSF2 in a native state. Alternatively, other changes in the intracellular environment, such as changes in the levels of nonprotein components induced in cells during recovery from heat shock, may be instrumental in preventing HSF2 inactivation, and thus further investigation would be required to identify the critical cellular factors responsible for protection.
DISCUSSION
The data presented here demonstrate that two members of the HSF gene family, HSF1 and HSF2, are activated differentially in response to diverse cellular stresses. We suggest that the presence of different combinations of activated HSF1 and HSF2 expand substantially the range of kinetics and duration of heat shock gene transcription beyond what was previously considered in situations where only HSF1 or HSF2 DNA binding activities were detected. Our observations additionally suggest an unexpected and intriguing relationship that exists between HSF1 and HSF2 only in cells exposed to heat shock and results in the inactivation of HSF2. We show that HSF2 has the biochemical properties of a temperature-sensitive protein that is rendered inactive in cells exposed to elevated temperature and sequestered to the cytoplasm as a perinuclear ring. As such, HSF2 cannot interfere with HSF1 for occupancy of heat shock gene promoters and the regulation of the heat shock response. In thermotolerant cells expressing higher levels of heat shock proteins, HSF2 is no longer thermolabile and is maintained in the soluble fraction in a DNA binding state.
Coactivation of HSF1 and HSF2.
Until recently, the activities of HSF1 and HSF2 have been investigated under conditions where either one or the other factor was predominantly detected. Studies in which proteasome inhibitors have been used to study activation of heat shock genes have revealed that both HSFs are activated, with substantial differences observed among cell lines. Reexamination of a subset of the classical stress conditions, as shown in this study, reveals that coactivation of HSF1 and HSF2 is indeed a common feature of stress, with heat shock alone resulting in sole activation of HSF1.
The stress specificity observed for the inactivation of HSF2 raises a number of interesting questions. As many of the stress conditions that induce HSF1 activity affect the flux of proteins to the degradative pool, one would also expect to observe the activation of HSF2 (22). For example, exposure to the heavy metal cadmium, the amino acid analog azetidine (25, 41), or the proteasome inhibitor MG132 results in the activation of both HSF1 and HSF2, as demonstrated by acquisition of both DNA binding activities and translocation of the HSFs to the nucleus. Whereas in previous studies only general statements on coactivation of HSF1 and HSF2 were possible, the ORIGEN HSF binding assay provides a quantitative assessment of the relative contributions of HSF1 and HSF2 and the ability, for the first time, to assess total HSF DNA binding activity. Based on these studies, we conclude that the involvement of HSF2 in the stress response is more widespread than previously suggested, contributing to regulation of heat shock gene expression under cellular conditions associated with changes in protein degradation. The implementation of the ORIGEN assay also reveals that both HSF1 and HSF2 DNA binding activities are present constitutively at low levels in tissue culture cells grown under normal unstressed conditions.
Rationale for HSF2 inactivation by heat and characteristics of the inactivated HSF2.
The heat shock-specific insolubilization and relocalization of HSF2 are intriguing and reveal an unexpected specificity associated with heat shock-induced stress signaling. A distinct feature specific to heat shock, among cellular stresses, is the rapid kinetics of the heat shock response, a consequence of the nearly instantaneous effects of elevated temperatures on native proteins and the appearance of misfolded proteins (24). In contrast to the effects of heat shock, other inducers of the heat shock response such as heavy metals or amino acid analogues exhibit much slower kinetics of heat shock gene induction, often requiring hours to activate a response that typically does not achieve the same magnitude (25, 41). Based on these observations, the presence of other HSFs would be predicted to have dominant-negative effects on HSF1 with deleterious consequences for the survival of cells exposed to heat shock. The lability of HSF2 in heat-shocked cells affords a means to ensure that such competition does not occur.
The heat-induced inactivation of HSF2 had been suggested from previous observations on the effects of heat shock on HSF2 DNA binding activity (26, 40). The inactivation by elevated temperature represents an intrinsic feature of HSF2 that may be due to a temperature-induced conformational change independent of its DNA binding state. The conversion of HSF2 to an insoluble state is defined by our inability to extract HSF2 using ionic conditions and detergents commonly employed to extract membrane-associated proteins or histones or to disrupt weak noncovalent interactions. Extraction of inactive HSF2 required denaturing detergents capable of disrupting noncovalent interactions. These observations resemble the reported effects of heat shock on other intracellular proteins, including various endogenous mammalian proteins (6, 7, 8) as well as reporter enzymes (luciferase and β-galactosidase) transfected as heterologous genes into mammalian cells (29), which all undergo a heat shock-induced transition into a Triton X-100-insoluble state.
The temperature threshold for HSF2 inactivation in human cells examined seems to differ from that observed in murine cells, since only partial insolubilization or inactivation of HSF2, if any, is observed in K562 cells at the temperatures used (Fig. 2B, data not shown, and L. Sistonen, personal communication). This difference may reflect intrinsic variation in the proteins themselves or differences in the intracellular milieu of the cells.
Heat shock proteins as positive regulators of HSF2.
The inactivation of HSF2 during heat shock does not appear to be an intrinsic feature of the protein, as HSF2 remains soluble in conditioned cells previously exposed to a priming heat shock and allowed to accumulate high levels of heat shock proteins. This suggests that the biochemical properties of HSF2 are influenced by other cellular events, in particular the appearance of proteins that accumulate following heat shock. Overexpression of individual chaperones (Hsp90, Hsc70, Hsp70, and Hdj1) using conditional promoters (tetracycline regulation) yielded only partial protection (data not shown), suggesting either that other proteins are involved or that HSF2 may interact with a specific cohort of heat shock proteins rather than be influenced by a single heat shock protein alone. From these data, we can conclude that HSF2 is regulated posttranslationally in a manner distinct from that for HSF1, whose activity is negatively regulated by specific heat shock proteins. For example, Hsp90 has been shown to affect HSF1 activation and trimerization (1, 61), and elevated levels of Hsp70 can negatively regulate the transcriptional activity of HSF1 (48).
The behavior of HSF2 during recovery from heat shock has some features in common with that of the reporter enzymes luciferase and β-galactosidase, which have been used to demonstrate chaperone-dependent protein refolding of thermosensitive enzymes expressed in mammalian cells. As observed with HSF2, reappearance of soluble protein is observed during recovery from heat shock; in the case of luciferase, 50% of its enzymatic activity could be recovered (35). Involvement of heat shock proteins in this recovery process was suggested by the observations that inactivation of the reporter enzymes was enhanced by ATP depletion (30), that a priming heat shock treatment to induce thermotolerance attenuated enzyme inactivation (29), and that overexpression of Hsp70 was sufficient to protect the activity of reporter enzymes (31). Induction of thermotolerance was also shown to prevent the perinuclear collapse of the vimentin network (56), suggesting common modes of regulation between cytoskeletal reorganization and HSF2 relocalization.
Cross talk of HSF activities.
In conclusion, the effects of heat shock and other stresses on HSF activities suggest that the differential regulation of HSF1 and HSF2 affords a fine-tuning mechanism to modulate the transcriptional responses to different stresses. Such regulation is achieved not only by differential activation of these transcription factors but also by their selective inactivation. Our data support the intriguing concept of cross talk between HSF family members as a means to coordinate the functions of the multiple HSF proteins expressed in vertebrate cells (27, 28, 38, 40, 46) and plants (5, 13, 14, 43, 44).
The communication between HSF family members promises to be varied and complex, ranging from the proposed negative regulation of HSF1 activity by HSF2 and the associated inactivation of HSF2 by heat shock to their regulated coactivation, as well as a positive requirement of HSF3 for heat shock-inducible HSF1 activity in avian cells (52) and the role of the plant HsfA1 to facilitate nuclear transport of HsfA2 (45). This cross talk is likely to be influenced by the expression of heat shock proteins, which may have distinct and opposing effects on individual HSF family members, as suggested here for HSF1 and HSF2. Such an interplay between HSFs and the heat shock proteins whose expression they mediate enables the cell to integrate various stress signals to respond appropriately to the particular stresses encountered and ultimately to maintain cellular protein homeostasis.
ACKNOWLEDGMENTS
These studies were supported by NIH grant GM38109, The Gollub Foundation, and the Daniel F. and Ada L. Rice Foundation. A.M. is a Fellow of the American Heart Association, Chicago Affiliate; C. Jolly was supported by the Association pour la Recherche contre le Cancer and a fellowship from the Daniel F. and Ada L. Rice Foundation; and S.K.M. was supported by a U.S. Army Breast Cancer Training Grant.
We thank R. A. Lamb for use of the confocal microscope, members of our laboratory for comments on the manuscript, and Kate Veraldi for manuscript preparation.
REFERENCES
- 1.Ali A, Bharadwaj S, O'Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18:4949–4960. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol. 1993;13:2486–2496. doi: 10.1128/mcb.13.4.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerji S S, Theodorakis N G, Morimoto R I. Heat shock-induced translational control of HSP70 and globin synthesis in chicken reticulocytes. Mol Cell Biol. 1984;4:2437–2448. doi: 10.1128/mcb.4.11.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cotto J, Fox S S, Morimoto R. HSF1 granules: a novel stress-induced nuclear compartment of human cells. J Cell Sci. 1997;110:2925–2934. doi: 10.1242/jcs.110.23.2925. [DOI] [PubMed] [Google Scholar]
- 5.Czarnecka-Verner E, Yuan C X, Fox P C, Gurley W B. Isolation and characterization of six heat shock transcription factor cDNA clones from soybean. Plant Mol Biol. 1995;29:37–51. doi: 10.1007/BF00019117. [DOI] [PubMed] [Google Scholar]
- 6.Dubois M F, Hovanessian A G, Bensaude O. Heat-shock-induced denaturation of proteins. Characterization of the insolubilization of the interferon-induced p68 kinase. J Biol Chem. 1991;266:9707–9711. [PubMed] [Google Scholar]
- 7.Dubois M F, Vincent M, Vigneron M, Adamczewski J, Egly J M, Bensaude O. Heat-shock inactivation of the TFIIH-associated kinase and change in the phosphorylation sites on the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 1997;25:694–700. doi: 10.1093/nar/25.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubois M F, Marshall N F, Nguyen V T, Dahmus G K, Bonnet F, Dahmus M E, Bensaude O. Heat shock of HeLa cells inactivates a nuclear protein phosphatase specific for dephosphorylation of the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 1999;27:1338–1344. doi: 10.1093/nar/27.5.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eriksson M, Jokinen E, Sistonen L, Leppa S. Heat shock factor 2 is activated during mouse heart development. Int J Dev Biol. 2000;44:471–477. [PubMed] [Google Scholar]
- 10.Fish W W, Reynolds J A, Tanford C. Gel chromatography of proteins in denaturing solvents. Comparison between sodium dodecyl sulfate and guanidine hydrochloride as denaturants. J Biol Chem. 1970;245:5166–5168. [PubMed] [Google Scholar]
- 11.Fort P, Marty L, Piechaczyk M, el Sabrouty S, Dani C, Jeanteur P, Blanchard J M. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman B C, Morimoto R I. The human cytosolic molecular chaperones hsp90, hsp70 (hsc70) and hdj-1 have distinct roles in recognition of a non-native protein and protein refolding. EMBO J. 1996;15:2969–2979. [PMC free article] [PubMed] [Google Scholar]
- 13.Gagliardi D, Breton C, Chaboud A, Vergne P, Dumas C. Expression of heat shock factor and heat shock protein 70 genes during maize pollen development. Plant Mol Biol. 1995;29:841–856. doi: 10.1007/BF00041173. [DOI] [PubMed] [Google Scholar]
- 14.Hubel A, Schoffl F. Arabidopsis heat shock factor: isolation and characterization of the gene and the recombinant protein. Plant Mol Biol. 1994;26:353–362. doi: 10.1007/BF00039545. [DOI] [PubMed] [Google Scholar]
- 15.Jakob U, Lilie H, Meyer I, Buchner J. Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase. Implications for heat shock in vivo. J Biol Chem. 1995;270:7288–7294. doi: 10.1074/jbc.270.13.7288. [DOI] [PubMed] [Google Scholar]
- 16.Johns E W. The preparation and characterization of histones. In: Phillips D M P, editor. Histones and nucleohistones. New York, N.Y: Plenum Publishing Corporation; 1971. pp. 1–45. [Google Scholar]
- 17.Jolly C, Morimoto R, Robert-Nicoud M, Vourc'h C. HSF1 transcription factor concentrates in nuclear foci during heat shock: relationship with transcription sites. J Cell Sci. 1997;110:2935–2941. doi: 10.1242/jcs.110.23.2935. [DOI] [PubMed] [Google Scholar]
- 18.Landry J, Chretien P, Bernier D, Nicole L M, Marceau N, Tanguay R M. Thermotolerance and heat shock proteins induced by hyperthermia in rat liver cells. Int J Radiat Oncol Biol Phys. 1982;8:59–62. doi: 10.1016/0360-3016(82)90385-6. [DOI] [PubMed] [Google Scholar]
- 19.Laszlo A, Li G C. Heat-resistant variants of Chinese hamster fibroblasts altered in expression of heat shock protein. Proc Natl Acad Sci USA. 1985;82:8029–8033. doi: 10.1073/pnas.82.23.8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legagneux V, Mezger V, Quelard C, Barnier J V, Bensaude O, Morange M. High constitutive transcription of HSP86 gene in murine embryonal carcinoma cells. Differentiation. 1989;41:42–48. doi: 10.1111/j.1432-0436.1989.tb00730.x. [DOI] [PubMed] [Google Scholar]
- 21.Li G C, Werb Z. Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts. Proc Natl Acad Sci USA. 1982;79:3218–3222. doi: 10.1073/pnas.79.10.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathew A, Mathur S K, Morimoto R I. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway Mol. Cell Biol. 1998;18:5091–5098. doi: 10.1128/mcb.18.9.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mezger V, Rallu M, Morimoto R I, Morange M, Renard J P. Heat shock factor 2-like activity in mouse blastocysts. Dev Biol. 1994;166:819–822. doi: 10.1006/dbio.1994.1361. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto R I. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 25.Mosser D D, Theodorakis N G, Morimoto R I. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol Cell Biol. 1988;8:4736–4744. doi: 10.1128/mcb.8.11.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy S P, Gorzowski J J, Sarge K D, Phillips B. Characterization of constitutive HSF2 DNA-binding activity in mouse embryonal carcinoma cells. Mol Cell Biol. 1994;14:5309–5317. doi: 10.1128/mcb.14.8.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakai A, Morimoto R I. Characterization of a novel chicken heat shock transcription factor, heat shock factor 3, suggests a new regulatory pathway. Mol Cell Biol. 1993;13:1983–1997. doi: 10.1128/mcb.13.4.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai A, Tanabe M, Kawazoe Y, Inazawa J, Morimoto R I, Nagata K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol. 1997;17:469–481. doi: 10.1128/mcb.17.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen V T, Morange M, Bensaude O. Protein denaturation during heat shock and related stress. Escherichia coli beta-galactosidase and Photinus pyralis luciferase inactivation in mouse cells. J Biol Chem. 1989;264:10487–10492. [PubMed] [Google Scholar]
- 30.Nguyen V T, Bensaude O. Increased thermal aggregation of proteins in ATP-depleted mammalian cells. Eur J Biochem. 1994;220:239–246. doi: 10.1111/j.1432-1033.1994.tb18619.x. [DOI] [PubMed] [Google Scholar]
- 31.Nollen E A, Brunsting J F, Roelofsen H, Weber L A, Kampinga H H. In vivo chaperone activity of heat shock protein 70 and thermotolerance. Mol Cell Biol. 1999;19:2069–2079. doi: 10.1128/mcb.19.3.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohlenbusch H H, Olivera B M, Tuan D, Davidson N. Selective dissociation of histones from calf thymus nucleoprotein. J Mol Biol. 1967;25:299–315. doi: 10.1016/0022-2836(67)90143-x. [DOI] [PubMed] [Google Scholar]
- 33.Parsell D A, Kowal A S, Singer M A, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 34.Paslaru L, Rallu M, Manuel M, Davidson S, Morange M. Cyclosporin A induces an atypical heat shock response. Biochem Biophys Res Commun. 2000;269:464–469. doi: 10.1006/bbrc.2000.2295. [DOI] [PubMed] [Google Scholar]
- 35.Pinto M, Morange M, Bensaude O. Denaturation of proteins during heat shock. In vivo recovery of solubility and activity of reporter enzymes. J Biol Chem. 1991;266:13941–13946. [PubMed] [Google Scholar]
- 36.Pirkkala L, Alastalo T P, Nykanen P, Seppa L, Sistonen L. Differentiation lineage-specific expression of human heat shock transcription factor 2. FASEB J. 1999;13:1089–1098. doi: 10.1096/fasebj.13.9.1089. [DOI] [PubMed] [Google Scholar]
- 37.Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- 38.Rabindran S K, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci USA. 1991;88:6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynolds J A, Tanford C. The gross conformation of protein-sodium dodecyl sulfate complexes. J Biol Chem. 1970;245:5161–5165. [PubMed] [Google Scholar]
- 40.Sarge K D, Zimarino V, Holm K, Wu C, Morimoto R I. Cloning and characterization of two mouse heat shock factors with distinct inducible and constitutive DNA-binding ability. Genes Dev. 1991;5:1902–1911. doi: 10.1101/gad.5.10.1902. [DOI] [PubMed] [Google Scholar]
- 41.Sarge K D, Murphy S P, Morimoto R I. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarge K D, Park S O, Kirby J D, Mayo K E, Morimoto R I. Expression of heat shock factor 2 in mouse testis: potential role as a regulator of heat-shock protein gene expression during spermatogenesis. Biol Reprod. 1994;50:1334–1343. doi: 10.1095/biolreprod50.6.1334. [DOI] [PubMed] [Google Scholar]
- 43.Scharf K D, Rose S, Zott W, Schoffl F, Nover L, Schoff F. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF. EMBO J. 1990;9:4495–4501. doi: 10.1002/j.1460-2075.1990.tb07900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scharf K D, Rose S, Thierfelder J, Nover L. Two cDNAs for tomato heat stress transcription factors. Plant Physiol. 1993;102:1355–1356. doi: 10.1104/pp.102.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scharf K D, Heider H, Hohfeld I, Lyck R, Schmidt E, Nover L. The tomato Hsf system: HsfA2 needs interaction with HsfA1 for efficient nuclear import and may be localized in cytoplasmic heat stress granules. Mol Cell Biol. 1998;18:2240–2251. doi: 10.1128/mcb.18.4.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuetz T J, Gallo G J, Sheldon L, Tempst P, Kingston R E. Isolation of a cDNA for HSF2: evidence for two heat shock factor genes in humans. Proc Natl Acad Sci USA. 1991;88:6911–6915. doi: 10.1073/pnas.88.16.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi Y, Kroeger P E, Morimoto R I. The carboxyl-terminal transactivation domain of heat shock factor 1 is negatively regulated and stress responsive. Mol Cell Biol. 1995;15:4309–4318. doi: 10.1128/mcb.15.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y, Mosser D D, Morimoto R I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sistonen L, Sarge K D, Phillips B, Abravaya K, Morimoto R I. Activation of heat shock factor 2 during hemin-induced differentiation of human erythroleukemia cells. Mol Cell Biol. 1992;12:4104–4111. doi: 10.1128/mcb.12.9.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sistonen L, Sarge K D, Morimoto R I. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol Cell Biol. 1994;14:2087–2099. doi: 10.1128/mcb.14.3.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subjeck J R, Sciandra J J, Chao C F, Johnson R J. Heat shock proteins and biological response to hyperthermia. Br J Cancer Suppl. 1982;45:127–131. [PMC free article] [PubMed] [Google Scholar]
- 52.Tanabe M, Kawazoe Y, Takeda S, Morimoto R I, Nagata K, Nakai A. Disruption of the HSF3 gene results in the severe reduction of heat shock gene expression and loss of thermotolerance. EMBO J. 1998;17:1750–1758. doi: 10.1093/emboj/17.6.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Why S K, Kim S, Geibel J, Seebach F A, Kashgarian M, Siegel N J. Thresholds for cellular disruption and activation of the stress response in renal epithelia. Am J Physiol. 1999;277:F227–F234. doi: 10.1152/ajprenal.1999.277.2.F227. [DOI] [PubMed] [Google Scholar]
- 54.Weber K, Osborn M. Proteins and sodium dodecyl sulfate: molecular weight determination on polyacrylamide gels and related procedures. In: Neurath H, Hill R L, editors. The proteins. I. New York, N.Y: Academic Press, Inc.; 1975. pp. 179–223. [Google Scholar]
- 55.Welch W J, Suhan J P. Morphological study of the mammalian stress response: characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol. 1985;101:1198–1211. doi: 10.1083/jcb.101.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Welch W J, Mizzen L A. Characterization of the thermotolerant cell. II. Effects on the intracellular distribution of heat-shock protein 70, intermediate filaments, and small nuclear ribonucleoprotein complexes. Cell Biol. 1988;106:1117–1130. doi: 10.1083/jcb.106.4.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiech H, Buchner J, Zimmermann R, Jakob U. Hsp90 chaperones protein folding in vitro. Nature. 1992;358:169–170. doi: 10.1038/358169a0. [DOI] [PubMed] [Google Scholar]
- 58.Wu B, Hunt C, Morimoto R. Structure and expression of the human gene encoding major heat shock protein HSP70. Mol Cell Biol. 1985;5:330–341. doi: 10.1128/mcb.5.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshima T, Yura T, Yanagi H. Function of the C-terminal transactivation domain of human heat shock factor 2 is modulated by the adjacent negative regulatory segment. Nucleic Acids Res. 1998;26:2580–2585. doi: 10.1093/nar/26.11.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshima T, Yura T, Yanagi H. Heat shock factor 1 mediates hemin-induced hsp70 gene transcription in K562 erythroleukemia cells. J Biol Chem. 1998;273:25466–25471. doi: 10.1074/jbc.273.39.25466. [DOI] [PubMed] [Google Scholar]
- 61.Zou J, Guo Y, Guettouche T, Smith D F, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 62.Zuo J, Baler R, Dahl G, Voellmy R. Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol Cell Biol. 1994;14:7447–7468. doi: 10.1128/mcb.14.11.7557. [DOI] [PMC free article] [PubMed] [Google Scholar]