

Summary
Bacteriophages play key roles in bacterial ecology and evolution and are potential antimicrobials. However, the determinants of phage-host specificity remain elusive. Here, we isolate 46 phages to challenge 138 representative clinical isolates of Klebsiella pneumoniae, a widespread opportunistic pathogen. Spot tests show a narrow host range for most phages, with <2% of 6,319 phage-host combinations tested yielding detectable interactions. Bacterial capsule diversity is the main factor restricting phage host range. Consequently, phage-encoded depolymerases are key determinants of host tropism, and depolymerase sequence types are associated with the ability to infect specific capsular types across phage families. However, all phages with a broader host range found do not encode canonical depolymerases, suggesting alternative modes of entry. These findings expand our knowledge of the complex interactions between bacteria and their viruses and point out the feasibility of predicting the first steps of phage infection using bacterial and phage genome sequences.
Keywords: bacteriophage, Klebsiella, host range, bacterial capsule, depolymerase, microbial evolution, horizontal gene transfer, genomics
Graphical abstract
Highlights
-
•
Klebsiella capsular diversity restricts the host range of most Klebsiella phages
-
•
Phage-encoded depolymerase domains can predict capsular tropism
-
•
Capsular tropism predictability is limited by post-adsorptive resistance mechanisms
-
•
Phages lacking capsule dependency and depolymerases exhibit broader host ranges
Beamud et al. analyze the host tropism of bacteriophages infecting Klebsiella pneumoniae, a nosocomial pathogen of global concern. They identify phage sequence domains that predict capsular tropism, the main determinant of infectivity. This work demonstrates how phenotypic and genomic data can be combined to better understand virus-host interactions.
Introduction
Bacteriophages are extremely abundant and diverse biological entities, and, as such, they are key ecosystem actors.1 Yet, the mechanisms that govern phage-bacteria interactions are not well understood since most studies have focused on small sets of virus-host pairs or have not followed a systematic whole-genome approach.2,3 Advances in high-throughput sequencing and metagenomics have allowed linking phages to their hosts via genomic signatures.4,5 Although very useful, these methods do not provide individual information about phage-bacteria interactions. Thus, isolation of phages using strains of interest6,7,8 along with genome screenings9 can complement these studies to better understand phage-bacteria interactions.
The determinants of phages’ host range are complex. Typically, a phage infects only a subset of strains of a given bacterial species.10 However, some phages can infect different bacterial species8,11 or even distinct genera.12,13 Available tools can predict phages’ host range with moderately good accuracies at the family and genus levels but perform worse at lower taxonomical ranks.14,15,16 Bacteria deploy a variety of mechanisms that can restrict phage infection, ranging from modification of surface receptors to a continuously expanding number of immunity systems, such as regularly interspaced short palindromic repeats (CRISPRs), restriction-modification (RM) systems, abortive infection systems, and prophage super-immunity, among many others,17,18 complicating our understanding of phage-host interactions.
Bacterial capsules are a major determinant of phage tropism as they can protect bacteria from infection by masking receptors19 or being used for phage attachment.6,20 Indeed, most environmental and opportunistic bacteria present an extracellular capsule.21 Capsules protect the cell from the immune system of multicellular organisms being important virulence factors.22 To overcome the bacterial capsule, some phages encode depolymerases (Dpos) capable of digesting specific oligosaccharide bonds.23,24 Most phages contain one or two Dpos and, as a result, infect one or a few cross-reactive capsular types.25 Dpos have a modular structure with potential module swapping between phages through recombination.26,27 However, how binding and depolymerization determine successful phage infection is not well understood.28,29
Klebsiella pneumoniae is a ubiquitous opportunistic Gram-negative Enterobacteriaceae and is included in the ESKAPE pathogen group.30 K. pneumoniae offers an excellent model for studying the role of bacterial capsules in phage infectivity because it exhibits a remarkable capsular diversity, with 77 serotypes and over 180 capsular locus types (CLTs) described so far,31 in addition to a large repertoire of other phage defense mechanisms.32 Given the limited treatments for emerging multidrug-resistant strains, K. pneumoniae is a major target of ongoing research on phage therapy.33,34,35 Most Klebsiella phages infect a few CLTs, but their association with phage Dpo sequences remains elusive20,36 or is restricted to a few phages.37,38 Instead, the typical procedure involves extensive host-range testing along with the expression and purification of the candidate enzymes.39 Additionally, some phages without Dpos have been described, but their determinants of host-range remain elusive.20,35,40
Here, we used a comprehensive collection of sequenced clinical K. pneumoniae strains (n = 138) and diverse environmental phages (n = 46) to study the relative importance of different bacteria and phage genetic traits as determinants of phages’ host range and infection outcome. We tested all phage-bacterium pairs by spot testing and confirmed positive results by bactericidal, progeny production, capsule depolymerization, and adsorption assays.
Results
Host tropism of isolated Klebsiella phages
We used water and soil samples near a sewage plant in Valencia (Spain) to isolate 70 plaques on 36 different K. pneumoniae clinical strains. Phages were amplified, purified, and sequenced, yielding 46 distinct genomes (Figure S1; Table S1). Phages belonged to 13 groups (A1–A3, P4, M5, S6, D7, S8–S11, M12, and S13) according to pairwise intergenomic similarity (IGS ≥ 45%). We named each virus with the first letter of the associated viral family, the number of the phylogenetic group, and a letter to identify each species (IGS ≥ 95%) (Figure 1). All phages lacked integrases, although several had lysogeny marker genes such as transcriptional repressors or parAB genes (Figure 1; Table S1). Transmission electron microscopy showed different morphologies of representative phages for each group (Figure S2). To determine their taxonomic classification, we retrieved 478 phage genomes from databases exhibiting at least 30% coverage and 70% average nucleotide identity (ANI) with our phages. According to pairwise IGS values, the 46 phages represented five families of the Caudovirales order (Autographiviridae, Drexlerviridae, Myoviridae, Siphoviridae, and Podoviridae) and a variety of genera (Drulisvirus, Przondovirus, Webervirus, Gamaleyavirus, Mydovirus, Yonseivirus, Jedunavirus, and Roufvirus; Figure 1; Table S1). They encompassed most families and subfamilies of previously described Klebsiella phages (Figure S3). All the phages except one exhibited <95% IGS with available sequences, suggesting that they represented unknown species. Eight phages were related to unclassified phages, which led us to suggest three subfamilies and five genera (Table S1; Figure S4).
Figure 1.

Diversity of isolated Klebsiella phages
IGS values were used to define 13 similarity groups and to classify phages in the closest viral family, subfamily (if applicable), and genus after comparison with database sequences. Phages were named after the first letter of the associated viral family, the number of the phylogenetic group, and a letter to identify each species or strain. A, Autographiviridae; D, Drexlerviridae; M, Myoviridae; P, Podoviridae; S, Siphoviridae. The genome organization of each phage is shown. Arrows represent coding sequences (CDSs) and are colored based on functional categories of PHROGS.
See also Figures S1–S4 and Table S1.
To examine the host range of isolated phages, we selected 138 sequenced K. pneumoniae clinical isolates from the Valencia region of 59 different CLTs, 14 O locus types (OLTs), and 76 sequence types (STs; Table S2). Of them, 16 were used for the initial isolation of phages, along with 20 additional strains not included in the collection (Table S2). The 138 strains were diverse in terms of antibiotic susceptibility, presence of prophages, and anti-phage defenses such as CRISPR-Cas and RM systems (Figure S5). The resulting collection represented the overall species’ diversity, as shown by comparison with the main clonal groups of K. pneumoniae (Figure S6).
To determine host tropism, we performed triplicate spot tests for all the 46 × 138 = 6,348 phage-bacteria pairs (6,319 after removing the phage-bacteria combinations used for initial phage isolation to avoid sampling bias). Only 124 combinations yielded reproducible spots, suggesting that, on average, a Klebsiella phage can infect less than 2% of host strains (Figure 2; Table S3). Host breadth ranged from 0 to 19 of the 138 strains (excluding the isolation strain), with two distinct patterns: most phages (42/46) infected one or a few strains (median: 1, mean: 1.79; 1–3 CLTs), but the four phages in groups 8 and 9 (S8/S9 phages) exhibited a much broader host range (median: 11.5, mean: 12.25; 4–16 CLTs, Figure 2; Table S3).
Figure 2.

Host tropism of isolated Klebsiella phages
Each row represents a phage (n = 46) and each column a bacterial strain (n = 138). For phages, the dendrogram constructed from IGS values is shown, whereas the maximum likelihood (ML) core phylogeny is shown for bacteria. Original host-phage pairs in which each phage was primarily isolated are indicated in purple. The black histogram and associated numbers indicate the host breadth of each phage in the 138 total bacterial strains analyzed, excluding isolation hosts. Three independent replicates were performed for all phage-bacteria pairs (n = 6,348). For a subset of interactions (n = 1,242), three additional replicates were performed using a different concentration of phage (Table S1). Finally, a subset of phage-bacteria of interest was reassayed twice by spotting several dilutions (n = 95). A given bacterial strain was considered to be susceptible to a given phage (green boxes) if at least two-thirds of the replicates of the spot assay were positive (clear spot, turbid spot, or single plaques).
See also Figures S5 and S6 and Tables S1, S2, and S3.
Predictability of phage-host interactions based on bacterial capsular type
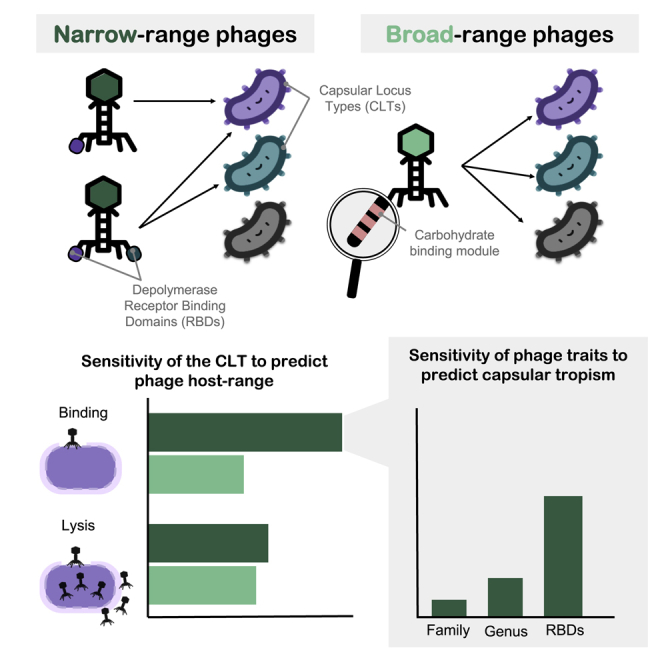
To investigate the determinants of host tropism, we first focused on the 42 non-S8/S9 phages. We assessed which host features predicted spotting by phages (true positive rate [TPR]). For each trait, we sampled random reference hosts and the phages’ spot outcomes and used this information to predict the ability of phages to spot other bacterial strains sharing the same trait. This showed that CLT was the host trait that best predicted tropism. Specifically, a phage spotting in a given strain had a 92% probability of also spotting in other strains of the same CLT (TPR = 0.92 ± 0.001 for non-S8/S9 phages; Figure 3A). Bacterial ST was another good predictor of host tropism (TPR = 0.60 ± 0.01), albeit it is correlated to CLT in our dataset (Cramér’s V = 0.9). In contrast, OLT and 45 additional proteins encoding putative phage receptors were poorly associated with host tropism (TPR = 0.04–0.09). This confirmed that the bacterial capsule is a major determinant of host tropism in Klebsiella phages.
Figure 3.

Predictability of phage-host interactions based on bacterial capsular type
(A) Sensitivity or true positive rate (TPR) of different host traits to predict positive spot tests for non-S8/S9 phages and phages from groups S8/S9. Three independent calculations were performed. Bars represent the standard error (n = 3). Asterisks represent the mean TPR values obtained with randomized data (null expectation). CLT, capsular locus type; OLT, O-antigen locus type; ST, sequence type. Other: presence of 45 secondary receptors (pooled).
(B) Dpo activity of halo-producing non-S8/S9 phages and non-halo-producing S8/S9 phages relative to non-inoculated controls. Each point represents the mean activity of two biological replicates of a given phage-bacterium combination. Four technical replicates were performed, and the median obtained. Asterisks indicate statistical significance (t test: p < 0.001).
(C) Predicted architecture of TSPs for non-S8/S9 phages with or without Dpos. The primary TSP is attached via an N-terminal anchor and the secondary TSP via a conserved short peptide. Top: podoviruses. Bottom: sipho- and myoviruses. Only one TSP copy is shown for simplicity.
Phages infecting encapsulated bacteria can encode Dpo domains in their receptor-binding proteins (RBPs) that specifically digest capsular polysaccharides to provide access to the cell surface.23,24 We found RBPs with putative Dpos in 38 of the 42 non-S8/S9 phage genomes. These were in the same open reading frames (ORFs) as tail proteins and were therefore referred as tail spike proteins (TSPs). Most of these TSPs should be functional because plaques were typically surrounded by haloes (Table S1), an indicator of Dpo activity. Supporting this, quantitation of capsule polysaccharide levels showed a strong reduction (t test: p < 0.001) in 14/14 bacterial strains inoculated with halo-producing phages relative to non-inoculated controls (Figure 3B). Failure to detect TSPs in phage genomes is common.20,33 For instance, three of the four non-S8/S9 phages without detectable TSPs showed haloes and significant activity against the capsule, suggesting Dpo activity (Figure 3B). However, in 20 phages, the number of TSPs encoded was larger than the number of susceptible CLTs (Table S1), suggesting inactivity of some TSPs or specificity of action toward CLTs not included in this work.
Inactivity of some TSPs could be the result of the enzymatic domain being truncated or not incorporated into the phage particle. To assess this, we determined the TSP architecture of each phage following a previous nomenclature.24 We found 13 podoviruses with two TSPs. Six of them only had a Dpo domain because the first TSP was truncated (Figure 3C). Additionally, we found two Dpos in phages A1l and A1q without the structural domains, suggesting that they could be soluble enzymes (Figure S7A). Interestingly, some podoviruses encoded adhesins (n = 3) and acetyl xylan esterases (n = 2), also involved in host recognition.41,42
All siphoviruses showed one TSP with a Dpo domain (Figure 3C). As an exception, Klebsiella phage D7a presented an esterase domain, which agrees with the lack of plaque haloes (Table S1).
Finally, the two myoviruses showed a more complex TSP architecture, as expected from their larger genomes (>100 kb).24 Even though we detected several structural domains typical of TSPs, phage M5a presented two Dpos and phage M5b just one (Table S1; Figure S7A).
In summary, five non-S8/S9 phages had no detected Dpos, 28 phages encoded one, eight phages encoded two domains, and the phage A1q encoded three (Figure 3C). This resulted in 27 phages with as many Dpos as susceptible CLTs, nine phages with less Dpos than susceptible CLTs, and six phages with more Dpos than CLTs.
Prediction of phage capsular tropism based on sequence analysis of receptor-binding domains
The capsular type of bacteria can be obtained from their genome sequence,22 but at present, it is not possible to predict the capsular tropism of phages in a similar way. To address this, we first expanded our dataset by including 64 additional Klebsiella phages from the literature with a known tropism. We then focused on the central and C-terminal regions of TSPs with Dpos, which we referred to as receptor-binding domains (RBDs). To avoid the inclusion of truncated TSPs from literature phages, we only retained those with at least 500 aa. We extracted 46 RBDs from the 42 non-S8/S9 phages and 117 RBDs from additional Klebsiella phages. The 163 RBDs grouped into 35 clusters with at least two RBDs sharing >40% sequence coverage and >50% amino acid identity (Table S4). We found that RBD clusters had a mean TPR of 58.1% ± 4% on phage tropism (Figure 4A, left panel). Remarkably, these values exceeded the TRP of phage phylogenetic markers. For instance, for any phage-CLT pair, other phages of the same phylogenetic group had only an 18% chance of infecting the same bacterial capsular type (Figure 4A, left panel). The accuracy of RBD-based CLT tropism prediction was variable, as 14 RBD clusters had a perfect association with capsular tropism (TPR = 100%), whereas nine showed no association (<10%; Figure 4A, right panel). We expected this considering that (1) many phages contained several Dpos, some of which could be cross-reactive against multiple capsular types, and (2) we could not experimentally test all the existing capsular types. Despite these limitations, we found 25 RBD clusters with a consensus capsular tropism, defined as clusters of RBD sequences in which at least two-thirds of the sequences belonged to phages spotting in the same CLT. These 25 clusters were composed of 79 RBDs of 65 phages and covered 19 distinct CLTs. RBD clusters involved phages from the same phylogenetic group (69.1%) but also from distinct genera and even families (22.7%; Figure 4B), showing horizontal gene transfer of RBDs across large evolutionary scales.
Figure 4.

RBD-based prediction of phage capsular tropism
(A) Left panel: sensitivity or TPR of different phage traits to predict infections in capsular types of bacteria. Three independent calculations were performed. For RBDs, the average of the 35 RBD clusters across calculations is shown. Asterisks represent the mean TPR values obtained with randomized data (null expectation). Right panel: TPR for each individual RBD cluster. Bars represent the standard error of independent calculations (n = 3).
(B) Representation of the 35 RBD similarity clusters obtained. RBDs are named by the phage name followed by the phage protein in which they were detected. Clusters labeled in red are those for which a consensus capsular tropism could be obtained. Colors of nodes indicate phage taxonomic families. Sequences obtained in the present work are shown as triangles and previous published sequences as circles. Edge weight (width) represents amino acid identity between RBDs within the same cluster. Each RBD is annotated with the corresponding phage CLT tropism. White labels represent tropisms that did not match the consensus CLT of an RBD cluster.
See also Table S4.
Validation of RBD-dependent phage tropism using prophage sequences
To further analyze the predictive power of RBD clustering, we extracted a representative sequence from the 25 RBD clusters showing a consensus capsular tropism (Figure 4B). We then interrogated the RefSeq database of bacterial genomes with RBD sequences in search of prophages with these motifs. We predicted the capsular tropism of the corresponding prophages by retrieving the CLT sequence of the host bacteria and inferring the capsular phenotype.43 Then, we compared this prediction with the consensus CLT of the RBD cluster. We made enough predictions (>30) for 18 of the 24 RBD clusters (79%), which encompassed 15 distinct CLTs (KL2/13, KL3, KL22/37, KL23, KL25, KL30, KL35, KL47, KL57, KL63, KL64, KL102, KL106, KL126, and KL140). For the remaining clusters, we did not get enough hits with a confident CLT, showing that these RBDs are rare in the prophages of typed bacteria. Overall, we made ∼10,000 predictions, distributed in 3,824 RefSeq bacterial genomes. The 18 RBD clusters had an average predictive power on CLT tropism of 35%, although this value varied widely, from 0% to 100% depending on the cluster. We found a statistically significant capsular tropism prediction for 13 clusters (72%). Predictabilities >98% were obtained for four RBD-CLT associations (KL2/13, KL30, KL57, and KL63; Figure 5A). Interestingly, it was possible to obtain accurate predictions (>99%) even beyond the Klebsiella genus (e.g., Escherichia/Shigella, for CLTs KL2/13, KL30, KL35, KL47, KL57, and KL63; 4/4, 90/91, 6/6, 43/43, 80/81, and 23/23 correct predictions, respectively). Also, accurate predictions were found across a wide range of RBD identities (Figure 5B).
Figure 5.

Validation of RBD-based capsular tropism prediction using prophage sequences
(A) Ability of the consensus RBD clusters to predict the CLT tropism of prophages obtained from the RefSeq database as described in the main text. The percentage of total and matching predictions by bacterial species is shown. The n values indicate the total predictions for each RBD. “Other” refers to spp. within Klebsiella sp. and also from other genera. Ns denotes no significance by Fisher’s exact test (p > 0.05).
(B) Distribution of RBD coverage and identity for prophage predictions. Colors indicate whether the CLT of the lysogenized host matched the consensus CLT of the RBD cluster used as a query. Hits with ≤40% RBD cover were not considered.
For the five RBDs in which the predicted CLT based on prophages showed no significant association with the consensus CLT of the cluster analysis, we analyzed the distribution of hits to determine which factors might explain this failure. First, for RBD1_KL22/37, most hits (56.36%) were assigned to KL25 instead of to KL22/37. Interestingly, capsule recombination between both CLTs has been identified previously.44 Second, for RBD8_KL102, we found that 55% of the predictions pointed to bacteria from the KL38. In this case, the RBD_KL102 representative sequence showed 35% amino acid identity and 84% query coverage with the Dpo of phage φKp34, which infects KL38,45 suggesting cross-reactivity or an evolutionary association between the abilities to infect both CLTs. Next, clusters RB64 and RBD65 were both present in phages infecting KL23, but only prophages with identity to RBD65 were found in bacteria from KL23, suggesting that the latter was the active Dpo. The same situation was observed in the RBD83_KL140. Finally, for the RBD87 cluster, the predicted CLT based on prophages was KL14 (72%) instead of KL126, but the link between these two CLTs remains unknown.
Productive infection is less predictable than phage spotting
Spot tests provide a fast method for examining host tropism, but they do not reveal productive infection.38,46 To address this, for each of the 124 positive spot tests, we determined phage virulence by measuring optical density in liquid cultures. We detected a significant reduction in host density relative to non-inoculated controls in 94 of the 124 assays (76%). Of the 30 negative pairs, five provided evidence of productive lytic infection by the progeny assay and hence were also classified as virulent. Then, we reexamined host tropism by considering only the 99, out of 6,319 (1.6%), phage-host pairs yielding virulent infections in liquid culture (Table S3). Host breadth now ranged from 0 to 17 of the 138 tested strains. Again, the 42 non-S8/S9 phages were restricted to one or a few strains (median: 1, mean: 1.33), whereas S8-S9 phages exhibited a much broader range (median: 10.5, mean: 10.75). Host CLT was again the single factor that best predicted virulent infection but with a considerably lower TPR than for spot tests (0.53 ± 0.01 for non-S8/S9 phages) (Figure 6A).
Figure 6.

Non-depolymerase determinants of host tropism
(A) Summary of host ranges for phages that showed both virulent and avirulent infections. Virulent infections were considered when the phage was able to produce a spot and to reduce bacterial density or produce progeny. Avirulence was considered when despite spot formation, no productive infection, and no significant effect on host density were observed.
(B) Quantitation of Dpo activity and adsorption in 25 avirulent phage-host pairs. Dpo activity was quantified by comparing capsule polysaccharide levels in phage-treated versus untreated controls. Adsorption was quantified by measuring phage titer following inoculation relative to cell-free mock cultures (eclipse phase). Each point represents the mean of 3 independent replicates with 4 and 3 technical replicates for Dpo activity and adsorption, respectively. Bars represent the standard deviation between biological replicates.
(C) Differential probability of resistance (dPR) for each phage-defense system. For avirulent host-phage combinations, only those in which adsorption was observed (B) were considered. Values of 1 indicate that the defense system was exclusively found in avirulent combinations. On the contrary, negative values indicate that the defense system was overrepresented in virulent combinations and thus probably did not contribute to the observed resistant phenotype.
(D) Genomic comparison of the RBPs of S8/S9 phages. Domains found with InterProScan5 are shown. Pident shows the aa percentage identity after blastp comparison. See also Table S5.
(E) Spotting of serial dilutions of S8/S9 phages (10–104 PFUs) as a function of the fraction of acapsular bacteria (Cap−) in the plate. Three independent replicates were performed, and a representative image was chosen. WT, wild type bacteria only; WT:Cap−, mix of WT-Cap− (1:1); Cap−, Cap only. Combinations CU630-S8c and NTUH-S9a were omitted as spots were not observed with these concentrations.
Therefore, spot tests seemed to better reflect capsule dependency than productive infection tests. To better understand this, we focused on the 25 phage-host combinations that yielded no virulent infection despite the phages producing virulent infections in other strains (Figure 6B). As expected, non-S8/S9 phages induced capsule digestion in the 13/13 phage-host combinations in which the phage produced a spot, despite being avirulent (t test: p ≤ 0.06; Figure 6B), showing the reliability of spot tests for assessing phage-induced depolymerization and capsule-dependent tropism. Interestingly, we observed successful adsorption for 16/25 combinations tested, of which 14/19 included non-S8/S9 phages (Figure 6B). Adsorption rates were close to 100% in many cases, strongly suggesting the presence of a functional receptor.
To investigate the causes of avirulence despite efficient phage adsorption, we searched genomes for known anti-phage systems. We found no CRISPR spacers or resident prophages with significant identity to the adsorbed phages, suggesting no involvement of these forms of immunity. To perform a more systematic analysis, for each phage, we calculated the frequency of known bacterial defense systems in the different strains. We found 39 phage-defense system combinations that were more frequent in resistant (phage avirulence) than in susceptible strains (differential probability of resistance [dPR] >0). Of them, 14 were exclusive of resistant bacteria (dPR = 1; Figure 6C). Overall, we found BREX, RexAB, Druantia, Hachiman, and dGTPase defense systems to be more frequently associated with resistant strains across phages (total dPR ≥ 1). Thus, even if non-S8/S9 phages stripped capsules and were adsorbed, post-adsorptive mechanisms might contribute to explain the lower ability of CLT analysis to predict productive infection than to predict spotting.
Broad-range S8/S9 phages show reduced CLT dependency and Dpo-independent entry
Whereas in our dataset, non-S8/S9 phages infecting a specific strain had a 92% probability of spotting in other strains of the same CLT, this association dropped for the broad-range S8/S9 phages (TPR = 0.39 ± 0.01; Figure 3A). Hence, the tropism of S8/S9 phages was weakly determined by CLT. Also, S8/S9 phages did not produce plaque haloes (Table S1). This suggests that S8/S9 phages use alternative mechanisms for penetrating the bacterial capsule. To verify this, we experimentally analyzed the Dpo activity of 36 different host-phage pairs involving S8/S9 phages. In contrast to the strong capsular polysaccharide digestion activity shown by the other phages, S8 phages exhibited low depolymerization activity, and the S9a phage showed none (Figure 3B). However, we found some capsule-degrading activity for some strains. For instance, phage S8b depolymerized strain KL39-CU293 but did not show Dpo activity in KL39-CU616. Interestingly, we found the opposite pattern in phage S8c. Thus, some proteins found in the genomes of S8 phages might be active against carbohydrates.
To explore the S8/S9 mechanisms of host recognition, we analyzed their putative RBPs. We found that S8 phages encoded two RBPs each, while the S9a phage had four (Table S5). Interestingly, the S8a and S8b first RBPs had >1,500 aa (2,309 and 3,201 aa, respectively), suggesting that they might represent a polyprotein.47,48 The first RBP of S8 phages showed 2–3 carbohydrate-binding modules (CBMs), while the second RBP showed a chaperone domain of endosialidase (Figure 6D; Table S5). However, no catalytic domains were found nor the typical structure of Dpos with parallel β-sheets when modeled with AlphaFold2 (Figure S7B). In the case of the phage S9a, only two RBPs showed >500 aa, suggesting that the other two might not have host recognition activity. For the remaining two RBPs, one showed an SGNH hydrolase-type esterase domain, suggestive of endolysin activity rather than Dpo activity.49
Finally, the ability of the CLT to predict productive infection increased for S8/S9 phages (spotting TPR: 0.39; productive infection TPR: 0.46; Figure 3A). In total, there were six S8 phage-bacteria combinations that showed a positive spot but that did not yield a productive infection in liquid (Figure 6A). These spots were very turbid, which may suggest phage replication in a subset of bacterial cells50 (Figure 6E). We thus hypothesized that S8/S9 turbid spots might be the result of phage replication in a subset of acapsular or low-capsular-producer bacteria.51 To examine this possibility, we compared phage spots in wild-type encapsulated bacteria, a spontaneous acapsular mutant (Cap−), and in a 1:1 mixture of both. For 4/6 combinations, we observed that S8 phage spots became clearer when increasing the concentration of the Cap− mutant in the agar plate (Figure 6E). This suggests that for some hosts, S8/S9 phages use alternative modes of entry that remain uncharacterized but are likely independent of capsular carbohydrate binding.
Discussion
Despite the relevance of phage-bacteria interactions, their study is limited by the low number of representative collections of bacteria and phages with a complete link between phenotypic and genotypic data.3 Here, we have performed a thorough analysis of the interactions between K. pneumoniae and its phages using a larger phage-host interaction matrix than in previous studies.20,33,36 All the analyzed phages but one were unknown and encompassed different families and undescribed taxonomical entities.52 Specifically, we found 44 phage species and 11 phage genera previously unknown, as they exhibited genomic similarity values with database sequences below the cutoffs of 95% and 70%, respectively.53 Rarefaction curves show no saturation in the identification of phage species and genera in the databases.54 This, together with our findings, shows the importance of carrying out more extensive samplings to uncover phage diversity.7,8,11
Given the tight relationship between phages and capsules, it is tempting to predict phage-bacteria interactions based on the CLT of the host and phage Dpo sequences.37,55 We found similar Dpo domains between distant phages with overlapping CLT specificity, suggesting a sizable diversity of Dpos for each CLT. Effective switching of tail spike domains between phages has been shown under laboratory conditions26 or concrete phages.27 Our results showed that tail spike domain swapping is frequent in nature, even across viral families. As a result, phage taxonomy was a poor predictor of host tropism, in contrast to some previous findings,37,56 but agrees with recent work showing that phage host range is largely independent of phage taxonomy11 or morphotype.8 Our results hint at the possibility of predicting phage capsular tropism only from Dpo sequence information, a goal that we achieved partially and that should be further addressed in the future. Machine-learning approaches57,58 in combination with deep mutational scanning studies to identify specificity regions in tail fibers59 could help achieve this goal.
To test the ability of Dpo RBD sequence analysis to predict capsular tropism, we used external data of prophage RBD sequences and the CLT of their hosts. It was thought that the Dpo sequences of temperate phages exhibited limited identity to those of lytic phages,20 but our analysis suggests extensive sharing of Dpo domains between both lifestyles. This may explain the high rate of recombination and suggests that temperate phages could be an important source of Dpo domains for incoming lytic phages, as observed with other genes.60 We predicted the capsular tropism for 13 CLTs (KL2, KL3, KL13, KL23, KL25, KL30, KL35, KL47, KL57, KL63, KL64, KL106, and KL140). These correspond to around one-third (n = 2,563 sequences) of the CLT abundance in the global dataset of K. pneumoniae (https://kleborate.erc.monash.edu/; accessed December 2, 2021; n = 8,366 sequences). The use of prophage RBD sequences to predict capsular tropism has several limitations. First, the host could undergo capsule swaps after prophage integration,44 leading to false associations between the phage RBD sequence and the observed host CLT. Second, establishing actual RBD-CLT associations can be complex in prophages containing multiple RBDs. Third, some CLTs can be cross-reactive.25 Fourth, we should keep in mind that despite successful capsule binding, degradation, and adsorption, intracellular mechanisms can further restrict phage infection. The use of extended host collections with a common capsular type might help to determine post-adsorptive defense mechanisms, as observed with closely related strains of Vibrio.61
Establishing associations between RBDs and pre-adsorptive phage activity might be useful for phage therapy applications because this would aid in the selection of phages prior to laboratory testing. However, this could lead to select some phages that digest capsules but that do not lyse their targets, which is usually not desirable for therapy. Still, depolymerization activity may aid the immune system or increase antibiotic susceptibility.62,63 Finally, understanding RBD-dependent capsular tropism can help to engineer previously characterized phages with other specificities. The modular structure of Dpos makes this possible, and this approach has been shown to work in Klebsiella phages.26
Regarding broad-range S8/S9 phages, their host tropism appeared to be largely independent of canonical Dpo domains. Instead, they possessed several carbohydrate-binding domains, which might aid them to access secondary receptors using their long tails, as observed in phages infecting Gram-positive bacteria.64,65 Hence, these siphophages might be able to overcome the capsule without the need for a catalytic-degrading activity. It can also be speculated that S8/S9 phages might preferentially encounter receptors in the equator of the cell surface, where the capsule is thinner,66 or in bacteria with lower capsule production levels.40,67
Overall, our results point to the feasibility of predicting the first steps of phage infection in an encapsulated host of priority public-health concern such as K. pneumoniae. The results obtained here could be extended to other encapsulated pathogenic bacteria such as Escherichia coli and Acinetobacter baumannii, as their phages also contain Dpos.23,68 More generally, comprehensive datasets with verified interactions similar to the data presented here are necessary to improve our understanding and the predictability of phage-bacteria interactions in nature and also for phage applications.
Limitations of the study
Given the specificity of phages for capsules, the use of different capsular types for isolation might have resulted in other susceptible CLTs. Another important limitation is the presentation of qualitative data based on subjective assays (spotting and killing curves), rather than quantifying the strength of each interaction. This may show differences in adsorption efficiencies based on receptors or even a more prominent role of intracellular defenses. We detected a subset of bacteria with post-adsorptive resistance to phages, but we did not verify that DNA ejection occurred. Future work should address this, as well as tropism of broad-range phages, which might be affected by the heterogeneous production of the capsule.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Phage Strains, see Table S1 | This work | N/A |
| Bacterial Strains, see Table S2 | This work | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| ADNasa TURBO™ | TermoFisher Scientific | Cat#AM2238 |
| Benzonase® Nuclease | Sigma | Cat#70664–3 |
| Micrococcal nuclease | New England Biolabs | Cat#M0247S |
| Proteinase K Solution (20 mg/mL) | Fisher Scientific | Cat#10259184 |
| Zwittergent 3–10 | Sigma | Cat#693021 |
| Citric acid monohydrate | Sigma | Cat#C1909 |
| Sodium tetraborate | Sigma | Cat#221732 |
| 3-hydroxybiphenyl | Sigma | Cat#262250 |
| Glucuronic acid | Sigma | Cat#G5269 |
| Critical commercial assays | ||
| Amicon Ultra-15 centrifugal filter units | Sigma | Cat#UFC910024 |
| DNA Clean & Concentrator 5-Kit | Zymo | Cat#D4004 |
| Invitrogen Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Cat#Q32851 |
| Illumina Nextera XT DNA kit | Illumina | Cat#FC-131-1096 |
| MiSeq Reagent Kit v2 | Illumina | Cat#MS-102-2002 |
| Deposited data | ||
| Phage killing curves | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| Database of depolymerase sequences from literature | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| List of phage receptors | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| Review tropism of Klebsiella phages | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| PDB files of phage RBPs from AlphaFold2 | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| Scripts for TPR calculation | This work; Mendeley Data | https://doi.org/10.17632/c696dvvynf.2 |
| Software and algorithms | ||
| PRINSEQ | Schmieder and Edwards69 | https://prinseq.sourceforge.net/ |
| Unicycler v0.4.7 | Wick et al.70 | https://github.com/rrwick/Unicycler |
| SPAdes v.3.14 | Bankevich et al.71 | https://github.com/ablab/spades |
| apc script | N/A | https://github.com/tseemann/apc/blob/master/apc.pl |
| MIRA4 | N/A | https://sourceforge.net/projects/mira-assembler/ |
| FastANI | Jain et al.72 | https://github.com/ParBLiSS/FastANI |
| VIRIDIC | Moraru et al.73 | http://rhea.icbm.uni-oldenburg.de/VIRIDIC/ |
| ape package | Paradis and Schliep74 | http://ape-package.ird.fr/ |
| Prokka v1.14 | Seemann75 | https://github.com/tseemann/prokka |
| Prokaryotic Virus Remote Homologous Groups (PHROGS) | Terzian et al.76 | http://millardlab.org/2021/11/21/phage-annotation-with-phrogs/ |
| genoplotR | Guy et al.77 | https://genoplotr.r-forge.r-project.org/ |
| Blast+ | NCBI | ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+ |
| inphared | Cook et al.54 | https://github.com/RyanCook94/inphared |
| bactaxR | Carroll et al.78 | https://github.com/lmc297/bactaxR |
| ViPTreeGen | Nishimura et al.79 | https://github.com/yosuken/ViPTreeGen |
| Kleborate | Wyres et al.43 | https://github.com/katholt/Kleborate |
| Snippy | Seemann | https://github.com/tseemann/snippy |
| trimAl | Capella-Gutiérrez et al.80 | http://trimal.cgenomics.org/ |
| IQ-TREE2 | Minh et al.81 | https://github.com/iqtree/iqtree2 |
| PhageBoost | Sirén et al.82 | https://github.com/ku-cbd/PhageBoost |
| CRISPRCasTyper | Russel et al.83 | https://crisprcas.i2bc.paris-saclay.fr/Home/Download |
| defense-finder | Tesson et al.32 | https://github.com/mdmparis/defense-finder |
| PIRATE | Bayliss et al.84 | https://github.com/SionBayliss/PIRATE |
| HMMER 3.1 | Eddy85 | http://hmmer.org/download.html |
| HH-suite3 | Steinegger et al.86 | https://github.com/soedinglab/hh-suite |
| InterProScan5 | Jones et al.87 | https://github.com/ebi-pf-team/interproscan |
| Phyre2 | Kelley et al.88 | http://www.sbg.bio.ic.ac.uk/∼phyre2/html/page.cgi?id=index |
| NGLVieweR | Rose et al.89 | https://github.com/nvelden/NGLVieweR |
| Gget AlphaFold2 | Luebbert and Pachter90 | https://github.com/pachterlab/gget |
| MAFFT v7 | Katoh and Standley91 | https://mafft.cbrc.jp/alignment/software/ |
| gggenomes | N/A | https://github.com/thackl/gggenomes |
| Gblocks | Castresana92 | https://anaconda.org/bioconda/gblocks |
| seqtk | N/A | https://github.com/lh3/seqtk |
Resource availability
Lead contact
Requests should be directed to the lead contact, Rafael Sanjuán (rafael.sanjuan@uv.es).
Materials availability
All bacterial and phage strains generated in this study are available from the lead contact without restriction.
Experimental model and subject details
Phage strains
All phages used in the study are detailed in Table S1.
Bacterial strains
All bacterial strains used in the study are detailed in Table S2.
Method details
Phage isolation, purification, and sequencing
Environmental samples (n = 9) were collected near a sewage water plant in Valencia (Spain) from soil and water. Isolation of candidate plaques was done as described previously38 using 36 clinical isolates of K. pneumoniae among which 19 had complete genome sequences (Table S2). Phages were amplified in 5 mL of LB broth for 3 h (150 rpm). After centrifugation (13,000 × g, 5 min), supernatants were passed through a 0.22 μm syringe filter to remove bacteria. Lysates were loaded into the upper reservoir of an Amicon filter device (100 kDa) and purified phages were recovered in 200 μL of SM buffer after two washing steps. Phages were ten-fold diluted in Turbo DNAse Buffer 1X (final volume of 100 μL) and 2 μL DNAse Turbo, benzonase, and micrococcal nuclease were added. The digestion was incubated for 1 h at 37°C and 1 μL of DNase Turbo was added during another hour. To inactivate DNases, 15 mM EDTA was used and incubated at 75°C for 10 min. To digest phage capsids, 10 μL of SDS 10% and 5 μL of proteinase K (20 mg/mL) were added and incubated for 45 min at 55°C. DNA Clean and Concentrator 5-Kit (Zymo) was used to extract and purify the DNA. Sequencing libraries were prepared using the Illumina Nextera XT DNA kit and paired-end reads (2x250 bp) were generated in the Illumina MiSeq platform (MiSeq Reagent Kit v2). For transmission electron microscopy, a drop from purified phages was deposited onto a carbon-coated Formvar supported by a 300 mesh copper grid and air-dried for 30 min. Phages were negatively stained with 2% phosphotungstic acid and visualized under Jeol JEM-1010.
Phage genome assembly and annotation
Raw reads were filtered with prinseq69 (-min_len 20 -min_qual_mean 20 -ns_max_n 30 -trim_qual_right 20 -derep 14) and assembled with Unicycler v0.4.7.70 Alternatively, SPAdes v.3.1471 or MIRA4 (https://sourceforge.net/projects/mira-assembler/) were used with the apc script (https://github.com/tseemann/apc/blob/master/apc.pl) to check genome circularity. Pairwise average nucleotide identity (ANI) values were obtained using FastANI72 (fraglen = 50). After removing phage duplicates (ANI and alignment coverage, >99%), intergenomic similarity (IGS) values were determined with VIRIDIC.73 These were used to build a Neighbor-Joining tree with the ape package.74 Phage genomes were annotated with Prokka v1.1475 using the database of Prokaryotic Virus Remote Homologous Groups (PHROGS)76 as suggested (http://millardlab.org/2021/11/21/phage-annotation-with-phrogs/). For visualization of phage genomes, phages were arbitrarily permuted at the small/large terminase subunit and genoplotR77 was used to draw blastn comparisons (-evalue 1e-5). Temperate behavior was inferred by searching annotated lysogeny markers (integrases, excisionases, recombinase, transposase, transcriptional regulators/repressors and parAB genes).
Phage classification
Phage genomes available at NCBI (PhageDB) were obtained with inphared54 (10/02/2021, n = 14,037) and compared against isolated phages with FastANI (fraglen = 500). PhageDB sequences with at least 70% ANI and 30% alignment coverage were kept for computing IGS values with our phages as above. The ANI.dendrogram function of the bactaxR package78 was used to define species and group clusters with IGS thresholds of 95% and 45%, respectively. ViPTreeGen79(https://github.com/yosuken/ViPTreeGen) was used to compare phage genomes with all Klebsiella phages from the PhageDB.
K. pneumoniae collection and genome analyses
The genome sequences of 1,145 K pneumoniae isolates were considered.93 Raw reads were trimmed with prinseq69 and Unicycler v.0.4.770 was used for assembly. Isolates of each capsular locus type (CLT) were chosen based on assembly quality, confidence of the CLT serotyping, and the O antigen and ST diversity obtained with Kleborate.43 Selected strains (160) were regrown and double colony-purified. To discard contamination, the purified isolates were typed by the wzi sequencing method94 and Sanger sequences were compared to the ones previously available from Illumina. This resulted in 138 bona fide strains including NTUH-K204495 (Table S2). These were compared against representative sequences of the K. pneumoniae clonal complexes.20,96 To do so, Snippy (https://github.com/tseemann/snippy) (--ctgs option) was used to obtain a core genome alignment of all sequences using NTUH-K2044 as reference. Positions with gaps in at least 10% of sequences were excluded with trimAl.80 IQ-TREE281 was used to infer a core ML phylogeny under the best fitting model.97 PhageBoost82 was used to extract prophages. CRISPRCasTyper83 was used to detect the presence of CRISPR systems. For the rest of bacterial defense systems, we used Defense-finder32 with default options. Finally, to determine the presence/absence of putative phage receptors, the genomes were annotated with Prokka v1.14.75 Proteins of phage receptors were obtained98 and a fasta file generated. This file as well as the 138 bacteria proteomes were used as input for PIRATE84 to obtain a presence/absence matrix. Additionally, a semantic search was done in order to obtain divergent or additional outer membrane proteins (see Data availability).
Phage host tropism (spotting)
Phage stocks (ranging from 108 to 1011 PFU/mL) were ten-fold diluted in a 96-deep well plate using a chessboard pattern. Spot tests were performed by adding a 2 μL drop of each diluted phage to the bacterial lawn of each K. pneumoniae strain in 0.3% top-agar LB media.99 This resulted in ≥ 104 PFU/spot (up to 107 PFU, Table S1), assessed in triplicate. Plates were incubated at 37°C overnight and a case was considered positive when at least two replicates showed a positive interaction (clear spot, turbid spot, or single plaques). As such, the lowest detection limit of this assay was an efficiency of phage plaquing 0.0001. Additionally, we evaluated 1,242 phage-bacteria interactions with two concentrations (Table S1), resulting in 4 discrepant cases only (0.003%). These, as well as a 95 phage-bacteria interactions of interest (that is, positive spots that did not lead to productive infections and differential spots across related phages), were re-tested by spotting 2 μL of serially diluted phage (10−1 to 10−8 from 109 PFU/mL stocks) on bacterial lawns in duplicate. We discarded spots that were not observed using the lowest dilution of this assay (105 PFU/spot).
Phage host tropism (virulent)
Every positive phage-bacteria interaction in the spotting assay was confirmed by the planktonic killing assay. This choice was based on the inability of some phages to produce plaques46 and sensitivity/time constraints.100 Briefly, bacteria were streaked from glycerol stocks and grown to an exponential phase. Adjusted exponential cultures and phage stocks were used to inoculate 200 μL of LB in a 96-well plate, resulting in 107 CFU/mL and 108 PFU/mL, respectively. Concentrations were chosen to maximize the detection of phage lysis from optical density (OD) measurements.101 Plates were incubated at 37°C with shaking (60 rpm) in a Tecan microplate reader Infinite 200 and OD600 was recorded every 20′ for at least 16h. A positive bactericidal effect was assigned when phage-treated bacteria showed impaired growth, that is, the growth curve of the phage-treated bacteria was below the control at any point of the time course, excluding the stationary phase. This was determined by direct observation of the combined plot of both curves in two independent replicates. If doubts, the interaction was classified as negative. Finally, to exclude false negatives of the killing assay due to low phage virulence or high bacterial resistance, we measured phage-progeny after 3 h of co-incubation (see quantification of free phage). A positive progeny production was considered if phage exceeded 10-fold over free-cells control. Positive cases of progeny production were also confirmed by observing phage plaques when spotting several phage dilutions as above.
Quantification of free phage
Three individual colonies were picked and grown in LB broth until they reached 107 CFU/mL. Bacteria were then infected with ∼105 PFU/mL and incubated for 3 h at 37°C with orbital shaking (250 rpm). Then, plates were centrifuged (4000 x g, 10 min) and serial dilutions of supernatants (10°-10−3) were used to titrate phage progeny in the amplification strain of each phage by the spotting assay. From these results, progeny production was calculated using the following expression: , being A and Ao the phage PFU/mL with or without bacteria, respectively. For combinations where phage was not totally adsorbed after 3 h, the incubation time was shortened to 30 min to avoid interference with newly produced particles. The same procedure was used to determine irreversible adsorption but, after co-incubation of phage and bacteria, the mixture was vigorously vortexed and centrifuged (13,000 x g, 5 min). Positive adsorption was considered when the mean free-particle titer dropped >80% compared to host-free controls.
True positive rate (TPR) calculation
To predict the effect of bacterial and phage genetic traits on positive infections, the statistical sensitivity (i.e. TPR) was calculated. For bacteria, the infection matrix resulting from the spotting assay was used as input. Briefly, for each host trait (e.g. CLT of bacteria) and host trait status (e.g. KL2), a reference strain was selected and its pattern of infection determined. This infection profile was compared with the pattern of infection of other bacteria with the same trait status (e.g. KL2). The selection of the reference bacteria was repeated 100 times to avoid biases. The resulting contingency table of positive and negative infections was used to calculate the TPR as follows: , being S|SR sensitivity to phages given sensitivity in the reference bacteria and R|SR resistance to phages given sensitivity in the reference bacteria. To determine the null TPR distribution of each trait, the reference strain was randomly selected within the whole dataset (e.g. any CLT). When the host trait represented binary data (e.g. presence/absence of a receptor), the TPR was estimated from the presence level. This process was repeated 3 times to estimate the standard error. For the TPR of phage traits, the CLT infection matrix was considered instead of individual phage-bacteria pairs. We considered that a phage was able to infect a CLT if at least one strain of the corresponding CLT was infected by the spotting assay or was referenced in the literature. Therefore, for each phage trait (family, group, or presence of a certain RBD), a reference phage was selected and the TPR calculated as above.
Phage depolymerase activity
Aliquots of 2 mL of overnight bacterial cultures were treated with 10 μL of phage (MOI ≥1) or 10 μL of SM buffer. After ON incubation at 37°C with moderate shaking (150 rpm), cultures were centrifuged (18,000 x g, 5 min) and washed twice with PBS. Then, the capsule was extracted as described previously102 with modifications. Washed cells in 0.5 mL of PBS were mixed with 100 μL of capsule extraction buffer (500 mM citric acid pH 2.0, 1% Zwittergent 3–10) and vortexed. The mix was heated at 56°C for 20 min and cellular debris was removed by centrifugation (18,000 x g, 5 min). To precipitate the capsule, the supernatant was mixed with ethanol to a final concentration of 80% (v/v) and incubated for 30 min at 4°C. The precipitates were collected by centrifugation (18,000 x g, 20 min, 4°C), air-dried, and resuspended in 200 μL of water. CPS was incubated for 2 h at 56°C103 before quantification by the uronic acid method.104 The capsule suspension was mixed with 1,200 μL of borax solution (12.5 mM disodium tetraborate in H2SO4), kept on ice for 10 min, followed by incubation at 95°C for 10 min, and immediate cooling on ice for another 10 min. The absorbance of the sample at 520 nm was measured after the addition of 20 μL of 3-hydroxybiphenyl in 0.5% NaOH. A standard curve using glucuronic acid was used to calculate uronic acid concentrations for each quantification batch. Capsule reduction (%) was calculated by comparing the uronic acid content of phage-treated versus untreated bacteria. This procedure was repeated at least twice for each phage-bacteria combination tested. Phage depolymerization activity was calculated using the following expression: , being C and Co the amount of bacterial glucuronic acid (μg/200μL) with or without phage, respectively. As positive controls of this assay, several phage-bacteria combinations in which capsule reduction was checked by microscopy were used.38 The presence of haloes was checked by visual inspection of phage plaques after overnight incubation in at least one representative bacteria of a CLT.
Search for depolymerase sequences
To enlarge our dataset of depolymerases analyzed, Klebsiella phages from the literature with known tropism were included (see Data availability). For those in which the proteins were not available at the NCBI, the phage genomes were assembled and annotated as described above. All phage proteins (>100 aa) were compared against well-characterized23,24 or experimentally validated depolymerase sequences (see Data availability) with blastp (e-value threshold of 10−5). Candidate proteins were verified to have both the N-terminal anchor and the central/C-terminal enzymatic domain using structural alignments. For the N-terminal domain, hmmsearch (e-value: 0.01 and coverage 0.01) was used against a database of HMMs N-terminal profiles obtained from the literature.24,48 For the C-terminal enzymatic domain, hhblits (database PDB70),86 InterProScan5,87 and Phyre288 were used. Proteins that structural aligned with profiles that contained the terms ‘depolymerase’, ‘pectin’, ‘pectate’, ‘sialidase’, ‘levanase’, ‘xylosidase’, ‘rhamnosidase’, ‘dextranase’, ‘alginate’, ‘hyaluronidase’, ‘hydrolase’, ‘lyase’, ‘lipase’ and ‘spike AND beta-helix|hydrolase’ were kept for further analyses.23,24 Additionally, hhblits were executed for every phage CDS. No additional Dpos apart from those obtained with the blastp search were found. Proteins with seemingly enzymatic activity were confirmed to have the characteristic β-helical domain using gget90 AlphaFold2105 (See Data availability). Protein structures were visualized with NGLVieweR.89
Phage RBP architecture
To determine how RBPs were incorporated into phage particles, a previously established nomenclature was used.24 When an N-terminal domain was identified in the RBP, this was assigned as the ‘primary RBP’. When a conserved peptide was found, the RBP was assigned as the ‘secondary RBP’. Both corresponded to ‘anchor-attached’ RBPs. If the structural domains were missing or could not be identified, the RBP was classified as ‘unknown’. This might represent a putative soluble protein. If a structural domain was present but the Dpo was absent, the RBP was classified as ‘truncated’. If the enzymatic domain present was different from a typical Dpo, this was also specified. Structural and enzymatic domains previously detected were used for this classification. Additionally, anchor domains and conserved peptides were identified based on multiple sequence alignments of RBPs from related phages (RBPs-MSAs). RBPs-MSAs were built as follows: Each RBP was compared against all phage proteins of the PhageDB and proteins of our phages with blastp (-evalue 1e-5 -max_hsps 1 -length 100). Hits considered to be intra-genus were used to build a sequence alignment with MAFFT (--adjustdirection).91 An intra-genus hit was considered when the corresponding phages scored at least 70% ANI in 40% of their genome with FastANI. The S8/S9 genomic context of RBPs was visualized with gggenomes (https://github.com/thackl/gggenomes) after blastp comparison and annotation with InterproScan5.87
Delimitation of receptor-binding domains (RBDs)
To focus on regions driving host tropism, anchor domains of depolymerases were removed. These domains are conserved among related phages and do not confer specificity.24 To do so, the conserved region of each RBPs-MSAs was removed. This region was detected with Gblocks (-b3 = 4)92 and extracted with subseq seqtk (https://github.com/lh3/seqtk). If more than one block was selected, the first block was considered as the anchor domain. If a conserved block could not be assigned or was <20 aa, the whole protein was retained. For eight cases, the anchor domain was delimited after visual inspection of the alignment. The remaining length of the tail spike was referred to as the receptor-binding domain (RBD). To determine clusters of RBDs, all versus all blastp comparisons were performed (-evalue 1e-5 -max_hsps 1 -qcov >40) and the bactaxR package used78 as above (identity threshold = 50).
Cross-validation of predicted tropisms using prophage sequence data
RBD clusters with capsular-tropism signal, that is, clusters where at least two-thirds of the sequences were in phages with shared CLT tropism, were extracted. Representative sequences of clusters were used to perform a search against the complete RefSeq protein database of bacteria with blastp (-evalue 1e-5 -max_hsps 1 -qcovs 0.4) to obtain putative prophage regions. Protein accessions of hits were used to retrieve the bacterial assembly accession and the corresponding taxonomy with efetch. Bacterial assemblies were downloaded from the NCBI ftp site and subjected to Kleborate43 to determine the CLT of each assembly. Bacterial assemblies with ‘None’ or ‘Low’ confidence were removed from the analyses. Then, the CLT assigned to each RBD from the cluster analysis was compared to the CLT of the bacteria with the residing prophage. To test for statistical significance, a random CLT was assigned to each bacterial assembly with probability equal to the frequency of each CLT in the K. pneumoniae RefSeq database (accessed: 27/04/2021). Then Fisher’s exact test was used to compare the proportion of correct predictions made in randomized versus actual data.
Mechanisms of avirulent infections
The adsorbed phages were compared by blastn with bacteria residing prophages (e-value 1e-5, min identity 80%, and alignment length ≥ 99 nt) to determine possible superinfection exclusion. CRISPRCasTyper83 was used to identify CRISPR arrays which were compared against phages with blastn-short (coverage and identity thresholds of 90% and e-value 1e-5).20 The presence of each defense system (DF, n = 42) was compared with the outcome of phage virulence (V) or avirulence (A) in that particular strain. Given this, the differential probability of resistance (dPR) for each phage-defense system was calculated as follows: , with values close to 1 indicating an overrepresentation of the DF in resistant strains (phage avirulence). On the contrary, negative values indicate that the DF was found overrepresented in susceptible strains (phage virulence) and they probably do not contribute to the observed resistant phenotype. Spontaneous acapsular bacteria were isolated as described previously,106 streak-purified to confirm the stability of the acapsular phenotype, and grown in LB broth. Serial dilutions of phage (from 10 to 104 PFU) were spotted over bacterial lawns from 500 μL of overnight WT bacteria, of WT:acapsular bacteria or acapsular bacteria only as previously.
Quantification and statistical analysis
Statistical analyses were performed using R version 3.6. Details of the statistical tests used are found in the methods and/or figure legends.
Acknowledgments
We thank the Networked Laboratory for Surveillance of Antimicrobial Resistance (NLSAR) of Comunitat Valenciana, Ana Djukovic, Beatriz Herrera, Carles Úbeda, and InfectERA Consortium for providing bacterial strains and access to genome sequences. We thank Professor Jin Town Wang for the NTUH-K2044 strain. We thank David Saiz and Lourdes Tordera for help with strain collection and depolymerization assays, respectively; Concha Hueso, Nuria Jiménez, Loles Catalán, and Alejandro Sanz Carbonell for technical assistance; and the Centro de Investigación Príncipe Felipe microscopy service for TEM. We acknowledge Dimi Boeckaerts, Olaya Rendueles, and Marta Lourenço for helpful discussions. This research was funded by ERC grants 724519 - Vis-a-Vis and 101019724 - EVADER to R.S.; projects PID2020-118602RB-I00 (Spanish MICINN) and BFU2017-89594R (Spanish MICINN) to F.G.-C.; project PROMETEO2016-122 (Generalitat Valenciana) to F.G.-C. and R.S.; and ESCMID Research Grant 20200063, project PID2020-112835RA-I00, funded by MCIN/AEI/10.13039/501100011033, and project SEJIGENT/2021/014 funded by Conselleria d’Innovació, Universitats, Ciència i Societat Digital (Generalitat Valenciana) to P.D.-C. P.D.-C. was financially supported by a Ramón y Cajal contract RYC2019-028015-I funded by MCIN/AEI/10.13039/501100011033, ESF Invest in your future. B.B. was funded by a PhD fellowship from Spanish MCIU FPU16/02139.
Author contributions
B.B. contributed to conceiving the project, designing research, and performing the experiments/computational analyses, data analysis, and manuscript writing. N.G.-G. contributed to computational analyses, resources, and visualization. M.G.-O. contributed to resources. F.G.-C. contributed to design research, resources, manuscript revision, and supervision. P.D.-C. isolated the phages and contributed to conceiving the project, designing research, revising the manuscript, and supervision. R.S. contributed to conceiving the project, designing research, analyzing data, writing and revising the manuscript, and supervision.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 6, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112048.
Contributor Information
Fernando González-Candelas, Email: fernando.gonzalez@uv.es.
Pilar Domingo-Calap, Email: pilar.domingo@uv.es.
Rafael Sanjuan, Email: rafael.sanjuan@uv.es.
Supplemental information
Phage ID, phylogenetic classification in 13 groups, genome size and circularity, sequencing depth, GC %, CDS, putative lysogeny genes, taxonomic family, subfamily, and genus according to IGS values with the closest phage are provided. Additionally, concentration of phages for the spot assay, formation of plaque haloes, susceptible CLTs, RBP system and number of depolymerase domains (Dpos) are also shown. REP: transcription repressor/regulator, PAR: parAB genes, REC: recombinases. TSP: Tail spike protein. CBM: carbohydrate-binding module. Taxonomy ranks marked with a star can be reclassified based on relationships showed in this study. CLTs marked with a star indicate that bacteria were acapsular. Dpos marked with a star indicate putative soluble Dpos.
The strain, sequencing details, virulence and antibiotic resistance scores, wzi allele, CLT and OLT inference, number of prophages, CRISPR loci, and presence of defense systems and secondary receptors are provided. The presence of a capsule was determined by visual inspection of colonies using light microscopy.
The strain used for phage isolation is indicated. Spotting: positive by the spot-assay. Virulent: positive by the killing planktonic or progeny assay.
RBDs are identified by the phage name followed by the phage protein in which the domain was detected, and the positions considered after the removal of the anchor domain.
Data and code availability
-
•
The FASTQ files of the 70 Klebsiella phages sequenced are deposited in the European Nucleotide Archive (ENA) under Bioproject PRJEB46367.
-
•
The genome sequences of the 46 phages detailed in this study are deposited in NCBI GenBank according to the Accession numbers provided in Table S1.
-
•
The sequences of the bacterial strains used are deposited in the ENA with Accession numbers provided in Table S2.
-
•
Raw data of phage killing curves, databases of depolymerases, list of phage receptors and Klebsiella phages from the literature, as well as protein databank files of modeled RBPs and scripts used for True positive rate (TPR) calculations are available at the Mendeley Data of this paper: https://doi.org/10.17632/c696dvvynf.2
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Abedon S.T. Cambridge University Press; 2008. Bacteriophage Ecology: Population Growth, Evolution, and Impact of Bacterial Viruses. [Google Scholar]
- 2.Chevallereau A., Pons B.J., van Houte S., Westra E.R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 2022;20:49–62. doi: 10.1038/s41579-021-00602-y. [DOI] [PubMed] [Google Scholar]
- 3.Lamy-Besnier Q., Brancotte B., Ménager H., Debarbieux L. Viral Host Range database, an online tool for recording, analyzing and disseminating virus-host interactions. Bioinformatics. 2021;37:2798–2801. doi: 10.1093/bioinformatics/btab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards R.A., McNair K., Faust K., Raes J., Dutilh B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016;40:258–272. doi: 10.1093/femsre/fuv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roux S., Hallam S.J., Woyke T., Sullivan M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. Elife. 2015;4:e08490. doi: 10.7554/eLife.08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porter N.T., Hryckowian A.J., Merrill B.D., Fuentes J.J., Gardner J.O., Glowacki R.W.P., Singh S., Crawford R.D., Snitkin E.S., Sonnenburg J.L., Martens E.C. Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol. 2020;5:1170–1181. doi: 10.1038/s41564-020-0746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maffei E., Shaidullina A., Burkolter M., Heyer Y., Estermann F., Druelle V., Sauer P., Willi L., Michaelis S., Hilbi H., et al. Systematic exploration of Escherichia coli phage–host interactions with the BASEL phage collection. PLoS Biol. 2021;19:e3001424. doi: 10.1371/journal.pbio.3001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauffman K.M., Chang W.K., Brown J.M., Hussain F.A., Yang J., Polz M.F., Kelly L. Resolving the structure of phage–bacteria interactions in the context of natural diversity. Nat. Commun. 2022;13:372. doi: 10.1038/s41467-021-27583-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mutalik V.K., Adler B.A., Rishi H.S., Piya D., Zhong C., Koskella B., Kutter E.M., Calendar R., Novichkov P.S., Price M.N., et al. High-throughput mapping of the phage resistance landscape in E. coli. PLoS Biol. 2020;18:e3000877. doi: 10.1371/journal.pbio.3000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jonge P.A., Nobrega F.L., Brouns S.J.J., Dutilh B.E. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol. 2019;27:51–63. doi: 10.1016/j.tim.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Göller P.C., Elsener T., Lorgé D., Radulovic N., Bernardi V., Naumann A., Amri N., Khatchatourova E., Coutinho F.H., Loessner M.J., Gómez-Sanz E. Multi-species host range of staphylococcal phages isolated from wastewater. Nat. Commun. 2021;12:6965. doi: 10.1038/s41467-021-27037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamdi S., Rousseau G.M., Labrie S.J., Tremblay D.M., Kourda R.S., Ben Slama K., Moineau S. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 2017;7:40349. doi: 10.1038/srep40349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gambino M., Nørgaard Sørensen A., Ahern S., Smyrlis G., Gencay Y.E., Hendrix H., Neve H., Noben J.-P., Lavigne R., Brøndsted L. Phage S144, A new polyvalent phage infecting Salmonella spp. and Cronobacter sakazakii. Int. J. Mol. Sci. 2020;21:5196. doi: 10.3390/ijms21155196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young F., Rogers S., Robertson D.L. Predicting host taxonomic information from viral genomes: a comparison of feature representations. PLoS Comput. Biol. 2020;16:e1007894. doi: 10.1371/journal.pcbi.1007894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarroel J., Kleinheinz K.A., Jurtz V.I., Zschach H., Lund O., Nielsen M., Larsen M.V. HostPhinder: a phage host prediction tool. Viruses. 2016;8 doi: 10.3390/v8050116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutinho F.H., Zaragoza-Solas A., López-Pérez M., Barylski J., Zielezinski A., Dutilh B.E., Edwards R., Rodriguez-Valera F. RaFAH: host prediction for viruses of Bacteria and Archaea based on protein content. Patterns. 2021;2:100274. doi: 10.1016/j.patter.2021.100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampton H.G., Watson B.N.J., Fineran P.C. The arms race between bacteria and their phage foes. Nature. 2020;577:327–336. doi: 10.1038/s41586-019-1894-8. [DOI] [PubMed] [Google Scholar]
- 18.Bernheim A., Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol. 2020;18:113–119. doi: 10.1038/s41579-019-0278-2. [DOI] [PubMed] [Google Scholar]
- 19.Scholl D., Adhya S., Merril C. Escherichia coli K1’s capsule is a barrier to bacteriophage T7. Appl. Environ. Microbiol. 2005;71:4872–4874. doi: 10.1128/AEM.71.8.4872-4874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Sousa J.A.M., Buffet A., Haudiquet M., Rocha E.P.C., Rendueles O. Modular prophage interactions driven by capsule serotype select for capsule loss under phage predation. ISME J. 2020;14:2980–2996. doi: 10.1038/s41396-020-0726-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rendueles O., Garcia-Garcerà M., Néron B., Touchon M., Rocha E.P.C. Abundance and co-occurrence of extracellular capsules increase environmental breadth: implications for the emergence of pathogens. PLoS Pathog. 2017;13:e1006525. doi: 10.1371/journal.ppat.1006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mostowy R.J., Holt K.E. Diversity-generating machines: genetics of bacterial sugar-coating. Trends Microbiol. 2018;26:1008–1021. doi: 10.1016/j.tim.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pires D.P., Oliveira H., Melo L.D.R., Sillankorva S., Azeredo J. Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016;100:2141–2151. doi: 10.1007/s00253-015-7247-0. [DOI] [PubMed] [Google Scholar]
- 24.Latka A., Leiman P.G., Drulis-Kawa Z., Briers Y. Modeling the architecture of depolymerase-containing receptor binding proteins in Klebsiella phages. Front. Microbiol. 2019;10:2649. doi: 10.3389/fmicb.2019.02649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pieroni P., Rennie R.P., Ziola B., Deneer H.G. The use of bacteriophages to differentiate serologically cross-reactive isolates of Klebsiella pneumoniae. J. Med. Microbiol. 1994;41:423–429. doi: 10.1099/00222615-41-6-423. [DOI] [PubMed] [Google Scholar]
- 26.Latka A., Lemire S., Grimon D., Dams D., Maciejewska B., Lu T., Drulis-Kawa Z., Briers Y. Engineering the modular receptor-binding proteins of Klebsiella phages switches their capsule serotype specificity. mBio. 2021;12:e00455–e00521. doi: 10.1128/mbio.00455-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scholl D., Adhya S., Merril C.R. Bacteriophage SP6 is closely related to phages K1-5, K5, and K1E but encodes a tail protein very similar to that of the distantly related P22. J. Bacteriol. 2002;184:2833–2836. doi: 10.1128/JB.184.10.2833-2836.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelkonen S., Aalto J., Finne J. Differential activities of bacteriophage depolymerase on bacterial polysaccharide: binding is essential but degradation is inhibitory in phage infection of K1-defective Escherichia coli. J. Bacteriol. 1992;174:7757–7761. doi: 10.1128/jb.174.23.7757-7761.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Born Y., Fieseler L., Klumpp J., Eugster M.R., Zurfluh K., Duffy B., Loessner M.J. The tail-associated depolymerase of Erwinia amylovora phage L1 mediates host cell adsorption and enzymatic capsule removal, which can enhance infection by other phage. Environ. Microbiol. 2014;16:2168–2180. doi: 10.1111/1462-2920.12212. [DOI] [PubMed] [Google Scholar]
- 30.Pendleton J.N., Gorman S.P., Gilmore B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 31.Lam M.M.C., Wick R.R., Judd L.M., Holt K.E., Wyres K.L. Kaptive 2.0: updated capsule and lipopolysaccharide locus typing for the Klebsiella pneumoniae species complex. Microb. Genom. 2022;8:000800. doi: 10.1099/mgen.0.000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tesson F., Hervé A., Mordret E., Touchon M., d'Humières C., Cury J., Bernheim A. Systematic and quantitative view of the antiviral arsenal of prokaryotes. Nat. Commun. 2022;13:2561. doi: 10.1038/s41467-022-30269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend E.M., Kelly L., Gannon L., Muscatt G., Dunstan R., Michniewski S., Sapkota H., Kiljunen S.J., Kolsi A., Skurnik M., et al. Isolation and characterization of Klebsiella phages for phage therapy. Phage. 2021;2:26–42. doi: 10.1089/phage.2020.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eskenazi A., Lood C., Wubbolts J., Hites M., Balarjishvili N., Leshkasheli L., Askilashvili L., Kvachadze L., van Noort V., Wagemans J., et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022;13:302. doi: 10.1038/s41467-021-27656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majkowska-Skrobek G., Markwitz P., Sosnowska E., Lood C., Lavigne R., Drulis-Kawa Z. The evolutionary trade-offs in phage-resistant Klebsiella pneumoniae entail cross-phage sensitization and loss of multidrug resistance. Environ. Microbiol. 2021 doi: 10.1111/1462-2920.15476. [DOI] [PubMed] [Google Scholar]
- 36.Venturini C., Ben Zakour N.L., Bowring B., Morales S., Cole R., Kovach Z., Branston S., Kettle E., Thomson N., Iredell J.R. Fine capsule variation affects bacteriophage susceptibility in Klebsiella pneumoniae ST258. FASEB J. 2020;34:10801–10817. doi: 10.1096/fj.201902735R. [DOI] [PubMed] [Google Scholar]
- 37.Sørensen A.N., Woudstra C., Sørensen M.C.H., Brøndsted L. Subtypes of tail spike proteins predicts the host range of Ackermannviridae phages. Comput. Struct. Biotechnol. J. 2021;19:4854–4867. doi: 10.1016/j.csbj.2021.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domingo-Calap P., Beamud B., Mora-Quilis L., González-Candelas F., Sanjuán R. Isolation and characterization of two Klebsiella pneumoniae phages encoding divergent depolymerases. Int. J. Mol. Sci. 2020;21:3160. doi: 10.3390/ijms21093160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Y.-J., Lin T.-L., Chen C.-C., Tsai Y.-T., Cheng Y.-H., Chen Y.-Y., Hsieh P.-F., Lin Y.-T., Wang J.-T. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 2017;91:e02457–e02516. doi: 10.1128/JVI.02457-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lourenço M., Osbelt L., Passet V., Gravey F., Strowig T., Rodrigues C., Brisse S. Phages against non-capsulated Klebsiella pneumoniae: broader host range, slower resistance. bioRxiv. 2022 doi: 10.1101/2022.08.04.502604. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson N.A., Cingolani G. A tail of phage adhesins. Structure. 2018;26:1565–1567. doi: 10.1016/j.str.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Prokhorov N.S., Riccio C., Zdorovenko E.L., Shneider M.M., Browning C., Knirel Y.A., Leiman P.G., Letarov A.V. Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol. Microbiol. 2017;105:385–398. doi: 10.1111/mmi.13710. [DOI] [PubMed] [Google Scholar]
- 43.Wyres K.L., Wick R.R., Gorrie C., Jenney A., Follador R., Thomson N.R., Holt K.E. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. 2016;2:e000102. doi: 10.1099/mgen.0.000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haudiquet M., Buffet A., Rendueles O., Rocha E.P.C. Interplay between the cell envelope and mobile genetic elements shapes gene flow in populations of the nosocomial pathogen Klebsiella pneumoniae. PLoS Biol. 2021;19:e3001276. doi: 10.1371/journal.pbio.3001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonilla E., Costa A.R., van den Berg D.F., van Rossum T., Hagedoorn S., Walinga H., Xiao M., Song W., Haas P.-J., Nobrega F.L., Brouns S.J.J. Genomic characterization of four novel bacteriophages infecting the clinical pathogen Klebsiella pneumoniae. DNA Res. 2021;28:dsab013. doi: 10.1093/dnares/dsab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyman P., Abedon S.T. Advances in Applied Microbiology. Academic Press; 2010. Chapter 7 - bacteriophage host range and bacterial resistance; pp. 217–248. [DOI] [PubMed] [Google Scholar]
- 47.Crépin T., Swale C., Monod A., Garzoni F., Chaillet M., Berger I. Polyproteins in structural biology. Curr. Opin. Struct. Biol. 2015;32:139–146. doi: 10.1016/j.sbi.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boeckaerts D., Stock M., De Baets B., Briers Y. Identification of phage receptor-binding protein sequences with hidden markov models and an extreme gradient boosting classifier. Viruses. 2022;14 doi: 10.3390/v14061329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latka A., Maciejewska B., Majkowska-Skrobek G., Briers Y., Drulis-Kawa Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017;101:3103–3119. doi: 10.1007/s00253-017-8224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daubie V., Chalhoub H., Blasdel B., Dahma H., Merabishvili M., Glonti T., De Vos N., Quintens J., Pirnay J.-P., Hallin M., Vandenberg O. Determination of phage susceptibility as a clinical diagnostic tool: a routine perspective. Front. Cell. Infect. Microbiol. 2022;12:1000721. doi: 10.3389/fcimb.2022.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roach D.R., Sjaarda D.R., Castle A.J., Svircev A.M. Host exopolysaccharide quantity and composition impact Erwinia amylovora bacteriophage pathogenesis. Appl. Environ. Microbiol. 2013;79:3249–3256. doi: 10.1128/AEM.00067-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herridge W.P., Shibu P., O’Shea J., Brook T.C., Hoyles L. Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J. Med. Microbiol. 2020;69:176–194. doi: 10.1099/jmm.0.001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner D., Kropinski A.M., Adriaenssens E.M. A roadmap for genome-based phage taxonomy. Viruses. 2021;13 doi: 10.3390/v13030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cook R., Brown N., Redgwell T., Rihtman B., Barnes M., Clokie M., Stekel D.J., Hobman J., Jones M.A., Millard A. INfrastructure for a PHAge REference database: identification of large-scale biases in the current collection of cultured phage genomes. Phage. 2021;2:214–223. doi: 10.1089/phage.2021.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hryckowian A.J., Merrill B.D., Porter N.T., Van Treuren W., Nelson E.J., Garlena R.A., Russell D.A., Martens E.C., Sonnenburg J.L. Bacteroides thetaiotaomicron-infecting bacteriophage isolates inform sequence-based host range predictions. Cell Host Microbe. 2020;28:371–379.e5. doi: 10.1016/j.chom.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gencay Y.E., Gambino M., Prüssing T.F., Brøndsted L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019;21:2095–2111. doi: 10.1111/1462-2920.14597. [DOI] [PubMed] [Google Scholar]
- 57.Boeckaerts D., Stock M., Criel B., Gerstmans H., De Baets B., Briers Y. Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 2021;11:1467. doi: 10.1038/s41598-021-81063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lood C., Boeckaerts D., Stock M., De Baets B., Lavigne R., van Noort V., Briers Y. Digital phagograms: predicting phage infectivity through a multilayer machine learning approach. Curr. Opin. Virol. 2022;52:174–181. doi: 10.1016/j.coviro.2021.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Huss P., Meger A., Leander M., Nishikawa K., Raman S. Mapping the functional landscape of the receptor binding domain of T7 bacteriophage by deep mutational scanning. Elife. 2021;10:e63775. doi: 10.7554/eLife.63775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moura de Sousa J.A., Pfeifer E., Touchon M., Rocha E.P.C. Causes and consequences of bacteriophage diversification via genetic exchanges across lifestyles and bacterial taxa. Mol. Biol. Evol. 2021;38:2497–2512. doi: 10.1093/molbev/msab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hussain F.A., Dubert J., Elsherbini J., Murphy M., VanInsberghe D., Arevalo P., Kauffman K., Rodino-Janeiro B.K., Gavin H., Gomez A., et al. Rapid evolutionary turnover of mobile genetic elements drives bacterial resistance to phages. Science. 2021;374:488–492. doi: 10.1126/science.abb1083. [DOI] [PubMed] [Google Scholar]
- 62.Gordillo Altamirano F., Forsyth J.H., Patwa R., Kostoulias X., Trim M., Subedi D., Archer S.K., Morris F.C., Oliveira C., Kielty L., et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021;6:157–161. doi: 10.1038/s41564-020-00830-7. [DOI] [PubMed] [Google Scholar]
- 63.Fang Q., Feng Y., McNally A., Zong Z. Characterization of phage resistance and phages capable of intestinal decolonization of carbapenem-resistant Klebsiella pneumoniae in mice. Commun. Biol. 2022;5:48. doi: 10.1038/s42003-022-03001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayes S., Vincentelli R., Mahony J., Nauta A., Ramond L., Lugli G.A., Ventura M., van Sinderen D., Cambillau C. Functional carbohydrate binding modules identified in evolved dits from siphophages infecting various Gram-positive bacteria. Mol. Microbiol. 2018;110:777–795. doi: 10.1111/mmi.14124. [DOI] [PubMed] [Google Scholar]
- 65.Lavelle K., Goulet A., McDonnell B., Spinelli S., van Sinderen D., Mahony J., Cambillau C. Revisiting the host adhesion determinants of Streptococcus thermophilus siphophages. Microb. Biotechnol. 2020;13:1765–1779. doi: 10.1111/1751-7915.13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phanphak S., Georgiades P., Li R., King J., Roberts I.S., Waigh T.A. Super-resolution fluorescence microscopy study of the production of K1 capsules by Escherichia coli: evidence for the differential distribution of the capsule at the Poles and the equator of the cell. Langmuir. 2019;35:5635–5646. doi: 10.1021/acs.langmuir.8b04122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rendueles O., de Sousa J.A.M., Rocha E.P.C. Competition between phage-resistance mechanisms determines the outcome of bacterial co-existence. bioRxiv. 2022 doi: 10.1101/2022.07.11.499539. Preprint at. [DOI] [Google Scholar]
- 68.Oliveira H., Costa A.R., Konstantinides N., Ferreira A., Akturk E., Sillankorva S., Nemec A., Shneider M., Dötsch A., Azeredo J. Ability of phages to infect Acinetobacter calcoaceticus-Acinetobacter baumannii complex species through acquisition of different pectate lyase depolymerase domains. Environ. Microbiol. 2017;19:5060–5077. doi: 10.1111/1462-2920.13970. [DOI] [PubMed] [Google Scholar]
- 69.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wick R.R., Judd L.M., Gorrie C.L., Holt K.E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moraru C., Varsani A., Kropinski A.M. VIRIDIC-A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses. 2020;12 doi: 10.3390/v12111268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paradis E., Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 75.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 76.Terzian P., Olo Ndela E., Galiez C., Lossouarn J., Pérez Bucio R.E., Mom R., Toussaint A., Petit M.-A., Enault F. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 2021;3:lqab067. doi: 10.1093/nargab/lqab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guy L., Kultima J.R., Andersson S.G.E. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. 2010;26:2334–2335. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carroll L.M., Wiedmann M., Kovac J. Proposal of a taxonomic nomenclature for the Bacillus cereus group which reconciles genomic definitions of bacterial species with clinical and industrial phenotypes. mBio. 2020;11 doi: 10.1128/mBio.00034-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishimura Y., Yoshida T., Kuronishi M., Uehara H., Ogata H., Goto S. ViPTree: the viral proteomic tree server. Bioinformatics. 2017;33:2379–2380. doi: 10.1093/bioinformatics/btx157. [DOI] [PubMed] [Google Scholar]
- 80.Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sirén K., Millard A., Petersen B., Gilbert M.T.P., Clokie M.R.J., Sicheritz-Pontén T. Rapid discovery of novel prophages using biological feature engineering and machine learning. NAR Genom. Bioinform. 2021;3:lqaa109. doi: 10.1101/2020.08.09.243022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russel J., Pinilla-Redondo R., Mayo-Muñoz D., Shah S.A., Sørensen S.J. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas loci. CRISPR J. 2020;3:462–469. doi: 10.1089/crispr.2020.0059. [DOI] [PubMed] [Google Scholar]
- 84.Bayliss S.C., Thorpe H.A., Coyle N.M., Sheppard S.K., Feil E.J. PIRATE: a fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. GigaScience. 2019;8:giz119. doi: 10.1093/gigascience/giz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlić A., Rose P.W. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–3758. doi: 10.1093/bioinformatics/bty419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luebbert L., Pachter L. Efficient querying of genomic reference databases with gget. Bioinformatics. 2023;39:btac836. doi: 10.1101/2022.05.17.492392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 93.García-González N., Beamud B., Fuster B., Giner S., Domínguez M.V., Sánchez A., Sevilla J., Coque T., Gimeno C., González-Candelas F. Tracking the emergence and dissemination of a blaNDM-23 gene in a multi-drug resistance plasmid of Klebsiella pneumoniae. bioRxiv. 2022 doi: 10.1101/2022.07.05.498915. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brisse S., Passet V., Haugaard A.B., Babosan A., Kassis-Chikhani N., Struve C., Decré D. Wzi Gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J. Clin. Microbiol. 2013;51:4073–4078. doi: 10.1128/JCM.01924-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu K.-M., Li L.-H., Yan J.-J., Tsao N., Liao T.-L., Tsai H.-C., Fung C.-P., Chen H.-J., Liu Y.-M., Wang J.-T., et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J. Bacteriol. 2009;191:4492–4501. doi: 10.1128/JB.00315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wyres K.L., Wick R.R., Judd L.M., Froumine R., Tokolyi A., Gorrie C.L., Lam M.M.C., Duchêne S., Jenney A., Holt K.E. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15:e1008114. doi: 10.1371/journal.pgen.1008114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Z., Yu F., Zou Y., Qiu Y., Wu A., Jiang T., Peng Y. Phage protein receptors have multiple interaction partners and high expressions. Bioinformatics. 2020;36:2975–2979. doi: 10.1093/bioinformatics/btaa123. [DOI] [PubMed] [Google Scholar]
- 99.Kauffman K.M., Polz M.F. Streamlining standard bacteriophage methods for higher throughput. MethodsX. 2018;5:159–172. doi: 10.1016/j.mex.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haines M.E.K., Hodges F.E., Nale J.Y., Mahony J., van Sinderen D., Kaczorowska J., Alrashid B., Akter M., Brown N., Sauvageau D., et al. Analysis of selection methods to develop novel phage therapy cocktails against antimicrobial resistant clinical isolates of bacteria. Cold Spring Harbor Laboratory. 2020 doi: 10.1101/2020.10.13.337345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajnovic D., Muñoz-Berbel X., Mas J. Fast phage detection and quantification: an optical density-based approach. PLoS One. 2019;14:e0216292. doi: 10.1371/journal.pone.0216292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Domenico P., Schwartz S., Cunha B.A. Reduction of capsular polysaccharide production in Klebsiella pneumoniae by sodium salicylate. Infect. Immun. 1989;57:3778–3782. doi: 10.1128/iai.57.12.3778-3782.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buffet A., Rocha E.P.C., Rendueles O. Nutrient conditions are primary drivers of bacterial capsule maintenance in Klebsiella. Proc. Biol. Sci. 2021;288:20202876. doi: 10.1098/rspb.2020.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blumenkrantz N., Asboe-Hansen G. New method for quantitative determination of uronic acids. Anal. Biochem. 1973;54:484–489. doi: 10.1016/0003-2697(73)90377-1. [DOI] [PubMed] [Google Scholar]
- 105.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chiarelli A., Cabanel N., Rosinski-Chupin I., Zongo P.D., Naas T., Bonnin R.A., Glaser P. Diversity of mucoid to non-mucoid switch among carbapenemase-producing Klebsiella pneumoniae. BMC Microbiol. 2020;20:325. doi: 10.1186/s12866-020-02007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phage ID, phylogenetic classification in 13 groups, genome size and circularity, sequencing depth, GC %, CDS, putative lysogeny genes, taxonomic family, subfamily, and genus according to IGS values with the closest phage are provided. Additionally, concentration of phages for the spot assay, formation of plaque haloes, susceptible CLTs, RBP system and number of depolymerase domains (Dpos) are also shown. REP: transcription repressor/regulator, PAR: parAB genes, REC: recombinases. TSP: Tail spike protein. CBM: carbohydrate-binding module. Taxonomy ranks marked with a star can be reclassified based on relationships showed in this study. CLTs marked with a star indicate that bacteria were acapsular. Dpos marked with a star indicate putative soluble Dpos.
The strain, sequencing details, virulence and antibiotic resistance scores, wzi allele, CLT and OLT inference, number of prophages, CRISPR loci, and presence of defense systems and secondary receptors are provided. The presence of a capsule was determined by visual inspection of colonies using light microscopy.
The strain used for phage isolation is indicated. Spotting: positive by the spot-assay. Virulent: positive by the killing planktonic or progeny assay.
RBDs are identified by the phage name followed by the phage protein in which the domain was detected, and the positions considered after the removal of the anchor domain.
Data Availability Statement
-
•
The FASTQ files of the 70 Klebsiella phages sequenced are deposited in the European Nucleotide Archive (ENA) under Bioproject PRJEB46367.
-
•
The genome sequences of the 46 phages detailed in this study are deposited in NCBI GenBank according to the Accession numbers provided in Table S1.
-
•
The sequences of the bacterial strains used are deposited in the ENA with Accession numbers provided in Table S2.
-
•
Raw data of phage killing curves, databases of depolymerases, list of phage receptors and Klebsiella phages from the literature, as well as protein databank files of modeled RBPs and scripts used for True positive rate (TPR) calculations are available at the Mendeley Data of this paper: https://doi.org/10.17632/c696dvvynf.2
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.