

Abstract
Congenital muscular dystrophy type 1A (MDC1A), the most common congenital muscular dystrophy in Western countries, is caused by recessive mutations in LAMA2, the gene encoding laminin alpha 2. Currently, no cure or disease modifying therapy has been successfully developed for MDC1A. Examination of patient muscle biopsies revealed altered distribution of lysosomes. We hypothesized that this redistribution was a novel and potentially druggable aspect of disease pathogenesis. We explored this hypothesis using candyfloss (caf), a zebrafish model of MDC1A. We found that lysosome distribution in caf zebrafish was also abnormal. This altered localization was significantly associated with fiber detachment and could be prevented by blocking myofiber detachment. Overexpression of transcription factor EB, a transcription factor that promotes lysosomal biogenesis, led to increased lysosome content and decreased fiber detachment. We conclude that genetic manipulation of the lysosomal compartment is able to alter the caf zebrafish disease process, suggesting that lysosome function may be a target for disease modification.
Introduction
Congenital muscular dystrophies (CMDs) are a group of congenital onset genetic diseases that share the common features of neonatal hypotonia, extremity muscle weakness and developmental motor dysfunction (1–5). As a group, CMDs are associated with severe lifelong disabilities, often including both ventilator and wheelchair dependence (6). Improvements in general care management, including physical, respiratory, speech and occupational therapies, together with corrective eye and cardiac surgeries, result in increased survival and longevity in patients with CMD (7–9). Recently, biomarkers and several therapeutic approaches have been established in preclinical models for some CMDs (5,10,11). However, there is currently no cure, nor any disease modifying therapy, for any CMD subtype; thus, there is a critical need to identify new treatment strategies for these devastating disorders.
CMDs are clinically and genetically diverse disorders (12,13). The three most common subtypes are merosin-deficient congenital muscular dystrophy (MDC1A), collagen VI-related CMD and the dystroglycanopathies (12,14). Mutations that cause these disorders arise in genes that encode components of the muscle attachment apparatus, which normally performs the functions of linking the myofiber cytoskeleton to the extracellular matrix (ECM) and providing mechanical stability to the muscle during contraction (12).
MDC1A is the most common form of CMD (15–17). It is caused by recessive mutations in laminin-α2 (LAMA2), which leads to the loss of laminin-211 expression (18), the primary laminin isoform found in the basal lamina of skeletal muscle and Schwann cells (18–21). Laminin-211 regulates muscle membrane stability and mediates cell signaling through interactions with the cell surface receptors α-dystroglycan and α7β1-integrin, both of which play a role in muscle force-production (22,23). α-dystroglycan is specifically implicated in mediating attachment between ECM and the actin cytoskeleton (24,25), while α7β1-integrin has been implicated in cell signaling pathways (26,27).
While the primary pathomechanism of MDC1A in skeletal muscle is detachment of muscle fibers from the basal membrane (28,29), dystrophic fibers in this condition maintain their sarcolemmal integrity (30) and are long-lived, undergoing remodeling before initiating a delayed cell death process (29) that leads to muscle wasting. Vertebrate models of the disease have suggested that abnormalities in degradation pathways contribute to the disease pathology. For example, increased activity of the ubiquitin-proteasome system (UPS), increased apoptosis and aberrant autophagy have been observed in mouse models of MDC1A, and these seem to exacerbate the dystrophic pathology (31–33). Furthermore, inhibiting these pathways provides partial improvement of the murine disease phenotype (32–37). Of note, while UPS and autophagy are distinct processes, there is considerable crosstalk between them, particularly in relation to muscle wasting, an important component of MDC1A (33,38–41). Lysosomes are critical common downstream mediators for aspects of these pathways (42,43) and have been implicated in membrane repair in various muscle disorders (44–46), making them an attractive subcellular compartment for drug targeting.
Zebrafish offer unique advantages for the study of MDC1A (47). The optical clarity of the zebrafish, along with the ease of genetic manipulation, enables the assessment of pathways in live embryos (48). Zebrafish also readily absorb small molecules and are thus ideal for testing the effects of chemical modifiers (49). Lastly, zebrafish have proven to accurately model the key features of MDC1A (50–52). The main zebrafish model of MDC1A is candyflossteg15a (caf), which harbors a mutation in the lama2 gene that leads to a premature stop codon and absence of LAMA2 expression (50). In this model, muscle fibers detach and retract from the myosepta beginning at 2 days post-fertilization (dpf), followed thereafter by progressive myofiber detachment and impaired motor function that ultimately leads to death between 9 and 14 dpf (50).
In this study, we explored roles for lysosome biogenesis and its regulatory mechanisms in identifying potential new therapeutic strategies for MDC1A. We observed alterations in the expression and distribution of lysosomes in muscle biopsies from MDC1A patients. We validated the altered localization in the caf zebrafish model for MDC1A and tested the impact of modulation of lysosomal function on the zebrafish phenotype. We observed significant changes in caf muscle fiber detachment with expression of the lysosomal biogenesis-promoting transcription factor EB (TFEB). On the other hand, we did not detect improvement in dystrophic changes or overall motor function with chemical manipulation of lysosomes. We also showed that, similar to mammalian models of MDC1A, the UPS is significantly upregulated in caf mutants (32) and confirmed the ability of the UPS inhibitor MG-132 to modify the MDC1A phenotype. In all, we identified lysosomal redistribution as a novel feature of MDC1A pathology and provide data showing that increasing lysosomal number can improve an MDC1A phenotype in a pre-clinical model of the disease.
Results
Abnormal distribution of lysosomes in muscle biopsies from MDC1A patients
We examined lysosomal distribution in muscle biopsies from muscular dystrophy patients using an antibody to the endo-lysosomal protein Lamp1. In non-dystrophic biopsy samples, as well as in cases of muscular dystrophy not associated with LAMA2 mutation, we observed the typical sparse punctate expression of Lamp1. However, in a subset of fibers from MDC1A biopsies, we saw a clear re-distribution of Lamp1 staining around the myofiber membrane (Fig. 1).
Figure 1 .
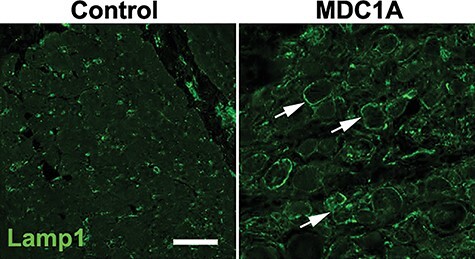
Lysosome distribution in muscle biopsies. Cross section of muscle samples from control and MDC1A patients, stained with antibody against Lamp1. Compared with the control sample, lysosomes appear to redistribute within muscle fibers and congregate at the membrane (n = 3 for each).
Abnormal distribution of lysosomes in a zebrafish model of MDC1A
We next sought to validate the observed change in lysosome expression in a zebrafish model of MDC1A (candyfloss or caf), which harbors a recessive mutation in lama2. To visualize lysosomes, we used a heat shock-inducible Lamp1-RFP cDNA construct injected into one-cell stage embryos. We performed live image analysis to examine the lysosome distribution at 3 and 5 dpf (Fig. 2). At both time points, wild-type (WT) clutch mates demonstrated a punctate pattern of Lamp1 expression consistent with the predicted cytoplasmic distribution of lysosomes (Fig. 2A and B). In caf mutants, however, we observed expression as large puncta both in the cytoplasm and at the myoseptal membrane (Fig. 2C–F). Membrane localization of lysosomes was detected in caf mutants starting at 3 dpf and became more pronounced by 5 dpf. The intensity of Lamp1-RFP at the myosepta was significantly higher in caf mutants than WT embryos (Fig. 2G and H). It was also seen in both attached and detached fibers (Fig. 2C–F).
Figure 2 .
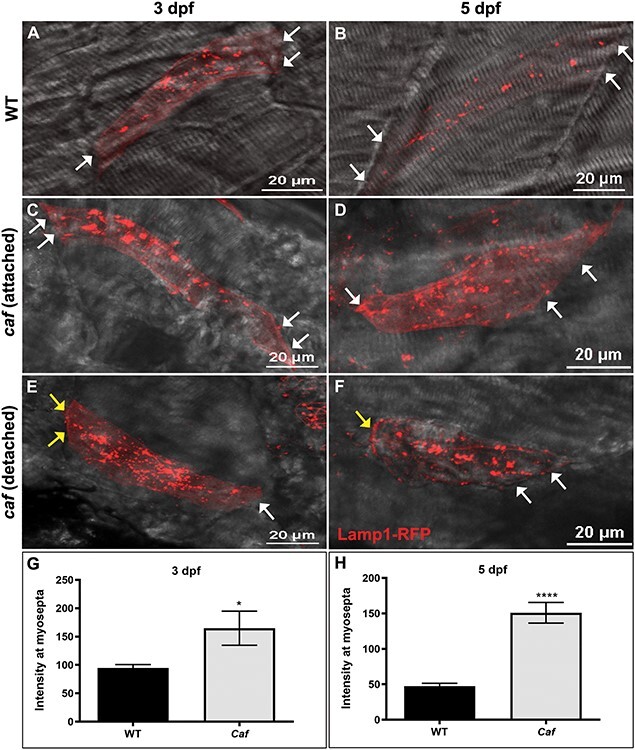
Lysosome distribution in caf mutants. (A–B) Lamp1-RFP expression in WT embryos shows that lysosomes are distributed throughout the cytoplasm in myofibers but not at myosepta (white arrows) at 3 and 5 dpf. (C–F) Lysosomes redistribute partially to the myosepta (white arrows) in caf mutants at 3 dpf (n = 10 fibers) and 5 dpf (n = 25 fibers), in both attached and detached fibers. Yellow arrows indicate where fibers have detached from the myosepta. (G–H) Lamp1-RFP fluorescence intensity at the myosepta was significantly increased in caf mutants at 3 dpf (*, P = 0.035) and 5 dpf (****, P < 0.0001). Bars represent mean ± SEM.
In order to determine whether or not lysosome mislocalization to the myosepta is dependent upon muscle fiber detachment in caf mutants, we repeated Lamp1-RFP visualization in caf embryos immobilized in N-benzyl-p-toluene sulphonamide (BTS), an inhibitor of myosin ATPase and actin-myosin interaction (53), which has been shown previously to delay muscle fiber detachment and degeneration (50). We first verified that we prevented myofiber detachment by examining muscle birefringence (a measure of myofiber organization) and found, as previously reported (50), that immobilized caf zebrafish muscle resembles that of WTs (data not shown). We then heat shocked embryos at 3 dpf to induce Lamp1-RFP expression and allowed for a 24-h recovery period. After heat shock induction, we found no significant difference in Lamp1-RFP localization in immobilized caf larvae as compared with WT, with little RFP signal detected at the myosepta (Fig. 3A and B). We quantified Lamp1-RFP expression at the myosepta from immobilized larvae and found no statistical difference in expression in immobilized caf mutants compared with their WT siblings (Fig. 3C). This suggests that the redistribution of lysosomes to the myosepta is dependent on muscle fiber detachment.
Figure 3 .
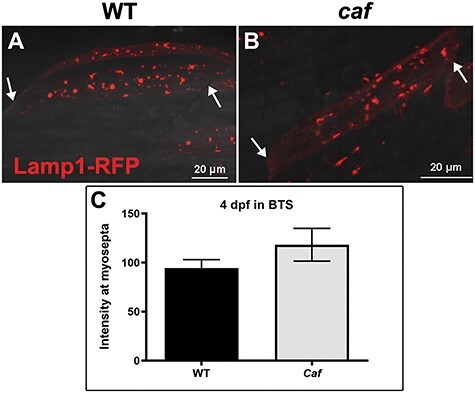
Lysosome distribution in immobilized caf mutants. (A–B) Lysosome distribution in BTS-immobilized WT and caf embryos is similar; lysosomes do not redistribute to the myosepta in paralyzed caf mutants. White arrows indicate the side of the fiber attached to the myosepta. (C) Lamp1-RFP fluorescence intensity at the myosepta was not significantly different in immobilized caf mutants at 4 dpf (P = 0.2591, n = 36 fibers) compared with WT. Bars represent mean ± SEM.
Lysosome re-distribution to myosepta is not observed in a dystrophin-deficient zebrafish model
In order to determine whether lysosome redistribution is a feature common to muscular dystrophies, we injected Lamp1-RFP in sapje (sap) embryos, a zebrafish model of Duchenne muscular dystrophy (54,55). After manual dechorionation at 24 h post-fertilization (hpf), larvae were heat shocked at 3 dpf and left to recover for 24 h. At 4 dpf, after the onset of fiber detachment in sap mutants, we observed Lamp1-positive staining in large cytoplasmic puncta, with increased aggregation in the detached fibers (Fig. 4), similar to caf mutants. However, we did not observe a redistribution of lysosomes to the myosepta (Fig. 4B and C). Consistent with this observation, we also did not detect a difference in the intensity of Lamp1-RFP at myosepta in sap myofibers (Fig. 4D), implying that the altered expression and localization seen in caf mutants is not a feature common to other muscular dystrophy zebrafish models.
Figure 4 .
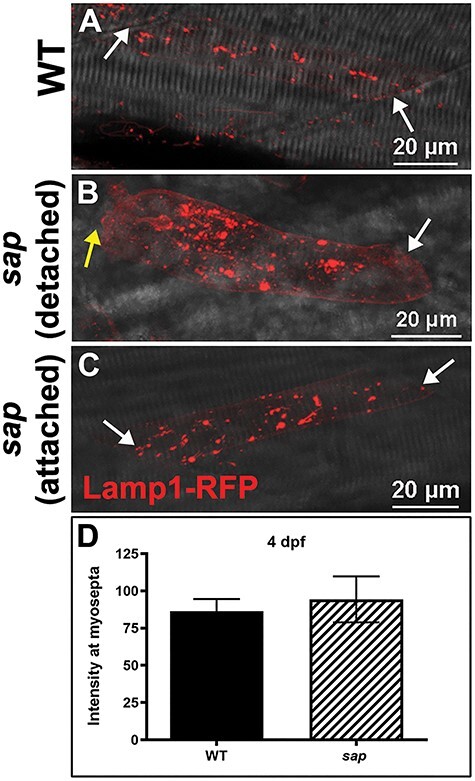
Lysosome distribution in sap mutants. (A) Lamp1-RFP expression in WT embryos shows that lysosomes are distributed throughout the cytoplasm in myofibers but not at myosepta (white arrows) at 4 dpf. (B–C) There is no significant redistribution of lysosomes to the myosepta (white arrows) in sap mutants at 4 dpf (n = 14 fibers), in either attached nor detached fibers. Yellow arrows indicate where fibers have detached from the myosepta. (D) Lamp1-RFP fluorescence intensity at the myosepta was not significantly different in sap mutants at 4 dpf (P = 0.6814, n = 14 fibers) compared with WT. Bars represent mean ± SEM.
There is an overall increase in lysosome number in caf mutants
In addition to localization, we wanted to see if the overall lysosome abundance was increased in caf larval muscle. We therefore quantified Lamp1-RFP expression in the entire myofiber (Fig. 5). There is an increase in overall Lamp1-RFP expression in muscle fibers of caf mutants at 3 and 5 dpf, which is significant at 5 dpf (Fig. 5A). However, there is no difference in whole-fiber lysosome abundance in immobilized caf mutants compared with their WT siblings, although overall expression is upregulated in immobilized embryos (Fig. 5B). This suggests that, in addition to a redistribution of lysosomes to the myosepta, there is an overall upregulation of lysosome biogenesis in caf mutants.
Figure 5 .
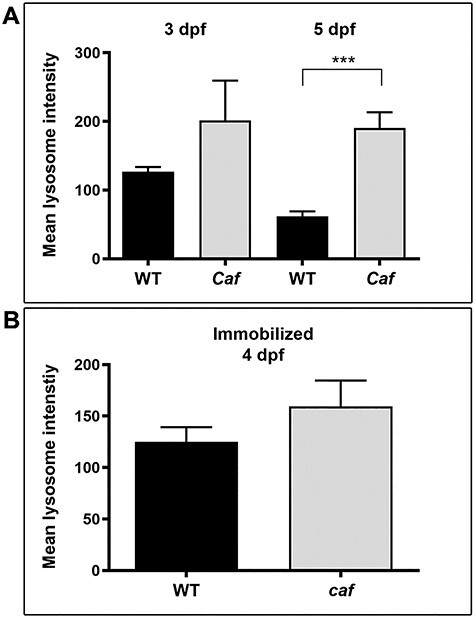
Mean overall Lamp1-RFP fluorescence intensity in muscle fibers. (A) There is an overall increase in Lamp1-RFP expression at 3 dpf (n = 10 fibers) and 5 dpf (n = 25 fibers) in caf mutants compared with WT siblings that is not significant at 3dpf (P = 0.4431) but it is significant at 5 dpf (P = 0.0026). (B) There is no overall difference in Lamp1-RFP expression in immobilized caf mutants compared with immobilized WT siblings (P = 0.3164, n = 36 fibers), nor in sap mutants compared with their WT siblings (P = 0.789, n = 14 fibers). Bars represent mean ± SEM.
Increased lysosomal biogenesis moderately protects against myofiber detachment in caf zebrafish
Given our observation of altered lysosome expression, we hypothesized that increasing the amount of lysosomes would be protective for myofibers in caf mutants by preventing detachment, via membrane repair. To increase lysosomal quantity, we over-expressed TFEB (56,57). Using co-expression with Lamp1-RFP, we first determined that TFEB expression in myofibers increased the number of lysosomes in caf myofibers (Fig. 6A). We then examined whether this increase led to a protection from myofiber detachment by expressing in skeletal muscle either TFEB-GFP or GFP attached to a fragment of phospholipase Cδ (PLCδ-PH-GFP) as control, along with the membrane marker mcherry-CAAX (58) to mark myofibers. We then counted detached versus undetached fibers expressing GFP (Fig. 6B and C). In WT embryos, the number of detached fibers was very minimal and was similar under both conditions (Supplementary Material, Table S1). In caf mutant embryos, after counting more than 250 fibers across 15 embryos from each experimental group, we found that the proportion of detached fibers was lower in mutant embryos expressing TFEB-GFP (33.9%) compared with those expressing PLCδ-PH-GFP (45.4%). This difference between the two experimental groups was significant (P = 0.0065) as indicated by a chi-square test (Table 1). These results suggest that increasing lysosomal biogenesis has a protective effect on caf myofibers. Of note, swimming behavior was not improved in TFEB-expressing caf embryos, likely because of the relatively small number of fibers with positive TFEB expression (Supplementary Material, Fig. S1).
Figure 6 .
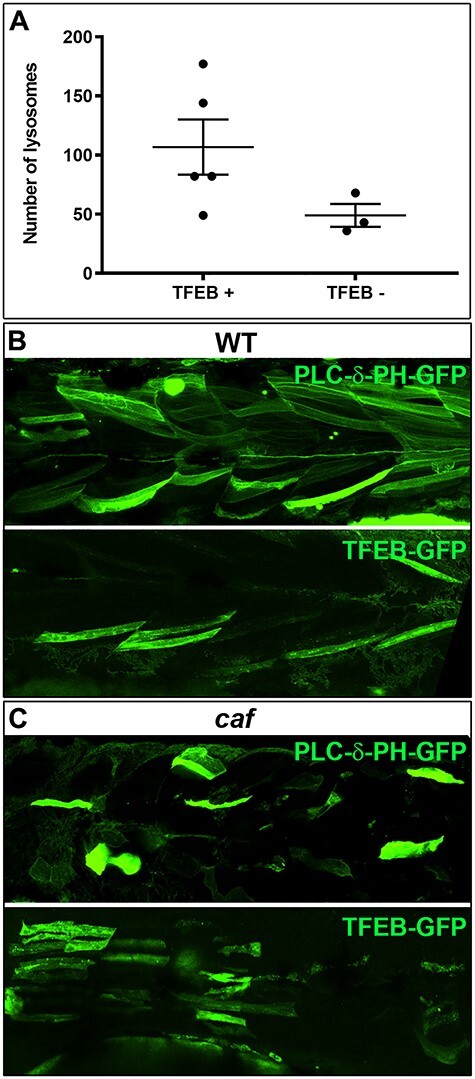
Increasing lysosomal biogenesis moderately protects against myofiber detachment in caf zebrafish. (A) Lysosomal biogenesis transcription factor TFEB increases the number of lysosomes in myofibers, however not significantly (P = 0.1200). (B) Confocal micrographs illustrating GFP-expressing myofibers in WT embryos. (C) Confocal micrographs illustrating GFP-expressing myofibers in caf mutant embryos (detached fibers: TFEB-GFP = 33.9%; PLCδ-PH-GFP = 45.4%; P = 0.0065).
Table 1.
Increasing lysosomal biogenesis protects against myofiber detachment. Number of fibers expressing the GFP constructs were counted and data were analyzed for Chi-square with Yates’ correction (Chi-square = 7.402; P = 0.0065)
| caf mutant | Detached fibers | Undetached fibers | Total |
|---|---|---|---|
| TFEB-GFP | 103 | 201 | 304 |
| PLCδ-PH-GFP | 122 | 147 | 269 |
| Total | 225 | 348 | 573 |
Treatment with the lysosomal agonist MLSA1 marginally improves swimming behavior but does not alter the dystrophic phenotype of caf mutants
We next wanted to chemically target lysosomes in order to determine whether we could improve or exacerbate the MDC1A zebrafish phenotype. Mucolipin synthetic activator 1 (MLSA1) is an activator of the lysosomal transient potential receptor mucolipin 1 (ML1) channel (43,59,60), which is a Ca2+ channel localized primarily on the lysosome membrane (61,62). ML1 is involved in lysosomal Ca2+ release, an important source of intracellular Ca2+, in addition to the sarcoplasmic reticulum (60,63), and was shown to mediate the Ca2+-dependent membrane delivery to damaged sarcolemma (46). Recently, it was shown that ML1 KO mice develop an early-onset muscular dystrophy due to a reduction in sarcolemma repair and that overexpressing the channel in a DMD mouse model (mdx) reduces its dystrophic phenotype (43). We treated zebrafish embryos with increasing concentrations of MLSA1 and found that 8 μM MLSA1 is the maximum concentration that is not toxic. We added the drug at two different developmental times (24 hpf and at 3 dpf) and then assessed the swimming behavior at 3 dpf (Supplementary Material, Fig. S2) and 5 dpf (Fig. 7). At 3 dpf, we did not observe significant differences between treated and untreated caf mutants. We observed, however, a significant decrease in distance traveled by WT-treated compared with WT-untreated embryos (Supplementary Material, Fig. S2B). At 5 dpf, we did observe a positive change with MLSA1 in swim behavior of caf mutants. Specifically, treatment of caf mutants with 8 μM MLSA1 significantly increased their time spent moving (Fig. 7A). There were, however, no significant improvements in the total distance traveled or average velocity in treated compared with untreated caf mutants (Fig. 7A).
Figure 7 .

Treatment with the lysosomal agonist MLSA1 partially improves swimming behavior of caf mutants at 5 dpf. (A) MLSA1 reduces swimming behavior of WT zebrafish at 5dpf in a dose-dependent manner. Conversely, in caf mutants, treatment with 5 or 8 μM significantly increases the time spent moving. However, there was no significant improvement in the total distance traveled or average velocity in treated versus untreated caf mutants at any dose tested (n = min 56). Bars represent mean ± SD. (B) Confocal micrographs of whole-mount embryos stained with anti-Lamp1 antibody (green), Phalloidin (magenta) and DAPI (blue). There are no differences in Lamp1 distribution between treated and untreated groups at 5 dpf, and no evidence of increased fiber integrity or decreased detachment.
To determine whether MLSA1 had any impact on the dystrophic phenotype of caf mutants, we quantified the intensity of birefringence in treated versus untreated caf mutants and their WT siblings at 3 dpf. We found no significant improvement of the dystrophic phenotype of caf mutants compared with the untreated mutants (Supplementary Material, Fig. S2D). To determine whether lysosome distribution is altered by MLSA1 treatment, we did immunostaining with anti-Lamp1 antibodies on whole-mount embryos at 5 dpf. We found no differences in Lamp1 localization between treated and untreated groups (Fig. 7B). Overall, these data suggest that MLSA1 has only marginal benefit in caf mutants, though an important caveat is that we were not able to verify drug-target engagement.
Inhibition of lysosome-membrane fusion with Vacuolin does not exacerbate the swimming behavior or the dystrophic phenotype of caf mutants
Vacuolin blocks Ca2+-dependent fusion of lysosomes to the cell membrane, thereby inhibiting cellular exocytosis (64). We hypothesized that this chemical would exacerbate the caf phenotype because it interferes with proper lysosomal function. We first treated zebrafish embryos with increasing concentrations of Vacuolin and found that 15 μM Vacuolin is the maximum non-toxic concentration. As with MLSA1, we added the drug at two different developmental times (24 hpf and at 3 dpf) and assessed swimming behavior at 3 dpf (Supplementary Material, Fig. S2) and 5 dpf (Supplementary Material, Fig. S3). We did not observe a significant change in swimming behavior at 3 or 5 dpf with any concentration of Vacuolin in caf mutants (Supplementary Material, Figs S2 and S3). We did observe, however, a dose-dependent worsening of swimming behavior in WT embryos (Supplementary Material, Fig. S3). There was no significant worsening of the dystrophic phenotype of treated caf mutants compared with the untreated mutants, as assessed by birefringence at 3dpf (Supplementary Material, Fig. S2). In all, our data did not support an exacerbation of the dystrophic mutant phenotype with lysosomal inhibition, again with the caveat of lack of confirmation of drug-target engagement.
Class III PI3 kinase inhibitors do not alter the phenotype of caf mutants
Targeting autophagy has been shown to be therapeutically beneficial in an MDC1A mouse model (33). Specifically, systemic treatment with 3-MA, a class III PI3K inhibitor that acts to prevent autophagosome formation (65–67), results in a moderate improvement to the dystrophic phenotype and walking time of the Lama2 mutant mice dy3k/dy3k (33). To assess autophagy levels in caf mutants, we performed immunostaining and western blot analysis using anti-LC3 antibody, an autophagy marker (68–70). We found that caf mutants have increased levels of LC3 (Fig. 8 A and B). To determine whether 3-MA treatment affects autophagy in zebrafish embryos, we looked at LC3 levels in WT embryos. We found that autophagy is reduced in WT-treated zebrafish compared with untreated WT group (Fig. 8C). We were interested to determine if autophagy inhibition leads to an improvement in the phenotype of caf zebrafish. We thus tested 3-MA at several doses, including 10 mM, the highest non-toxic concentration. Regardless of the dosage, we saw no change in any aspect of swimming behavior of caf mutants treated with 3-MA as compared with control mutants (Fig. 8D–F).
Figure 8 .
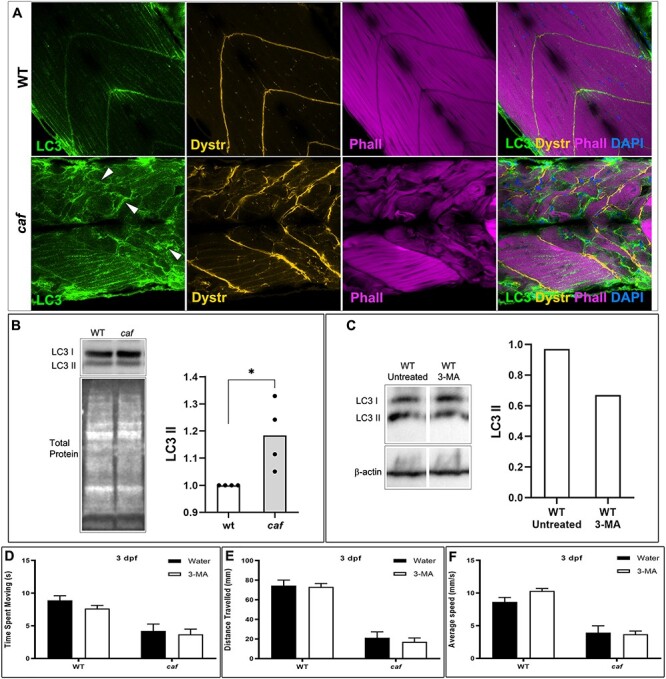
Autophagy is upregulated in caf mutants but 3-MA treatment does not improve the swimming behavior or dystrophic phenotype of caf mutants. (A) Confocal micrographs of whole-mount embryos stained with anti-LC3 (green), anti-Dystrophin (orange), Phalloidin (magenta) and DAPI (blue). LC3 localization and abundance are affected in caf mutants at 5 dpf; LC3 accumulates at sites of fiber detachment (white arrowheads). (B) Western blot analysis showing increased levels of LC3 II in caf mutants. LC3 II levels were normalized against total protein. (C) Western blot analysis showing decreased levels of LC3 II in WT zebrafish after 3-MA treatment. LC3 II levels were normalized against β-actin. (D–F) Swimming behavior analysis. There was no significant change at 3 dpf in time spent moving (D), total distance traveled (E) or average velocity (F) in caf mutants after treatment with 10 mM 3-MA (n = 165). Bars represent mean ± SEM.
Given that 3-MA is not entirely specific as a class III PI3 kinase inhibitor (67), we additionally tested a more specific class III PI3K inhibitor, Vps34-IN1 (71,72). Vps34, a class III PI3K involved in generating PI3P, is essential for phagosome formation and maturation and for completion of phagocytosis (73,74). We observed a small but not statistically significant improvement in treated caf mutants in time spent moving (Fig 9A) and total distance traveled (Fig. 9B). We detected no changes in the dystrophic phenotype in caf mutants after Vps34-IN1 treatment, as assessed by birefringence (Fig. 9D).
Figure 9 .
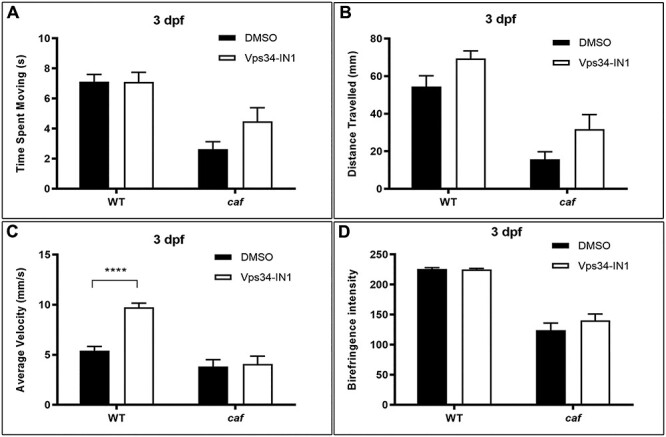
Vps34-IN1 treatment does not significantly improve the swimming behavior and dystrophic phenotype of caf mutants. (A and B) There is a small magnitude, non-significant, increase in time spent moving and total distance traveled in caf mutants after treatment with 100 nM Vps34-IN1 (n = 96). (C) There is no change in average velocity in caf mutants after treatment with 100 nM Vps34-IN1 (n = 96). (D) Birefringence intensity measurements show no significant improvement to the dystrophic phenotype of caf mutants after Vps34-IN1 treatment (n = 16). ****P < 0.0001. Bars represent mean ± SEM.
Inhibition of UPS significantly improves the phenotype of caf mutants
To determine whether the UPS is also upregulated in the zebrafish model of MDC1A, similar to the mouse model (32), we measured the levels of Polyubiquitinylated (PolyU) conjugates in zebrafish embryos at 5 dpf. We found that PolyU levels are significantly increased in caf mutants (Fig. 10A and B). Previous studies showed that pharmacological inhibition of the proteasome successfully rescues aspects of the muscle phenotype in several muscular dystrophies (32,75–77). Of these, the MG-132 proteasome inhibitor was shown to improve the dystrophic phenotype and lifespan of the dy3K/dy3K mouse model of MDC1A (32) and the sap zebrafish model of DMD (78). We therefore tested MG-132 in our caf zebrafish. Corroborating the findings in the mouse, we found that 10 μM MG-132 treatment improves the swimming behavior of caf mutants at 3 dpf (Fig. 10C–F). While there was no significant improvement in time spent moving (Fig. 10C), treated embryos had significant improvement in distance traveled (Fig. 10D) and average velocity (Fig. 10E). We also looked at the extent of muscle fiber detachment and degeneration with birefringence, and we found that MG-132 improves the dystrophic phenotype of caf zebrafish at 3 dpf, although the chemical does not restore muscle integrity to WT levels (Fig. 10F). The therapeutic benefit of MG-132 in the caf mutants did not extend beyond 3 dpf, as there was no improvement in swimming behavior of caf larvae at 4 dpf (Supplementary Material, Fig. S4A–C). Furthermore, there was no survival benefit with 10 μM MG-132 (Supplementary Material, S4D). These data suggest that MG-132 promote modest but significant benefit in the caf mutants, consistent with the data from MDC1A mice (32).
Figure 10 .
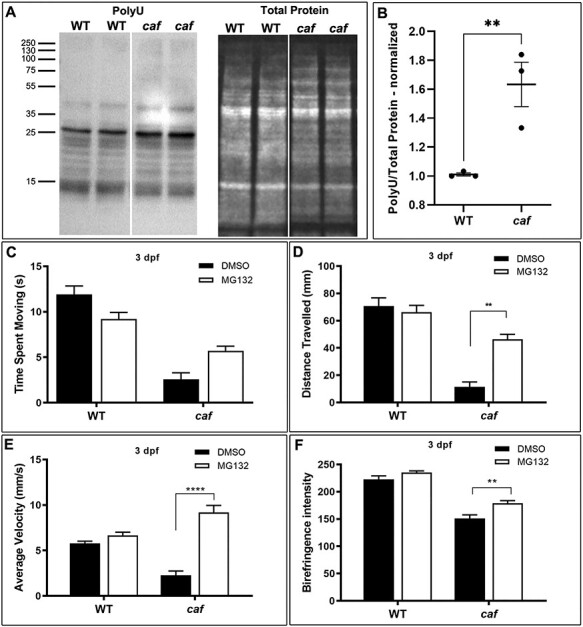
UPS is upregulated and MG-132 treatment improves the swimming behavior and dystrophic phenotype of caf mutants. (A and B) Western blot analysis showing increased levels of Polyubiquitinylated proteins (PolyU) in caf mutants. (C–E) Swimming behavior analysis. (C) There is no significant improvement in time spent swimming in caf mutants treated with 10 μM MG132 (n = 166). (D and E) There are significant improvements in distance traveled and average velocity in caf mutants at 3 dpf after treatment with 10 μM MG132. (F) Treatment with 10 μM MG132 improves the dystrophic phenotype of caf mutants at 3 dpf but does not restore muscle integrity to WT levels (n = 43). **P < 0.0075; ****P < 0.0001. Bars represent mean ± SEM.
Discussion
Normal development and functioning of the skeletal muscle depends on tightly regulated protein synthesis and degradation events (39). The autophagy-lysosome pathway and the UPS play essential roles in protein degradation and quality control of skeletal muscle proteins (39). In many muscle disorders, these proteolytic systems are misregulated (79,80). MDC1A, a form of CMD, is characterized by generalized muscle weakness and wasting (81). In this study, we used genetic and pharmacological approaches to study the lysosome, autophagy and UPS systems in an MDC1A zebrafish model.
Our results show that lysosome distribution is altered in MDC1A and this feature is common to both MDC1A patient muscle biopsies and to a lama2 muscular dystrophy zebrafish model. We found that, in addition to their cytoplasmic localization, lysosomes were distributed at the myofiber membranes in the human muscle and at the myosepta in zebrafish muscle. There are several potential explanations for our observation of increased lysosomal concentration at the sarcolemmal membrane of MDC1A myofibers. The most enticing one is that the lysosomes are redirected to sites of impaired membrane integrity in an effort to prevent and/or repair membrane damage. This is supported in part by our observation of significantly reduced myofiber detachment with TFEB expression. When we increased lysosome numbers by expressing TFEB, we observed an overall decrease in number of detached fibers in caf zebrafish mutants. Based on these results, we hypothesize that this re-distribution implies a therapeutic potential for treatments that improve lysosome number or membrane repair function.
However, we did not see more than minimal improvement in the phenotype of caf mutants with MLSA-1, a drug known to improve lysosome-mediated sarcolemmal membrane repair, nor did we observe worsening of the phenotype with Vacuolin, an inhibitor of lysosomal fusion. These negative results obtained with chemical manipulation of the lysosomal system could mean that these pathways might not play an essential role in the pathomechanisms associated with MDC1A. There are, however, important caveats associated with pharmacological approaches in zebrafish, especially related to the inability to accurately measure target engagement to be assured the drugs truly alter lysosomal function, to measure the final concentration of the drug in the muscle tissue or to assess low-level toxicity of the drugs (82). In addition, it is quite possible that the lysosomal function(s) enhanced with TFEB expression are not recapitulated fully by these chemical treatments.
Another hypothesis for increased lysosomal concentration at the sarcolemmal membrane is that lysosomes are directed to the sites of fiber detachment from the ECM to limit and ‘clean up’ the cellular damage that occurs with detachment or membrane injury. ECM–myofiber interactions are critical for normal muscle function and homeostasis (18,83,84). Autophagic flux is altered in response to cellular detachment from the ECM in a variety of biological systems (33,85–90), and accumulation of autophagosomes at these stress sites directly correlates with lysosome accumulation, which are responsible for the final step in degradation of autophagic vesicles (91–93). These observations fit with our data showing a connection between Lamp1 redistribution and fiber detachment and are supported by the fact that manipulating proteolytic pathways, particularly the UPS, appear to have a positive effect on the caf phenotype. However, while we did see a change in autophagic markers in untreated caf mutant zebrafish, inhibiting autophagy with class III PI3K inhibitors did not significantly improve swimming behavior and dystrophic phenotype (71,72,94). This lack of effect may reflect incomplete target engagement (though we did observe changes in autophagic flux with 3-MA) or instead suggest that blocking autophagy is not able to prevent myofiber detachment or otherwise modify the MDC1A phenotype. Alternatively, it is conceivable that other autophagy pathways might be activated, compensating for the Vps34 inhibition, similarly to results previously observed in myotubes (95).
Additional evidence in support of this latter hypothesis is the more robust improvement in swimming behavior and dystrophic phenotype we observed when we treated caf zebrafish with the proteasome inhibitor MG-132. These results are similar to those described in other animal models of muscular dystrophies (32,75–78,96). Increased protein ubiquitination and upregulation of the proteasome complex were shown to be hallmarks of the dystrophic muscle (96,97), and inhibition of proteasome activity was shown to rescue the dystrophic phenotype (32,76–78). Of note, MG-132 is the only compound we found that improves both swim behavior and muscle integrity.
Overall, our results reveal new pathophysiological features of MDC1A and contribute to an improved understanding of the advantages and limitations of pharmacological treatments of MDC1A. Based on our results, we conclude that lysosome redistribution in MDC1A is an intriguing phenomenon with uncertain therapeutic implications that warrants further investigations.
Material and Methods
Zebrafish husbandry and strains
We adhered to established zebrafish husbandry and all protocols used in this study were approved by the Animal Care Committee at the Peter Gilgan Centre for Research and Learning at The Hospital for Sick Children (Protocol #: 1000052731). The caf and sap zebrafish strains were obtained from University of Tubingen (Tubingen, Germany). Heterozygous carriers were genotyped using genomic DNA extracted from tail clips and specially designed Taqman PCR protocols (Thermo Fischer Scientific) we optimized for each strain. Heterozygous incrosses provided caf and sap homozygous mutants, and all embryos that were not raised were sacrificed in accordance with our established protocols.
Microinjections and DNA constructs
Embryos from a heterozygous caf or sap incross were isolated at 20 min post-fertilization. Lamp1-RFP heat shock-inducible DNA construct was injected into one-cell stage embryos at 15 pg concentration. These were then incubated in egg water (system water with methylene blue) at 28.5°C, manually dechorionated at 24 hpf and either placed back in the incubator for observation at a later time point or immediately placed in 10 mL of system water (without methylene blue) in a 60 × 15 mm Petri dish, which was then placed in a 37°C water bath for one hour. After heatshock, embryos were returned to the 28.5°C incubator for a 24-h recovery period. Upon recovery, larvae were immobilized with 0.04% ethyl 3-aminobenzoate methanesulfonate salt (tricaine) and embedded in 1% agarose diluted in system water on a MatTek 35 mm glass bottom dish (No. 1) (MatTek Corp., Ashland, MA, USA). PLC-δ-PH-GFP (Tobias Meyer Lab, Stanford University, CA), mcherry-CAAX (58) and TFEB-GFP (98) (Shawn Ferguson Lab; Addgene plasmid # 38119; http://n2t.net/addgene:38119; RRID:Addgene_38 119) constructs were injected at 10 pg concentration each.
Muscle biopsies
Muscle biopsy samples were obtained from three unrelated MDC1A patients with complete merosin deficiency.
Immobilization of caf embryos and larvae
Embryos in the 75%-epiboly-stage (99) were manually dechorionated with forceps and incubated in 50 μM BTS (Sigma-Aldrich #S949760) in egg water at 28.5°C, which was changed daily.
Drug treatment
Drugs were added at two developmental timepoints: either at 24 hpf or at 3 dpf. The embryos treated at 24 hpf were first manually dechorionated at 24 hpf and placed into groups of 20 in 24-well plates. The chemicals [3-MA (Sigma; #M9281); Vps34-IN1 (EMD Millipore; #532628); MG-132 (Sigma; #M7499); MLSA1 (Tocris; #4746); Vacuolin (Santa Cruz; #351986–85-1)] were first dissolved in either double-distilled water (3-MA) or DMSO (all the other drugs). Then, they were diluted in fresh system water to their working concentrations and paired with either water or DMSO controls. Final concentration of double-distilled water or DMSO was 0.1%. Solutions were changed every 24 h until analysis of birefringence, survival or movement. The drugs were used at the following concentrations: 10 mM 3-MA; 100 nM Vps34-IN1; 10 μM MG-132; 2.5, 5 and 8 μM MLSA1; and 5, 10 and 15 μM Vacuolin.
Birefringence
At 3 or 5 dpf, larvae were anaesthetized with 0.04% tricaine and muscle integrity was assessed by birefringence using an Olympus SZX7 stereoscope equipped with two polarizing filters. Images were acquired with a Firefly camera (Belmont, MA, USA). Measurements of birefringence intensity were made with ImageJ (ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA, https://imagej.nih.gov/ij/).
Survival analysis
Embryos were manually dechorionated at 24 hpf and placed into groups of 20 in a 24-well plate. The chemical or control solution was added at this stage and the solution was changed every 24 h. At 5 dpf, the larvae were transferred to the fish facility system daily for feeding with a glass pipette. Larvae remained on the system for feeding for 7 h and were then retrieved with a glass pipette and returned to treatment or control conditions in a 60 × 15 mm Petri dish and incubated at 28.5°C. Larvae were counted before and after transfer to the system for feeding, upon which dead embryos were removed.
Swimming behavior
To assess motor function of the larvae with and without chemical treatment, our lab developed a photochemical movement assay using the optovin analogue 6b8 (ChemBridge, #5707191) (48,100). Optovin is a reversible transient receptor protein (TRP) A1 (TRPA1) ligand that elicits motor excitation following exposure to light. The 6b8 analogue stock was prepared to 10 mM concentration in DMSO then to 10 μM working concentration by adding the stock into system water in a 1:1000 ratio. Larvae were placed into 96-well plates with a glass pipette using surface tension and excess water was removed. The larvae would remain covered in minimal water that contributed negligibly to the overall volume of the well. After 150 μL of the 6b8 solution was added to each well, the plates were covered with aluminum foil to protect the larvae from light and incubated at 28.5°C for 5 min prior to the analysis of motor behavior. After incubation, movement of the larvae was monitored for 30 s using the Zebrabox platform (Viewpoint Behaviour Technology, Lyon, France). We designed the parameters of the assay as follows: 10 s light off; 10 s light on and 10 s light off. Time spent moving, total distance traveled and average velocity during the 30 s cycle were used to compare groups of treated versus untreated larvae for all chemicals tested, and the data collected was treated as continuous. We also used this assay to determine the baseline function of caf mutants compared with their WT sibling in normal conditions.
Westerns
Protein samples were isolated from zebrafish embryos in RIPA lysis buffer supplemented with protease and phosphatase inhibitors. Total protein was stained with Revert Total Protein Stain (LI-COR Biotechnology; Lincoln, NE, USA) and visualized with Odyssey FX Imaging system using Image Studio software (LI-COR Biotechnology). Membranes were blocked in TBST (TBS with 0.1% Tween-20) with 5% BSA, for 1 h, at RT, and probed with primary antibodies, overnight, at 4°C. The following dilutions were used: rabbit anti-LC3B (1:1000; Novus Biologicals; NB600–1384), mouse IgM anti-PolyU (1:1000; Enzo Life Sciences; BML-PW8805–0500) and rabbit anti-β-actin (1:1000; Cell Signaling Technology #4967). After rinsing with TBST (3 × 10 min), membranes were incubated for 1 h at RT with goat anti-rabbit or rabbit anti-mouse IgM HRP-conjugate secondary antibodies (ThermoFisher Scientific), diluted at 1:5000 in blocking solution. ECL detection (ThermoFisher Scientific) was performed on Bio-Rad Gel Doc System using Image Lab software (Bio-Rad Laboratories, Hercules, CA, USA). Blot quantification analysis was made using Image J (ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA, https://imagej.nih.gov/ij/).
Microscopic preparations and immunofluorescence staining
Muscle biopsy microscopic preparations were processed for immunostaining as previously described (84). Anti-Lamp1 rat antibody (1DB4, Developmental Studies Hybridoma Bank) was used at 1:100 dilution.
For whole-mount microscopic preparations, zebrafish embryos were fixed overnight in 4% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA, USA), rinsed 3 × 5 min with PBST (PBS + 0.3% Triton X), washed once in ddH2O for 5 min, followed by 10 min treatment with 500 μL pre-chilled acetone at −20°C. Then, embryos were washed gently in ddH2O for 5 min, rinsed once in PBST for 5 min and blocked in PBSTB (PBST with 5% BSA) for 2 h at room temperature (RT). Embryos were incubated with primary antibodies diluted in PBSTB, overnight, at 4°C. The following dilutions were used: rabbit anti-Lamp1 (1:200; Abcam; ab24170), rabbit anti-LC3B (1:200; Novus Biologicals; NB600–1384) and mouse anti-Dystrophin (1:10; Developmental Studies Hybridoma Bank; 7A10). Next, embryos were incubated with secondary antibodies or fluorescent dyes diluted in PBSTB, at RT, for 2–3 h. The following dilutions were used: Alexa Fluor 488 goat anti-rabbit (1:250; ThermoFisher Scientific), Alexa Fluor 555 goat anti-mouse (1:250; ThermoFisher Scientific), Alexa Fluor 647 goat anti-mouse (1:250; ThermoFisher Scientific), Rhodamine-Phalloidin (1:200; Molecular Probes) and Alexa Fluor 633 Phalloidin (1:200; ThermoFisher Scientific). After rinsing with PBST 3 × 10 min, the embryos were mounted in ProLong Gold Antifade Mountant with DAPI (ThermoFisher Scientific).
Microscopy and image analysis
Larvae were visualized with a Nikon A1R laser confocal microscope, using a 40× or 63× oil-immersion lens. Images were acquired with NIS Elements software (Nikon Instruments Inc., Melville, NY, USA). Fluorescence intensity at myosepta and lysosome abundance in the entire myofiber were measured in regions of interest drawn either along the myosepta or along selected regions in the myofiber (i.e. central region, sarcolemma) using Volocity 6.3 software (PerkinElmer, Inc., Waltham, MA, USA). Micrographs were edited using NIS Elements software and/or Adobe Photoshop (Adobe Inc., San Jose, CA, USA).
Statistical analysis
All statistical analyses (ANOVA, Chi-square and non-parametric t-tests) were performed using GraphPad Prism 6.0 and 8.0.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors gratefully thank all Dowling Lab members for guidance, useful discussions and insightful suggestions; SickKids Zebrafish Facility for housing and taking care of our fish lines; University of Tubingen (Germany) for providing the caf and sap zebrafish lines; Paul Paroutis and Kimberley Lau (SickKids Imaging Facility) for technical support, and Developmental Studies Hybridoma Bank for the Lamp1 and Dystrophin antibodies. Human muscle biopsy samples were obtained from the Iowa Wellstone Muscular Dystrophy Specialized Research Center funded by NINDS, NS053672, which also provides support to S.A.M.
Conflict of Interest statement. The authors declare no conflict of interest.
Contributor Information
Sarah J Smith, Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 1A8, Canada; Program for Genetics & Genome Biology, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada; Department of Family Medicine, University of Calgary, Calgary T2R 0X7, Alberta.
Lacramioara Fabian, Program for Genetics & Genome Biology, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada.
Adeel Sheikh, Program for Genetics & Genome Biology, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada; Schulich School of Medicine and Dentistry, Western University, London, ON N6A 5C1, Canada.
Ramil Noche, Program for Genetics & Genome Biology, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada; Zebrafish Genetics and Disease Models Core Facility, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada.
Xiucheng Cui, Zebrafish Genetics and Disease Models Core Facility, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada.
Steven A Moore, Department of Pathology, University of Iowa Medical Center, Iowa City, IA, USA.
James J Dowling, Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 1A8, Canada; Program for Genetics & Genome Biology, Hospital for Sick Children, Toronto, ON M5G 0A4, Canada; Division of Neurology, Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; Department of Paediatrics, University of Toronto, Toronto, ON M5G 1X8, Canada.
Funding
CureCMD; Natural Sciences and Engineering Research Council (NSERC); Mogford Campbell Family Chair at the Hospital for Sick Children.
References
- 1. Mendell, J.R., Sahenk, Z. and Prior, T.W. (1995) The childhood muscular dystrophies: diseases sharing a common pathogenesis of membrane instability. J. Child Neurol., 10, 150–159. [DOI] [PubMed] [Google Scholar]
- 2. Emery, A.E. (2002) Muscular dystrophy into the new millennium. Neuromuscul. Disord., 12, 343–349. [DOI] [PubMed] [Google Scholar]
- 3. Bonnemann, C.G., Wang, C.H., Quijano-Roy, S., Deconinck, N., Bertini, E., Ferreiro, A., Muntoni, F., Sewry, C., Beroud, C., Mathews, K.D. et al. (2014) Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord., 24, 289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falsaperla, R., Pratico, A.D., Ruggieri, M., Parano, E., Rizzo, R., Corsello, G., Vitaliti, G. and Pavone, P. (2016) Congenital muscular dystrophy: from muscle to brain. Ital. J. Pediatr., 42, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Datta, N. and Ghosh, P.S. (2020) Update on muscular dystrophies with focus on novel treatments and biomarkers. Curr. Neurol. Neurosci. Rep., 20, 14. [DOI] [PubMed] [Google Scholar]
- 6. Sparks, S.E., Quijano-Roy, S., Harper, A., Rutkowski, A., Gordon, E., Hoffman, E.P. and Pegoraro, E. (1993) In Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 20301295. [Google Scholar]
- 7. Quijano-Roy, S., Sparks, S.E. and Rutkowski, A. (1993) In Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 20301295. [Google Scholar]
- 8. Birnkrant, D.J., Bushby, K., Bann, C.M., Apkon, S.D., Blackwell, A., Colvin, M.K., Cripe, L., Herron, A.R., Kennedy, A., Kinnett, K. et al. (2018) Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol., 17, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gieron-Korthals, M. and Fernandez, R. (2020) New developments in diagnosis, treatment, and Management of Duchenne Muscular Dystrophy. Adv. Pediatr. Infect. Dis., 67, 183–196. [DOI] [PubMed] [Google Scholar]
- 10. Wojtal, D., Kemaladewi, D.U., Malam, Z., Abdullah, S., Wong, T.W., Hyatt, E., Baghestani, Z., Pereira, S., Stavropoulos, J., Mouly, V. et al. (2016) Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet., 98, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koutsoulidou, A. and Phylactou, L.A. (2020) Circulating biomarkers in muscular dystrophies: disease and therapy monitoring. Mol Ther Methods Clin Dev, 18, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mercuri, E. and Muntoni, F. (2012) The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol., 72, 9–17. [DOI] [PubMed] [Google Scholar]
- 13. Mercuri, E. and Muntoni, F. (2013) Muscular dystrophies. Lancet, 381, 845–860. [DOI] [PubMed] [Google Scholar]
- 14. Butterfield, R.J. (2019) Congenital muscular dystrophy and congenital myopathy. Continuum (Minneap Minn), 25, 1640–1661. [DOI] [PubMed] [Google Scholar]
- 15. Durbeej, M. (2015) Laminin-alpha2 chain-deficient congenital muscular dystrophy: pathophysiology and development of treatment. Curr. Top. Membr., 76, 31–60. [DOI] [PubMed] [Google Scholar]
- 16. Mohassel, P., Foley, A.R. and Bonnemann, C.G. (2018) Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol., 71–72, 188–204. [DOI] [PubMed] [Google Scholar]
- 17. Mercuri, E., Bonnemann, C.G. and Muntoni, F. (2019) Muscular dystrophies. Lancet, 394, 2025–2038. [DOI] [PubMed] [Google Scholar]
- 18. Holmberg, J. and Durbeej, M. (2013) Laminin-211 in skeletal muscle function. Cell Adhes. Migr., 7, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helbling-Leclerc, A., Zhang, X., Topaloglu, H., Cruaud, C., Tesson, F., Weissenbach, J., Tome, F.M., Schwartz, K., Fardeau, M., Tryggvason, K. et al. (1995) Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet., 11, 216–218. [DOI] [PubMed] [Google Scholar]
- 20. Sztal, T.E., Sonntag, C., Hall, T.E. and Currie, P.D. (2012) Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum. Mol. Genet., 21, 4718–4731. [DOI] [PubMed] [Google Scholar]
- 21. Previtali, S.C. and Zambon, A.A. (2020) LAMA2 neuropathies: human findings and pathomechanisms from mouse models. Front. Mol. Neurosci., 13, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tunggal, P., Smyth, N., Paulsson, M. and Ott, M.C. (2000) Laminins: structure and genetic regulation. Microsc. Res. Tech., 51, 214–227. [DOI] [PubMed] [Google Scholar]
- 23. Pozzi, A., Yurchenco, P.D. and Iozzo, R.V. (2017) The nature and biology of basement membranes. Matrix Biol., 57–58, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lisi, M.T. and Cohn, R.D. (2007) Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim. Biophys. Acta, 1772, 159–172. [DOI] [PubMed] [Google Scholar]
- 25. Han, R., Kanagawa, M., Yoshida-Moriguchi, T., Rader, E.P., Ng, R.A., Michele, D.E., Muirhead, D.E., Kunz, S., Moore, S.A., Iannaccone, S.T. et al. (2009) Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of alpha-dystroglycan. Proc. Natl. Acad. Sci. U. S. A., 106, 12573–12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohn, R.D., Herrmann, R., Sorokin, L., Wewer, U.M. and Voit, T. (1998) Laminin alpha2 chain-deficient congenital muscular dystrophy: variable epitope expression in severe and mild cases. Neurology, 51, 94–100. [DOI] [PubMed] [Google Scholar]
- 27. Burkin, D.J. and Kaufman, S.J. (1999) The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res., 296, 183–190. [DOI] [PubMed] [Google Scholar]
- 28. Straub, V., Rafael, J.A., Chamberlain, J.S. and Campbell, K.P. (1997) Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol., 139, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hall, T.E., Wood, A.J., Ehrlich, O., Li, M., Sonntag, C.S., Cole, N.J., Huttner, I.G., Sztal, T.E. and Currie, P.D. (2019) Cellular rescue in a zebrafish model of congenital muscular dystrophy type 1A. N.P.J. Regen. Med., 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith, S.J., Horstick, E.J., Davidson, A.E. and Dowling, J. (2015) Analysis of zebrafish larvae skeletal muscle integrity with evans blue dye. J. Vis. Exp., (105), 53183. doi: 10.3791/53183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bentzinger, C.F., Barzaghi, P., Lin, S. and Ruegg, M.A. (2005) Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J., 19, 934–942. [DOI] [PubMed] [Google Scholar]
- 32. Carmignac, V., Quere, R. and Durbeej, M. (2011) Proteasome inhibition improves the muscle of laminin alpha2 chain-deficient mice. Hum. Mol. Genet., 20, 541–552. [DOI] [PubMed] [Google Scholar]
- 33. Carmignac, V., Svensson, M., Korner, Z., Elowsson, L., Matsumura, C., Gawlik, K.I., Allamand, V. and Durbeej, M. (2011) Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet., 20, 4891–4902. [DOI] [PubMed] [Google Scholar]
- 34. Girgenrath, M., Dominov, J.A., Kostek, C.A. and Miller, J.B. (2004) Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest., 114, 1635–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dominov, J.A., Kravetz, A.J., Ardelt, M., Kostek, C.A., Beermann, M.L. and Miller, J.B. (2005) Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum. Mol. Genet., 14, 1029–1040. [DOI] [PubMed] [Google Scholar]
- 36. Erb, M., Meinen, S., Barzaghi, P., Sumanovski, L.T., Courdier-Fruh, I., Ruegg, M.A. and Meier, T. (2009) Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J. Pharmacol. Exp. Ther., 331, 787–795. [DOI] [PubMed] [Google Scholar]
- 37. Girgenrath, M., Beermann, M.L., Vishnudas, V.K., Homma, S. and Miller, J.B. (2009) Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann. Neurol., 65, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sandri, M. (2010) Autophagy in skeletal muscle. FEBS Lett., 584, 1411–1416. [DOI] [PubMed] [Google Scholar]
- 39. Sandri, M. (2013) Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol., 45, 2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sandri, M., Coletto, L., Grumati, P. and Bonaldo, P. (2013) Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci., 126, 5325–5333. [DOI] [PubMed] [Google Scholar]
- 41. Kocaturk, N.M. and Gozuacik, D. (2018) Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol., 6, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malicdan, M.C. and Nishino, I. (2012) Autophagy in lysosomal myopathies. Brain Pathol., 22, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng, X., Zhang, X., Gao, Q., Ali Samie, M., Azar, M., Tsang, W.L., Dong, L., Sahoo, N., Li, X., Zhuo, Y. et al. (2014) The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med., 20, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J.A., Annunziata, F., Zare, H., Polishchuk, R., Puertollano, R., Parenti, G. et al. (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med., 5, 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barzilai-Tutsch, H., Dewulf, M., Lamaze, C., Butler Browne, G., Pines, M. and Halevy, O. (2018) A promotive effect for halofuginone on membrane repair and synaptotagmin-7 levels in muscle cells of dysferlin-null mice. Hum. Mol. Genet., 27, 2817–2829. [DOI] [PubMed] [Google Scholar]
- 46. Yu, L., Zhang, X., Yang, Y., Li, D., Tang, K., Zhao, Z., He, W., Wang, C., Sahoo, N., Converso-Baran, K. et al. (2020) Small-molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci. Adv., 6, eaaz2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fabian, L. and Dowling, J.J. (2020) Zebrafish models of LAMA2-related congenital muscular dystrophy (MDC1A). Front. Mol. Neurosci., 13, 122. doi: 10.3389/fnmol.2020.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gibbs, E.M., Horstick, E.J. and Dowling, J.J. (2013) Swimming into prominence: the zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J., 280, 4187–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Volpatti, J.R., Endo, Y., Knox, J., Groom, L., Brennan, S., Noche, R., Zuercher, W.J., Roy, P., Dirksen, R.T. and Dowling, J.J. (2020) Identification of drug modifiers for RYR1 related myopathy using a multi-species discovery pipeline. elife, 9, e52946. doi: 10.7554/eLife.52946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hall, T.E., Bryson-Richardson, R.J., Berger, S., Jacoby, A.S., Cole, N.J., Hollway, G.E., Berger, J. and Currie, P.D. (2007) The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A., 104, 7092–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupta, V.A., Kawahara, G., Myers, J.A., Chen, A.T., Hall, T.E., Manzini, M.C., Currie, P.D., Zhou, Y., Zon, L.I., Kunkel, L.M. et al. (2012) A splice site mutation in laminin-alpha2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PLoS One, 7, e43794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith, S.J., Wang, J.C., Gupta, V.A. and Dowling, J.J. (2017) A novel early onset phenotype in a zebrafish model of merosin deficient congenital muscular dystrophy. PLoS One, 12, e0172648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kagawa, M., Sato, N. and Obinata, T. (2006) Effects of BTS (N-benzyl-p-toluene sulphonamide), an inhibitor for myosin-actin interaction, on myofibrillogenesis in skeletal muscle cells in culture. Zool. Sci., 23, 969–975. [DOI] [PubMed] [Google Scholar]
- 54. Granato, M., van Eeden, F.J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C.P., Jiang, Y.J. et al. (1996) Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development, 123, 399–413. [DOI] [PubMed] [Google Scholar]
- 55. Bassett, D.I., Bryson-Richardson, R.J., Daggett, D.F., Gautier, P., Keenan, D.G. and Currie, P.D. (2003) Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development, 130, 5851–5860. [DOI] [PubMed] [Google Scholar]
- 56. Sardiello, M., Palmieri, M., di Ronza, A., Medina, D.L., Valenza, M., Gennarino, V.A., Di Malta, C., Donaudy, F., Embrione, V., Polishchuk, R.S. et al. (2009) A gene network regulating lysosomal biogenesis and function. Science, 325, 473–477. [DOI] [PubMed] [Google Scholar]
- 57. Gatto, F., Rossi, B., Tarallo, A., Polishchuk, E., Polishchuk, R., Carrella, A., Nusco, E., Alvino, F.G., Iacobellis, F., De Leonibus, E. et al. (2017) AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe disease. Sci. Rep., 7, 15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao, M., Smith, L., Volpatti, J., Fabian, L. and Dowling, J.J. (2019) Insights into wild-type dynamin 2 and the consequences of DNM2 mutations from transgenic zebrafish. Hum. Mol. Genet., 28, 4186–4196. [DOI] [PubMed] [Google Scholar]
- 59. Shen, D., Wang, X., Li, X., Zhang, X., Yao, Z., Dibble, S., Dong, X.P., Yu, T., Lieberman, A.P., Showalter, H.D. et al. (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun., 3, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu, M., Li, X., Walsh, S.W., Zhang, Y., Abais, J.M., Boini, K.M. and Li, P.L. (2013) Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am. J. Physiol. Cell Physiol., 304, C458–C466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. LaPlante, J.M., Ye, C.P., Quinn, S.J., Goldin, E., Brown, E.M., Slaugenhaupt, S.A. and Vassilev, P.M. (2004) Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV. Biochem. Biophys. Res. Commun., 322, 1384–1391. [DOI] [PubMed] [Google Scholar]
- 62. Nilius, B. and Owsianik, G. (2011) The transient receptor potential family of ion channels. Genome Biol., 12, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kiselyov, K., Chen, J., Rbaibi, Y., Oberdick, D., Tjon-Kon-Sang, S., Shcheynikov, N., Muallem, S. and Soyombo, A. (2005) TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J. Biol. Chem., 280, 43218–43223. [DOI] [PubMed] [Google Scholar]
- 64. Cerny, J., Feng, Y., Yu, A., Miyake, K., Borgonovo, B., Klumperman, J., Meldolesi, J., McNeil, P.L. and Kirchhausen, T. (2004) The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep., 5, 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Seglen, P.O. and Gordon, P.B. (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A., 79, 1889–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ito, S., Koshikawa, N., Mochizuki, S. and Takenaga, K. (2007) 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int. J. Oncol., 31, 261–268. [PubMed] [Google Scholar]
- 67. Wu, Y.T., Tan, H.L., Shui, G., Bauvy, C., Huang, Q., Wenk, M.R., Ong, C.N., Codogno, P. and Shen, H.M. (2010) Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem., 285, 10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fodor, E., Sigmond, T., Ari, E., Lengyel, K., Takacs-Vellai, K., Varga, M. and Vellai, T. (2017) Methods to study autophagy in zebrafish. Methods Enzymol., 588, 467–496. [DOI] [PubMed] [Google Scholar]
- 69. Johansen, T. and Lamark, T. (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy, 7, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ruparelia, A.A., McKaige, E.A., Williams, C., Schulze, K.E., Fuchs, M., Oorschot, V., Lacene, E., Meregalli, M., Lee, C., Serrano, R.J. et al. (2020) Metformin rescues muscle function in BAG3 myofibrillar myopathy models. Autophagy, 1–17. doi: 10.1080/15548627.2020.1833500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bago, R., Malik, N., Munson, M.J., Prescott, A.R., Davies, P., Sommer, E., Shpiro, N., Ward, R., Cross, D., Ganley, I.G. et al. (2014) Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J., 463, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Marsh, T. and Debnath, J. (2015) Ironing out VPS34 inhibition. Nat. Cell Biol., 17, 1–3. [DOI] [PubMed] [Google Scholar]
- 73. Jaber, N., Dou, Z., Chen, J.S., Catanzaro, J., Jiang, Y.P., Ballou, L.M., Selinger, E., Ouyang, X., Lin, R.Z., Zhang, J. et al. (2012) Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U. S. A., 109, 2003–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Backer, J.M. (2016) The intricate regulation and complex functions of the class III phosphoinositide 3-kinase Vps34. Biochem. J., 473, 2251–2271. [DOI] [PubMed] [Google Scholar]
- 75. Bonuccelli, G., Sotgia, F., Schubert, W., Park, D.S., Frank, P.G., Woodman, S.E., Insabato, L., Cammer, M., Minetti, C. and Lisanti, M.P. (2003) Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am. J. Pathol., 163, 1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bonuccelli, G., Sotgia, F., Capozza, F., Gazzerro, E., Minetti, C. and Lisanti, M.P. (2007) Localized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle, 6, 1242–1248. [DOI] [PubMed] [Google Scholar]
- 77. Gazzerro, E., Assereto, S., Bonetto, A., Sotgia, F., Scarfi, S., Pistorio, A., Bonuccelli, G., Cilli, M., Bruno, C., Zara, F. et al. (2010) Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am. J. Pathol., 176, 1863–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Winder, S.J., Lipscomb, L., Angela Parkin, C. and Juusola, M. (2011) The proteasomal inhibitor MG132 prevents muscular dystrophy in zebrafish. PLoS Curr., 3, RRN1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bonaldo, P. and Sandri, M. (2013) Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech., 6, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bodine, S.C. and Baehr, L.M. (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab., 307, E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Allamand, V. and Guicheney, P. (2002) Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur. J. Hum. Genet., 10, 91–94. [DOI] [PubMed] [Google Scholar]
- 82. Gut, P., Reischauer, S., Stainier, D.Y.R. and Arnaout, R. (2017) Little fish, big data: zebrafish as a model for cardiovascular and metabolic disease. Physiol. Rev., 97, 889–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Carmignac, V. and Durbeej, M. (2012) Cell-matrix interactions in muscle disease. J. Pathol., 226, 200–218. [DOI] [PubMed] [Google Scholar]
- 84. Gawlik, K.I. and Durbeej, M. (2020) A family of Laminin alpha2 chain-deficient mouse mutants: advancing the research on LAMA2-CMD. Front. Mol. Neurosci., 13, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bernardi, P. and Bonaldo, P. (2008) Dysfunction of mitochondria and sarcoplasmic reticulum in the pathogenesis of collagen VI muscular dystrophies. Ann. N. Y. Acad. Sci., 1147, 303–311. [DOI] [PubMed] [Google Scholar]
- 86. Debnath, J. (2009) Detachment-induced autophagy in three-dimensional epithelial cell cultures. Methods Enzymol., 452, 423–439. [DOI] [PubMed] [Google Scholar]
- 87. Fung, C., Lock, R., Gao, S., Salas, E. and Debnath, J. (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell, 19, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Grumati, P., Coletto, L., Sandri, M. and Bonaldo, P. (2011) Autophagy induction rescues muscular dystrophy. Autophagy, 7, 426–428. [DOI] [PubMed] [Google Scholar]
- 89. Hernandez-Caceres, M.P., Munoz, L., Pradenas, J.M., Pena, F., Lagos, P., Aceiton, P., Owen, G.I., Morselli, E., Criollo, A., Ravasio, A. et al. (2021) Mechanobiology of autophagy: the unexplored side of cancer. Front. Oncol., 11, 632956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lock, R. and Debnath, J. (2008) Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol., 20, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ballabio, A. and Bonifacino, J.S. (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol., 21, 101–118. [DOI] [PubMed] [Google Scholar]
- 92. Jahreiss, L., Menzies, F.M. and Rubinsztein, D.C. (2008) The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic, 9, 574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yu, L., Chen, Y. and Tooze, S.A. (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy, 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dowdle, W.E., Nyfeler, B., Nagel, J., Elling, R.A., Liu, S., Triantafellow, E., Menon, S., Wang, Z., Honda, A., Pardee, G. et al. (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol., 16, 1069–1079. [DOI] [PubMed] [Google Scholar]
- 95. Bilanges, B., Alliouachene, S., Pearce, W., Morelli, D., Szabadkai, G., Chung, Y.L., Chicanne, G., Valet, C., Hill, J.M., Voshol, P.J. et al. (2017) Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun., 8, 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Assereto, S., Stringara, S., Sotgia, F., Bonuccelli, G., Broccolini, A., Pedemonte, M., Traverso, M., Biancheri, R., Zara, F., Bruno, C. et al. (2006) Pharmacological rescue of the dystrophin-glycoprotein complex in Duchenne and Becker skeletal muscle explants by proteasome inhibitor treatment. Am. J. Physiol. Cell Physiol., 290, C577–C582. [DOI] [PubMed] [Google Scholar]
- 97. Briguet, A., Erb, M., Courdier-Fruh, I., Barzaghi, P., Santos, G., Herzner, H., Lescop, C., Siendt, H., Henneboehle, M., Weyermann, P. et al. (2008) Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J., 22, 4190–4200. [DOI] [PubMed] [Google Scholar]
- 98. Roczniak-Ferguson, A., Petit, C.S., Froehlich, F., Qian, S., Ky, J., Angarola, B., Walther, T.C. and Ferguson, S.M. (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal, 5, ra42. [DOI] [PMC free article] [PubMed]
- 99. Kimmel, C.B., Ballard, W.W., Kimmel, S.R., Ullmann, B. and Schilling, T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 100. Kokel, D., Cheung, C.Y., Mills, R., Coutinho-Budd, J., Huang, L., Setola, V., Sprague, J., Jin, S., Jin, Y.N., Huang, X.P. et al. (2013) Photochemical activation of TRPA1 channels in neurons and animals. Nat. Chem. Biol., 9, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.