

Abstract
Background
An estimated 60% of pharmacological randomised trials use placebo control interventions to blind (i.e. mask) participants. However, standard placebos do not control for perceptible non‐therapeutic effects (i.e. side effects) of the experimental drug, which may unblind participants. Trials rarely use active placebo controls, which contain pharmacological compounds designed to mimic the non‐therapeutic experimental drug effects in order to reduce the risk of unblinding. A relevant improvement in the estimated effects of active placebo compared with standard placebo would imply that trials with standard placebo may overestimate experimental drug effects.
Objectives
We aimed to estimate the difference in drug effects when an experimental drug is compared with an active placebo versus a standard placebo control intervention, and to explore causes for heterogeneity. In the context of a randomised trial, this difference in drug effects can be estimated by directly comparing the effect difference between the active placebo and standard placebo intervention.
Search methods
We searched PubMed, CENTRAL, Embase, two other databases, and two trial registries up to October 2020. We also searched reference lists and citations and contacted trial authors.
Selection criteria
We included randomised trials that compared an active placebo versus a standard placebo intervention. We considered trials both with and without a matching experimental drug arm.
Data collection and analysis
We extracted data, assessed risk of bias, scored active placebos for adequacy and risk of unintended therapeutic effect, and categorised active placebos as unpleasant, neutral, or pleasant. We requested individual participant data from the authors of four cross‐over trials published after 1990 and one unpublished trial registered after 1990. Our primary inverse‐variance, random‐effects meta‐analysis used standardised mean differences (SMDs) of active versus standard placebo for participant‐reported outcomes at earliest post‐treatment assessment. A negative SMD favoured the active placebo. We stratified analyses by trial type (clinical or preclinical) and supplemented with sensitivity and subgroup analyses and meta‐regression. In secondary analyses, we investigated observer‐reported outcomes, harms, attrition, and co‐intervention outcomes.
Main results
We included 21 trials (1462 participants). We obtained individual participant data from four trials. Our primary analysis of participant‐reported outcomes at earliest post‐treatment assessment resulted in a pooled SMD of −0.08 (95% confidence interval (CI) −0.20 to 0.04; I2 = 31%; 14 trials), with no clear difference between clinical and preclinical trials. Individual participant data contributed 43% of the weight of this analysis. Two of seven sensitivity analyses found more pronounced and statistically significant differences; for example, in the five trials with low overall risk of bias, the pooled SMD was −0.24 (95% CI −0.34 to −0.13). The pooled SMD of observer‐reported outcomes was similar to the primary analysis. The pooled odds ratio (OR) for harms was 3.08 (95% CI 1.56 to 6.07), and for attrition, 1.22 (95% CI 0.74 to 2.03). Co‐intervention data were limited. Meta‐regression found no statistically significant association with adequacy of the active placebo or risk of unintended therapeutic effect.
Authors' conclusions
We did not find a statistically significant difference between active and standard placebo control interventions in our primary analysis, but the result was imprecise and the CI compatible with a difference ranging from important to irrelevant. Furthermore, the result was not robust, because two sensitivity analyses produced a more pronounced and statistically significant difference. We suggest that trialists and users of information from trials carefully consider the type of placebo control intervention in trials with high risk of unblinding, such as those with pronounced non‐therapeutic effects and participant‐reported outcomes.
Plain language summary
Do treatment effects in randomised trials differ when using active placebo compared to standard placebo?
Key messages
1. We found no clear difference in effect between active and standard placebos, but we are very uncertain about the results.
2. We suggest that researchers carefully consider the type of placebo when investigating medicines with clear side effects.
What are standard placebos and active placebos?
Blinding (or masking) is an important part of randomised trials and ensures that participants and healthcare providers are not influenced by knowing whether the participant is receiving the experimental treatment. If they had this knowledge, they might unintentionally overestimate (or underestimate) the effect of the treatment. One method of blinding is to give the non‐treated group of participants a placebo, which looks like the medicine (e.g. a tablet of similar shape, colour, smell, taste, and texture) but does not contain its active ingredients.
Blinding with a placebo may not always be successful. The treatment may have side effects that distinguish it from the placebo so that the treatment effect is still not measured accurately. For this reason, some trials use active placebos, which mimic some of the side effects of the experimental medicine. However, it is unclear whether the choice of placebo type actually makes a difference to the treatment effects.
What did we want to find out?
We wanted to find out whether treatment effects in randomised trials differ when using active placebos compared to standard placebos.
What did we do?
We collected and analysed trials that directly compared the two types of placebo, or that compared both placebos with an experimental medicine. If people receiving active placebo experience better results than those receiving standard placebo, this difference might be due to them believing that they are receiving the experimental medicine. It would also mean that in trials that compare a medicine with standard placebo, the beneficial effect of the medicine is exaggerated. This is important if the measured benefits of the treatment are small or moderate and thus especially sensitive to changes in the methods used for the trial.
What did we find?
We included 21 trials in this review, covering subjects such as pain and psychiatry. We found no clear difference between the two types of placebo in participant‐reported outcomes (such as pain intensity). However, because the result was uncertain, the possible range of this result included both no difference and a potentially important difference in favour of active placebo. When we limited our analysis to higher‐quality trials, active placebos were more beneficial than standard placebos, but these trials were not typical clinical trials and might not be applicable to clinical scenarios.
Background
Blinding (sometimes called 'masking') is considered a cornerstone in protecting against bias in randomised trials (Hróbjartsson 2011; Schulz 2002). In pharmacological trials, investigators can blind participants and personnel by using a standard placebo control intervention, which is identical to the experimental intervention with respect to external properties such as size, shape, colour, texture, smell, and taste (Bello 2016; Boutron 2006). However, some drugs have clearly perceptible non‐therapeutic effects (i.e. side effects), such as sedation or dry mouth, that may unblind trial participants to the nature of their allocated intervention and potentially bias experimental intervention effect estimates (Jensen 2017).
The number of placebo‐controlled drug trials at risk of unblinding due to perceptible side effects is unknown, but is likely to be considerable. We estimate that around 80% of drug trials use blinding (Chan 2005; Hopewell 2010), and among these, around 75% are placebo‐controlled trials (Boutron 2006). This would mean that approximately 60% of drug trials use a placebo. In absolute numbers, we note that from 2010 to 2020, PubMed indexed approximately 47,000 randomised trials that had the term placebo in the title, abstract or keywords. Different authors have highlighted the problem of unblinding due to side effects for specific drugs, such as selective serotonin reuptake inhibitors (SSRIs; Greenberg 1994), tricyclic antidepressants (Moncrieff 2004), methylphenidate (Storebø 2015), and ketamine (Aan Het Rot 2012). Furthermore, unblinding may be a particular issue in trials with participant‐reported outcome measures, which are used in up to half of all trials (Mercieca‐Bebber 2018).
Description of the methods being investigated
To reduce risk of unblinding, some trials have used active placebo control interventions. These are considered therapeutically inactive, but contain pharmacological compounds that mimic some of the perceptible non‐therapeutic effects of the experimental drugs (Jensen 2017). One example of an active placebo is atropine, which simulates the anticholinergic effects of tricyclic antidepressants (e.g. dry mouth; Thomson 1982a). Risk of unblinding in trials is also relevant to the risk of bias assessment, which is a standard procedure in systematic reviews. In some cases, review authors may consider the risk of unblinding as high when a trial does not use an active placebo control intervention (Jakobsen 2017; Storebø 2015). This is consistent with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a), and the revised Cochrane tool for assessing risk of bias in randomised trials (RoB 2; Sterne 2019).
Why it is important to do this review
Less than 1% of placebo‐controlled trials use active placebos (Boutron 2006; Jensen 2017). The advantages and disadvantages of using active placebo control interventions have been the focus of intermittent debate since the introduction of the randomised trial, although the central question in most cases is related to the evidence base for a specific intervention, such as antidepressants (Moncrieff 2004; Salamone 2000). The findings of one methodological overview suggest that the use of active placebos is clustered in certain areas and does not follow logically from the side effect profile of the experimental drug. For example, several pain trials have compared SSRIs with active placebo, but depression trials have compared the same type of drug with standard placebo (Jensen 2017). This may be partly due to tradition, and to the fact that for many interventions without clear and obvious non‐therapeutic effects, there is little reason to use an active placebo. Furthermore, active placebos are more challenging to design than standard placebos; may have unintended therapeutic effects; and induce an ethical challenge, because participants in the control group experience (potentially unnecessary) side effects (Jensen 2017; Gaudiano 2005; Colagiuri 2010).
Empirical comparisons between active and standard placebo interventions could help trialists decide on appropriate control interventions, or help users of trial reports reflect on the risk of unblinding. Unfortunately, few empirical studies have compared the two types of placebo, and these studies have focused on specific fields and have relied on observational data with a high risk of confounding (Moncrieff 2003; Thomson 1982a; Wilkinson 2019). The ideal study design to address the question is a meta‐analysis of trials from various fields that randomise participants to both active placebo and standard placebo interventions.
A relevant improvement in the estimated effects of active versus standard placebo in such a meta‐analysis would imply that trials with standard placebo may overestimate experimental drug effects.
Objectives
We aimed to estimate the difference in drug effects when an experimental drug is compared with an active placebo versus a standard placebo control intervention, and to explore causes for heterogeneity. In the context of a randomised trial, this difference in drug effects can be estimated by directly comparing the effect difference between the active placebo and standard placebo intervention.
Methods
Criteria for considering studies for this review
Types of studies
We included clinical and preclinical randomised trials that allocated participants to an active placebo intervention and to a standard placebo intervention. We included trials with a parallel‐group or cross‐over design and excluded split‐body and cluster randomised trials.
Types of data
We included data on estimated intervention effects, using both continuous and dichotomous variables.
Types of methods
We considered an active placebo to be any intervention described as such or designed to imitate the perceptible non‐therapeutic effects of an experimental intervention (e.g. anticholinergic effects of tricyclic antidepressants), but without any known or suspected benefit on the outcomes under investigation. We considered a standard placebo to be any intervention described as such or designed to mimic only the external properties of the experimental intervention (or the active placebo), such as appearance, smell, taste, and texture (e.g. lactose placebo or saline placebo). We also included two‐arm trials without a matching experimental drug arm (i.e. trials where the active placebo might mimic a hypothetical experimental intervention).
We excluded trials that had a hypothesis of harm, such as those investigating experimentally induced memory deficits using drugs, or investigating effects other than clinical therapeutic effects, including physiological effects such as impact on taste.
Appendix 1 provides further details regarding eligibility criteria.
Types of outcome measures
We included the following outcomes.
Participant‐reported outcomes at earliest post‐treatment assessment (primary outcome)
Participant‐reported outcomes at latest follow‐up
Blinded observer‐reported outcomes earliest post‐treatment assessment
Blinded observer‐reported outcomes at latest follow‐up
Any harm outcome at any time point (including predictable effects of the active placebo)
Attrition rates at earliest post‐treatment assessment
Co‐intervention rates at any time point (e.g. co‐intervention received or not)
Mean co‐intervention use at any time point (e.g. mean dose)
Search methods for identification of studies
We searched for publications of eligible trials in a previous review of active placebo‐controlled trials (Jensen 2017). We also updated the electronic search from that review in PubMed (on 11 March 2019) and Cochrane Central Register of Controlled Trials (CENTRAL; on 11 March 2019), both restricted to records added after 1 January 2015. We searched Ovid Embase with no publication date restrictions (on 20 March 2019).
We searched for full texts in Google Scholar (in June 2020 and July 2020) and ProQuest (on 21 October 2020) and for trial records in ClinicalTrials.gov (clinicaltrials.gov; on 7 September 2020) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/clinical-trials-registry-platform; on 20 October 2020). We applied no time restrictions for these databases.
We screened reference lists and citations of relevant publications as well as publications from the first and last authors. We also screened references in key publications and contacted trial authors and experts within the field.
Appendix 1 provides further details regarding the searches.
Data collection and analysis
Selection of studies
One review author (DRTL) screened the titles and abstracts of the records retrieved from the database searches, then two review authors (DRTL and CHN/ADF/AH) independently screened the full‐text reports of potentially relevant studies against our eligibility criteria. We resolved any disagreements by discussion or, if necessary, by consulting an arbiter (AH). For the full text and trial register searches, one review author (DRTL) screened text excerpts and full texts if needed, and removed those that were obviously ineligible. When it was unclear whether the trial authors considered the intervention in question to be an active placebo (e.g. if they described it as such post hoc in the discussion of the trial report), or when we considered the intervention was not a satisfactory active placebo, we classified the trial as nearly eligible and excluded it from our analysis.
Data extraction and management
We extracted the following data.
Publication and author information
Trial characteristics (e.g. trial type: clinical or preclinical; trial design: parallel‐group or cross‐over)
Active placebo characteristics (e.g. component and dose)
Information on trial conduct (e.g. duration of treatment)
Participant numbers (e.g. number of participants randomised)
From each trial, we selected one of each of the outcomes listed in Types of outcome measures. When selecting participant‐reported and observer‐reported outcomes, we used the following order of preference.
Continuous outcomes, otherwise dichotomous outcomes
Primary outcomes of the study, if noted, otherwise the most clinically relevant outcomes (e.g. mentioned first in objectives or in methods)
Absolute values, otherwise change scores.
Then, for participant‐reported outcomes, we applied the following scheme.
Symptom‐specific outcomes (e.g. pain), otherwise global outcomes (e.g. quality of life score)
Private outcomes (e.g. pain), otherwise potentially observable outcomes (e.g. emesis)
For observer‐reported outcomes, we preferred subjective interactive outcomes (e.g. rating scales with participant contact), otherwise purely observational subjective outcomes (e.g. assessments without participant contact) or objective outcomes (e.g. blood samples). Principles of outcome selection for harm, co‐intervention, and attrition outcomes are presented in Appendix 1.
For participant‐reported and observer‐reported outcomes, we selected the earliest available post‐treatment time point and the latest follow‐up time point. For attrition, we preferred the same early time point as for the participant‐reported outcomes. For harms and co‐intervention outcomes, we chose the latest available time point.
For each outcome measure of interest, we extracted basic information (e.g. type: continuous; domain and instrument: pain on visual analogue scale (VAS); directionality: lower is better) and time point of assessment (e.g. two weeks post‐treatment).
We extracted the summary data from the active placebo and standard placebo intervention groups (e.g. mean, standard deviation (SD), and number of participants for continuous data; number of events and number of participants for dichotomous data). If only an effect estimate was available (e.g. mean difference (MD), standardised mean difference (SMD), or odds ratio (OR)), we extracted this and the corresponding standard error (SE) or other measure of variation (e.g. 95% confidence interval (CI)).
For each outcome, we preferred the most complete analysis (e.g. intention‐to‐treat analysis rather than per‐protocol analysis). For trials reported in multiple publications, we used data from all the sources. We had planned to contact trial authors in case of discrepancies (for trials published after 1990), but none arose.
We scored the adequacy of each active placebo on a scale of 1 to 5 (higher score for better matching), considering the likeness to the experimental intervention in terms of quality, intensity, and rapidness of perceptible effects. For example, we would assign a score of 1 for no matching of perceptible effects, 3 for adequate matching of many important effects, and 5 for perfect or almost perfect matching of effects. For this assessment, our primary source of information was the trial publication, followed by relevant references, and then informal probing of the literature. We also scored the risk of the active placebo having an unintended therapeutic effect on a scale of 1 to 5 (higher score for a higher risk), considering elements such as the type of evidence (e.g. animal research), the relevance (e.g. data on acute pain but not chronic pain), and pharmacological properties (e.g. short duration of effect compared to outcome time point). For example, we would assign a score of 1 for no indication of a therapeutic effect in the literature, 3 for mixed or indirect evidence of a therapeutic effect, and 5 for clear evidence of a therapeutic effect. We used the same sources of information as for adequacy scoring. Third, we assessed whether the effects of the active placebo intervention could be considered unpleasant, neutral, or pleasant, based on the literature and blinded to the trial results. We developed all three assessments ourselves for this review, as we could not find any relevant tools in the literature for these purposes.
Two review authors (DRTL and CHN/ADF/ASP) independently collected data, selected outcomes, and scored the active placebos. We resolved any disagreements by discussion or by consulting an arbiter (AH). A clinical pharmacologist performed the classification of unpleasantness (MRH). Appendix 1 provides further details regarding data collection.
Obtaining individual participant data
For four cross‐over trials (published after 1990) with continuous outcomes but no reported correlation coefficient, we asked the trial authors whether they would be willing to share individual participant data, so that we could adjust effect estimates for the correlation. These data would also allow us to impute a correlation coefficient in any cross‐over trials published before 1990 or without successful author contact. For one trial without published data (registered after 1990), we contacted the trialist for individual participant data, so that the trial could contribute to our meta‐analyses.
We requested individual participant data for all potentially relevant outcomes as well as clarifying information regarding trial conduct. We then chose outcomes for analysis based on similar principles as those listed above (Appendix 1).
Assessment of risk of bias in included studies
Two review authors (DRTL and CHN) independently assessed risk of bias in two domains from the RoB 2 tool for participant‐reported outcomes at earliest post‐treatment assessment: risk of bias arising from the randomisation process and risk of bias in selection of the reported results (Sterne 2019). We did not assess the other three domains regarding blinding and attrition (risk of bias due to deviations from the intended interventions, risk of bias due to missing outcome data, and risk of bias in measurement of the outcome) because they relate directly to the bias under investigation in our review.
Assessment of heterogeneity
We assessed heterogeneity with the I2 statistic and calculated prediction intervals for the primary analysis.
Assessment of reporting biases
We assessed small‐study effects by visual inspection of the funnel plot for the primary analysis.
Data synthesis
We conducted inverse‐variance, random‐effects meta‐analyses of our eight outcomes of interest for active placebo versus standard placebo, stratified by type of trial (clinical or preclinical trials). Our analyses were based on the following effect measures.
Participant‐reported outcomes at earliest post‐treatment assessment: SMD
Participant‐reported outcomes at latest follow‐up: SMD
Blinded observer‐reported outcomes at earliest post‐treatment assessment: SMD
Blinded observer‐reported outcomes at latest follow‐up: SMD
Any harm outcome at any time point: OR
Attrition rates at earliest post‐treatment assessment: OR
Co‐intervention rates at any time point: OR
Mean co‐intervention use at any time point: SMD
For continuous outcomes, we calculated an estimate of the SMD (Hedges' g) and its SE. For dichotomous outcomes, we calculated the OR and its 95% CI. For participant‐reported and observer‐reported outcomes, we then converted any ORs to SMDs (Chinn 2000). For harms, we converted any SMDs to ORs. For cross‐over trials, we included continuous outcome results from all cross‐over periods and calculated the SMD and its SE adjusted for correlation (Appendix 1). We standardised direction of effect estimates, such that an SMD below 0 and an OR below 1 favoured the active placebo intervention (i.e. the active placebo was more beneficial, had a lower occurrence of harms, had fewer dropouts, or had less co‐intervention use than the standard placebo). For trials with individual participant data, we calculated the effect estimates directly based on the data set (Appendix 1).
If we were unable to include a trial in the primary analysis due to missing data, we summarised the results qualitatively.
Subgroup analysis and investigation of heterogeneity
For the primary analysis, we performed a meta‐regression analysis with our scores for adequacy of the active placebo and unintended therapeutic effect as continuous variables. We also performed the following post‐hoc subgroup analyses of the primary analysis.
Parallel‐group versus cross‐over trials
Trials with versus without a matching experimental intervention
Unpleasant versus pleasant/neutral active placebo intervention
Peer‐reviewed publications versus grey literature (e.g. theses, posters, currently unpublished trial results)
For harms, we performed the following post‐hoc subgroup analysis.
Predictable effects of the active placebo versus other harms.
Sensitivity analysis
We performed the following sensitivity analyses of the primary analysis.
Excluding dichotomous outcome data converted to SMD
Adding nearly eligible trials
Including only trials at low risk of bias
Reanalysing the results with a fixed‐effect model
The first two analyses were explicitly prespecified in the protocol (Laursen 2020), and the last two were not, but are considered standard procedures for systematic reviews (Deeks 2021). We performed the following three additional post‐hoc sensitivity analyses.
Adding trials with only observer‐reported outcomes
Using change scores instead of absolute values (if both were available)
Adjusting for baseline as a covariate in trials with individual participant data
Appendix 1 provides further details regarding data analysis.
Results
Description of studies
Results of the search
We screened 5352 records by title and abstract from PubMed, CENTRAL, Embase, and Jensen 2017. After exclusions, we read 162 full‐text publications and included 12 relevant trials in this review (Adelson 1962; Berna 2017; Bjørkedal 2011; Dellemijn 1997; Greenbaum 1987; Malmstrom 2006; Prosenz 2017; Rief 2012; Shader 1964; Solomon 1960; Wang 2008a; Woodrow 1988). From other databases and sources, we included 9 trials (Casey 1960; Fangman 1963; Kurland 1961; Letemendia 1959; Lipman 1966; McKinley 2016; Parkes 2015; Werner 2019a; Werner 2019b), yielding a total of 21 trials (Figure 1). We obtained individual participant data for two of the four cross‐over trials for which we had requested data (Bjørkedal 2011; Prosenz 2017). We also obtained data for an unpublished parallel‐group trial (Werner 2019b) and for a trial presented as an abstract, (Werner 2019a), from the same trial author.
1.
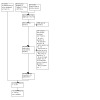
Study flow diagram.
Included studies
The number of participants in the active and standard placebo groups in each study ranged from four to 351 (median 50), and the total number of participants was 1462. One trial was unpublished (Werner 2019b), and the other trials were published between 1959 and 2019 (median 1997).
Among 10 clinical trials (929 participants), there were six parallel‐group and four cross‐over trials. The most common clinical areas were psychiatry (N = 5) and pain (N = 2). The most frequent active placebo components in the clinical trials were atropine (N = 2) and phenobarbital (N = 2). Antipsychotic drugs of the phenothiazine drug class (e.g. chlorpromazine) were the most frequent matching experimental drug (N = 3). Duration of study participation ranged from six hours to eight months (counting only one treatment period for cross‐over trials).
Among 11 preclinical trials (533 participants), there were five parallel‐group trials and six cross‐over trials. In these trials, experimental pain was the most frequent clinical area (N = 7), and the most frequent active placebos were atropine (N = 3) and capsaicin (N = 3). Eight trials used an active placebo without a matching experimental drug arm. Duration of study participation ranged from 20 minutes to four days.
Among the 11 trials with a matching experimental treatment, the median score for adequacy of the active placebo was 2 on a scale of 1 to 5 (range 1 to 3). The median score among all 21 trials for risk of unintended therapeutic effect was 2 on a scale of 1 to 5 (range 1 to 4). Eighteen trials had active placebos with unpleasant effects. The effects were considered neutral in three trials, and there were no trials with pleasant effects.
Details regarding the individual trials are presented in Table 1 and Characteristics of included studies.
1. Characteristics of included trials.
| Trial | Condition | Design | Experimental intervention(s) | Active placebo | Standard placebo | Participant‐reported outcome | Number of participantsa |
| Clinical | |||||||
| Adelson 1962 | Schizophrenia | Parallel | 4 different phenothiazines | Scopolamine and chlorprophenpyridamine maleate | Lactose | None | 96 |
| Casey 1960 | Schizophrenia | Parallelb | 2 different phenothiazines | Phenobarbital | Lactose | None | 351 |
| Dellemijn 1997 | Continuous unilateral neuropathic pain | Parallelc | Fentanyl | Diazepam | Saline | Peak pain intensity difference | 50 |
| Greenbaum 1987 | Irritable bowel syndrome | Cross‐over | Desipramine | Atropine | Not described | Pain index | 28 |
| Kurland 1961 | Need for tranquillizers (mainly schizophrenia) | Parallel | 6 different phenothiazines | Phenobarbital | Saline and lactose | None | 52 |
| Letemendia 1959 | Schizophrenia or depression | Parallel | None | Nicotinic acid | Lactose | None | 28 |
| Lipman 1966 | Psychoneurotic complaints | Parallel | Noned | Atropine | Similar capsules | Symptom checklist | 203 |
| Malmstrom 2006 | Pain after removal of third molars | Parallel | Lidocainee | Diphenhydramine | Saline | Weighted total pain relief | 19 |
| Solomon 1960 | Postmenopausal osteoporosis or Paget's disease | Cross‐over | Methallenestril and fluoxymesterone | Dextroamphetamine and amobarbital | Lactose | Questionnaire covering 10 categories | 12 |
| Werner 2019b | Insomnia | Parallel | None | Beetroot extract | Lactose fibres as filler | Insomnia severity index | 90 |
| Preclinical | |||||||
| Berna 2017 | Heat‐induced pain | Parallel | Nonef | Atropine | Microcrystalline cellulose | Pain intensity | 100 |
| Bjørkedal 2011 | Laser‐induced heat pain | Cross‐over | None | Caffeine in grapefruit juice | Grapefruit juice | Pain intensity | 20 |
| Fangman 1963 | Radiant heat pain and ischaemic muscle pain | Cross‐over | Morphine | Phenobarbital | Water | Radiant heat pain threshold | 4 |
| McKinley 2016 | Memory and breathing | Parallel | None | Capsaicin | Saline | Perceived efficacy on memory | 80 |
| Parkes 2015 | Breathing | Parallel | None | Capsaicin | Saline and sesame oil | Breathing sensation scale | 80 |
| Prosenz 2017 | Contact heat, electrical pain, pressure pain | Cross‐over | Fentanyl | Midazolam | Saline | Suprathreshold heat pain | 24 |
| Rief 2012 | Heat‐induced pain | Parallel | None | Capsaicin | Sesame oil | Pain threshold | 114 |
| Shader 1964 | Psychiatric symptoms in healthy participants | Cross‐over | None | Phenobarbital and atropine sulphate | Lactose | Revised Clyde Mood Scale (self‐sort) | 20 |
| Wang 2008a | Capsaicin‐induced pain | Cross‐over | Pregabalin and morphine | Diphenhydramine hydrochloride | Not described | Area of hyperalgesia | 20 |
| Werner 2019a | Sleep | Parallel | None | Beetroot extract | Lactose fibres as filler | Insomnia severity index | 57 |
| Woodrow 1988 | Pressure tolerance pain | Cross‐over | Morphine and alcohol | Atropine | Saline | Pain tolerance | 14 |
aOnly counting participants in the placebo groups. Participants in cross‐over trials counted only once. bSpecial cross‐over design where not all participants crossed over in the second period. Trial publication only presents useful data for the first period. We used these first period data and treated the study as a parallel‐group study in the analysis. cSpecial cross‐over design where participants were randomised in parallel to either a cross‐over subtrial with experimental intervention and active placebo, or to a cross‐over subtrial with experimental treatment and standard placebo. Thus, placebo comparisons are compared in parallel, and we considered the study to be a parallel‐group study in our analyses. dTrial has a chlordiazepoxide hydrochloride drug arm, apparently not matching the active placebo. eTrial also has an oxycodone/acetaminophen arm. fTrial has a diclofenac arm, apparently not matching the active placebo.
Risk of bias in included studies
Of the 14 trials with a relevant participant‐reported outcome for the primary meta‐analysis, five trials were at overall low risk of bias due to low risk of bias in both domains (Berna 2017; Bjørkedal 2011; Prosenz 2017; Werner 2019a; Werner 2019b), and the other nine had some concerns for overall risk of bias (Dellemijn 1997; Greenbaum 1987; Lipman 1966; Malmstrom 2006; McKinley 2016; Parkes 2015; Rief 2012; Wang 2008a; Woodrow 1988). There were some concerns related to selection bias for all nine of these trials, and some concerns related to randomisation for six of them. No trials were at high risk of bias (Figure 2; Figure 3).
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effect of methods
Main analyses
For the primary analysis of participant‐reported outcomes at earliest post‐treatment assessment, SMDs from 14 trials with 899 participants ranged from −0.44 to 0.41, with a pooled average of −0.08 (95% CI −0.20 to 0.04; I2 =31%; 95% prediction interval (PI) −0.38 to 0.22; Analysis 1.1, 3 other trials included in the analysis did not contribute data). For five clinical trials (390 participants), the pooled SMD was −0.02 (95% CI −0.21 to 0.16; I2 = 0%; 95% PI −0.33 to 0.28) and for nine preclinical trials (509 participants), the pooled SMD was −0.08 (95% CI −0.24 to 0.07, I2 = 38%, 95% PI −0.46 to 0.30). Four trials with individual participant data (191 participants) contributed 43% of the weight of the meta‐analysis.
1.1. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 1: Main analysis
Three older trials also investigated participant‐reported outcomes, but did not report sufficient data to be included in the meta‐analysis (Fangman 1963; Shader 1964; Solomon 1960). In general, the results were inconclusive regarding direction and potential size of impact (Appendix 2). Four trials had no participant‐reported outcomes (Adelson 1962; Casey 1960; Kurland 1961; Letemendia 1959).
Malmstrom 2006 also reported an SMD for a participant‐reported outcome at a late follow‐up time point (SMD 0.05, 95% CI −0.85 to 0.95; Analysis 2.1, 2 other trials included in the analysis did not contribute data). For observer‐reported outcomes at earliest post‐treatment assessment, the pooled SMD for 11 trials was −0.06 (95% CI −0.18 to 0.07; I2 = 12%; Analysis 3.1, 2 other trials included in the analysis did not contribute data). For clinical trials the pooled SMD was 0.01 (95% CI −0.11 to 0.13; I2 = 0%) and for preclinical trials the pooled SMD was −0.29 (95% CI −0.53 to −0.05; I2 = 0%: test for subgroup differences P = 0.03). For observer‐reported outcomes at latest follow‐up, one trial reported an SMD of −0.27 (95% CI −0.67 to 0.13; Analysis 4.1; Adelson 1962).
2.1. Analysis.

Comparison 2: Patient‐reported outcomes at latest follow‐up, Outcome 1: Main analysis
3.1. Analysis.

Comparison 3: Observer‐reported outcomes at earliest post‐treatment assessment, Outcome 1: Main analysis
4.1. Analysis.

Comparison 4: Observer‐reported outcomes at latest follow‐up, Outcome 1: Main analysis
ORs for harms ranged from 0.43 to 121 in 10 trials, and the pooled OR was 3.08 (95% CI 1.56 to 6.07: I2 = 65%; Analysis 5.1, 3 other trials included in the analysis did not contribute data). The OR for attrition ranged from 0.30 to 6.50 in eight trials, and the pooled OR was 1.22 (95% CI 0.74 to 2.03; I2 = 0%; Analysis 6.1). Seven trials did not contribute to the analysis because no attrition occurred for either placebo intervention, and six trials did not have sufficient data on attrition.
5.1. Analysis.

Comparison 5: Harms, Outcome 1: Main analysis
6.1. Analysis.

Comparison 6: Attrition, Outcome 1: Main analysis
There was insufficient data on co‐intervention use for meaningful meta‐analyses. In one trial, all participants received the co‐intervention, which was rescue pain medication (Malmstrom 2006). The median time to rescue medication in this trial for active placebo was 1.2 hours (95% CI 1.0 to 1.3) versus 1.4 hours (95% CI 1.3 to 1.6) for standard placebo. Another trial reported instances of concomitant medication use: 25 instances among 28 participants for active placebo and 22 instances among 28 participants for standard placebo (Greenbaum 1987).
See Table 2 for all pooled estimates of the main analyses.
2. Pooled estimates for primary and secondary meta‐analyses.
| Analysis | Measure | Clinical trials | Preclinical trials | All trials | ||||||
| Estimate with 95% CIa | I2 | N | Estimate with 95% CI | I2 | N | Estimate with 95% CI | I2 | N | ||
| Participant‐reported outcomes at earliest post‐treatment assessment (primary) | SMD | −0.02 (−0.21 to 0.16) | 0% | 5 | −0.08 (−0.24 to 0.07) | 38% | 9 | −0.08 (−0.20 to 0.04) | 31% | 14 |
| Participant‐reported outcomes at latest follow‐up | SMD | 0.05 (−0.85 to 0.95) | NA | 1 | NA | NA | NA | 0.05 (−0.85 to 0.95) | NA | 1 |
| Blinded observer‐reported outcomes at earliest post‐treatment assessment | SMD | 0.01 (−0.11 to 0.13) | 0% | 7 | −0.29 (−0.53 to −0.05) | 0% | 4 | −0.06 (−0.18 to 0.07) | 12% | 11 |
| Blinded observer‐reported outcomes at latest follow‐up | SMD | −0.27 (−0.67 to 0.13) | NA | 1 | NA | NA | NA | −0.27 (−0.67 to 0.13) | NA | 1 |
| Harms at any time point | OR | 1.97 (0.85 to 4.55) | 47% | 6 | 5.48 (1.82 to 16.48) | 75% | 4 | 3.08 (1.56 to 6.07) | 65% | 10 |
| Attrition rates at earliest post‐treatment assessment | OR | 1.03 (0.55 to 1.96) | 0% | 5 | 1.74 (0.54 to 5.57) | 48% | 3 | 1.22 (0.74 to 2.03) | 0% | 8 |
| Co‐intervention rates at any time point | OR | NA | NA | 0 | NA | NA | 0 | NA | NA | 0 |
| Mean co‐intervention use at any time point | SMD | NA | NA | 0 | NA | NA | 0 | NA | NA | 0 |
aSMD < 0 or OR < 1 favoured the active placebo (i.e. active placebo was more beneficial, had a lower occurrence of harms, had fewer dropouts, or had less co‐intervention use than the standard placebo) CI: confidence interval; N: number of trials; NA: not applicable; OR: odds ratio; SMD: standardised mean difference.
Sensitivity analyses
For the primary analysis, we did not need to convert OR to SMD for any trials. A fixed‐effect model yielded a pooled SMD of −0.15 (95% CI −0.23 to −0.07; I2 =31%; 14 trials; 3 other trials included in the analysis did not contribute data). When restricted to trials with low risk of bias, the pooled SMD was −0.24 (95% CI −0.34 to 0.13; I2 = 0%; 5 trials; Analysis 1.4). The remaining sensitivity analyses did not materially change the main result (Table 3; Appendix 2). One trial that was nearly eligible for the review investigated experimental pain, but did not provide sufficient information for inclusion in the sensitivity meta‐analysis and was summarised qualitatively (Gracely 1987; Appendix 2).
1.4. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 4: Sensitivity analysis – low risk of bias
3. Sensitivity analyses of the primary meta‐analysis (standardised mean difference of participant‐reported outcomes at earliest post‐treatment assessment).
| Analysis | Clinical trials | Preclinical trials | All trials | ||||||
| SMD with 95% CI* | I2 | N | SMD with 95% CI | I2 | N | SMD with 95% CI | I2 | N | |
| Excluding dichotomous data converted to SMD | −0.02 (−0.21 to 0.16) | 0% | 5 | −0.08 (−0.24 to 0.07) | 38% | 9 | −0.08 (−0.20 to 0.04) | 31% | 14 |
| Adding nearly eligible trials | −0.02 (−0.20 to 0.16) | 0% | 6 | −0.05 (−0.22 to 0.11) | 41% | 10 | −0.06 (−0.19 to 0.06) | 30% | 16 |
| Trials with low risk of bias | −0.08 (−0.49 to 0.33) | NA | 1 | −0.25 (−0.35 to −0.14) | 0% | 4 | −0.24 (−0.34 to −0.13) | 0% | 5 |
| Fixed‐effect model | −0.02 (−0.21 to 0.16) | 0% | 5 | −0.18 (−0.28 to −0.09) | 38% | 9 | −0.15 (−0.23 to −0.07) | 31% | 14 |
| Adding trials with only observer‐reported outcomes | −0.01 (−0.14 to 0.12) | 1% | 10 | −0.08 (−0.24 to 0.07) | 38% | 9 | −0.06 (−0.17 to 0.05) | 33% | 19 |
| Preferring change scores | −0.07 (−0.27 to 0.14) | 11% | 5 | −0.08 (−0.26 to 0.09) | 42% | 9 | −0.09 (−0.23 to 0.04) | 35% | 14 |
aSMD < 0 indicated that the active placebo was more beneficial than the standard placebo. CI: confidence interval; N: number of trials; NA: not applicable; SMD: standardised mean difference.
Investigation of heterogeneity
In the meta‐regression model for the primary analysis, the coefficient for the adequacy score was 0.13 (95% CI −0.27 to 0.54) and the coefficient for the risk of therapeutic effect was −0.36 (95% CI −0.80 to 0.08; residual I2 = 0%, R2 = 100%). Thus, there was no statistically significant association between the difference between active and standard placebo and our scores for adequacy and risk of therapeutic effect of the active placebos (Appendix 2).
The impact of active placebo was not substantially statistically significantly different for cross‐over versus parallel‐group trials, for trials with versus without a matching experimental intervention, or for trials with unpleasant versus pleasant/neutral effects (Table 4).
4. Subgroup analyses of the primary meta‐analysis (standardised mean difference of participant‐reported outcomes at earliest post‐treatment assessment).
| Analysis | Clinical trials | Preclinical trials | All trials | |||||||
| SMD with 95% CIa | I2 | N | SMD with 95% CI | I2 | N | SMD with 95% CI | I2 | N | ||
| Trial design | Parallel‐group | −0.04 (−0.28 to 0.21) | 17% | 4 | −0.07 (−0.26 to 0.12) | 0% | 5 | −0.04 (−0.18 to 0.10) | 0% | 9 |
| Cross‐over | −0.08 (−0.52 to 0.35) | NA | 1 | −0.04 (−0.33 to 0.25) | 65% | 4 | −0.07 (−0.30 to 0.17) | 55% | 5 | |
| Matching experimental drug armb | Yes | −0.21 (−0.53 to 0.11) | 0% | 3 | 0.09 (−0.21 to 0.39) | 23% | 3 | −0.04 (−0.24 to 0.17) | 6% | 6 |
| No | 0.07 (−0.16 to 0.30) | 0% | 2 | −0.18 (−0.32 to −0.04) | 16% | 6 | −0.09 (−0.25 to 0.06) | 42% | 8 | |
| Active placebo effects | Unpleasant | −0.04 (−0.28 to 0.21) | 17% | 4 | −0.07 (−0.24 to 0.10) | 45% | 8 | −0.07 (−0.21 to 0.07) | 41% | 12 |
| Pleasant or neutral | −0.08 (−0.49 to 0.33) | NA | 1 | −0.11 (−0.63 to −0.41) | NA | 1 | −0.09 (−0.42 to 0.23) | 0% | 2 | |
| Publication status | Peer‐reviewed | −0.04 (−0.28 to 0.21) | 17% | 4 | −0.13 (−0.31 to 0.05) | 42% | 6 | −0.10 (−0.25 to 0.05) | 41% | 10 |
| Grey literature | −0.08 (−0.49 to 0.33) | NA | 1 | 0.07 (−0.21 to 0.35) | 0% | 3 | 0.03 (−0.21 to 0.26) | 0% | 4 | |
aSMD < 0 indicated that the active placebo was more beneficial than the standard placebo. bWhether the two placebo interventions are compared directly to an experimental intervention. CI: confidence interval; N: number of trials; NA: not applicable; SMD: standardised mean difference.
For the subgroup analysis of harms, the pooled OR for predictable effects of the active placebo was 4.64 (95% CI 1.60 to 13.45; I2 = 77%) and the pooled OR for other harms was 2.07 (95% CI 0.81 to 5.30; I2 = 48%; test for subgroup differences P = 0.26; Analysis 5.2).
5.2. Analysis.

Comparison 5: Harms, Outcome 2: Subgroup analysis – predictable effects versus other harms
Selective publication or reporting
The funnel plot for the primary analysis did not reveal clear asymmetry (Figure 4). We included four trials not published in peer‐reviewed journals, but as theses or a poster (Fangman 1963; McKinley 2016; Parkes 2015; Werner 2019a). We identified one unpublished trial in a registry, and we were able to obtain data from the trial author (Werner 2019b). The impact of active placebo was not statistically significantly different for trials reported in peer‐reviewed and grey literature (Table 4). We had access to primary data from four trials (Bjørkedal 2011; Prosenz 2017; Werner 2019a; Werner 2019b) and to the registered planned analysis for a fifth trial (Berna 2017).
4.

Funnel plot of Comparison 1. Patient‐reported outcomes at earliest post‐treatment assessment; Outcome 1.1. Main analysis.
Discussion
Summary of main results
In our primary meta‐analysis of 17 randomised trials, of which 14 trials and 899 participants contributed data, we found no statistically significant difference between active and standard placebo interventions on participant‐reported outcomes at post‐treatment (pooled SMD −0.08; 95% CI −0.20 to 0.04; Analysis 1.1). However, the result was imprecise and the CI compatible with an average difference ranging from important to irrelevant to users of trial results.
The difference was more pronounced, and statistically significant, in some sensitivity analyses. For example, five trials with low overall risk of bias had a pooled SMD of −0.24 (95% CI −0.34 to −0.13; Analysis 1.4). For observer‐reported outcomes at post‐treatment, the pooled SMD was −0.06 (95% CI −0.18 to 0.07), but with a larger difference for preclinical trials (pooled SMD −0.29; 95% CI −0.53 to −0.05) than for clinical trials (pooled SMD 0.01; 95% CI −0.11 to 0.13). The remaining secondary analyses of SMDs were imprecise. Participants in the active placebo group reported harms more frequently (pooled OR 3.08, 95% CI 1.56 to 6.07; Analysis 5.1), but there was no statistically significant difference in attrition between the two groups (pooled OR 1.22, 95% CI 0.74 to 2.03; Analysis 6.1).
Potential biases in the review process
The novelty and strength of our review is that we included trials that directly randomised participants to the two placebo types, thus avoiding the considerable risk of confounding from observational studies. Based on known potential candidates in the early stages of our review, we had expected to identify few trials, and were surprised when we identified 21. Despite our comprehensive search, we may have missed some relevant trials, especially if the trial abstract did not use the term 'active placebo', and we would appreciate contact from any reader with knowledge of such trials.
Authors of four trials with 191 participants shared their individual participant data with us, which enabled more precise intervention effect estimates and allowed us to use unpublished results. These four trials contributed more than 40% of the weight of our primary analysis. With these primary data, we were also able to calculate a more realistic estimate of the correlation coefficient in the remaining cross‐over trials.
Our study sample included a variety of trials: both clinical and preclinical trials, with cross‐over and parallel‐group design, with and without a matching experimental drug arm. We had expected considerable statistical heterogeneity, but I2 values showed modest heterogeneity (about 30%), although the span of point estimates in the prediction interval included a considerable difference between active and standard placebo in either direction. We consider it unlikely that this span is entirely due to random error, but more trials are needed to confirm or refute this.
We consider our main result to be at low risk of non‐reporting bias. The funnel plot was symmetrical and there was no evidence of an effect difference between trials in peer‐reviewed and grey literature. Furthermore, we had access to individual participant data in four trials (one unpublished) and access to the intended analysis from a trial registry for one trial.
The main limitation of our review is imprecision. Our main result could be interpreted both as 'no statistically significant difference' and as 'insufficient statistical power to detect a methodologically important difference'. Assuming, for example, a true SMD of −0.10, the number of participants needed in a single trial to push the upper confidence limit below zero would be approximately 1900.
Another limitation is that our primary analysis was not robust to routine methodological choices in a meta‐analysis. We planned for a limited number of sensitivity and subgroup analyses in the protocol because we expected to include few trials. Notwithstanding the risk of spurious results in the supplementary analyses, we consider it noteworthy that two such analyses resulted in more pronounced and statistically significant differences. Of particular note is the analysis restricted to the five trials with low overall risk of bias (SMD −0.24; 95% CI −0.34 to −0.13; Analysis 1.4).
It is also challenging to determine the generalisability of our results. The included clinical trials were mostly in psychiatry. We consider the clinical setting less important than the profile and quality of the non‐therapeutic effects of the active placebo in question; however, we do note the absence of trials from many clinical fields. Also, we were surprised by a potentially smaller impact of active placebo in clinical trials than in preclinical trials, especially for observer‐reported outcomes, whereas the difference was not statistically significant for participant‐reported outcomes and seemed to be driven by one clinical trial of atropine active placebo from 1966 (Lipman 1966).
Agreements and disagreements with other studies or reviews
We are aware of three reviews that have compared the impact of active placebo versus standard placebo interventions. Two investigated an overlapping sample of active placebo‐controlled trials of tricyclic antidepressants and compared them with standard placebo‐controlled trials (Moncrieff 2003; Thomson 1982a). Thomson 1982a reported that the experimental drug was more often superior in standard placebo‐controlled trials compared to active‐placebo controlled trials. Moncrieff 2003 used multivariate meta‐regression analysis to investigate heterogeneity in the estimated effect of tricyclic antidepressants (measured in SMD), where the coefficient for active placebo versus standard placebo control was −0.18 (SE 0.21). The third review investigated the effect of ketamine in active placebo‐controlled and in standard placebo‐controlled trials of depression and bipolar disorder (Wilkinson 2019). The effect size with active placebo was −0.7 (95% CI −0.9 to −0.4) versus −1.8 (95% CI −2.2 to −1.4) with standard placebo, corresponding to a difference of −1.1 (we recoded the sign direction to match ours). All three reviews should be interpreted with considerable caution as they depend on potentially confounded observational comparisons. Nevertheless, the direction of these results is in line with our point estimate, even though the large difference between effect sizes in Wilkinson 2019 is striking.
These three earlier reviews addressed the risk of unblinding (in blinded trials). Several other studies have addressed lack of blinding (in non‐blinded trials). One systematic review of 12 trials (of complementary/alternative interventions) that had randomised participants to a blinded or a non‐blinded subtrial, found a pooled difference in effect sizes of −0.56 with a 95% CI of −0.71 to −0.41 (Hróbjartsson 2014). The considerable difference between studies that investigate lack of blinding and studies that investigate unblinding is coherent with the theoretical assumption that unblinding rarely affects all participants, and that in general, bias due to unblinding is less pronounced than bias due to lack of blinding. Interestingly, some meta‐epidemiological studies have not found a statistically significant impact of participant blinding (Moustgaard 2020).
Meaning and mechanisms
The focus of our review was the impact of choice of placebo control intervention on the estimated effect of an experimental drug intervention. Therefore, we interpret our SMD as a difference between SMDs (dSMD), not as a clinical intervention effect. Taken at face value, and ignoring the statistical imprecision, the point estimate implies that using standard placebo controls leads to overestimation of the effect of experimental interventions by an average of 0.08 SDs compared to using active placebo controls.
Considering the pattern of our result, including the statistically significant sensitivity analyses, our cautious interpretation is that active placebo controls likely reduce risk of bias in randomised trials of pharmacological interventions with clear side effects and participant‐reported outcomes, but that this needs to be confirmed or refuted in further studies.
Users of our review should take three key aspects into account. First, the analysis of predictable effects of the active placebo (i.e. the intended perceptible non‐therapeutic effects) showed a substantial difference between the groups, with a large pooled OR (4.64, 95% CI 1.60 to 13.45; Analysis 5.2). Therefore, trial participants noticed the difference between active placebo and standard placebo in the included trials. From a primary methodological perspective, trial participants were unblinded. The interesting secondary question that follows is the degree to which this unblinding translates into differences in effect estimates.
Second, the 95% CI of the average difference (SMD) between active and standard placebo ranged from −0.20 to 0.04, which we interpret as covering both important and irrelevant differences. In contrast to clinical effect sizes, there is no common classification of dSMD impact sizes. However, some previous methodological studies have pragmatically considered 0.10 as a cutoff point for an important change in SMD (Gøtzsche 2007). For example, in randomised drug trials with small to medium estimated effects sizes of −0.10 to −0.50 (Cohen 1988), an exaggeration of 0.10 units (for example) would mean that the real drug effect would be reduced by a considerable fraction (20% to 100%) and would, potentially, be negligible.
Furthermore, researchers often investigate the impact of other trial design features for dichotomous outcomes using ratios of odds ratios (ROR). Converting our dSMD estimate to ROR yields 0.86, with a 95% CI of 0.70 to 1.08 (Chinn 2000), which is similar to RORs of other potential biases that are considered important, such as bias due to incomplete allocation concealment (Dechartres 2016; Page 2016).
Third, our primary analysis may underestimate the true impact of active placebos, because the sensitivity analysis restricted to trials with low overall risk of bias gave a statistically significant SMD of −0.24. We used a modified version of RoB 2 to assess risk of bias, focusing on bias arising from the randomisation process and bias in selection in the reported result. We stress that there is a risk of confounding in such assessment, and that the association between low risk of bias and large effect size may be spurious. In particular, the trials at low risk of bias were almost all preclinical and without a matching experimental intervention; therefore, it is uncertain whether their results apply to clinical trials with active placebos more directly matched against experimental interventions. Also, the direction of the apparent bias is unusual, because the standard scenario is larger estimated effects in trials with high risk of bias.
Theoretically, the impact of active placebo could be more pronounced in cross‐over trials, where participants are exposed to both types of placebo. However, our subgroup analysis did not support this assumption. Moreover, the impact of active placebo interventions may differ depending on whether it is used in a three‐arm trial (with a matching experimental intervention) or in a two‐arm trial (with an implicit hypothetical experimental group), but this was also not supported by our subgroup analysis. We considered that the impact from the active placebo might differ depending on whether its effects are neutral/pleasant or unpleasant, but subgroup analysis did not confirm this.
When we planned the review, we carefully considered the adequacy and risk of unintended therapeutic effect of the active placebo, but our meta‐regression could not confirm these factors as important. The analysis of adequacy may have been limited by the fact that we did not give a score higher than 3 to any trial. Apparently, no trials had any formal pretrial validation of the active placebos with regard to adequacy and risk of therapeutic effect (apart from citation of previous literature). It is unclear if any trials conducted such a validation without reporting it. On the other hand, if the trial objective is to validate the active placebo, and not solely to investigate the therapeutic effect of an experimental drug, pretrial validation may be irrelevant. This was the case for at least three included three‐arm trials and one nearly eligible three‐arm trial (Eriksson 2019; Fangman 1963; Prosenz 2017; Wang 2008a). To our knowledge, there is no formal guidance for developing, validating, and assessing active placebos in randomised trials.
The ethical basis for using active placebos is that the expected benefit for future patients is considered more important than the additional unpleasantness inflicted on participants in the active placebo control group. Future research into the difference between unpleasant and neutral/pleasant active placebos may be useful because there are fewer ethical challenges for the latter.
Ideally, an active placebo should fully match the non‐therapeutic effects of the experimental drug. In practice, this is difficult to achieve, and it is unclear whether an active placebo that is a perfect match is more effective at maintaining blinding than a partial match or a credible non‐match. We addressed this question in our meta‐regression of active placebo adequacy and found no clear association between scores for active placebo adequacy and SMD. This analysis – albeit limited – may support the hypothesis that inducing some credible effect through an active placebo is more important than perfect matching.
Authors' conclusions
Implication for systematic reviews and evaluations of healthcare.
Our findings neither strongly support nor refute use of active placebo controls. Based on an overall assessment of the issue, we suggest trialists should consider active placebo control interventions where the experimental drug has strong perceptible non‐therapeutic effects, particularly in trials with participant‐reported outcomes. Where the experimental intervention has no obvious non‐therapeutic effects, we suggest using standard placebo control interventions.
The risk of unblinding is a concern for users of trial information. Readers of trial reports, or researchers formally assessing risk of bias in trials included in a systematic review, will often consider risk of unblinding. Based on an overall assessment of the issue, we consider that use of an active placebo control group in the scenarios mentioned above reflects a reduced risk of bias due to unblinding.
Implication for methodological research.
Further trials with both standard and active placebos are needed to address the main limitation of this review, namely imprecision. It may be worthwhile to include an active placebo group in large randomised trials that fit the scenarios mentioned above. As we noted in Potential biases in the review process, a large trial with almost 2000 participants would be needed to push our main result beyond statistical significance. However, the number and size of the trials needed will differ depending on type of trial and underlying true difference between active and standard placebo, so if particularly sensitive scenarios can be identified, the number needed might be lower.
An alternative approach is to conduct a meta‐epidemiological study (Sterne 2002). A meta‐epidemiological study includes meta‐analyses with both active‐placebo controlled trials and standard placebo‐controlled trials. Such comparisons within a meta‐analysis between trials using active placebos and standard placebo are observational by nature. However, the risk of confounding is lower than that of standard observational studies, because type of participants, interventions, and outcomes are comparable. We have planned such a meta‐epidemiological study, and have identified approximately 25 potentially eligible meta‐analyses (up to December 2021).
In summary, we did not find a statistically significant difference between active and standard placebo control interventions in our primary analysis, but the result was imprecise and the confidence interval was compatible with a difference ranging from important to irrelevant. Moreover, the result was not robust, as the difference was more pronounced and statistically significant in sensitivity analyses of trials at low risk of bias and when using a fixed‐effect model. We suggest that trialists and users of information from trials carefully consider the type of placebo control intervention in trials with high risk of unblinding, such as those that investigate drugs with pronounced non‐therapeutic effects and that include participant‐reported outcomes.
History
Protocol first published: Issue 7, 2020
Acknowledgements
We thank Karsten Juhl Jørgensen for commenting on the review protocol, Lasse Østengaard for commenting on earlier version of the search strategy, Jakob Solgaard Jensen for providing a detailed reference list of the 89 active placebo trials, Jennifer Gewandter for providing a list of potentially relevant trials from their review, Barbara Kitchenham for guidance in the calculation of effect sizes for cross‐over trials, and Klaus Linde for commenting on a draft of the paper.
Appendices
Appendix 1. Additional methods
Eligibility criteria
We defined 'randomised trial' as any trial denoted explicitly as such, or with a design that could be assumed to have a random allocation schedule (e.g. Latin squares design).
We defined 'clinical trial' as any trial investigating therapeutic effects on people with disease or at risk of disease. We defined 'preclinical trial' as any trial investigating therapeutic effects in healthy participants, (e.g. experimentally induced conditions such as laser‐induced pain or capsaicin‐induced pain).
If the active placebo was adequately described as an active placebo but was not called an active placebo in the trial report, we accepted other terms, such as 'active control' or 'active control treatment'.
We only considered active placebo interventions intended as active placebos a priori (i.e. before obtaining trial results). We excluded trials where an intervention designed to be experimental was reinterpreted as an active placebo (e.g. when reflecting on the results in the discussion section of a trial report). We also excluded trials where the authors stated that the potential active placebo intervention was not an adequate active placebo. Finally, we excluded trials where it was probable from the reporting that the so‐called 'active placebo' was meant as a therapeutically active control treatment.
We excluded trials where the active placebo and standard placebo clearly did not match in external characteristics, or when the participants in the two intervention groups received different information.
Search
The updated search was based on the following search strategy from Jensen 2017, performed in PubMed and Cochrane Central Register of Controlled Trials (CENTRAL) for records from 1 January 2015 onward (run on 11 March 2019):
"active placebo" OR "active placebos" OR "active control treatment" OR "active control" OR "sham diphenhydramine" OR "diphenhydramine placebo" OR "sham atropine" OR "atropine placebo" OR "sham benztropine" OR "sham benzodiazepine" OR "histamine placebo" OR "benztropine placebo" OR "benzodiazepine placebo"
The restricted search was in part inspired by Jensen 2017. We used the following search strategy for Ovid Embase (including Embase Classic, from inception (1947), run on 20 March 2019):
| # | |
| 1 | active placebo*.af. |
| 2 | active control*.af. |
| 3 | (inert or inactive or standard or sham or true placebo* or lactose or saline).af. |
| 4 | exp Placebos/ or exp PLACEBO EFFECT/ |
| 5 | 2 and (3 or 4) |
| 6 | (diphenhydramine or atropine or benztropine or benzodiazepine* or midazolam or diazepam or lorazepam or histamine).af. |
| 7 | (inert placebo* or inactive placebo* or standard placebo* or true placebo*).af. |
| 8 | 6 and 7 |
| 9 | (sham diphenhydramine or diphenhydramine placebo* or sham atropine or atropine placebo* or sham benztropine or benztropine placebo* or sham benzodiazepine or benzodiazepine placebo* or sham diazepam or diazepam placebo* or sham histamine or histamine placebo* or sham midazolam or midazolam placebo* or sham lorazepam or lorazepam placebo*).af. |
| 10 | 1 or 5 or 8 or 9 |
We searched the first 100 hits for the following strings in Google Scholar, sorted by relevance (different searches run in June and July 2020):
| # | |
| 1 | "active placebo" inert |
| 2 | "active placebo" inactive |
| 3 | "active placebo" saline |
| 4 | "active placebo" lactose |
In ProQuest, we used the following search string (run on 21 October 2020):
(("active placebo" OR "active placebos") AND ("inert placebo" OR "inactive placebo")) AND stype.exact("Dissertations & Theses")
We used a combination of Web of Science, Scopus, and Google Scholar to screen citations of relevant publications. We used the same sources to search for other publications from the first and last authors. The search string was "active placebo" OR "active placebos" (with quotes) in ClinicalTrials.gov (clinicaltrials.gov; run on 7 September 2020), and active placebo* (without quotes) in the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/clinical-trials-registry-platform; run on 20 October 2020).
Data collection
We extracted the following data from each study.
Publication and author information
Trials characteristics (clinical or preclinical, clinical area, country, trial design, information on funding and conflicts of interest, type of participants or condition, interventions)
Characteristics of interventions used, including the active placebo (component, dose, design, administration, terminology)
Information on trial conduct (duration of treatment and follow‐up, description of randomisation, blinding status, co‐interventions)
Participant flow numbers (number of participants screened, enrolled, and randomised)
For harm outcomes, co‐intervention outcomes and attrition, we first applied the following principles for selection (followed by the principles for participant‐reported outcomes described in Data extraction and management).
For outcomes of harm, we preferred predictable effects of the active placebo, then participant‐reported outcomes, and then observer‐reported outcomes.
For co‐intervention outcomes, we selected both continuous and dichotomous outcomes.
For attrition, we used the number of participants for whom the participant‐reported outcome was available (or alternatively, the observer‐reported outcome), compared to the number of randomised participants.
For trials with missing data on the standard deviation (SD), we looked for studies that reported the SD on the same scale used for similar populations. We then calculated and used the median of all the reported estimates. If a single paper described multiple populations, we only used the relevant ones (e.g. clinical population) and calculated a median for that paper's populations before calculating the median for that publication. We did this for two trials using the MSRPP (Multi‐Dimensional Scale for Rating Psychiatric Patients; Casey 1960; Kurland 1961), where two other studies provided data for the imputed SD estimate; and for one trial using HSCL‐64 (64‐item Hopkins Symptom Checklist; Lipman 1966) where seven publications provided data.
For trials that only reported useful data in figures, we used WebPlotDigitizer to extract the data (automeris.io/WebPlotDigitizer).
Data analysis
In trials with a factorial design, we analysed all participants receiving active placebo in one group and all participants receiving standard placebo in another group, disregarding factors other than placebo type.
Cross‐over trials
We treated two cross‐over trials as parallel‐group trials: in Casey 1960, the useful data for analysis came from the first period only, and in Dellemijn 1997, the placebo groups were in separate parallel arms, each with a cross‐over design. For dichotomous outcomes in cross‐over trials (including harms and attrition), we used the data naively, ignoring any correlation between the groups.
For the two cross‐over trials from which we obtained individual participant data (Bjørkedal 2011; Prosenz 2017), we calculated the mean differences (MDs) and SDs ourselves, as well as the correlation coefficients. For Bjørkedal 2011, we used a linear mixed model in STATA with drug and information as fixed components, and participant as a random component. For each intervention group, the pain outcome consisted of 20 measurements, so we used the individual measurements rather than the aggregated data, to improve precision. We accepted the slight imbalance between the sequences used in the trial. For Prosenz 2017, the pain outcome consisted of two measurements for each participant for each placebo intervention. Therefore, we entered the individual measurements in a mixed model with drug as a fixed component and participant as a random component. The trial was uniform, which accounted for any potential period effects (four participants in each of the six possible sequences of three interventions).
For trials without individual participant data, we used the data from the paper to calculate MD and a relevant SD, for within‐participant variation (SD_within), total participant variation (SD_total) or the difference (SD_diff), whichever was available. We imputed the correlation coefficient using the median of correlation coefficients calculated from the two trials with individual participant data.
For simple cross‐over designs with active placebo versus standard placebo, we used the effect size formula from the Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions to calculate the independent‐group standardised mean difference (SMD; Higgins 2021b).
For the two trials with individual participant data, where there were repeated measurements within each placebo group (Bjørkedal 2011; Prosenz 2017), we used the following approximation formulae of the effect size, called g_IG, with small‐sample size correction and independent group adjustment. Estimates were obtained from the mixed model as before: MD based on the drug parameter, within‐participant SD (SD_within), between‐participant SD (SD_between), r (intraclass correlation), t (the t statistic for the drug effect parameter), and number of participants (N).
First, we calculated the repeated measures effect size and its variance:
d_RM = MD/SD_within
A = d_RM/t
var_d_RM = A^2*(1+t^2/(2*N))
Then, we calculated the independent groups effect size and its variance:
SD_total = sqrt(SD_between^2+SD_within^2)
d_IG = d_RM*sqrt(1‐r) = MD/SD_total
var_d_IG = var_d_RM*(1‐r)
Finally, we added the small sample‐size adjustment:
c(N‐1) = 1‐3/(4*(N‐1)‐1)
g_IG = d_IG*c(N‐1)
var_g_IG = var_d_IG*c(N‐1)^2
SE_g_IG=sqrt(var_g_IG)
We then used g_IG and SE_g_IG in the inverse‐variance meta‐analysis. For further background regarding effect sizes for repeated measurements, see Johnson 1940, Madeyski 2018, and Morris 2002.
Parallel‐group trials with individual participant data
For two parallel‐group trials where we obtained individual participant data from the trial author (Werner 2019a; Werner 2019b), we calculated the results for use in our meta‐analysis. For the participant‐reported outcome, we used all available cases, whereas for the observer‐reported outcome (sleep duration), we used the per‐protocol population where the trial author had verified the sleep data. The observer‐reported outcome consisted of measurements from multiple nights, so we used a mixed model similar to those above, with intervention as a fixed component and participants as a random component. With the estimates from the model, we calculated SMD and its standard error (SE) using the repeated measurement formula set out above (in the 'Cross‐over trials' section of Appendix 1). We calculated attrition based on the participant flow diagrams supplied by the trial author.
Sensitivity analyses
For the sensitivity analysis of change from baseline scores, in three of the trials with individual participant data, each measurement had a corresponding baseline value, so we simply used the difference as the change score and analysed as before (Bjørkedal 2011; Werner 2019a; Werner 2019b). For the fourth trial with individual participant data, there was only one common baseline session for all cross‐over periods, so we used that to calculate the change score for both intervention groups (Prosenz 2017). We used the average of the two measurements for both interventions.
For all four trials with individual participant data, we also conducted a sensitivity analysis with adjustment for baseline scores, where we added the baseline as a fixed factor in the linear regression models, using the individual baselines where possible (Bjørkedal 2011; Werner 2019a; Werner 2019b), and the common average baseline otherwise (Prosenz 2017). The results of this analysis are presented in Appendix 2.
Heterogeneity
Trials without a matching experimental drug did not contribute to the combined meta‐regression of the primary analysis because we did not assess adequacy of their active placebo. We therefore also conducted separate meta‐regression analyses for adequacy scores and for risk of unintended therapeutic effect scores.
Appendix 2. Additional results
Description of studies
Two publications mentioned two other trials, but we were unable to obtain sufficient information to ascertain their eligibility for our review, even after attempts to contact the authors of the more recent trial, published after 1990 (Thomson 1982a; Wang 2008a). Their sample sizes are unknown, but the standard placebos in both trials appeared to be more beneficial than the active placebos.
We excluded several trials where we considered the active placebo interventions not to be active placebos according to our criteria, but rather active controls. One trial investigated the effect of reserpine on acute mental disturbance (Abse 1956). The trial authors stated they had used powdered opium as an active placebo, but elsewhere they called it an active drug. We deemed it unlikely that opium could be regarded a therapeutically inactive placebo. In another trial, the effect of degraded carrageenin on peptic ulcer was compared to aluminium sucrose sulphate, called active placebo (Yamagata 1973). Elsewhere in the text, this was described as an antipepsin agent already in clinical use, used in this trial as the control agent. In a Japanese paper (for which we based our assessment on the English abstract), researchers investigated the effects of a drug on urolithiasis, describing hyoscine‐N‐butylbromide as an active placebo (Yuge 1973). However, this drug is used to treat abdominal pain associated with cramps; it was approved for medical use in 1951 in Germany and later became available worldwide (Tytgat 2007). Another trial investigated the effects of cannabis extract premedication on anaesthetic depth and postoperative pain (Ibera 2018). The trial report was in Hebrew, which we read using Google Translate, but we also identified the likely corresponding record on ClinicalTrials.gov, where the active placebo was stated to be midazolam and paracetamol. We were unable to receive a clarifying response from the trial authors and so excluded the trial.
Main analyses
Three trials with participant‐reported outcomes did not report sufficient data to be included in the primary analysis. Shader 1964 included 20 healthy participants and aimed to identify an appropriate active placebo against phenothiazines. We were unable to determine even a direction of the impact from this publication, due to the nature of the outcome used (Clyde Mood Scale). Solomon 1960 investigated the effects of oestrogen and androgen in 12 osteoporotic women who responded to questionnaires covering 10 categories, including pain. The four drugs were ranked from worst to best (1 to 4); active placebo received an average ranking of 2.7 and standard placebo 3.1. The trial did not report whether this difference was significant. Regarding change over time, the active placebo improved in 7/9 categories and standard placebo in 4/9 categories. Fangman 1963 was a cross‐over trial on experimental pain. The trial authors measured three outcomes in four participants. The active placebo was significantly better in 4/12 cases, significantly worse in 1/12 cases, and there was no significant difference in 7/12 cases. We could not determine the size or precision of a potential impact.
Sensitivity analyses
We considered three trials to be nearly eligible studies: we identified Gracely 1987 in the primary search, and we found Eriksson 2019 and Max 1988 in other databases and sources (Figure 1). Eriksson 2019 compared an experimental treatment (2.0 % lidocaine injection), active placebo (0.1 % lidocaine) and standard placebo (saline) for experimental piercing pain in the mouth. The trial authors determined that the active placebo did not meet the requirements of a good active placebo, in part because it did not elicit the experience of being anaesthetised. We included the trial in a sensitivity analysis (Analysis 1.3). Gracely 1987 investigated magnitude of painful tooth pulp stimulation before and after administration of fentanyl, diazepam, both in combination, or neither. The trial authors did not use the term active placebo for diazepam directly, but rather reflected on it post hoc in their discussion. There was insufficient data to include the trial in the sensitivity meta‐analysis, so we summarised the results qualitatively. In two‐way ANOVAs, painfulness responses did not change after standard placebo, but did drop after active placebo. Visual inspection of the study figure appeared to show some improved response in active placebo compared to standard placebo groups. Max 1988 investigated the effects of amitriptyline and lorazepam for postherpetic neuralgia. The trial authors only described lorazepam as an active placebo post hoc in the discussion. We included the trial in a sensitivity analysis, where we dichotomised pain relief categories into worse/none/slight versus moderate/a lot/complete, as the trial authors had done (Analysis 1.3).
1.3. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 3: Sensitivity analysis – adding nearly eligible trials
In another sensitivity analysis adjusting for baseline as a covariate for trials with individual participant data, the pooled SMD was −0.09 (95% CI −0.22 to 0.05; I2 =40%; 14 trials). For clinical trials alone, the pooled SMD was −0.05 (95% C −0.25 to 0.1; I2 = 5%; 5 trials), and for preclinical trials alone, −0.09 (95% C −0.25 to 0.10; I2 = 47%; 9 trials) for preclinical trials.
Investigation of heterogeneity
In separate meta‐regression analyses for each score, the coefficients were unchanged in direction for adequacy (0.17, 95 CI −0.26 to 0.59; residual I2 = 14%; R2 =0%) and for risk of therapeutic effect (−0.13, 95% CI −0.29 to 0.03; residual I2 =20%; R2 = 44%). This was similar to the results in our meta‐regression with both scores together.
Data and analyses
Comparison 1. Participant‐reported outcomes at earliest post‐treatment assessment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Main analysis | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.1.1 Clinical | 6 | 442 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.21, 0.16] |
| 1.1.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.24, 0.07] |
| 1.2 Sensitivity analysis – excluding dichotomous data | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.2.1 Clinical | 6 | 442 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.21, 0.16] |
| 1.2.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.24, 0.07] |
| 1.3 Sensitivity analysis – adding nearly eligible trials | 20 | 1251 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.19, 0.06] |
| 1.3.1 Clinical | 7 | 507 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.20, 0.16] |
| 1.3.2 Preclinical | 13 | 744 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.22, 0.11] |
| 1.4 Sensitivity analysis – low risk of bias | 17 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.4.1 Clinical: low risk of bias | 1 | 90 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.49, 0.33] |
| 1.4.2 Clinical: some concern | 5 | 352 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.28, 0.21] |
| 1.4.3 Clinical: high risk of bias | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 1.4.4 Preclinical: low risk of bias | 4 | 285 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.35, ‐0.14] |
| 1.4.5 Preclinical: some concern | 7 | 398 | Std. Mean Difference (IV, Random, 95% CI) | 0.08 [‐0.14, 0.29] |
| 1.4.6 Preclinical: high risk of bias | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 1.4.7 All trials: low risk of bias | 5 | 375 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.34, ‐0.13] |
| 1.4.8 All trials: some concern | 12 | 750 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.12, 0.18] |
| 1.4.9 All trials: high risk of bias | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 1.5 Sensitivity analysis – fixed‐effect model | 17 | 1125 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.15 [‐0.23, ‐0.07] |
| 1.5.1 Clinical | 6 | 442 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐0.21, 0.16] |
| 1.5.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.28, ‐0.09] |
| 1.6 Sensitivity analysis – adding observer‐reported outcomes | 21 | 1645 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.17, 0.05] |
| 1.6.1 Clinical | 10 | 962 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.14, 0.12] |
| 1.6.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.24, 0.07] |
| 1.7 Sensitivity analysis – preferring change scores | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.23, 0.04] |
| 1.7.1 Clinical | 6 | 442 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.27, 0.14] |
| 1.7.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.26, 0.09] |
| 1.8 Sensitivity analysis – adjusting trials with individual participant data for baseline | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.22, 0.05] |
| 1.8.1 Clinical | 6 | 442 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.25, 0.14] |
| 1.8.2 Preclinical | 11 | 683 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.25, 0.10] |
| 1.9 Subgroup analysis – trial design | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.9.1 Clinical: parallel | 4 | 362 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.28, 0.21] |
| 1.9.2 Clinical: crossover | 2 | 80 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.52, 0.35] |
| 1.9.3 Preclinical: parallel | 5 | 431 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.26, 0.12] |
| 1.9.4 Preclinical: crossover | 6 | 252 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.33, 0.25] |
| 1.10 Subgroup analysis – experimental arm or no experimental arm | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.10.1 Clinical: with experimental arm | 4 | 149 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.53, 0.11] |
| 1.10.2 Clinical: no experimental arm | 2 | 293 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.16, 0.30] |
| 1.10.3 Preclinical: with experimental arm | 4 | 132 | Std. Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.21, 0.39] |
| 1.10.4 Preclinical: no experimental arm | 7 | 551 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.32, ‐0.04] |
| 1.11 Subgroup analysis – active placebo effects | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.11.1 Clinical: unpleasant | 5 | 352 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.28, 0.21] |
| 1.11.2 Clinical: neutral or pleasant | 1 | 90 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.49, 0.33] |
| 1.11.3 Preclinical: unpleasant | 10 | 626 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.24, 0.10] |
| 1.11.4 Preclinical: neutral or pleasant | 1 | 57 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐0.63, 0.41] |
| 1.12 Subgroup analysis – publication status | 17 | 1125 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.12.1 Clinical: peer‐reviewed publication | 5 | 352 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.28, 0.21] |
| 1.12.2 Clinical: grey literature | 1 | 90 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.49, 0.33] |
| 1.12.3 Preclinical:peer‐reviewed publication | 7 | 450 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.31, 0.05] |
| 1.12.4 Preclinical: grey literature | 4 | 233 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.21, 0.35] |
| 1.13 Data for main analysis – continuous data | 9 | 793 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.18, 0.10] |
| 1.13.1 Parallel | 9 | 793 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.18, 0.10] |
| 1.14 Data for main analysis – dichotomous data | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.15 Data for main analysis – continuous crossover data | 5 | 252 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.30, 0.17] |
| 1.15.1 Individual data | 2 | 128 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.39, ‐0.09] |
| 1.15.2 Imputed correlation coefficient | 3 | 124 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.17, 0.42] |
| 1.16 Data for sensitivity analysis – preferring change scores – continuous data | 3 | 247 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.47, 0.03] |
| 1.16.1 Parallel | 3 | 247 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.47, 0.03] |
| 1.17 Data for sensitivity analysis – adding nearly eligible studies – continuous data | 1 | 21 | Std. Mean Difference (IV, Random, 95% CI) | 0.52 [‐0.35, 1.40] |
| 1.17.1 Parallel | 1 | 21 | Std. Mean Difference (IV, Random, 95% CI) | 0.52 [‐0.35, 1.40] |
| 1.18 Data for sensitivity analysis – adding nearly eligible studies – dichotomous data | 1 | 65 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.27, 4.28] |
| 1.18.1 Crossover | 1 | 65 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.27, 4.28] |
| 1.19 Not in meta‐analysis | 7 | Other data | No numeric data | |
| 1.19.1 Insufficient data | 3 | Other data | No numeric data | |
| 1.19.2 No relevant data | 4 | Other data | No numeric data |
1.2. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 2: Sensitivity analysis – excluding dichotomous data
1.5. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 5: Sensitivity analysis – fixed‐effect model
1.6. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 6: Sensitivity analysis – adding observer‐reported outcomes
1.7. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 7: Sensitivity analysis – preferring change scores
1.8. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 8: Sensitivity analysis – adjusting trials with individual participant data for baseline
1.9. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 9: Subgroup analysis – trial design
1.10. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 10: Subgroup analysis – experimental arm or no experimental arm
1.11. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 11: Subgroup analysis – active placebo effects
1.12. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 12: Subgroup analysis – publication status
1.13. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 13: Data for main analysis – continuous data
1.14. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 14: Data for main analysis – dichotomous data
1.15. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 15: Data for main analysis – continuous crossover data
1.16. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 16: Data for sensitivity analysis – preferring change scores – continuous data
1.17. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 17: Data for sensitivity analysis – adding nearly eligible studies – continuous data
1.18. Analysis.

Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 18: Data for sensitivity analysis – adding nearly eligible studies – dichotomous data
1.19. Analysis.
Comparison 1: Participant‐reported outcomes at earliest post‐treatment assessment, Outcome 19: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| Fangman 1963 | Pricking pain threshold in terms of seconds for 12 spots (and 2 other outcomes: pricking pain at spot C, and ischaemic pain) | Uncertainty on how to use in meta‐analysis. Reports individual patient point estimates, individual baseline standard deviations and F‐score from ANOVA tests. For 12 cases (3 outcomes among 4 participants), active placebo was better in 4 cases, worse in 1 case and not significantly different in the remaining cases. We were not able to determine the size and precision of a potential effect. |
| Shader 1964 | Revised Clyde Mood Scales (subjects) | Not enough data for meta‐analysis. However, it is also unclear if the outcome has an obvious direction of benefit. |
| Solomon 1960 | Questionnaire covering 10 categories (household activities, social activities, resting, motor performance, sleeping, pain, psychological distress, well‐being, interest and participation in environment, and libidinal stimulation) | 12 patients were analysed. The 4 drugs were ranked from worst to best (1 to 4) in the categories where active placebo received an average ranking of 2.7 and standard placebo 3.1. The trial did not report whether this difference was significant. Looking at the change over time, the active placebo improved in 7/9 categories and the standard placebo in 4/9 categories |
| No relevant data | ||
| Adelson 1962 | No indication of a relevant outcome measured | |
| Casey 1960 | No indication of a relevant outcome measured | |
| Kurland 1961 | No indication of a relevant outcome measured | |
| Letemendia 1959 | No indication of a relevant outcome measured | |
Comparison 2. Patient‐reported outcomes at latest follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Main analysis | 3 | 176 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.85, 0.95] |
| 2.1.1 Clinical | 1 | 19 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.85, 0.95] |
| 2.1.2 Preclinical | 2 | 157 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 2.2 Data for main analysis – continuous data | 1 | 19 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.85, 0.95] |
| 2.2.1 Parallel | 1 | 19 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.85, 0.95] |
| 2.3 Not in meta‐analysis | 20 | Other data | No numeric data | |
| 2.3.1 Insufficient data | 2 | Other data | No numeric data | |
| 2.3.2 No relevant data | 18 | Other data | No numeric data |
2.2. Analysis.

Comparison 2: Patient‐reported outcomes at latest follow‐up, Outcome 2: Data for main analysis – continuous data
2.3. Analysis.
Comparison 2: Patient‐reported outcomes at latest follow‐up, Outcome 3: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| McKinley 2016 | Perceived efficacy on memory performance | "There was no significant main effect for condition on perceived efficacy of the medication for memory performance at 24‐hour follow‐up, F(3, 79) = 2.01, P = 0.120." |
| Parkes 2015 | Breathing sensation | Probably measured, but not reported |
| No relevant data | ||
| Adelson 1962 | No indication of a relevant outcome measured | |
| Berna 2017 | No relevant follow‐up time point for participant‐reported outcome | |
| Bjørkedal 2011 | No relevant follow‐up time point for participant‐reported outcome | |
| Casey 1960 | No indication of a relevant outcome measured | |
| Dellemijn 1997 | Peak value used for early time point. Late time point not used. | |
| Fangman 1963 | No relevant follow‐up time point for participant‐reported outcome | |
| Greenbaum 1987 | No relevant follow‐up time point for participant‐reported outcome | |
| Kurland 1961 | No indication of a relevant outcome measured | |
| Letemendia 1959 | No indication of a relevant outcome measured | |
| Lipman 1966 | No relevant follow‐up time point for participant‐reported outcome | |
| Prosenz 2017 | No relevant follow‐up time point for patient‐reported outcome | |
| Rief 2012 | No relevant follow‐up time point for participant‐reported outcome | |
| Shader 1964 | No relevant follow‐up time point for participant‐reported outcome | |
| Solomon 1960 | No relevant follow‐up time point for participant‐reported outcome | |
| Wang 2008a | Time‐summed value used for early time point. Late time point not used. | |
| Werner 2019a | No relevant follow‐up time point for participant‐reported outcome | |
| Werner 2019b | No relevant follow‐up time point for participant‐reported outcome | |
| Woodrow 1988 | No indication of a relevant outcome measured | |
Comparison 3. Observer‐reported outcomes at earliest post‐treatment assessment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Main analysis | 13 | 1113 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.18, 0.07] |
| 3.1.1 Clinical | 8 | 863 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.11, 0.13] |
| 3.1.2 Preclinical | 5 | 250 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.53, ‐0.05] |
| 3.2 Data for main analysis – continuous data | 5 | 659 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.35, 0.10] |
| 3.2.1 Parallel | 5 | 659 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.35, 0.10] |
| 3.3 Data for main analysis – dichotomous data | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.26, 8.23] |
| 3.3.1 Parallel | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.26, 8.23] |
| 3.4 Data for main analysis – continuous cross‐over data | 5 | 197 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.19, 0.15] |
| 3.4.1 Individual data | 2 | 84 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.30, 0.17] |
| 3.4.2 Imputed correlation coefficient | 3 | 113 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.31, 0.45] |
| 3.5 Not in meta‐analysis | 10 | Other data | No numeric data | |
| 3.5.1 Insufficient data | 2 | Other data | No numeric data | |
| 3.5.2 No relevant data | 8 | Other data | No numeric data |
3.2. Analysis.

Comparison 3: Observer‐reported outcomes at earliest post‐treatment assessment, Outcome 2: Data for main analysis – continuous data
3.3. Analysis.

Comparison 3: Observer‐reported outcomes at earliest post‐treatment assessment, Outcome 3: Data for main analysis – dichotomous data
3.4. Analysis.

Comparison 3: Observer‐reported outcomes at earliest post‐treatment assessment, Outcome 4: Data for main analysis – continuous cross‐over data
3.5. Analysis.
Comparison 3: Observer‐reported outcomes at earliest post‐treatment assessment, Outcome 5: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| Lipman 1966 | Target symptoms (doctor‐ and other observer‐reported outcomes) | Not enough data for meta‐analysis. Reports means and number of participants only. Standard deviation unclear. Qualitatively, no strong direction of benefit (active placebo: mean = 2.15, N = 99, standard placebo: mean = 2.17, N = 90). |
| Shader 1964 | Revised Clyde Mood Scales (observer) | Not enough data for meta‐analysis. However, unclear if the relevant outcome reported in the trial has a clear directionality. |
| No relevant data | ||
| Berna 2017 | No indication of a relevant outcome measured | |
| Bjørkedal 2011 | No indication of a relevant outcome measured | Measured laser‐evoked potentials with EEG recordings, including N2 and P2 amplitudes and latencies, to rule out the possible effects of response bias. We were uncertain whether they could be interpreted as clinically relevant observer‐reported outcomes of benefit and therefore did not use them for analysis. |
| Dellemijn 1997 | No indication of a relevant outcome measured | |
| Fangman 1963 | No indication of a relevant outcome measured | |
| Malmstrom 2006 | No indication of a relevant outcome measured | |
| Prosenz 2017 | No indication of a relevant outcome measured | Measured sedation using bispectral index (BIS) and Richmond Agitation‐Sedation Scale (RASS) as well as psychomotor and executive functions using the Groton Maze Learning Test Task and the Detection Task. We decided that they were not clinically relevant observer‐reported outcomes of benefit and therefore did not use them for analysis. |
| Rief 2012 | No indication of a relevant outcome measured | |
| Woodrow 1988 | No indication of a relevant outcome measured | |
Comparison 4. Observer‐reported outcomes at latest follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Main analysis | 1 | 96 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.67, 0.13] |
| 4.1.1 Clinical | 1 | 96 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.67, 0.13] |
| 4.2 For main analysis – continuous data | 1 | 96 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.67, 0.13] |
| 4.2.1 Parallel | 1 | 96 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.67, 0.13] |
| 4.3 Not in meta‐analysis | 20 | Other data | No numeric data | |
| 4.3.1 No relevant data | 20 | Other data | No numeric data |
4.2. Analysis.

Comparison 4: Observer‐reported outcomes at latest follow‐up, Outcome 2: For main analysis – continuous data
4.3. Analysis.
Comparison 4: Observer‐reported outcomes at latest follow‐up, Outcome 3: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| No relevant data | ||
| Berna 2017 | No indication of a relevant outcome measured | |
| Bjørkedal 2011 | No relevant follow‐up time point for observer‐reported outcome | |
| Casey 1960 | No relevant follow‐up time point for observer‐reported outcome | |
| Dellemijn 1997 | No indication of a relevant outcome measured | |
| Fangman 1963 | No indication of a relevant outcome measured | |
| Greenbaum 1987 | No relevant follow‐up time point for observer‐reported outcome | |
| Kurland 1961 | No relevant follow‐up time point for observer‐reported outcome | |
| Letemendia 1959 | No relevant follow‐up time point for observer‐reported outcome | |
| Lipman 1966 | No relevant follow‐up time point for observer‐reported outcome | |
| Malmstrom 2006 | No indication of a relevant outcome measured | |
| McKinley 2016 | No relevant follow‐up time point for observer‐reported outcome | |
| Parkes 2015 | No relevant follow‐up time point for observer‐reported outcome | |
| Prosenz 2017 | No indication of a relevant outcome measured | Measured sedation using bispectral index (BIS) and Richmond Agitation‐Sedation Scale (RASS) as well as psychomotor and executive functions using the Groton Maze Learning Test Task and the Detection Task. We decided that they were not clinically relevant observer‐reported outcomes of benefit and therefore did not use them for analysis. |
| Rief 2012 | No indication of a relevant outcome measured | |
| Shader 1964 | No relevant follow‐up time point for observer‐reported outcome | |
| Solomon 1960 | No relevant follow‐up time point for observer‐reported outcome | |
| Wang 2008a | Time‐summed value used for early time point. Late time point not used. | |
| Werner 2019a | No relevant follow‐up time point for observer‐reported outcome | |
| Werner 2019b | No relevant follow‐up time point for observer‐reported outcome | |
| Woodrow 1988 | No indication of a relevant outcome measured | |
Comparison 5. Harms.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Main analysis | 13 | 788 | Odds Ratio (IV, Random, 95% CI) | 3.08 [1.56, 6.07] |
| 5.1.1 Clinical | 8 | 422 | Odds Ratio (IV, Random, 95% CI) | 1.97 [0.85, 4.55] |
| 5.1.2 Preclinical | 5 | 366 | Odds Ratio (IV, Random, 95% CI) | 5.48 [1.82, 16.48] |
| 5.2 Subgroup analysis – predictable effects versus other harms | 13 | Odds Ratio (IV, Random, 95% CI) | Subtotals only | |
| 5.2.1 Clinical: predictable effects | 2 | 118 | Odds Ratio (IV, Random, 95% CI) | 2.00 [0.69, 5.85] |
| 5.2.2 Clinical: other harms | 6 | 304 | Odds Ratio (IV, Random, 95% CI) | 1.75 [0.42, 7.35] |
| 5.2.3 Preclinical: predictable effects | 3 | 206 | Odds Ratio (IV, Random, 95% CI) | 8.51 [1.59, 45.44] |
| 5.2.4 Preclinical: other harms | 2 | 160 | Odds Ratio (IV, Random, 95% CI) | 2.57 [1.14, 5.76] |
| 5.2.5 All trials: predictable effects | 5 | 324 | Odds Ratio (IV, Random, 95% CI) | 4.64 [1.60, 13.45] |
| 5.2.6 All trials: other harms | 8 | 464 | Odds Ratio (IV, Random, 95% CI) | 2.07 [0.81, 5.30] |
| 5.3 For main analysis – dichotomous data | 8 | 434 | Odds Ratio (M‐H, Random, 95% CI) | 3.74 [1.49, 9.39] |
| 5.3.1 Parallel | 6 | 330 | Odds Ratio (M‐H, Random, 95% CI) | 2.69 [1.22, 5.95] |
| 5.3.2 Crossover – unadjusted | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 17.10 [0.42, 691.72] |
| 5.4 For main analysis – continuous data | 2 | 170 | Std. Mean Difference (IV, Random, 95% CI) | 0.34 [0.01, 0.67] |
| 5.4.1 Parallel | 2 | 170 | Std. Mean Difference (IV, Random, 95% CI) | 0.34 [0.01, 0.67] |
| 5.5 Not in meta‐analysis | 11 | Other data | No numeric data | |
| 5.5.1 Insufficient data | 4 | Other data | No numeric data | |
| 5.5.2 No relevant data | 7 | Other data | No numeric data |
5.3. Analysis.

Comparison 5: Harms, Outcome 3: For main analysis – dichotomous data
5.4. Analysis.

Comparison 5: Harms, Outcome 4: For main analysis – continuous data
5.5. Analysis.
Comparison 5: Harms, Outcome 5: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| Adelson 1962 | Only reported harms as count data. Adverse experiences also reported as dichotomous outcome, but not for active placebo and standard placebo. |
Number of side‐effects: Active placebo: 187 among 52 participants Standard placebo: 161 among 52 participants |
| Casey 1960 | Side effects | 9 participants in active placebo experienced side effects 2 participants in standard placebo experienced side effects Unclear how many randomised to each |
| Fangman 1963 | Qualitative summary effect and side‐effect experiences by each participant. Not useful for meta‐analysis. | |
| McKinley 2016 | Side effects | Registered, but not clearly reported for use in meta‐analysis |
| No relevant data | ||
| Bjørkedal 2011 | No clear mention of adverse effects. | |
| Lipman 1966 | No clear mention of adverse effects. | |
| Rief 2012 | No clear mention of adverse effects. | |
| Shader 1964 | Adverse experiences monitored, but not reported. | |
| Solomon 1960 | No clear mention of adverse effects. | |
| Wang 2008a | Adverse experiences monitored but not reported. | |
| Woodrow 1988 | No clear mention of adverse effects. | |
Comparison 6. Attrition.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Main analysis | 15 | 864 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.74, 2.03] |
| 6.1.1 Clinical | 7 | 394 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.55, 1.96] |
| 6.1.2 Preclinical | 8 | 470 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.54, 5.57] |
| 6.2 Not in meta‐analysis | 6 | Other data | No numeric data | |
| 6.2.1 Insufficient data | 6 | Other data | No numeric data |
6.2. Analysis.
Comparison 6: Attrition, Outcome 2: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Outcome data |
| Insufficient data | ||
| Casey 1960 | Attrition | Unclear number of randomised participants between groups |
| Kurland 1961 | Attrition | Unclear number of randomised participants between groups: Indication of larger dropout for active placebo compared to standard placebo. Number of subjects at each evaluation point: Active placebo: 37 referred, 29 initially, 23 after 2 weeks, 8 after 4 weeks, 2 after 6 weeks Standard placebo: 37 referred, 30 initially, 29 after 2 weeks, 16 after 4 weeks, 9 after 6 weeks |
| Lipman 1966 | Attrition | Unclear number of randomised participants between groups |
| Parkes 2015 | Attrition | Unclear number of randomised participants between groups. However, for the 80 out of 81 randomised participants accounted for, attrition was 0/40 participants in each placebo group for the primary evaluation time point. |
| Rief 2012 | Attrition | Unclear number of randomised participants between groups |
| Shader 1964 | Attrition | Unclear number of participants for whom an outcome was measured |
Comparison 7. Co‐interventions, dichotomous.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Main analysis | 1 | 19 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 7.1.1 Clinical | 1 | 19 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 7.1.2 Preclinical | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 7.2 Not in meta‐analysis | 20 | Other data | No numeric data | |
| 7.2.1 Insufficient data | 3 | Other data | No numeric data | |
| 7.2.2 No relevant data | 17 | Other data | No numeric data |
7.1. Analysis.

Comparison 7: Co‐interventions, dichotomous, Outcome 1: Main analysis
7.2. Analysis.
Comparison 7: Co‐interventions, dichotomous, Outcome 2: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| Adelson 1962 | Co‐interventions | Some co‐interventions were used, but unclear if registered. |
| Casey 1960 | Co‐interventions | Co‐interventions were allowed (including conventional hypnotics, except barbiturates). Only individual and group psychotherapy, shock therapy, and interward transfer were restricted during the study. Unclear if these were registered |
| Greenbaum 1987 | Co‐interventions | Only count data: Instances of concomitant medication use: Active placebo: 25 among 28 participants Standard placebo: 22 among 28 participants |
| No relevant data | ||
| Berna 2017 | No indication of a relevant outcome measured | |
| Bjørkedal 2011 | No indication of a relevant outcome measured | |
| Dellemijn 1997 | No indication of a relevant outcome measured | |
| Fangman 1963 | No indication of a relevant outcome measured | |
| Kurland 1961 | No indication of a relevant outcome measured | |
| Letemendia 1959 | No indication of a relevant outcome measured | |
| Lipman 1966 | No indication of a relevant outcome measured | |
| McKinley 2016 | No indication of a relevant outcome measured | |
| Parkes 2015 | No indication of a relevant outcome measured | |
| Prosenz 2017 | No indication of a relevant outcome measured | |
| Rief 2012 | No indication of a relevant outcome measured | |
| Shader 1964 | No indication of a relevant outcome measured | |
| Solomon 1960 | No indication of a relevant outcome measured | |
| Wang 2008a | No indication of a relevant outcome measured | |
| Werner 2019a | No indication of a relevant outcome measured | |
| Werner 2019b | No indication of a relevant outcome measured | |
| Woodrow 1988 | No indication of a relevant outcome measured | |
Comparison 8. Co‐interventions, continuous.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Main analysis | 4 | 522 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 8.1.1 Clinical | 4 | 522 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 8.1.2 Preclinical | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | Not estimable |
| 8.2 Not in meta‐analysis | 19 | Other data | No numeric data | |
| 8.2.1 Insufficient data | 4 | Other data | No numeric data | |
| 8.2.2 No relevant data | 15 | Other data | No numeric data |
8.1. Analysis.

Comparison 8: Co‐interventions, continuous, Outcome 1: Main analysis
8.2. Analysis.
Comparison 8: Co‐interventions, continuous, Outcome 2: Not in meta‐analysis
| Not in meta‐analysis | ||
| Study | Outcome | Notes |
| Insufficient data | ||
| Adelson 1962 | Co‐interventions | Some co‐interventions were used, but unclear if registered. |
| Casey 1960 | Co‐interventions | Co‐interventions were allowed (including conventional hypnotics, except barbiturates). Only individual and group psychotherapy, shock therapy, and interward transfer were restricted during the study. Unclear if these were registered |
| Greenbaum 1987 | Co‐interventions | Only count data: Instances of concomitant medication use: Active placebo: 25 among 28 participants Standard placebo: 22 among 28 participants |
| Malmstrom 2006 | Co‐interventions | Only time‐to‐event data: Median time to rescue medication in hours: Active placebo: 1.4 (95% CI 1.3 to 1.6; 10 participants) Standard placebo: 1.2 (95% CI 1.0 to 1.3; 9 participants) |
| No relevant data | ||
| Berna 2017 | No indication of a relevant outcome measured | |
| Bjørkedal 2011 | No indication of a relevant outcome measured | |
| Dellemijn 1997 | No indication of a relevant outcome measured | |
| Fangman 1963 | No indication of a relevant outcome measured | |
| Kurland 1961 | No indication of a relevant outcome measured | |
| Letemendia 1959 | No indication of a relevant outcome measured | |
| Lipman 1966 | No indication of a relevant outcome measured | |
| McKinley 2016 | No indication of a relevant outcome measured | |
| Parkes 2015 | No indication of a relevant outcome measured | |
| Prosenz 2017 | No indication of a relevant outcome measured | |
| Rief 2012 | No indication of a relevant outcome measured | |
| Shader 1964 | No indication of a relevant outcome measured | |
| Solomon 1960 | No indication of a relevant outcome measured | |
| Wang 2008a | No indication of a relevant outcome measured | |
| Woodrow 1988 | No indication of a relevant outcome measured | |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adelson 1962.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel Objective: to investigate the effectiveness of 4 phenothiazine derivatives Treatment duration: 4.5 months Follow‐up duration (including treatment duration): 8 months Washout between cross‐over periods: NA |
|
| Data | Country: USA Condition: schizophrenia Number of people randomised: 48 to each placebo group (288 in total) Number of people analysed: 48 in each placebo group (288 in total) |
|
| Comparisons | Active placebo: scopolamine (maximum daily dose 1.50 mg) and chlorprophenpyridamine maleate (maximum daily dose 30 mg) Standard placebo: lactose Experimental intervention: 4 phenothiazines (doses in brackets are maximum daily doses): chlorpromazine (3000 mg), triflupromazine (750 mg), prochlorperazine (450 mg), perphenazine (240 mg) Administration: oral |
|
| Outcomes | Participant‐reported outcome: none Observer‐reported outcome: MRSPP (Multidimensional Scale for Rating Psychiatric Patients, Hospital Form; also called Lorr scale); assessed at 4 months, 5.5 months (earliest post‐treatment assessment), and 8 months (latest follow‐up) Harm outcome: number of side effects Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 3 Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding, but drugs supplied by the industry. Conflicts of interest not reported. | |
| Notes | 15 people dropped out and were replaced with randomly selected substitutes: 2 on active placebo and 2 on inactive placebo (i.e. keeping number of participants in the groups at 48). | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Stagger system. Allocation concealment not clearly described, but assignment carried out by a person not involved in the study. Study staff had no knowledge of the assignment. No indication of baseline differences. Replacements brought in for dropouts (n = 15, 2 for each placebo control intervention). |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Berna 2017.
| Study characteristics | ||
| Methods | Type: preclinical Design: parallel and 2 × 2 factorial design with double dummy, as follows.
For analysis, we combined 1 with 3 (all active placebo groups) and 2 with 4 (all standard placebo groups). Objective: to test the analgesic effects of diclofenac, an NSAID, with and without an induced side effect (dry mouth) produced by the addition of atropine Treatment duration: instant (single oral dose) Follow‐up duration (including treatment duration): 60 minutes Washout between cross‐over periods: NA |
|
| Data | Country: USA Condition: experimental heat‐induced pain Number of people randomised: 25 to each group Number of people analysed: 25 in each group |
|
| Comparisons | Active placebo: atropine (1.2 mg) Standard placebo: microcrystalline cellulose Experimental intervention: no matched experimental intervention. However, diclofenac (100 mg) can be seen as an add‐on to the comparison between active placebo and standard placebo. Administration: Oral |
|
| Outcomes | Participant‐reported outcome: pain intensity (100 mm VAS), assessed at 60 minutes Observer‐reported outcome: none Harm outcome: number of participants with dry mouth Co‐intervention outcome: none |
|
| Terminology for active placebo | Atropine to induce side effects. Report uses the term "active placebo" in the discussion. We interpreted this to refer to atropine a priori. | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | No industry funding. Trial authors declared no conflicts of interest. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Random numbers generator. Apparently remote pharmacy randomisation. Baseline differences did not suggest problem. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Outcome analysed seems to match with ClinicalTrials.gov record. Both post scores and change scores reported. No indication of selection on the basis of the result. |
Bjørkedal 2011.
| Study characteristics | ||
| Methods | Type: preclinical Design: 4‐period cross‐over and 2 × 2 factorial design (balanced placebo design), as follows.
For analysis, we combined 1 with 2 (all standard placebo groups) and 3 with 4 (all active placebo groups). Objective: to test whether side effects of drugs (atropine) can enhance expectancies and placebo responses Treatment duration: instant (single oral dose) Follow‐up duration (including treatment duration): 30 minutes per period Washout between cross‐over periods: minimum 24 hours |
|
| Data | Country: Norway Condition: experimental laser‐induced heat pain Number of people randomised: 23 (to a sequence of 4 interventions, i.e. 92 units). Number of people analysed: 20 (80 units), as 3 subjects did not find the stimulus painful. |
|
| Comparisons | Active placebo: caffeine (4 mg/kg) in grapefruit juice Standard placebo: grapefruit juice Experimental intervention: no matched experimental treatment. However, placebo intervention supplied with information explaining it was a placebo or a painkiller. Administration: oral |
|
| Outcomes | Participant‐reported outcome: pain on NRS of 0–10, assessed at 30 minutes Observer‐reported outcome: laser‐evoked potentials with EEG recordings, including N2 and P2 amplitudes and latencies, assessed at 30 minutes Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori. However, in some cases the trial authors also refer to"active placebo" as the combination of active placebo drug with information that they are receiving a painkiller (group 4). | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | No industry funding. Trial authors declared no conflicts of interest. | |
| Notes | Access to individual participant data with 20 measurements for each participant for each intervention | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Information from trial author: random draw of sealed, opaque envelopes containing an ID. Remote telephone contact with trial author who matched the ID with sequence. 23/24 possible treatment sequences used. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Trial author provided access to all study data. |
Casey 1960.
| Study characteristics | ||
| Methods | Type: clinical Design: special 2‐period cross‐over design, not all participants crossed over in the second period. Report only presents useful data for the first period. We used these first period data and treated the study as a parallel‐group study in the analysis. Objective: to determine the efficacy of tranquilising drugs for people with schizophrenia Treatment duration: 12 weeks per cross‐over period Follow‐up duration (including treatment duration): 12 weeks per cross‐over period Washout between cross‐over periods: none |
|
| Data | Country: USA Condition: schizophrenia Number of people randomised: 805 in total (unclear how many in placebo groups) Number of people analysed: 692 in total in the first period (173 and 178 in the 2 placebo groups) |
|
| Comparisons | Active placebo: phenobarbital (200 mg daily) Standard placebo: lactose Experimental intervention: 2 experimental groups: chlorpromazine (400 mg daily); promazine (400 mg daily) Administration: oral |
|
| Outcomes | Participant‐reported outcome: none Observer‐reported outcome: MSRPP (Multi‐Dimensional Scale for Rating Psychiatric Patients), assessed at 12 weeks (and 24 weeks) Harm outcome: number of participants with side effects Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" in secondary methods publication | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding not reported, but study received drug from industry. Conflicts of interest not reported. | |
| Notes | SD for MSRPP scale imputed from other studies | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Allocation concealment is not sufficiently described. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Outcomes reported matched the outcomes prespecified in the protocol. Specific analyses (final score or change score) not described. Outcome likely not selected on the basis of results. |
Dellemijn 1997.
| Study characteristics | ||
| Methods | Type: clinical Design: special cross‐over design. Participants randomised in parallel to either a cross‐over subtrial with experimental treatment and active placebo, or to a cross‐over subtrial with experimental treatment and standard placebo. Thus, placebo comparisons were compared in parallel, and we considered the study to be parallel‐group study in our analyses. Objective: to assess the efficacy of IV opioids in neuropathic pain Treatment duration: 5 hours per cross‐over period Follow‐up duration (including treatment duration): 8 hours per cross‐over period Washout between cross‐over periods: 7 days |
|
| Data | Country: Netherlands Condition: continuous unilateral neuropathic pain Number of people randomised: 27 to active placebo‐controlled subtrial, 26 to standard placebo‐controlled subtrial Number of people analysed: 26 in active placebo‐controlled subtrial, 24 in standard placebo‐controlled subtrial |
|
| Comparisons | Active placebo: diazepam (0.2 mg/mL, adjusted for bodyweight (1 mL/kg/hour) Standard placebo: saline Experimental intervention: fentanyl (5 μg/mL, adjusted for bodyweight 1 mL/kg/hour) Administration: IV |
|
| Outcomes | Participant‐reported outcome: peak pain intensity difference (difference between pain intensity rated on NRS of 0–10 during infusions and at baseline) Observer‐reported outcome: none Harm outcome: number of participants who had infusions stopped because of side effects Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 3 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding and conflicts of interest not reported | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Sequence generation not described. Remote randomisation, sealed envelopes. No indication of baseline differences. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Eriksson 2019 (sensitivity).
| Study characteristics | ||
| Methods | NA | |
| Data | NA | |
| Comparisons | NA | |
| Outcomes | NA | |
| Terminology for active placebo | NA | |
| Scores and unpleasantness of the active placebo | NA | |
| Funding and conflicts of interest | NA | |
| Notes | Only used in a sensitivity analysis | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Computer generated block randomisation with block size 12 (and 3 interventions). |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Fangman 1963.
| Study characteristics | ||
| Methods | Type: preclinical Design: 12‐period cross‐over design (4 repetitions of standard placebo, 2 repetitions of 2 doses of active placebo, 2 repetitions of 2 doses of experimental drug) Objective: to investigate the effects of morphine on radiant heat pain thresholds and ischaemic muscle pain thresholds Treatment duration: instant (single bolus per cross‐over period) Follow‐up duration (including treatment duration): 2 hours per cross‐over period Washout between cross‐over periods: each cross‐over treatment probably on separate days |
|
| Data | Country: USA Condition: experimental pain (radiant heat pain and ischaemic muscle pain) Number of people randomised: 4 randomised to a sequence of interventions Number of people analysed: 4 |
|
| Comparisons | Active placebo: phenobarbital (2 groups: 30 mg and 60 mg) Standard placebo: water Experimental intervention: morphine (2 groups: 7.5 mg and 10 mg) Administration: IV |
|
| Outcomes | Participant‐reported outcome: radiant heat pain threshold (pricking pain) and ischaemic muscle pain threshold (both probably assessed during the 2‐hour cross‐over period) Observer‐reported outcome: none Harm outcome: qualitative summary effect and side‐effect experienced by each participant Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 3
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding and conflicts of interest not reported | |
| Notes | Not in meta‐analysis for any outcome | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Gracely 1987 (sensitivity).
| Study characteristics | ||
| Methods | NA | |
| Data | NA | |
| Comparisons | NA | |
| Outcomes | NA | |
| Terminology for active placebo | NA | |
| Scores and unpleasantness of the active placebo | NA | |
| Funding and conflicts of interest | NA | |
| Notes | Only experiment II. Only for sensitivity analysis, however insufficient data for meta‐analysis so summarised qualitatively. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Greenbaum 1987.
| Study characteristics | ||
| Methods | Type: clinical Design: 3‐period cross‐over design Objective: to ascertain the effects of placebo, atropine, and desipramine on multiple somatic, psychological, and physiological attributes Treatment duration: 6 weeks per cross‐over period Follow‐up duration (including treatment duration): 6 weeks per cross‐over period Washout between cross‐over periods: none, but first 2 weeks in each period not analysed. |
|
| Data | Country: USA Condition: irritable bowel syndrome Number of people randomised: 41 (to a sequence of 3 interventions, i.e. 123 units in total) Number of people analysed: 28 (i.e. 84 units in total) |
|
| Comparisons | Active placebo: atropine (increasing from 0.4 mg to 1.2 mg daily) Standard placebo: placebo tablet not described Experimental intervention: desipramine (increasing from 50 mg to 150 mg daily) Administration: oral |
|
| Outcomes | Participant‐reported outcome: pain index assessed at 6 weeks Observer‐reported outcome: Brief Psychiatric Rating Scale (BPRS) assessed at 6 weeks Harm outcome: number of participants with adverse events Co‐intervention outcome: instances of concomitant medication use (number of events) |
|
| Terminology for active placebo | Atropine to induce symptom (mouth dryness), called "placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 3 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding, but drugs supplied by the industry. Conflicts of interest not reported. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Kurland 1961.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel Objective: to determine differential effects of various phenothiazine derivatives on morbidity and severity of illness Treatment duration: 6 weeks Follow‐up duration (including treatment duration): 6 weeks Washout between cross‐over periods: NA |
|
| Data | Country: USA Condition: indication for tranquillisers, e.g. anxiety, agitation, restlessness Number of people randomised: 37 "referred" to active placebo and 37 to standard placebo (277 for all arms) Number of people analysed: 23 in active placebo group and 29 in standard placebo group with at least 1 assessment (at 2 weeks or later; 187 for all arms) |
|
| Comparisons | Active placebo: phenobarbital (IV: minimum daily dose 195 mg; oral: minimum daily dose 97.5 mg) Standard placebo: saline and lactose Experimental intervention: 6 arms with different phenothiazines (doses in brackets are minimum daily doses): promazine (IV: 150 mg; oral: 300 mg), chlorpromazine (IV: 75 mg; oral: 300 mg), mepazine (IV: 75 mg; oral: 75 mg), triflupromazine (IV: 75 mg; oral: 75 mg), prochlorperazine (IV: 15 mg; oral: 30 mg), and perphenazine (IV: 15 mg; oral: 24 mg) Administration: IV for the first 2 days, then oral |
|
| Outcomes | Participant‐reported outcome: none Observer‐reported outcome: Multidimensional Scale for Rating Psychiatric Patients (MSRPP) at the latest available assessment (2 weeks or later) Harm outcome: side effects causing termination of treatment Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding unclear (funded by non‐commercial organisations, but several pharmaceutical companies are acknowledged). Conflicts of interest not reported. | |
| Notes | SD for MSRPP scale imputed from other studies. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | Outcome probably not selected on the basis of results. No other information. |
Letemendia 1959.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel Objective: to investigate the effect of the physical side effects of treatment on the assessment of its results, by answering the following questions.
Treatment duration: 9 weeks Follow‐up duration (including treatment duration): 9 weeks Washout between cross‐over periods: NA |
|
| Data | Country: UK Condition: chronic psychosis in males Number of people randomised: 14 to each placebo Number of people analysed: 14 in each placebo group |
|
| Comparisons | Active placebo: nicotinic acid (100 mg daily) Standard placebo: lactose Experimental intervention: none Administration: oral |
|
| Outcomes | Participant‐reported outcome: none Observer‐reported outcome: nurses assessments of mental state (improved, versus no change or worse), assessed at 9 weeks Harm outcome: number of participants with flushing, cumulative total at 9 weeks Co‐intervention outcome: none |
|
| Terminology for active placebo | Nicotinic acid for somatic effect, no therapeutic effect. Report does not use the term "active placebo". | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: neutral |
|
| Funding and conflicts of interest | Non‐industry funding. Conflicts of interest not declared. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Participants matched and allocated at random to 2 groups. One group chosen to receive active placebo by coin toss. No other information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Lipman 1966.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel and 2 × 2 × 2 factorial design, with the following factors.
Multicentre trials with 3 centres. For our analysis, all groups receiving active placebo were combined and all groups receiving standard placebo were combined. Objective: to test the hypothesis that the difference in therapeutic improvement between people receiving atropine medications and non‐atropine medications under the positive therapeutic treatment would be reliably greater than the magnitude of the difference in therapeutic response between people receiving atropine and non‐atropine medications under the neutral therapeutic treatment Treatment duration: 1 week Follow‐up duration (including treatment duration): 1 week Washout between cross‐over period: NA |
|
| Data | Country: USA Condition: people with anxiety neurosis Number of people randomised: not reported Number of people analysed: 104 for active placebo and 99 for standard placebo (with or without experimental drug and with either positive or neutral expectation) |
|
| Comparisons | Active placebo: atropine (1.5 mg daily) Standard placebo: similar capsules Experimental intervention: no matched experimental intervention. However, chlordiazepoxide hydrochloride (30 mg daily) can be seen as an add‐on to the comparison between active placebo and standard placebo. Administration: oral |
|
| Outcomes | Participant‐reported outcome: participant‐reported symptom checklist (SCL, 64 items) Observer‐reported outcome: doctor‐assessed target symptoms Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | Atropine to produce dry mouth (a priori). Called "active placebo" in discussion. We interpret it to mean an active placebo a priori. | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding, but drugs supplied by the industry. Conflicts of interest not reported. | |
| Notes | SD for symptom checklist scale imputed from other studies. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | No information on sequence generation. Remote pharmacy randomisation. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Malmstrom 2006.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel Objective: to assess analgesic effect of intravenous lidocaine on pain following the removal of impacted third molars. Treatment duration: 1 hour Follow‐up duration (including treatment duration): 6 hours Washout between cross‐over periods: NA |
|
| Data | Country: USA Condition: pain after removal of third molars Number of people randomised: 10 to each placebo (40 in total to all study arms) Number of people analysed: 10 for active placebo and 9 for standard placebo |
|
| Comparisons | Active placebo: diphenhydramine (bolus of 10 mg, continuous infusion of 40 mg) Standard placebo: saline infusion Experimental intervention: likely matched intervention: lidocaine (1.25 mg/kg bolus and 2.75 mg/kg infusion). The fourth intervention arm was oral oxycodone/acetaminophen (10 mg/650 mg; matched by placebo tablets in the other arms) Administration: IV |
|
| Outcomes | Participant‐reported outcome: TOPAR1 and TOPAR6 (weighted total of pain relief scores over the first hour and the first 6 hours). Trial's own primary time point was TOPAR2, which was not the earliest post‐treatment time point. Observer‐reported outcome: none Harm outcome: number of participants with 1 or more adverse experiences. Paper also reports number of each particular side effect. Co‐intervention outcome: number of people requesting rescue medication, and median time to rescue medication (acetaminophen/hydrocodone 500/5 mg) |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Industry funded. All 7 trial authors appear to be employed by Merck. | |
| Notes | CIs were truncated at 0 for the lower bound; we recalculated and used the untruncated interval. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Computer‐generated schedule. No information on allocation concealment. Baseline differences do not suggest a problem. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Max 1988 (sensitivity).
| Study characteristics | ||
| Methods | NA | |
| Data | NA | |
| Comparisons | NA | |
| Outcomes | NA | |
| Terminology for active placebo | NA | |
| Scores and unpleasantness of the active placebo | NA | |
| Funding and conflicts of interest | NA | |
| Notes | Only used in a sensitivity analysis. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
McKinley 2016.
| Study characteristics | ||
| Methods | Type: preclinical Design: parallel Objective: to investigate to what extent the frequency of reported side effects influences the perceived efficacy of a medication Treatment duration: instant administration Follow‐up duration (including treatment duration): 24 hours Washout between cross‐over periods: NA |
|
| Data | Country: New Zealand Condition: memory and breathing (healthy participants) Number of people randomised: 20 to each group (80 in total to all study arms) Number of people analysed: all randomised participants |
|
| Comparisons | Active placebo: capsaicin in sesame oil (3 doses: 0.0001%, 0.00005%, 0.000025%) Standard placebo: saline Experimental intervention: none Administration: intranasal spray |
|
| Outcomes | Participant‐reported outcome: perceived efficacy on memory (0–10) Observer‐reported outcome: FEV1% Harm outcome: modified GASE scale Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding sources not reported; study possibly received drugs from industry. Conflicts of interest not reported. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Sequence generation not described. Medicine in sealed envelopes, probably after randomisation process. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Parkes 2015.
| Study characteristics | ||
| Methods | Type: preclinical Design: parallel and 2 × 2 factorial design with the following factors.
Objective: to investigate how the price of a placebo medication influences placebo and nocebo effects. Additionally, to investigate the effect of using an active placebo on placebo and nocebo effects, and whether using an active placebo has an impact on the price effect. Treatment duration: instant administration Follow‐up duration (including treatment duration): 24 hours Washout between cross‐over periods: NA |
|
| Data | Country: New Zealand Condition: breathing (healthy participants) Number of people randomised: 81 in total, of which 1 excluded at baseline (unclear which group) Number of people analysed: 40 for active placebo and 40 for standard placebo |
|
| Comparisons | Active placebo: capsaicin (0.00014%) in sesame oil Standard placebo: saline and sesame oil Experimental intervention: none Administration: intranasal |
|
| Outcomes | Participant‐reported outcome: breathing sensation scale (10 cm VAS) Observer‐reported outcome: FEV1% (% of predicted) Harm outcome: modified GASE scale (number of side effects) Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 1
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Funding and conflicts of interest not reported | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Sequence generation not described. Medicine placed in sealed envelopes, unclear whether before or after randomisation. Unclear if several baseline differences arose by chance. Difference in sex distribution noted by trial authors and used post hoc as a covariate in analysis. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Prosenz 2017.
| Study characteristics | ||
| Methods | Type: preclinical Design: cross‐over Objective: to investigate the use of midazolam as an active placebo. To determine whether midazolam exerts analgesic effects at a clinically moderate sedative dose. Treatment duration: 1 minute (as bolus) per cross‐over period Follow‐up duration (including treatment duration): 120 minutes per cross‐over period Washout between cross‐over periods: 5 days |
|
| Data | Country: Austria Condition: experimental pain (contact heat, electrical pain, and pressure pain) Number of people randomised: 24 participants randomised to a sequence of 3 interventions. Number of people analysed: 24 for each placebo. |
|
| Comparisons | Active placebo: midazolam (0.06 mg/kg) Standard placebo: saline Experimental intervention: fentanyl (1 μg/kg) Administration: IV |
|
| Outcomes | Participant‐reported outcome: suprathreshold heat pain (0–100) Observer‐reported outcome: sedation using bispectral index (BIS) and Richmond Agitation‐Sedation Scale (RASS) as well as psychomotor and executive functions using the Groton Maze Learning Test Task and the Detection Task Harm outcome: number of participants with adverse events (predictable of the active placebo, e.g. somnolence, nausea) Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 3 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Mixed funding; received study material from industry. Declares no conflicts of interest. | |
| Notes | Trial author provided individual participant data. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Computer‐based randomisation. Sealed, opaque envelopes. Equal proportions to different cross‐over sequences, no obvious baseline differences between cross‐over sequences. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Study data received from trial author Data analysis appears in accordance with plan from ClinicalTrials.gov. No indication of selection of outcome on the basis of result from multiple outcomes or analyses. |
Rief 2012.
| Study characteristics | ||
| Methods | Type: preclinical Design: parallel and 2 × 3 + 1 factorial design with a separate no treatment group as well as 6 groups separated by the following factors.
Objective: to investigate the effects of manipulating the expectation of receiving an active drug, and of medication‐associated bodily sensations, to estimate the influence of these RCT‐typical features. Treatment duration: instant (single dose) Follow‐up duration (including treatment duration): 20 minutes Washout between cross‐over periods: NA |
|
| Data | Country: Germany Condition: experimental heat pain Number of people randomised: 144 volunteers included, unclear how many were randomised to each group. 57 randomised to each placebo type. Number of people analysed: 57 for each placebo type |
|
| Comparisons | Active placebo: capsaicin (0.014 %) in sesame oil Standard placebo: sesame oil Experimental intervention: none Administration: intranasal |
|
| Outcomes | Participant‐reported outcome: pain threshold (in degrees centigrade) Observer‐reported outcome: none Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding. Trial authors declared no conflicts of interest. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No indication of problems with baseline differences. No other information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Shader 1964.
| Study characteristics | ||
| Methods | Type: preclinical Design: cross‐over Objective: to find an active control substance that could later be used in a psychotic population under double‐blind conditions with an effective phenothiazine Treatment duration: 4 days per cross‐over period Follow‐up duration (including treatment duration): 4 days per cross‐over period Washout between cross‐over periods: 9 days |
|
| Data | Country: USA Condition: psychiatric symptoms in healthy participants Number of people randomised: 20 Number of people analysed: 20 in each placebo group |
|
| Comparisons | Active placebo: phenobarbital (50 mg) and atropine sulphate (0.3 mg) 4 times daily Standard placebo: lactose Experimental intervention: none Administration: oral |
|
| Outcomes | Participant‐reported outcome: Revised Clyde Mood Scale (self‐sort) Observer‐reported outcome: Revised Clyde Mood Scale (observer‐sort) Harm outcome: symptoms and side effects questionnaire Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active control" a priori (also uses the term "active placebo" elsewhere in the introduction) | |
| Scores and unpleasantness of the active placebo | Adequacy: 3
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding. Conflicts of interest not reported. | |
| Notes | This is a 2‐phase study; we were only interested in phase I. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Solomon 1960.
| Study characteristics | ||
| Methods | Type: clinical Design: cross‐over Objective: to investigate psychologic and osteometabolic responses to sex hormones in elderly osteoporotic women Treatment duration: 6 weeks per cross‐over period Follow‐up duration (including treatment duration): 6 weeks per cross‐over period Washout between cross‐over periods: not stated explicitly, but probably none |
|
| Data | Country: USA Condition: women with postmenopausal osteoporosis or Paget's disease Number of people randomised: 12 randomised to a cross‐over sequence Number of people analysed: all 12 completed, but unclear if all 12 analysed for each placebo |
|
| Comparisons | Active placebo: dextroamphetamine and amobarbital (5 mg, twice daily) Standard placebo: lactose Experimental intervention: 2 groups: methallenestril (3 mg twice daily) and fluoxymesterone (5 mg twice a day) Administration: oral |
|
| Outcomes | Participant‐reported outcome: questionnaire covering 10 categories (household activities, social activities, resting, motor performance, sleeping, pain, psychological distress, well‐being, interest and participation in environment, and libidinal stimulation) Observer‐reported outcome: 24 hour urinary calcium excretion Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 2
Risk of therapeutic effect: 4 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Mixed; received drugs from companies. Conflicts of interest not reported. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | Completed Latin squares design. No other information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Wang 2008a.
| Study characteristics | ||
| Methods | Type: preclinical Design: cross‐over Objective: to determine whether pregabalin and morphine could inhibit secondary hyperalgesia and allodynia induced by ID capsaicin compared with the active placebo diphenhydramine hydrochloride and true placebo; and to evaluate the value of diphenhydramine as a control. Treatment duration: instant (single dose) Follow‐up duration (including treatment duration): 240 minutes from capsaicin; 265 minutes from randomised IV treatment/315 minutes from randomised oral treatment; 330 minutes from first assessment; per cross‐over period (separate days) Washout between cross‐over periods: crossover periods were on separate days, no further information |
|
| Data | Country: USA (presumably) Condition: experimental capsaicin‐induced pain Number of people randomised: 20 Number of people analysed: 20 for each placebo type |
|
| Comparisons | Active placebo: diphenhydramine hydrochloride (50 mg) Standard placebo: not described Experimental intervention: 2 groups: pregabalin (300 mg) and morphine (10 mg) Administration: oral (except IV morphine) |
|
| Outcomes | Participant‐reported outcome: area of hyperalgesia Observer‐reported outcome: area of flare Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 3
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Industry funded. All trial authors had conflicts of interest. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Computer‐generated allocation schedule. Described as blinded to investigators and participants. Probably concealment of allocation |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
Werner 2019a.
| Study characteristics | ||
| Methods | Type: preclinical Design: parallel Objective: to evaluate a new model of an active placebo eliciting side effects Treatment duration: 7 days Follow‐up duration (including treatment duration): 7 days Washout between cross‐over periods: NA |
|
| Data | Country: Australia Condition: sleep Number of people randomised: 30 to active placebo, 28 to standard placebo, 27 to no treatment Number of people analysed: 20 per protocol for active placebo, 26 for standard placebo, 25 for no treatment |
|
| Comparisons | Active placebo: beetroot extract E162 (500 mg) and oxalic acid (250 mg), four times daily Standard placebo: lactose fibres as filler (4 capsules daily) Experimental intervention: none (other arm: no treatment) Administration: oral |
|
| Outcomes | Participant‐reported outcome: Insomnia Severity Index (ISI) after 7 days of treatment Observer‐reported outcome: sleep duration using actigraphy during the 7 days of treatment Harm outcome: urine colouration (0–3) > 0 at any time during the 7 days of treatment Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: neutral |
|
| Funding and conflicts of interest | Non‐industry funding. Trial authors declared no conflicts of interest. | |
| Notes | Trial author provided individual participant data. The author's PhD thesis, which also described the trial, was published just before publication of this review and was not used. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Random number from envelope, off‐site allocation. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Study data received from trial author. |
Werner 2019b.
| Study characteristics | ||
| Methods | Type: clinical Design: parallel Objective: to test whether participants who are given a placebo under 'double‐blind' conditions are more likely to believe that they are receiving a real medication if they receive an active placebo eliciting side effects, compared with a benign placebo that only contains lactose fibres; and to test whether side effects increase the effectiveness of an otherwise inactive placebo treatment. Treatment duration: 7 days Follow‐up duration (including treatment duration): 7 days Washout between cross‐over periods: NA |
|
| Data | Country: Australia Condition: insomnia Number of people randomised: 47 to active placebo, 44 to standard placebo, 25 to no treatment Number of people analysed: 38 per protocol for active placebo, 38 for standard placebo, 21 for no treatment |
|
| Comparisons | Active placebo: beetroot extract E162 (500 mg) and oxalic acid (250 mg), four times daily Standard placebo: lactose fibres as filler (4 capsules daily) Experimental intervention: none (other arm: no treatment) Administration: Oral |
|
| Outcomes | Participant‐reported outcome: Insomnia Severity Index (ISI) after 7 days of treatment Observer‐reported outcome: sleep duration using actigraphy during the 7 days of treatment Harm outcome: mean urine colouration (0–100) during the 7 days of treatment Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: NA
Risk of therapeutic effect: 1 Unpleasant/neutral/pleasant: neutral |
|
| Funding and conflicts of interest | Non‐industry funding. Trial authors declared no conflicts of interest. | |
| Notes | Trial author provided individual participant data. The author's PhD thesis, which also described the trial, was published just before publication of this review and was not used. | |
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Yes | Central randomisation by computer. |
| Free from risk of bias in selection of the reported result All outcomes | Yes | Trial author provided study data. |
Woodrow 1988.
| Study characteristics | ||
| Methods | Type: preclinical Design: cross‐over Objective: to compare ethyl alcohol to a narcotic analgesic in experimentally induced pain Treatment duration: instant (single dose on separate occasions at weekly intervals) Follow‐up duration (including treatment duration): 30 minutes (90 minutes for alcohol) on separate occasions at weekly intervals Washout between cross‐over periods: crossover periods were on separate weeks |
|
| Data | Country: USA Condition: experimental pain (pressure tolerance) Number of people randomised: 18 Number of people analysed: 14 for each placebo (4 not analysed because they missed their alcohol session) |
|
| Comparisons | Active placebo: atropine (0.4 mg) Standard placebo: saline (0.5 ml) Experimental intervention: 2 groups: morphine (0.17 mg/kg) and ethyl alcohol (2 mg/kg) Administration: subcutaneous except for alcohol (oral) |
|
| Outcomes | Participant‐reported outcome: pain tolerance (study also reports pain threshold) Observer‐reported outcome: none Harm outcome: none Co‐intervention outcome: none |
|
| Terminology for active placebo | "Active placebo" a priori | |
| Scores and unpleasantness of the active placebo | Adequacy: 1
Risk of therapeutic effect: 2 Unpleasant/neutral/pleasant: unpleasant |
|
| Funding and conflicts of interest | Non‐industry funding. Conflicts of interest not reported. | |
| Notes | ||
| Risk of bias | ||
| Item | Authors' judgement | Support for judgement |
| Free from risk of bias arising from the randomisation process | Unclear | No information. |
| Free from risk of bias in selection of the reported result All outcomes | Unclear | No information. |
CI: confidence interval; EEG: electroencephalogram; GASE: General Assessment of Side Effects; ID: intradermal; IV: intravenous; NA: not applicable; NRS: numeric rating scale; NSAID: nonsteroidal anti‐inflammatory drug; RCT: randomised clinical trial; SD: standard deviation; VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abse 1956 | Not active placebo (see Appendix 2). |
| Eriksson 2019 | Described as a bad active placebo. Included in a sensitivity analysis. |
| Gracely 1987 | Described as active placebo post hoc (after results). Included in a sensitivity analysis. |
| Ibera 2018 | Not active placebo (see Appendix 2). |
| Max 1988 | Potential active placebo implied post hoc (after results). Included in a sensitivity analysis. |
| Yamagata 1973 | Not active placebo (see Appendix 2). |
| Yuge 1973 | Not active placebo (see Appendix 2). |
Characteristics of studies awaiting classification [ordered by study ID]
Thomson 1982b.
| Methods | |
| Data | |
| Comparisons | |
| Outcomes | |
| Notes | Cited in Thomson 1982a: "Thomson and Chute (in preparation) found that an active placebo (nicotinic acid) induced significantly greater depression in normal volunteers than did an inert (lactose) placebo." We have not been able to identify any publications for this study. |
Wang 2008b.
| Methods | |
| Data | |
| Comparisons | |
| Outcomes | |
| Notes | Cited in Wang 2008a: "Interestingly, we observed in a second study similarly an increase of area of hyperalgesia by diphenhydramine than true placebo (unpublished data)." We contacted the trial author and Merck for further information regarding this study, but no information was available. |
Differences between protocol and review
See Laursen 2020 (protocol for review).
Changes to specified protocol content
For harms, due to the lack of data for available multiple time points, we decided to use any one time point in one analysis, preferring the latest available, rather than extracting both an early post‐treatment and a late follow‐up time point. We also decided to combine continuous and dichotomous outcomes of harms into one analysis, due to the paucity of continuous data, rather than extracting both continuous harms and dichotomous harms separately. We decided to measure attrition at the time of the primary outcome (early post‐treatment assessment, or average data or similar), so that the two analyses corresponded to each other in time.
After publication of our protocol, we decided to use the revised version of Cochrane risk of bias tool (RoB 2) rather than the first version of the tool.
Additions to content
We included many more trials in our review than expected; consequently, we decided to add the following post‐hoc sensitivity analyses of our primary analysis.
Adding trials with only observer‐reported outcomes
Using change scores instead of absolute values
Adjusting for baseline as a covariate in trials with individual participant data (this analysis is described in Appendix 1).
We also added the following subgroup analyses.
Parallel‐group versus cross‐over trials
Trials with versus without a matching experimental intervention
Unpleasantness versus pleasant/neutral active placebo intervention
Peer‐reviewed publications versus grey literature
Clarifications of protocol content in more detail
We clarified our intention to not include active placebo trials where the study hypothesis was one of harm, since the impact of active placebo may be different in such trials.
We conducted the following sensitivity analyses, not explicitly defined in the protocol, but standard practise for meta‐analyses.
Including only trials at low risk of bias
Reanalysing the results with a fixed‐effect model
We also clarified that eligible clinical and preclinical trials investigated clinical therapeutic effects and not physiological, pharmacodynamic or sociological effects, as we were interested in the clinical impact.
We clarified the choice of time point for different outcomes: we used the earliest post‐treatment time point unless the trial used another metric in its primary analysis (e.g. peak or time‐weighted average; Appendix 1). For trials with repeated measurements of individual participant data, we used data from all the measurements in mixed models for improved precision.
For co‐interventions, we clarified that we accepted any available time point, preferably the latest follow‐up.
Contributions of authors
DRTL and AH developed the protocol. DRTL, CHN, and ADF performed the search and screening. DRTL, CHN, ADF, MRH, and ASP did the data collection and assessments. EB, JP, and CPW provided individual participant data. DRTL performed the statistical analyses. DRTL and AH wrote the first draft of the review. All authors contributed to the drafting of the review. DRTL is the guarantor.
Sources of support
Internal sources
-
University of Southern Denmark (SDU), Denmark
Scholarship from the SDU, which funds one year of DRTL’s PhD employment
-
Cochrane Denmark, Denmark
Funds the remainder of DRTL's PhD employment. Cochrane Denmark is funded by the Danish state.
External sources
No sources of support provided
Declarations of interest
MRH has received grants from Pfizer, paid to his employer, outside the submitted work, has received speaking fees from Novartis outside the submitted work, and owns stocks in Novo Nordisk. During the editorial process, MRH changed employment from Odense University Hospital and University of Southern Denmark to Novo Nordisk on 1 February 2022; this was after submission of the first review draft and response to peer review comments, but before the copy editing. All three affiliations are therefore listed.
JP, EB and CPW are co‐authors of three of the included studies and were not involved in the study inclusion, data extraction, and risk of bias assessment of any studies.
The remaining authors (DRTL, CHN, ADF, ASP, AH) declare no financial or non‐financial conflicts of interest.
New
References
References to studies included in this review
Adelson 1962 {published data only}
- Adelson D, Epstein LJ. A study of phenothiazines with male and female chronically ill schizophrenic patients. Journal of Nervous and Mental Disease 1962;134(6):543. [DOI: 10.1097/00005053-196206000-00008] [PMID: ] [DOI] [PubMed] [Google Scholar]
Berna 2017 {published data only}
- Berna C, Kirsch I, Zion SR, Lee YC, Jensen KB, Sadler P, et al. Side effects can enhance treatment response through expectancy effects: an experimental analgesic randomized controlled trial. Pain 2017;158(6):1014-20. [DOI: 10.1097/j.pain.0000000000000870] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bjørkedal 2011 {published and unpublished data}
- Bjørkedal E, Flaten MA. Interaction between expectancies and drug effects: an experimental investigation of placebo analgesia with caffeine as an active placebo. Psychopharmacology 2011;215(3):537-48. [DOI: 10.1007/s00213-011-2233-4] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkedal E, Nermo T, Aslaksen PM, Flaten MA. The effect of an active placebo on pain-induced event related potentials in a balanced placebo design. Psychophysiology 2009;46:S127. [DOI: 10.1111/j.1469-8986.2009.00920.x] [DOI] [Google Scholar]
- Bjørkedal E. Active placebo – the relation of treatment expectancies to active analgesic treatments. The Arctic University of Norway (UiT) 2016. [hdl.handle.net/10037/10017]
- Flaten M, Aslaksen P, Bjørkedal E. Event-related potentials to painful stimulation after caffeine and placebo in a balanced placebo design. Journal of Pain 2010;11(4):S56. [DOI: 10.1016/j.jpain.2010.01.233] [DOI] [Google Scholar]
Casey 1960 {published data only}
- Casey JF, Bennett IF, Lindley CJ, Hollister LE, Gordon MH, Springer NN. Drug therapy in schizophrenia. A controlled study of the relative effectiveness of chlorpromazine, promazine, phenobarbital, and placebo. A.M.A. Archives of General Psychiatry 1960;2(2):210-20. [DOI: 10.1001/archpsyc.1960.03590080086012] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Lindley CJ, editor(s). Transactions of the Second (1957) Research Conference on Chemotherapy in Psychiatry. Vol. 2. Washington, D.C.: Veterans Administration, 1958. [Google Scholar]
Dellemijn 1997 {published data only}
- Dellemijn PL, Vanneste JA. Randomised double-blind active-placebo-controlled crossover trial of intravenous fentanyl in neuropathic pain. Lancet (London, England) 1997;349(9054):753-8. [DOI: 10.1016/S0140-6736(96)09024-1] [PMID: ] [DOI] [PubMed] [Google Scholar]
Eriksson 2019 (sensitivity) {published data only}
- Eriksson L, Asadi J. Intraoral injections of active/non-active local anesthetic agents – normal response and response in relation to perceived numbness. [Intraoral injektion av aktiv och icke-aktiv lokalanestetika – normalt gensvar och gensvar i relation till upplevd bedövningskänsla]. Malmö universitet/Odontologiska fakulteten 2019. [DIVA, ID: diva2:1479798]
Fangman 1963 {published data only}
- Fangman JJ. The effects of morphine on two forms of experimental pain. ETD Collection for Fordham University 1963:1-81. [https://research.library.fordham.edu/dissertations/AAI6305590]
Gracely 1987 (sensitivity) {published data only}
- Gracely RH, Dubner R. Reliability and validity of verbal descriptor scales of painfulness. Pain 1987;29(2):175-85. [DOI: 10.1016/0304-3959(87)91034-7] [PMID: ] [DOI] [PubMed] [Google Scholar]
Greenbaum 1987 {published data only}
- Greenbaum DS, Mayle JE, Vanegeren L, Jerome J, Mayor J, Greenbaum R, et al. Effects of desipramine, atropine and placebo on irritable bowel syndrome (IBS). Gastroenterology 1984;86(5):1098. [DOI: 10.1016/S0016-5085(84)80002-5] [DOI] [PubMed] [Google Scholar]
- Greenbaum DS, Mayle JE, Vanegeren LE, Jerome JA, Mayor JW, Greenbaum RB, et al. Effects of desipramine on irritable bowel syndrome compared with atropine and placebo. Digestive Diseases and Sciences 1987;32(3):257-66. [DOI: 10.1007/BF01297051] [PMID: ] [DOI] [PubMed] [Google Scholar]
Kurland 1961 {published data only}
- Kurland AA, Hanlon TE, Tatom MH, Ota KY, Simopoulos AM. The comparative effectiveness of six phenothiazine compounds, phenobarbital, and inert placebo in treatment of acutely ill patients: global measures of severity of illness. Journal of Nervous and Mental Disease 1961;133(1):1-18. [DOI: 10.1097/00005053-196107000-00001] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Kurland AA, Hanlon TE, Tatom MH, Simopoulos AM. Comparative studies of the phenothiazine tranquilizers: methodological and logistical considerations. Journal of Nervous and Mental Disease 1961;132:61-74. [PMID: ] [PubMed] [Google Scholar]
- Kurland AA, Michaux MH, Hanlon TE, Ota KY, Simopoulos AM. The comparative effectiveness of six phenothiazine compounds, phenobarbital and inert placebo in the treatment of acutely ill patients: personality dimensions. Journal of Nervous and Mental Disease 1962;134(1):48-60. [PMID: ] [PubMed] [Google Scholar]
- Kurland AA, Sutherland GF. The phenothiazine tranquilizers – their neurological complications and significance. Psychosomatics 1960;1(4):192-4. [DOI: 10.1016/S0033-3182(60)72984-0] [DOI] [Google Scholar]
Letemendia 1959 {published data only}
- Letemendia F, Harris AD. The right kind of control. Lancet 1959;273(7076):785-6. [DOI: 10.1016/S0140-6736(59)91863-X] [DOI] [Google Scholar]
- Letemendia FJ, Harris AD. The influence of side-effects on the reporting of symptoms. Psychopharmacologia 1959;1(1):39-47. [DOI: 10.1007/BF00408110] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Letemendia FJ, Harris AD. The influence of side-effects on the reporting of symptoms – reply. Psychopharmacologia 1960;1(4):354-6. [DOI: 10.1007/BF00404232] [DOI] [PubMed] [Google Scholar]
Lipman 1966 {published data only}
- Lipman RS, Park LC, Rickels K. Paradoxical influence of a therapeutic side-effect interpretation. Archives of General Psychiatry 1966;15(5):462-74. [DOI: 10.1001/archpsyc.1966.01730170014004] [PMID: ] [DOI] [PubMed] [Google Scholar]
Malmstrom 2006 {published data only}
- Malmstrom K, Kotey P, McGratty M, Ramakrishnan R, Gottesdiener K, Reicin A, et al. Dental impaction pain model as a potential tool to evaluate drugs with efficacy in neuropathic pain. Journal of Clinical Pharmacology 2006;46(8):917-24. [DOI: 10.1177/0091270006289847] [PMID: ] [DOI] [PubMed] [Google Scholar]
Max 1988 (sensitivity) {published data only}
- Max MB, Schafer SC, Culnane M, Smoller B, Dubner R, Gracely RH. Amitriptyline, but not lorazepam, relieves postherpetic neuralgia. Neurology 1988;38(9):1427-32. [DOI: 10.1212/wnl.38.9.1427] [PMID: ] [DOI] [PubMed] [Google Scholar]
McKinley 2016 {published data only}
- McKinley M. The influence of side effects on the nocebo and placebo response. University of Auckland 2016. [hdl.handle.net/2292/34346]
Parkes 2015 {published data only}
- Parkes B. The effect of medication price on placebo and nocebo responding. University of Auckland 2015. [hdl.handle.net/2292/29075]
Prosenz 2017 {published and unpublished data}
- Prosenz J, Gustorff B. Midazolam as an active placebo in 3 fentanyl-validated nociceptive pain models. Pain 2017;158(7):1264-71. [DOI: 10.1097/j.pain.0000000000000910] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rief 2012 {published data only}
- Rief W, Glombiewski JA. The hidden effects of blinded, placebo-controlled randomized trials: an experimental investigation. Pain 2012;153(12):2473-7. [DOI: 10.1016/j.pain.2012.09.007] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Rief W. The relevance of placebo effects in clinical trials. Journal of Psychosomatic Research 2012;72(6):498. [DOI: 10.1016/j.jpsychores.2012.03.004] [DOI] [Google Scholar]
Shader 1964 {published data only}
- Shader RI, Cohler J, Elashoff R, Grinspoon L. Phenobarbital and atropine in combination, an active control substance for phenothiazine research. Journal of Psychiatric Research 1964;2(3):169-83. [DOI: 10.1016/0022-3956(64)90018-4] [PMID: ] [DOI] [PubMed] [Google Scholar]
Solomon 1960 {published data only}
- Gordan GS, Vaughan C. Prevention and treatment of postmenopausal osteoporosis. In: Pasetto N, Paoletti R, Ambrus JL, editors(s). The menopause and postmenopause: the proceedings of an international symposium held in Rome, June 1979. Dordrecht: Springer Netherlands, 1980:179-93. [DOI: 10.1007/978-94-011-7230-1_18] [DOI] [Google Scholar]
- Solomon GF, Dickerson WJ, Eisenberg E. Psychologic and osteometabolic responses to sex hormones in elderly osteoporotic women. Geriatrics 1960;15:46-60. [PMID: ] [PubMed] [Google Scholar]
Wang 2008a {published data only}
- Wang H, Bolognese J, Calder N, Baxendale J, Kehler A, Cummings C, et al. Effect of morphine and pregabalin compared with diphenhydramine hydrochloride and placebo on hyperalgesia and allodynia induced by intradermal capsaicin in healthy male subjects. Journal of Pain 2008;9(12):1088-95. [DOI: 10.1016/j.jpain.2008.05.013] [PMID: ] [DOI] [PubMed] [Google Scholar]
Werner 2019a {published and unpublished data}
- Werner C, Faasse K, Sharpe L, Colagiuri B. The development and evaluation of a new model of an active placebo. In: 2nd official Society for Interdisciplinary Placebo Studies (SIPS) conference. Leiden, 2019.
- Werner C. The development and evaluation of a new experimental model of an active placebo to investigate how the experience of side effects influences the placebo effects. University of Sydney 2022. [https://hdl.handle.net/2123/27394]
Werner 2019b {unpublished data only}
- Unpublished data. Trial data obtained from trial author.
- Werner C. The development and evaluation of a new experimental model of an active placebo to investigate how the experience of side effects influences the placebo effects. The University of Sydney 2022. [https://hdl.handle.net/2123/27394]
Woodrow 1988 {published data only}
- Woodrow KM, Eltherington LG. Comments on "Feeling no pain: alcohol as an analgesic". Pain 1991;46(3):343. [DOI: 10.1016/0304-3959(91)90117-G] [DOI] [PubMed] [Google Scholar]
- Woodrow KM, Eltherington LG. Feeling no pain: alcohol as an analgesic. Pain 1988;32(2):159-63. [DOI: 10.1016/0304-3959(88)90064-4] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Abse 1956 {published data only}
- Abse DW, Curtis TE, Dahlstrom WG, Hawkins DR, Toops TC. The use of reserpine in the management of acute mental disturbance on an in-patient service: preliminary report. Journal of Nervous and Mental Disease 1956;124(3):239-45. [DOI: 10.1097/00005053-195609000-00003] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Abse DW, Dahlstrom WG, Tolley AG. Evaluation of tranquilizing drugs in the management of acute mental disturbance. American Journal of Psychiatry 1960;116:973-80. [DOI: 10.1176/ajp.116.11.973] [PMID: ] [DOI] [PubMed] [Google Scholar]
Eriksson 2019 {published data only}
- Eriksson L, Asadi J. Intraoral injections of active/non-active local anesthetic agents – normal response and response in relation to perceived numbness. [Intraoral injektion av aktiv och icke-aktiv lokalanestetika – normalt gensvar och gensvar i relation till upplevd bedövningskänsla]. Malmö universitet/Odontologiska fakulteten 2019. [DIVA, ID: diva2:1479798]
Gracely 1987 {published data only}
- Gracely RH, Dubner R. Reliability and validity of verbal descriptor scales of painfulness. Pain 1987;29(2):175-85. [DOI: 10.1016/0304-3959(87)91034-7] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ibera 2018 {published data only}
- Ibera C, Shalom B, Saifi F, Shruder J, Davidson E. Effects of cannabis extract premedication on anesthetic depth. Harefuah 2018;157(3):162-6. [PMID: ] [PubMed] [Google Scholar]
Max 1988 {published data only}
- Max MB, Schafer SC, Culnane M, Smoller B, Dubner R, Gracely RH. Amitriptyline, but not lorazepam, relieves postherpetic neuralgia. Neurology 1988;38(9):1427-32. [DOI: 10.1212/wnl.38.9.1427] [PMID: ] [DOI] [PubMed] [Google Scholar]
Yamagata 1973 {published data only}
- Yamagata S, Ishimori A, Sato H, Okabe H, Ishihara K. Clinical evaluation of pharmacotherapy for peptic ulcer with antipepsin agents by double blind technique – multicenter clinical study. Tohoku Journal of Experimental Medicine 1973;110(4):377-404. [DOI: 10.1620/tjem.110.377] [PMID: ] [DOI] [PubMed] [Google Scholar]
Yuge 1973 {published data only}
- Yuge J, Tsukada O, Nito H, Saito T, Fukuda S, Nishimura Y, et al. A clinical trial on analgesic effect of 1,3,5-trihydroxybenzene (Dilospan inj.) – Simple double blind test by the cases of urolithiasis [1,3,5-trihydroxybenzene(Dilospan注)の鎮痛効果に関する臨床実験 –尿路結石症を対象とした単純二重盲検実験]. Hinyokika Kiyo 1973;19(1):119-25. [hdl.handle.net/2433/121473] [Google Scholar]
References to studies awaiting assessment
Thomson 1982b {unpublished data only}
Wang 2008b {unpublished data only}
Additional references
Aan Het Rot 2012
- Aan Het Rot M, Zarate CA, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biological Psychiatry 2012;72(7):537-47. [DOI: 10.1016/j.biopsych.2012.05.003] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bello 2016
- Bello S, Wei M, Hilden J, Hróbjartsson A. The matching quality of experimental and control interventions in blinded pharmacological randomised clinical trials: a methodological systematic review. BMC Medical Research Methodology 2016;16:18. [DOI: 10.1186/s12874-016-0111-9] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boutron 2006
- Boutron I, Estellat C, Guittet L, Dechartres A, Sackett DL, Hróbjartsson A, et al. Methods of blinding in reports of randomized controlled trials assessing pharmacologic treatments: a systematic review. PLOS medicine 2006;3(10):e425. [DOI: 10.1371/journal.pmed.0030425] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2005
- Chan AW, Altman DG. Epidemiology and reporting of randomised trials published in PubMed journals. Lancet (London, England) 2005;365(9465):1159-62. [DOI: 10.1016/S0140-6736(05)71879-1] [PMID: ] [DOI] [PubMed] [Google Scholar]
Chinn 2000
- Chinn S. A simple method for converting an odds ratio to effect size for use in meta-analysis. Statistics in Medicine 2000;19(22):3127-31. [DOI: 10.1002/1097-0258(20001130)19:22<3127::aid-sim784>3.0.co;2-m] [PMID: ] [DOI] [PubMed] [Google Scholar]
Cohen 1988
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. New York: Routledge, 1988. [Google Scholar]
Colagiuri 2010
- Colagiuri B. Participant expectancies in double-blind randomized placebo-controlled trials: potential limitations to trial validity. Clinical Trials (London, England) 2010;7(3):246-55. [DOI: 10.1177/1740774510367916] [PMID: ] [DOI] [PubMed] [Google Scholar]
Dechartres 2016
- Dechartres A, Trinquart L, Faber T, Ravaud P. Empirical evaluation of which trial characteristics are associated with treatment effect estimates. Journal of Clinical Epidemiology 2016;77:24-37. [DOI: 10.1016/j.jclinepi.2016.04.005] [PMID: ] [DOI] [PubMed] [Google Scholar]
Deeks 2021
- Deeks JJ, Higgins JP, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Gaudiano 2005
- Gaudiano BA, Herbert JD. Methodological issues in clinical trials of antidepressant medications: perspectives from psychotherapy outcome research. Psychotherapy and Psychosomatics 2005;74(1):17-25. [DOI: 10.1159/000082022] [PMID: ] [DOI] [PubMed] [Google Scholar]
Greenberg 1994
- Greenberg RP, Bornstein RF, Zborowski MJ, Fisher S, Greenberg MD. A meta-analysis of fluoxetine outcome in the treatment of depression. Journal of Nervous and Mental Disease 1994;182(10):547-51. [DOI: 10.1097/00005053-199410000-00003] [PMID: ] [DOI] [PubMed] [Google Scholar]
Gøtzsche 2007
- Gøtzsche PC, Hróbjartsson A, Maric K, Tendal B. Data extraction errors in meta-analyses that use standardized mean differences. JAMA 2007;298(4):430-7. [DOI: 10.1001/jama.298.4.430] [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2021a
- Higgins JP, Savovic J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Higgins 2021b
- Higgins JP, Eldridge S, Li T. Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.1.
Hopewell 2010
- Hopewell S, Dutton S, Yu LM, Chan AW, Altman DG. The quality of reports of randomised trials in 2000 and 2006: comparative study of articles indexed in PubMed. BMJ (Clinical research ed.) 2010;340:c723. [DOI: 10.1136/bmj.c723] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hróbjartsson 2011
- Hróbjartsson A, Boutron I. Blinding in randomized clinical trials: imposed impartiality. Clinical Pharmacology and Therapeutics 2011;90(5):732-6. [DOI: 10.1038/clpt.2011.207] [PMID: ] [DOI] [PubMed] [Google Scholar]
Hróbjartsson 2014
- Hróbjartsson A, Emanuelsson F, Skou Thomsen AS, Hilden J, Brorson S. Bias due to lack of patient blinding in clinical trials. A systematic review of trials randomizing patients to blind and nonblind sub-studies. International Journal of Epidemiology 2014;43(4):1272-83. [DOI: 10.1093/ije/dyu115] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jakobsen 2017
- Jakobsen JC, Katakam KK, Schou A, Hellmuth SG, Stallknecht SE, Leth-Møller K, et al. Selective serotonin reuptake inhibitors versus placebo in patients with major depressive disorder. A systematic review with meta-analysis and Trial Sequential Analysis. BMC psychiatry 2017;17(1):58. [DOI: 10.1186/s12888-016-1173-2] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jensen 2017
- Jensen JS, Bielefeldt AØ, Hróbjartsson A. Active placebo control groups of pharmacological interventions were rarely used but merited serious consideration: a methodological overview. Journal of Clinical Epidemiology 2017;87:35-46. [DOI: 10.1016/j.jclinepi.2017.03.001] [PMID: ] [DOI] [PubMed] [Google Scholar]
Johnson 1940
- Johnson NL, Welch BL. Applications of the non-central t-distribution. Biometrika 1940;31(3-4):362-89. [DOI: 10.1093/biomet/31.3-4.362] [DOI] [Google Scholar]
Laursen 2020
- Laursen DR, Hansen C, Paludan-Müller AS, Hróbjartsson A. Active placebo versus standard placebo control interventions in pharmacological randomised trials. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: MR000055. [DOI: 10.1002/14651858.MR000055] [DOI] [Google Scholar]
Madeyski 2018
- Madeyski L, Kitchenham B. Effect sizes and their variance for AB/BA crossover design studies. Empirical Software Engineering 2018;23(4):1982-2017. [DOI: 10.1007/s10664-017-9574-5] [DOI] [Google Scholar]
Mercieca‐Bebber 2018
- Mercieca-Bebber R, Williams D, Tait MA, Roydhouse J, Busija L, Sundaram CS, et al. Trials with patient-reported outcomes registered on the Australian New Zealand Clinical Trials Registry (ANZCTR). Quality of Life Research 2018;27(10):2581-91. [DOI: 10.1007/s11136-018-1921-5] [PMID: ] [DOI] [PubMed] [Google Scholar]
Moncrieff 2003
- Moncrieff J. A comparison of antidepressant trials using active and inert placebos. International Journal of Methods in Psychiatric Research 2003;12(3):117-27. [DOI: 10.1002/mpr.148] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moncrieff 2004
- Moncrieff J, Wessely S, Hardy R. Active placebos versus antidepressants for depression. Cochrane Database of Systematic Reviews 2004, Issue 1. Art. No: CD003012. [DOI: 10.1002/14651858.CD003012.pub2] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Morris 2002
- Morris SB, DeShon RP. Combining effect size estimates in meta-analysis with repeated measures and independent-groups designs. Psychological Methods 2002;7(1):105-25. [DOI: 10.1037/1082-989x.7.1.105] [PMID: ] [DOI] [PubMed] [Google Scholar]
Moustgaard 2020
- Moustgaard H, Clayton GL, Jones HE, Boutron I, Jørgensen L, Laursen DR, et al. Impact of blinding on estimated treatment effects in randomised clinical trials: meta-epidemiological study. BMJ (Clinical research ed.) 2020;368:l6802. [DOI: 10.1136/bmj.l6802] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Page 2016
- Page MJ, Higgins JP, Clayton G, Sterne JA, Hróbjartsson A, Savovic J. Empirical evidence of study design biases in randomized trials: systematic review of meta-epidemiological studies. PLOS One 2016;11(7):e0159267. [DOI: 10.1371/journal.pone.0159267] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Salamone 2000
- Salamone JD. A critique of recent studies on placebo effects of antidepressants: importance of research on active placebos. Psychopharmacology 2000;152(1):1-6. [DOI: 10.1007/s002130000509] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schulz 2002
- Schulz KF, Grimes DA. Blinding in randomised trials: hiding who got what. Lancet (London, England) 2002;359(9307):696-700. [DOI: 10.1016/S0140-6736(02)07816-9] [PMID: ] [DOI] [PubMed] [Google Scholar]
Sterne 2002
- Sterne JA, Jüni P, Schulz KF, Altman DG, Bartlett C, Egger M. Statistical methods for assessing the influence of study characteristics on treatment effects in 'meta-epidemiological' research. Statistics in Medicine 2002;21(11):1513-24. [DOI: 10.1002/sim.1184] [PMID: ] [DOI] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ (Clinical research ed.) 2019;366:l4898. [DOI: 10.1136/bmj.l4898] [PMID: ] [DOI] [PubMed] [Google Scholar]
Storebø 2015
- Storebø OJ, Ramstad E, Krogh HB, Nilausen TD, Skoog M, Holmskov M, et al. Methylphenidate for children and adolescents with attention deficit hyperactivity disorder (ADHD). Cochrane Database of Systematic Reviews 2015, Issue 11. Art. No: CD009885. [DOI: 10.1002/14651858.CD009885.pub2] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thomson 1982a
- Thomson R. Side effects and placebo amplification. Journal of Mental Science 1982;140:64-8. [DOI: 10.1192/bjp.140.1.64] [PMID: ] [DOI] [PubMed] [Google Scholar]
Tytgat 2007
- Tytgat GN. Hyoscine butylbromide: a review of its use in the treatment of abdominal cramping and pain. Drugs 2007;67(9):1343-57. [DOI: 10.2165/00003495-200767090-00007] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wilkinson 2019
- Wilkinson ST, Farmer C, Ballard ED, Mathew SJ, Grunebaum MF, Murrough JW, et al. Impact of midazolam vs. saline on effect size estimates in controlled trials of ketamine as a rapid-acting antidepressant. Neuropsychopharmacology 2019;44(7):1233-8. [DOI: 10.1038/s41386-019-0317-8] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]