

Abstract
High levels of lipoprotein(a) [Lp(a)], an apoB100-containing lipoprotein, are an independent and causal risk factor for atherosclerotic cardiovascular diseases through mechanisms associated with increased atherogenesis, inflammation, and thrombosis. Lp(a) is predominantly a monogenic cardiovascular risk determinant, with ≈70% to ≥90% of interindividual heterogeneity in levels being genetically determined. The 2 major protein components of Lp(a) particles are apoB100 and apolipoprotein(a). Lp(a) remains a risk factor for cardiovascular disease development even in the setting of effective reduction of plasma low-density lipoprotein cholesterol and apoB100. Despite its demonstrated contribution to atherosclerotic cardiovascular disease burden, we presently lack standardization and harmonization of assays, universal guidelines for diagnosing and providing risk assessment, and targeted treatments to lower Lp(a). There is a clinical need to understand the genetic and biological basis for variation in Lp(a) levels and its relationship to disease in different ancestry groups. This scientific statement capitalizes on the expertise of a diverse basic science and clinical workgroup to highlight the history, biology, pathophysiology, and emerging clinical evidence in the Lp(a) field. Herein, we address key knowledge gaps and future directions required to mitigate the atherosclerotic cardiovascular disease risk attributable to elevated Lp(a) levels.
Keywords: AHA Scientific Statements; apolipoprotein B100; atherosclerotic cardiovascular disease; cholesterol, low-density lipoprotein; lipoprotein(a)
Cardiovascular disease (CVD) is the leading cause of death and disability worldwide.1 Advances over the past 70 years have led to the identification of common and novel CVD risk factors, and the introduction of many pharmacological interventions for use in primary and secondary prevention, as well. Despite significant progress, there remains substantial residual CVD risk, even among well-treated groups.2 The role of apolipoprotein B100 (apoB) containing lipoproteins as the central determinants of atherogenesis and risk for CVD is well established.3 The apoB concentration in plasma is a marker of both cardiovascular risk and disease severity.4 Lipoprotein(a) [Lp(a)] is an apoB-containing lipoprotein bound to a hydrophilic, highly glycosylated protein called apolipoprotein(a) [apo(a)]5,6 (Figure, see location a).
Figure. Lp(a) structure, properties, regulation, and relation to disease.
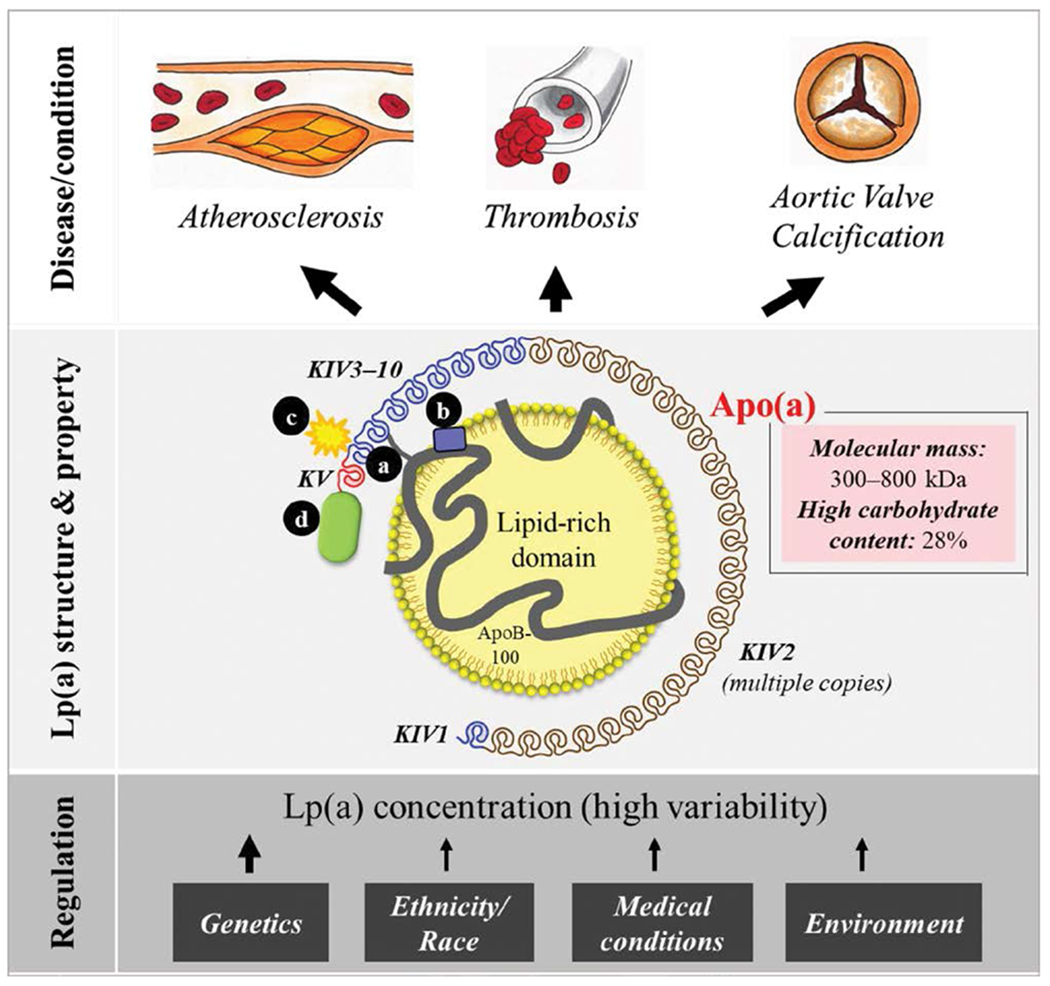
Lipoprotein(a) [Lp(a)] consists of a lipid-rich domain, primarily cholesteryl esters, and apolipoprotein(a) [apo(a)]. Apo(a) binds to apolipoprotein B100 (apoB) via a single disulfide bond (a) at a location close the low-density lipoprotein receptor binding site of apoB (b). Apo(a) contains repeated kringle (K) structures (KIV and KV), comparable with those in plasminogen. There are 10 different subtypes of apo(a) KIV, where type 2 is present in multiple copies, resulting in a highly variable molecular mass (300–800 kDa). Apo(a) is compositionally unique among apolipoproteins with a high carbohydrate content (≈28%). Proinflammatory and proatherogenic oxidized phospholipids bind to apo(a) KIV type 10 (c) and can also be found in the lipid phase. Apo(a) contains a protease domain (d) that lacks enzymatic activity. The Lp(a) concentration is heterogeneous and, to a major extent, controlled by genetics, inversely related to the copy number variation in the LPA gene. Other factors such as ethnicity and race and medical and environmental conditions also play roles in Lp(a) regulation. Lp(a) has been associated with increased risks of atherosclerosis, thrombosis, and aortic valve calcification.
Epidemiological, genome-wide association, and Mendelian randomization data7–11 provide clear support for a causal role for elevated Lp(a) in the development of atherosclerotic cardiovascular disease (ASCVD).12 What is defined as high Lp(a) levels can differ, depending on (1) the assay and units of measurement (milligrams per deciliter versus nanomoles per liter) used; (2) the population ancestry; and (3) the underlying disease and clinical characteristics of the cohort. These factors have made it difficult to establish universal thresholds for clinical use.13,14 Our current ability to lower Lp(a) with approved apoB and low-density lipoprotein cholesterol (LDL-C)–lowering therapies15 may not be optimal for reducing the cardiovascular risk associated with high Lp(a) levels.16
Novel therapies for Lp(a) lowering that target hepatic synthesis of apo(a) are in various phases of clinical trials (NCT04606602-SLN360, NCT04023552-TQJ230, and NCT04270760-AMG890). In addition, outcome studies using lipoprotein apheresis to remove Lp(a) and other apoB-containing lipoproteins from plasma are ongoing (NCT02791802). The completion of these studies will provide critical insight into the cardiovascular benefits of lowering Lp(a) and provide further evidence supporting or refuting its role as a causal risk factor.
This consensus statement, written by a multidisciplinary group of experts, will highlight the established and emerging biology, pathophysiology, and clinical epidemiology of Lp(a). It will identify key gaps in our understanding of the role of Lp(a) in ASCVD. The overall goal is to provide a rationale for targeted research efforts that can provide clinical direction for risk reduction, encourage appropriate screening strategies, and highlight the need for further studies of Lp(a) biology.
HISTORICAL PERSPECTIVE
In 1963, the geneticist Kåre Berg identified a unique antigen in the low-density lipoprotein (LDL) fraction of human serum that he called apolipoprotein(a).5 Studying families, Berg soon determined the strong genetic control of Lp(a) levels, and by 1974, he had linked the presence of Lp(a) to coronary heart disease (CHD).17 Confirmation of the association of Lp(a) with CHD required improvements in the assays to measure Lp(a),18 but by the mid-1980s, results from numerous small to modest retrospective and cross-sectional studies supported Berg’s initial observation.19 Results from some early prospective studies suggested a central role for Lp(a) in CVD,20,21 whereas others did not.22 Strong genetic evidence supporting Lp(a) as an independent and potentially causal risk factor for ASCVD was reported in large studies published in 2009,9,23 in support of earlier smaller studies demonstrating associations of Lp(a) phenotypes/genotypes with CVD.24,25
In parallel with the early population-based studies, efforts by several groups resulted in the isolation and purification of Lp(a),26 which provided key insights into the structural and biochemical characteristics of this lipoprotein. In the late 1980s, the cloning and sequencing of a cDNA corresponding to the gene encoding apo(a) (now designated LPA) demonstrated that LPA had evolved through duplication of the plasminogen (PLG) gene, providing important clues to some of the proposed pathophysiological mechanisms of Lp(a).27,28 Although PLG encodes 5 kringles (80–90 amino acid–long triple-looped protein structures) and a fibrinolytic protease region, LPA in humans lacks sequences encoding PLG kringles I to III and encodes 10 kringle IV subtypes (KIV1 through KIV10) similar to PLG kringle IV, followed by 1 PLG kringle V–like domain, and an inactive protease region (Figure, locations a and d).28
DETERMINANTS OF PLASMA Lp(a) LEVELS: GENETICS, PRODUCTION, AND CLEARANCE
Plasma Lp(a) levels arise from codominant expression of 2 LPA alleles. As such, in any given individual, the Lp(a) plasma level represents the sum of levels contributed by each LPA allele. Most individuals have 2 detectable circulating Lp(a) isoforms, each arising from a differently sized apo(a); the smaller isoform is usually present at higher levels in plasma. Small amounts of non–apoB-bound apo(a) fragments in plasma and in urine have been reported, and their physiological relevance remains unknown.29–33
Lp(a) levels are ≈70% to ≥90% genetically determined. The KIV2 copy number variant is inversely related to the Lp(a) concentration and is estimated to associate with 19% to 69% of interindividual heterogeneity in Lp(a) concentrations.34 In addition, numerous single nucleotide polymorphisms (SNPs) in the LPA locus strongly associate with Lp(a) levels.16 Although some are in linkage disequilibrium with the KIV2 copy number variant, SNPs independently associated with both high and low levels of Lp(a) have been reported.35
Outside the LPA gene locus, the APOEε2 allele associates with lower Lp(a) levels, explaining an estimated 0.5% of the Lp(a) concentration variation.35 Recent genome-wide association studies have also pointed to additional relationships of Lp(a) levels with APOH36; this gene codes (β2-glycoprotein 1 that has been found to be associated with PCSK9 (proprotein convertase subtilisin kexin type 9) and binds to apo(a) KIV2.37 β2-Glycoprotein 1 is a known participant in coagulation.36,38 The contribution of other genes to the regulation of Lp(a) levels requires further investigation. In the absence of various clinical conditions,39 levels of Lp(a) have not been shown to substantially change across the life course, although some variability occurs, as documented by intraindividual temporal variability in serial measurements from placebo-treated subjects in clinical trials.40 Results from the latter study demonstrate intraindividual biological variability of Lp(a) up to 20% suggesting that, for some patients, a mean of 2 Lp(a) determinations be obtained at different times to refine ASCVD risk stratification. The distribution of Lp(a) levels reported in large cohorts varies >100-fold,16 with the highest variation observed in populations of European descent in which Lp(a) levels are highly positively skewed. In general, population Lp(a) levels are reported as median values because they are not normally distributed.
It is well established that Lp(a) levels differ across self-reported racial and ethnic groups. Black individuals of African descent and South Asian populations have higher median Lp(a) levels than White or East Asian individuals.39 The difference between Black individuals of African descent and White individuals may result primarily from higher levels of Lp(a) associated with medium apo(a) alleles in people of African descent compared with White individuals. In White individuals, high Lp(a) levels are associated with small alleles.41,42 For several SNPs that are in linkage disequilibrium with LPA allele sizes, the associations with Lp(a) levels in plasma differ between different ancestry groups.43,44 Recently published data from diverse cohorts, such as the ARIC study (Atherosclerosis Risk in Communities),45,46 the MESA study (Multi-Ethnic Study of Atherosclerosis),47 and the MASALA study (Mediators of Atherosclerosis in South Asians Living in America),48 link high plasma Lp(a) levels to increased ASCVD risk in various populations. However, there is large variability between studies in the methods used to measure Lp(a), making it difficult to compare findings across different populations.
The assembly of Lp(a) from apo(a) and apoB expressed in hepatocytes and the pathways for Lp(a) removal from the circulation are not completely defined. Several mechanisms may contribute to the efficiency of Lp(a) synthesis.49 The best studied is apo(a) isoform size-dependent variation in the secretion rate of apo(a). Large apo(a) isoforms are retained longer in the endoplasmic reticulum, despite being folded at the same rate as smaller isoforms and are subjected to increased degradation by the proteasome. This mechanism contributes to the general inverse correlation between apo(a) isoform size and plasma Lp(a) levels.49 However, other mechanisms may contribute to plasma Lp(a) levels through isoform-dependent or independent mechanisms, including the modulation of LPA expression, mRNA stability of differently sized LPA transcripts, and isoform size–dependent regulation of apo(a) translation efficiency. The assembly of Lp(a) particles involves a 2-step process whereby initial lysine-dependent noncovalent interactions between apo(a) and apoB precede the formation of covalent disulfide bonds between apo(a) and apoB that result in Lp(a) particles.49 The covalent bonding of apo(a) to apoB-containing lipoproteins occurs extracellularly (on, or proximal to, the plasma membrane), possibly using an oxidase-like enzyme secreted from the cells.50,51 The domains involved in the noncovalent association between apo(a) and apoB have been identified: respectively, weak lysine binding sites in apo(a) and lysine-containing sequences in the N-terminal domain of apoB, suggesting the potential for inhibition of this first step in Lp(a) assembly using small molecules targeted to the lysine-binding domains of apo(a).49 There is also ongoing controversy about the nature of the apoB-containing lipoprotein particle that binds to apo(a) to create Lp(a): whether apo(a) during its lifespan in circulation may exchange between more than 1 apoB-containing lipoprotein particle, and the role of internalized and recycled apo(a) and apoB-containing lipoproteins.52 However, to date, a significant measurable pool of free apo(a) in plasma has not been identified, which suggests that free circulating apo(a) and recycling of apo(a) within the circulation may have a minor role in Lp(a) metabolism.52
Initial metabolic studies in humans to elucidate the regulation of Lp(a) were performed by various laboratories using radiolabeled apo(a).6 Combined with recent studies using stable isotopes, human studies show evidence for both clearance and production of apo(a) contributing to plasma levels of Lp(a).52 The liver has been identified as the primary site of Lp(a) catabolism; however, the receptors involved await definitive identification.53,54 The LDL receptor may play a role in Lp(a) uptake under certain circumstances, such as the use of statins with a PCSK9 inhibitor in patients with elevated Lp(a).55 However, results from earlier turnover studies showed similar rates of radiolabeled Lp(a) clearance in homozygous LDL receptor deficiency compared to those with normal LDL receptor activity.56 A role for plasminogen receptors including Plg-RKT has also been suggested,57 although human data are lacking. Additional hepatic receptors such as SR-B1 (scavenger receptor class B type 1), LRP1, and LRP8 (low-density lipoprotein receptor–related protein-1 and -8)54 could also play a role in Lp(a) clearance, although their relative contributions to Lp(a) catabolism in humans are unknown. Nonetheless, levels of circulating Lp(a), apo(a) isoform size, Lp(a)-associated proteins, and Lp(a) lipid content may modulate selectivity for particular receptors. Data from recently published studies suggest that additional proteins associated with Lp(a) (ApoH, ApoCIII, ApoE) may also play a role in the clearance of the Lp(a) particle.36,58,59
The restricted species distribution of Lp(a) (orthologues of apo(a) are only found in humans, Old World monkeys, apes, and hedgehogs) poses challenges to the design and interpretation of animal model data. Of note, only human Lp(a) contains a strong lysine binding site in apo(a) KIV10 which may limit the applicability of findings about the assembly of Lp(a) from other species. The lack of a proper animal model has hindered elucidation of the true pathophysiological mechanisms of Lp(a) and, hence, the identification of therapeutic targets. In Table 1, we highlight some priorities for ongoing knowledge gaps.
Table 1.
Priorities to Address Current Gaps in Knowledge
| Determine how the genetic architecture of LPA accounts for differences in Lp(a) levels in different ancestry groups. Studies using properly processed samples and reliable methods for determining Lp(a) levels and apolipoprotein(a) isoform size will be required for this purpose. |
| Develop a complete understanding of apolipoprotein(a) synthesis and Lp(a) particle assembly by using sophisticated technologies, such as cryogenic electron microscopy. |
| Determine the mechanism(s) of Lp(a) clearance from the circulation and identify the key liver and extrahepatic receptors that are relevant to humans. |
Lp(a) indicates lipoprotein(a).
QUANTIFICATION OF Lp(a) IN HUMAN PLASMA
Determination of Lp(a) levels in clinical chemistry laboratories is performed by immunoassays using antibody specific to apo(a) with 2 major problems affecting the accuracy of Lp(a) results and their clinical interpretation. The first problem, related to the size variability of apo(a), results in under- or overestimation of Lp(a) levels measured by different immunoassays as clearly elucidated using an ELISA method based on a monoclonal antibody that does not recognize the variably repeated KIV2 motifs of apo(a).60 However, recent commercially available methods based on the use of 5 independent calibrators with a large range of Lp(a) levels and a suitable distribution of apo(a) isoforms are able to quantify Lp(a) with a reduced impact of apo(a) size if the values of the assay calibrators are well validated.18 The use of this methodology on automated analyzers results in high precision although the assays are not able to fully eliminate the impact of apo(a) size variability in all evaluated samples. However, considering the large distribution of Lp(a) levels in populations, the use of Lp(a) levels measured by these assays for risk stratification should not result in high numbers of misclassifications. Studies specifically addressing this issue need to be performed.
The second problem is that, at present, there are 2 approaches to immunoassay calibration resulting in 2 different units for reporting Lp(a) results. The first highly sensitive assay for measuring Lp(a) was reported in the 1970s by Albers et al61 before the structure of Lp(a) was elucidated. The authors purified Lp(a) from plasma of a single donor and the protein, lipid, and carbohydrate components were individually measured. The sum of all the components of this purified Lp(a) were assigned a value in milligrams per deciliter and used as the assay calibrator. All the subsequently developed immunoassays were calibrated in milligrams per deciliter of total Lp(a) mass although the assays only measured the apo(a) component of Lp(a). This historic approach to assay calibration, which assumed that the mass of the individual Lp(a) components was constant in all the individuals, is unacceptable, in particular, considering the well-known extreme size variability of apo(a).
The gold standard ELISA method60 was calibrated in nanomoles per liter of apo(a), thus reflecting the number of Lp(a) particles. Recognizing the scientific validity of this approach, a value in nanomoles per liter was assigned to the reference material SRM-2B developed by the International Federation of Clinical Chemistry. Following the National Heart, Lung, and Blood Institute recommendations for expressing Lp(a) results in nanomoles per liter,12,62 several commercially available methods have been introduced with values in nanomoles per liter traceable to the International Federation of Clinical Chemistry reference material. Several Lp(a) guidelines and statements have recommended reporting Lp(a) values in nanomoles per liter.63–65 The existence of 2 different units for expressing Lp(a) levels is confusing to clinicians and patients and there is no unbiased conversion factor from milligrams per deciliter to nanomoles per liter or vice versa. Improvements in methods for measuring Lp(a), the traceability of the measured value to a common reference material, and the reporting of values in nanomoles per liter are essential steps toward crucially needed harmonization of Lp(a) assays. A recently published article66 describes the development and validation of a mass spectrometry method as a proposed candidate reference method for Lp(a) standardization. The high correlation of values with those obtained by the gold standard ELISA and the excellent recovery of the reference material indicate that no significant changes in calibration are expected for methods that are traceable to the World Health Organization/International Federation of Clinical Chemistry Reference Material SRM-2B.
The cholesterol content of Lp(a) is included in all clinical assays that quantify LDL-C, including the reference method β-quantification.18 Therefore, some studies have tried to account for this by subtracting, from LDL-C, 30% of the mass concentration (milligrams per deciliter) of Lp(a). Results from a recent study in a small population of subjects with high Lp(a) demonstrated a significant variability in the amount of cholesterol in isolated Lp(a) particles.67 These recent data confirm that estimating the cholesterol content of Lp(a) by a fixed percentage of measured Lp(a) in milligrams per deciliter requires several assumptions that limit its use in clinical practice. Lp(a) is measured by immunochemical methods and therefore even assays reporting Lp(a) values in milligrams per deciliter only estimate the total mass of the particle. The concentration of apoB and apoB-containing lipoprotein particles, including LDL, are more highly associated with ASCVD risk than mass of LDL-C or size of LDL particles.3 Moreover, a recently completed study showed that, in statin-treated patients, elevated apoB and non–high-density lipoprotein cholesterol, but not LDL-C, were associated with residual risk of all-cause mortality and myocardial infarction.68 Additional work is needed to encourage the use of standardized methods to measure Lp(a). In Table 2, we provide clinical use considerations for Lp(a) measurements, and in Table 3, we provide insight on the impact of Lp(a) to the plasma pool of apoB. It is important to note that Lp(a) is an ASCVD risk modifier and contributes to residual ASCVD risk at all levels of LDL-C and ApoB.
Table 2.
Clinical Use of Lipoprotein(a) Measurements
| Why would a clinician measure Lp(a)? |
|
|
| Elevated Lp(a) is a common independent atherosclerotic cardiovascular disease risk factor that is not measured in the majority of affected patients. |
| The only currently available method to know if someone has elevated Lp(a) is to measure Lp(a) with a simple blood test that is relatively inexpensive. |
| Awareness of the presence of elevated Lp(a) is important, because high Lp(a) increases atherosclerotic cardiovascular disease risk and could inform clinical decision-making regarding risk management. |
| Cascade screening of family members of patients with elevated Lp(a) may identify additional individuals with elevated Lp(a) because of its autosomal codominant inheritance pattern. |
|
|
| How should one measure Lp(a)? |
| Lp(a) should be measured with: |
| An isoform-insensitive assay |
| Assay that is traceable to the internationally accepted calibrator (World Health Organization/International Federation of Clinical Chemistry Reference Material SRM-2B) |
| Assay that is reported in nanomoles per liter (nmol/l). |
| If measurements are not uniformly calibrated, one cannot compare measurements generated by different assays. |
Lp(a) indicates lipoprotein(a).
Table 3.
How Can a Clinician Gauge the Proportion of apoB-Containing Lipoproteins That Is Actually Lp(a)?
| 1. ApoB levels have been shown to be better predictors than low-density lipoprotein cholesterol to estimate incident and residual cardiovascular risk.3,68 |
| 2. By measuring total apoB in blood, the clinicians can calculate the proportion of apoB-containing lipoproteins attributable to Lp(a). |
| 3. Unlike Lp(a), it is easy to convert apoB levels from milligrams per deciliter (mg/dL) to nanomoles per liter (nmol/L). |
| 4. When apoB levels are available, clinicians can calculate how much of the plasma apoB is Lp(a) by converting the total apoB levels from milligrams per deciliter to nanomoles per liter by multiplying by 20. |
| The clinician can then divide the plasma Lp(a) levels in nanomoles per liter by the plasma apoB levels in nanomoles per liter and derive the percent of apoB that is in Lp(a). This conversion allows the clinician to understand the contribution of Lp(a) to apoB levels. |
|
|
| Example: If apoB is 100 mg/dL, this is ≈2000 nmol/L of apoB. Therefore, if the reported Lp(a) concentration is 20 nmol/L (the median level in the UK BioBank69), Lp(a)-apoB comprises 1% of total plasma apoB. |
| If the Lp(a) concentration is 200 nmol/L, Lp(a) is 10% of total apoB. If Lp(a) is 600 nmol/L, Lp(a)-apoB is 30% of total apoB. |
| If total apoB is lowered by treatments without a change in Lp(a), the latter comprises a greater percent of total apoB. |
apo(B) indicates apolipoprotein B100; and Lp(a), lipoprotein(a).
GENETIC APPROACHES TO ASCERTAINING Lp(a) ATTRIBUTABLE RISK
Its highly genetically determined levels make Lp(a) an excellent candidate for Mendelian randomization studies. In recent years, studies examining associations of LPA genotypes with Lp(a) levels and disease risk have provided supporting evidence for a causal relationship between high Lp(a) levels and ASCVD.16 Large genetic epidemiological studies have documented strong, graded associations of high Lp(a) levels and corresponding LPA risk genotypes with increased risk of CHD and calcific aortic valvular disease (CAVD). In key studies including a large classic Mendelian randomization study (N>40000) with data on Lp(a) levels and KIV2 genotypes, each 2-fold higher level of genetically determined Lp(a) levels was associated with 22% greater risk of myocardial infarction.23 In a large case-control study including 3100 CHD cases genotyped for ≈49000 genetic variants in 2100 candidate genes, 2 LPA SNPs had the strongest association with risk of CHD of all SNPs tested; SNP carriers versus noncarriers had higher Lp(a) levels and a lower number of KIV2 repeats.9 Of note, LPA SNPs associated with Lp(a) levels independently of apo(a) isoform size have been associated with CHD.35
In 2013, a genome-wide association study demonstrated an association of a particular SNP in the LPA locus with CAVD with an odds ratio of 2.05 per allele,70 an even greater risk than that reported for CHD.9 Subsequent prospective data from 2 combined Danish Mendelian randomization studies71 and the EPIC-Norfolk study (European Prospective Investigation into Cancer Norfolk)72 cohorts supported these results. A post hoc analysis of the ASTRONOMER randomized controlled trial (Effects of Rosuvastatin on Aortic Stenosis Progression), in which statin therapy did not slow progression of aortic stenosis, found accelerated progression of aortic stenosis in the top tertile of Lp(a) levels.73 LPA remains the only monogenic risk factor identified for aortic stenosis.
Associations of Lp(a) with cerebrovascular disease and stroke are less clear. Although a recent Mendelian randomization analysis of data from 2 combined Danish cohorts suggests that Lp(a) is a risk factor for stroke (albeit not nearly as strong as for coronary disease or CAVD),74 previous analyses of smaller cohorts have not found an association.75 However, a role for Lp(a) in risk for stroke is further supported by a large genetic study (N>100000) in which 4 LPA SNPs, associated with low Lp(a) levels, were associated with 13% lower risk of stroke per 1 SD genetically lower Lp(a) levels. This same study observed ≈30% to 40% lower risk of CHD, peripheral artery disease, and CAVD.76 In other studies, Lp(a) has been suggested as an independent risk factor for prevalent lower extremity atherosclerotic disease,75,77 with risk alleles predictive of peripheral artery disease as well.76,78 Recent studies have also described a greater risk of cardiovascular and all-cause mortality for the highest Lp(a) levels/lowest number of LPA KIV2 repeats.79
The association of LPA genotypes that determine circulating Lp(a) levels with risk of ASCVD represents strong evidence of causality, because genotype-disease associations in homogeneous populations, in general, are unconfounded and cannot result from reverse causality. Nonetheless, final proof of causality awaits randomized cardiovascular outcome trial data. In Table 4, we provide some priorities to address our existing gaps in knowledge.
Table 4.
Priorities to Address Current Gaps in Knowledge
| Decode mechanisms by which genes outside LPA regulate Lp(a) levels (eg, APOE, APOH). |
| Fully characterize the role of single nucleotide polymorphisms not associated with KIV2. |
| Identify bona fide quantitative trait locuses in LPA and determine their mechanism of action. |
| Determine the role of ancestry on the magnitude of risk associated with Lp(a) including examination of the role of apolipoprotein(a) size, independent of Lp(a) levels, in different ancestry groups. |
| Develop tools to identify which patients with elevated Lp(a) levels may not have increased atherosclerotic cardiovascular disease risk, and elucidate potential mechanisms, as well. |
Lp(a) indicates lipoprotein(a).
CARDIOVASCULAR RISKS ASSOCIATED WITH Lp(a) AND ISSUES OF EFFECT MODIFICATION BY apo(a) SIZE AND ANCESTRY
Some LPA SNPs have been associated with apo(a) size and ASCVD risk.35 Although the evidence of an association between Lp(a) levels and ASCVD is robust, as underscored in a large population-based study of ancestry groups with different Lp(a) level distributions that showed similar ASCVD risks by a given incremental increase in Lp(a) levels,69 the question whether apo(a) sizes might be associated with ASCVD has attracted much interest. Several early studies reported that the presence of a small apo(a) or the combination of a high Lp(a) level and a small apo(a) isoform was associated with ASCVD. The close association between high Lp(a) levels and small apo(a) sizes in samples from non-Black individuals, and the methodological difficulties in assessing allele-specific apo(a) levels, as well, have made it challenging to dissect this issue. In many recent studies, assessment of either the sum of the 2 combined apo(a) allele sizes or the dominant apo(a) size in a given individual have been used. Studies have collectively reported both the presence and the absence of any association of apo(a) size with ASCVD.11,80 Although a large Mendelian randomization study in a South Asian cohort identified an independent causal role in ASCVD for both Lp(a) levels and apo(a) isoform size,11 a definitive role for apo(a) size independent of Lp(a) levels in conveying ASCVD risk remains to be firmly established. For the latter, appropriate genetic tools in a variety of cohorts must be used, as up to now studies have used analytical techniques that might not necessarily be appropriate to answer this question. If smaller isoform sizes are independently associated with increased risk, this implies that certain pathogenic mechanisms mediated by apo(a) (see later in this scientific statement) are influenced by isoform size. At present, measurements of the Lp(a) molar concentration are believed to be sufficient for clinical assessment of ASCVD risk because the addition of apo(a) size data does not further discriminate risk in most patients.
A recent analysis from the UK Biobank provided the largest study to date examining the risk of ASCVD associated with Lp(a) in a broad and diverse sample of 460 000 individuals.69 Patel et al69 reported Lp(a) levels measured in nanomoles per liter using an immunoturbidometric assay with excellent concordance with the World Health Organization/International Federation of Clinical Chemistry reference material. Median Lp(a) levels were 19.6 nmol/L overall, and were 19, 31, 75, and 16 nmol/L among self-identified White, South Asian, Black, and Chinese participants. Lp(a) levels were also somewhat higher among women (22 nmol/L) than among men (17 nmol/L). The risk of ASCVD associated with Lp(a) levels was log-linear for levels above the median, with increasing risk for higher Lp(a) levels. The standardized risk for ASCVD was 11% higher for each increment of 50 nmol/L (hazard ratio 1.11 per 50 nmol/L [95% CI, 1.10–1.12]), independent of adjustment for traditional risk factors, and with similar effect estimates in all race and ethnicity groups. These findings support the current American College of Cardiology/American Heart Association cholesterol and primary prevention guidelines’ recommendation to use Lp(a) as a risk-enhancing factor that, if measured, would favor statin initiation among individuals at borderline (5%–7.4%) or intermediate (7.5%–19.9%) 10-year predicted risk for ASCVD. Further guidance on how to use the risk information from Lp(a) levels is provided in Table 5.
Table 5.
Clinical Implementation of Lp(a) Levels in Risk Assessment for Primary Prevention of Atherosclerotic Cardiovascular Disease
| Current American College of Cardiology/American Heart Association guidelines15,81,82 recommend that risk assessment for primary prevention of atherosclerotic cardiovascular disease should begin with 10-y risk estimation using the Pooled Cohort Equations (or similar well-validated equation for the patient population). |
| If the patient is in the borderline (5%–7.4%) or intermediate (7.5%–19.9%) 10-y risk group, personalization and recalibration of the risk estimate should be attempted during a patient-clinician discussion that considers risk-enhancing factors, including family history of premature atherosclerotic cardiovascular disease, chronic kidney disease, and other chronic conditions. |
| If measured, the Lp(a) level can be used as a risk-enhancing factor in this scenario. Based on the data from Patel et al,69 the clinician could adjust the 10-y risk estimate based on the following formula to provide an approximate updated 10-y risk estimate: Predicted 10-y risk×[1.11(patient’s Lp(a) level in nmol/L/50)] |
| Patient example: For a patient with 10-y risk estimate of 10.0%, who has an Lp(a) level of 250 nmol/L, the updated predicted risk estimate would be 16.9%: 10.0 %×1.11(250/50)=10.0%×1.115=10.0%×1.69=16.9% |
Lp(a) indicates lipoprotein(a).
CURRENT UNDERSTANDING OF THE PATHOPHYSIOLOGY OF Lp(a) AND ASCVD
Initial hypotheses based on Lp(a) composition suggested that it could contribute to both atherosclerosis (through the lipoprotein moiety) and thrombosis (through the plasminogen-like apo(a) moiety; Figure, locations a–d). However, results from recent studies have yielded appreciation of the unique roles that confer specific pro-inflammatory and procalcific effects to Lp(a). The apoB moiety of Lp(a) is expected to be atherogenic because of its similarity to LDL. Furthermore, although there are many fewer circulating plasma Lp(a) particles than LDL particles, even in the presence of very high Lp(a) levels, Lp(a) may be selectively retained in the arterial wall through binding of apo(a) to extracellular matrix proteins.16,83 Lp(a) also carries oxidized phospholipids (OxPLs) (Figure [b and c]), which are covalently bound to apo(a) and present in the lipid phase of Lp(a). OxPLs are endogenous danger-associated molecular patterns recognized by the innate immune system, triggering sterile inflammation and calcification84 with relevance to ASCVD and CAVD pathogenesis. As a consequence of these effects, the OxPL content of Lp(a) may modulate the atherogenicity of Lp(a). Increased18 fluorodeoxyglucose-positron emission tomography imaging demonstrates enhanced arterial wall inflammation in patients with elevated Lp(a) and supports the role of Lp(a) in promoting atherosclerosis.85
Apo(a) interacts with fibrin/fibrinogen and endothelial cells through its strong lysine-binding site and is found in human atheromas and calcified aortic valves.86–88 Lp(a), through its OxPL component, upregulates endothelial adhesion molecule and cytokine expression and facilitates monocyte transmigration in vitro.89 Moreover, Lp(a) is a strong chemoattractant for monocytes and upregulates monocyte cytokine expression.85 Although inflammation is a key feature of atherosclerosis and early CAVD, progression of the latter is primarily dependent on aortic valve mineralization and ossification. Patients with elevated Lp(a) or OxPL-apoB have enhanced18 F-NaF aortic valve uptake representative of calcifying activity, faster aortic valve calcification progression on serial computed tomography scans, and worse clinical outcomes.73,90 Lp(a) components, including apo(a), OxPL, and autotaxin, colocalize in diseased human CAVD adjacent to valvular calcification.88 Upregulation of procalcific and osteogenic genes in human valvular interstitial cells by Lp(a) and apo(a) were partially dependent on OxPL.90 Moreover, Lp(a)-treated valvular interstitial cells resulted in hydroxyapatite mineral deposition.91 Autotaxin and its enzymatic product lysophosphatidic acid have been also implicated in the procalcific Lp(a) phenotype in vitro and in an animal model of CAVD, respectively.88
Whether Lp(a) contributes to atherothrombotic disease by directly inhibiting fibrinolysis, promoting thrombosis, or because of its role in atherogenesis remains the subject of investigation.92 Apo(a) inhibits plasmin-mediated fibrinolysis in vitro. However, ex vivo clot lysis times were unchanged after Lp(a) lowering with apo(a) antisense oligonucleotide treatment.93 The extensive homology between apo(a) and plasminogen has prompted investigation of a possible link between elevated Lp(a) levels and venous thromboembolism. Although some early reports suggested a positive association of Lp(a) level and risk of venous thromboembolism,94,95 other data are mixed96,97 and genetic data do not support a meaningful association,78,98 except at very high Lp(a) levels. Therefore, the antifibrinolytic properties of apo(a) may be masked when covalently associated with apoB in the Lp(a) particle. Even if Lp(a) is not inherently antifibrinolytic, the impact of Lp(a) on formation of thrombosis, that is, through effects on platelets and the coagulation cascade, has yet to be thoroughly investigated. The need remains to develop in vivo models of elevated Lp(a) levels that can be translated to humans.
Lp(a)-LOWERING THERAPIES: EXISTING AND INVESTIGATIONAL
We currently lack definitive proof that specific pharmacological lowering of Lp(a) reduces adverse cardiovascular outcomes. However, data from several lines of genetic evidence support this notion.16,99,100 Accordingly, many clinicians have the secondary goal of lowering Lp(a) in addition to lowering LDL-C and apoB in high-risk patients, in particular, when recurrent ASCVD events occur despite aggressive LDL-C lowering. Results from studies of dietary intervention show very modest effects on Lp(a) levels.101
The most effective clinically available intervention for Lp(a) lowering is lipoprotein apheresis. It is typically done every 2 weeks as Lp(a) levels return to their high levels over this interval.102 It is performed every 2 weeks in the United States, but often weekly in Germany. Lipoprotein apheresis is Food and Drug Administration approved for lowering LDL in patients with functional familial hypercholesterolemia and coronary artery disease who have LDL-C >100 mg/dL [regardless of the level of Lp(a)], while also receiving maximally tolerated lipid-lowering treatments and lifestyle intervention. The Food and Drug Administration approval specifically for Lp(a) lowering requires Lp(a) >60 mg/dL. During the course of a single 3- to 4-hour treatment, the Lp(a) concentration is acutely lowered by ≈50% to 85%, in association with comparable reductions in oxidized phospholipids. In addition to lowering Lp(a), lipoprotein apheresis lowers LDL concentrations by 60% to 85%.103,104 Limited clinical trial data suggest that Lp(a) lowering with lipoprotein apheresis may reduce the risk of ASCVD events,105 but definitive studies are needed.
Standard LDL-C and apoB lowering treatments have minimal Lp(a)-lowering efficacy, with some statins minimally increasing Lp(a) levels.106,107 Of note, data from trials of monoclonal antibodies directed against PCSK9 demonstrated dramatic LDL-C lowering by an average of 50% to 60%, but also modest Lp(a) lowering of 25% to 30%. The results of a recent analysis suggested that alirocumab-mediated Lp(a) lowering independently contributed to major adverse cardiovascular event reduction.108
Moreover, in patients with recent acute coronary syndrome on optimized statin therapy and LDL-C <70 mg/dL, alirocumab only lowered major adverse cardiovascular events in patients with mildly elevated (>13.7 mg/dL) Lp(a); there was no such interaction between Lp(a) levels and alirocumab benefit when LDL-C was ≥70 mg/dL.109 Niacin may dose-dependently lower Lp(a) up to 25% to 40%, but the cardiovascular benefit of this intervention is unknown, and the adverse side effect profile of niacin in the setting of statins may be a concern.110,111
Several experimental therapies targeting the apo(a) moiety of Lp(a) are in development. An antisense oligonucleotide for apo(a), pelacarsen (formerly known as IONIS-APO(a)-LRx, AKCEA-APO(a)-LRx, and TQJ230), lowers Lp(a) 80% at a dose of 20 mg subcutaneously weekly, with seemingly good tolerance.112 The administration of potent and specific Lp(a)-lowering antisense oligonucleotides to patients reduces the inflammatory gene expression profile in circulating monocytes and their ex vivo transendothelial migration capacity.113 A 7682-person placebo-controlled randomized clinical outcomes trial to assess the effects of pelacarsen on ASCVD outcomes is in progress. Two investigational small interfering ribonucleic acid molecules targeting apo(a) RNA are in phase II (ARO-LPA [AMG890]) and phase I (SLN-360) testing as of 2020.
SUMMARY
Epidemiological data consistently indicate a direct and dose-dependent risk association of Lp(a) with ASCVD and calcific aortic valvular disease. On the basis of mechanistic, observational, and genetic data, a strong case can be made that elevated Lp(a) is causal for ASCVD. Lp(a) levels are largely determined by genetic factors, with minimal influence from dietary or other behavioral factors. Further work is needed to understand the mechanistic links between apo(a) isoforms and risk for ASCVD; pathways for Lp(a) synthesis, regulation, and metabolism; and Lp(a)-associated risk in diverse genetic and environmental contexts. Although Lp(a) is a common, genetically inherited, and clinically important ASCVD risk factor that can be measured with a simple blood test, Lp(a) is not measured in most patients before or even after they have an ASCVD event. International standards for measurement of Lp(a) need to be established and codified to allow for consistent measurement, using assays expressing results in nanomoles per liter, and a common protocol is needed for monitoring of assay performance to ensure comparable results between laboratories. This will be especially important because targeted Lp(a)-lowering trials are conducted with different agents and repeated measurement of Lp(a) as a biomarker of therapeutic efficacy becomes possible.
At present, the evidence in favor of screening for Lp(a) is the strongest for those with a family or personal history of ASCVD, with consideration of cascade screening in appropriate individuals. Various organizations have proposed to obtain a level once in every adult.64,65,114 Once the issues with Lp(a) measurement are resolved, which should be in the near future, a reassessment of broader population-based screening should be considered. The current best approach to lower overall ASCVD risk in patients with high Lp(a) is to target LDL-C/apoB with statin and adjunctive medications as initial therapy to lower risk for ASCVD. Additional information is needed on whether newer therapies for apoB lowering reduce ASCVD risk in part through effects on Lp(a).108,115,116 Novel therapeutics that directly target apo(a) production are in clinical development. Further trials will indicate whether these therapies not only potently lower Lp(a), but also reduce ASCVD events. Positive results of such trials would firmly establish Lp(a) as modifiable causal risk factor and add to our therapeutic armamentarium to combat ASCVD and potentially CAVD.
Acknowledgment
The authors would like to acknowledge Dr Enkhmaa Byambaa, Department of Internal Medicine, School of Medicine, University of California Davis, for her efforts in designing and developing the figure.
Disclosures
Writing Group Disclosures
| Writing group member | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Gissette Reyes-Soffer | Columbia University | Amgen, Inc. (responsible for regulatory IRB for CUIMC [site])* | None | None | None | None | None | None |
| Marlys L. Koschinsky | University of Western Ontario, Robarts Research Insitute (Canada) | Canadian Institutes of Health Research†; Natural Sciences and Engineering Research Council†; Abcentra* | None | Novartis Canada* | None | None | Ayma Therapeutics* | University of Western Ontario (professor)† |
| Lars Berglund | University of California, Davis | None | None | None | None | None | None | None |
| P. Barton Duell | Oregon Health and Science University, Knight Cardiovascular Institute | None | None | None | None | None | None | None |
| Henry N. Ginsberg | Columbia University Irving Institute | Amgen (Site PI on a phase 2a/b trial of Amgen’s siRNA against apo(a). The grant from Amgen supports the nurse practitioner and study costs. He does not receive any salary or other types of funds from the research grant.)† | None | None | None | None | Amgen*; Silence Therapeutics* | None |
| Sean P. Heffron | New York University School of Medicine | None | None | None | None | None | None | None |
| Pia R. Kamstrup | Herlev and Gentofte Hospital, Copenhagen University Hospital (Denmark) | None | None | None | None | None | None | None |
| Donald M. Lloyd-Jones | Northwestern University, Feinberg School of Medicine | None | None | None | None | None | None | None |
| Santica M. Marcovina | Medpace Reference Laboratories | None | None | Novartis* | None | None | Denka-Seiken*; Roche Diagnostics* | None |
| Calvin Yeang | University of California San Diego | None | None | None | None | None | None | None |
This table represents the relationships of writing group members that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all members of the writing group are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Reviewer Disclosures
| Reviewer | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board |
|---|---|---|---|---|---|---|---|
| Shobha Ghosh | VCU Medical Center | None | None | None | None | None | None |
| Florian Kronenberg | Medical University of Innsbruck (Austria) | None | None | Amgen*; Novartis* | None | None | Kaneka* |
| Peter Libby | Brigham and Women’s Hospital | Novartis (IL-1 biology - nothing to do with lipids)† | NIH (Research on erosion - nothing to do with lipids)† | None | None | None | Novartis*; Amgen* |
| Neil J. Stone | Northwestern University | None | None | None | None | None | None |
This table represents the relationships of reviewers that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all reviewers are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Footnotes
The International Atherosclerosis Society endorses this statement.
The American Heart Association makes every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
Publisher's Disclaimer: This statement was approved by the American Heart Association Science Advisory and Coordinating Committee on August 26, 2021, and the American Heart Association Executive Committee on October 11, 2021. A copy of the document is available at https://professional.heart.org/statements by using either “Search for Guidelines & Statements” or the “Browse by Topic” area. To purchase additional reprints, call 215-356-2721 or Meredith.Edelman@wolterskluwer.com
The American Heart Association requests that this document be cited as follows: Reyes-Soffer G, Ginsberg HN, Berglund L, Duell PB, Heffron SP, Kamstrup PR, Lloyd-Jones DM, Marcovina SM, Yeang C, Koschinsky ML; on behalf of the American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease. Lipoprotein (a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2021;41:e•••–e•••. doi: 10.1161/ATV.0000000000000147
Contributor Information
Gissette Reyes-Soffer, Columbia University.
Henry N. Ginsberg, Columbia University Irving Institute.
Lars Berglund, University of California, Davis.
P. Barton Duell, Oregon Health and Science University, Knight Cardiovascular Institute.
Sean P. Heffron, New York University School of Medicine.
Pia R. Kamstrup, Herlev and Gentofte Hospital, Copenhagen University Hospital (Denmark).
Donald M. Lloyd-Jones, Northwestern University, Feinberg School of Medicine.
Santica M. Marcovina, Medpace Reference Laboratories.
Calvin Yeang, University of California San Diego.
Marlys L. Koschinsky, University of Western Ontario, Robarts Research Insitute (Canada).
REFERENCES
- 1.Xu J, Murphy SL, Kockanek KD, Arias E. Mortality in the United States, 2018. NCHS Data Brief. 2020;355:1–8. [PubMed] [Google Scholar]
- 2.Dhindsa DS, Sandesara PB, Shapiro MD, Wong ND. The evolving understanding and approach to residual cardiovascular risk management. Front Cardiovasc Med. 2020;7:88. doi: 10.3389/fcvm.2020.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4:1287–1295. doi: 10.1001/jamacardio.2019.3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grundy SM, Stone NJ. Elevated apolipoprotein B as a risk-enhancing factor in 2018 cholesterol guidelines. J Clin Lipidol. 2019;13:356–359. doi: 10.1016/j.jacl.2019.05.009 [DOI] [PubMed] [Google Scholar]
- 5.Berg K. A new serum type system in man–the LP system. Acta Pathol Microbiol Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x [DOI] [PubMed] [Google Scholar]
- 6.Kostner KM, Kostner GM. Lipoprotein (a): a historical appraisal. J Lipid Res. 2017;58:1–14. doi: 10.1194/jlr.R071571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennet A, Di Angelantonio E, Erqou S, Eiriksdottir G, Sigurdsson G, Woodward M, Rumley A, Lowe GD, Danesh J, Gudnason V. Lipoprotein(a) levels and risk of future coronary heart disease: large-scale prospective data. Arch Intern Med. 2008;168:598–608. doi: 10.1001/archinte.168.6.598 [DOI] [PubMed] [Google Scholar]
- 9.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, et al. ; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604 [DOI] [PubMed] [Google Scholar]
- 10.Afshar M, Kamstrup PR, Williams K, Sniderman AD, Nordestgaard BG, Thanassoulis G. Estimating the population impact of Lp(a) lowering on the incidence of myocardial infarction and aortic stenosis-brief report. Arterioscler Thromb Vasc Biol. 2016;36:2421–2423. doi: 10.1161/ATVBAHA.116.308271 [DOI] [PubMed] [Google Scholar]
- 11.Saleheen D, Haycock PC, Zhao W, Rasheed A, Taleb A, Imran A, Abbas S, Majeed F, Akhtar S, Qamar N, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5:524–533. doi: 10.1016/S2213-8587(17)30088-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, Moriarty PM, Rader DJ, Remaley AT, Reyes-Soffer G, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71:177–192. doi: 10.1016/j.jacc.2017.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12:1313–1323. doi: 10.1016/j.jacl.2018.07.003 [DOI] [PubMed] [Google Scholar]
- 14.Cegla J, Neely RDG, France M, Ferns G, Byrne CD, Halcox J, Datta D, Capps N, Shoulders C, Qureshi N, et al. ; HEART UK Medical, Scientific and Research Committee. HEART UK consensus statement on lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70. doi: 10.1016/j.atherosclerosis.2019.10.011 [DOI] [PubMed] [Google Scholar]
- 15.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140:e596–e646. doi: 10.1161/CIR.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamstrup PR. Lipoprotein(a) and cardiovascular disease. Clin Chem. 2021;67:154–166. doi: 10.1093/clinchem/hvaa247 [DOI] [PubMed] [Google Scholar]
- 17.Berg K, Dahlén G, Frick MH. Lp(a) lipoprotein and pre-beta1-lipoprotein in patients with coronary heart disease. Clin Genet. 1974;6:230–235. doi: 10.1111/j.1399-0004.1974.tb00657.x [DOI] [PubMed] [Google Scholar]
- 18.Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57:526–537 doi: 10.1194/jlr.R061648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Djurovic S, Berg K. Epidemiology of Lp(a) lipoprotein: its role in atherosclerotic/thrombotic disease. Clin Genet. 1997;52:281–292. doi: 10.1111/j.1399-0004.1997tb04345.x [DOI] [PubMed] [Google Scholar]
- 20.Seman LJ, DeLuca C, Jenner JL, Cupples LA, McNamara JR, Wilson PW, Castelli WP, Ordovas JM, Schaefer EJ. Lipoprotein(a)-cholesterol and coronary heart disease in the Framingham Heart Study. Clin Chem. 1999;45:1039–1046. [PubMed] [Google Scholar]
- 21.Bostom AG, Cupples LA, Jenner JL, Ordovas JM, Seman LJ, Wilson PW, Schaefer EJ, Castelli WP. Elevated plasma lipoprotein(a) and coronary heart disease in men aged 55 years and younger. A prospective study. JAMA. 1996;276:544–548. doi: 10.1001/jama.1996.03540070040028 [DOI] [PubMed] [Google Scholar]
- 22.Ridker PM, Hennekens CH, Stampfer MJ. A prospective study of lipoprotein(a) and the risk of myocardial infarction. JAMA. 1993;270:2195–2199. [PubMed] [Google Scholar]
- 23.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801 [DOI] [PubMed] [Google Scholar]
- 24.Sandholzer C, Saha N, Kark JD, Rees A, Jaross W, Dieplinger H, Hoppichler F, Boerwinkle E, Utermann G. Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler Thromb. 1992;12:1214–1226. doi: 10.1161/01.atv.12.10.1214 [DOI] [PubMed] [Google Scholar]
- 25.Geethanjali FS, Luthra K, Lingenhel A, Kanagasaba-Pathy AS, Jacob J, Srivastava LM, Vasisht S, Kraft HG, Utermann G. Analysis of the apo(a) size polymorphism in Asian Indian populations: association with Lp(a) concentration and coronary heart disease. Atherosclerosis. 2003;169:121–130. doi: 10.1016/s0021-9150(03)00143-6 [DOI] [PubMed] [Google Scholar]
- 26.Albers JJ, Hazzard WR. Immunochemical quantification of human plasma Lp(a) lipoprotein. Lipids. 1974;9:15–26. doi: 10.1007/BF02533209 [DOI] [PubMed] [Google Scholar]
- 27.Eaton DL, Fless GM, Kohr WJ, McLean JW, Xu QT, Miller CG, Lawn RM, Scanu AM. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci USA. 1987;84:3224–3228. doi: 10.1073/pnas.84.10.3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137 doi: 10.1038/330132a0 [DOI] [PubMed] [Google Scholar]
- 29.Kostner KM, Maurer G, Huber K, Stefenelli T, Dieplinger H, Steyrer E, Kostner GM. Urinary excretion of apo(a) fragments. Role in apo(a) catabolism. Arterioscler Thromb Vasc Biol. 1996;16:905–911. doi: 10.1161/01.atv.16.8.905 [DOI] [PubMed] [Google Scholar]
- 30.Fortunato JE, Bassiouny HS, Song RH, Kocharian H, Glagov S, Edelstein C, Scanu AM. Apolipoprotein (a) fragments in relation to human carotid plaque instability. J Vasc Surg. 2000;32:555–563. doi: 10.1067/mva.2000.107757 [DOI] [PubMed] [Google Scholar]
- 31.Mooser V, Marcovina SM, White AL, Hobbs HH. Kringle-containing fragments of apolipoprotein(a) circulate in human plasma and are excreted into the urine. J Clin Invest. 1996;98:2414–2424. doi: 10.1172/JCI119055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mooser V, Seabra MC, Abedin M, Landschulz KT, Marcovina S, Hobbs HH. Apolipoprotein(a) kringle 4-containing fragments in human urine. Relationship to plasma levels of lipoprotein(a). J Clin Invest. 1996;97:858–864. doi: 10.1172/JCI118487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gries A, Nimpf J, Nimpf M, Wurm H, Kostner GM. Free and Apo B-associated Lpa-specific protein in human serum. Clin Chim Acta. 1987;164:93–100 doi: 10.1016/0009-8981(87)90110-0 [DOI] [PubMed] [Google Scholar]
- 34.Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57:1339–1359. doi: 10.1194/jlr.R067314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mack S, Coassin S, Rueedi R, Yousri NA, Seppälä I, Gieger C, Schönherr S, Forer L, Erhart G, Marques-Vidal P, et al. ; KORA-Study Group. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J Lipid Res. 2017;58:1834–1844. doi: 10.1194/jlr.M076232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoekstra M, Chen HY, Rong J, Dufresne L, Yao J, Guo X, Tsai MY, Tsimikas S, Post WS, Vasan RS, et al. Genome-wide association study highlights APOH as a novel locus for lipoprotein(a) levels—brief report. Arterioscler Thromb Vasc Biol. 2021;41:458–464. doi: 10.1161/ATVBAHA.120.314965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Köchl S, Fresser F, Lobentanz E, Baier G, Utermann G. Novel interaction of apolipoprotein(a) with beta-2 glycoprotein I mediated by the kringle IV domain. Blood. 1997;90:1482–1489. [PubMed] [Google Scholar]
- 38.Nimpf J, Wurm H, Kostner GM. Interaction of beta 2-glycoprotein-I with human blood platelets: influence upon the ADP-induced aggregation. Thromb Haemost. 1985;54:397–401. [PubMed] [Google Scholar]
- 39.Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57:1111–1125. doi: 10.1194/jlr.R051904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcovina SM, Viney NJ, Hughes SG, Xia S, Witztum JL, Tsimikas S. Temporal variability in lipoprotein(a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J Clin Lipidol. 2018;12:122–129 e2. [DOI] [PubMed] [Google Scholar]
- 41.Enkhmaa B, Anuurad E, Zhang W, Kim K, Berglund L. Heritability of apolipoprotein (a) traits in two-generational African-American and Caucasian families. J Lipid Res. 2019;60:1603–1609. doi: 10.1194/jlr.P091249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcovina SM, Albers JJ, Wijsman E, Zhang Z, Chapman NH, Kennedy H. Differences in Lp[a] concentrations and apo[a] polymorphs between black and white Americans. J Lipid Res. 1996;37:2569–2585. [PubMed] [Google Scholar]
- 43.Lee SR, Prasad A, Choi YS, Xing C, Clopton P, Witztum JL, Tsimikas S. LPA gene, ethnicity, and cardiovascular events. Circulation. 2017;135:251–263. doi: 10.1161/CIRCULATIONAHA.116.024611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schachtl-Riess JF, Kheirkhah A, Grüneis R, Di Maio S, Schoenherr S, Streiter G, Losso JL, Paulweber B, Eckardt KU, Köttgen A, et al. ; GCKD Investigators. Frequent LPA KIV-2 variants lower lipoprotein(a) concentrations and protect against coronary artery disease. J Am Coll Cardiol. 2021;78:437–449. doi: 10.1016/j.jacc.2021.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehta A, Virani SS, Ayers CR, Sun W, Hoogeveen RC, Rohatgi A, Berry JD, Joshi PH, Ballantyne CM, Khera A. Lipoprotein(a) and family history predict cardiovascular disease risk. J Am Coll Cardiol. 2020;76:781–793. doi: 10.1016/j.jacc.2020.06.040 [DOI] [PubMed] [Google Scholar]
- 46.Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, Coresh J, Mosley TH, Morrisett JD, Catellier DJ, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125:241–249. doi: 10.1161/CIRCULATIONAHA.111.045120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steffen BT, Thanassoulis G, Duprez D, Stein JH, Karger AB, Tattersall MC, Kaufman JD, Guan W, Tsai MY. Race-based differences in lipoprotein(a)-associated risk of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:523–529. doi: 10.1161/ATVBAHA.118.312267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makshood M, Joshi PH, Kanaya AM, Ayers C, Budoff M, Tsai MY, Blaha M, Michos ED, Post WS. Lipoprotein (a) and aortic valve calcium in South Asians compared to other race/ethnic groups. Atherosclerosis. 2020;313:14–19. doi: 10.1016/j.atherosclerosis.2020.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borrelli MJ, Youssef A, Boffa MB, Koschinsky ML. New frontiers in Lp(a)-targeted therapies. Trends Pharmacol Sci. 2019;40:212–225. doi: 10.1016/j.tips.2019.01.004 [DOI] [PubMed] [Google Scholar]
- 50.Koschinsky ML, Marcovina SM Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15:167–174. doi: 10.1097/00041433200404000-00009 [DOI] [PubMed] [Google Scholar]
- 51.Becker L, Nesheim ME, Koschinsky ML. Catalysis of covalent Lp(a) assembly: evidence for an extracellular enzyme activity that enhances disulfide bond formation. Biochemistry. 2006;45:9919–9928. doi: 10.1021/bi060283t [DOI] [PubMed] [Google Scholar]
- 52.Reyes-Soffer G, Ginsberg HN, Ramakrishnan R. The metabolism of lipoprotein (a): an ever-evolving story. J Lipid Res. 2017;58:1756–1764. doi: 10.1194/jlr.R077693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoover-Plow J, Huang M. Lipoprotein(a) metabolism: potential sites for therapeutic targets. Metabolism. 2013;62:479–491. doi: 10.1016/j.metabol.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCormick SPA, Schneider WJ. Lipoprotein(a) catabolism: a case of multiple receptors. Pathology. 2019;51:155–164. doi: 10.1016/j.pathol.2018.11.003 [DOI] [PubMed] [Google Scholar]
- 55.Vuorio A, Watts GF, Schneider WJ, Tsimikas S, Kovanen PT. Familial hypercholesterolemia and elevated lipoprotein(a): double heritable risk and new therapeutic opportunities. J Intern Med. 2020;287:2–18. doi: 10.1111/joim.12981 [DOI] [PubMed] [Google Scholar]
- 56.Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech LA, Hoeg JM, Davignon J, Lupien P, Grossman M. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest. 1995;95:1403–1408. doi: 10.1172/JCI117794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma M, Redpath GM, Williams MJ, McCormick SP. Recycling of Apolipoprotein(a) after PlgRKT-mediated endocytosis of lipoprotein(a). Circ Res. 2017;120:1091–1102. doi: 10.1161/CIRCRESAHA.116.310272 [DOI] [PubMed] [Google Scholar]
- 58.Croyal M, Blanchard V, Ouguerram K, Chétiveaux M, Cabioch L, Moyon T, Billon-Crossouard S, Aguesse A, Bernardeau K, Le May C, et al. VLDL (very-low-density lipoprotein)-apo E (apolipoprotein E) may influence Lp(a) (lipoprotein [a]) synthesis or assembly. Arterioscler Thromb Vasc Bioi. 2020;40:819–829. doi: 10.1161/ATVBAHA.119.313877 [DOI] [PubMed] [Google Scholar]
- 59.Capoulade R, Torzewski M, Mayr M, Chan KL, Mathieu P, Bossé Y, Dumesnil JG, Tam J, Teo KK, Burnap SA, et al. ApoCIII-Lp(a) complexes in conjunction with Lp(a)-OxPL predict rapid progression of aortic stenosis. Heart. 2020;106:738–745. doi: 10.1136/heartjnl-2019-315840 [DOI] [PubMed] [Google Scholar]
- 60.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995;41:246–255. [PubMed] [Google Scholar]
- 61.Albers JJ, Adolphson JL, Hazzard WR. Radioimmunoassay of human plasma Lp(a) lipoprotein. J Lipid Res. 1977;18:331–338. [PubMed] [Google Scholar]
- 62.Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute workshop on lipoprotein(a) and cardiovascular disease: recent advances and future directions. Clin Chem. 2003;49:1785–1796. doi: 10.1373/clinchem.2003.023689 [DOI] [PubMed] [Google Scholar]
- 63.Raygor V, Khera A. New recommendations and revised concepts in recent guidelines on the management of dyslipidemias to prevent cardiovascular disease: the 2018 ACC/AHA and 2019 ESC/EAS Guidelines. Curr Cardiol Rep. 2020;22:87. doi: 10.1007/s11886-020-01331-z [DOI] [PubMed] [Google Scholar]
- 64.Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, et al. ; ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. doi: 10.1093/eurheartj/ehz455 [DOI] [PubMed] [Google Scholar]
- 65.Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, Orringer CE. Use of lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13:374–392. doi: 10.1016/j.jacl.2019.04.010 [DOI] [PubMed] [Google Scholar]
- 66.Marcovina SM, Clouet-Foraison N, Koschinsky ML, Lowenthal MS, Orquillas A, Boffa MB, Hoofnagle AN, Vaisar T. Development of an LC-MS/MS proposed candidate reference method for the standardization of analytical methods to measure lipoprotein(a). Clin Chem. 2021;67:490–499. doi: 10.1093/clinchem/hvaa324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein(a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res. 2021;62:100053. doi: 10.1016/j.jlr.2021.100053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johannesen CDL, Mortensen MB, Langsted A, Nordestgaard BG. Apolipoprotein B and non-HDL cholesterol better reflect residual risk than LDL cholesterol in statin-treated patients. J Am Coll Cardiol. 2021;77:1439–1450. doi: 10.1016/j.jacc.2021.01.027 [DOI] [PubMed] [Google Scholar]
- 69.Patel AP, Wang M, Pirruccello JP, Ellinor PT, Ng K, Kathiresan S, Khera AV. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler Thromb Vasc Biol. 2021;41:465–474. doi: 10.1161/ATVBAHA.120.315291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, et al. ; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038 [DOI] [PubMed] [Google Scholar]
- 72.Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume E, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400 [DOI] [PubMed] [Google Scholar]
- 73.Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, Tam JW, Teo KK, Mahmut A, Yang X, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66:1236–1246. doi: 10.1016/j.jacc.2015.07.020 [DOI] [PubMed] [Google Scholar]
- 74.Langsted A, Nordestgaard BG, Kamstrup PR. Elevated lipoprotein(a) and risk of ischemic stroke. J Am Coll Cardiol. 2019;74:54–66. doi: 10.1016/j.jacc.2019.03.524 [DOI] [PubMed] [Google Scholar]
- 75.Gurdasani D, Sjouke B, Tsimikas S, Hovingh GK, Luben RN, Wainwright NW, Pomilla C, Wareham NJ, Khaw KT, Boekholdt SM, et al. Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: the EPIC-Norfolk prospective population study. Arterioscler Thromb Vasc Biol. 2012;32:3058–3065. doi: 10.1161/ATVBAHA.112.255521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Emdin CA, Khera AV, Natarajan P, Klarin D, Won HH, Peloso GM, Stitziel NO, Nomura A, Zekavat SM, Bick AG, et al. ; CHARGE–Heart Failure Consortium; CARDIoGRAM Exome Consortium. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68:2761–2772. doi: 10.1016/j.jacc.2016.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Volpato S, Vigna GB, McDermott MM, Cavalieri M, Maraldi C, Lauretani F, Bandinelli S, Zuliani G, Guralnik JM, et al. Lipoprotein(a), inflammation, and peripheral arterial disease in a community-based sample of older men and women (the InCHIANTI study). Am J Cardiol. 2010;105:1825–1830. doi: 10.1016/j.amjcard.2010.01.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Helgadottir A, Gretarsdottir S, Thorleifsson G, Holm H, Patel RS, Gudnason T, Jones GT, van Rij AM, Eapen DJ, Baas AF, et al. Apolipoprotein(a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J Am Coll Cardiol. 2012;60:722–729. doi: 10.1016/j.jacc.2012.01.078 [DOI] [PubMed] [Google Scholar]
- 79.Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40:2760–2770. doi: 10.1093/eurheartj/ehy902 [DOI] [PubMed] [Google Scholar]
- 80.Paré G, Çaku A, McQueen M, Anand SS, Enas E, Clarke R, Boffa MB, Koschinsky M, Wang X, Yusuf S; INTERHEART Investigators. Lipoprotein(a) levels and the risk of myocardial infarction among 7 ethnic groups. Circulation. 2019;139:1472–1482. doi: 10.1161/CIRCULATIONAHA.118.034311 [DOI] [PubMed] [Google Scholar]
- 81.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lloyd-Jones DM, Braun LT, Ndumele CE, Smith SC Jr, Sperling LS, Virani SS, Blumenthal RS. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American Heart Association and American College of Cardiology. Circulation. 2019;139:e1162–e1177. doi: 10.1161/CIR.0000000000000638 [DOI] [PubMed] [Google Scholar]
- 83.Nielsen LB Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis. 1999;143:229–243. doi: 10.1016/s0021-9150(99)00064-7 [DOI] [PubMed] [Google Scholar]
- 84.Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16:485–497. doi: 10.1038/nri.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–624. doi: 10.1161/CIRCULATIONAHA.116.020838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boonmark NW, Lou XJ, Yang ZJ, Schwartz K, Zhang JL, Rubin EM, Lawn RM. Modification of apolipoprotein(a) lysine binding site reduces atherosclerosis in transgenic mice. J Clin Invest. 1997;100:558–564. doi: 10.1172/JCI119565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hughes SD, Lou XJ, Ighani S, Verstuyft J, Grainger DJ, Lawn RM, Rubin EM. Lipoprotein(a) vascular accumulation in mice. In vivo analysis of the role of lysine binding sites using recombinant adenovirus. J Clin Invest. 1997;100:1493–1500. doi: 10.1172/JCI119671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. 2019;16:305–318. doi: 10.1038/s41569-018-0153-2 [DOI] [PubMed] [Google Scholar]
- 89.Schnitzler JG, Hoogeveen RM, Ali L, Prange KHM, Waissi F, van Weeghel M, Bachmann JC, Versloot M, Borrelli MJ, Yeang C, et al. Atherogenic lipoprotein(a) increases vascular glycolysis, thereby facilitating inflammation and leukocyte extravasation. Circ Res. 2020;126:1346–1359. doi: 10.1161/CIRCRESAHA.119.316206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, White AC, Timmers NKLM, Hjortnaes J, Rogers MA, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73:2150–2162. doi: 10.1016/j.jacc.2019.01.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu B, Hafiane A, Thanassoulis G, Ott L, Filwood N, Cerruti M, Gourgas O, Shum-Tim D, Al Kindi H, de Varennes B, et al. Lipoprotein(a) induces human aortic valve interstitial cell calcification. JACC Basic Transl Sci. 2017;2:358–371. doi: 10.1016/j.jacbts.2017.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–757. doi: 10.1194/jlr.R060582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boffa MB, Marar TT, Yeang C, Viney NJ, Xia S, Witztum JL, Koschinsky ML, Tsimikas S. Potent reduction of plasma lipoprotein (a) with an antisense oligonucleotide in human subjects does not affect ex vivo fibrinolysis. J Lipid Res. 2019;60:2082–2089. doi: 10.1194/jlr.P094763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.von Depka M, Nowak-Göttl U, Eisert R, Dieterich C, Barthels M, Scharrer I, Ganser A, Ehrenforth S. Increased lipoprotein (a) levels as an independent risk factor for venous thromboembolism. Blood. 2000;96:3364–3368. [PubMed] [Google Scholar]
- 95.Nowak-Göttl U, Junker R, Hartmeier M, Koch HG, Münchow N, Assmann G, von Eckardstein A. Increased lipoprotein(a) is an important risk factor for venous thromboembolism in childhood. Circulation. 1999;100:743–748. doi: 10.1161/01.cir.100.7743 [DOI] [PubMed] [Google Scholar]
- 96.Marcucci R, Liotta AA, Cellai AP, Rogolino A, Gori AM, Giusti B, Poli D, Fedi S, Abbate R, Prisco D. Increased plasma levels of lipoprotein(a) and the risk of idiopathic and recurrent venous thromboembolism. Am J Med. 2003;115:601–605. doi: 10.1016/j.amjmed.2003.06.005 [DOI] [PubMed] [Google Scholar]
- 97.Vormittag R, Vukovich T, Stain M, Lehr S, Minar E, Pabinger I. Lipoprotein (a) in patients with spontaneous venous thromboembolism. Thromb Res. 2007;120:15–20. doi: 10.1016/j.thromres.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 98.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1732–1741. doi: 10.1161/ATVBAHA.112.248765 [DOI] [PubMed] [Google Scholar]
- 99.Tsimikas S A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69:692–711. doi: 10.1016/j.jacc.2016.11.042 [DOI] [PubMed] [Google Scholar]
- 100.Nordestgaard BG, Langlois MR, Langsted A, Chapman MJ, Aakre KM, Baum H, Borén J, Bruckert E, Catapano A, Cobbaert C, et al. ; European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Joint Consensus Initiative. Quantifying atherogenic lipoproteins for lipid-lowering strategies: consensus-based recommendations from EAS and EFLM. Atherosclerosis. 2020;294:46–61. doi: 10.1016/j.atherosclerosis.2019.12.005 [DOI] [PubMed] [Google Scholar]
- 101.Enkhmaa B, Petersen KS, Kris-Etherton PM, Berglund L. Diet and Lp(a): does dietary change modify residual cardiovascular risk conferred by Lp(a)? Nutrients. 2020;12:E2024. doi: 10.3390/nu12072024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kroon AA, van’t Hof MA, Demacker PN, Stalenhoef AF. The rebound of lipoproteins after LDL-apheresis. Kinetics and estimation of mean lipoprotein levels. Atherosclerosis. 2000;152:519–526. doi: 10.1016/s0021-9150(00)00371-3 [DOI] [PubMed] [Google Scholar]
- 103.Pokrovsky SN, Afanasieva OI, Ezhov MV. Therapeutic apheresis for management of Lp(a) hyperlipoproteinemia. Curr Atheroscler Rep. 2020;22:68. doi: 10.1007/s11883-020-00886-0 [DOI] [PubMed] [Google Scholar]
- 104.Waldmann E, Parhofer KG. Lipoprotein apheresis to treat elevated lipoprotein (a). J Lipid Res. 2016;57:1751–1757. doi: 10.1194/jlr.R056549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P, Leebmann J, Lehmacher W, Kamstrup PR, Nordestgaard BG, et al. ; Pro(a) LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36:2019–2027. doi: 10.1161/ATVBAHA.116.307983 [DOI] [PubMed] [Google Scholar]
- 106.Fras Z Increased cardiovascular risk associated with hyperlipoproteinemia (a) and the challenges of current and future therapeutic possibilities. Anatol J Cardiol. 2020;23:60–69. doi: 10.14744/AnatolJCardiol.2019.56068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoogeveen RC, Ballantyne CM. Residual cardiovascular risk at low LDL: remnants, lipoprotein(a), and inflammation. Clin Chem. 2021;67:143–153. doi: 10.1093/clinchem/hvaa252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bittner VA, Szarek M, Aylward PE, Bhatt DL, Diaz R, Edelberg JM, Fras Z, Goodman SG, Halvorsen S, Hanotin C, et al. ; ODYSSEY OUTCOMES Committees and Investigators. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75:133–144. doi: 10.1016/j.jacc.2019.10.057 [DOI] [PubMed] [Google Scholar]
- 109.Schwartz GG, Szarek M, Bittner VA, Diaz R, Goodman SG, Jukema JW, Landmesser U, López-Jaramillo P, Manvelian G, Pordy R, et al. ; ODYSSEY Outcomes Committees and Investigators. Lipoprotein(a) and benefit of PCSK9 inhibition in patients with nominally controlled LDL cholesterol. J Am Coll Cardiol. 2021;78:421–433. doi: 10.1016/j.jacc.2021.04.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, et al. ; HPS Thrive Collaborative Group. Effects of extended-release niacin with laro-piprant in high-risk patients. N Engl J Med. 2014;371:203–212. doi: 10.1056/NEJMoa1300955 [DOI] [PubMed] [Google Scholar]
- 111.Albers JJ, Slee A, O’Brien KD, Robinson JG, Kashyap ML, Kwiterovich PO Jr, Xu P, Marcovina SM. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62:1575–1579. doi: 10.1016/j.jacc.2013.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, Shapiro MD, Stroes ES, Moriarty PM, Nordestgaard BG, et al. ; AKCEA-APO(a)-LRx Study Investigators. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239 [DOI] [PubMed] [Google Scholar]
- 113.Stiekema LCA, Prange KHM, Hoogeveen RM, Verweij SL, Kroon J, Schnitzler JG, Dzobo KE, Cupido AJ, Tsimikas S, Stroes ESG, et al. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur Heart J. 2020;41:2262–2271. doi: 10.1093/eurheartj/ehaa171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pearson GJ, Thanassoulis G, Anderson TJ, Barry AR, Couture P, Dayan N, Francis GA, Genest J, Grégoire J, Grover SA, et al. 2021 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in adults. Can J Cardiol. 2021;37:1129–1150. doi: 10.1016/j.cjca.2021.03.016 [DOI] [PubMed] [Google Scholar]
- 115.O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, Im K, Lira Pineda A, Wasserman SM, Češka R, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–1492. doi: 10.1161/CIRCULATIONAHA.118.037184 [DOI] [PubMed] [Google Scholar]
- 116.Szarek M, Bittner VA, Aylward P, Baccara-Dinet M, Bhatt DL, Diaz R, Fras Z, Goodman SG, Halvorsen S, Harrington RA, et al. ; ODYSSEY OUTCOMES Investigators. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur Heart J. 2020;41:4245–4255. doi: 10.1093/eurheartj/ehaa649 [DOI] [PMC free article] [PubMed] [Google Scholar]