

Abstract
To meet its high energy demands, the brain mostly utilizes glucose. However, the brain has evolved to exploit additional fuels, such as ketones, especially during prolonged fasting. With aging and neurodegenerative diseases (NDDs), the brain becomes inefficient at utilizing glucose due to changes in glia and neurons that involve glucose transport, glycolytic and Krebs cycle enzyme activities, and insulin signaling. Positron emission tomography and magnetic resonance spectroscopy studies have identified glucose metabolism abnormalities in aging, Alzheimer’s disease (AD) and other NDDs in vivo. Despite glucose hypometabolism, brain cells can utilize ketones efficiently, thereby providing a rationale for the development of therapeutic ketogenic interventions in AD and other NDDs. This review compares available ketogenic interventions and discusses the potential of the potent oral Ketone Ester for future therapeutic use in AD and other NDDs characterized by inefficient glucose utilization.
1. Introduction
The brain is a very demanding organ in terms of its energy requirements. While it represents only 2% of the total body mass, it can spend approximately 20–23% of the total body energy, corresponding to ~110–140g of glucose per day (Cunnane et al., 2016; Owen et al., 1967). It spends this amount of energy maintaining resting potentials, generating action potentials and post-synaptic potentials, regulating pre-synaptic calcium levels, and recycling neurotransmitters, especially glutamate (Attwell and Laughlin, 2001).
In the “fed state,” the brain utilizes almost solely glucose, but during fasting it begins to co-utilize ketone bodies [mainly β-hydroxybutyrate (BHB) and secondarily acetoacetate] (Cahill, 2006). A less recognized energy source for the brain is lactate produced by anaerobic glycolysis and utilized at basal and, most importantly, hyperlactatemic conditions (van Hall et al., 2009).
Aging and neurodegenerative processes are accompanied by alterations in the utilization of energy substrates, especially glucose, by the entire brain and regionally. These alterations could be the cause and/or result of aging and neurodegenerative processes. Understanding the mechanisms implicated in normal and abnormal brain metabolism is crucial for the development of anti-aging strategies and treatments for neurodegenerative diseases (NDDs) (Kapogiannis and Mattson, 2011).
This review investigates the brain metabolism of glucose and ketones, two of the brain’s most quantitatively important energy substrates, in the context of aging and neurodegeneration, with a particular focus on Alzheimer’s disease (AD). First, we present evidence from observational imaging studies on human brain metabolism and then discuss the alterations that occur at the level of membrane receptors, intracellular cascades, and biochemical pathways in different brain regions and cell types. We believe that the accumulated evidence from preclinical and clinical studies on brain metabolism has reached a critical mass that powerfully motivates the use of metabolic interventions for AD and other NDDs as potential therapeutic strategies (Kapogiannis and Mattson, 2011).
2. Brain glucose metabolism in aging, Alzheimer’s disease and other neurodegenerative diseases
2.1. Aging
2.1.1. Brain glucose metabolism in aging: Evidence from imaging studies in humans
[18F]-fluorodeoxyglucose positron-emission tomography (FDG-PET) studies on aging conducted before the year 2000 presented a dichotomy of results; about half of the studies concluded that brain glucose hypometabolism is associated with aging, while the rest found no such associations (Cunnane et al., 2011). The heterogeneity of the age of patients included in these studies, poorly defined age groups, inadequate control of cofounders (e.g., cognitive status and other health parameters), sub-optimal PET scan conditions, and no correction for brain atrophy during statistical analysis may partially account for this inconsistency (Cunnane et al., 2011).
More recent studies have consistently shown a decrease in brain glucose metabolism with aging in humans. While most studies report region-specific hypometabolism, some studies report hypometabolism affecting the entire gray matter (Kalpouzos et al., 2009; Nugent et al., 2014a, 2014b, 2016). The frontal lobes, for example, are repeatedly found to be affected in studies comparing ~72-year old with ~25-year old cognitively intact individuals (Kalpouzos et al., 2009; Nugent et al., 2014a, 2014b, 2016). The observed frontal cortex hypometabolism corresponds to the executive dysfunction and frontal-type memory impairment observed in non-demented, older adults (Buckner, 2004). Other brain areas found to display glucose hypometabolism are the temporal lobes (Nugent et al., 2014b), parietal cortex (Nugent et al., 2016), cingulate (Nugent et al., 2014b), and subcortical regions, i.e., the caudate (Nugent et al., 2014a, 2016), putamen (Nugent et al., 2014b), and thalamus (Nugent et al., 2014b). From an evolutionary perspective, it is possible that brain structures that evolved earlier (e.g., the amygdala, hippocampus, thalamus, and cingulate gyrus) are more metabolically resilient, while those that evolved later (e.g., the frontal and parietal cortex) are more vulnerable to aging-associated glucose hypometabolism and atrophy (Grieve et al., 2005).
In addition to FDG-PET scans, we have used magnetic resonance spectroscopy (MRS) to measure glucose concentration (normalized to creatine or water) in the posterior cingulate/precuneus. These brain MRS studies have revealed a stepwise increase in glucose concentrations from younger cognitively normal, to older cognitively normal, to individuals with AD. We interpreted this finding as a manifestation of decreased glucose utilization, which leads to increased concentration of remaining intra- and/or extracellular glucose (Mullins et al., 2018). Data reanalysis from cognitively normal individuals exclusively showed a correlation between age and MRS glucose concentration, where higher glucose concentrations that reflect glucose hypometabolism are associated with aging (Fig. 1).
Fig. 1.
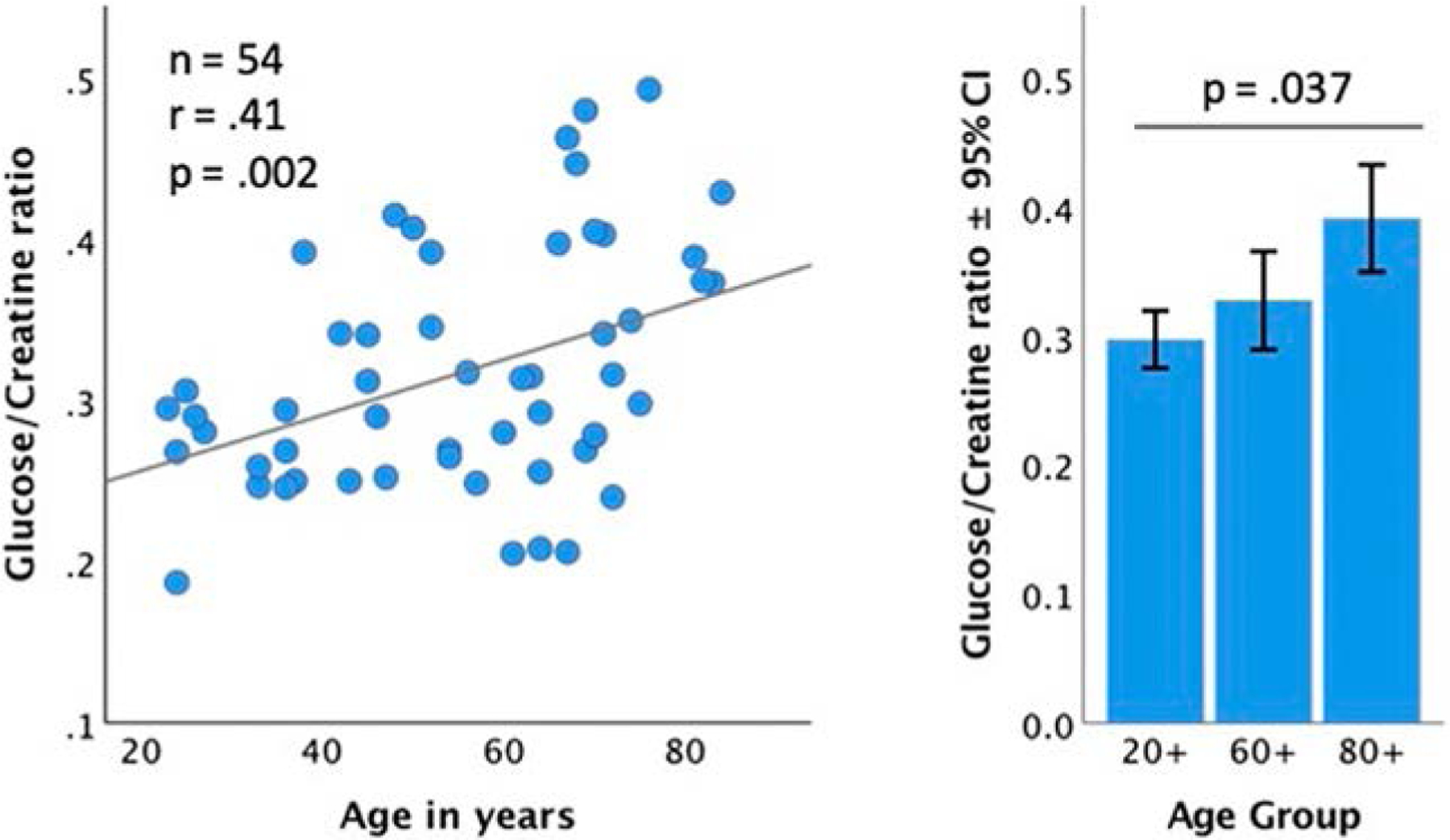
The stepwise increase in the precuneus glucose to creatine ratio associated with age and by age group. Reanalysis of data included in Mullins et al. (2018).
2.1.2. Brain glucose transport in aging
Glucose transporters are necessary for glucose transfer into brain cells since cell membranes are impermeable to glucose. There are two types of glucose transporters in the brain: the glucose transporter proteins (GLUTs) that transport glucose through facilitative diffusion (a form of passive transport), and sodium-dependent glucose transporters (SGLTs) that use an energy-coupled mechanism (active transport). Given the minor role of SGLTs for brain glucose utilization (Szablewski, 2017), we will focus our review mainly on GLUTs.
The GLUT family consists of at least 14 members (Scheepers et al., 2004). Most GLUTs are expressed by various brain cell types; for example, astrocytes express GLUT-1/2, oligodendrocytes express GLUT-2, microglia express GLUT-5, and neurons express GLUT-1/2/3/4 (Szablewski, 2017). GLUT-3 is considered the principal neuronal glucose transporter. Importantly, GLUT-1 is also expressed by the endothelial cells of the blood-brain barrier (BBB) (Szablewski, 2017). Topographically, GLUTs are expressed in brain areas that are characteristically affected by NDDs (such as the hippocampus and the frontal lobes), but also in brain regions considered resilient to NDDs (such as the cerebellum) (Szablewski, 2017).
GLUT-3 has high affinity for glucose to ensure adequate glucose transport into neurons even when glucose concentration is low (Thorens and Mueckler, 2010). Conversely, GLUT-2 has low affinity for glucose. Interestingly, GLUT-1 is acutely upregulated whenever the concentration of glucose is low (Thorens and Mueckler, 2010). Finally, GLUT-4 is regulated by insulin (Thorens and Mueckler, 2010). The diversity of GLUT transporters likely corresponds to differences in functionality. For example, at the tripartite synapse, glutamate stimulates astrocytic GLUT-1 activity to promote glucose uptake by astrocytes, whereas neuronal GLUT-3 activity is suppressed to decrease glucose influx and promote lactate uptake by adjacent astrocytes (the astrocyte-neuron lactate shuttle hypothesis) (Barros and Deitmer, 2010). It is clear that energy utilization in the brain is a process that requires communication between different cell types, and GLUTs are important components of this cellular “cross-talk.” Thus, any alteration in the number and/or function of GLUTs that occurs with aging or disease could have serious implications not only for individual cells, but also for their functional assemblies (i.e., microcircuits and networks) (Kapogiannis and Mattson, 2011).
The expression of GLUTs mRNA and protein in the brain changes with aging (Ding et al., 2013; Mooradian and Shah, 1997). Studies in rodents have revealed age-related decreases in the expression of cerebral and hippocampal GLUT-1 and GLUT-3 (Ding et al., 2013; Mooradian and Shah, 1997; Souza et al., 2015). Increasing age has been associated with decreased expression of GLUT-1 at the BBB (Ding et al., 2013) and in astrocytes (Souza et al., 2015), and importantly with decreased expression of GLUT-3 in rat neurons (Ding et al., 2013). GLUT-3 is the insulin-insensitive, principal neuronal isoform, and its age-associated reduction in the hippocampus of aged rodents (Ding et al., 2013; Fattoretti et al., 2002) is coupled to a decline in brain glucose uptake measured with PET (Ding et al., 2013). In one rat study, there was an increase of the insulin-sensitive GLUT-4 in the membrane despite the age-associated reduction in GLUT-3, although total GLUT-4 levels did not change (Ding et al., 2013). This suggests a potential compensatory mechanism for the observed GLUT-3 reduction. No change in GLUT-5 levels was observed in this study (Ding et al., 2013). The molecular mechanisms responsible for reduced neuronal glucose transport in aging remain to be established, but are likely diverse and may involve amyloid beta (Aβ) peptide induced membrane lipid peroxidation, which may lead to GLUT-3 dysfunction, decreased ATP levels and neuronal death (Mark et al., 1997).
2.1.3. The role of insulin in aging
Glucose utilization in the brain has been considered to be largely independent of the action of insulin (Banks et al., 2012). However, the fact that insulin-sensitive GLUT-4 is expressed in the brain increases the likelihood that glucose metabolism in the brain is at least partially regulated by insulin (Szablewski, 2017). An in vitro study has shown that direct infusion of insulin into neuronal cultures promotes translocation of the insulin-insensitive GLUT-3 to the plasma membrane, although it does not promote its fusion to the membrane (Uemura and Greenlee, 2006). However, once the neuron is depolarized, GLUT-3 fuses with the membrane. Moreover, neurons pretreated with insulin express higher levels of GLUT-3 at their membranes (Uemura and Greenlee, 2006), suggesting that insulin may indirectly facilitate glucose uptake through additional GLUTs besides GLUT-4. In the rat brain, GLUT-4 immunoreactive cells co-localize with neurons expressing GLUT-3 (Apelt et al., 1999). This physical proximity of GLUT-3- and GLUT-4-expressing cells further suggests that insulin plays an important role in regulating brain glucose. Finally, there is evidence that GLUT-8, which is expressed by hippocampal and other rat brain neurons, is also insulin-sensitive, but that insulin modulates its function at the subcellular level (i.e., transfer of glucose from intracellular organelles to the cytoplasm) (McEwen and Reagan, 2004). The finding that insulin partially regulates brain glucose uptake through these mechanisms has provided rationale for testing intranasal insulin to treat neurological conditions (Avgerinos et al., 2018).
Aging is associated with chronic hyperinsulinemia combined with reduced expression of insulin receptors and altered insulin signaling cascades (i.e., insulin resistance) in the brain (Steculorum et al., 2014; Zhao et al., 2004). Since glucose transportation in the brain is partially modulated by insulin, it is plausible that insulin resistance may contribute to the altered brain glucose metabolism associated with aging. Interestingly, in aged individuals without cognitive impairment, peripheral insulin resistance (measured by Homeostatic Assessment of Insulin Resistance, HOMA-IR) has been associated with reduced brain glucose metabolism in a regional pattern similar to that of AD (Baker et al., 2011; Willette et al., 2015a).
In the absence of biomarkers for central insulin resistance, earlier investigations have correlated peripheral insulin resistance measures (such as HOMA-IR) with brain function, under the assumption that central and peripheral insulin resistance partly overlap. This has changed since the development of methodologies to derive neuronal-enriched extracellular vesicles (NEVs) from peripheral blood and the introduction of NEV biomarkers. The NEV biomarkers, such as phosphorylated forms of insulin receptor substrate 1 (IRS-1) and downstream mediators, may reflect neuronal insulin signaling. Studies have shown that these biomarkers can differentiate between cognitively normal individuals (with and without diabetes) and individuals with AD (Kapogiannis et al., 2015). They also predict preclinical AD (Kapogiannis et al., 2019) and are associated with gray matter atrophy in AD (Mullins et al., 2017b). Furthermore, they differ between drug-naı¨ve individuals with schizophrenia and control subjects (Goetzl et al., 2019). Importantly, NEVs show a priori-hypothesized and pathway-specific responses to experimental interventions targeting neuronal glucose metabolism, such as (i) dietary protein restriction in cancer (Eitan et al., 2017), (ii) exenatide in Parkinson’s disease (PD) (Athauda et al., 2019), and (iii) intranasal insulin in AD (Mustapic et al., 2019). The NEV biomarkers contain signaling molecules related to multiple cellular pathways and may provide a “window to the brain” with a simple blood draw (Mustapic et al., 2017).
2.1.4. Biochemistry of glucose metabolism in aging
Glucose in brain cells can be metabolized to ATP by either oxidative metabolism or non-oxidative metabolism. Oxidative metabolism refers to the final pathway of glucose metabolism via oxidative phosphorylation, after its conversion to pyruvate and passage through the Krebs cycle (Demetrius et al., 2014). On the other hand, non-oxidative metabolism refers to the metabolism of glucose into lactate, and can occur in the presence or absence of oxygen (aerobic or anaerobic glycolysis, respectively) (Demetrius et al., 2014). Although non-oxidative metabolism of glucose produces far less ATP than oxidative phosphorylation, it is often favored by the brain because it also provides substrates for myelination, synaptogenesis, axonal elongation, and produces less reactive oxygen andspecies than oxidative phosphorylation (Bauernfeind et al., 2014; Goyal et al., 2014). Even in the presence of oxygen, glucose is not completely metabolized via the oxidative pathway; some glucose is always being metabolized via the non-oxidative pathway (aerobic glycolysis), which reveals its importance. Notably, PET studies have shown that non-oxidative metabolism of glucose through aerobic glycolysis gradually decreases with aging reaching zero by age 60 (Goyal et al., 2017). This reduction is responsible for the total glucose use reduction associated with aging, while oxidative glucose use remains unaffected (Goyal et al., 2017).
As discussed, glucose handling is a well-orchestrated process between adjacent brain cells of different types. Glial cells take up glucose mainly via GLUT-1 for energy production. During this process, they also produce lactate that can be shuttled into neurons (Jha and Morrison, 2018) and enter the Krebs cycle. Neuronal GLUT-3s also take up glucose, which is used for glycolysis and the pentose phosphate pathway, the latter being necessary for the biosynthesis of molecules that protect neurons against oxidative stress (Benarroch, 2014; Bolanos and Almeida, 2010). In aged mice, the intercellular metabolic “cross-talk” between glia and neurons is disrupted (Drulis-Fajdasz et al., 2018). It is possible that as the lactate shuttle is impaired with aging, neurons rely primarily on their own glycolysis and oxidation for energy (Drulis-Fajdasz et al., 2018) and thus have diminished capacity for energy generation.
In addition to extracellular glucose, brain cells (mainly astrocytes, and to some extent neurons) store glycogen to readily mobilize glucose as an energy source (Magistretti and Allaman, 2007). Acting as an energy reservoir, astrocytic and neuronal glycogen plays an important role in memory formation and learning (Alberini et al., 2018). On the other hand, aging in mice and Drosophila has been associated with increased accumulation of glycogen-based aggregates in the brain. These aggregates resemble the neuronal polyglucosan bodies found in Lafora’s disease, an aggressive neurodegenerative disease (Sinadinos et al., 2014). It is possible that the accumulation of these glycogen aggregates with aging may interfere with normal neuronal function (Sinadinos et al., 2014). Interestingly, targeted reduction of glycogen synthesis in aged mice and Drosophila leads to reduced levels of these aggregates and consequently improved neurological function and life extension (Sinadinos et al., 2014). So, although glycogen may play a role in normal cognitive function, its mobilization may be disrupted in aging, resulting in aggregates that exert negative effects on brain function.
2.2. Alzheimer’s disease
2.2.1. Brain glucose metabolism in Alzheimer’s disease: Evidence from imaging studies
Glucose hypometabolism is a consistent finding in FDG-PET studies in AD. The most severely affected areas are the hippocampus (Mosconi et al., 2005), lateral and medial temporal lobes (Castellano et al., 2015; Croteau et al., 2018a), and posterior cingulate/precuneus (Croteau et al., 2018a). These regions are widely known to be vulnerable to AD pathology (Braak and Del Tredici, 2012, 2013; Gefen et al., 2018) and are nodes of the default mode network of the brain (Kapogiannis and Mattson, 2011; Seeley et al., 2009). Regional brain glucose hypometabolism is closely linked to tau deposition and clinical manifestations of AD (Ossenkoppele et al., 2016; Sintini et al., 2019).
Using MRS, we have shown higher glucose concentrations in the precuneus of AD patients compared to younger and older cognitively normal individuals (Mullins et al., 2018) and attributed this to glucose hypometabolism. Our in vivo study replicated a post-mortem study that also showed glucose elevation in the brains of AD patients (An et al., 2018). Interestingly, conditions that increase the risk for AD, such as carrying the ApoE4 allele (Reiman et al., 1996, 2004) or being prediabetic/diabetic and elderly (Baker et al., 2011), cause AD-like reductions of brain glucose metabolism even without any clinical manifestation of the disease. Mild cognitive impairment (MCI) is the prodrome of AD and manifests brain glucose hypometabolism in areas similar to those in AD, but to a lesser degree (Croteau et al., 2018a; Mosconi et al., 2005). We have shown that in AD, as in normal aging, FDG hypometabolism is positively associated with HOMA-IR (Baker et al., 2011; Willette et al., 2015a). Interestingly, insulin resistance in MCI is associated with a paradoxical, transient, and perhaps pathogenic increase in hippocampal glucose metabolism (Willette et al., 2015). Similarly to MCI, individuals with Down Syndrome have an increased risk of developing AD and are characterized by glucose hypometabolism in some areas vulnerable to AD (precuneus), but potentially compensatory hypermetabolism in other areas (inferior temporal/entorhinal cortex) (Haier et al., 2003).
2.2.2. Brain glucose transport in Alzheimer’s disease
The formation of ATP in the brain is reduced by ~20% in early AD, and an even greater reduction occurs in advanced stages of the disease (Hoyer, 1992). This decrease in energy production can be partially explained by the inadequate cellular uptake of glucose due to a decreased number of astrocytic, BBB and neuronal GLUTs (An et al., 2018; Liu et al., 2008; Simpson et al., 1994). It is not clear, however, if these decreases are the cause or result of AD.
Of the various GLUTs expressed in the brain, isoforms 1–4 have been most studied in the context of AD. Post-mortem studies in individuals with AD have shown reductions in GLUT-1 (expressed by astrocytes and BBB endothelial cells), and especially GLUT-3 (primarily expressed by neurons), which are more evident in frontal, parietal, occipital, temporal cortices, the caudate nucleus and the hippocampus (An et al., 2018; Liu et al., 2008; Simpson et al., 1994). These reductions occur independently of neuronal loss (Liu et al., 2008) and in the same brain regions that appear hypometabolic in FDG-PET scans (Simpson et al., 1994). They are also associated with reduced glycolytic flux (An et al., 2018). Importantly, GLUT reductions strongly correlate to tau-phosphorylation and subsequent increase of the number of hyperphosphorylated tau-containing neurofibrillary tangles (NFTs) (Liu et al., 2008) that are a pathological hallmark of AD.
We have shown that the normal regional expression of GLUTs, as well as insulin signaling mediators, are inversely associated with the density of NFTs and (to a lesser extent) Aβ-containing neuritic plaques (NPs) in AD. In other words, brain areas that normally show a low expression of GLUT-1 and GLUT-4 (as well as IRS-1) are more likely to develop tau pathology in AD (Mullins et al., 2017a) (Fig. 2). Reductions in GLUT-3 levels have also been associated with the formation of plaques in other autopsy studies (An et al., 2018).
Fig. 2.
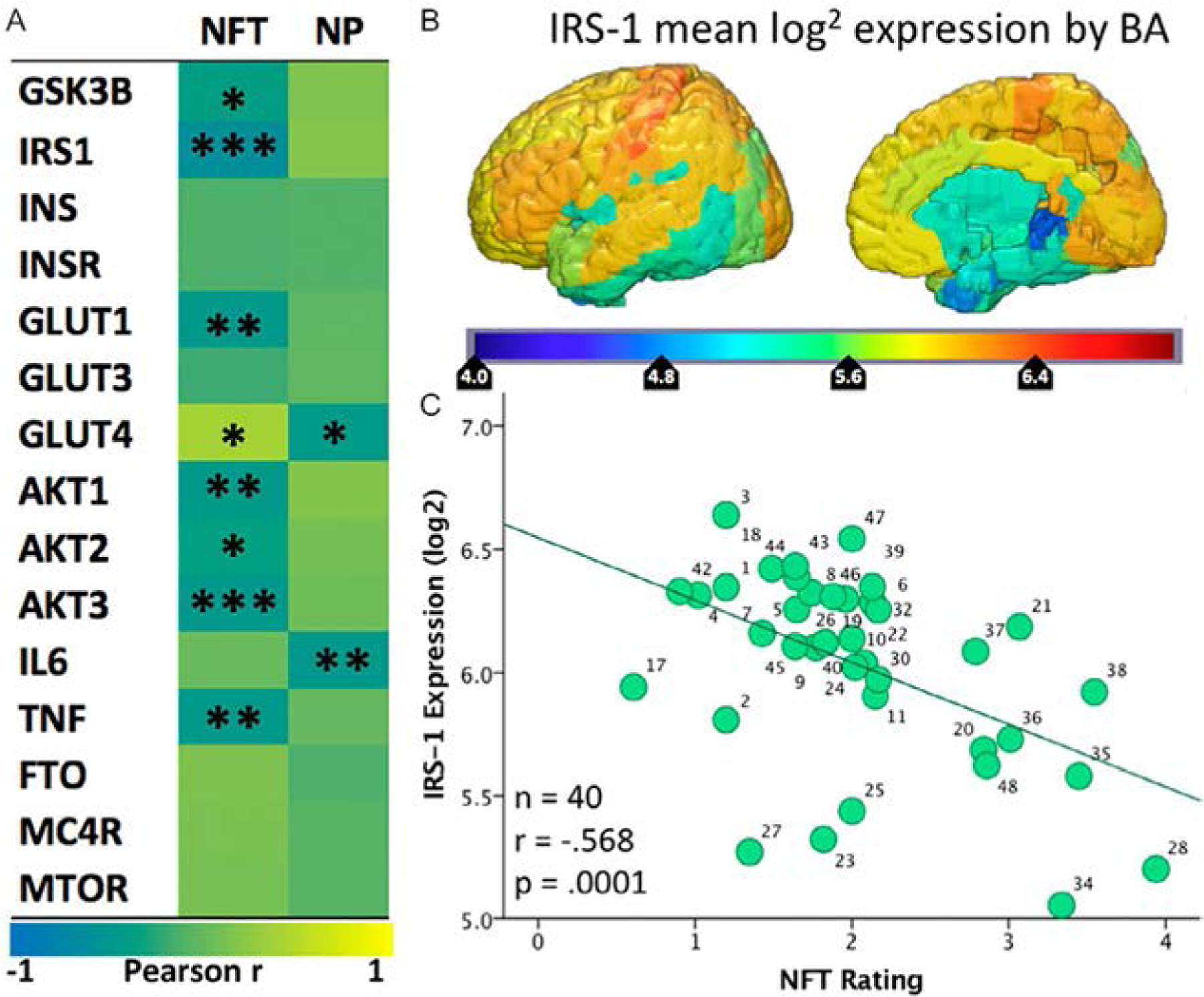
(A) Heatmap (“jet”: red high, blue low) of the spatial correlation between levels of expression of various genes from the Allen Human Brain Atlas and the density of NFTs or NPs in AD brains reported by Arnold et al. (1991). Asterisks (*/**/***) represent P values of <0.05/0.01/0.001, respectively. (B) Map of mean IRS-1 log2 expression in the six healthy human specimens included in the Allen Human Brain Atlas. (C) Scatter plot of the mean IRS-1 log2 expression from the Allen Human Brain Atlas and NFT density reported by Arnold et al. (1991). Each of the 40 data points corresponds to a Brodmann Area for which both gene expression levels and tangle density ratings were available. Previously published in Mullins et al. (2017a).
In an amyloidogenic mouse model (APP/PS1), hippocampal DG and CA1 areas have been characterized by decreased GLUT-1 levels at 18 months (Hooijmans et al., 2007). This finding supports the hypothesis of an amyloid-driven GLUT-1 decrease (Hooijmans et al., 2007) rather than the possibility that reductions in GLUT-1 lead to increased amyloid burden. In an in vitro study, hippocampal neurons from rats were treated with Aβ, resulting in decreased glucose uptake, increased GLUT-3 receptor transcription and translocation, but decreased fusion of GLUT-3 with the plasma membrane (Prapong et al., 2002). It was hypothesized that Aβ induces functional alterations to GLUT-3, which result in decreased glucose uptake despite increased GLUT-3 expression (Prapong et al., 2002). Additionally, it has been shown that Aβ-induced decrease in glucose uptake by cultured hippocampal/cortical rat neurons may be mediated by lipid peroxidation of the neuronal membrane and inactivation of GLUT-3 (Mark et al., 1997).
These findings are generally compatible with the amyloid hypothesis, which posits that an increased Aβ burden results in tau hyperphosphorylation and deposition, and ultimately neurodegeneration. Aβ-induced reduction of GLUT-1,3 number and functionality leads to decreased glucose uptake and decreased protein O-GlcNAcylation; the latter is associated with tau hyperphosphorylation (Liu et al., 2008).
Notably, brain GLUT-2 overexpression, but not GLUT-4, has been reported in AD (Liu et al., 2008). Over-expression of GLUT-2 in AD brain may be due to astrocytic activation, since increased GLUT-2 expression was associated with increased expression of the astrocytic marker, glial fibrillary acidic protein (GFAP). It is possible that the brain attempts to compensate for the energetic deficit due to the neuronal GLUT-1,3 reductions, by inducing reactive astrogliosis to provide additional number of astrocytic GLUTs (Liu et al., 2008), and ultimately shuttle energy to neurons through the astrocytes.
2.2.3. The role of insulin in Alzheimer’s disease
Since insulin partially regulates glucose handling in neuronal and glial cells, alterations in the insulin cascades are likely to be implicated in glucose hypometabolism associated with AD. In terms of mechanistic evidence, Aβ oligomers decrease plasma membrane insulin receptors and induce oxidative stress and synaptic spine deterioration in hippocampal neuronal cultures (De Felice et al., 2009). These effects are rescued by insulin (De Felice et al., 2009).
We have associated peripheral insulin resistance with glucose hypometabolism in AD (Willette et al., 2015a, 2015b), as central insulin resistance is believed to partly overlap with peripheral insulin resistance. In studies using novel NEV biomarkers of insulin signaling and insulin resistance, we have been able to differentiate between cognitively normal individuals and those with AD (Kapogiannis et al., 2015), predict preclinical AD (Kapogiannis et al., 2019), and show an association of these biomarkers with cortical atrophy in AD (Mullins et al., 2017b). The areas of atrophy associated with NEV biomarkers of insulin resistance were very similar to the areas of glucose hypometabolism in clinical AD. These areas are also affected in the brains of individuals with a genetic risk of developing AD despite absence of symptoms (Reiman et al., 2004). This reveals a connection between aberrations in central insulin signaling, glucose brain pathology, and glucose under-utilization in AD.
An additional parameter is insulin-degrading enzyme (IDE), a protease that degrades insulin and also plays a significant role in Aβ degradation (Kurochkin et al., 2018). Metabolic Syndrome (MetS) and Type 2 Diabetes Mellitus (T2DM) are conditions associated with increased risk of AD. In human and rodent tissues, both elevated glucose and Aβ have been associated with increased S-nitrosylation and subsequent inhibition of IDE, revealing “cross-talk” between MetS/T2DM and AD (Akhtar et al., 2016). An alternative hypothesis is that increased insulin levels in MetS/T2DM may competitively prevent the binding of IDE with Aβ and thereby reducing its degradation (Kurochkin et al., 2018).
2.2.4. Biochemistry of glucose metabolism in Alzheimer’s disease
In addition to alterations in GLUT, diminished glycolysis has been shown in AD. Decreased enzymatic activity in several rate-controlling enzymes of glycolysis (An et al., 2018) and in the Krebs cycle occur and constitute a “metabolic block” (Mamelak, 2012). Importantly, these abnormalities in glycolysis are coupled with AD pathology in frontal (Aβ deposition) and temporal areas (tau accumulation) (An et al., 2018). Interestingly, an autopsy study revealed elevation of glycolytic enzymes in AD, although this was attributed to survival bias, i.e., brain cells that survived neurodegeneration may have upregulated rather than downregulated glycolysis (Soucek et al., 2003). This finding corresponds well with in vitro studies on Aβ-resistant neurons that show an upregulation of pyruvate kinase and lactate dehydrogenase, the major regulators of aerobic glycolysis (Newington et al., 2011; Soucek et al., 2003).
Upregulation of glycolysis is associated with reduced levels of reactive oxygen species (Newington et al., 2011), while the inhibition of key glycolytic enzymes results in sensitization to Aβ toxicity (Newington et al., 2011). Furthermore, Aβ treatment of cortical neurons in culture leads to the production of nicotinamide adenine dinucleotide (NADH) and hypoxia-inducible factor (HIF)-1, both of which are the result of upregulated glycolysis and may mediate neuroprotection (Soucek et al., 2003). It has been suggested that an increased reliance on glycolysis and suppression of mitochondrial respiration (the Warburg effect) confers increased resistance and survival to neurons (Newington et al., 2011). Upregulation of glycolysis in neurons may act as a compensatory mechanism against AD pathology. Although this compensation might be helpful, it ultimately fails as the disease progresses and the brain manifests prominent glucose hypometabolism, even in preclinical stages of AD.
In addition to autopsy studies showing alterations in glycolytic enzymes, brain imaging studies in patients with AD reveal changes of glucose metabolism in vivo. FDG-PET scans reflect glycose utilization as a whole and do not capture the distinction between oxidative phosphorylation and aerobic glycolysis (i.e., the non-oxidative portion of glucose metabolism). In a FDG-PET study where aerobic glycolysis was calculated by subtracting the oxidative fraction of cerebral metabolic rate of glucose (CMRglc) from total CMRglc, it was found that in individuals with high Aβ burden, reductions in aerobic glycolysis facilitated tau deposition (Vlassenko et al., 2018). Thus, reductions in aerobic glycolysis occur not only in aging (see Section 2.1.4) but also in AD, where they are associated with AD pathology.
2.3. Brain glucose metabolism aberrations in other neurodegenerative diseases
Dysregulated glucose metabolism, including changes in FDG metabolism, alterations in GLUTs, and evidence of central insulin resistance have been observed in other NDDs besides AD. Individuals with PD and intact cognition show FDG hypometabolism in occipital lobes and hypermetabolism in frontal regions (Firbank et al., 2017). In PD with impaired cognition, FDG hypometabolism extends to parietal and temporal cortices (Firbank et al., 2017), predicting atrophy in these regions (Gonzalez-Redondo et al., 2014). At the same time, neuronal insulin resistance is implicated in the pathogenesis of PD and constitutes a target for PD treatment (Athauda and Foltynie, 2016).
In Huntington’s disease (HD), glucose hypometabolism in frontal and temporal lobes and the striatum occurs even in asymptomatic disease (Ciarmiello et al., 2006). In HD, the cortex and caudate expresses decreased GLUT-1 and GLUT-3, especially in late disease (Gamberino and Brennan, 1994). Interestingly, in areas of neuronal loss yet seemingly normal glia, glial GLUT-1 and GLUT-3 were under-expressed and may indicate that remaining glia no longer need to support neuronal glucose metabolism (Gamberino and Brennan, 1994). The importance of GLUTs in HD has been also shown in a large human cohort, in which an increased copy number of the gene SLC2A3 encoding GLUT-3 was associated with delayed HD symptomatology (Vittori et al., 2014).
Brain insulin resistance has also been detected in brain autopsy studies of animal models and humans with Multiple System Atrophy (MSA), localized mostly in putaminal oligodendrocytes bearing α-synuclein aggregates (Bassil et al., 2017).
Evidence that glucose dysmetabolism is present in a spectrum of NDDs reveals the central role of proper glucose handling and metabolism for brain health. It also points toward the possibility of employing similar therapeutic strategies to improve brain energy metabolism across diseases.
3. Brain ketones metabolism in aging, Alzheimer’s disease and other neurodegenerative diseases
3.1. Aging
3.1.1. Brain ketones metabolism in aging: Evidence from imaging studies
In a dual tracer PET study comparing glucose and acetoacetate utilization in aged and young rats, the overall cerebral metabolic rate for glucose (CMRglc) and acetoacetate (CMRAcAc) was similar in 21- and 4-month rats (Roy et al., 2012). In a similar study in humans using both glucose and acetoacetate tracers, it was shown that (i) glucose hypometabolism occurred with aging (shown both for the CMRglc and the uptake rate constant for glucose, Kglc); and, (ii) there was no difference in CMRAcAc with aging, but KAcAc was reduced at both the whole brain level and regionally (Nugent et al., 2014b). The CMRAcAc varies as a function of peripheral ketone concentration, such that increases in peripheral ketone concentration can increase CMRAcAc (Cunnane et al., 2016). In contrast, the uptake rate constant for AcAc (KAcAc) expresses an inherent property of brain tissue and is independent of peripheral acetoacetate levels (Nugent et al., 2014b). These findings suggest that, although the capacity of the aged brain for ketone uptake is less than that of the young brain, increased peripheral levels of ketones can increase brain ketone metabolism.
3.1.2. Transport and utilization of brain ketones in aging
Monocarboxylate transporters (MCTs) are a family of 14 receptors. Most are expressed on the membrane of brain cells and facilitate passive transport of lactate, pyruvate and ketone bodies into the brain (Perez-Escuredo et al., 2016). In the hippocampus of aged female mice, the expression and glycolytic capacity of GLUTs is decreased, but astrocytic MCT-4 expression is increased with a trend toward increased neuronal MCT-2 expression also (Ding et al., 2013). This rise in MCTs suggests that the aging brain favors ketone metabolism, perhaps as compensatory response to glucose hypometabolism. This is further supported by the observation that the elevation of ketones in aged rat brain coincides with the elevation in markers of mitochondrial dysfunction (Klosinski et al., 2015). It may be that due to the energy deficit, the aged brain tries to meet its energetic needs by utilizing ketones (Klosinski et al., 2015). At the same time, a trend for decreased MCT-1 expression in the BBB (Ding et al., 2013) indicates a diminished capacity for ketone transport from the periphery to the brain with aging.
Under normal circumstances, the liver is the main source of ketones for peripheral organs and the brain. However, brain lipids derived from myelin catabolism can also serve as a source of ketones when peripherally-produced ketones are not available to the brain, as occurs with decreased ketone transport to the brain with aging (Klosinski et al., 2015). The aging brain may compensate for its energy deficit by maladaptively catabolizing lipid-rich myelin to produce fatty acids that can be converted to ketones, inadvertently causing white matter degeneration (Klosinski et al., 2015). Specifically, the age-induced decline in mitochondrial respiration and production of H2O2 may activate the cPLA2-sphingomyelinase pathway to catabolize myelin lipids into fatty acids. Those fatty acids may be further catabolized by astrocytes to produce ketone bodies that can enter into neurons via MCTs. This phenomenon may contribute to the demyelination observed in AD and have pathogenic importance (Bartzokis, 2011).
Recently, we have developed extracellular vesicle (EV)-based biomarkers to assess ketone transport by brain cells. We probed MCT-1 and MCT-2 in NEVs and astrocytic-origin EVs expressing L1 cell adhesion molecule (L1CAM) and the astrocytic amino acid transporter (GLAST), respectively. We demonstrated differential expression (enrichment) in these two types of EVs corresponding to their cellular origin, i.e., neurons and astrocytes (Fig. 3) and propose that these novel biomarkers can be used as outcome measures in interventions that aim to boost brain ketone utilization.
Fig. 3.
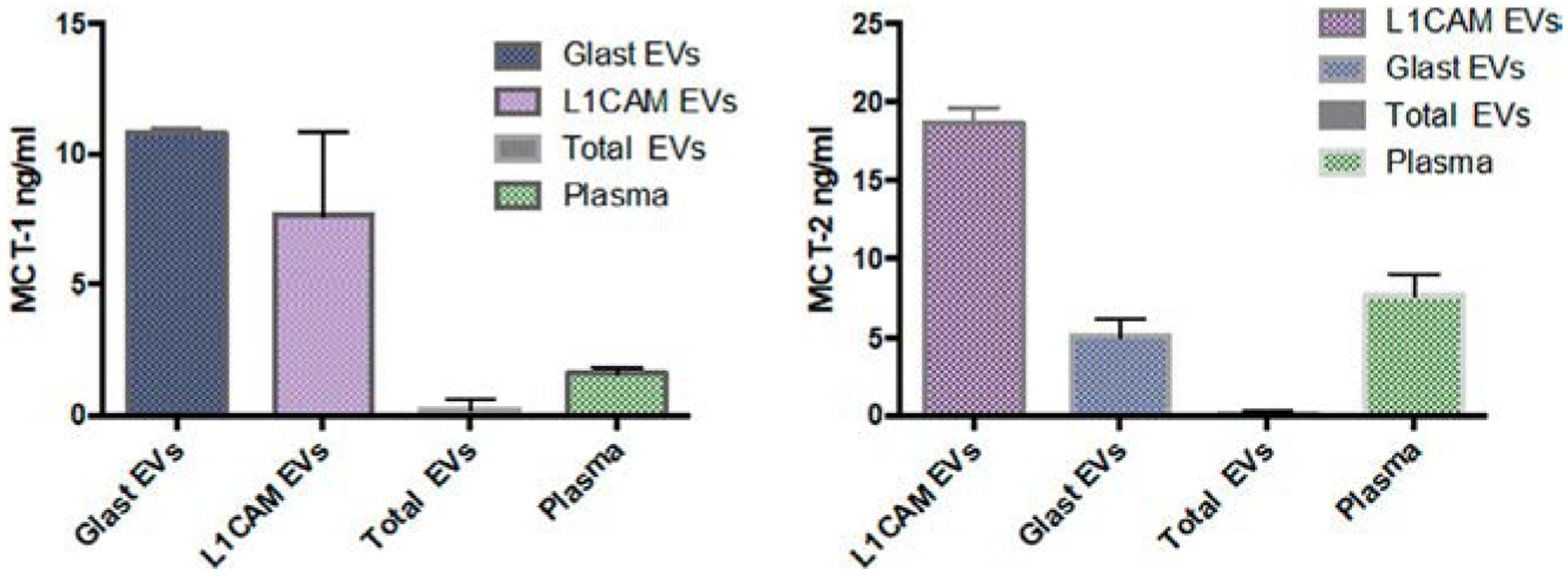
MCT-1 and MCT-2 in L1CAM and GLAST EVs. Laboratory of Clinical Investigation, NIA, NIH.
3.2. Alzheimer’s disease
3.2.1. Brain ketones metabolism in Alzheimer’s disease: Evidence from imaging studies
Dual tracer PET studies in MCI and AD individuals have shown that brain regions that are hypometabolic for glucose (i.e., show decreased CMRglc and Kglc) in parietal, temporal and frontal lobes, and the posterior cingulate, hippocampus, precuneus brain regions, metabolize acetoacetate normally (i.e., unchanged CMRAcAc and KAcAc) (Castellano et al., 2015; Croteau et al., 2018a). Brain ketone uptake is proportional to peripheral levels of et al., ketones; as this ability is conserved in the AD brain, it suggests that normal ketone metabolism occurs in AD brain (Cunnane et al., 2016). Interventions to elevate ketones in periphery, such as exercise and administration of medium-chain triglycerides (ketogenic supplement) result in a direct increase of brain ketone utilization, even in individuals with AD (Castellano et al., 2017; Croteau et al., 2018b).
3.2.2. Brain ketones transport and utilization in Alzheimer’s disease
As with aging mice, the development of pathology in AD model mice (3xTgAD) results in alteration of hippocampal MCTs (Ding et al., 2013). In AD, glial MCT-1 and MCT-4 protein expression decreases, but neuronal MCT-2 protein expression increases (Ding et al., 2013). These changes are associated with reductions in GLUT-1 and GLUT-3 protein expression (Ding et al., 2013), suggesting a compensatory mechanism to tackle glucose hypometabolism.
In female 3xTgAD mice, the expression of glycolytic enzymes, such as pyruvate dehydrogenase (PDH), decreases in parallel with increases in enzymes that metabolize ketones, such as succinyl-CoA:3-ketoacid coenzyme A transferase (SCOT) (Yao et al., 2010). Such shifts in enzymatic activity indicate a bioenergetic shift away from glucose and toward ketones as metabolic substrates in AD (Yao et al., 2010). During this bioenergetic shift, the brain may utilize ketones produced by liver ketogenesis. In addition, evidence from mice studies show that myelin may serve as a source of fatty acids that are converted to ketone bodies (especially when peripheral ketone bodies are suppressed or cannot cross the BBB) causing white matter degeneration (Yao et al., 2011). Notably, the latter is an early hallmark of AD (Bartzokis, 2011; Kuczynski et al., 2010). Alternatively, it may be that ketone bodies transported from periphery to the AD brain are mostly spent covering the energetic deficit rather than in the repair of myelin degeneration (Morris, 2005).
3.3. Brain ketone metabolism in other neurodegenerative diseases
The ability of the brain to utilize ketones in other NDDs, such as PD, HD or frontotemporal lobar degenerations (FTLD), has not been studied thoroughly. However, it has been shown in human studies that, as with AD, most NDDs present with brain glucose hypometabolism, albeit with different regional distributions specific to their pathology (Ciarmiello et al., 2006; Firbank et al., 2017; Foster et al., 2007). As with aging and AD, these diseases may be associated with preserved metabolism of ketones, which could compensate for the energy deficit related to glucose hypometabolism. This is supported by the positive effects of ketogenic interventions in experimental models of PD and HD (Chen et al., 2016; Tieu et al., 2003).
4. Interplay between glucose and ketone brain metabolism: Insights and therapeutic strategies for Alzheimer’s disease and other neurodegenerative diseases
4.1. Ketones are an alternative brain fuel to glucose
Glucose is the main energy substrate for the brain in the “fed state”; during fasting, however, ketone bodies (primarily BHB and secondarily, acetoacetate and acetone) become important for brain preservation (Cahill, 2006; Cahill and Veech, 2003). In addition, lactate produced by muscle during physical exercise can also serve as a supplemental energy fuel for the brain (Dienel, 2019).
It has been hypothesized that BHB may have conveyed evolutionary advantages that helped large, human brains to survive during periods of prolonged fasting (Cahill, 2006; Cahill and Veech, 2003). β-Hydroxybutyrate is an efficient fuel for brain cells (Cahill, 2006; Cahill and Veech, 2003; Veech et al., 2001). In experiments where the main energy fuel is BHB, the reactants at one step of the mitochondrial chain (NAD+/NADH) become more reduced, while those at the next step (Coenzyme Q/Coenzyme QH2) become more oxidized. Thus, increased availability of BHB widens the energetic difference between these two steps, and this increased energetic gap is associated with greater ATP production (Pawlosky et al., 2017). In an experiment where BHB was perfused into hearts of rats, there was an increase in the hydraulic work of heart, despite decreased oxygen consumption (Kashiwaya et al., 1994; Veech et al., 2001). Similarly, BHB-treated sperm increased its mobility, again, despite decreased oxygen consumption (Veech et al., 2001). More pertinently, neurons in models of AD and PD, as well as cells under conditions of low oxygen availability, survive longer if supplied with BHB (Cahill, 2006; Kashiwaya et al., 2000). However, it should be emphasized that the brain never relies solely on ketones and that glucose is important not only as an energy substrate, but also plays additional roles for normal brain function, for example, by promoting expression of genes regulating synaptic plasticity (Goyal et al., 2014).
Many clinicians may have underestimated the importance of ketones because they link the term “ketosis” with the dangerous medical condition termed “Diabetic Ketoacidosis” (DKA) (Cahill and Veech, 2003) that occurs only with complete insulin deficiency (Veech et al., 2001). Quantitatively, during the “fed state” ketones cover almost none of the brain’s needs, but they can suffice for 60% or more of the energy demands during a state of starvation (Cahill, 2006; Cahill and Veech, 2003; Cunnane et al., 2011). Evidence suggests that in the presence of both glucose and ketones, the brain preferentially metabolizes ketones (Cahill and Veech, 2003).
The PDH complex links glycolysis with the Krebs cycle, and has been found to decrease with aging and AD. Interestingly, preservation of PDH complex activity has been shown to increase life span (Liu et al., 2009). Ketones bypass this block by their direct conversion into acetyl-CoA and thus direct entry into the Krebs cycle. Blockade of PDH in AD also causes decreased levels citrate, which is the precursor of acetylcholine (Veech et al., 2001). An acetylcholine deficit is one of the main neurotransmitter deficits in AD and may be ameliorated by ketones (Veech et al., 2001).
Decreased insulin signaling may result from a metabolic switch from glucose to ketones, leading to increased forkhead box O (FOXO) proteins and decreased activation of the mammalian target of rapamycin (mTOR) pathway (Edwards et al., 2014; Veech et al., 2017). In animal models undergoing ketogenic interventions, these factors have been proposed as mechanisms that mediate life span extension (Edwards et al., 2014; Veech et al., 2017). In a mouse model of AD, ketones blocked the intracellular accumulation of pathogenic Aβ42 protein and rescued the activity of the mitochondrial complex 1 (restoring normal energy production by the mitochondria), which was impaired by Aβ42 (Yin et al., 2016). Furthermore, ketones reduced oxidative stress and improved synaptic plasticity (Yin et al., 2016).
In summary, the brain demonstrates a decreased capacity for glucose utilization, while ketone metabolism is relatively spared in NDDS. However, brain ketone utilization is dependent on plasma ketone levels, which are usually suppressed by normal or even increased insulin levels, in NDDs. The challenge is to effectively elevate brain ketone utilization as a therapeutic strategy for AD and other NDDs.
4.2. Ketogenic interventions
Strategies that elevate brain ketone utilization are directed toward inducing peripheral ketosis. These strategies are collectively known as ketogenic interventions and are relatively well known from their long-standing use in the treatment of epilepsy. From the least to the most robust, ketogenic interventions include: short-term fasting (12–24h) (Castellano et al., 2017); moderate aerobic exercise (Castellano et al., 2017); the intake of medium-chain triglycerides at doses of 20–30g/day while on usual diet (Avgerinos et al., 2019); a low carbohydrate diet (<50g carbs/day) for several weeks (Boden et al., 2005); a high fat (>150g fat/day) combined with low carbohydrate (<20g carbs/day) diet for several weeks (Phillips et al., 2018); and oral intake of ketone salts and esters (Fischer et al., 2018; Stubbs et al., 2017). Oral intake of ketone ester [KE; chemical name: (R)-3-hydroxybutyl (R)-3-hydroxybutyrate] is considered to be a more robust way to induce ketosis (Soto-Motaand et al., 2019; Stubbs et al., 2017). After its ingestion, KE is rapidly hydrolyzed by gut enzymes into d-β-hydroxybutyrate, which is readily absorbed, and 1,3-butanediol, which is also absorbed and converted to d-β-hydroxybutyrate in the liver (Cox et al., 2016). Absorption of both d-β-hydroxybutyrate and 1,3-butanediol results in a rapid rise in BHB blood concentration, which becomes available to the various tissues and brain (Cox et al., 2016).
A single oral dose of around 25–30g KE can elevate plasma BHB up to 4mM which is comparable to the BHB levels induced after complete starvation for 2 or more weeks (Cahill, 2006; Cahill and Veech, 2003); it is also four times greater than the levels induced after several weeks on a low carbohydrate/high-fat diet (Phillips et al., 2018); 10 times greater than a single high dose (~30g) of oral medium-chain triglycerides (Avgerinos et al., 2019) and 40 times more than the levels reached after overnight fasting (Castellano et al., 2017). In terms of safety, a single dose of KE induces less than half the elevation of BHB seen in DKA (BHB in DKA reaches ~9–10mM) and BHB levels return back to normal within a 4-h time period. In contrast, ketones in DKA continue to rise uncontrollably unless insulin is provided.
Factors that should be considered when selecting a candidate ketogenic intervention to restore brain energy in AD or other NDDs are: (a) its potency, i.e., its ability to induce peripheral and thus central ketosis; (b) its risk profile; and, (c) the convenience of the intervention, which is vital, especially for individuals with cognitive impairment who have difficulty following complex instructions, and for their caregivers. Fortunately, the most robust ketogenic intervention, the oral supplement KE, is associated with minimal side effects and is considered to be convenient for daily intake. A recent 28-day study in healthy individuals taking the KE showed that it produces robust peripheral ketosis and infrequent mild gastrointestinal side effects, mainly nausea (Soto-Mota et al., 2019). Multiple other studies have reported good tolerability and safety of KE supplementation, however no study has been conducted in individuals with MCI or AD so far (Clarke et al., 2012; Cox et al., 2016; Dearlove et al., 2019; Faull et al., 2019; Holdsworth et al., 2017; Myette-Cote et al., 2018; Soto-Mota et al., 2019; Stubbs et al., 2017, 2018, 2019).
In contrast, long-term restrictive (low carbohydrate) and high-fat diets may be very difficult for AD patients to comply with and may be associated with a negative long-term impact on cardiovascular health (e.g., atherogenesis). Supplementation with medium-chain triglycerides is associated with frequent and intolerable side effects, such as diarrhea (Courchesne-Loyer et al., 2017). In addition, both ketogenic diets and medium-chain triglycerides produce less robust ketosis than KE (Avgerinos et al., 2019; Boden et al., 2005; Phillips et al., 2018). Furthermore, ketone salts are also less robust than KE (Stubbs et al., 2017) and linked to more frequent gastrointestinal effects, perhaps due to their salt content (Stubbs et al., 2019).
4.3. The therapeutic potential of the ketone ester in Alzheimer’s disease and other neurodegenerative diseases
So far, only medium-chain triglycerides have been extensively studied in AD and while small cognitive benefits have been reported, overall results remain inconsistent (Avgerinos et al., 2019; Henderson et al., 2009). We speculate that the inconsistent cognitive benefit may be due to the mildness of BHB elevation as a result of this intervention (~0.3–0.5mM) (Avgerinos et al., 2019; Henderson et al., 2009). In comparison, KE administration induces multi-fold higher elevation of peripheral BHB (~3–4mM) (Soto-Mota et al., 2019). Nevertheless, KE has never been tested for its therapeutic effects in AD despite preclinical evidence to support this approach.
In a study involving healthy rats that consumed KE vs. isocaloric placebo for 5 days as part of their diet (30% of calories), rats in the KE group made more correct decisions on a eight-arm radial maze test, suggesting improved cognition (Murray et al., 2016). Moreover, when chronic KE administration was compared with isocaloric placebo in triple-transgenic AD (3xTgAD) mice, KE was shown to improve learning and spatial memory while reducing Aβ and hyperphosphorylated tau brain deposits (Kashiwaya et al., 2013). In another study using 3xTgAD mice, KE administration increased hippocampal d-β-hydroxybutyrate and decreased oxidative stress (Pawlosky et al., 2017).
β-Hydroxybutyrate infusion confers resistance to dopaminergic neurodegeneration in animal models of PD, in vitro and in vivo (Kashiwaya et al., 2000; Tieu et al., 2003). In HD mouse models, BHB improves electrophysiological measures, attenuates motor symptoms, reduces microgliosis and striatal lesions, and extends lifespan (Chen et al., 2016; Lim et al., 2011). These studies suggest that ketones induce multiple beneficial cellular effects besides providing energy as “another fuel,” and may even mimic the life extending properties of caloric restriction (Veech et al., 2017).
5. Conclusion
In summary, metabolic dysregulation and especially that associated with glucose is a common feature of NDDs, such as AD, PD, HD, and FTLD. In contrast, the capacity for ketone metabolism in these diseases appears to be relatively preserved. Preclinical and limited clinical data suggest that boosting ketone levels has therapeutic potential for these diseases, although the mechanisms by which this strategy may be effective are not completely understood. Unfortunately, only a small percentage of clinical trials in AD explicitly aim to improve metabolism (Cummings et al., 2019). We view this situation as an unfortunate translational gap from animal to human studies that needs to be addressed, especially since dominant therapeutic strategies that sought to reduce Aβ burden (the amyloid hypothesis) have largely failed, and a new direction may be valuable.
Acknowledgment
This research was supported entirely by the Intramural Research Program of the National Institute on Aging, NIH.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- ATP
adenosine triphosphate
- BBB
blood-brain barrier
- BHB
β-hydroxybutyrate
- CMRAcAc
cerebral metabolic rate for acetoacetate
- CMRglc
cerebral metabolic rate for glucose
- DKA
diabetic ketoacidosis
- EV
extracellular vesicle
- FDG-PET
[18F]-fluorodeoxyglucose positron-emission tomography
- FOXO
forkhead box O
- FTLD
frontotemporal lobar degenerations
- GFAP
glial fibrillary acidic protein
- GLAST
glutamate aspartate transporter
- GLUTs
glucose transporter proteins
- H2O2
hydrogen peroxide
- HD
Huntington’s disease
- HIF
hypoxia-inducible factor
- HOMA-IR
homeostatic assessment of insulin resistance
- IDE
insulin-degrading enzyme
- IRS-1
insulin receptor substrate-1
- KAcAc
uptake rate constant for acetoacetate
- KE
ketone ester
- Kglc
uptake rate constant for glucose
- L1CAM
L1 cell adhesion molecule
- MCI
mild cognitive impairment
- MCTs
monocarboxylate transporters
- MetS
metabolic syndrome
- MRS
magnetic resonance spectroscopy
- MSA
multiple system atrophy
- mTOR
mammalian target of rapamycin
- NADH
nicotinamide adenine dinucleotide
- NDDs
neurodegenerative diseases
- NEVs
neuronal-enriched extracellular vesicles
- NFTs
neurofibrillary tangles
- NPs
neuritic plaques
- PD
Parkinson’s disease
- PDH
pyruvate dehydrogenase
- SCOT
succinyl-CoA:3-ketoacid coenzyme A transferase
- SGLTs
sodium-dependent glucose transporters
- T2DM
type 2 diabetes mellitus
References
- Akhtar MW, Sanz-Blasco S, Dolatabadi N, Parker J, Chon K, Lee MS, et al. (2016). Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nature Communications, 7, 10242. 10.1038/ncomms10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Cruz E, Descalzi G, Bessieres B, & Gao V (2018). Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia, 66(6), 1244–1262. 10.1002/glia.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, et al. (2018). Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s & Dementia, 14(3), 318–329. 10.1016/j.jalz.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelt J, Mehlhorn G, & Schliebs R (1999). Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. Journal of Neuroscience Research, 57(5), 693–705. . [DOI] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, & Van Hoesen GW (1991). The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cerebral Cortex, 1(1), 103–116. [DOI] [PubMed] [Google Scholar]
- Athauda D, & Foltynie T (2016). Insulin resistance and Parkinson’s disease: A new target for disease modification? Progress in Neurobiology, 145–146, 98–120. 10.1016/j.pneurobio.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, et al. (2019). Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: A secondary analysis of the exenatide-PD trial. JAMA Neurology, 76(4), 420–429. 10.1001/jamaneurol.2018.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, & Laughlin SB (2001). An energy budget for signaling in the grey matter of the brain. Journal of Cerebral Blood Flow and Metabolism, 21(10), 1133–1145. 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Avgerinos KI, Egan JM, Mattson MP, & Kapogiannis D (2019). Medium chain triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Research Reviews, 58, 101001. 10.1016/j.arr.2019.101001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgerinos KI, Kalaitzidis G, Malli A, Kalaitzoglou D, Myserlis PG, & Lioutas VA (2018). Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. Journal of Neurology, 265(7), 1497–1510. 10.1007/s00415-018-8768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, & Craft S (2011). Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Archives of Neurology, 68(1), 51–57. 10.1001/archneurol.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Owen JB, & Erickson MA (2012). Insulin in the brain: There and back again. Pharmacology & Therapeutics, 136(1), 82–93. 10.1016/j.pharmthera.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros LF, & Deitmer JW (2010). Glucose and lactate supply to the synapse. Brain Research Reviews, 63(1–2), 149–159. 10.1016/j.brainresrev.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Bartzokis G (2011). Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiology of Aging, 32(8), 1341–1371. 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassil F, Canron MH, Vital A, Bezard E, Li Y, Greig NH, et al. (2017). Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain, 140(5), 1420–1436. 10.1093/brain/awx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind AL, Barks SK, Duka T, Grossman LI, Hof PR, & Sherwood CC (2014). Aerobic glycolysis in the primate brain: Reconsidering the implications for growth and maintenance. Brain Structure & Function, 219(4), 1149–1167. 10.1007/s00429-013-0662-z. [DOI] [PubMed] [Google Scholar]
- Benarroch EE (2014). Brain glucose transporters: Implications for neurologic disease. Neurology, 82(15), 1374–1379. 10.1212/WNL.0000000000000328. [DOI] [PubMed] [Google Scholar]
- Boden G, Sargrad K, Homko C, Mozzoli M, & Stein TP (2005). Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Annals of Internal Medicine, 142(6), 403–411. 10.7326/0003-4819-142-6-200503150-00006. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, & Almeida A (2010). The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life, 62(1), 14–18. 10.1002/iub.280. [DOI] [PubMed] [Google Scholar]
- Braak H, & Del Tredici K (2012). Alzheimer’s disease: Pathogenesis and prevention. Alzheimer’s & Dementia, 8(3), 227–233. 10.1016/j.jalz.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Braak H, & Del Tredici K (2013). Evolutional aspects of Alzheimer’s disease pathogenesis. Journal of Alzheimer’s Disease, 33(Suppl. 1), S155–S161. 10.3233/JAD-2012-129029. [DOI] [PubMed] [Google Scholar]
- Buckner RL (2004). Memory and executive function in aging and AD: Multiple factors that cause decline and reserve factors that compensate. Neuron, 44(1), 195–208. 10.1016/j.neuron.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Cahill GF Jr. (2006). Fuel metabolism in starvation. Annual Review of Nutrition, 26, 1–22. 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- Cahill GF Jr., & Veech RL (2003). Ketoacids? Good medicine? Transactions of the American Clinical and Climatological Association, 114, 149–161. discussion 162–143. [PMC free article] [PubMed] [Google Scholar]
- Castellano CA, Nugent S, Paquet N, Tremblay S, Bocti C, Lacombe G, et al. (2015). Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. Journal of Alzheimer’s Disease, 43(4), 1343–1353. 10.3233/JAD-141074. [DOI] [PubMed] [Google Scholar]
- Castellano CA, Paquet N, Dionne IJ, Imbeault H, Langlois F, Croteau E, et al. (2017). A 3-month aerobic training program improves brain energy metabolism in mild Alzheimer’s disease: Preliminary results from a neuroimaging study. Journal of Alzheimer’s Disease, 56(4), 1459–1468. 10.3233/JAD-161163. [DOI] [PubMed] [Google Scholar]
- Chen JY, Tran C, Hwang L, Deng G, Jung ME, Faull KF, et al. (2016). Partial amelioration of peripheral and central symptoms of Huntington’s disease via modulation of lipid metabolism. Journal of Huntingtons Disease, 5(1), 65–81. 10.3233/JHD-150181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, et al. (2006). Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. Journal of Nuclear Medicine, 47(2), 215–222. [PubMed] [Google Scholar]
- Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, et al. (2012). Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regulatory Toxicology and Pharmacology, 63(3), 401–408. 10.1016/j.yrtph.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne-Loyer A, Lowry CM, St-Pierre V, Vandenberghe C, Fortier M, Castellano CA, et al. (2017). Emulsification increases the acute ketogenic effect and bioavailability of medium-chain triglycerides in humans: Protein, carbohydrate, and fat metabolism. Current Developments in Nutrition, 1(7). e000851. 10.3945/cdn.117.000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, et al. (2016). Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metabolism, 24(2), 256–268. 10.1016/j.cmet.2016.07.010. [DOI] [PubMed] [Google Scholar]
- Croteau E, Castellano CA, Fortier M, Bocti C, Fulop T, Paquet N, et al. (2018a). A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Experimental Gerontology, 107, 18–26. 10.1016/j.exger.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Croteau E, Castellano CA, Richard MA, Fortier M, Nugent S, Lepage M, et al. (2018b). Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. Journal of Alzheimer’s Disease, 64(2), 551–561. 10.3233/JAD-180202. [DOI] [PubMed] [Google Scholar]
- Cummings J, Lee G, Ritter A, Sabbagh M, & Zhong K (2019). Alzheimer’s disease drug development pipeline: 2019. Alzheimers & Dementia (New York, N. Y.), 5, 272–293. 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Courchesne-Loyer A, St-Pierre V, Vandenberghe C, Pierotti T, Fortier M, et al. (2016). Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Annals of the New York Academy of Sciences, 1367(1), 12–20. 10.1111/nyas.12999. [DOI] [PubMed] [Google Scholar]
- Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, et al. (2011). Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition, 27(1), 3–20. 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. (2009). Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proceedings of the National Academy of Sciences of the United States of America, 106(6), 1971–1976. 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearlove DJ, Faull OK, Rolls E, Clarke K, & Cox PJ (2019). Nutritional ketoacidosis during incremental exercise in healthy athletes. Frontiers in Physiology, 10, 290. 10.3389/fphys.2019.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrius LA, Magistretti PJ, & Pellerin L (2014). Alzheimer’s disease: The amyloid hypothesis and the inverse Warburg effect. Frontiers in Physiology, 5, 522. 10.3389/fphys.2014.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA (2019). Brain glucose metabolism: Integration of energetics with function. Physiological Reviews, 99(1), 949–1045. 10.1152/physrev.00062.2017. [DOI] [PubMed] [Google Scholar]
- Ding F, Yao J, Rettberg JR, Chen S, & Brinton RD (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: Implication for bioenergetic intervention. PLoS One, 8(11), e79977. 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drulis-Fajdasz D, Gizak A, Wojtowicz T, Wisniewski JR, & Rakus D (2018). Aging-associated changes in hippocampal glycogen metabolism in mice. Evidence for and against astrocyte-to-neuron lactate shuttle. Glia, 66(7), 1481–1495. 10.1002/glia.23319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C, Canfield J, Copes N, Rehan M, Lipps D, & Bradshaw PC (2014). D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging (Albany NY), 6(8), 621–644. 10.18632/aging.100683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitan E, Tosti V, Suire CN, Cava E, Berkowitz S, Bertozzi B, et al. (2017). In a randomized trial in prostate cancer patients, dietary protein restriction modifies markers of leptin and insulin signaling in plasma extracellular vesicles. Aging Cell, 16(6), 1430–1433. 10.1111/acel.12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattoretti P, Bertoni-Freddari C, Casoli T, Di Stefano G, Solazzi M, & Giorgetti B (2002). Decreased expression of glucose transport protein (Glut3) in aging and vitamin E deficiency. Annals of the New York Academy of Sciences, 973, 293–296. 10.1111/j.1749-6632.2002.tb04653.x. [DOI] [PubMed] [Google Scholar]
- Faull OK, Dearlove DJ, Clarke K, & Cox PJ (2019). Beyond RPE: The perception of exercise under normal and ketotic conditions. Frontiers in Physiology, 10, 229. 10.3389/fphys.2019.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firbank MJ, Yarnall AJ, Lawson RA, Duncan GW, Khoo TK, Petrides GS, et al. (2017). Cerebral glucose metabolism and cognition in newly diagnosed Parkinson’s disease: ICICLE-PD study. Journal of Neurology, Neurosurgery, and Psychiatry, 88(4), 310–316. 10.1136/jnnp-2016-313918. [DOI] [PubMed] [Google Scholar]
- Fischer T, Och U, Klawon I, Och T, Gruneberg M, Fobker M, et al. (2018). Effect of a sodium and calcium DL-beta-hydroxybutyrate salt in healthy adults. Journal of Nutrition and Metabolism, 2018, 9812806. 10.1155/2018/9812806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, et al. (2007). FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain, 130(Pt. 10), 2616–2635. 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- Gamberino WC, & Brennan WA Jr. (1994). Glucose transporter isoform expression in Huntington’s disease brain. Journal of Neurochemistry, 63(4), 1392–1397. 10.1046/j.1471-4159.1994.63041392.x. [DOI] [PubMed] [Google Scholar]
- Gefen T, Papastefan ST, Rezvanian A, Bigio EH, Weintraub S, Rogalski E, et al. (2018). Von Economo neurons of the anterior cingulate across the lifespan and in Alzheimer’s disease. Cortex, 99, 69–77. 10.1016/j.cortex.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Nogueras-Ortiz C, Mustapic M, Mullins RJ, Abner EL, Schwartz JB, et al. (2019). Deficient neurotrophic factors of CSPG4-type neural cell exosomes in Alzheimer disease. The FASEB Journal, 33(1), 231–238. 10.1096/fj.201801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Redondo R, Garcia-Garcia D, Clavero P, Gasca-Salas C, Garcia-Eulate R, Zubieta JL, et al. (2014). Grey matter hypometabolism and atrophy in Parkinson’s disease with cognitive impairment: A two-step process. Brain, 137(Pt. 8), 2356–2367. 10.1093/brain/awu159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, & Raichle ME (2014). Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metabolism, 19(1), 49–57. 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, et al. (2017). Loss of brain aerobic glycolysis in normal human aging. Cell Metabolism, 26(2). 10.1016/j.cmet.2017.07.010. 353–360.e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieve SM, Clark CR, Williams LM, Peduto AJ, & Gordon E (2005). Preservation of limbic and paralimbic structures in aging. Human Brain Mapping, 25(4), 391–401. 10.1002/hbm.20115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haier RJ, Alkire MT, White NS, Uncapher MR, Head E, Lott IT, et al. (2003). Temporal cortex hypermetabolism in Down syndrome prior to the onset of dementia. Neurology, 61(12), 1673–1679. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, & Costantini LC (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & Metabolism (London), 6, 31. 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth DA, Cox PJ, Kirk T, Stradling H, Impey SG, & Clarke K (2017). A ketone ester drink increases postexercise muscle glycogen synthesis in humans. Medicine and Science in Sports and Exercise, 49(9), 1789–1795. 10.1249/MSS.0000000000001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooijmans CR, Graven C, Dederen PJ, Tanila H, van Groen T, & Kiliaan AJ (2007). Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Research, 1181, 93–103. 10.1016/j.brainres.2007.08.063. [DOI] [PubMed] [Google Scholar]
- Hoyer S (1992). Oxidative energy metabolism in Alzheimer brain. Studies in early-onset and late-onset cases. Molecular and Chemical Neuropathology, 16(3), 207–224. 10.1007/bf03159971. [DOI] [PubMed] [Google Scholar]
- Jha MK, & Morrison BM (2018). Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Experimental Neurology, 309, 23–31. 10.1016/j.expneurol.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalpouzos G, Chetelat G, Baron JC, Landeau B, Mevel K, Godeau C, et al. (2009). Voxel-based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiology of Aging, 30(1), 112–124. 10.1016/j.neurobiolaging.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Kapogiannis D, Boxer A, Schwartz JB, Abner EL, Biragyn A, Masharani U, et al. (2015). Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. The FASEB Journal, 29(2), 589–596. 10.1096/fj.14-262048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, & Mattson MP (2011). Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurology, 10(2), 187–198. 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Mustapic M, Shardell MD, Berkowitz ST, Diehl TC, Spangler RD, et al. (2019). Association of extracellular vesicle biomarkers with Alzheimer disease in the baltimore longitudinal study of aging. JAMA Neurology, 76(11), 1340–1351. 10.1001/jamaneurol.2019.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiology of Aging, 34(6), 1530–1539. 10.1016/j.neurobiolaging.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, et al. (1994). Control of glucose utilization in working perfused rat heart. The Journal of Biological Chemistry, 269(41), 25502–25514. [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, & Veech RL (2000). D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 97(10), 5440–5444. 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosinski LP, Yao J, Yin F, Fonteh AN, Harrington MG, Christensen TA, et al. (2015). White matter lipids as a ketogenic fuel supply in aging female brain: Implications for Alzheimer’s disease. eBioMedicine, 2(12), 1888–1904. 10.1016/j.ebiom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski B, Targan E, Madison C, Weiner M, Zhang Y, Reed B, et al. (2010). White matter integrity and cortical metabolic associations in aging and dementia. Alzheimer’s & Dementia, 6(1), 54–62. 10.1016/j.jalz.2009.04.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkin IV, Guarnera E, & Berezovsky IN (2018). Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends in Pharmacological Sciences, 39(1), 49–58. 10.1016/j.tips.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Lim S, Chesser AS, Grima JC, Rappold PM, Blum D, Przedborski S, et al. (2011). D-beta-hydroxybutyrate is protective in mouse models of Huntington’s disease. PLoS One, 6(9), e24620. 10.1371/journal.pone.0024620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. (2009). Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. The Journal of Biological Chemistry, 284(45), 31484–31492. 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, & Gong CX (2008). Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Letters, 582(2), 359–364. 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, & Allaman I (2007). Glycogen: A Trojan horse for neurons. Nature Neuroscience, 10(11), 1341–1342. 10.1038/nn1107-1341. [DOI] [PubMed] [Google Scholar]
- Mamelak M (2012). Sporadic Alzheimer’s disease: The starving brain. Journal of Alzheimer’s Disease, 31(3), 459–474. 10.3233/JAD-2012-120370. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, & Mattson MP (1997). Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. The Journal of Neuroscience, 17(3), 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, & Reagan LP (2004). Glucose transporter expression in the central nervous system: Relationship to synaptic function. European Journal of Pharmacology, 490(1–3), 13–24. 10.1016/j.ejphar.2004.02.041. [DOI] [PubMed] [Google Scholar]
- Mooradian AD, & Shah GN (1997). Age-related changes in glucose transporter-one mRNA structure and function. Proceedings of the Society for Experimental Biology and Medicine, 216(3), 380–385. 10.3181/00379727-216-44185. [DOI] [PubMed] [Google Scholar]
- Morris AA (2005). Cerebral ketone body metabolism. Journal of Inherited Metabolic Disease, 28(2), 109–121. 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, et al. (2005). Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology, 64(11), 1860–1867. 10.1212/01.WNL.0000163856.13524.08. [DOI] [PubMed] [Google Scholar]
- Mullins RJ, Diehl TC, Chia CW, & Kapogiannis D (2017a). Insulin resistance as a link between amyloid-beta and Tau pathologies in Alzheimer’s disease. Frontiers in Aging Neuroscience, 9, 118. 10.3389/fnagi.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RJ, Mustapic M, Goetzl EJ, & Kapogiannis D (2017b). Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Human Brain Mapping, 38(4), 1933–1940. 10.1002/hbm.23494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins R, Reiter D, & Kapogiannis D (2018). Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Annals of Clinical Translational Neurology, 5(3), 262–272. 10.1002/acn3.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, et al. (2016). Novel ketone diet enhances physical and cognitive performance. The FASEB Journal, 30(12), 4021–4032. 10.1096/fj.201600773R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapic M, Eitan E, Werner JK Jr., Berkowitz ST, Lazaropoulos MP, Tran J, et al. (2017). Plasma extracellular vesicles enriched for neuronal origin: A potential window into brain pathologic processes. Frontiers in Neuroscience, 11, 278. 10.3389/fnins.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapic M, Tran J, Craft S, & Kapogiannis D (2019). Extracellular vesicle biomarkers track cognitive changes following intranasal insulin in Alzheimer’s disease. Journal of Alzheimer’s Disease, 69(2), 489–498. 10.3233/JAD-180578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myette-Cote E, Neudorf H, Rafiei H, Clarke K, & Little JP (2018). Prior ingestion of exogenous ketone monoester attenuates the glycaemic response to an oral glucose tolerance test in healthy young individuals. The Journal of Physiology, 596(8), 1385–1395. 10.1113/JP275709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newington JT, Pitts A, Chien A, Arseneault R, Schubert D, & Cumming RC (2011). Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS One, 6(4), e19191. 10.1371/journal.pone.0019191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent S, Castellano CA, Bocti C, Dionne I, Fulop T, & Cunnane SC (2016). Relationship of metabolic and endocrine parameters to brain glucose metabolism in older adults: Do cognitively-normal older adults have a particular metabolic phenotype? Biogerontology, 17(1), 241–255. 10.1007/s10522-015-9595-7. [DOI] [PubMed] [Google Scholar]
- Nugent S, Castellano CA, Goffaux P, Whittingstall K, Lepage M, Paquet N, et al. (2014a). Glucose hypometabolism is highly localized, but lower cortical thickness and brain atrophy are widespread in cognitively normal older adults. American Journal of Physiology. Endocrinology and Metabolism, 306(11), E1315–E1321. 10.1152/ajpendo.00067.2014. [DOI] [PubMed] [Google Scholar]
- Nugent S, Tremblay S, Chen KW, Ayutyanont N, Roontiva A, Castellano CA, et al. (2014b). Brain glucose and acetoacetate metabolism: A comparison of young and older adults. Neurobiology of Aging, 35(6), 1386–1395. 10.1016/j.neurobiolaging.2013.11.027. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain, 139(Pt. 5), 1551–1567. 10.1093/brain/aww027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, & Cahill GF Jr. (1967). Brain metabolism during fasting. The Journal of Clinical Investigation, 46(10), 1589–1595. 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlosky RJ, Kemper MF, Kashiwaya Y, King MT, Mattson MP, & Veech RL (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. Journal of Neurochemistry, 141(2), 195–207. 10.1111/jnc.13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Escuredo J, Van Hee VF, Sboarina M, Falces J, Payen VL, Pellerin L, et al. (2016). Monocarboxylate transporters in the brain and in cancer. Biochimica et Biophysica Acta, 1863(10), 2481–2497. 10.1016/j.bbamcr.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MCL, Murtagh DKJ, Gilbertson LJ, Asztely FJS, & Lynch CDP (2018). Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial. Movement Disorders, 33(8), 1306–1314. 10.1002/mds.27390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prapong T, Buss J, Hsu WH, Heine P, West Greenlee H, & Uemura E (2002). Amyloid beta-peptide decreases neuronal glucose uptake despite causing increase in GLUT3 mRNA transcription and GLUT3 translocation to the plasma membrane. Experimental Neurology, 174(2), 253–258. 10.1006/exnr.2001.7861. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. (1996). Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. The New England Journal of Medicine, 334(12), 752–758. 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proceedings of the National Academy of Sciences of the United States of America, 101(1), 284–289. 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Nugent S, Tremblay-Mercier J, Tremblay S, Courchesne-Loyer A, Beaudoin JF, et al. (2012). The ketogenic diet increases brain glucose and ketone uptake in aged rats: A dual tracer PET and volumetric MRI study. Brain Research, 1488, 14–23. 10.1016/j.brainres.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Scheepers A, Joost HG, & Schurmann A (2004). The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. JPEN Journal of Parenteral and Enteral Nutrition, 28(5), 364–371. 10.1177/0148607104028005364. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, & Greicius MD (2009). Neurodegenerative diseases target large-scale human brain networks. Neuron, 62(1), 42–52. 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Chundu KR, Davies-Hill T, Honer WG, & Davies P (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Annals of Neurology, 35(5), 546–551. 10.1002/ana.410350507. [DOI] [PubMed] [Google Scholar]
- Sinadinos C, Valles-Ortega J, Boulan L, Solsona E, Tevy MF, Marquez M, et al. (2014). Neuronal glycogen synthesis contributes to physiological aging. Aging Cell, 13(5), 935–945. 10.1111/acel.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintini I, Schwarz CG, Martin PR, Graff-Radford J, Machulda MM, Senjem ML, et al. (2019). Regional multimodal relationships between tau, hypometabolism, atrophy, and fractional anisotropy in atypical Alzheimer’s disease. Human Brain Mapping, 40(5), 1618–1631. 10.1002/hbm.24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Mota A, Vansant H, Evans RD, & Clarke K (2019). Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regulatory Toxicology and Pharmacology, 109, 104506. 10.1016/j.yrtph.2019.104506. [DOI] [PubMed] [Google Scholar]
- Soucek T, Cumming R, Dargusch R, Maher P, & Schubert D (2003). The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron, 39(1), 43–56. 10.1016/s0896-6273(03)00367-2. [DOI] [PubMed] [Google Scholar]
- Souza DG, Bellaver B, Raupp GS, Souza DO, & Quincozes-Santos A (2015). Astrocytes from adult Wistar rats aged in vitro show changes in glial functions. Neurochemistry International, 90, 93–97. 10.1016/j.neuint.2015.07.016. [DOI] [PubMed] [Google Scholar]
- Steculorum SM, Solas M, & Bruning JC (2014). The paradox of neuronal insulin action and resistance in the development of aging-associated diseases. Alzheimer’s & Dementia, 10(1 Suppl), S3–11. 10.1016/j.jalz.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Stubbs BJ, Cox PJ, Evans RD, Cyranka M, Clarke K, & de Wet H (2018). A ketone ester drink lowers human Ghrelin and appetite. Obesity (Silver Spring), 26(2), 269–273. 10.1002/oby.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, et al. (2017). On the metabolism of exogenous ketones in humans. Frontiers in Physiology, 8, 848. 10.3389/fphys.2017.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs BJ, Cox PJ, Kirk T, Evans RD, & Clarke K (2019). Gastrointestinal effects of exogenous ketone drinks are infrequent, mild and vary according to ketone compound and dose. International Journal of Sport Nutrition and Exercise Metabolism, 29(6), 1–23. 10.1123/ijsnem.2019-0014. [DOI] [PubMed] [Google Scholar]
- Szablewski L (2017). Glucose transporters in brain: In health and in Alzheimer’s disease. Journal of Alzheimer’s Disease, 55(4), 1307–1320. 10.3233/JAD-160841. [DOI] [PubMed] [Google Scholar]
- Thorens B, & Mueckler M (2010). Glucose transporters in the 21st century. American Journal of Physiology. Endocrinology and Metabolism, 298(2), E141–E145. 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. The Journal of Clinical Investigation, 112(6), 892–901. 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura E, & Greenlee HW (2006). Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Experimental Neurology, 198(1), 48–53. 10.1016/j.expneurol.2005.10.035. [DOI] [PubMed] [Google Scholar]
- van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, et al. (2009). Blood lactate is an important energy source for the human brain. Journal of Cerebral Blood Flow and Metabolism, 29(6), 1121–1129. 10.1038/jcbfm.2009.35. [DOI] [PubMed] [Google Scholar]
- Veech RL, Bradshaw PC, Clarke K, Curtis W, Pawlosky R, & King MT (2017). Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life, 69(5), 305–314. 10.1002/iub.1627. [DOI] [PubMed] [Google Scholar]
- Veech RL, Chance B, Kashiwaya Y, Lardy HA, & Cahill GF Jr. (2001). Ketone bodies, potential therapeutic uses. IUBMB Life, 51(4), 241–247. 10.1080/152165401753311780. [DOI] [PubMed] [Google Scholar]
- Vittori A, Breda C, Repici M, Orth M, Roos RA, Outeiro TF, et al. (2014). Copy-number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington’s disease. Human Molecular Genetics, 23(12), 3129–3137. 10.1093/hmg/ddu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Gordon BA, Goyal MS, Su Y, Blazey TM, Durbin TJ, et al. (2018). Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiology of Aging, 67, 95–98. 10.1016/j.neurobiolaging.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willette AA, Bendlin BB, Starks EJ, Birdsill AC, Johnson SC, Christian BT, et al. (2015a). Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurology, 72(9), 1013–1020. 10.1001/jamaneurol.2015.0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willette AA, Modanlo N, Kapogiannis D, & Alzheimer’s Disease Neuroimaging Initiative. (2015b). Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes, 64(6), 1933–1940. 10.2337/db14-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Hamilton RT, Cadenas E, & Brinton RD (2010). Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochimica et Biophysica Acta, 1800(10), 1121–1126. 10.1016/j.bbagen.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Rettberg JR, Klosinski LP, Cadenas E, & Brinton RD (2011). Shift in brain metabolism in late onset Alzheimer’s disease: Implications for biomarkers and therapeutic interventions. Molecular Aspects of Medicine, 32(4–6), 247–257. 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JX, Maalouf M, Han P, Zhao M, Gao M, Dharshaun T, et al. (2016). Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiology of Aging, 39, 25–37. 10.1016/j.neurobiolaging.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Chen H, Quon MJ, & Alkon DL (2004). Insulin and the insulin receptor in experimental models of learning and memory. European Journal of Pharmacology, 490(1–3), 71–81. 10.1016/j.ejphar.2004.02.045. [DOI] [PubMed] [Google Scholar]