

SUMMARY
p53 is a key tumor suppressor that is frequently mutated in human tumors. In this study, we investigated how p53 is regulated in precancerous lesions prior to mutations in the p53 gene. Analyzing esophageal cells in conditions of genotoxic stress that promotes development of esophageal adenocarcinoma, we find that p53 protein is adducted with reactive isolevuglandins (isoLGs), products of lipid peroxidation. Modification of p53 protein with isoLGs diminishes its acetylation and binding to the promoters of p53 target genes causing modulation of p53-dependent transcription. It also leads to accumulation of adducted p53 protein in intracellular amyloid-like aggregates that can be inhibited by isoLG scavenger 2-HOBA in vitro and in vivo. Taken together, our studies reveal a posttranslational modification of p53 protein that causes molecular aggregation of p53 protein and its non-mutational inactivation in conditions of DNA damage that may play an important role in human tumorigenesis.
Graphical abstract
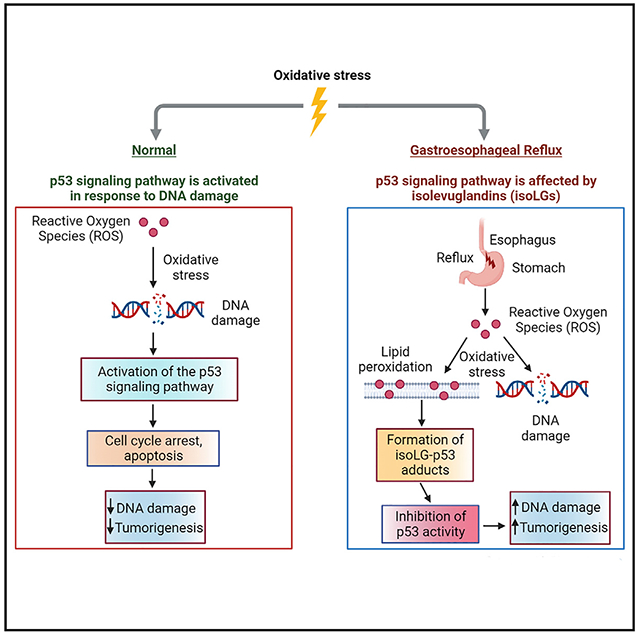
In brief
Caspa Gokulan et al. describe a non-mutational mechanism of inhibition of p53 protein during esophageal tumor progression through its adduction with active isolevuglandins. Adduction of p53 leads to inhibition of its biological activity that can be prevented with isoLG scavenger, 2-hydroxybenzylamine.
INTRODUCTION
Certain human tumors are promoted by factors that are not normally considered to be carcinogenic. One interesting example is esophageal adenocarcinoma (EAC).1 EAC is the most common histological subtype of esophageal malignancy in the United States. and many Western countries.2 The unique etiology links EAC to gastroesophageal reflux disease (GERD), a common digestive disorder that affects approximately 20% of the adult population in North America and is considered to be one of the strongest risk factors for EAC and Barrett’s esophagus (BE), a metaplasia that is prone to malignant transformation.3,4 In GERD and BE patients, epithelial cells, which line the esophagus, are exposed to gastric acid frequently mixed with bile. Among reflux components, hydrochloric acid and bile salts are the two most studied irritants responsible for pathological consequences in the esophagus. Bile salts deserve special attention, as multiple studies suggested that exposure of esophageal cells to these body metabolites is a significant contributing factor to esophageal tissue injury and EAC.5 Studies of the esophageal refluxates in GERD and BE patients have shown that esophageal cells are typically exposed to a high concentration of bile salts that can reach up to the millimolar range.6–9 Hydrochloric acid and bile salts induce significant damage of the esophageal epithelial lining and inflammation, which can further exacerbate tissue injury. Although the specific molecular mechanisms of reflux-induced tumorigenesis remain poorly understood, one important contributing factor is reflux-induced DNA damage. Several studies including ours have found that acidic bile salts (ABS) induce reactive oxygen species (ROS) and strong DNA damage that increases the mutation rate and promotes genomic instability.10–12 Due to persistent DNA damage induced by reflux, EAC is characterized by high frequencies of genomic alterations.13
Under DNA damage conditions, cells normally trigger a surveillance mechanism termed the DNA Damage Response (DDR), which senses damaged DNA and engages various tumor suppression mechanisms. One of the critical branches of the DDR is controlled by the p53 tumor suppressor. This protein regulates cell-cycle progression and determines whether cells proceed with DNA damage repair or undergo cell-cycle arrest or apoptosis. p53 tumor suppressor is one of the most mutated proteins in human tumors. Mutations in the TP53 gene commonly arise at late stages of the multistep tumorigenic process. In high-grade esophageal dysplasia and EAC, frequencies of p53 mutations can reach 50%–70% of cases, whereas in esophageal precancerous lesions, BE, and low-grade dysplasia, TP53 mutations are relatively rare.14,15 How p53 protein is regulated in precancerous conditions is largely unknown.
Our recent studies revealed that exposure of esophageal cells to reflux components increases levels of ROS leading to the formation of isolevuglandins (isoLG), products of lipid peroxidation, that form multiple protein adducts in esophageal epithelial cells in GERD patients.16 The entire spectrum of isoLG-affected proteins generated by reflux remains currently unknown. Our preliminary screening analysis suggested that among adducted proteins is p53 tumor suppressor.16 However, it is unclear how reflux-generated isoLGs affect p53 or other adducted proteins.
IsoLGs are a family of γ-ketoaldehydes generated by free radical-induced peroxidation of lipids. Several isoLG stereoisomers are also produced as byproducts of enzymatic reactions catalyzed by cyclooxygenase 2.17 The interest in isoLGs originates from their highly reactive nature. IsoLGs are able to adduct biomolecules with significantly greater avidity than other reactive aldehydes.18 IsoLGs can react with free amine group of lysine and other amino acid residues to form covalent protein adducts and protein-protein crosslinks.18 A better understanding of the biological role of isoLG protein adducts has only recently started to emerge, but it is already clear that they have a broad pathological effect.19,20
In this study, we analyzed, how isoLGs affect the cellular DDR with a specific emphasis on the p53 protein.
RESULTS
Genotoxic effect of reflux
To investigate the regulation of the DDR in reflux conditions, we first explored DNA damage in our experimental model of interest. We used non-transformed esophageal epithelial cell lines CP-A, BAR-T, and EPC-2, which express wild-type p53 protein, and were isolated from Barrett’s (CP-A, BAR-T) and normal squamous epithelium (EPC-2). In order to reproduce effect of reflux in vitro, these cells were exposed to acidic growth medium (pH 4.0), supplemented with 100 μM bile salts cocktail (ABS) for 10 min and left to recover as discussed in the STAR Methods section and previous publications.11,12,16 The composition, total bile salts concentration, and pH were selected based on clinical analyses of the refluxates in GERD and BE patients.7,8,11,21 Cells were then analyzed for DNA damage using comet and γH2AX phosphorylation assays. As a control, cells were treated with DNA-damaging agent camptothecin (10 μM) for 10 min. Consistent with previous reports, levels of DNA damage in ABS- and camptothecin-treated cells were found be comparable (Figures 1A and 1B). ABS-induced DNA damage in EPC2 cells is shown in Figure S1A.
Figure 1. Induction of DNA damage by reflux.
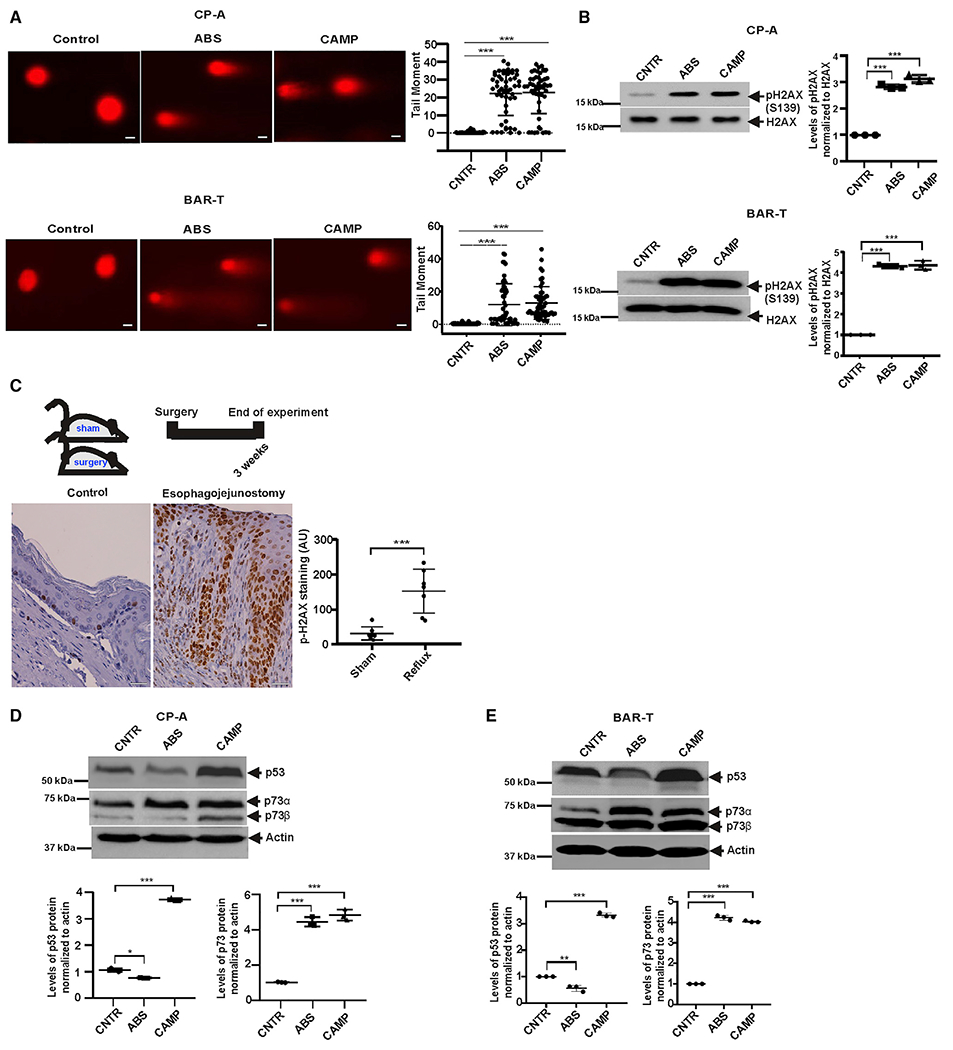
(A) Analysis of DNA damage by comet assay in CP-A and BAR-T cells. Cells were treated with 100 μM bile salts cocktail, pH 4.0 or 10 μM camptothecin (CAMP) for 10 min and analyzed for DNA damage 8 h after treatment. Representative images are shown (scale bar, 5 μm). Graph shows the comet tail moment analysis (control versus ABS, ***p < 0.001; control versus CAMP, ***p < 0.001; n = 3; Mann-Whitney test).
(B) The same as (A) but DNA damage was assessed by western blotting with p-H2AX antibody (control versus ABS, ***p < 0.001; control versus CAMP, ***p < 0.001; n = 3; Tukey’s multiple comparison).
(C) Representative images of immunohistochemical staining for p-H2AX in esophageal tissues collected from sham control mice (n = 8) and ones with esophagojejunostomy (n = 7). Significant increase of H2AX protein phosphorylation was observed in esophageal tissues collected from animals with reflux compared with sham controls (***p < 0.001; Tukey’s multiple comparison). Scale bar, 10 μm.
(D) Western blot analyses of cell extracts collected from CP-A cells treated with ABS or CAMP, as discussed in the Results section. In contrast to p53 protein, levels of p73 were higher in ABS- and CAMP-treated compared with control untreated cells (control versus ABS, ***p < 0.001, control versus CAMP, ***p < 0.001; n = 3).
(E) The same as (D) but BAR-T cells were analyzed (p53 control versus ABS, **p < 0.01; control versus CAMP, ***p < 0.001 and p73 control versus ABS, ***p < 0.001; control versus CAMP, ***p < 0.001). Expression of p53 and p73 proteins were analyzed by densitometry. Expression levels in control untreated cells were arbitrarily set at 1. All results are expressed as mean ± SD. See also Figure S1.
To investigate DNA damage in vivo, we used mice in which reflux was induced by esophagojejunostomy as described in the STAR Methods section and Figure S1B. Using immunohistochemistry with γH2AX antibody, levels of DNA damage were compared in esophageal specimens collected from reflux mice and control animals with sham surgery (seven mice/group) 2 weeks after recovery from surgery. Our analyses show that reflux caused significant DNA damage (p < 0.001) in esophageal epithelium of reflux mice, but not in sham controls (Figure 1C). Combined, our data show that reflux induces strong DNA damage in vivo and in vitro.
Exposure of esophageal cells to reflux components affects p53 protein activity
Since p53 protein plays a key role in the regulation of the DDR and is known to be activated by DNA damage,22 ABS-treated and control cells described above were analyzed for expression of p53 protein. Our studies revealed that despite the presence of DNA damage, p53 was not significantly affected by ABS, while camptothecin (or other DNA-damaging drugs such as cisplatin and etoposide) strongly upregulated p53 protein suggesting that ABS may affect the DDR and p53 (Figures 1D and 1E). These findings were consistent with previous reports showing inhibition and downregulation of p53 by ABS.23,24 To test whether reflux components have an impact on other proteins induced by DNA damage, we analyzed p73 protein, which has significant structural and functional similarities to p53.25 In contrast to p53, DNA damage induced by ABS led to a strong upregulation of p73 protein in the same cells, confirming a selective inhibitory effect of ABS on p53 (Figures 1D and 1E). To confirm that p73 plays a role in the regulation of p53 target genes in ABS-treated cells, we assessed expression of CDKN1A(p21) and BBC3(PUMA) mRNA in CP-A cells stably transfected with either p73 small hairpin RNA (shRNA) or control shRNA. These p53 target genes were selected based on their important role in the regulation of cell cycle, apoptosis, and the DDR. Consistent with previous report,26 we found that inhibition of p73 leads to significant decrease in mRNA expression of these target genes, showing that p73 is responsible in their upregulation (Figure S2A).
Thus, to assess the specific role of p53 and exclude effect of p73 protein, which is known to transactivate the overlapping set of p53 target genes,27 our analysis was conducted in p73-deficient CP-A cells, (Figure 2A; upper panel). We used the RT2 Profiler PCR array, which includes 84 genes regulating the p53 signaling. We found that ABS has a profound effect on the p53 pathway, significantly inhibiting expression of multiple genes as shown on the heatmap (Figure 2A). We repeated this experiment in the presence of 2-hydroxybenzylamine (2-HOBA), a scavenger of reactive isolevuglandins,28 which efficiently reacts with isoLGs preventing their interaction with cellular biomolecules.28,29 Treatment with 2-HOBA recovered the transcription profile of the p53 signaling pathway, suggesting that isoLGs are involved in the regulation of p53 (Figures 2A and S2B). Notably, we did not find any effects of 2-HOBA on pH or other experimental parameters.
Figure 2. Acidic bile salts inhibit activity of p53.
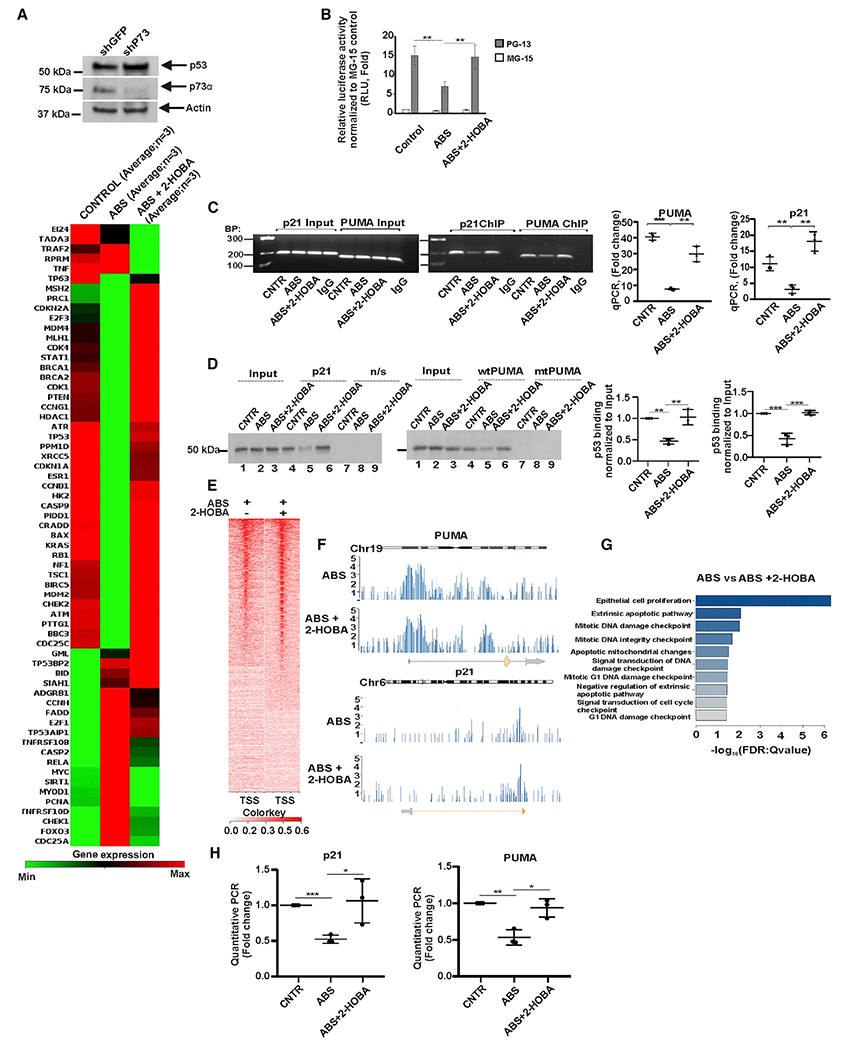
(A) Analysis of the p53 signaling pathway by PCR focus array in p73-deficient CP-A cells. The heatmap represents mRNA expression of 84 genes regulating the p53 signaling pathway (n = 3). Upper panel shows expression of p73 and p53 proteins.
(B) Analysis of p53 transcription activity in CP-A cells using the dual-luciferase reporter assay. PG13-Luc and control MG15-Luc p53 reporters were used. The endogenous p53 activity was significantly reduced by ABS treatment (control versus ABS, **p < 0.01) and 2-HOBA prevents effect of ABS (ABS versus 2-HOBA, **p < 0.01).
(C) ChIP analysis of p53 binding to the promoters of the CDKN1A(p21) (control versus ABS, **p < 0.01; ABS versus ABS+2-HOBA, **p < 0.01) and BBC3(PUMA) (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, **p < 0.01) genes in CP-A cells treated with ABS alone or in combination with 2-HOBA (n = 3; Student’s t test). The non-specific immunoglobulin (Ig)Gs were used as negative controls.
(D) DAI analyses of the p53 protein binding to the promoters of its target genes, CDKN1A(p21) (control versus ABS, **p < 0.01; ABS versus ABS+2-HOBA, **p < 0.01) and BBC3(PUMA) (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001; n = 3; Tukey’s multiple comparison; n/s, non-specific DNA probe). Binding of p53 protein in control cells was arbitrarily set at 1.
(E) Next generation sequencing (NGS) plot shows ChIP-seq binding profile in ABS and ABS+2-HOBA treated CP-A cells. TSS, transcription start site.
(F) ChIP-seq binding peaks in ABS- and ABS+2-HOBA-treated CP-A cells.
(G) The gene ontology analysis of biological processes using the aforementioned ChIP-seq data.
(H) Real-time qPCR analysis of p21 and PUMA mRNA expression in p73-deficient CP-A cells treated with ABS. Levels of p21 and PUMA mRNAs were decreased after ABS treatment, p21 mRNA (control versus ABS, ***p < 0.001) and PUMA mRNA (control versus ABS, **p < 0.01), whereas 2-HOBA prevented the effect of ABS treatment (n = 3; Student’s t test). Expression of p53 mRNA in untreated cells was arbitrarily set at 1. All results are expressed as mean ± SD. See also Figure S2A.
To further investigate regulation of p53 activity, we used PG13-Luc p53 luciferase reporter, which contains several repeats of the p53 binding site within its promoter.30 As a control, mutant p53 reporter MG15-Luc was used. Analyzing activity of endogenous p53 protein with the dual-luciferase reporter assay, we found that ABS significantly inhibits p53 activity, while 2-HOBA alleviates ABS-induced inhibition (Figure 2B).
To corroborate our findings, we analyzed binding of p53 protein to the promoters of p53 target genes, CDKN1A(p21) and BBC3(PUMA), using chromatin immunoprecipitation (ChIP). ChIP analysis revealed that ABS robustly inhibit binding of p53 protein to the p53 target gene promoters, while isoLG scavenger 2-HOBA counteracts this inhibitory effect and restore the promoter binding of p53 (Figure 2C). This inhibitory effect was further verified using DNA affinity immunoblotting.31 Cellular extracts collected from ABS-treated and control CPA cells were incubated with biotinylated DNA probes containing the p53 binding sites from the p21(CDKN1A) and PUMA(BBC3) genes.32 The corresponding probes with mutated p53 binding sites (n/s) were used as controls. Consistent with ChIP analysis, DAI demonstrated that the specific DNA binding of p53 protein is suppressed in ABS-treated cells (Figure 2D) and this inhibitory effect can be reversed by 2-HOBA treatment, which efficiently restores the promoter binding of p53 (Figure 2D; compare lanes 4, 5, and 6).
We also used ChIP-sequencing (ChIP-seq) to assess the genome-wide effect of isoLG adduction of p53. The next generation sequencing (NGS) plots show that 2-HOBA restores the p53 binding in CP-A cells treated with ABS including that of the p21(CDKN1A) and PUMA(BBC3) genes (Figures 2E and 2F). The recovery of DNA damage signaling pathways by 2-HOBA was uncovered using gene ontology analysis (Figure 2G). These data were also consistent with expression analysis of p21 and PUMA mRNA, which levels were measured by qPCR in p73-deficient cells in the same experimental conditions (Figure 2H). Collectively, our analyses demonstrate that isoLGs modulate DNA binding of p53 protein and its transcription activity.
IsoLGs form adducts with p53 protein
Given the highly reactive nature of isoLGs and their ability to form covalent protein adducts,18 we next explored whether isoLGs react with p53 protein in reflux conditions. Using the p53 immunocapture kit (Abcam), p53 protein was immunoprecipitated from CP-A and EPC2 cells treated with ABS alone or in combination with 2-HOBA. The immunoprecipitated p53 protein was then analyzed for isoLG adducts by western blotting with D11 ScFv antibody. The D11 ScFv is a single-chain fragment antibody that has been generated by screenings of phage-display libraries and tested to specifically recognize isoLG-lysyl protein adducts (products of reaction of isoLG with lysine residues) independently of surrounding protein amino acid sequences.17 The gel loading was normalized to the total amount of p53 protein detected with p53 (D01) antibody. We found that ABS treatment significantly increases levels of p53-isoLG protein adducts, whereas 2-HOBA significantly inhibits their formation (Figures 3A and 3B). Using the same approach, p53 adduction with isoLG was further confirmed by treating of CP-A cells with chemically synthesized isoLGs (0.5 μM), as described in the STAR Methods section. Similar to ABS, synthetic isoLGs was found forming p53-isoLG adducts (Figure 3C).
Figure 3. Acidic bile salts increase the formation of isoLG-p53 adducts.
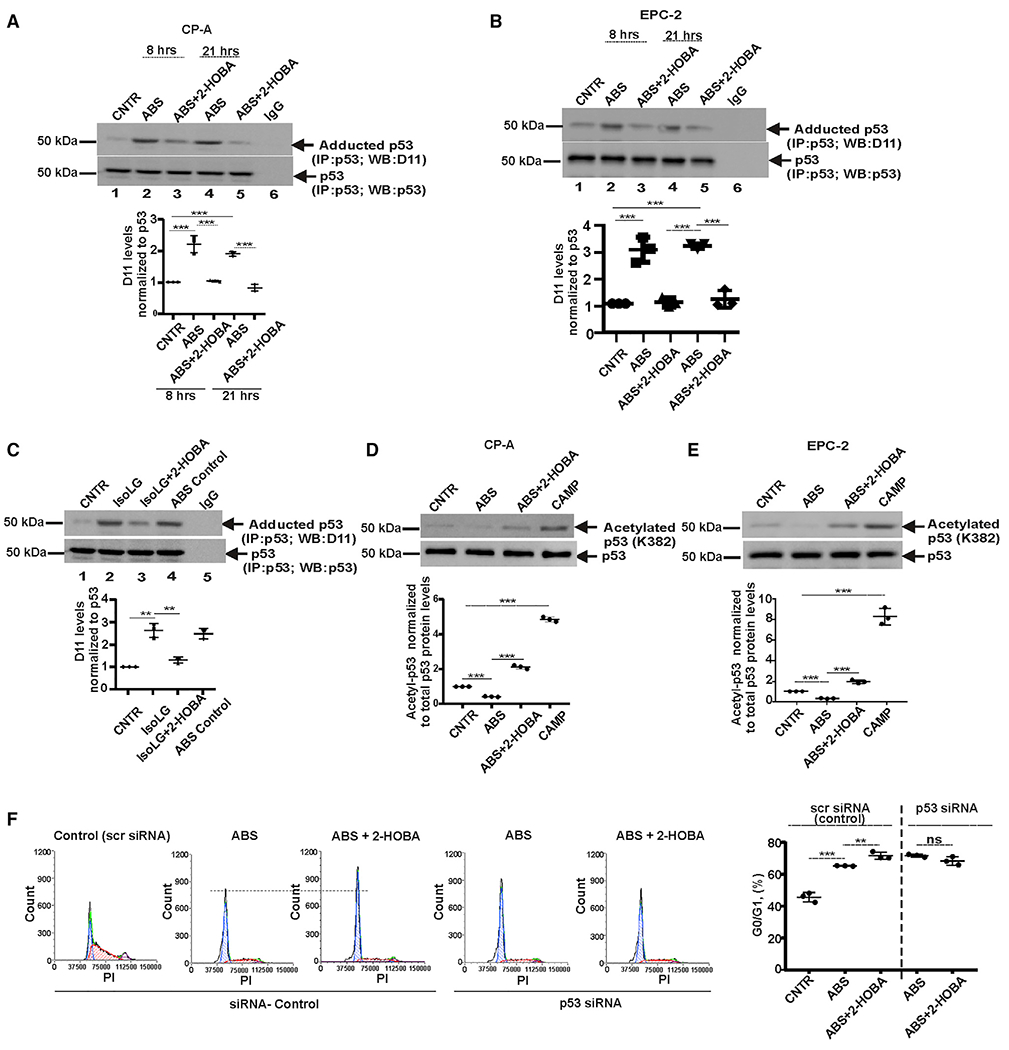
(A) p53 protein was immunoprecipitated from CP-A cells and analyzed for adduction of p53 protein with D11 scFv antibody by western blotting. ABS treatment increases levels of isoLG-p53 protein adducts, while 2-HOBA counteracts this effect by preventing the isoLG adduction of p53 protein (n = 3; Tukey’s multiple comparison) at 8 h (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001) and 21 h (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001). Levels of isoLG: p53 adducts was normalized to total levels of p53 protein, which were analyzed with p53(D01) antibody. Levels of p53 protein adduction in control cells was arbitrarily set at 1.
(B) The same as (A) but the p53 protein adduction was analyzed in EPC-2 cells at 8 h (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001) and 21 h time points (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001) after treatment.
(C) CP-A cells were treated with 0.5 μM synthetic isoLGs and analyzed for the adduction of p53 protein (control versus isoLG, **p < 0.01; isoLG versus isoLG+2-HOBA, **p < 0.01; n = 3; Tukey’s multiple comparison).
(D) Analyses of p53 protein acetylation in CP-A cells by western blotting with antibody recognizing acetylated p53 at Lys382 (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001; n = 3; Tukey’s multiple comparison).
(E) The same as (D) but acetylation of p53 protein was analyzed in EPC-2 cells (control versus ABS, ***p < 0.001; ABS versus ABS+2-HOBA, ***p < 0.001; n = 3; Tukey’s multiple comparison).
(F) Cell-cycle analysis in CP-A cells transfected with either p53 siRNA or scrambled siRNA and treated with ABS alone or in combination with 2-HOBA. Cell cycle was analyzed by flow cytometry and compared between groups (scr siRNA: ABS versus ABS+2-HOBA, **p < 0.01; p53 siRNA: ABS versus ABS+2-HOBA; NS, not significant; n = 3; Tukey’s multiple comparison). All results are expressed as mean ± SD.
These findings led us to another hypothesis. Since isoLGs react with lysine residues,18 the formation of p53-isoLG adducts may affect the lysine acetylation of p53 protein. This question is particularly important because p53 acetylation at lysine residues is indispensable for activation of p53.33 To test this hypothesis, acetylation of p53 protein was analyzed in CPA and EPC2 cells by western blotting using the specific antibody recognizing acetylated p53 at Lys382. Our studies found that despite DNA damage induced by ABS, p53 acetylation is not significantly increased compared with control untreated cells. This was contrasted with camptothecin (CAMP), which strongly increased acetylation of p53 protein. 2-HOBA was found to reverse inhibitory effects of ABS and increases p53 acetylation in conditions of ABS-induced DMA damage, suggesting that isoLG adduction may affect acetylation of p53 protein (Figures 3D and 3E).
Next, we assessed whether isoLG adduction affects p53 biological activity. Since p53 is an important regulator of the cell cycle that can induce G1/G0 cell-cycle arrest,34 we performed the cell-cycle analysis in CP-A treated with ABS. To assess the specific contribution of p53, this analysis was conducted in control and p53-deficient CP-A cells, transfected with either scrambled siRNA (scr siRNA) or p53 siRNA, respectively (Figure 3F). Consistent with data shown above, we found that 2-HOBA leads to significant increase in G1/G0 cell-cycle arrest in p53-expressing cells, whereas it does not occur in p53-deficient cells, suggesting that 2-HOBA activates p53. Interestingly, when we repeat these experiments in cells treated with camptothecin, 2-HOBA did not significantly increase p53 activity (Figure S3B), plausibly reflecting differences in the levels of isoLG adduction in ABS- and CAMP-treated cells.
Taken together, we found that exposure of esophageal cells to ABS or chemically synthesized isoLGs leads to posttranslational modification of p53 protein with isoLGs and its inhibition.
isoLGs affect subcellular localization of p53 protein
Accumulation of p53 protein in the nucleus is critically important for its transcription function in conditions of cellular stress.35 To analyze how isoLGs affect the p53 subcellular localization, we investigated p53 protein using immunofluorescence (IF) with p53 (D01) antibody. We found that following treatment with ABS, p53 is localized to large intracellular aggregates that are formed in the cytoplasm and nucleus of esophageal cells. Control untreated cells showed diffuse p53 staining, primarily in the nucleus and no visible aggregation. Similar to control cells, no visible p53 aggregates were observed in cells treated with ABS in combination with 2-HOBA (Figure 4A).
Figure 4. p53 adduction leads to p53 protein aggregation.
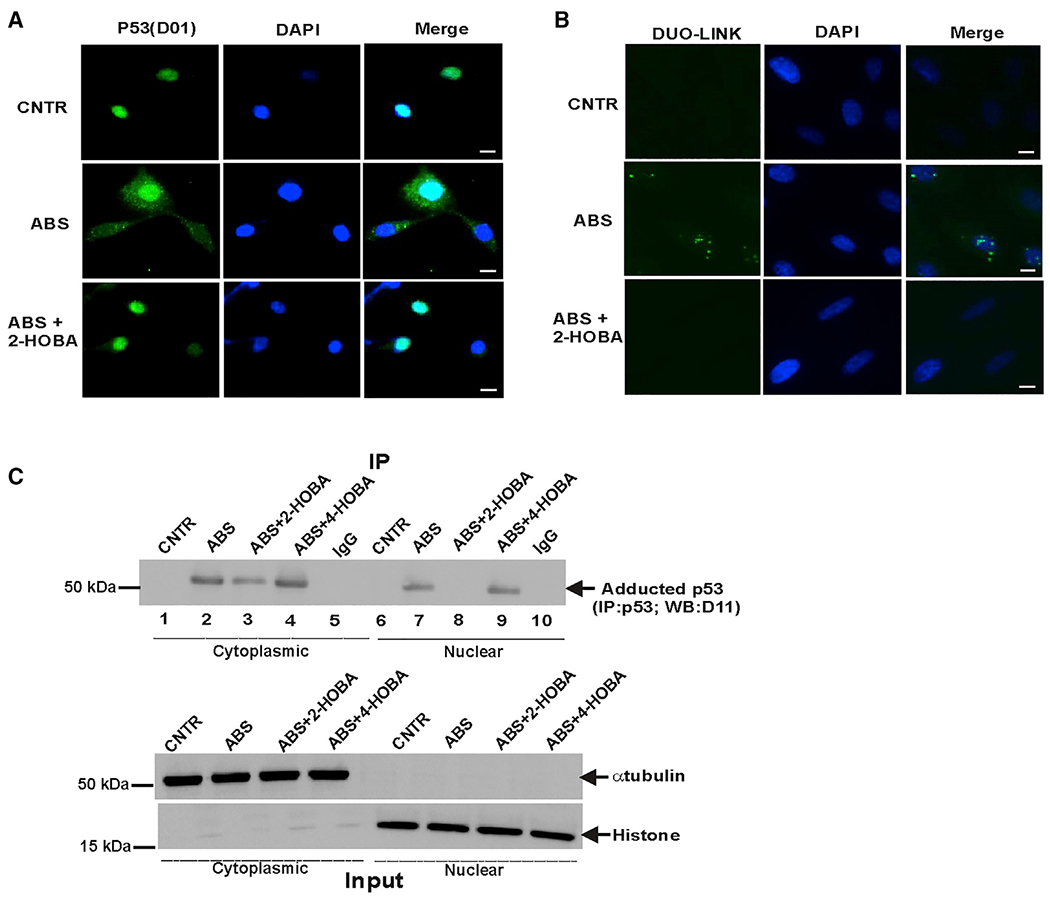
(A) Representative images of p53 protein aggregates in CP-A cells (scale bar, 5 μm). Immunofluorescence staining was performed with p53(D0-1) antibody in cells treated with ABS alone or in combination with 2-HOBA. 2-HOBA prevented aggregation of p53 protein (n = 3).
(B) Representative images of Duolink PLA in CP-A cells treated with ABS. PLA was performed using D0-1 and D11: E-tag primary antibodies (scale bar, 5 μm). The isoLG-p53 adducts were detected in large cellular aggregates in cells treated with ABS. The adduct-specific PLA signals were not observed in cells treated with 2-HOBA (n = 3).
(C) p53 protein adduction was analyzed in the cytoplasmic and nuclear fractions of CP-A cells after ABS treatment for 8 h. Equal amount of total protein (10 μg) was loaded in each well. Bottom panel shows experimental inputs.
To further analyze the subcellular distribution of adducted p53 protein, we performed subcellular fractionation of CP-A cells using the Subcellular Protein Fractionation Kit (ThermoFisher Scientific) and collected cytoplasmic and nuclear fractions after ABS treatment. In each fraction, p53 was immunoprecipitated and analyzed for isoLG-adduction with isoLGs using D11 antibody as discussed above. Our data confirmed that ABS treatment causes adduction of p53 protein in the nuclear and cytoplasmic fractions, suggesting that p53 is inhibited in both compartments (Figure 4C).
The subcellular localization of p53 protein was also analyzed using the Duolink proximity ligation assay (PLA), which has been used for analyses of protein interactions in situ.36,37 This approach is based on detection of a fluorescence signal from two interacting proteins when they are positioned close together. In this study, we used two primary antibodies: p53(D01) and D11 ScFv, which recognize p53 protein and isoLG-lysyl adducts, respectively. p53 protein adducts were found to be localized to granular aggregates in the nucleus and cytoplasm (Figure 4B). No detectable isoLG and p53 positive aggregates were found in control and ABS+2-HOBA-treated cells (Figure 4B). Additional PLA controls are shown in Figure S4.
ABS and isoLGs affect p53 molecular conformation
Due to hydrophobic nature of isoLGs and their ability to react with proteins, they may alter structural and functional properties of adducted proteins.38 Therefore, we next investigated whether isoLGs affect structural characteristics of the p53 molecules. We took advantage of the existing conformation-specific p53 antibody PAb 240 (Abcam), which recognizes an evolutionary conserved epitope (aa 211–217) of the p53 protein, and used as a marker for misfolded p53 protein.39 This epitope is normally hidden within the protein structure, but exposed after changes in the tertiary structure of the p53 molecule. Lysates generated from CP-A and EPC2 cells, which were treated with ABS alone or in combination with 2-HOBA, were spotted onto nitrocellulose membrane, washed, and incubated with two p53 antibodies: PAb 240, which recognizes the hidden p53 epitope, and D01, which recognizes a surface-exposed epitope at the N-terminus of the p53 protein.39 We found that while D01 antibody detects p53 protein in ABS-treated and control untreated cells, the conformation-specific PAb 240 antibody only recognizes p53 in ABS-treated cells (Figures 5A–5C). Notably, 2-HOBA was found to prevent conformational changes in the p53 molecule in CP-A and EPC2 cells, suggesting that ABS induces conformational changes exposing the intramolecular PAb 240 epitope of p53 (Figures 5A–5C). The same conclusion can be drawn from western blot analyses of p53 in non-denaturing conditions. When p53 protein was analyzed on native blue gels (gradient 4%–12%),40 we found that p53 protein forms high molecular weight aggregates, which are recognized by PAb 240 (Figure 5D) or D01 antibodies (Figure S3C). Their molecular weights were found to be higher than p53 monomers, dimers, or tetramers present in untreated cells. High molecular weight p53 protein aggregates were also found in the CP-A cells treated with ABS and isoLGs and analyzed using p53(D01) antibody and SDS-PAGE (Figures S5A and S5B). No protein aggregates were found in cells treated with ABS in combination with 2-HOBA.
Figure 5. ABS treatment causes conformation change of the p53 protein molecule.
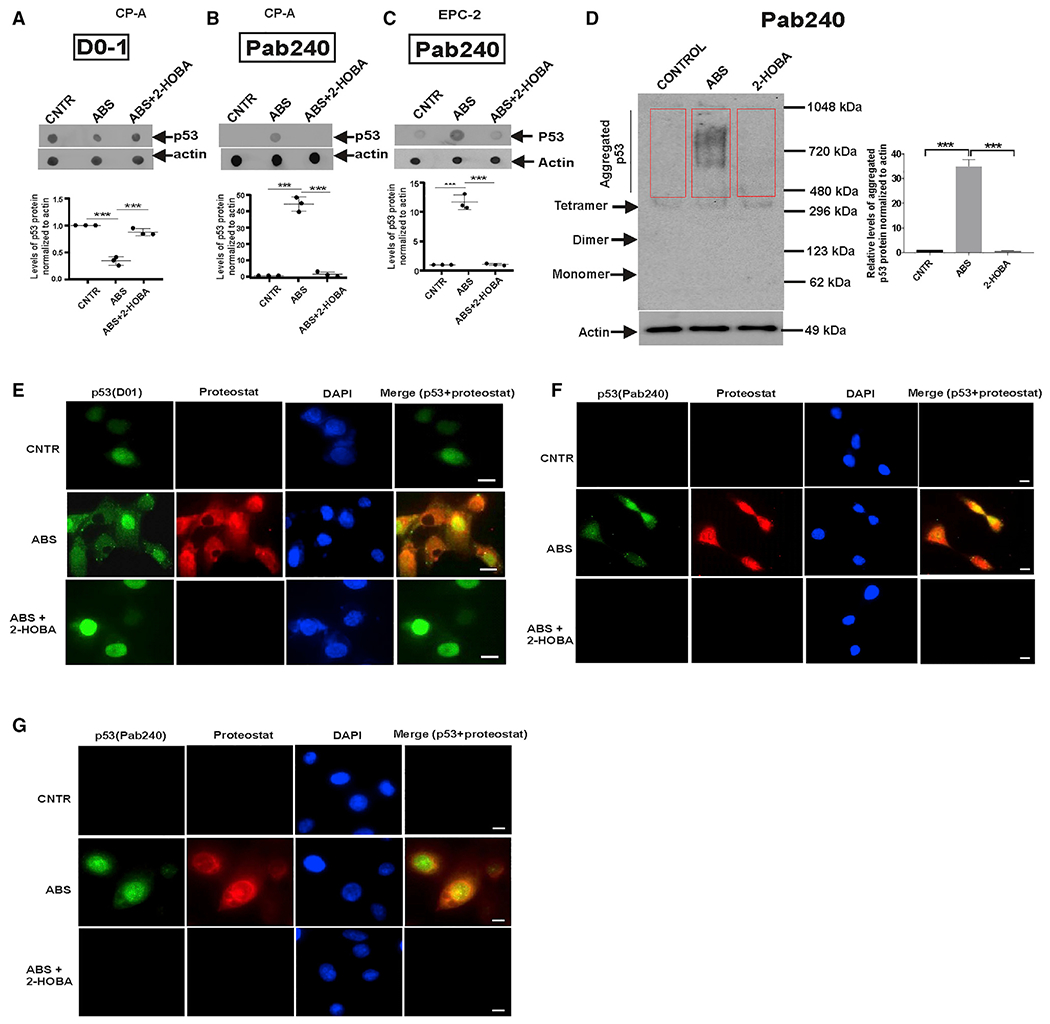
(A) Dot blot analysis of CP-A cell extracts using D01 antibody.
(B) The same as (A) but p53 (PAb 240) antibody was used. Staining was higher in cells treated with ABS than in control or 2-HOBA-treated cells (***p < 0.001; n = 3; Tukey’s multiple comparison).
(C) The same as (B) but EPC-2 cells were studied (ABS versus ABS+2-HOBA, ***p < 0.001; n = 3; Tukey’s multiple comparison).
(D) Native blue PAGE demonstrates misfolding and aggregation of p53 protein in CP-A cells collected after treatment with ABS. The graph shows the densitometric measurement of p53 aggregation detected by p53 (PAb 240) antibody (control versus ABS, ***p < 0.001; ABS versus 2-HOBA, ***p < 0.001; n = 3; Tukey’s multiple comparison). Aggregated p53 proteins are indicated by a red box. Protein expression in control cells was arbitrarily set at 1.
(E) Representative images show co-localization of p53 protein with ProteoStat-positive aggregates (scale bar, 5 μm). p53 was analyzed with p53(D0-1) and ProteoStat dye was used to identify protein aggregates in CP-A cells treated with ABS alone or in combination with 2-HOBA (n = 3).
(F) The same as (E) but PAb 240 antibody was used for detection of misfolded p53 protein (scale bar, 5 μm).
(G) The same as (F) but the p53 misfolding was analyzed in EPC-2 cells. All results are expressed as mean ± SD.
Protein aggregation may affect physicochemical properties of the p53 molecules. Indeed, our previous studies have shown that ABS treatment significantly reduces solubility of p53 protein.41 As a result, the p53 protein was found to be accumulated in the insoluble cellular fraction that was generated by centrifugation of total cell lysates at 16,000 × g for 20 min. The scheme illustrating the collection of soluble and insoluble cellular fractions is shown Figure S1C. 2-HOBA decreased accumulation of p53 protein in the insoluble fraction, suggesting that this process is mediated by isoLGs (Figure S1D). Interestingly, inhibition of ROS or COX2, which are responsible for isoLG generation, with N-Acetyl L-Cysteine (NAC), Tempol, and NS-398 (10 μM) were also effective in maintaining p53 protein solubility (Figures S3D and S3E). However, 2-HOBA treatment was more potent than that of other inhibitors, plausibly because 2-HOBA directly scavenges isoLGs, while others drugs inhibit upstream regulators of isoLGs ROS and COX2.
Reflux causes the formation of amyloid-like aggregates
To further investigate the formation of p53 aggregates, we used the ProteoStat molecular-rotor dye that undergoes a fluorescent enhancement upon binding to aggregates.42 We reproducibly observed a strong ProteoStat signal in esophageal cells treated with ABS (Figure 5E) that is co-localized with p53 protein detected with p53(D01) and p53(PAb 240) immunostainings in CP-A cells (Figures 5E and 5F). The same co-localization was also observed in EPC-2 cells (Figure 5G). 2-HOBA efficiently suppressed protein aggregation and PAb 240 immunostaining induced by reflux (Figures 5E–5G).
To characterize the type of aggregates produced by isoLG adduction, we used amyloid fibrils OC antibody (Millipore), which recognizes generic epitopes common to many amyloid fibrils and oligomers.43 An increased formation of amyloid-like aggregates in ABS-treated cells was detected with dot blot technique (Figures 6A and 6B) and immunofluorescence with PAb 240 antibody (Figure 6C), revealing that p53 protein is co-localized with amyloid-like aggregates. Inhibition of isoLGs with 2-HOBA prevented the formation of amyloid-like aggregates positive for p53 (Figure 6C).
Figure 6. IsoLGs form amyloid-like p53 aggregates.
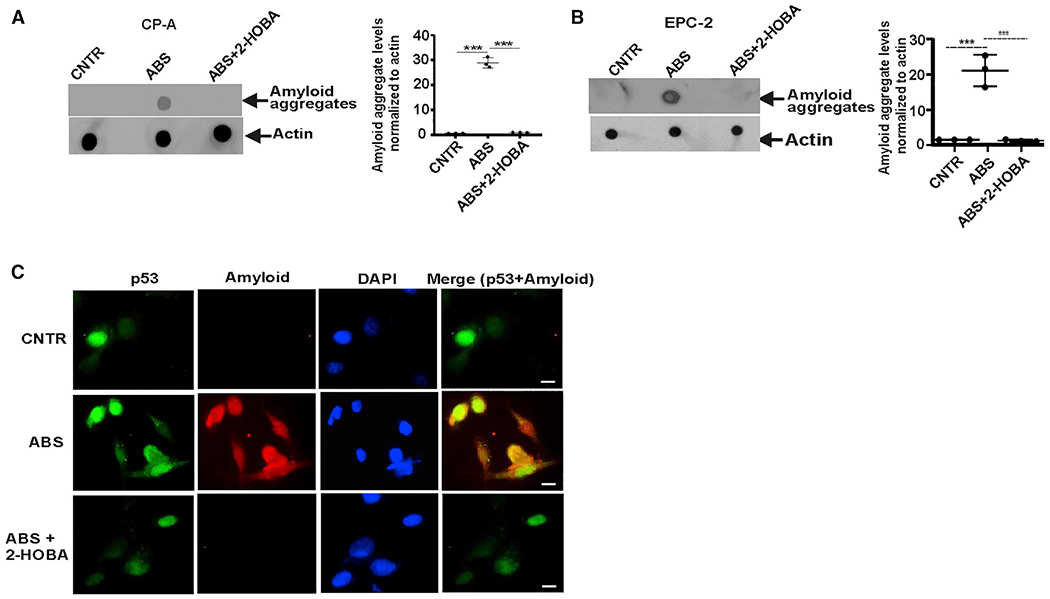
(A) Dot blot analysis of CP-A cell extracts using anti-amyloid OC antibody. Protein aggregation levels were found to be higher in CP-A cells treated with ABS than in control untreated cells (***p < 0.001; n = 3; Tukey’s multiple comparison). 2-HOBA prevented amyloid aggregation (***p < 0.001; n = 3; Tukey’s multiple comparison).
(B) The same as (A) but the formation of amyloid aggregates was analyzed in EPC-2 cells.
(C) Representative images of co-localization of p53 protein with amyloid fibrils detected with anti-amyloid OC antibody in CP-A cells (n = 3) (scale bar, 5 μm). All results are expressed as mean ± SD.
To investigate aggregation of p53 protein in vivo, we used esophageal specimens collected from mice in which reflux was induced by esophagojejunostomy. The experimental design is shown in Figure 7A. Using OC antibody, protein aggregates were revealed in esophageal epithelium of reflux mice, but not sham control animals and were co-localized with misfolded p53 protein, which was detected with p53(PAb 240) antibody in nucleus and cytoplasm in the same tissues (Figure 7B). Treatment of animals with 2-HOBA in drinking water (8 mM) for 10 days hindered accumulation of protein aggregates and misfolded p53 protein (Figures 7B, 7C, and S5C).
Figure 7. IsoLGs induce misfolding of p53 protein in vivo.
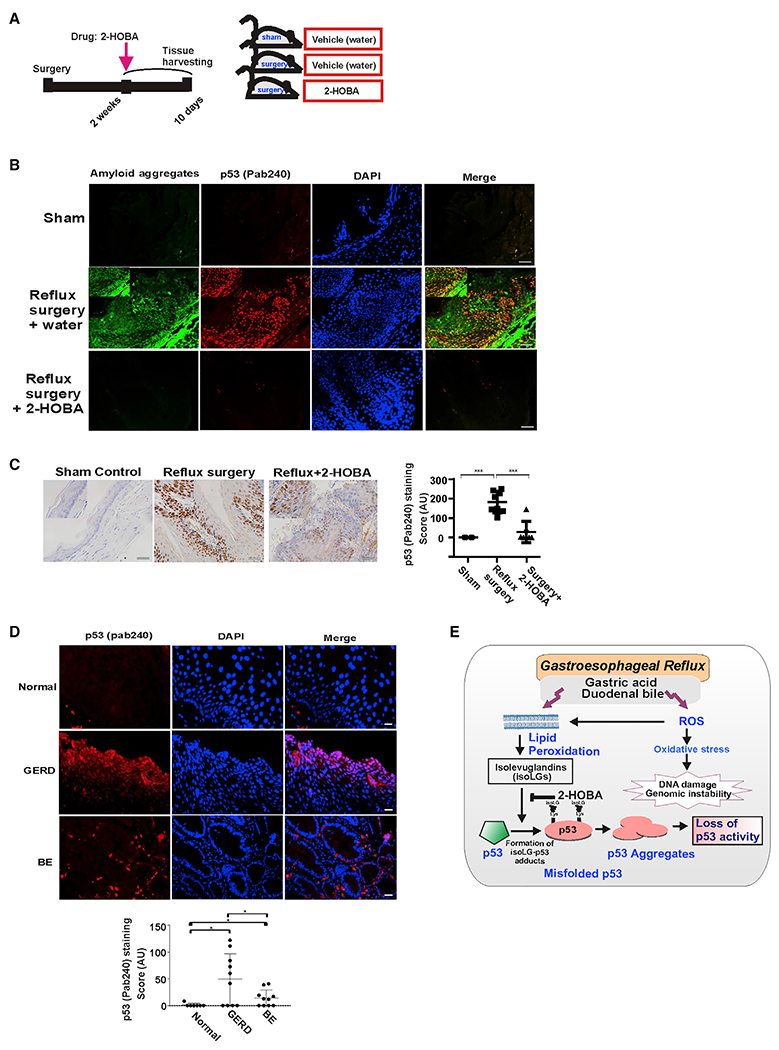
(A) The schematic representation of animal experiment.
(B) Representative images of p53 protein co-localization with amyloid fibrils detected using amyloid OC antibody in murine esophageal tissues. Mice received 2-HOBA dissolved in water (1 g/L) 2 weeks after surgery. Control mice received vehicle (water). Esophageal tissues were collected after drug treatments for 10 days and analyzed for misfolding of p53 protein. Co-localization of p53 and amyloid staining was observed in mice with reflux surgery. The staining was not found in mice treated with 2-HOBA. Inset: high magnification image. Scale bar, 10 μm.
(C) Representative immunohistochemical staining of murine esophageal tissues with p53 (PAb 240) antibody. Insets show high magnification images. Graph shows the IHC scores (sham control versus reflux mice, ***p < 0.001; reflux mice versus reflux mice treated with 2-HOBA, ***p < 0.001; Tukey’s multiple comparison). Mice with sham surgery were used as a control. Scale bar, 10 μm.
(D) Representative images of p53 protein misfolding in the human esophagus. The human esophageal tissues from healthy subjects, GERD and BE patients were analyzed using p53 (PAb 240) antibody. Misfolded p53 protein was observed in the esophagus of GERD and BE patients, but not in normal (no GERD) esophageal epithelium (scale bar, 5 μm). Graph shows the immunofluorescence scores (normal versus GERD, *p < 0.01; normal versus BE, *p < 0.01).
(E) Graphical representation of p53 protein regulation by isoLGs. All results are expressed as mean ± SD.
To analyze whether p53 misfolding occurs in human tissues, we analyzed esophageal tissues of healthy individuals and patients with GERD and BE. Esophageal tissues collected from human patients with EAC served as a positive control (Figure S5D). Using immunofluorescence with p53(PAb 240) antibody, we found that misfolded p53 protein was present in 6 of 10 (60%) GERD and BE patients. However, intensity of p53 staining was significantly lower in esophageal tissues collected from BE patients. Specimens collected from healthy subjects (n = 7) did not show significant positive staining (Figure 7D). Taken together, our studies revealed that reflux components promote p53 misfolding in vitro and in vivo in the murine and human esophagi.
DISCUSSION
Our studies provide evidence that p53 protein is modified with reactive isolevuglandins. Adduction of p53 protein with isoLGs interferes with the p53 binding to the promoters of its target genes and modulates p53-dependent transcription and biological activity. Our studies also revealed that isoLGs facilitate the formation of p53 protein aggregates that produce large intracellular granular structures with amyloid-like properties. Another interesting finding is that 2-HOBA, which is an isoLG scavenger but not an antioxidant,44 is able to inhibit aggregation of p53 protein and restore its activity.
It has been previously reported that p53 protein is misfolded in several types of tumors forming amyloid aggregates.45–47 p53 mutations or other factors were found to destabilize the native protein structure allowing p53 to aggregate. In our experiments, we used non-transformed cell lines, which express functional wild-type p53. These studies demonstrate that misfolding of p53 and its aggregation can occur without mutations and is driven by isoLG protein adduction. We detected misfolded p53 in esophageal cells in vitro, reflux animals, and human GERD and BE patients showing that p53 is inhibited in cancerogenic conditions associated with esophageal reflux.
Using D11 ScFv antibody, which recognizes isoLG-lysyl adducts essentially independent of adjacent amino acid residues,48 we showed that lysine residues on the p53 molecule are adducted with isoLGs. It is plausible that posttranslational modification of lysine residues with isoLGs may interfere with their proper acetylation, which is required for full activation of p53. Consistent with this notion, our data show that ABS inhibits acetylation of p53 at lysine residues even in conditions of DNA damage induced by ABS and this process is prevented by isoLG scavenger 2-HOBA. These findings are also consistent with previous reports showing inhibition of p53 by bile salts.23,24
Interestingly, despite significant structural and functional similarities between p53 and p73 proteins, they show different sensitivity to isoLG adduction. While p73 is known to be induced and activated by ABS,26 p53 is adducted and inhibited. Further studies are needed to investigate the nature of these differences.
In our study, we investigated reflux, as a model for isoLG protein adduction. It was previously shown that acid and bile salts induce ROS, lipid peroxidation, oxidative stress, and activation of the COX2 pathway. These factors are plausibly responsible for production of isoLGs in GERD conditions. Reflux has also a genotoxic/mutagenic effect.10,49–53 Although the entire spectrum of reflux-induced DNA lesions is currently unknown, it has been shown that the exposure of esophageal cells to ABSs promotes the formation of single-, double-strand breaks, oxidized and nitrated DNA, resulting in highly cytotoxic and mutagenic effects.2 Our current studies show that reflux not only induces DNA damage but also inhibits/modulates the p53 response required for an appropriate resolution of genotoxic stresses. This occurs due to adduction of p53 protein with isoLGs that can be prevented with 2-HOBA. Yet, we cannot exclude that besides p53, which is the main of focus these studies, adduction of other proteins may contribute to inhibition of the DDR. We also cannot exclude that other reactive aldehydes may contribute to p53 inhibition.54,55 Further studies are needed to elucidate their relative contribution.
p53 mutations are rarely found in GERD and BE patients, implying that non-mutational inactivation of p53 by isoLGs occurs during early stages of neoplastic transformation that may lead to accumulation of tumorigenic alterations caused by reflux in the esophagus.56
Our data also suggest that p53 adduction may not limited to the esophagus. p53 may be adducted in other cancer types or other diseases that are characterized by free radical-induced peroxidation of lipids and activation of the COX2 pathway.19,20 Indeed, one such example is Helicobacter pylori infection that is considered to be one of the strongest risk factors for gastric cancer. It has been recently reported that gastric infection with H. pylori is characterized by strong induction of ROS and isolevuglandins in the stomach.57 In our studies, we investigated p53 protein adduction in gastric epithelial cells (GES-1 and SNU-1 cell lines) co-cultured with H. pylori tumorigenic strain 7.13.58 We found that H. pylori infection results in adduction of p53 protein with isoLGs that can be inhibited with isoLG scavenger 2-HOBA (Figure S3A). Further studies are needed to elucidate this mechanism.
In summary, this study demonstrates inactivation and aggregation of p53 protein by isoLG adduction that may potentially lead to carcinogenic alterations. This process can be prevented by isoLG scavenger, 2-HOBA, that efficiently inhibits the formation of isoLG protein adducts and restores p53 (Figure 7E).
Limitations of the study
We demonstrated the mechanism of inhibition of p53 mediated by isoLG adduction. However, we cannot exclude contribution of other reactive aldehydes, which may also inhibit p53. Another limitation of our studies is a relatively small number of analyzed human specimens that need to be increased in the future.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Alexander Zaika (axz353@med.miami.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data supporting the findings of this study are available within the main manuscript and the supplementary files provided. The ChIP-seq data is available on NCBI Sequence Read Archive (SRA) with the accession details provided in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-p53 (D0-1) | Millipore | Cat# OP43 |
| Rabbit anti-acetyl-p53 (K382) | Cell Signaling Technology | Cat#2525 |
| Rabbit anti-histone H2AX | Cell Signaling Technology | Cat#2595 |
| Rabbit anti-phospho-histone H2AX (Ser139) | Cell Signaling Technology | Cat#9718 |
| Rabbit anti-p53 antibody | Bethyl Laboratories Inc, | Cat#A300-247A |
| Mouse anti-p53 (pab240) | Abcam | Cat# AB26 |
| Mouse anti-p53 (pab240) | Novus Biologicals | Cat# NB200-103 |
| Rabbit anti-p73 | Bethyl laboratories | Cat# A300-126A |
| Rabbit anti-amyloid OC fibrils | Millipore | Cat#AB2286 |
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A5441 |
| Goat anti-mouse IgG HRP | Promega | Cat# PAW4021 |
| D-11 scFv antibody | A gift from Dr. Raymond Mernaugh, Vanderbilt University, USA | N/A |
| Rabbit anti- E:tag antibody | Abcam | Cat# AB3415 |
| Bacterial and virus strains | ||
| H.pylori 7.13 | A gift from Dr. Richard Peek, Vanderbilt University Medical Center, USA | N/A |
| Biological samples | ||
| Human Tissue | Department of Pathology, University of Miami, USA | N/A |
| Mouse samples | University of Miami, USA | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PI/RNase staining buffer | BD Biosciences | Cat# 550825 |
| Coomassie Brilliant Blue R-250 | Biorad | Cat# 1610400 |
| NAC (N-acetylcysteine) | Sigma-Aldrich | Cat# A0737 |
| TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperydine-1-oxyl) | Enzo Lifesciences | Cat# ALX-430-081-G001 |
| NS-398 | Cayman Chemical | Cat#70590 |
| 2-HOBA | Cayman Chemical | Cat#25357 |
| Critical commercial assays | ||
| p53 human immunocapture kit | Abcam | Cat# AB154470 |
| RNeasy Mini Kit | QIAGEN Inc | Cat#74134 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| RT2 Profiler PCR Array, Human p53 signaling pathway | QIAGEN Inc | Cat# 330231 |
| Magna ChIP A/G kit | Millipore | Cat# 17-10085 |
| Subcellular Protein Fractionation Kit | ThermoFisher Scientific | Cat#78840 |
| Proteostat Protein Aggregation Assay Kit | Enzo Lifesciences | Cat#ENZ-51023-KP050 |
| Dual Luciferase Reporter Assay Kit | Promega | E1910 |
| Deposited data | ||
| ChIP-Seq data | This manuscript | NCBI SRA (Sequence Read Archive); PRJNA912777 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA912777?reviewer=6cnca4108s5od672ijo7001e36) |
| Experimental models: Cell lines | ||
| BAR-T (human Barrett’s epithelial cells) | A kind gift from Dr. Souza, Baylor University Medical Center, USA | N/A |
| CP-A (human Barrett’s epithelial cells) | ATCC (American Type Culture Collection) | Cat#CRL-4027 |
| EPC-2 (human normal epithelial cells) | A gift from Dr. Claudia Andl, University of Central Florida, USA | N/A |
| GES-1 (human normal gastric epithelial cells) | A kind gift from Dr. El-rifai, University of Miami USA | N/A |
| SNU-1 (human gastric epithelial cancer cells) | ATCC (American Type Culture Collection) | Cat# CRL-5971 |
| Experimental models: Organisms/strains | ||
| 129/sv mice | University of Miami | N/A |
| Oligonucleotides | ||
| HPRT Forward: 5′-TTGGAAAGGG TGTTTATTCCTCA-3′ Reverse: 5′- TCCAGCAGGTCAGCAAAGAA-3′ |
Integrated DNA Technologies (IDT) | N/A |
| p21 Forward: 5′-CTGGAGACTC TCAGGGTCGAAA-3′ Reverse: 5′-GA TTAGGGCTTCCTCTTGGAGAA-3′ |
Millipore Sigma | N/A |
| PUMA Forward: 5′-ACGACCTCAA CGCACAGTACG-3′ Reverse: 5′-TCC CATGATGAGATTGTACAGGAC-3′ |
Integrated DNA Technologies (IDT) | N/A |
| 5′-Biotinylated p21; 5-biotin-AG CCTCCCTCCATCCCTAT-3 and 5′-CCCTTCCTCACCTGAAAACA-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| 5′-Biotinylated PUMA; 5′-biotin-CCC AGTCAGTGTGTGTGTCC-3′ and 5′-CCCCCGCGTGACGCTAC-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| Non-specific probe,n/s, 5′-biotin-TA GCTGGGAAGCTGGGACTA-3′ and 5′-GGTTTCCTTGCCCTAAAAGG-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| ChIP qPCR; p21 (5′-ACCTTTCA CCATTCCCCTAC-3′, 5′-GCCC AAGGACAAAATAGCCA-3′ |
Vilgelm et al., 2010; Millipore Sigma | N/A |
| ChIP qPCR; PUMA (5′-TGTCCA TGGTGTGGATTTGCG-3′, 5′-AGAC ACCGGGACAGTCGGACA-3′ |
Vilgelm et al., 2010; Millipore Sigma | N/A |
| Human p53 siRNA | Santa Cruz Biotechnology | Cat#sc-29435 |
| Software and algorithms | ||
| GeneGlobe Data Analysis Center provided by Qiagen Inc. | QIAGEN Inc. | https://geneglobe.qiagen.com/us/analyze |
| Open Comet software | Open Comet | https://cometbio.org/ |
| FCS express 7 | De Novo software | https://denovosoftware.com/ |
| GraphPad Prism 8.0.0 | GraphPad Software | https://www.graphpad.com/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human samples
The human tissues were obtained from the de-identified tissue blocks from the Department of Pathology, University of Miami after getting clearance from the University of Miami ethics approval committee (No: 20210591).
Murine samples
Esophagojejunostomy was performed on 6-week-old 129/SV mice according to the protocol approved by the University of Miami Animal Care and Use Committee26. Both male and female mice were used in this study.
METHOD DETAILS
Cell lines
BAR-T and CP-A cells were isolated from human Barrett’s esophageal epithelium and EPC-2 cells were derived from human normal esophagus. CP-A cell line was purchased from ATCC (Manassas, VA), Dr. Souza (Baylor University Medical Center, Dallas) kindly gifted BAR-T (h-TERT immortalized human Barrett’s esophageal cells) and EPC-2 cells were provided by Dr. Claudia Andl (University of Central Florida, FL). Morphology, karyotyping and PCR-based approaches were used to confirm the identity of cell lines. Both cell lines were cultured in keratinocyte media (SFM) supplemented with 1.0 ng/ml epidermal growth factor, 40 μg/mL bovine pituitary extract (Life Technologies, Carlsbad, CA), and 5% fetal bovine serum.
Human immortalized gastric epithelial cell line, GES-1 was a gift from Dr. El-Rifai (University of Miami), and human gastric epithelial cancer cell line SNU1 was purchased from ATCC (Manassas, VA). Cells were grown in RPMI (GES1) media or Ham’s F12 (SNU1), supplemented with 10% Fetal Bovine Serum (Thermo Fisher Scientific, Waltham, MA) at 37°C in a humidified atmosphere containing 5% of CO2.
Treatment with acidic bile salt cocktail (ABS), IsoLGs and 2-HOBA. Co-culture with H. pylori
ABS was prepared with 20 μM equimolar mixture of glycodeoxycholic, glycochenodeoxycholic, glycocholic, taurocholic, and deoxycholic sodium salts (Sigma-Aldrich, St. Louis, MO). The total bile salt concentration was 100 μM. The ABS cocktail was diluted in Dulbecco’s modified Eagle’s media (Life Technologies, Carlsbad, CA) and pH was adjusted to 4.0.
ABS treatment was done as follows: CP-A, BAR-T and EPC-2 cells were treated with ABS for 10 min. After incubation with ABS, acidic medium was removed by washing with fresh media and cells were incubated in keratinocyte SFM media for an additional 8 hours.
IsoLG treatment was done as follows: CP-A and EPC-2 cells were incubated with keratinocyte SFM media containing chemically synthesized isoLGs (provided by Dr. Boutaud, Vanderbilt University, TN) at a concentration of 0.5 μM for 1 hour. The cells were collected for analysis immediately after treatment. The IsoLG scavenger 2-HOBA and COX-2 inhibitor NS-398 were from Cayman Chemical Company (Ann Arbor, MI). NAC (N-acetylcysteine) and TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperydine-1-oxyl) were purchased from Sigma-Aldrich, St. Louis, MO, and Enzo Biochem, Farmingdale, NY, respectively.
Cells were pre-treated with 2-HOBA, 4-HOBA, NAC, TEMPOL, NS-398 for 1 hour at a final concentration of 20 μM. Drugs were removed by washing with fresh media, and treated with ABS or isoLGs as discussed above. After treatment with ABS, the culture media were reconstituted with 2-HOBA.
p53 protein was immunoprecipitated using the p53 human immunocapture kit (Abcam, Cambridge, MA; cat# AB154470) and analyzed by Western blotting with D11 scFv antibody. The analyzed membranes were stripped with the Restore™ Western Blot Stripping Buffer (ThermoFisher Scientific, Waltham, MA) for 1 hour at 50°C, blocked with 5% nonfat dried milk, and re-blotted with p53 (D0-1) antibody (Millipore, Burlington, MA; cat# OP43).
Gastric epithelial cells were co-culture with H. pylori strain 7.13 for 24 hours and analyzed for p53 protein adduction.59,60
Comet assay
To determine levels of DNA damage, comet assay was performed in alkaline conditions. Briefly, 3000–5000 cells were mixed with LM Agarose (Trevigen, Gaithersburg, MD) and allowed to solidify on flare comet slides (Trevigen, Gaithersburg, MD) at 4°C. Then, the slides were incubated in lysis buffer for 2 hours at 4°C. Electrophoresis was performed on slides in alkaline conditions. After washing, the slides were fixed with 100% cold ethanol, stained with ethidium bromide, and visualized with fluorescence microscope (Olympus, Pittsburg, PA). A minimum of 50–70 cells were analyzed using the Open Comet software.
RNA extraction, Quantitative-PCR and focus array
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN Inc., Germantown, MD, USA) according to the manufacturer’s protocol. The reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit as described by the manufacturer (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using the iCycler (Bio-Rad, Hercules, CA).30 Each sample was assayed in triplicate and data were normalized to the housekeeping gene HPRT (hypoxanthine guanine phosphoribosyl transferase). The conditions for thermal cycling included an initial heat-denaturing step at 95°C for 3 min, followed by 30 cycles at 95°C for 10 s and 60°C for 30 s.
The mRNA expression profiles of the p53 signaling pathway genes were analyzed in p73 deficient CP-A cells using the RT2 Profiler PCR Array, Human p53 signaling pathway (QIAGEN Inc., Germantown, MD, USA). Eighty-four key genes regulating the p53 signaling pathway were assessed. The results were analyzed at GeneGlobe Data Analysis Center provided by Qiagen Inc. (QIAGEN Inc., Germantown, MD, USA).
Luciferase reporter assay
To determine the integral transcription activity of the p53 family, CPA cells were co-transfected with a reporter (pRL-TK) expressing the Renilla luciferase and either PG13 or MG15 reporter plasmids at a molar ratio of PG13/MG15 to (pRL-TK) = 9:1 for 24 h. Cells were next treated with 100 μM (ABS) and 20 μM (2-HOBA). Results were normalized to Renilla luciferase activity. The results were averaged from three independent experiments and expressed as mean values ±standard deviation (SD).
DNA affinity immunoblotting (DAI)
DAI was performed in CP-A esophageal cell.30 In brief, treated or untreated cells were collected after 8 hours of treatment, lysed (500 μg of total protein) and incubated with 20 ng of 5′-biotinylated DNA probes for 30 min at room temperature. DNA-protein complexes were precipitated with NeutrAvidin agarose beads (ThermoFisher Scientific, Waltham, MA), washed, eluted and analyzed by Western blotting with p53 (D0-1) antibody (Millipore, Burlington, MA; cat# OP43).
Chromatin immunoprecipitation (ChIP) and ChIP-seq analysis
ChIP was performed in CP-A cells according to the manufacturer’s protocol (Magna ChIP A/G kit; Millipore, Burlington, MA) with mouse nonspecific IgG as a control. The samples were collected after 8 hours of ABS treatment. The cells were fixed with formaldehyde and sonicated on ice. The chromatins were then immunoprecipitated with p53 (D0-1) antibody. The antibody-bound complexes were isolated using beads, crosslinked at 65°C for 30 min, and analyzed for p53 target genes by qPCR. The ChIP-seq analysis was done in CP-A cells treated with ABS alone and ABS with 2-HOBA. The three different experimental samples were pooled for ChIP-sequencing and rabbit polyclonal p53 antibody (A300-247A; Bethyl Laboratories Inc, Montgomery, TX) was used. The ChIP sequencing was done according to the manufacturer’s protocol (Active Motif, Carlsbad, CA). All ChIP-seq data generated in this study were analyzed according to the following methodology: The quality of these fastq files were checked with FastQC (ver0.11.4) and MultiQC (ver1.7). Alignment of the single end raw reads to hg 38 UCSC genomes was performed using BWA aligner (ver0.7.17). The p53 binding peaks were called from MACS2 (ver2.1.1) against mock sample as input control. The differential peaks were produced from DiffBind (ver1.20). The ChIPseq quality control was performed with ChIPQC (ver1.18.2). The peaks were annotated with goldmine (ver1.0). Genome peak profiles where visualized using UCSC Genome Browser (https://genome.ucsc.edu/). The bed files from up-regulated peaks (at least 1.5-fold increase) were used as inputs for GREAT online tool for GO Biological Process activation analysis. The common pathways with the HyperFdrQ value in both samples less than 0.05 were obtained for plotting. The bed files from down-regulated peaks (at least 1.5-fold reduction) were used as inputs for GREAT online tool for the GO Biological Process suppression analysis. NO enrichment results were obtained for these samples.
Dot blot analysis
Dot blot analysis was performed in CP-A and EPC-2 cells to investigate conformation changes in the p53 molecule. Two microliters of a protein sample were applied to a nitrocellulose membrane, dried, blocked with 5% nonfat dried milk for 1 hour and incubated overnight with primary antibodies at 4°C. After incubation, membranes were washed three times with PBS and then incubated with the respective secondary antibodies for 1 hour at room temperature, washed and developed with ECL chemiluminescence kit (Millipore, Burlington, MA).
Native PAGE
Cells were rinsed with ice-cold PBS and lysed using lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10% glycerol, protease inhibitors). The cell lysates were mixed with loading dye (240mM Tris, 30% glycerol and 0.02% Coomassie R-250) and loaded onto native 4–12% Bis-Tris gradient gels. Electrophoresis was performed using a running buffer containing 25mM Tris, 190mM Glycine, and 0.02% Coomassie R-250 in the cathode buffer for 120 mins at 4°C. The protein marker bands were cut, fixed with 8% acetic acid for 20 min and stained with Coomassie R-250 to show molecular-weight markers (NativeMark, Invitrogen, Carlsbad, CA). Proteins were transferred to PVDF membranes, blocked with 5% nonfat dried milk, and analyzed with p53 antibodies.
Immunofluorescence and protein aggregation
Cells growing on chamber slides were fixed with methanol and acetone mixture at a 1:1 ratio (v/v), permeabilized with 0.1% Triton X-100 and blocked with 10% normal goat serum (Life Technologies, Carlsbad, CA). The fixed cells were incubated with the primary p53 (D01; 1:300) antibody in a humidified chamber overnight at 4°C and the secondary antibody conjugated with FITC (Invitrogen, Carlsbad, CA) for 1 hour at room temperature. After washing with PBS, the cells were mounted using DAPI-containing media (ThermoFisher Scientific, Waltham, MA) and examined under a fluorescence microscope.
For co-localization analysis of p53 protein with amyloid fibrils, cells were incubated with a mixture of p53 (D0-1; 1:300) and amyloid fibrils (OC; 1:500) antibodies overnight at 4°C. Then, cells were incubated with a mixture of two secondary antibodies, conjugated with FITC to detect p53 protein and Alexa Fluor 568 to detect amyloid fibrils (Invitrogen, Carlsbad, CA)for 1 hour at room temperature. The cells were mounted with DAPI-containing media and examined under a fluorescence microscope.
Protein aggregation was assessed using ProteoStat aggregation dye (Enzo Life Sciences, Farmingdale, NY). The analyzed cells were incubated with p53 (D01) or p53 (Pab240) antibodies overnight at 4°C, followed by incubation with the corresponding secondary antibodies conjugated with FITC (Invitrogen, Carlsbad, CA) for 1 hour at room temperature. Then, cells were incubated with ProteoStat dye (1:10000) for 30 mins at room temperature, washed with PBS and mounted with DAPI-containing media. The cells were examined under a fluorescence microscope.
Subcellular fractionation and cell cycle analysis
Cytoplasmic and nuclear fractions were collected according to the protocol provided with the Subcellular Protein Fractionation Kit (ThermoFisher scientific, Waltham, MA). P53 protein was immunoprecipitated and analyzed for the formation of isoLG adduction using D11 antibody. CP-A cells transfected with either p53 siRNA or scrambled siRNA, were treated with ABS alone or in combination with 2-HOBA and analyzed 24 hours after treatment. Cells were fixed with 70% ethanol overnight at −20 C, washed with ice-cold PBS and analyzed by FACS using PI/RNase buffer according to the manufacturer’s protocol (BD Biosciences, Franklin Lakes, NJ). The cell cycle histograms were generated using FCS express 7.0 software.
Animal model of reflux and immunohistochemistry
The DNA damage was assessed in the esophageal tissues collected from surgery mice (n = 7) and control mice with sham surgery (n = 8) using immunohistochemical staining with p-H2AX antibody (1:500). All animal studies have been approved by the University of Miami Animal Care and Use Committee. To investigate misfolding and aggregation of p53 in vivo, immunohistochemical staining was performed using p53 (PAb 240; 1:100) antibody. p53 protein misfolding was compared in esophageal tissues collected from surgical mice and control mice with sham surgery. The intensity of staining was graded as 0 (negative), 1 (weak), 2 (moderate) or 3 (strong). Total scores were calculated by multiplying the intensity score by the percentage of positive cells. As a negative control, primary antibody was omitted.
Analysis of protein aggregation in human tissues
The human esophageal specimens from healthy adults (n = 7) and GERD and BE patients (n = 10) was used to investigate the p53 aggregation. Immunofluorescence staining was done with p53 (pab240) antibody. The esophageal adenocarcinoma tissues were used as positive controls. The intensity of staining was graded as 0 (negative), 1 (weak), 2 (moderate) or 3 (strong). The percentage of positive cells was used to grade the staining frequency. Total scores were calculated by multiplying the intensity score by the percentage of positive cells. As a negative control, primary antibody was omitted.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments had a minimum of three independent repeats. Each experiment produced similar results with low variability between experiments. Statistical analyses were performed using 1-way ANOVA followed by Tukey’s multiple comparison test, Mann-Whitney test and 2-tailed Student’s t-test, depending on the dataset. Results are expressed as mean ± SD, if not specifically indicated. Results were considered significant if p < 0.05.
Supplementary Material
Highlights.
Active isolevuglandins react with p53 protein to form adducts
Adduction causes conformational changes in p53 and amyloid-like aggregation
Adduction of p53 protein leads to alteration of its biological activity
IsoLG scavengers inhibit adduction of p53 and restore its function
ACKNOWLEDGMENTS
This work was supported by grants from the National Cancer Institute P01 CA268991, RO1 206564, and R01 138833, Department of Veterans Affairs BX002115, and Sylvester Comprehensive Cancer Center P30CA24013. Other support includes NIH R01HL144943 and RO1HL157583 grants. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the Department of Veterans Affairs, National Institutes of Health, or University of Miami. We thank Dr. Raymond Mernaugh for help with reagents. We thank BioRender.com for generation of the graphical abstract.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112024.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- 2.Caspa Gokulan R, Garcia-Buitrago MT, and Zaika AI (2019). From genetics to signaling pathways: molecular pathogenesis of esophageal adenocarcinoma. Biochim. Biophys. Acta, Rev. Cancer 1872, 37–48. 10.1016/j.bbcan.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Serag HB, Sweet S, Winchester CC, and Dent J (2014). Update on the epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut 63, 871–880. 10.1136/gutjnl-2012-304269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubenstein JH, and Shaheen NJ (2015). Epidemiology, diagnosis, and management of esophageal adenocarcinoma. Gastroenterology 149, 302–317.e1. 10.1053/j.gastro.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQuaid KR, Laine L, Fennerty MB, Souza R, and Spechler SJ (2011). Systematic review: the role of bile acids in the pathogenesis of gastro-oesophageal reflux disease and related neoplasia. Aliment. Pharmacol. Ther 34, 146–165. 10.1111/j.1365-2036.2011.04709.x. [DOI] [PubMed] [Google Scholar]
- 6.Iftikhar SY, Ledingham S, Steele RJ, Evans DF, Lendrum K, Atkinson M, and Hardcastle JD (1993). Bile reflux in columnar-lined Barrett’s oesophagus. Ann. R. Coll. Surg. Engl 75, 411–416. [PMC free article] [PubMed] [Google Scholar]
- 7.Nehra D, Howell P, Williams CP, Pye JK, and Beynon J (1999). Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity. Gut 44, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauer WK, Peters JH, DeMeester TR, Feussner H, Ireland AP, Stein HJ, and Siewert RJ (1997). Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery 122, 874–881. [DOI] [PubMed] [Google Scholar]
- 9.Gotley DC, Morgan AP, and Cooper MJ (1988). Bile acid concentrations in the refluxate of patients with reflux oesophagitis. Br. J. Surg 75, 587–590. [DOI] [PubMed] [Google Scholar]
- 10.Bhardwaj V, Horvat A, Korolkova O, Washington MK, El-Rifai W, Dikalov SI, and Zaika AI (2016). Prevention of DNA damage in Barrett’s esophageal cells exposed to acidic bile salts. Carcinogenesis 37, 1161–1169. 10.1093/carcin/bgw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A, et al. (2007). Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett’s oesophagus. Gut 56, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, Wang JY, Cheng E, Meyer F, Wang DH, et al. (2011). Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-kappaB activation in benign Barrett’s epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol 301, G278–G286, ajpgi.00092.2011 [pii]. 10.1152/ajpgi.00092.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, et al. (2013). Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet 45, 478–486. 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reid BJ, Prevo LJ, Galipeau PC, Sanchez CA, Longton G, Levine DS, Blount PL, and Rabinovitch PS (2001). Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am. J. Gastroenterol 96, 2839–2848. 10.1111/j.1572-0241.2001.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz-Summercorn A, Anand S, Ingledew S, Huang Y, Roberts T, Galeano-Dalmau N, O’Donovan M, Liu H, and Fitzgerald RC (2017). Application of a multi-gene next-generation sequencing panel to a non-invasive oesophageal cell-sampling device to diagnose dysplastic Barrett’s oesophagus. J. Pathol. Clin. Res 3, 258–267. 10.1002/cjp2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caspa Gokulan R, Adcock JM, Zagol-Ikapitte I, Mernaugh R, Williams P, Washington KM, Boutaud O, Oates JA Jr., Dikalov SI, and Zaika AI (2019). Gastroesophageal reflux induces protein adducts in the esophagus. Cell. Mol. Gastroenterol. Hepatol 7, 480–482.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan HP, Roberts LJ, Davies SS, Pohlmann P, Parl FF, Estes S, Maeng J, Parker B, and Mernaugh R (2017). Isolevuglandins as a gauge of lipid peroxidation in human tumors. Free Radic. Biol. Med 106, 62–68. 10.1016/j.freeradbiomed.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi W, Jang GF, Zhang L, Crabb JW, Laird J, Linetsky M, and Salomon RG (2016). Molecular structures of isolevuglandin-protein crosslinks. Chem. Res. Toxicol 29, 1628–1640. 10.1021/acs.chemrestox.6b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Govindarajan B, Junk A, Algeciras M, Salomon RG, and Bhattacharya SK (2009). Increased isolevuglandin-modified proteins in glaucomatous astrocytes. Mol. Vis 15, 1079–1091. [PMC free article] [PubMed] [Google Scholar]
- 20.Salomon RG, and Bi W (2015). Isolevuglandin adducts in disease. Antioxidants Redox Signal. 22, 1703–1718. 10.1089/ars.2014.6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theisen J, Nehra D, Citron D, Johansson J, Hagen JA, Crookes PF, DeMeester SR, Bremner CG, DeMeester TR, and Peters JH (2000). Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J. Gastrointest. Surg 4, 50–54. 10.1016/s1091-255x(00)80032-3. [DOI] [PubMed] [Google Scholar]
- 22.Reinhardt HC, and Schumacher B (2012).The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 28, 128–136. 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao D, Gaitonde SV, Qi W, and Martinez JD (2001). Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis 22, 957–964. 10.1093/carcin/22.6.957. [DOI] [PubMed] [Google Scholar]
- 24.Roman S, Pétré A, Thépot A, Hautefeuille A, Scoazec JY, Mion F, and Hainaut P (2007). Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am. J. Physiol. Gastrointest. Liver Physiol 293, G45–G53. 10.1152/ajpgi.00583. [DOI] [PubMed] [Google Scholar]
- 25.Ozaki T, and Nakagawara A (2005). p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 96, 729–737. 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaika E, Wei J, Yin D, Andl C, Moll U, El-Rifai W, and Zaika AI (2011). p73 protein regulates DNA damage repair. Faseb. J 25, 4406–4414, fj.11-192815 [pii]. 10.1096/fj.11-192815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levrero M, De Laurenzi V, Costanzo A, Gong J, Wang JY, and Melino G (2000). The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci 113, 1661–1670. [DOI] [PubMed] [Google Scholar]
- 28.May-Zhang LS, Kirabo A, Huang J, Linton MF, Davies SS, and Murray KT (2021). Scavenging reactive lipidsto prevent oxidative injury. Annu. Rev. Pharmacol. Toxicol 61, 291–308. 10.1146/annurev-pharmtox-031620-035348. [DOI] [PubMed] [Google Scholar]
- 29.Davies SS, Brantley EJ, Voziyan PA, Amarnath V, Zagol-Ikapitte I, Boutaud O, Hudson BG, Oates JA, and Roberts LJ, 2nd. (2006). Pyridoxamine analogues scavenge lipid-derived gamma-ketoaldehydes and protect against H2O2-mediated cytotoxicity. Biochemistry 45, 15756–15767. 10.1021/bi061860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vilgelm AE, Washington MK, Wei J, Chen H, Prassolov VS, and Zaika AI (2010). Interactions of the p53 protein family in cellular stress response in gastrointestinal tumors. Mol. Cancer Therapeut 9, 693–705. 10.1158/1535-7163.MCT-09-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng SX, Dai MS, Keller DM, and Lu H (2002). SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J. 21, 5487–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilgelm AE, Hong SM, Washington MK, Wei J, Chen H, El-Rifai W, and Zaika A (2010). Characterization of DeltaNp73 expression and regulation in gastric and esophageal tumors. Oncogene 29, 5861–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Y, Zhao W, Chen Y, Zhao Y, and Gu W (2008). Acetylation is indispensable for p53 activation. Cell 133, 612–626. 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakin ND, and Jackson SP (1999). Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655. 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 35.Ozaki T, and Nakagawara A (2011). Role of p53 in cell death and human cancers. Cancers 3, 994–1013. 10.3390/cancers3010994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouladan S, Trautmann M, Orouji E, Hartmann W, Huss S, Büttner R, and Wardelmann E (2015). Differential diagnosis of solitary fibrous tumors: a study of 454 soft tissue tumors indicating the diagnostic value of nuclear STAT6 relocation and ALDH1 expression combined with in situ proximity ligation assay. Int. J. Oncol 46, 2595–2605. 10.3892/ijo.2015.2975. [DOI] [PubMed] [Google Scholar]
- 37.Verset L, Tommelein J, Decaestecker C, De Vlieghere E, Bracke M, Salmon I, De Wever O, and Demetter P (2017). ADAM-17/FHL2 colocalisation suggests interaction and role of these proteins in colorectal cancer. Tumour Biol. 39, 1010428317695024. 10.1177/1010428317695024. [DOI] [PubMed] [Google Scholar]
- 38.May-Zhang LS, Yermalitsky V, Huang J, Pleasent T, Borja MS, Oda MN, Jerome WG, Yancey PG, Linton MF, and Davies SS (2018). Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J. Biol. Chem 293, 9176–9187. 10.1074/jbc.RA117.001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabapathy K, and Lane DP (2019). Understanding p53 functions through p53 antibodies. J. Mol. Cell Biol 11, 317–329. 10.1093/jmcb/mjz010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang-Hartwich Y, Soteras MG, Lin ZP, Holmberg J, Sumi N, Craveiro V, Liang M, Romanoff E, Bingham J, Garofalo F, et al. (2015). p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene 34, 3605–3616. 10.1038/onc.2014.296. [DOI] [PubMed] [Google Scholar]
- 41.Caspa Gokulan R, Adcock JM, Zagol-Ikapitte I, Mernaugh R, Williams P, Washington KM, Boutaud O, Oates JA Jr., Dikalov SI, and Zaika AI (2019). Gastroesophageal reflux induces protein adducts in the esophagus. Cell. Mol. Gastroenterol. Hepatol 7, 480–482.e7. 10.1016/j.jcmgh.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oshinbolu S, Shah R, Finka G, Molloy M, Uden M, and Bracewell DG (2018). Evaluation of fluorescent dyes to measure protein aggregation within mammalian cell culture supernatants. J. Chem. Technol. Biotechnol 93, 909–917. 10.1002/jctb.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, et al. (2007). Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener 2, 18. 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, et al. (2014). DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Invest 124, 4642–4656. 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh S, Salot S, Sengupta S, Navalkar A, Ghosh D, Jacob R, Das S, Kumar R, Jha NN, Sahay S, et al. (2017). p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis. Cell Death Differ. 24, 1784–1798. 10.1038/cdd.2017.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lasagna-Reeves CA, Clos AL, Castillo-Carranza D, Sengupta U, Guerrero-Muñoz M, Kelly B, Wagner R, and Kayed R (2013). Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun 430, 963–968. 10.1016/j.bbrc.2012.11.130. [DOI] [PubMed] [Google Scholar]
- 47.Levy CB, Stumbo AC, Ano Bom APD, Portari EA, Cordeiro Y, Silva JL, and De Moura-Gallo CV (2011). Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol 43, 60–64. 10.1016/j.biocel.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 48.Davies SS, Talati M, Wang X, Mernaugh RL, Amarnath V, Fessel J, Meyrick BO, Sheller J, and Roberts LJ, 2nd. (2004). Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic. Biol. Med 36, 1163–1174. 10.1016/j.freeradbiomed.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Hong J, Chen Z, Peng D, Zaika A, Revetta F, Washington MK, Belkhiri A, and El-Rifai W (2016). APE1-mediated DNA damage repair provides survival advantage for esophageal adenocarcinoma cells in response to acidic bile salts. Oncotarget 7, 16688–16702. 10.18632/oncotarget.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kubo N, Morita M, Nakashima Y, Kitao H, Egashira A, Saeki H, Oki E, Kakeji Y, Oda Y, and Maehara Y (2014). Oxidative DNA damage in human esophageal cancer: clinicopathological analysis of 8-hydroxydeoxyguanosine and its repair enzyme. Dis. Esophagus 27, 285–293. 10.1111/dote.12107. [DOI] [PubMed] [Google Scholar]
- 51.Li D, and Cao W (2014). Role of intracellular calcium and NADPH oxidase NOX5-S in acid-induced DNA damage in Barrett’s cells and Barrett’s esophageal adenocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol 306, G863–G872. 10.1152/ajpgi.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao L, Lin J, Huang M, Ajani JA, and Wu X (2005). Predictors of esophageal cancer risk: assessment of susceptibility to DNA damage using comet assay. Genes Chromosomes Cancer 44, 415–422. 10.1002/gcc.20254. [DOI] [PubMed] [Google Scholar]
- 53.Bhardwaj V, Gokulan RC, Horvat A, Yermalitskaya L, Korolkova O, Washington KM, El-Rifai W, Dikalov SI, and Zaika AI (2017). Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep 7, 9956. 10.1038/s41598-017-09620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nieva J, Song BD, Rogel JK, Kujawara D, Altobel L 3rd, Izharrudin A, Boldt GE, Grover RK, Wentworth AD, and Wentworth P Jr. (2011). Cholesterol secosterol aldehydes induce amyloidogenesis and dysfunction of wild-type tumor protein p53. Chem. Biol 18, 920–927. 10.1016/j.chembiol.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moos PJ, Edes K, and Fitzpatrick FA (2000). Inactivation of wild-type p53 tumor suppressor by electrophilic prostaglandins. Proc. Natl. Acad. Sci. USA 97, 9215–9220. 10.1073/pnas.160241897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fléjou JF, and Svrcek M (2007). Barrett’s oesophagus–a pathologist’s view. Histopathology 50, 3–14. 10.1111/j.1365-2559.2006.02569.x. [DOI] [PubMed] [Google Scholar]
- 57.Gobert AP, Boutaud O, Asim M, Zagol-Ikapitte IA, Delgado AG, Latour YL, Finley JL, Singh K, Verriere TG, Allaman MM, et al. (2021). Dicarbonyl electrophiles mediate inflammation-induced gastrointestinal carcinogenesis. Gastroenterology 160, 1256–1268.e9. 10.1053/j.gastro.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asim M, Chikara SK, Ghosh A, Vudathala S, Romero-Gallo J, Krishna US, Wilson KT, Israel DA, Peek RM Jr., and Chaturvedi R (2015). Draft genome sequence of gerbil-adapted carcinogenic helicobacter pylori strain 7.13. Genome Announc. 3, 641. 10.1128/genomeA.00641-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei J, Nagy TA, Vilgelm A, Zaika E, Ogden SR, Romero-Gallo J, Piazuelo MB, Correa P, Washington MK, El-Rifai W, et al. (2010). Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139, 1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei J, O’Brien D, Vilgelm A, Piazuelo MB, Correa P, Washington MK, El-Rifai W, Peek RM, and Zaika A (2008). Interaction of helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 134, 1412–1423. 10.1053/j.gastro.2008.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the main manuscript and the supplementary files provided. The ChIP-seq data is available on NCBI Sequence Read Archive (SRA) with the accession details provided in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-p53 (D0-1) | Millipore | Cat# OP43 |
| Rabbit anti-acetyl-p53 (K382) | Cell Signaling Technology | Cat#2525 |
| Rabbit anti-histone H2AX | Cell Signaling Technology | Cat#2595 |
| Rabbit anti-phospho-histone H2AX (Ser139) | Cell Signaling Technology | Cat#9718 |
| Rabbit anti-p53 antibody | Bethyl Laboratories Inc, | Cat#A300-247A |
| Mouse anti-p53 (pab240) | Abcam | Cat# AB26 |
| Mouse anti-p53 (pab240) | Novus Biologicals | Cat# NB200-103 |
| Rabbit anti-p73 | Bethyl laboratories | Cat# A300-126A |
| Rabbit anti-amyloid OC fibrils | Millipore | Cat#AB2286 |
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A5441 |
| Goat anti-mouse IgG HRP | Promega | Cat# PAW4021 |
| D-11 scFv antibody | A gift from Dr. Raymond Mernaugh, Vanderbilt University, USA | N/A |
| Rabbit anti- E:tag antibody | Abcam | Cat# AB3415 |
| Bacterial and virus strains | ||
| H.pylori 7.13 | A gift from Dr. Richard Peek, Vanderbilt University Medical Center, USA | N/A |
| Biological samples | ||
| Human Tissue | Department of Pathology, University of Miami, USA | N/A |
| Mouse samples | University of Miami, USA | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PI/RNase staining buffer | BD Biosciences | Cat# 550825 |
| Coomassie Brilliant Blue R-250 | Biorad | Cat# 1610400 |
| NAC (N-acetylcysteine) | Sigma-Aldrich | Cat# A0737 |
| TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperydine-1-oxyl) | Enzo Lifesciences | Cat# ALX-430-081-G001 |
| NS-398 | Cayman Chemical | Cat#70590 |
| 2-HOBA | Cayman Chemical | Cat#25357 |
| Critical commercial assays | ||
| p53 human immunocapture kit | Abcam | Cat# AB154470 |
| RNeasy Mini Kit | QIAGEN Inc | Cat#74134 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| RT2 Profiler PCR Array, Human p53 signaling pathway | QIAGEN Inc | Cat# 330231 |
| Magna ChIP A/G kit | Millipore | Cat# 17-10085 |
| Subcellular Protein Fractionation Kit | ThermoFisher Scientific | Cat#78840 |
| Proteostat Protein Aggregation Assay Kit | Enzo Lifesciences | Cat#ENZ-51023-KP050 |
| Dual Luciferase Reporter Assay Kit | Promega | E1910 |
| Deposited data | ||
| ChIP-Seq data | This manuscript | NCBI SRA (Sequence Read Archive); PRJNA912777 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA912777?reviewer=6cnca4108s5od672ijo7001e36) |
| Experimental models: Cell lines | ||
| BAR-T (human Barrett’s epithelial cells) | A kind gift from Dr. Souza, Baylor University Medical Center, USA | N/A |
| CP-A (human Barrett’s epithelial cells) | ATCC (American Type Culture Collection) | Cat#CRL-4027 |
| EPC-2 (human normal epithelial cells) | A gift from Dr. Claudia Andl, University of Central Florida, USA | N/A |
| GES-1 (human normal gastric epithelial cells) | A kind gift from Dr. El-rifai, University of Miami USA | N/A |
| SNU-1 (human gastric epithelial cancer cells) | ATCC (American Type Culture Collection) | Cat# CRL-5971 |
| Experimental models: Organisms/strains | ||
| 129/sv mice | University of Miami | N/A |
| Oligonucleotides | ||
| HPRT Forward: 5′-TTGGAAAGGG TGTTTATTCCTCA-3′ Reverse: 5′- TCCAGCAGGTCAGCAAAGAA-3′ |
Integrated DNA Technologies (IDT) | N/A |
| p21 Forward: 5′-CTGGAGACTC TCAGGGTCGAAA-3′ Reverse: 5′-GA TTAGGGCTTCCTCTTGGAGAA-3′ |
Millipore Sigma | N/A |
| PUMA Forward: 5′-ACGACCTCAA CGCACAGTACG-3′ Reverse: 5′-TCC CATGATGAGATTGTACAGGAC-3′ |
Integrated DNA Technologies (IDT) | N/A |
| 5′-Biotinylated p21; 5-biotin-AG CCTCCCTCCATCCCTAT-3 and 5′-CCCTTCCTCACCTGAAAACA-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| 5′-Biotinylated PUMA; 5′-biotin-CCC AGTCAGTGTGTGTGTCC-3′ and 5′-CCCCCGCGTGACGCTAC-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| Non-specific probe,n/s, 5′-biotin-TA GCTGGGAAGCTGGGACTA-3′ and 5′-GGTTTCCTTGCCCTAAAAGG-3′ |
Vilgelm et al., 2010; Integrated DNA Technologies (IDT) | N/A |
| ChIP qPCR; p21 (5′-ACCTTTCA CCATTCCCCTAC-3′, 5′-GCCC AAGGACAAAATAGCCA-3′ |
Vilgelm et al., 2010; Millipore Sigma | N/A |
| ChIP qPCR; PUMA (5′-TGTCCA TGGTGTGGATTTGCG-3′, 5′-AGAC ACCGGGACAGTCGGACA-3′ |
Vilgelm et al., 2010; Millipore Sigma | N/A |
| Human p53 siRNA | Santa Cruz Biotechnology | Cat#sc-29435 |
| Software and algorithms | ||
| GeneGlobe Data Analysis Center provided by Qiagen Inc. | QIAGEN Inc. | https://geneglobe.qiagen.com/us/analyze |
| Open Comet software | Open Comet | https://cometbio.org/ |
| FCS express 7 | De Novo software | https://denovosoftware.com/ |
| GraphPad Prism 8.0.0 | GraphPad Software | https://www.graphpad.com/ |