

Summary
Development of liver fibrosis is paralleled by contraction of hepatic stellate cells (HSCs), the main profibrotic hepatic cells. Yet, little is known about the interplay of neprilysin (NEP) and its substrate neuropeptide Y (NPY), a potent enhancer of contraction, in liver fibrosis. We demonstrate that HSCs are the source of NEP. Importantly, NPY originates majorly from the splanchnic region and is cleaved by NEP in order to terminate contraction. Interestingly, NEP deficiency (Nep−/−) showed less fibrosis but portal hypertension upon liver injury in two different fibrosis models in mice. We demonstrate the incremental benefit of Nep−/− in addition to AT1R blocker (ARB) or ACE inhibitors for fibrosis and portal hypertension. Finally, oral administration of Entresto, a combination of ARB and NEP inhibitor, decreased hepatic fibrosis and portal pressure in mice. These results provide a mechanistic rationale for translation of NEP-AT1R-blockade in human liver fibrosis and portal hypertension.
Keywords: portal hypertension, renin-angiotensin system, neuropeptide Y receptor 1, hepatic stellate cell, cirrhosis, neprilysin, neuropeptide Y
Graphical abstract
Highlights
-
•
Proteolysis of NPY by NEP promotes liver fibrosis via NPY1 receptor blockade in HSCs
-
•
In the liver, NPY enhances contraction induced by AT1R stimulation
-
•
Administration of Entresto decreases liver fibrosis and portal pressure
Neprilysin is upregulated in fibrosis and activated hepatic stellate cells (HSCs). Nep−/− reduced fibrosis due to increased levels of neuropeptide Y. Ortiz et al. describe a new player in fibrosis and deliver the molecular rationale for combined neprilysin and angiotensin inhibition as treatment of human liver fibrosis with portal hypertension.
Introduction
Fibrosis is a common pathological feature of most chronic diseases, and it has a major impact on morbidity and mortality.1 Progressive liver fibrosis is the main reason for development of portal hypertension and severe complications, such as ascites and variceal bleeding.2 Hepatic stellate cells (HSCs) are crucially involved in fibrosis and portal hypertension since activated HSCs deposit extracellular matrix and contract.3 Neurohumoral vasoconstrictor systems, such as the renin-angiotensin system4 and the endothelin system,5 can activate HSCs and aggravate fibrosis and portal hypertension.6 Angiotensin-converting enzyme (ACE) and angiotensin II (Ang II) levels, in particular, are dramatically upregulated7 and mainly involved in HSC activation and fibrosis.8,9 To date, strategies to blunt fibrosis or portal hypertension using Ang II receptor, type 1 (AT1R) blocker or ACE inhibitors have failed in the clinical setting, indicating the complexity of these two processes.10
Neuropeptide Y (NPY), a 36-amino-acid peptide, has been described as an enhancer of contraction mediated by AT1R and α1 adrenoceptor. Interestingly, NPY is also a potent enhancer of contraction in extrahepatic vessels in cirrhosis, which are known to be hypocontractile.11 However, NPY does not appear to elicit increased hepatic vasoconstriction in cirrhosis, possibly due to the degradation of NPY in the liver via endopeptidases, such as neprilysin (NEP).12 In addition dipeptidylpeptidase 4 (DPP4) and fibroblast activation protein (FAP) have been described to degrade NPY.13,14
The present study demonstrates that systemic levels of NPY increase with increased severity of liver disease and that NPY reaches the liver via portal circulation.15 However, in the presence of NEP, which also increases in the liver in line with liver fibrosis severity, NPY is degraded to fragments that block NPY1 receptor (Y1R) in HSCs and in turn induce fibrosis. NEP deficiency blunts fibrosis but aggravates portal hypertension, which can be decreased by AT1R blockers and ACE inhibitors. Thus, combinations of NEP inhibitors with AT1R blockers or ACE inhibitors decreased fibrosis as well as portal pressure. As a translational approach, oral administration of Entresto (sacubitril/valsartan) to two different mice models of metabolic liver fibrosis showed that simultaneous inhibition of NEP and AT1R decreases fibrosis as well as portal pressure in the liver.
Results
NPY protein is degraded in the liver, and its levels are associated with severity of chronic liver disease
Since the vasoconstrictor NPY may play a substantial role in the hyperdynamic circulation during cirrhosis,11 we analyzed NPY protein levels in different vascular compartments in human cirrhosis. First, ELISA analysis demonstrated that in the peripheral circulation, NPY serum levels were increased in cirrhotic patients compared with healthy individuals (Figure 1A). Moreover, serum NPY levels increased with increasing severity of liver disease, as indicated by the Child-Pugh score (Figure 1B). To investigate the source of NPY, we compared circulating NPY from the portal vein (PV) (inflow to the liver) and the hepatic vein (HV) (outflow out of the liver) in samples from both compartments taken simultaneously in 16 patients with alcoholic liver cirrhosis and found that, compared with HV levels, serum NPY levels were increased to levels found in PV (Figure 1C). Next, NPY levels from the hepatic artery (HA) were compared with levels found in the HV by analyzing samples also collected simultaneously in both compartments from a further 21 patients (Figure 1D). We found no significant difference in NPY levels between these two compartments. Overall, the gradient of NPY levels over liver (PV-HV) was significantly higher than the gradient of the rest of the body (HA-HV) (Figure 1E), suggesting that most of the NPY derives from the gastrointestinal tract (as described by Dietrich et al.16) and reaches the liver through the PV and that it may be degraded to some extent, as suggested by the lower levels in the liver outflow. Independently, in silico analysis of human liver tissue showed a highly statistically significant direct correlation between the mRNA levels of NPY and the fibrosis marker TGFB1 (Figure 1F).
Figure 1.

NPY levels correlate with liver disease in mice and in human hepatic cirrhosis
(A) Serum NPY ELISA from control and cirrhotic patients undergoing transjugular intrahepatic portosystemic shunt (TIPS) treatment, n = 7 (TIPS-group, cirrhotic patients) and n = 6 (controls), ∗∗p < 0.02 for cirrhotic patients vs. healthy individuals (absolute values).
(B) Serum NPY ELISA in cirrhotic patients shows increased NPY in Child-Pugh class C compared with Child-Pugh class A patients, n = 5 per group, ∗p < 0.05 for Child-Pugh class C vs. Child-Pugh class A cirrhotic patients. Results are expressed as mean ± standard error of the mean (SEM).
(C) Serum NPY ELISA from portal vein (PV, hepatic inflow) and hepatic vein (HV, hepatic outflow) in TIPS patients, n = 16 per group, ∗∗p < 0.02 for PV vs. HV.
(D) Serum NPY ELISA from hepatic vein (HV) and hepatic artery (HA) in n = 21 per group, n.s. (not significant).
(E) NPY levels compared with portal vein and hepatic artery, ∗∗∗p < 0.001.
(F) In silico analysis of hepatic mRNA NPY levels compared with TGFB1 expression in human liver tissues.
(G) Hepatic Nep and Col1A1 mRNA expression in patients with chronic liver disease; number of XY pairs = 17, r = 0.7472, p = 0.0006.
(H and I) Hepatic Nep mRNA expression from bile duct ligation (BDL)-treated and CCl4-treated WT (C57BL/6) mice for 2 and 4 weeks, respectively, ∗∗p < 0.02 for BDL- or CCl4-treated vs. corresponding control mice. Data were normalized to the expression of 18S RNA.
(J) Liver homogenates NPY ELISA from WT and Nep−/− mice, control vs. CCl4, ∗p < 0.05.
(K) Morphometric analysis of Sirius red staining of livers from WT mice compared with Nep−/− with and without NPYFL (left panel) and WT BDL mice vs. Nep−/− BDL with and without NPYFL (right panel). n = 5 per group, ∗∗p < 0.01, ∗∗∗p < 0.001, and n.s. (non-significant).
(L) Liver sections stain with Sirius red. Scale bar: 200 μm.
(M) Col1A1 mRNA expression of WT mice comparing non-treated controls vs. animals that were injected with full-length NPY, ∗∗p < 0.01. Col1A1 mRNA expression of Nep−/− mice after profibrotic stimuli (BDL) comparing control vs. Nep−/− after injecting full-length NPY, ∗∗p < 0.01. Data were normalized to the expression of 18S RNA.
NEP is described as a degrading enzyme of NPY.12 mRNA from hepatic Nep correlated with mRNA levels of Col1a1, the key marker of fibrosis, in a further 17 patients with different stages of liver fibrosis (r = 0.742, p = 0.0006), demonstrating that in humans, Nep expression is strongly associated with severity of liver fibrosis (Figure 1G). Further analysis of NEP levels and portal hypertension in 125 cirrhosis patients showed that the expression of NEP enzyme does not correlate with the portal pressure (Figure S1A). These results underline the fact that NEP may be involved in promoting distinct mechanisms toward fibrosis and portal hypertension.
Hepatic Nep expression could be reproduced in two well-known mouse models of liver fibrosis. Nep mRNA levels were significantly induced 2 weeks after bile duct ligation (BDL) or 4 weeks after CCl4 intoxication in wild-type (WT) mice compared with their respective controls (Figures 1H and 1I). Moreover, NPY protein levels were higher in the livers of Nep−/− compared with WT in control and fibrotic mice (Figures 1J and S1B). To investigate the role of NPY in fibrosis, we injected the full-length NPY (NPYFL) in WT mice and Nep−/− as well as WT and Nep−/− mice after BDL operation. Sirius red staining was performed to assess the fibrotic phenotype of these livers (Figures 1K and 1L). The differences on collagen induction were quantified showing important differences when NPYFL was administrated. Furthermore, we analyzed hepatic mRNA levels of Col1a1, which were significantly increased when NPYFL was administrated. On the contrary, when the NPYFL was administrated to Nep−/− mice after strong profibrotic stimuli (BDL), mRNA levels of Col1a1 were significantly reduced (Figure 1M).
Altogether, our results demonstrate that NPY, depending on the presence or absence of NEP enzyme, may control fibrosis in mice. Therefore, it is important to understand the hepatic cell type that expresses NEP.
NEP-mediated proteolysis of NPY uncouples the contractile and profibrotic response of HSCs
Cellular distribution of NEP in primary liver cells revealed that the hepatic expression of Nep is exclusively observed in HSCs, with a further increase after their activation in vivo (after BDL). In hepatocytes, Kupffer cells, or liver sinusoidal endothelial cells, NEP was not detected (Figure 2A). This is not surprising since HSCs are the main profibrotic and contractile cells in liver fibrosis. By contrast, the amount of NPY released by HSCs was negligible compared with levels in fetal calf serum (FCS) assessed by ELISA analysis in media conditions of quiescent or activated HSCs (Figure 2B), and it was much higher than in hepatocytes, as shown recently.17 Results showed a 500-fold increase of NPY levels in FCS compared with activated HSC-conditioned medium (FCM) and HSC quiescence-conditioned medium. Therefore, HSCs, whether quiescent or activated, do not express large amounts of NPY. These results paralleled the ones obtained in the Human Liver proteome database (Figure S3A). We analyzed the relative abundance of NPY protein among the different hepatic cell types, which confirms that the HSCs are the cells in which NPY is almost undetectable.
Figure 2.

Nep−/− shows more contraction when treated with NPY than in WT HSC
(A) Hepatic Nep mRNA expression in hepatocytes (HEPs), Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), and hepatic stellate cells (HSCs) from healthy (Ctrl) and BDL-treated WT mice. qPCR data were referred to HEPs and normalized to the expression of 18S RNA.
(B) NPY ELISA from fetal calf serum (FCS, n = 3), human serum control and cirrhosis (n = 5, n = 8), fibroblast-conditioned medium (FCM, n = 3), and supernatants of conditioned hepatic stellate cells (HSC, n = 3). Dulbecco’s Modified Eagle Medium (DMEM, 0% FCS) was used as negative control (Neg. Ctrl). Results are expressed as mean ± standard deviation (SD).
(C–F) Nep−/− and WT (C57BL/6) mice were used to isolate HSCs, and cells were treated with NPY 1nM. mRNA expression levels of Tgfβ1, Col1A1, and Acta2 were analyzed after 24 h treatment. Results are expressed as mean ± SEM; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(G) Contraction assay of primary HSCs cultured on collagen gels and treated with and without recombinant NPY 0–100 nM for 0–48 h. The relative area was calculated by recording the diameter change of gels at several time points with a digital camera at a fixed distance above the gels. Results are expressed in percentage of initial gel surface and as mean ± SEM; n = 3–6 per group. ∗p < 0.05 compared to control.
(H) Migration assay of WT and Nep−/− HSCs. Nep−/− migrated faster than the WT HSCs after 24 h. Results are expressed as mean ± SEM; ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
(I) In situ liver perfusion of cirrhotic animals. For pre-contraction of the livers, methoxamine (100 μM) was used, and then, increasing concentrations of full-length NPY and cleaved C-terminal fragments of NPY (cNPY) (as indicated) were used to measure the perfusion pressure of the livers (n = 3 per group). ∗p < 0.05 compared to MTX.
To investigate the role of NPY and its relationship with NEP, WT, and Nep−/−, HSCs were treated with recombinant NPY (Figure 2C) after dose-finding experiments (Figure S2A). The mRNA levels of fibrosis markers Tgfβ1 and Col1a1 increased in the presence of NPY in control WT HSCs. In the absence of NEP (Nep−/−), Tgfβ1 as well as Col1a1 mRNA levels decreased after incubation with NPY. This suggests that NPY acts in a profibrotic manner in the presence of NEP (Figures 2D and 2E). As described before in vivo, similar profibrotic phenotype was observed in vitro.
During fibrogenesis, activation of HSCs is accompanied by α-smooth muscle actin (α-SMA) expression. Therefore, we investigated the role of NPY cleavage by NEP in the contractile phenotype of HSCs by measuring Acta2 mRNA levels (Figure 2F). In the absence of NEP, NPY induced Acta2 expression compared with the WT control. Primary HSCs were cultured on collagen gels and treated with physiological concentrations of NPY.18 In primary mouse HSCs, NPY induced more HSC contraction in Nep−/− than in WT, as assessed by a decrease of gel surface (Figure 2G). To elucidate the signaling by which NPY increases contraction in Nep−/− HSC, we investigated one key player in this process,11,19 namely ROCK (regulator of contractile turn) and the readout for its activity, which is phosphorylated moesin (p-moesin), in primary WT and Nep−/− HSCs treated with NPY (Figure S3B). As such, ROCK protein and p-moesin expression detected by western blot were increased in Nep−/− and increased further after NPY incubation compared with WT cells. In the presence of NEP, NPY seems to induce not only contraction (Figure 2G) but also contractile phenotype (Figure S3B). To further characterize this increase, a migration assay was performed (Figure 2H). After 24 h, Nep−/− HSCs migrated faster than the WT. This result confirms that Nep−/− HSCs are, in addition to being contractile, more migrative, which explains why they express more α-SMA. Consequently, presence of NEP might prevent NPY-induced contraction and migration in HSC, which may be explained by proteolysis of NPY. Moreover, in order to test a direct intrahepatic vasoconstrictive effect of NPY and cNPY, we performed isolated in situ liver perfusion in cirrhotic rats. NPY was administrated at increasing doses after initial pre-contraction with the α-adrenergic agonist methoxamine (Figure 2I). Thus, NPY, but not cNPY, significantly increased hepatic vascular resistance in a dose-dependent fashion.
In conclusion, we can demonstrate that NPY induces contraction in vitro, as shown by HSC contraction assay, and in situ, as shown by isolated liver perfusion.
NPY cleavage mediated by NEP induces fibrogenesis in HSCs
It is tempting to assume that NPY cleavage by NEP generates profibrotic fragments acting on HSCs. Therefore, NPY in vitro cleavage by NEP was performed as previously described.18 Primary Nep−/− HSCs were treated with NPY or NEP-cleaved NPY (cNPY) (Figure 3A). We could confirm that cNPY, but not NPYFL, increases Col1a1 mRNA expression in HSCs and decreases Acta2 expression (Figure 3B), which was confirmed at protein level (Figure 3C).
Figure 3.

Synthetic NPY fragments derived from NEP proteolysis induce fibrogenesis in HSC
(A) Nep−/− mice were used to isolate primary HSCs and were treated with full-length NPY (1 nM) or their respective cleaved fragments (cNPY) (30 μM). After 24-h treatment, cells were used for further analysis.
(B) mRNA expression levels of Col1A1 and Acta2 were analyzed after 24-h treatment, ∗∗p < 0.01. Data were normalized to the expression of 18S RNA.
(C) Western blot analysis and quantifications of Col1a1 and α-SMA protein expression using GAPDH as a loading control.
(D–G) Nep−/− HSCs were fixed after treatment for immunofluorescence with collagen I and αSMA antibodies. Confocal microscopy was used to detect these proteins. Changes in collagen I protein were detected in cells treated with the cNPY fragments as well as the changes detected in αSMA protein in the cells treated with full-length NPY; ∗p < 0.05 and ∗∗p < 0.01 compared to control. Representative images and fluorescence quantification from three independent experiments are shown. Images were taken with the 20x confocal objective that corresponds to a scale bar of 50 μm.
Predicted NEP cleavage should generate N-terminal (1–20 and 1–30) fragments as well as the shorter NPY C-terminal (21–36 and 31–36) fragments (Figure S2B). Intensive efforts using liquid chromatography-mass spectrometry were performed to identify in vitro the short C-terminal NPY fragments released by NEP cleavage. Under the assay conditions, the amount of NPY was decreased when incubated with NEP. However, we were unable to identify the fragments, probably due to their small mass and volatile properties (Figures S4A–S4D). It has been shown that N-terminal fragments of NPY protein bind less strongly to the receptor than C-terminal NPY fragments.20 Therefore, we investigated by confocal microscopy the effects of NPY C-terminal fragments 21–36 and 31–36 regarding their profibrogenic potential on primary HSCs from Nep−/− mice and could confirm that both shorter fragments increase collagen expression in HSCs (Figures 3D and 3F). Moreover, NPYFL increased αSMA expression (Figures 3E and 3G) as already elucidated. To further investigate the role of NPY C-terminal fragments in vivo, we injected intraperitoneally (i.p.) either NPYFL or its fragments in WT mice (Figure S5A). Analysis of portal pressure in these animals showed an increase when both NPY variants were injected (Figure S5B). mRNA expression from Acta2 was significantly induced when NPYFL was injected and significantly reduced when NPY fragments were administrated (Figure S5C). mRNA expression of the main fibrotic markers, Col1a1 and Tgfβ1, was induced when NPYFL was injected, but the effect of NPY fragments aggravated the fibrotic phenotype of WT mice. Altogether, both in vitro and in vivo experiments confirm the dual role of NEP/NPY axis in fibrosis and contraction.
The question that arises is whether the effect of NPY fragments in the presence of NEP is receptor mediated or not.
NPY cleavage mediated by NEP induces fibrogenesis via Y1R in HSC
To analyze a possible receptor-mediated effect, we investigated hepatic expression of NPY receptors 1, 2, and 5 (Y1R, Y2R, and Y5R)21 in fibrotic mice and humans. We found hepatic upregulation of Y1R, Y2R, and Y5R mRNA expression in cirrhotic patients (Figure 4A), However, negligible Y2R and Y5R mRNA expressions were found in activated HSCs (Figure 4B), suggesting that only Y1R is involved in the NPY effect in HSC. Similarly, upregulation of hepatic Y1R mRNA expression was observed in fibrotic BDL or CCl4-Nep−/− mice (Figure 4C). Yet, Dpp4 and FAP may play a role in the processing of the different NPY fragments, mostly NPY (3–36).13,14,22 Hepatic mRNA expression in WT and Nep−/− fibrotic mice demonstrated that the expression of Dpp4 was highly reduced, and FAP was not significantly induced in Nep deficiency (Figure S6A). These results were confirmed using transcriptomic data from human primary HSCs (Figure S6B). Thus, Dpp4 or FAP expression during fibrogenesis seems to be rather stable and is not likely to influence our results.
Figure 4.

Cleavage of NPY by NEP shifts the physiological response of Y1R in HSC
(A) Hepatic Npy1r, -2r, and -5r mRNA levels in healthy and cirrhotic patients. Results are expressed as mean ± SEM; n = 5–10 per group, ∗∗∗p < 0.001 for cirrhotic vs. healthy patients.
(B) Npy1r, -2r, and -5r mRNA levels in WT mouse HSCs.
(C) Hepatic Npy1r mRNA levels in CCl4- and BDL-treated Nep−/− and WT mice. Results are expressed as mean ± SEM; ∗p < 0.05 for CCl4 vs. control and ⋅p < 0.05 for Nep−/− vs. WT mice.
(D and E) Structural models of the Y1R-NPY (21–36) and Y1R-NPY (31–36) complexes. Y1R is represented in blue and its interacting residues in yellow. The NPY fragments are shown in red, while the NPY aa residues are colored orange. The distances between residues are indicated by dashed lines.
(F) Primary Nep−/− HSCs were isolated and treated with NPY and cNPY. After 24 h, cAMP concentrations were analyzed in response to cNPY and full-length NPY. Results are expressed as pmol cAMP per mg of total protein. Results are expressed as mean ± SEM; n = 3–4 per group, ∗p < 0.05 for cNPY-treated vs. full-length NPY-treated Nep−/− HSC.
(G) Western blot analysis from Nep−/− HSC control, NPYFL, and cNPY and quantification of p-ERK and p-moesin protein expression using GAPDH as a loading control.
(H) Diagram of NPYFL (left) and cNPY (right) showing the activation of the receptor (NPY1R). The binding of NPY and cNPY activates the inhibitory effect of G protein and inhibits the enzyme adenylate cyclase (AD), which in turn decreases the production of cAMP. Downstream pathways regulated by cAMP, p-ERK, and p-moesin are downregulated or upregulated, depending on the presence of NPYFL (left) or cNPY (right). NPYFL (left) decreases fibrosis and increases contraction, and cNPY (right) increases fibrosis and decreases contraction in the absence of NEP.
Next, we investigated whether NPY C-terminal (21–36 and 31–36) fragments have profibrogenic potential on HSCs through Y1R modulation (Figure S2B), since N-terminal fragments of NPY protein bind less strongly to the receptor than C-terminal NPY fragments.17 Therefore, we further analyzed the biomolecular interaction between NPY fragments and Y1R. We docked each of the C-terminal fragments NPY (21–36) and NPY (31–36) to the receptor using the high ambiguity driven biomolecular docking (HADDOCK)23 program and compared the top best cluster results (see Table S1) with the structure of Y1R in complex with the well-known Y1R-antagonist BIBO3304. The docking results suggested that cNYP fragments act as partial antagonist of Y1R, contrary to the agonistic effect of NPYFL.16,23 In particular, both cNPY fragments were unable to establish an interaction between their C-terminal Tyr36 and the Y1R residue Gln120 (i.e., distance >3.0 Å, Figures 4D and 4E) when compared with the close interaction formed by these two residues in the NPY-Y1R complex (<3.0 Å).24 Gln120 has been proposed to be essential for receptor activation.24,25,26 Moreover, manual docking of the antagonist used in this work, BIBO3304 (Figure S7B), as well as the crystal structure of UR-MK29924 (another antagonist whose chemical structure mimics the cNPY residues Arg35 and Tyr36, see Figure S7A) also exhibited distances longer than 3.0 Å with Y1R-Gln120. All in all, the computational modeling further supports our hypothesis that cNPY fragments act as partial antagonists of Y1R.
We analyzed the cAMP generation in response to cNPY fragments and NPYFL in primary Nep−/− HSCs (Figure 4F). Importantly, activation of Y1R by NPYFL peptide activates the inhibitory G protein GI /Go family, which in turn inhibits effector enzyme adenylate cyclase and decreases production of cAMP.27 In our experiments, the cAMP ELISA results showed a reduction in the cAMP measurements when NPYFL and cNPY peptide interacts with the receptor. cAMP signals are translated into action by different effector proteins, and it shows that this pathway triggers cellular effects, at times with opposing functional outcomes. To further elaborate the downstream pathway of the Y1R after activation with either NPYFL or its fragments, we analyzed the phosphorylation of ERK and moesin (p-ERK and p-moesin), pathways that are regulated by cAMP.28,29 Nep−/− HSCs were treated with NPYFL and its C-terminal fragments and blotted against p-ERK and p-moesin (Figure 4G). We observed that p-ERK was induced in cells treated with the cNPY fragments, indicating the ability of cNPY to induce fibrosis. The opposite effect was detected with p-moesin, which showed a decrease when cells were treated with the cNPY.
In conclusion, the ability of the cNPY but not of NPYFL to block and induce fibrosis confirmed the involvement of Y1R in NPY signaling, indicating that cleavage of NPY mediated by NEP shifts the physiological response of Y1R from contraction to relaxation and from an antifibrogenic to a profibrogenic response in HSCs (Figure 4H).
Nep deletion protects against liver fibrosis in vivo in two different models of fibrosis in mice
In order to confirm our findings in vivo, we investigated the role of NEP deficiency in liver fibrosis and portal hypertension in two well-established models of liver fibrosis.30 Sirius red staining (Figures S8A and S8B) and hydroxyproline content measurement (Figure S8C) were performed to analyze hepatic fibrosis in BDL (2 weeks) and CCl4-intoxicated (4 weeks) mice and showed that Nep−/− mice developed less fibrosis than fibrotic WT mice.
Col1a1 mRNA expression (Figure S8D) confirmed these findings, despite similar Tgfβ1 mRNA expression (Figure S9A). In addition, we performed gene ontology analysis on human liver transplantation transcriptomics data to elaborate fibrotic pathways linked to Nep expression (Figure S9B). In liver with lower Nep expression, the collagen expression is downregulated. This was confirmed at the protein level of the downstream effectors of Tgfβ1 pathway, the p-SMAD2/3 proteins (Figure S10). Also, in Nep deletion, p-SMAD2/3 proteins were significantly decreased compared with WT mice. These results suggest that the Tgfβ pathway is involved in the effects observed in Nep−/− and confirms that Nep deficiency protects from fibrosis in two models of liver fibrosis in mice.
To further evaluate the in vivo role of Nep deletion in HSC activation, α-SMA protein and mRNA expression, a marker of HSC activation,31 were analyzed in fibrotic BDL and CCl4-Nep−/− and WT mice. Immunohistochemistry (IHC) analysis revealed higher α-SMA staining in the fibrous septae (Figure 5A), but not at perisinusoidal spaces of BDL- and CCl4-induced fibrosis Nep−/− mice compared with WT (Figure 5B). The hepatic upregulation of α-SMA staining in the fibrous septae was confirmed using qPCR and western blot analysis in Nep−/− mice, when compared with fibrotic BDL and CCl4-WT mice (Figures 5C–5E). As we did before for NPY, independent in silico analysis in human non-tumorous liver tissue confirmed this statistically significant inverse correlation between the decreasing mRNA levels of NEP and the induction of ACTA2 mRNA expression (Figure 5D).
Figure 5.

Nep deletion increases HSC activation in BDL- and CCl4-treated mice
(A and B) Hepatic αSMA IHC in BDL- and CCl4-treated Nep−/− mice and WT mice with their respective morphometric analysis. Scale bar: 200 μm.
(C) Hepatic Acta2 mRNA levels in BDL- and CCl4-treated Nep−/− mice and WT mice. All data were normalized to the expression of 18S RNA.
(D) In silico analysis of NEP (MME) levels compared with ACTA2 expression in human non-tumorous liver tissue samples.
(E) Western blot analysis from controls vs. CCl4-treated Nep−/− mice developed with αSMA antibody. The expression of GAPDH was used as a loading control. Results are expressed as mean ± SEM; n = 5 per group, ∗p < 0.05,∗∗p < 0.01 for BDL- or CCl4-treated vs. corresponding control mice.
Since HSC proliferation is a crucial process in the fibrotic liver, Ki67 IHC and proliferating cell nuclear antigen western blot analysis were performed in livers from fibrotic BDL and CCl4-Nep−/− mice (Figures S11A and S11B). Nep deletion decreased hepatic proliferation in Nep−/− mice compared with fibrotic CCl4 WT mice (Figure S11B). To analyze whether the observed effect was mediated by other effectors influenced by NEP, specific markers of the endothelin system and angiogenesis were characterized in these models (Figures S12A–S12E). There were no clear indications at the mRNA expression for an involvement of endothelin system or VEGFα. We also assessed the inflammatory response in these livers (Figure S13). We could not find any significant change in the inflammatory response in the absence of Nep.
We therefore conclude that while NEP deficiency prevents fibrosis via NPY-mediated effects, it may aggravate the pro-contractile phenotype as observed in vitro and as suggested by αSMA expression in vivo.
Aggravation of portal pressure in Nep−/− mice is hampered by AT1R antagonists or ACE inhibitors
To investigate the in vivo role of NEP in the degree of portal hypertension, we analyzed portal pressure in WT and Nep−/− mice and in BDL- and CCl4-induced fibrotic mice (Figure 6A). Similar to the in vitro phenotype, portal pressure was higher in control and fibrotic BDL and CCl4 Nep−/− mice compared with their respective WT animals, suggesting that the presence of NEP plays a role in lowering portal pressure.
Figure 6.

Nep deletion, AT1R blockage, or angiotensin-converting enzyme inhibition reduces portal hypertension
(A) Portal pressure is significantly increased in Nep−/− mice under basal conditions further enhanced in CCl4- and BDL-treated Nep−/−mice compared with WT mice. Results are expressed as mean ± SEM; n = 6 per group, ∗p < 0.05, ∗∗p < 0.01 for BDL- or CCl4-treated mice vs. corresponding control mice. All data were normalized to the expression of 18S RNA.
(B) During liver fibrosis, NPY is cleaved by NEP producing two different cNPY peptides, which induce a fibrogenic response mediated by Y1R (red arrows). Full-length NPY enhances contraction through activation of the Y1R and Gαi-adenylate cyclase (AD) (green arrows). Ang II, agonist of AT1R, results in contraction, further increased by the activation of Y1R. In the absence of NEP (gray) and AT1R blockage (losartan) or ACE inhibition (captopril) (red), full-length NPY will decrease fibrosis. ANG II, which results in contraction (gray), will not exert its function due to the administration of either captopril (red) or losartan (red). In the absence of NEP, full-length NPY will activate Y1R and decrease the fibrogenic response previously mediated by cNPY (green arrows). Deletion of NEP in combination with ACE inhibitor or AT1R blockage reduces portal pressure and fibrosis.
(C and D) Sirius red staining in captopril- and losartan-treated CCl4Nep−/− mice and their respective morphometric analysis. Scale bar: 200 μm.
(E and F) Portal pressure measurement (mm Hg) in captopril- and losartan-treated CCl4Nep−/− mice and hepatic Col1a1 mRNA levels. Results are expressed as mean ± SEM; n = 6 per group, ∗∗p < 0.01 for CCl4-treated mice vs. corresponding control mice and n = 6 per group, ∗p < 0.05, and ∗∗∗p < 0.001 for CCl4-treated mice vs. corresponding control mice.
We suspected the effect to be mediated by the increased levels of NPYFL, which is an enhancer of contraction mediated by Ang II via adenylate cyclase activation (Figure 6B). Therefore, we further evaluated the effect on portal pressure in addition to the decreased fibrosis in Nep−/− mice, and we administrated captopril (ACE inhibitor32) or losartan (AT1R blocker8) in fibrotic Nep−/− mice. As expected, portal pressure and Col1a1 mRNA expression were significantly reduced in captopril- and losartan-treated CCl4-intoxicated Nep−/− mice, compared with non-treated fibrotic Nep−/− mice (Figures 6E and 6F). Sirius red staining (Figures 6C and 6D) confirmed similar fibrosis in captopril- and losartan-treated Nep−/− mice, suggesting that the aggravation of portal hypertension associated with Nep deletion can be abrogated by AT1R antagonism.
NEP has a complex enzymatic profile33; it cleaves and reduces levels of angiotensin I, resulting in reduced production of Ang II. Thus, NEP may lower portal pressure via reduced stimulation of AT1R (Figure 6B). However, NEP cleaves NPY into cNPY fragments, which—via NPY1R—induce either contraction (NPY), enhancing the effect of AT1R, or fibrosis (cNPY).
Thus, we demonstrated that Nep−/− decreases fibrosis, while additional AT1R antagonism or ACE inhibition blunts portal hypertension. These data identify the combination of NEP inhibitors with anti-Ang II drugs as a powerful option for the treatment of fibrosis with portal hypertension.
Dual angiotensin II type I receptor neprilysin inhibition with Entresto in mice improves fibrosis and portal pressure in the liver
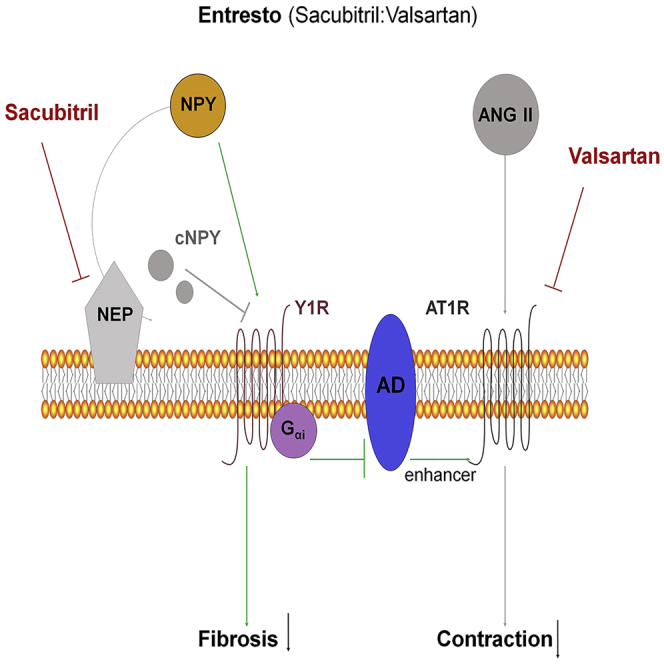
Entresto (sacubitril/valsartan) is an AT1R antagonist combined with NEP inhibitor, which is used to treat patients with chronic heart failure.34 To confirm our previous results, we investigated the therapeutical effect of oral administration of Entresto in models of liver fibrosis in mice. The inhibition of NEP by sacubitril will lead to increased levels of NPYFL, which in turn, will decrease fibrosis. Also, NPYFL bound to its receptor activates adenylate cyclase enhancing contraction via AT1R. Due to valsartan, AT1R inhibition will decrease contraction (Figure 7A).
Figure 7.

Oral administration of Entresto to fibrotic mice improves fibrosis and lowers portal hypertension
(A) Diagram of Entresto effects. Dual inhibition of NEP (sacubitril) (red) and AT1R (valsartan) (red) decreases fibrosis and contraction together with portal pressure. Although full-length NPY enhances contraction through activation of the Y1R and Gαi-adenylate cyclase (AD) (green arrows), the blockade of AT1R by valsartan cannot be enhanced by the action of AD, so contraction is decreased.
(B) Portal pressure measurements showed a significant reduction in both Entresto-treated mice groups. Results are expressed as mean ± SEM; n = 5 per group, ∗∗p < 0.01 for Entresto-treated mice vs. corresponding control mice.
(C) Hepatic Col1a1 mRNA levels in Entresto-treated mice compared with control mice, n = 5/6 per group, ∗∗p < 0.01. All data were normalized to the expression of 18S RNA.
(D) Liver sections stained with Sirius red and hepatic αSMA IHC with their respective morphometric analysis. Scale bar: 200 μm.
(E) Western blot analysis of Col1a1 and αSMA proteins, in Entresto-treated mice compared with their respective controls. The expression of GAPDH was used as a loading control. n = 3 per group, ∗p < 0.05,∗∗p < 0.01 for Col1a1 or αSMA vs. GAPDH.
Two different models were used: ethanol in water with CCl4 injection (i.p.) and Western diet with CCl4 (i.p.) for 7 weeks. At the end of the experiment, we measured the portal pressure of the animals (Figure 7B). Mice treated with Entresto showed a significant decrease in portal pressure compared with placebo group. To address the extent of liver fibrosis, we analyzed the hepatic Col1a1 mRNA expression using qPCR (Figure 7C). In both fibrosis models, Entresto significantly reduced the expression of Col1a1 when compared with their respective placebo animals. Sirius red staining confirmed the reduction of collagen induced by Entresto (Figure 7D). To analyze the activation of HSCs of the livers, we performed α-SMA staining (Figure 7D). IHC staining showed a significant decrease in α-SMA accumulation in the fibrotic septae treated with Entresto, suggesting that administration of Entresto can stop the progression of liver fibrosis and activation in HSCs.
Protein levels were analyzed to confirm the reduction in collagen and α-SMA (Figure 7E). Western blot results showed a significant decrease in collagen as well as α-SMA proteins in both models after treatment with Entresto. In order to compare the efficacy of Entresto treatment with the use of its single components, in vivo experiments were performed by treating the mice with valsartan or sacubitril respectively (Figures S14A–S14C). Neither valsartan nor sacubitril were able to significantly reduce portal pressure or stop the progression of liver fibrosis.
These data demonstrate that Entresto administration significantly reduces liver fibrosis, activation of HSCs, and portal pressure, thus confirming the results using Nep−/− and losartan or captopril (Figures 6B and 7A).
Discussion
In scarring tissues, myofibroblasts simultaneously show a profibrotic and contractile phenotype. This is also the case in chronic liver disease, where contraction and its clinically relevant form portal hypertension typically occur in parallel with progressive fibrosis. This study identifies NEP as one of the key players responsible for the simultaneous occurrence of these two mechanisms. In the presence of NEP, NPY degradation occurred and NPY fragments blocked NPY1 receptor and induced fibrosis. NEP deletion led to dissociation of fibrosis from contraction, via lack of NPY degradation into small fragments, but aggravation of contraction in the myofibroblastic cells in the liver, leading to aggravation of portal hypertension in vivo. Indeed, simultaneous NEP deletion and anti-Ang II strategies could reduce fibrosis as well as portal pressure in vivo.
Progressive fibrosis with portal hypertension is mostly maintained and aggravated by splanchnic vasodilation.35 While many vasoconstrictors, e.g., Ang II, fail to contract these vessels,36 NPY, as an enhancer of Ang II-mediated contraction, induces contraction in these hypocontractile vessels, as shown previously.11,16,37 In particular, we were able to identify a gradient of NPY levels over the liver with higher NPY levels in the PV when compared with the HV/HA, which renders this hypothesis more probable. However, an enhanced contraction in the liver would be deleterious in the progression of liver fibrosis, as it would seriously aggravate portal hypertension. Therefore, NEP is upregulated specifically in activated HSCs, which regulate the hepatic vascular tone, and it increases with severity of liver fibrosis.
Our in vitro results show that the presence of NEP shifts the physiological response of HSCs to NPY from contraction to relaxation and from an antifibrogenic to a profibrogenic response. In vivo, we were not only able to show increased contraction but also decreased fibrosis in Nep−/− mice. In addition to NEP deletion, AT1R blockade or ACE inhibition induces a decrease in portal pressure. This is confirmed by using Entresto, which induces a decrease in fibrosis and portal pressure. While clinical evidence is missing as the drug has not yet been tested in liver disease, Entresto clearly has translational potential.
NEP is a stalked membrane protein that can terminate neuropeptide signals, such as NPY,38 at the cell surface. Studies have shown that NEP cleaves NPY into C-terminal fragments, which are the most abundant fragments generated by NEP activity in vivo.39 The C terminus of NPY is crucial for receptor activation.24 Recent functional studies of the Y1R structure have provided insights into the binding determinants of NPY to its receptor.24
In fact, different NPY receptors have different affinities for NPY and behave differently toward truncations or mutations of the NPY peptide.40,41 In our study, Y1R seems to be the main target of cleaved NPY in HSC, enhancing contraction and modulating fibrogenesis. There is a shift in the physiological response of Y1R signaling depending on presence or absence of NEP. NPYFL induces collagen gel contraction and Acta2 mRNA but reduces Col1a1 and Tgfβ1 mRNA expression in Nep−/− HSC.
By contrast, and as expected, NPY enhanced contraction and portal pressure, presumably by enhancing the effect of AT1R. This is highly relevant for clinical translation, since oral administration of Entresto, a combination of AT1R and NEP inhibition, is in clinical use for heart failure.42
Limitations of the study
While this study reveals insights into NEP-dependent regulation of the NPY/Y1R axis in cirrhosis by shifting the physiological response of Y1R from contraction to relaxation and from profibrogenic to antifibrogenic, two aspects remain open that could be addressed in future research.
First, the temporal sequence of NPY cleavage and binding to the Y1R receptor was not examined in our study. Thus, we cannot conclude whether NPY binds to the receptor first and NEP is able to cleave bound NPY or vice versa. It is likely that NEP, as a proteolytic enzyme, first cleaves NPY to terminate neuropeptide signaling, and that in a further step, cleaved NPY fragments bind the receptor to exert its function.
Another aspect not addressed concerns the saturation of NEP in the cell and possible NEP saturation due to high NPY concentrations and resulting competitive and/or allosteric competition between the different NPY versions, if any. Work on the purified protein and enzyme kinetics studies could address these points and determine the affinity of the NPY fragments produced by NEP.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal to alpha smooth muscle Actin | Abcam | Cat#ab5694; RRID:AB_2223021 |
| Goat Anti-Type I Collagen-UNLB | SouthernBiotech | Cat#1310-01; RRID:AB_2753206 |
| Glyceraldehyde-3-phosphate dehydrogenase | Santa Cruz Biotechnology | Cat# sc-25778; RRID:AB_10167668 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat#9101; RRID:AB_331646 |
| Phospho (Thr558) Moesin | Santa Cruz Technology | Cat#sc-12895; RRID:AB_2148235 |
| Biological samples | ||
| Human liver biopsy | University of Bonn | 202/01; BioProject PRJNA844027 |
| Human portal and hepatic venous blood samples | University of Bonn | 029/13; 121/14 |
| Human hepatic venous and arterial blood samples | University of Copenhagen | H-1-2011-151 |
| Chemicals, peptides, and recombinant proteins | ||
| Captopril | Sigma-Aldrich | Cat#C4042 |
| Losartan | Sigma-Aldrich | Cat#SML3317 |
| Entresto | Novartis | PZN 11126514 |
| Sacubitril | MedChemExpress | Cat#HY-15407A/CS-2514.Batch n°:15499 |
| Valsartan | ChemCruz | Cat#sc-220362B |
| NPY recombinant protein | Abnova Corporation | Cat#H00004852-P02 |
| C-terminal peptide 1 NPY (20–36) | GenScript | Cat#SC1208 |
| C-terminal peptide 2 NPY (30–36) | GenScript | Cat#SC1208 |
| Critical commercial assays | ||
| Cyclic AMP ELISA kit | Thermo Fisher Scientific | Cat#4412182 |
| Human NPY ELISA kit | Millipore | Cat# EZHNPY-25K |
| Experimental models: Cell lines | ||
| Primary mouse hepatocytes | This paper | N/A |
| Primary mouse liver sinusoidal endothelial cells | This paper | N/A |
| Primary mouse hepatic stellate cells | This paper | N/A |
| Primary mouse Kupffer cells | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mice: WT C57Bl/6J | Charles River | RRID:IMSR_JAX:000664 |
| Mouse: Nep−/− (Lu et al.)43 | Kindly provided by Prof. Thomas Walther, University College Cork, Cork, Ireland, Great Britain | N/A |
| Oligonucleotides | ||
| Acta2 (Mm00725412_s1) | Thermo Fisher Scientific | Cat#4331182 |
| Col1a1 (Mm00801666_g1) | Thermo Fisher Scientific | Cat#4331182 |
| Nep (MME) (Mm00485028_m1) | Thermo Fisher Scientific | Cat#4331182 |
| Npyr1 (Mm00650798_g1) | Thermo fisher Scientific | Cat#4331182 |
| Npyr1 (Hs00702150_s1) | Thermo Fisher Scientific | Cat#4331182 |
| Software and algorithms | ||
| GraphPad Prism 9.5.0 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | ImageJ Software | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Prof. Dr. Jonel Trebicka (Jonel.Trebicka@ukmuenster.de).
Materials availability
All materials generated in this study will be available on request by the lead contact.
Experimental model and subject details
Mice
Nep−/− mice have been described44 and were originally generated by Lu et al.43 Heterozygous (Nep+/−) on a C57BL/6J genetic background were bred to generate WT (Nep +/+) and homozygous knockout (Nep−/−) mice. 10–12-week-old male and female Nep−/− and WT mice were used in all experiments. All experiments were performed in accordance with the German animal protection law and the guidelines of the animal care facility at our university (Haus für Experimentelle Therapie, University Clinics Bonn, Germany), and approved by the North Rhine-Westphalian State Agency for Nature, Environment, and Consumer Protection (LANUV, Germany; File Reference LANUV84-02.04.2014.A137).
Entresto® study
A total of 60 mice were used for this study. Male wildtype (WT, C57BL6/J) mice (10 weeks old) were purchased (Charles River Laboratories Research Model and Services Germany, Sulzfeld, Germany). The experiments were performed according to the guidelines and regulations approved by Regierungspräsidium Darmstad, the responsible committee for animal studies in the German federal state of Hesse (permission number FK/2005). Liver fibrosis was induced by CCl4 injection (i.p) 2μL/g (CCl4:Corn oil = 1:2) two times a week for seven weeks. CCl4 injections were combined either by addition of ethanol to the drinking water (4% during week 1, 8% during week 2, and 16% until animals were sacrificed) and normal chow (Ssniff, Soest, Germany) to induce ASH, or by additional high-fat cholesterol-rich diet without ethanol (WD; Ssniff) to induce NASH. Entresto® (49mg/51mg) was purchased from Novartis, Valsartan (sc-220362B) from Santa Cruz and Sacubitril (HY 15407) from MedChemExpress. One tablet of Entresto® was crushed and resuspended in 0.9% NaCl. Identical procedure was applied for the independent drugs. Oral administration (100 mg/kg) was determined based on Yang et al..45 Mice were treated with Entresto® and the respective drugs for 14 days until animals were sacrificed.
In situ perfusion
This protocol was performed according to previous publication.11 In order to test a direct vasoconstrictive effect, NPY was administrated at increasing doses (0.1nM to 5μM). In a different protocol, the C-terminal NPY fragments were administrated at the same concentrations as before, however, no significant contraction was detected.
Human samples and serum and plasma collection
This study included patients who were evaluated and regularly scheduled for implantation of a transjugular portosystemic shunt (TIPS). Inclusion criteria were the TIPS indications variceal bleeding (representing compensated cirrhosis) or ascites (representing decompensated cirrhosis) Basic clinical as well as biochemical markers and hemodynamics were assessed at TIPS implantation. Portal, hepatic, and central venous blood as well as peripheral blood was sampled at TIPS implantation (Total patient n°: 55, age range: 32–78 years, sex: male = 35 and female = 20) (Control patients: 6, age range: 27–56 years, sex: male = 2 and female = 4).The study was performed in accordance with the declaration of Helsinki and approved by the local ethics committee (Nr. 121/14) and capital region of Copenhagen (H-1-2011-151), all patients gave their written informed consent. Serum and plasma blood samples were processed within 1 h. Samples were centrifuged at 2.000xg for 5 min at 4°C. Serum and plasma were aliquoted and stored at −80°C until further use.
Human NPY was detected by sandwich ELISA (Cat. EZHNPY-25K EMD Millipore, St. Louis, Missouri, USA). The human NPY ELISA is specific for full-length NPY (100%), NPY 2–26 (67%), NPY 3–36 (68%), NPY 31–36 (41%), NPY 22–36 (0%) and NPY 1–24 (0%) according to the manufacturers’ description.
Induction of liver fibrosis
CCl4 and BDL models were used to induce liver fibrosis. In the CCl4 model, mice were exposed to 1L/min of CCl4 three times a week for four weeks. CCl4-intoxicated mice received phenobarbital (3.0 mg/kg/day) in their drinking water to induce cytochrome P450 metabolic activity. In two subsets of experiments, CCl4 mice were co-treated with losartan (5 mg/kg/day) or captopril (4 mg/kg/day) for four weeks. In the BDL model, mice underwent ligation of the common bile duct to induce cholestasis or sham operation (control) and were sacrificed after two weeks.
Method details
General methodology
Details on general methodology, such as induction of liver fibrosis, Sirius red staining, portal pressure measurement, qPCR and Western blot, are as previously described.9,30,46 The sources of commercially available antibodies can be found in Table S2. The qPCR primers used are shown in Table S3.
Histological staining and IHC
The left liver lobe was excised and fixed in 10% neutral-buffered formalin, embedded in paraffin and sectioned for hematoxylin and eosin (H&E), Sirius red staining (SR), and Ki67, αSMA, and F4/80 IHC. Images were captured using Panoramic Viewer (3DHistech, Budapest, Hungary). Histological analysis was performed using Histoquant (3DHistech, Budapest, Hungary) and large bile ducts and vessels were excluded following the principles of computational analysis as described previously(36).
Hydroxyproline content measurement
Hepatic hydroxyproline (HP) content was measured biochemically in 250–300 mg liver samples from two different lobes (representing >10% of the liver). Total hydroxyproline (μg/100 mg liver) was calculated based on individual liver weights and the corresponding relative hydroxyproline content.
Determination of cellular cAMP
Primary HSC isolated from WT and Nep−/− mice were grown in 6-well plates and serum starved before adding test agents. Cells were assayed for cAMP production by ELISA after 30 min incubation with full-length NPY 1nM and a mix of C-terminal NPY short fragments 30μM. Cyclic AMP was determined by competitive ELISA kit (Thermo Fisher Scientific Inc., Vienna, Austria).
Molecular docking
In order to provide a molecular insight into the antagonistic effect of NPY(31–36) and NPY(21–36), we built models of Y1R in complex with the C-terminal NPY fragments using the HADDOCK2.2 webserver.23,47 The receptor structure was taken from the recently solved X-ray structure of human Y1R in complex with the antagonist UR-MK299 (PDB code 5ZBQ),24 except for the N-terminal residues 18–30, which were removed following the protocol in.24 By contrast, the structure of the C-terminal NPY fragments was extracted from the NMR solution structure of full-length human NPY (PDB code 1RON).48 The receptor-peptide docking was guided using three sets of restraints: (i) unambiguous restraints based on experimental data,24 (ii) ambiguous restraints derived from a Fpocket prediction of the binding cavity residues,49 and (iii) an additional restraint between the centers of mass of the two molecules. In addition, for NPY (21–36), we included unambiguous restraints between the helical segment 21–32 and the extracellular loops ECL1 and ECL3 of NPY in line with the orientation predicted in.24 A complete list of the restraint sets is included in Table S4. The docking protocol consisted of three steps. First, 1000 structures were generated by rigid body docking. Next, the 200 best-ranked structures were submitted to a semi-flexible simulated annealing refinement, followed by further refinement in the presence of explicit solvent. The resulting 200 structures were clustered using the algorithm in ref. 50 with a root-mean-square deviation (RMSD) cutoff of 7.5 Å. Finally, we built a model of human Y1R in complex with BIBO3304, i.e. the antagonist used in the experiments in Figure 4. The chemical structure of BIBO3304 is designed to mimic the C-terminal NPY residues Arg35 and Tyr36 and shares the same scaffold with UR-MK299 (Figure S7). Hence, the model of the Y1R/BIBO3304 complex was built by manual modification of the experimental structure of the Y1R/UR-MK299 complex (PDB code 5ZBQ).24 In particular, the carbamoyl tail (in blue) in Figure S7 was removed from the guanidinium group and the hydroxyl group (in green) was replaced by a methylurea group.
Cell culture and treatments
In vitro studies were carried out with primary mouse HSC isolated as previously described.51 HSC were cultured at least one week before the experiments, i.e. they were fully activated. None of the buffers used contained traces of Zn and/or EDTA. HSC were seeded on 6-well plates (300,000 cells/well) in DMEM (1x) supplemented with 20% FBS, penicillin and streptomycin. The medium was replaced with serum-free medium 12 h before treatment with 1 nM of human recombinant NPY (rNPY) (Abcam ab112330, UK) or 10 nM of human recombinant NEP (rNEP) (R&D Systems 1182-ZNC-010, Minneapolis, MN). To induce NPY cleavage, 30 μM of rNPY and 2.5 μg/mL of rNEP were incubated at 37°C for 1 h.12
Collagen gel contraction assay
1 × 104 HSC from WT and Nep−/− mice were populated in 500 μL of 1 mg/mL collagen gels (BD Biosciences, Bedford MA) and placed on a 24-well dish in triplicate per group. Once solidified, gels were detached from the walls of the dish and incubated in 10%FBS/DMEM or NPY (1–100 nM). Media and treatments were changed every three days and collagen gel area was measured by ImageJ (NIH, Bethesda, MD).
Immunofluorescence and confocal microcopy
HSC were treated with full-length NPY and NPY C-short terminal synthetic peptides for 24 h. After treatment, cells were fixed with 4% paraformaldehyde for 10 min at 37°C. Permeabilization was done with 1% Triton X-100 in PBS1X for 10 min at 37°C. After washing with PBS1X, cells were blocked with 1% BSA in PBS1X four 1 h and incubated overnight at 4°C with collagen I diluted 1:50 (Col1a1, 1310-01) and α-smooth muscle actin antibody diluted 1:200 (α-SMA, ab5694) in blocking solution. On the next day, after extensive washing in PBS1X, secondary antibody (anti-rabbit A594, Thermo Fisher A32740 and anti-goat A488, Thermo Fisher A32814) was used at a dilution of 1:3000 and incubated for 2 h in the dark. For nuclear staining, DAPI (D9542) was diluted to 1:10.000 and incubate for 5 min in the dark. The cells were visualized by confocal microscope (Zeiss LSM 800) and further processed with the ImageJ program.
NEP-NPY degradation experiments
rhNEP used for NPY degradation experiments had a concentration of 10 ng/mL and was diluted with purified water as per manufacturer’s instructions. NPY (10−5 M) was incubated with rhNEP at 10 ng/m, as described above and diluted with 50mM Tris-HCl buffer pH 7.4 laced with 0.1% BSA to minimize adhesion of the peptide onto the tube walls, and 10−5 M candoxatrilat, a specific NEP inhibitor, was used to determine NEP specific activity. Incubations were allowed to proceed for 1 h for rhNEP samples. After the designated time, the reaction was stopped by addition of half volume of 350 mM HClO4. Samples were then centrifuged to remove any particles prior to LCMS analysis.
Liquid chromatography-mass spectrometry analysis (LCMS)
NPY analysis was performed by reversed phase chromatography using an LCMS-20/20 with UV detection from Shimadzu (Duisburg, Germany). Each sample was loaded onto an RP Jupiter Proteo C12 column (250 × 4.6 mm, 4 μm) from Phenomenex with a flow rate of 0.5 mL/min. The peptides were detected at 206 nm by a PDA detector before being analyzed by a coupled ESI-MS.
Quantification and statistical analysis
Data are expressed as mean ± SEM unless otherwise specified. Statistical comparisons among groups were performed by two-factor analysis of variance (ANOVA). For in vitro experiments, paired t-Student tests were used. All experiments were performed in triplicate at least four times.
In silico analysis
NEP (MME), NPY, TGFB1 and ACTA2 expression was analyzed in human non-disease liver tissue from the Genotype-Tissue Expression project (https://www.gtexportal.org/) or non-tumorous liver tissue from the cancer genome atlas (TCGA) (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) using the gene expression profiling and interactive analyses (GEPIA) web application.52
Transcriptomic analysis
For pairwise differential expression analysis we used OMICSBOX software. The TMM (trimmed mean of M values) was selected for normalization method by using exact test. The number of differentially expressed (DE) features (FDR <0.05) was 1363 (genes human liver transplant) and 2849 (genes human hepatic stellate cells (HSCs)).53,54
Acknowledgments
All the authors are very grateful to Gudrun Hack, Silke Bellinghausen, Christiane Esch, Dikra Zouiten, and Evelyn Süß for their excellent technical assistance and to Sabine Dentler for critical reading. The authors were supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57, CRC1382) and Eurostars (ID 12350). The MICROB-PREDICT, GALAXY, and LIVERHOPE projects have received funding from the European Union’s Horizon 2020 research and innovation program under grant agreements numbers 825694, 668031, and 731875, respectively. The manuscript reflects only the authors’ views, and the European Commission is not responsible for any use that may be made of the information it contains. The funders had no influence on study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
F.M., C.O., and J.T. drafted the manuscript. W.R., S.K., C.O., F.M., S.G., and M.A.-P. acquired, analyzed, and interpreted the data. C.O., R.S., F.U., C.S., T.W., P.D., and C.H. provided substantial material and methods and interpreted data. J.T. designed the original study, interpreted the data, supervised the study, and obtained financial support for the study. All authors reviewed the draft for important intellectual content and approved the final article for submission.
Declaration of interests
The authors have no conflict of interest.
Published: January 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112059.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any information required to reanalyse the data reported in this paper are available upon request.
References
- 1.Zhao X., Kwan J.Y.Y., Yip K., Liu P.P., Liu F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020;19:57–75. doi: 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- 2.de Franchis R., Baveno VI Faculty Expanding consensus in portal hypertension: report of the Baveno VI xonsensus workshop: stratifying risk and individualizing care for portal hypertension. J. Hepatol. 2015;63:743–752. doi: 10.1016/j.jhep.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 3.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 4.Grace J.A., Klein S., Herath C.B., Granzow M., Schierwagen R., Masing N., Walther T., Sauerbruch T., Burrell L.M., Angus P.W., et al. Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology. 2013;145:874–884.e5. doi: 10.1053/j.gastro.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 5.Moore K. Endothelin and vascular function in liver disease. Gut. 2004;53:159–161. doi: 10.1136/gut.2003.024703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grace J.A., Herath C.B., Mak K.Y., Burrell L.M., Angus P.W. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin. Sci. 2012;123:225–239. doi: 10.1042/CS20120030. [DOI] [PubMed] [Google Scholar]
- 7.Casey S., Schierwagen R., Mak K.Y., Klein S., Uschner F., Jansen C., Praktiknjo M., Meyer C., Thomas D., Herath C., et al. Activation of the alternate renin-angiotensin system correlates with the clinical status in human cirrhosis and corrects post liver transplantation. J. Clin. Med. 2019;8:419. doi: 10.3390/jcm8040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granzow M., Schierwagen R., Klein S., Kowallick B., Huss S., Linhart M., Mazar I.G.R., Görtzen J., Vogt A., Schildberg F.A., et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60:334–348. doi: 10.1002/hep.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein S., Rick J., Lehmann J., Schierwagen R., Schierwagen I.G., Verbeke L., Hittatiya K., Uschner F.E., Manekeller S., Strassburg C.P., et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut. 2017;66:145–155. doi: 10.1136/gutjnl-2015-309600. [DOI] [PubMed] [Google Scholar]
- 10.Abraldes J.G., Trebicka J., Chalasani N., D’Amico G., Rockey D.C., Shah V.H., Bosch J., Garcia-Tsao G. Prioritization of therapeutic targets and trial design in cirrhotic portal hypertension. Hepatology. 2019;69:1287–1299. doi: 10.1002/hep.30314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moleda L., Trebicka J., Dietrich P., Gäbele E., Hellerbrand C., Straub R.H., Sauerbruch T., Schoelmerich J., Wiest R. Amelioration of portal hypertension and the hyperdynamic circulatory syndrome in cirrhotic rats by neuropeptide Y via pronounced splanchnic vasoaction. Gut. 2011;60:1122–1132. doi: 10.1136/gut.2010.226407. [DOI] [PubMed] [Google Scholar]
- 12.Wagner L., Wolf R., Zeitschel U., Rossner S., Petersén Å., Leavitt B.R., Kästner F., Rothermundt M., Gärtner U.T., Gündel D., et al. Proteolytic degradation of neuropeptide Y (NPY) from head to toe: identification of novel NPY-cleaving peptidases and potential drug interactions in CNS and Periphery. J. Neurochem. 2015;135:1019–1037. doi: 10.1111/jnc.13378. [DOI] [PubMed] [Google Scholar]
- 13.Wilson J.R., Kerman S.J., Hubers S.A., Yu C., Nian H., Grouzmann E., Eugster P.J., Mayfield D.S., Brown N.J. Dipeptidyl peptidase 4 inhibition increases postprandial norepinephrine via substance P (NK1 receptor) during RAAS inhibition. J. Endocr. Soc. 2019;3:1784–1798. doi: 10.1210/js.2019-00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong P.F., Gall M.G., Bachovchin W.W., McCaughan G.W., Keane F.M., Gorrell M.D. Neuropeptide Y is a physiological substrate of fibroblast activation protein: enzyme kinetics in blood plasma and expression of Y2R and Y5R in human liver cirrhosis and hepatocellular carcinoma. Peptides. 2016;75:80–95. doi: 10.1016/j.peptides.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Møller S., Hobolth L., Winkler C., Bendtsen F., Christensen E. Determinants of the hyperdynamic circulation and central hypovolaemia in cirrhosis. Gut. 2011;60:1254–1259. doi: 10.1136/gut.2010.235473. [DOI] [PubMed] [Google Scholar]
- 16.Dietrich P., Moleda L., Kees F., Müller M., Straub R.H., Hellerbrand C., Wiest R. Dysbalance in sympathetic neurotransmitter release and action in cirrhotic rats: impact of exogenous neuropeptide Y. J. Hepatol. 2013;58:254–261. doi: 10.1016/j.jhep.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 17.Dietrich P., Wormser L., Fritz V., Seitz T., De Maria M., Schambony A., Kremer A.E., Günther C., Itzel T., Thasler W.E., et al. Molecular crosstalk between Y5 receptor and neuropeptide Y drives liver cancer. J. Clin. Invest. 2020;130:2509–2526. doi: 10.1172/JCI131919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigala B., McKee C., Soeda J., Pazienza V., Morgan M., Lin C.-I., Selden C., Vander Borght S., Mazzoccoli G., Roskams T., et al. Sympathetic nervous system catecholamines and neuropeptide Y neurotransmitters are upregulated in human NAFLD and modulate the fibrogenic function of hepatic stellate cells. PLoS One. 2013;8:e72928. doi: 10.1371/journal.pone.0072928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein S., Van Beuge M.M., Granzow M., Beljaars L., Schierwagen R., Kilic S., Heidari I., Huss S., Sauerbruch T., Poelstra K., et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J. Hepatol. 2012;57:1220–1227. doi: 10.1016/j.jhep.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 20.Leban J.J., Heyer D., Landavazo A., Matthews J., Aulabaugh A., Daniels A.J. Novel modified carboxy terminal fragments of neuropeptide Y with high affinity for Y2-type receptors and potent functional antagonism at a Y1-type receptor. J. Med. Chem. 1995;38:1150–1157. doi: 10.1021/jm00007a012. [DOI] [PubMed] [Google Scholar]
- 21.Abid K., Rochat B., Lassahn P.-G., Stöcklin R., Michalet S., Brakch N., Aubert J.-F., Vatansever B., Tella P., De Meester I., et al. Kinetic study of neuropeptide Y (NPY) proteolysis in blood and identification of NPY3-35: a new peptide generated by plasma kallikrein. J. Biol. Chem. 2009;284:24715–24724. doi: 10.1074/jbc.M109.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubers S.A., Wilson J.R., Yu C., Nian H., Grouzmann E., Eugster P., Shibao C.A., Billings F.T., Jafarian Kerman S., Brown N.J. DPP (dipeptidyl peptidase)-4 inhibition potentiates the vasoconstrictor response to NPY (neuropeptide Y) in humans during renin-angiotensin-aldosterone system inhibition. Hypertension. 2018;72:712–719. doi: 10.1161/HYPERTENSIONAHA.118.11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., van Dijk M., de Vries S.J., Bonvin A.M.J.J. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z., Han S., Keller M., Kaiser A., Bender B.J., Bosse M., Burkert K., Kögler L.M., Wifling D., Bernhardt G., et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature. 2018;556:520–524. doi: 10.1038/s41586-018-0046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 26.Filipek S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019;55:114–120. doi: 10.1016/j.sbi.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 27.Herzog H., Hort Y.J., Ball H.J., Hayes G., Shine J., Selbie L.A. Cloned human neuropeptide Y receptor couples to two different second messenger systems. Proc. Natl. Acad. Sci. USA. 1992;89:5794–5798. doi: 10.1073/pnas.89.13.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumaz N., Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft für Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 2005;272:3491–3504. doi: 10.1111/j.1742-4658.2005.04763.x. [DOI] [PubMed] [Google Scholar]
- 29.Hensel N., Baskal S., Walter L.M., Brinkmann H., Gernert M., Claus P. ERK and ROCK functionally interact in a signaling network that is compensationally upregulated in Spinal Muscular Atrophy. Neurobiol. Dis. 2017;108:352–361. doi: 10.1016/j.nbd.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Klein S., Klösel J., Schierwagen R., Körner C., Granzow M., Huss S., Mazar I.G.R., Weber S., van den Ven P.F.M., Pieper-Fürst U., et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab. Invest. 2012;92:1440–1450. doi: 10.1038/labinvest.2012.106. [DOI] [PubMed] [Google Scholar]
- 31.Asahina K., Tsai S.Y., Li P., Ishii M., Maxson R.E., Sucov H.M., Tsukamoto H. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology. 2009;49:998–1011. doi: 10.1002/hep.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreira de Macêdo S., Guimarães T.A., Feltenberger J.D., Sousa Santos S.H. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides. 2014;62:189–196. doi: 10.1016/j.peptides.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Oefner C., Roques B.P., Fournie-Zaluski M.-C., Dale G.E. Structural analysis of neprilysin with various specific and potent inhibitors. Acta Crystallogr. D Biol. Crystallogr. 2004;60:392–396. doi: 10.1107/S0907444903027410. [DOI] [PubMed] [Google Scholar]
- 34.McMurray J.J.V., Packer M., Desai A.S., Gong J., Lefkowitz M.P., Rizkala A.R., Rouleau J.L., Shi V.C., Solomon S.D., Swedberg K., et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014;371:993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 35.Hennenberg M., Trebicka J., Sauerbruch T., Heller J. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut. 2008;57:1300–1314. doi: 10.1136/gut.2007.144584. [DOI] [PubMed] [Google Scholar]
- 36.Schepke M., Raab P., Hoppe A., Schiedermaier P., Brensing K.A., Sauerbruch T. Comparison of portal vein velocity and the hepatic venous pressure gradient in assessing the acute portal hemodynamic response to propranolol in patients with cirrhosis. Am. J. Gastroenterol. 2000;95:2905–2909. doi: 10.1111/j.1572-0241.2000.03202.x. [DOI] [PubMed] [Google Scholar]
- 37.Hartl J., Dietrich P., Moleda L., Müller-Schilling M., Wiest R. Neuropeptide Y restores non-receptor-mediated vasoconstrictive action in superior mesenteric arteries in portal hypertension. Liver Int. 2015;35:2556–2563. doi: 10.1111/liv.12874. [DOI] [PubMed] [Google Scholar]
- 38.Turner A.J., Isaac R.E., Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Rose J.B., Crews L., Rockenstein E., Adame A., Mante M., Hersh L.B., Gage F.H., Spencer B., Potkar R., Marr R.A., et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J. Neurosci. 2009;29:1115–1125. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merten N., Lindner D., Rabe N., Römpler H., Mörl K., Schöneberg T., Beck-Sickinger A.G. Receptor subtype-specific docking of Asp6.59 with C-terminal arginine residues in Y receptor ligands. J. Biol. Chem. 2007;282:7543–7551. doi: 10.1074/jbc.M608902200. [DOI] [PubMed] [Google Scholar]
- 41.Pedragosa-Badia X., Stichel J., Beck-Sickinger A.G. Neuropeptide Y receptors: how to get subtype selectivity. Front. Endocrinol. 2013;4:5. doi: 10.3389/fendo.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velazquez E.J., Morrow D.A., DeVore A.D., Duffy C.I., Ambrosy A.P., McCague K., Rocha R., Braunwald E., PIONEER-HF Investigators Angiotensin-neprilysin inhibition in acute decompensated heart failure. N. Engl. J. Med. 2019;380:539–548. doi: 10.1056/NEJMoa1812851. [DOI] [PubMed] [Google Scholar]
- 43.Lu B., Gerard N.P., Kolakowski L.F., Bozza M., Zurakowski D., Finco O., Carroll M.C., Gerard C. Neutral endopeptidase modulation of septic shock. J. Exp. Med. 1995;181:2271–2275. doi: 10.1084/jem.181.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker M., Siems W.-E., Kluge R., Gembardt F., Schultheiss H.-P., Schirner M., Walther T. New function for an old enzyme: NEP deficient mice develop late-onset obesity. PLoS One. 2010;5:e12793. doi: 10.1371/journal.pone.0012793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang C.-C., Chen Y.-T., Chen C.-H., Li Y.-C., Shao P.-L., Huang T.-H., Chen Y.-L., Sun C.-K., Yip H.-K. The therapeutic impact of entresto on protecting against cardiorenal syndrome-associated renal damage in rats on high protein diet. Biomed. Pharmacother. 2019;116:108954. doi: 10.1016/j.biopha.2019.108954. [DOI] [PubMed] [Google Scholar]
- 46.Schierwagen R., Maybüchen L., Hittatiya K., Klein S., Uschner F.E., Braga T.T., Franklin B.S., Nickenig G., Strassburg C.P., Plat J., et al. Statins improve NASH via inhibition of RhoA and ras. Am. J. Physiol. Gastrointest. Liver Physiol. 2016;311:G724–G733. doi: 10.1152/ajpgi.00063.2016. [DOI] [PubMed] [Google Scholar]
- 47.de Vries S.J., van Dijk M., Bonvin A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 48.Monks S.A., Karagianis G., Howlett G.J., Norton R.S. Solution structure of human neuropeptide Y. J. Biomol. NMR. 1996;8:379–390. doi: 10.1007/BF00228141. [DOI] [PubMed] [Google Scholar]
- 49.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daura X., van Gunsteren W.F., Mark A.E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins. 1999;34:269–280. doi: 10.1002/(sici)1097-0134(19990215)34:3<269::aid-prot1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 51.Trebicka J., Hennenberg M., Schulze Pröbsting A., Laleman W., Klein S., Granzow M., Nevens F., Zaagsma J., Heller J., Sauerbruch T. Role of beta3-adrenoceptors for intrahepatic resistance and portal hypertension in liver cirrhosis. Hepatology. 2009;50:1924–1935. doi: 10.1002/hep.23222. [DOI] [PubMed] [Google Scholar]
- 52.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Home Omiqa Bioinformatics. https://www.omiqa.bio/.
- 54.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any information required to reanalyse the data reported in this paper are available upon request.