

Abstract
Background and aims:
Treatment of non-alcoholic steatohepatitis (NASH) is challenging, because suppressing fibrotic progression has not been achieved consistently by drug candidates currently in clinical trials. The aim of this study was to investigate the molecular interplays underlying NASH-associated fibrosis in a mouse NASH model and human specimens.
Methods:
mice were divided into 4 groups: Controls; NASH (high fat/Calorie diet plus high fructose and glucose in drinking water, HFCD-HF/G) for 16 weeks; HFCD-HF/G plus docosahexaenoic acid (DHA) for 16 or 8 weeks.
Results:
Along with NASH progression, fibrotic deposition was documented in HFCD-HF/G-fed mice. Liver succinate content was significantly increased along with decreased expression of succinate dehydrogenase-A (SDH-A) in these mice; whereas, GPR-91 receptor expression was much enhanced in histology compared to control mice, and co-localized histologically with hepatic stellate cells (HSCs). Succinate content was increased in fatty acid-overloaded primary hepatocytes with significant oxidant stress and lipotoxicity. Exposure to succinate led to up-regulation of GPR-91 receptor in primary and immortalized HSCs. In contrast, suppression of GPR-91 receptor expression abolished succinate stimulatory role in GPR-91 expression and extracellular matrix production in HSCs. All these changes were minimized or abrogated by DHA supplementation in vivo or in vitro. Moreover, GPR-91 receptor expression correlates with severity of fibrosis in human NASH biopsy specimens.
Conclusion:
Succinate accumulation in steatotoic hepatocytes may result in HSC activation through GPR-91 receptor signaling in NASH progression, and the cross-talk between hepatocytes and HSC through GPR-91 signaling is most likely to be the molecular basis of fibrogenesis in NASH.
Keywords: Nonalcoholic steatohepatitis, GPR-91 receptor, succinate, hepatic stellate cells, docosahexaenoic acid
LAY SUMMARY
Nonalcoholic steatohepatitis (NASH) is the active and progressive stage of nonalcoholic fatty liver disease (NAFLD), which affects 30–40% general population in the US. NASH-associated liver scaring (fibrosis) is a predictor for long-term survival and comorbidities in NAFLD, and is the major target for pharmacologic intervention in NASH. We have identified a new molecular target that is critical in mediation of NASH-associated fibrosis and could be valuable in the development of intervention in blocking NASH progression.
GRAPHICAL ABSTRACT
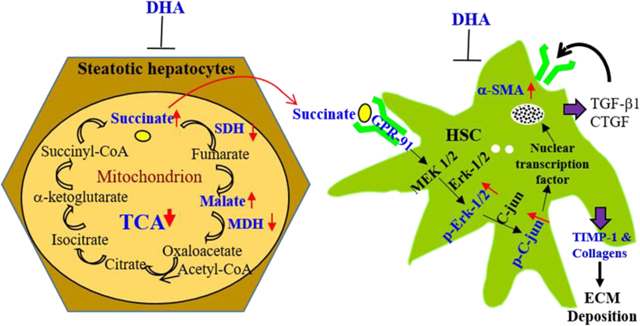
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a spectrum of conditions with fat accumulation in the liver, and ranges from nonalcoholic fatty liver (NAFL), nonalcoholic steatohepatitis (NASH), to its associated fibrosis/cirrhosis or liver cancer. NAFLD is pandemic in the Western countries, such as the US and Europe, and some Asian countries including China. It has become a global health threat in general population; and has been the major reason for a patient profile shift in hepatology clinics 1–3. NASH, an active and progressing stage requiring intensive intervention, may develop from one-fifth to one-fourth of NAFL. Liver fibrosis is the only histological variable that independently predicts future liver-related morbidity and mortality in NAFLD patients 4. As one of the major complications of diabetes, obesity and metabolic syndrome, NASH-associated end-stage liver disease has become the primary etiology for liver transplantation 5 and is responsible for the increase in liver cancer incidence in the US over the last two decades 6. The lack of FDA-approved medications for NASH treatment has resulted in the conduct of several clinical trials of potential therapeutics, such as the peroxisome proliferator-activated receptor (PPAR)-α/δ agonist Elafibranor 7, the farnesoid X receptor (FXR) agonist obeticholic acid 8, the C-C chemokine receptor (CCR)-2/5 antagonist Cenicriviroc 9, an apoptosis signal-regulating kinase-1 (ASK-1) inhibitor Selonsertib 10, all of which are in phase III. Additionally thyroid receptor β agonists, VK2809 (AASLD 2018 Abstract LB-4), MGL-3196 (AASLD 2018 Abstract 14) or stearoyl-CoA desaturase-1 (SCD-1) modulator, Aramchol (AASLD 2018 Abstract LB-5) are in the pipeline 11. In addition to the improvement of NAFLD activity score (NAS), the resolution of hepatic fibrosis is one of major endpoints in these phase II and III clinical trials.
Docosahexaenoic acid (DHA), a polyunsaturated fatty acid, is a nutritional supplement used for treating various conditions, such as hyperlipidemia, cardiovascular and neural disorders 12. Two clinical trials have demonstrated that DHA plus eicosapentaenoic acid (EPA) or choline plus vitamin E was effective in improving steatosis in adult or pediatric NAFLD patients after 15–18 months of treatment 13,14. Therefore, the present study aimed to investigate molecular interplays underlying hepatic fibrogenesis in a well-characterized murine NASH model 15 and human NASH specimens, and delineate the beneficial effects of DHA as a potential therapeutic candidate for NASH treatment. The findings of the study revealed that the accumulation of succinate in hepatocytes due to disrupted tricarboxylic acid (TCA) cycle may result in activation of hepatic stellate cells (HSCs) through the GPR-91 receptor signaling, and that DHA supplementation strikingly attenuated hepatic steatohepatitis, insulin resistance and fibrosis through its antioxidant effects, and led to improved mitochondrial metabolism, as well as reversed hepatic fibrosis through minimizing succinate production and HSC activation. These findings shed new lights on the molecular mechanisms of hepatic fibrogenesis in NASH and establish a molecular basis to interrupt GPR-91 receptor signaling as a novel target for intervention.
MATERIALS AND METHODS
Animals and diets
Male C57BL6J mice aged 6–8 weeks were purchased from Nanjing Biomedical Research Institute of Nanjing University, and housed in Fudan University Experimental Animal Center. Mice were randomly divided into four groups (n=5 per group) after a 2-week acclimatization: (1) the control group which received feeding of normal pellet diet and tap water; (2) the HFCD-HF/G group that was fed high fat/Calorie diet plus high fructose/glucose in drinking water; (3) the HFCD-HF/G plus DHA group in which mice were fed HFCD-HF/G plus DHA for 16 weeks (HFCD-HF/G+DHA (16W)); (4) the HFCD-HF/G plus DHA for 8 weeks (DHA supplementation between 9–16 weeks of HFCD-HF/G feeding) (HFCD-HF/G+DHA (8W)). The two groups with DHA supplementation were to reflect partially as prevention (concurrent use) or therapeutic potential (starting at 9 weeks). DHA (C22:6) was generously provided as LifesOmega™60 by Applied Human Evidence, DSM Nutritional Products Columbia, Maryland, USA, which contains 40% DHA and 15% EPA. DHA at 1.5% (gram/gram) was mixed with powder of HFCD from Research Diets (cat. no. D12492), New Brunswick, NJ, USA, and the resulting mixture was re-pelleted, and stored in −20°C until feeding. The timeline and groups of the animal experiment are illustrated in Fig. 1A. The experimental procedures were approved by the Animal Ethic Committee of Fudan University School of Basic Medical Sciences; and performed following the NIH Guidelines of Experimental Animal Handling and Use. Mice were sacrificed after 16 weeks of HFCD-HF/G feeding; the liver tissue was collected and stored at −80°C for histopathologic, biochemical and molecular biological analyses as reported in our previous study 15.
Fig. 1. Hepatic fibrogenesis in HFCD-HF/G-fed mice and effects of DHA on NASH-associated fibrosis.
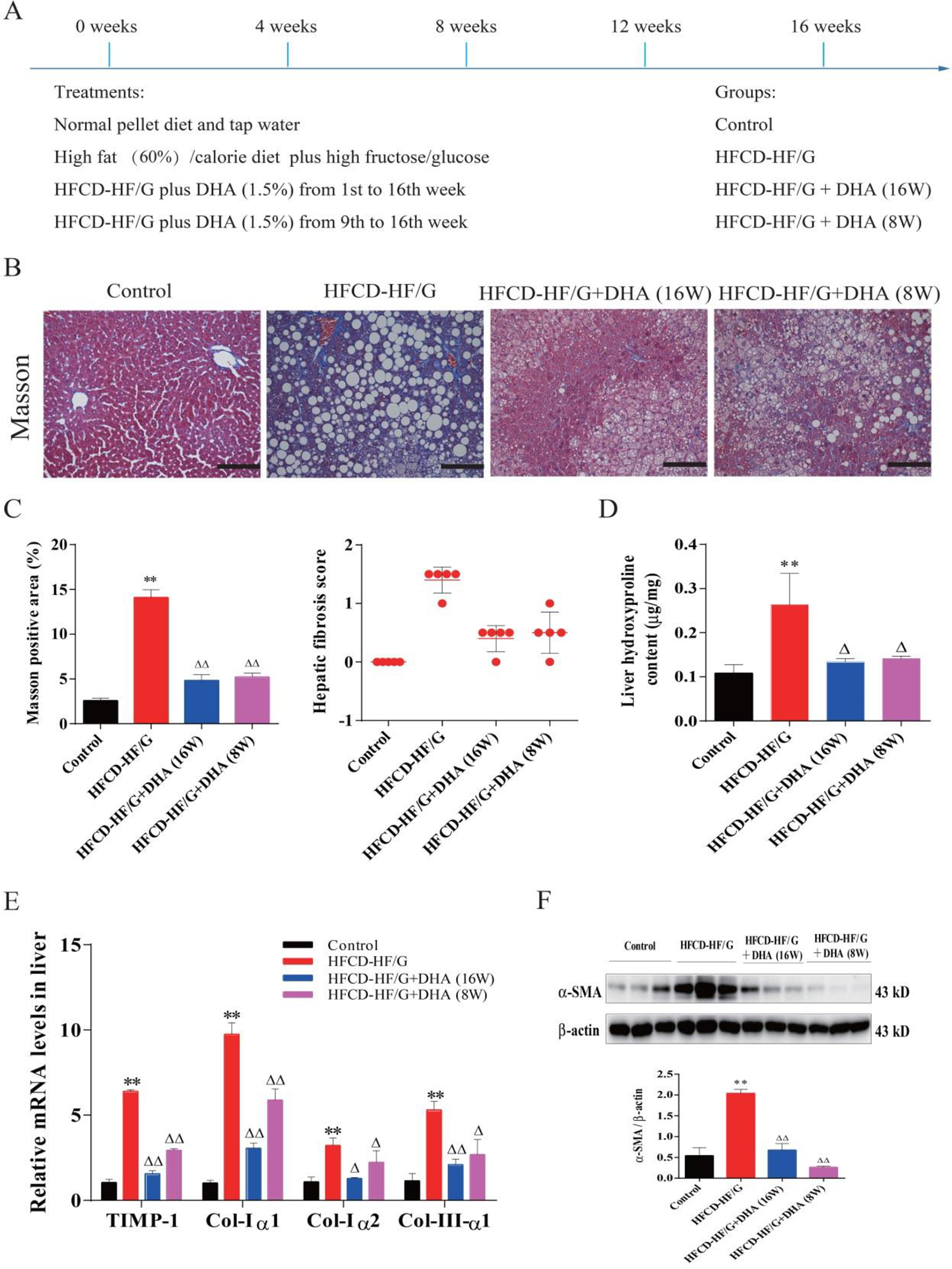
A. Illustration of animal experimental design, treatment and time line of DHA supplementation. B. Representative micrographs of Masson Trichrome staining. Images were taken at original magnification (200×). Scale bars = 100 μm. C. Morphometrical quantitation of Masson staining positive area is shown on the left panel. Semiquantitative score of hepatic fibrosis by a pathologist is shown on the right panel. D. Liver hydroxyproline content (μg/mg tissue). E. mRNA levels of procollagen type-I (Col-Iα1, Col-Iα2), procollagen type-III (Col-IIIα1) and tissue inhibitor of metalloproteinase-1 (TIMP-1) in mouse liver. F. Western blot analysis of smooth muscle α-actin (α-SMA) protein levels. The densitometric ratio of α-SMA over β-actin is shown on the below. N=5 in each group, except Western Blots (N=3). All data were expressed as mean ± SEM. ** p < 0.01 compared to the controls. △, △△ p < 0.05 and 0.01 compared to HFCD-HF/G. The ANOVA variance test was used to compare between groups, and the LSD test was used for multiple comparisons between two given groups.
Isolation of primary rat hepatocytes and hepatic stellate cells
Rat primary hepatocytes were isolated by two-step collagenase digestion as we reported previously [8]. Sprague-Dawley rats with body weight at 100–150g were anesthetized with pentobarbital (60 mg/kg, i.p.). Isolated hepatocytes were seeded on collagen type I-precoated culture dishes, and incubated in Williams E medium with supplements.
Primary hepatic stellate cells (HSCs) were isolated as we reported previously 16–18 using Sprague-Dawley rats with body weight at 500g and seeded on plastic dishes. Five days after isolation, they were ready for immunohistochemical staining (seeded on procollagen type I-precoated coverslips), or treated with succinate or succinate plus DHA. Total RNA or protein was extracted 12 and 48 hours after the treatment with sodium succinate (SUC, cat. no. O0625 from Sigma-Aldrich Chemical Co.) at a final concentration of 400 μM.
In vitro experiments
Palmitic acid (PA) and oleic acid (OA) were first dissolved in methanol, and DHA was first dissolved in ethanol, and three of them were separately diluted with serum-free medium containing 1% bovine serum albumin (BSA) free of fatty acids, added into culture medium at final concentrations of 200 μM (PA), 400 μM (OA) and 400 μM (DHA), respectively. A stock solution of N-acetyl-L-cysteine (NAC, an anti-oxidant agent) was added to culture medium at a final concentration of 2 mM. Primary hepatocytes were exposed to PA+OA plus/minus DHA or NAC in time frames indicated in the Results section.
Procurement and immunohistochemical staining of human liver biopsy sections
The use of the human tissue was approved by the Duke University Medical Center Internal Review Board under the project protocol (Pro00091516). The paraffin-embedded sections of liver biopsy specimens were obtained from the Dept. of Gastroenterology, NAFLD program, Duke University Medical Center with permission for research in a form of written consent obtained when biopsy was performed (Pro00005368). All patient identification was removed, and the sections were renumbered for record. For NAFLD activity score (NAS) and fibrosis, all sections were individually diagnosed by a board-certified pathologist according to the criteria published previously 19.
Paraffin-embedded liver sections were deparaffinized, re-hydrated, and incubated in 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. Antigen retrieval was performed by heating sections in 10 mmol/L sodium citrate buffer (pH 6.0) for 10 min. Sections were blocked in Dako protein block solution for 1 hour and incubated overnight at 4°C with anti-GRP91 primary antibodies. After vigorous washing with Tris-buffered saline with Tween 20 (TBST), EnVision System horseradish peroxidase (HRP)-labeled polymer anti-rabbit secondary antibodies from Dako were applied for 1 hour at room temperature, and Dako 3,3-Diaminobenzidine Substrate Chromogen System was used for color development as described in our previous study 20. Micrographs were taken under a microscope and positive area for GPR-91 staining was morphometrically quantitated with Image J software as previously reported by us 15, and numeric data were converted to relative number using an average value of G1 group as a reference. The relative number of each group was used for correlation coefficient (r2) and Univariate association analyses.
Statistical analysis
All data were expressed as mean ± SEM. Statistical analysis was performed with SPSS 17.0 software. The ANOVA variance test was used to compare between groups, and multiple comparisons between two given groups were completed by the LSD test. A value of p<0.05 was considered to be statistically significant.
RESULTS
Significant liver fibrotic progression in HFCD-HF/G-fed mice and effect of DHA
The extent of fibrosis was determined by Masson’s Trichrome staining of mouse liver sections, and subsequent by semi-quantitative scoring based on the well-accepted criteria. Collagenous fibrils were deposited in the portal triads and pericellular space in the HFCD-HF/G-fed mice. Collagen deposition was significantly increased in HFCD-HF/G-fed mice compared to the controls, and partially resolved in mice given DHA supplementation for 8 and 16 weeks as evidenced by semi-quantitative Trichrome staining and morphometric semi-quantitation of positive Masson’s staining (Fig. 1B/C). Quantitation of liver hydroxyproline content further verified the increased collagen deposition, and the effectiveness of DHA in minimizing this change to nearly normal levels (Fig. 1D). Accordingly, liver mRNA levels of procollagen type-I (Col-Iα1 and Col-Iα2), procollagen type-III (Col-IIIα1) and tissue inhibitor of metalloproteinase-1 (TIMP-1) supported much more production of collagenous fibrils and other extracellular matrices in HFCD-HF/G-fed mice compared to the controls, and confirmed inhibitory effects by DHA supplementation compared to HFCD-HF/G-fed mice (Fig. 1E). Moreover, elevated α-SMA protein levels provided additional evidence for the notion that there was active transformation to myofibroblast-like cells in HFCD-HF/G-fed mice; in contrast, DHA supplementation significantly reversed this phenomenon, suggesting its effective suppression on HSC activation (Fig. 1F). In summary, it is notable that progression of hepatic fibrosis was remarkable in HFCD-HF/G-fed mice as evidenced by worse histopathology, enhanced collagen synthesis and deposition, as well as elevated HSC activation. Obviously, DHA strikingly blocked these changes.
HFCD-HF/G feeding caused major abnormalities in key enzyme levels involved in the tricarboxylic acid cycle, and the effects of DHA supplementation
In order to further investigate metabolic disruption in HFCD-HF/G-fed mice, metabolites (succinate and malate) and enzymes participating in the tricarboxylic acid (TCA) cycle were investigated. We found that liver succinate content was remarkably increased (2.3-fold) in HFCD-HF/G-fed mice compared to the controls, along with elevated malate content (3.7-fold) (Fig. 2A). In contrast, mRNA and protein levels of succinate dehydrogenase complex subunit-A (SDH-A) and malate dehydrogenase-1/2 (MDH-1/2) were significantly decreased in HFCD-HF/G-fed mice compared to the controls (Fig. 2B/C). These findings indicate that the elevated succinate and malate levels were possibly due to suppressed mitochondrial SDH-A and MDH-1/2 enzyme levels and/or activity as a result of mitochondrial dysfunction. These changes were partially corrected by DHA supplementation (Fig. 2B/C). Consistent with this observation, the co-localization of mitochondrial cytochrome oxidase-IV (COX-IV) with SDH-A or MDH-2 in HFCD-HF/G-fed mice was less positive than the controls, however, it was restored by DHA supplementation (Fig. 2D). Interestingly, mRNA and total GPR-91 protein levels were slightly elevated (p>0.05) in the liver of HFCD-HF/G-fed mice (Fig. 2B), and suppressed by DHA supplementation. The protein levels did not see any significant changes in the liver of HFCD-HF/G-fed mice (Fig. 2C). The up-regulated GPR-91 receptor expression in the liver of HFCD-HF/G-fed mice was evidenced by co-immunohistochemical staining of α-SMA (Fig. 2E), indicating that there was enhanced GPR-91 activation in HSCs in these mice. More specifically, the GPR-91 receptor was co-localized with ATPase, a cytoplasmic membrane marker, and co-activated with α-SMA in primary rat HSCs by succinate stimulation (Fig. 4E). It is known that succinate is the ligand for the GPR-91 receptor, and therefore it is speculated that elevated succinate levels in hepatocytes may act as a strong ligand for HSC activation through the GPR-91 receptor signaling in NASH-associated fibrogenesis.
Fig. 2. Down-regulation of MDH2 and SDHA expression and activation GPR-91 receptor in HSCs in NASH.
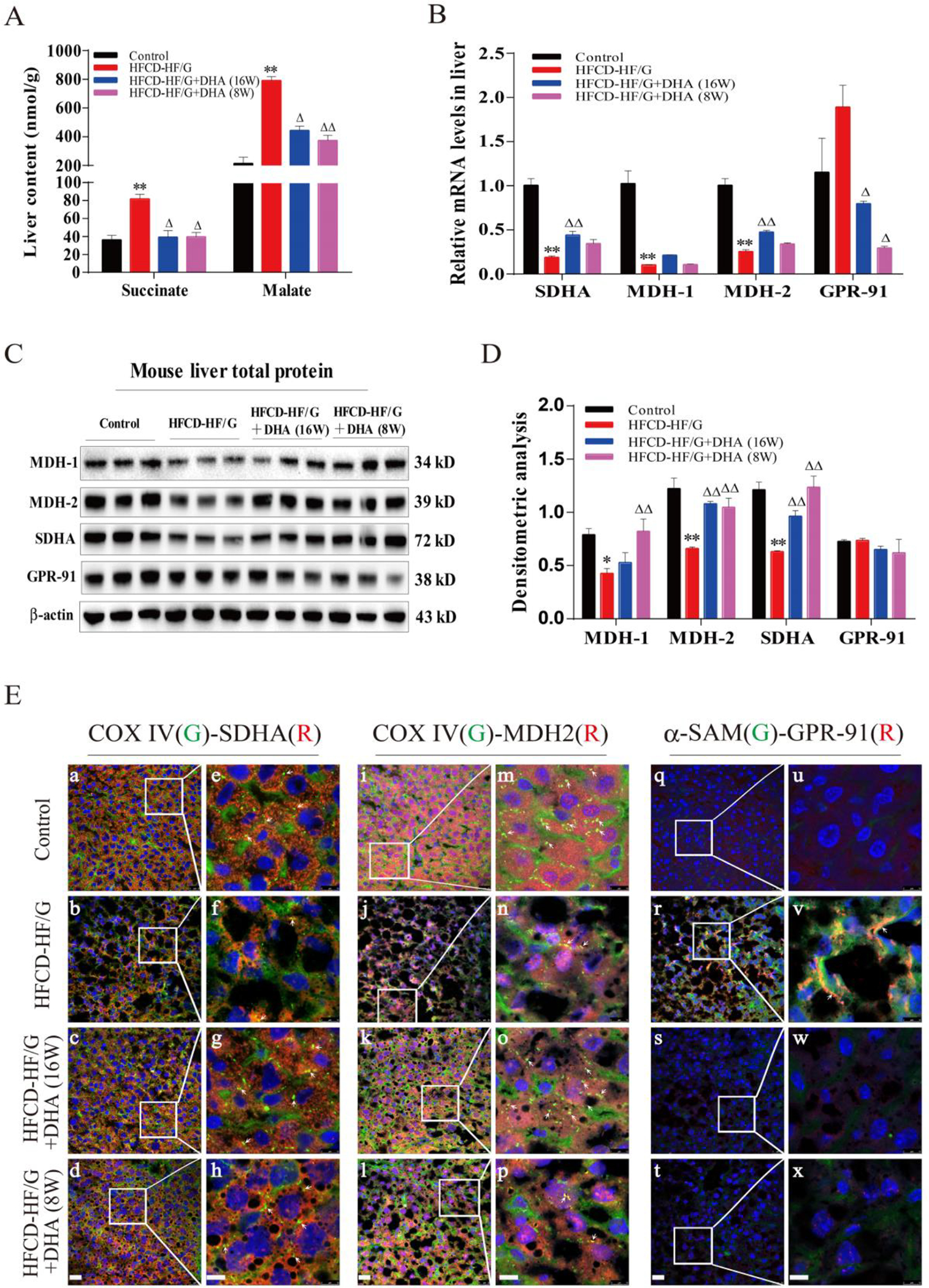
A. Liver content of succinate and malate. B. Relative mRNA levels of SDHA, MDH-1/2 and GPR-91 receptor in mouse liver were determined by quantitative RT-PCR analysis. C. Total protein levels of MDH-1, MDH-2, SDHA and GPR-91 in mouse liver were assayed by Western blot. D. Densitometric analysis of Western blot image. E. Representative confocal micrographs of double-staining. For a-h, SDHA was stained in red and COX IV was stained in green. For i-p, MDH2 was stained in red and COX IV was stained in green. For q-x, GPR-91 receptor was stained in red and α-SMA was stained in green. For a-d, i-l and q-t, images were taken at original magnification (400×), scale bars = 50 μm; for e-h, m-p and u-x, images were taken at original magnification (400×3), scale bars = 10 μm. N=5 in each group, except Western Blot (N=3). All data were expressed as mean ± SEM. *,** p < 0.05 and 0.01 compared to the controls. △, △△ p < 0.05 and 0.01 compared to HFCD-HF/G. The ANOVA variance test was used to compare between groups, and the LSD test was used for multiple comparisons between two given groups.
Fig. 4. Increased succinate levels were responsible for HSC activation through the GPR-91 signal pathway.

A. Total protein levels of -SMA, GPR-91, p-Erk1/2, p-c-Jun in immortalized rat BSCC-10 and human HSC cells were analyzed by Western blot analysis. B. Total protein levels of α-SMA, GPR-91, p-Erk1/2, p-c-Jun in primary rat hepatic stellate cells (HSCs). C. Relative mRNA levels of α-SMA, Coll-Iα1, CTGF and GPR-91 in immortalized rat BSCC-10 and human HSCs were determined by qRT-PCR. D. Relative mRNA levels of CTGF, TIMP-1, Coll-Iα1 and GPR-91 in primary HSCs were determined by qRT-PCR. E. Representative confocal micrographs of double-staining of the GPR-91 receptor with cytoplasmic α-SMA (a-l) or membrane ATPase (m-x) in primary hepatic stellate cells (HSCs). For a-l, GPR-91 was stained in red and β-SMA was stained in green. For m-x, GPR-91 was stained in red and ATPase was stained in green. Images were taken at original magnification (400×). Scale bars = 25 μm. DHA was first dissolved in ethanol and added to medium at the final concentration of 400 μM. Succinate (SUC) was dissolved in double distilled water and added to medium at the final concentration of 400 μM. DHA was added to medium 15 min before cells were exposed to SUC. The experiment was repeated at least 3 times. All data were expressed as mean ± SEM. *, ** p < 0.05 and 0.01 compared to BSA. △, △△ p < 0.05 and 0.01 compared to SUC. The ANOVA variance test was used to compare between groups, and the LSD test was used for multiple comparisons between two given groups.
In order to further investigate whether fatty acid overload affects mitochondrial enzymes involved in the TCA cycle, primary hepatocytes were exposed to a mixture of saturated fatty acids, palmitic acid (PA) and oleic acid (OA) at ratio of 1:2. The overload of fatty acids was used as an in vitro model to evaluate lipotoxicity, inflammasome activation, oxidant stress, as well as metabolic abnormalities. We found that the succinate content was strikingly increased (1.8-fold) in fatty acid-loaded hepatocytes compared to the controls, along with elevated malate content (1.4-fold) (Fig. 3A right panel). Accordingly, succinate content in culture medium for hepatocytes exposure to PA/OA for 24 hours was markedly elevated (p<0.01) and reversed by the addition of DHA in the medium (Fig. 3B). As shown in Fig. 3C, treatment with PA/OA significantly reduced hepatocellular SDH-A and MDH-1/2 protein levels in the mitochondrial fraction; whereas, when DHA was added at 400 μM, the decrease in these enzyme levels was partially restored. In total cell lysates, SDH-A had an expression pattern similar to the mitochondrial fraction. Consistent with this observation, the co-localization of mitochondrial SDH-A or MDH-2 with COX-IV in fatty acid-loaded hepatocytes was less positive than the controls, however it was restored by DHA supplementation (Fig. 3D/E). In summary, decreased mitochondrial SDH-A and MDH-1/2 protein levels were observed in HFCD-HF/G-fed mice and in fatty acid-loaded hepatocytes, and accompanied with markedly increased succinate content in cell lysates and culture medium; whereas, DHA supplementation significantly mitigated these TCA cycle abnormalities.
Fig. 3. Down-regulation of MDH2 and SDHA expression in primary hepatocytes exposed to fatty acid overload.
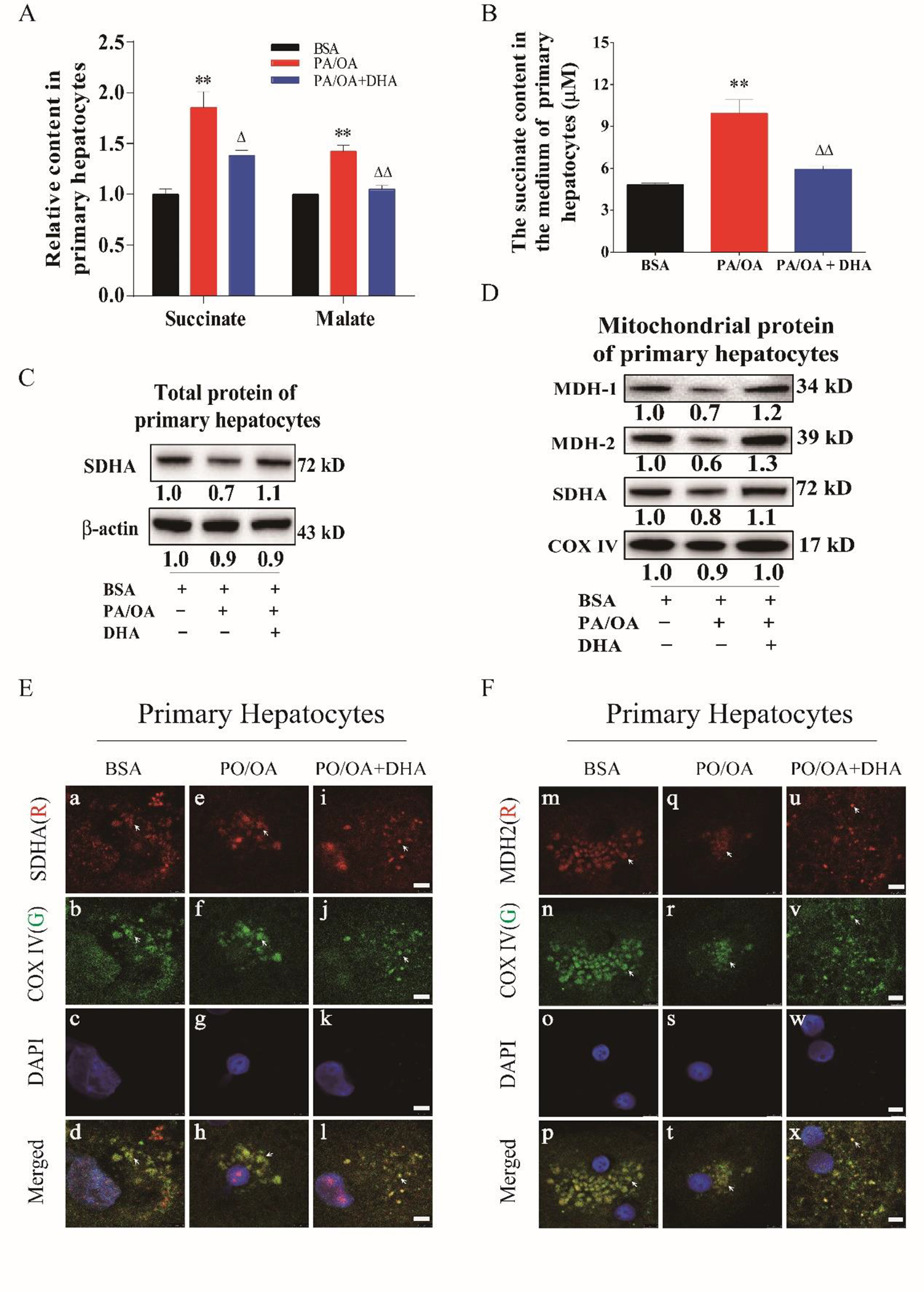
A. Succinate and malate content in primary hepatocytes. B. Succinate content in culture medium of primary hepatocytes exposed to PA/OA overload plus/minus DHA supplementation. C. Protein levels of SDHA in in primary hepatocytes in response to fatty acid overload. D. Protein levels of MDH1/2 in primary hepatocytes in response to fatty acid overload. E. Immunocytochemical staining of SDHA in primary hepatocytes in exposure to fatty acid overload. F. Immunocytochemical staining of MDH2 in primary hepatocytes in exposure to fatty acid overload. Primary hepatocytes were exposed to a combination overload of palmitic acid (PA) with oleic acid (OA). PA and OA was first dissolved in methanol and added to medium at the final concentration of 200μM and 400μM. DHA was first dissolved in ethanol and added to medium at the final concentration of 400 μM. DHA was added to medium 15 minutes before PA/OA. The experiment was repeated at least 3 times. All data were expressed as mean ± SEM. ** p < 0.01 compared to BSA. △, △△ p < 0.05 and 0.01 compared to PA/OA. The ANOVA variance test was used to compare between groups, and the LSD test was used for multiple comparisons between two given groups.
Caspase-3 activity was used to indicate apoptotic activity. As shown in Fig. S4C, it was elevated in primary hepatocytes after overload with the PA/OA mixture, and the increase was inhibited by addition of DHA or an antioxidant, N-acetyl cysteine (NAC) as a positive control. Lactate dehydrogenase (LDH) release is an indicator of cell viability loss, and 2’,7’-dichlorofluorescin (DCF) fluorescent intensity was used to indicate cytosolic reactive oxygen species (H2O2) production (Fig. S4D&E). Both LDH release and DCF intensity were elevated in fatty acid-loaded hepatocytes; the compromised cell viability and overwhelming oxidant stress were partially corrected by the addition of DHA, indicating that anti-oxidant property may be partially responsible for its beneficial effects in both in vitro and in vivo experiments (Fig. S4C,D&E).
Increased succinate promoted HSC activation through the GPR-91 signaling pathway in HFCD-HF/G-fed mice
There was a good correlation between elevated succinate levels with HSC activation in HFCD-HF/G-fed mice as demonstrated by liver metabolite analysis, immunohistochemical staining, and determination of GPR-91 receptor expression by counter-staining in the liver tissue. It is uncertain whether elevated succinate in hepatocytes is responsible for HSC activation. To answer this question, primary rat HSCs, immortalized human and rat hepatic stellate cells were treated with succinate. It was clear that succinate treatment resulted in the enhanced expression of α-SMA and GPR-91 receptors in primary HSCs, immortalized rat BSCC-10 cells and human HSCs as evidenced by the increased receptor protein levels, and these changes were diminished by addition of DHA (Fig. 4A&B). Activation of GPR-91 receptor and elevated α-SMA, Col-Iα1, connective tissue growth factor (CTGF) by succinate treatment were further confirmed at mRNA levels in primary rat HSCs, immortalized SBCC-10 cells and human HSCs (Fig. 4C&D). Moreover, it appeared that succinate treatment led to much stronger GPR-91 activation in primary HSCs along with α-SMA staining in primary rat HSCs than immortalized HSC lines (Fig. 4A&B). Erk-1/2 and c-Jun are the downstream signaling molecules for GPR-91 receptor activation. Succinate stimulation resulted in enhanced GPR-91 receptor expression in primary HSCs, SBCC-10 cells and human HSCs at the protein level; phosphorylation of Erk-1/2 and c-Jun was evident and was abrogated in DHA-treated HSCs (Fig. 4A/B).
To further confirm GPR-91 receptor-mediated HSC activation after succinate stimulation, lentiviral vector-mediated GPR-91 receptor silence was performed in immortalized rat and human HSCs. The transduction of lentiviral vector harboring shRNA against the GPR-91 receptor in rat and human HSCs resulted in a marked decrease in GPR-91 receptor at the protein levels (Fig. 5A&B). It appeared that inhibition of the GPR-91 receptor at the mRNA and protein levels resulted in a decrease in protein levels of α-SMA and phosphorylation of the downstream signaling molecules, Erk-1/2 and c-Jun in immortalized rat and human HSCs, indicating the loss of response to succinate stimulation (Fig. 5E&F). Procollagen synthesis and CTGF production (Fig. 5C&D) further confirmed less HSC activation upon succinate stimulation with GPR-91 receptor silence. In summary, we have demonstrated a strong link between the increase in succinate levels in hepatocytes and HSC activation through the GPR-91 receptor in a mouse NASH model and in vitro experiments with primary hepatocytes and HSCs; and DHA supplementation as an effective intervention remarkably blocked hepatic fibrogenesis by reducing cross-talk between the parenchymal hepatocytes and non-parenchymal HSCs, most likely via reduced succinate production with improved mitochondrial function and reduced lipotoxicity.
Fig. 5. Inhibition of GPR-91 receptor expression by an RNAi approach abrogated succinate-stimulated HSC activation.
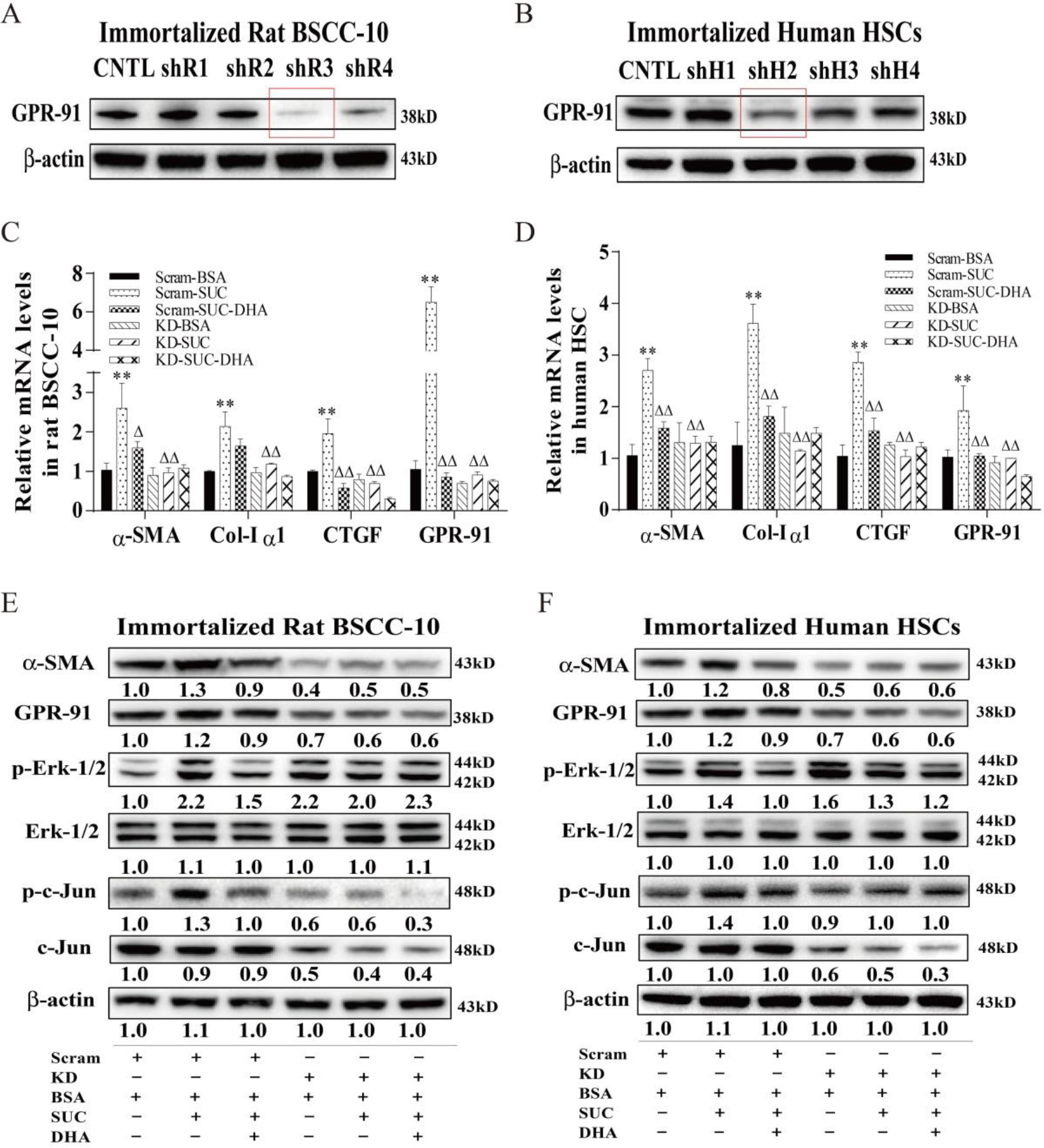
A&B. Total protein levels of the GPR-91 receptor in immortalized rat BSCC-10 and human HSC cells transduced with a lentivirus harboring scrambled shRNA (Control) or shRNA against the GPR-91 receptor (H-GPR91-KD, R-GPR91-KD) were analyzed by Western blot analysis. C&D. mRNA levels of α-SMA, Col-Iα1, CTGF and GPR-91 in immortalized rat BSCC-10 and human HSC cells transduced with a lentivirus harboring a scrambled shRNA (Control) or shRNA against GPR-91 (H-GPR91-KD, R-GPR91-KD). E&F. Total protein levels of α-SMA, GPR-91, p-Erk1/2, p-c-Jun in immortalized rat BSCC-10 and human HSC cells transduced with a lentivirus harboring a scrambled shRNA (Control) or shRNA against GPR-91 (H-GPR91-KD, R-GPR91-KD). Scram = Scrambled, KD = knockdown with shRNA. The experiment was repeated for at least 3 times. All data were expressed as mean ± SEM. ** p < 0.01 compared to Scram-BSA. △, △△ p < 0.05 and 0.01 compared to Scram-SUC. The ANOVA variance test was used to compare between groups, and the LSD test was used for multiple comparisons between two given groups.
GPR-91 receptor activation in human NASH biopsy specimens
In order to investigate whether our findings in cell culture and mouse model of NASH are relevant to NASH patients, liver biopsy sections of NAFLD patients were stained immunohistochemically with anti-GPR-91 antibodies, and the positively-stained area was semi-quantitated and correlated with NAS score and fibrotic scores. As shown in Fig. 6A, representative HE staining images denote G1 as NAFL (NAS score <4), and G2 as NASH with minimal fibrosis/F-1 or 2, and G3 as NASH with advanced fibrosis/F-3 or 4. It is evident that the GPR-91 staining positivity (most in the sinusoids) appears to be more intense along with severity of fibrotic infiltration (Fig. 6B), and there are positive correlations between GPR-91 positive area and NAS scores (r2=0.58, p=0.0002) or fibrosis scores (r2=0.55, p=0.0034) (Fig. 6C&D). Therefore, human liver biopsy staining verifies that GPR-91 receptor is highly expressed in NASH with fibrosis, and the positivity becomes more intense along with severity of NASH progression with fibrosis.
Fig. 6. GPR-91 receptor expression in human NASH biopsy specimens and illustration of cross-talks between hepatocytes and HSCs through the succinate-GPR-91 signaling pathway.
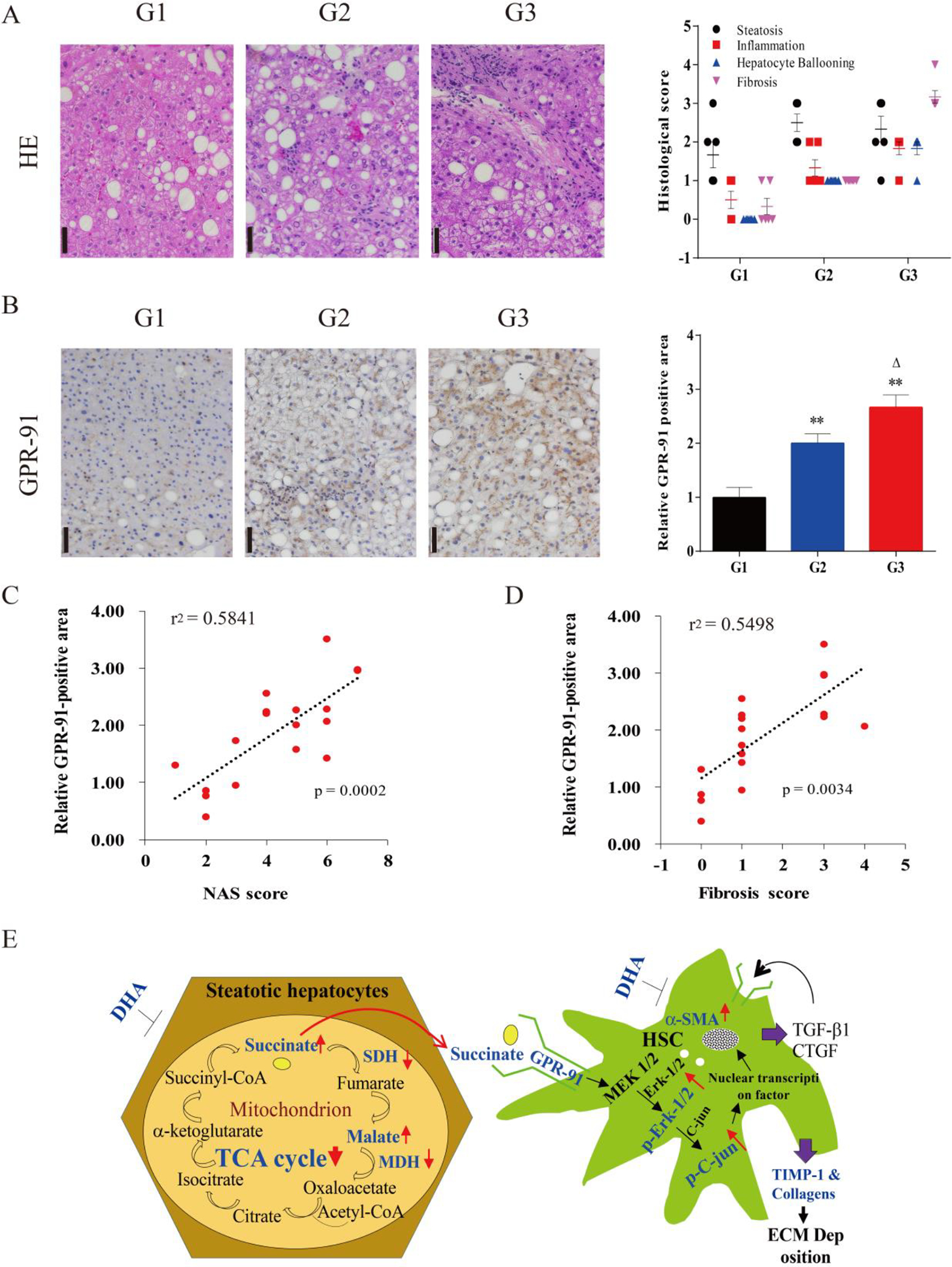
A. Representative micrographs of HE staining of liver biopsied specimens in different stages (G1-G3) of NASH progression with fibrosis and NAS score. B. Representative micrographs of immunohistochemical staining of GPR-91 receptor in NASH patient biopsied sections and morphometric quantitation of GPR-91 staining positive area (n=6). C&D. Univariate association analyses and correlation coefficient (r) analysis of GPR-91 staining-positive area with NAS score and fibrosis severity. E. Schematic illustration of increased succinate is responsible for HSC activation through the GPR-91 receptor signaling. DHA helped maintain mitochondrial function, increased the activity of SDHA and MDH1/2, resulting in lessened hepatocellular injury and less accumulation of succinate, and subsequently less hepatic stellate cell activation and deposition of extracellular matrix components through the GPR-91 receptor and it’s downstream phosphorylated of Erk1/2 and c-Jun.
DISCUSSION
The present study highlights a major advance in understanding the progression of hepatic fibrosis in NASH. Through metabolic measurements, the liver content of succinate, malate and 3-hydroxybutyrate (3-HB) in the HFCD-HF/G-fed mice was found to be much higher than the controls, due to down-regulated gene and protein levels of SDH-A and MDH-1/2 in both mouse liver tissue and primary hepatocytes treated with PA+OA. This finding indicates that there was a significant impairment in the TCA cycle due to mitochondrial damage and impaired mitophagy as a mechanism in clearance of damaged mitochondria as demonstrated in our recent study 21. In supporting our observation in a murine NASH model, elevated succinate levels were reported in NASH patients 22. At the same time, GPR-91, a G-protein-coupled receptor, was upregulated in HSCs of HFCD-HF/G-fed mice, HSCs with treatment of succinate in vitro and in human NASH specimens with fibrotic progression, demonstrating a positive correlation between NASH fibrosis severity and GPR-91 positive staining in liver biopsy specimens. GRP-91 receptor activation elicited phosphorylation of downstream signaling molecules, such as Erk and c-Jun, and production of more extracellular matrices (Procol-I, III, TIMP) and release of fibrogenic cytokines, such as CTGF. It has been previously shown that succinate is the ligand for the GPR-91 receptor in immortalized LX2 cells 23,24. The results of our in vitro study with primary HSCs or immortalized human and rat HSCs are in agreement with this finding, and specifically delineate the molecular basis for NASH-associated fibrosis progression in an animal model and human specimens. Notably, DHA supplementation in HFCD-HF/G-fed mice attenuated the accumulation of succinate, malate and 3-hydroxylbutryrate (3-HB), attenuated the down-regulation of SDHA and MDH-1/2, and hence reduced the succinate accumulation in hepatocytes and release into the extracellular space. Therefore, DHA supplementation indirectly decreased GPR-91 receptor protein levels in mouse liver through reduced succinate accumulation, and consequently abrogated progression of hepatic fibrosis. This scenario has been confirmed in our in vitro studies with primary hepatocytes, primary HSCs and immortalized HSCs. DHA protected hepatocellular injury in exposure to PA+OA through restoration of SDH-A, MDH-1/2 protein levels, and reversed HSC activation upon exposure to succinate stimulation by suppressing GPR-91 receptor activation and downstream signaling molecules, such as Erk and c-Jun phosphorylation. Therefore, it is reasonable to postulate that mitochondrial damage due to oxidant stress and/or impaired mitophagic machinery gives rise to a blockage in a late stage of the TCA cycle, and failure of energy transformation, as well as accumulation of succinate and malate in hepatocytes 1,21. The elevated succinate in hepatocytes is released into the extracellular space and neighboring HSCs as a critical cross-talk intermediate eliciting HSC activation through the GPR-91 receptor, and plays a pivotal role in the progression of hepatic fibrosis in NASH (Fig. 6E).
RNA interference (RNAi) was further employed to inhibit GPR-91 receptor expression in immortalized human and rat HSCs, and suppressing GPR-91 expression by lentiviral transduction led to less Erk and c-Jun phosphorylation in the GPR-91 receptor downstream signaling event as well as extracellular matrix production (Fig. 5C–F). Moreover, DHA improved liver fatty acid profiles and metabolism (Supplemental Table 5), and helped maintain mitochondrial function, renewal and energy transformation, resulting in reduced hepatocellular injury and less accumulation of succinate, and subsequently less HSC activation and deposition of extracellular matrix components. Taken together, the findings of the present study delineate the novel metabolic mechanism underlying hepatic fibrosis progression in NASH and a new mode of DHA action in this aspect as illustrated in a schematic cartoon (Fig. 6E).
We have previously demonstrated that DHA was effective in minimizing steatohepatitis and insulin resistance in a trans-fat-induced NASH in a short term 25. In order to assess overall effects of DHA on NASH in clinically accepted endpoints, we employed a well-characterized NASH model that was developed in normal mice, recapitulates the pathophysiologic changes of human NASH in a scalable and measurable fashion 15, and features marked fat accumulation, steatohepatitis, insulin resistance and fibrosis. With this model we were able to assess the preclinical efficacy of DHA supplementation in ameliorating fat accumulation (histology, liver TG and fatty acid content) (Supplemental Fig. 1), steatohepatitis (histology, TUNEL staining, ALT, AST) (Supplemental Fig. 2), insulin resistance, such as intraperitoneal glucose tolerant test (IGTT), 3-HB, fasting serum insulin levels, HOMA-IR, dephosphorylation of insulin receptor signaling molecules (IRS-1 and Akt) (Supplemental Fig. 3), as well as hepatic fibrosis (Masson’s staining, hydroxyproline content and procollagen I & III gene expression levels). Each category consists of quantitative, semi-quantitative or qualitative parameters to determine the effects of DHA supplementation. Our results demonstrate that DHA supplementation strikingly attenuated the marked steatosis and steatohepatitis, insulin resistance and hepatic fibrosis in HFCD-HF/G-fed mice, and the extent of the effects was over 50% for liver TG, NAS score, TUNEL-positive cell count, serum ALT and AST levels, fasting serum insulin content, 3-HB, semi-quantitative fibrosis score and hydroxyproline content in both HFCD-HF/G plus DHA 16W and 8W groups compared to the HFCD-HF/G group. Therefore, it is evident to claim that DHA supplementation was effective in improving major pathophysiological alterations in this well-characterized murine NASH model, although there were no significant changes in some variables of blood lipid panels, such as cholesterol levels, VLDL, LDL and HDL, body weight, and liver weight. The improvements appear to extend to the innate immunity response as evidenced in less activation of inflammasomes (NLRP1 and NLRP3) and its downstream pyroptotic cascade, including release of inflammatory cytokines, IL-1β, IL-18 and TNF-α, as well as adipokines, such as leptin and adiponectin (Fig. S5). In summary, this preclinical study further confirmed the beneficial effects of DHA supplementation in the treatment of NASH with significant improvements in major endpoints that are employed in clinical studies 7–10,26.
As one of major surrogates of metabolic alternations in NASH, an abnormal fatty acid profile, especially elevation of total fatty acids and saturated fatty acids, is a hallmark of NASH 27. As demonstrated by fatty acid measurement, HFCD-HF/G feeding caused an elevation of both saturated and unsaturated fatty acids in mouse liver and serum. DHA supplementation gave rise to a significant increase in unsaturated fatty acids, especially omega-3 (n3) polyunsaturated fatty acids, however, the extent of increase in omega-6 (n6) moieties was not as profound as for the omega-3 ones. In turn, the ratio of n3/n6 was remarkably increased in HFCD-HF/G plus DHA supplementation groups compared to the HFCD-HF/G group (Supplemental Table 5). Hence, this change are thought to be responsible for the beneficial role of DHA composite that contains 40% DHA and 15% EPA. The increased ratio of n3/n6 indicates the switch of the unsaturated fatty acid profile towards a healthier one since it is well accepted that omega-3 polyunsaturated fatty acids exert protective effects in NAFLD and other metabolic disorders 28,29. Therefore, the present study confers additional evidence in supporting additional benefits in individuals receiving DHA as a nutritional supplement and/or therapeutic intervention.
In conclusion, the findings of the present study uncovered a novel metabolic pathway that underscores the importance of mitochondrial impairment, and the crosstalk between damaged hepatocytes and hepatic stellate cells. Therefore, this study sheds new light on the molecular mechanisms of NASH-associated fibrosis, and establishes a molecular basis to interrupt GPR-91 receptor signaling as a novel target. Moreover, this preclinical study further verified the beneficial role of DHA supplementation as a therapeutic intervention in a well-characterized murine NASH model with the quantitative and qualitative end-points used in most clinical trials. Thus, DHA is a potential therapeutic candidate valuable to be added to current NASH treatment regimens.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Ms. Xiao Guo in the Joint Live Small Animal Imaging Laboratory of Fudan University Shanghai Medical College-PerkinElmer Company, for her technical support in the use of high resolution X-ray microCT scanning. Part of this work was presented in the 69th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD), Nov. 8–13, 2018, San Francisco, CA, USA, and published as an abstract in Hepatology 2018; 68:441A.
GRANT SUPPORT:
This work is supported by the Ministry of Science & Technology of China (#2016YFE0107400 to J.W.); the National Natural Science Foundation of China (NSFC #81272436, 81572356, 81871997 to J.W. and #81470857 to X-P. L.), and Shanghai Commission of Sciences and Technologies (#16140903700 to J.W.), as well as Chinese Academy of Sciences (XDB27020201 to C.Y.). Dr. Anna Mae Diehl is supported by the National Institutes of Health (NIH) grants (5R37AA010154, 5R01DK077794). Dr. Dr. Xue-Jing Liu was a recipient of the National Graduate Scholarship of China. Dr. Kuo Du is a recipient of the 2019 AASLD Afdhal/McHutchison LIFER Award. Li Xie is a recipient of Fudan University Daiichi Sankyo Company named scholarship.
Footnotes
DECLARATION OF CONFLICT OF INTEREST:
All authors in the present study declare that there is no conflict of interest in participation and conduct of research activity for this paper.
REFERENCES
- 1.Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med. 2017;377(21):2063–2072. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan JG, Kim SU, Wong VW. New trends on obesity and NAFLD in Asia. J Hepatol. 2017;67(4):862–873. [DOI] [PubMed] [Google Scholar]
- 4.Hagstrom H, Nasr P, Ekstedt M, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol. 2017;67(6):1265–1273. [DOI] [PubMed] [Google Scholar]
- 5.Wu J Utilization of animal models to investigate nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Oncotarget. 2016;7(27):42762–42776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degasperi E, Colombo M. Distinctive features of hepatocellular carcinoma in non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol. 2016;1(2):156–164. [DOI] [PubMed] [Google Scholar]
- 7.Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology. 2016;150(5):1147–1159 e1145. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loomba R, Lawitz E, Mantry PS, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2018;67(2):549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayiner M, Lam B, Golabi P, Younossi ZM. Advances and challenges in the management of advanced fibrosis in nonalcoholic steatohepatitis. Therap Adv Gastroenterol. 2018;11:1756284818811508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghasemi Fard S, Wang F, Sinclair AJ, Elliott G, Turchini GM. How does high DHA fish oil affect health? A systematic review of evidence. Crit Rev Food Sci Nutr. 2018:1–44. [DOI] [PubMed] [Google Scholar]
- 13.Scorletti E, Bhatia L, McCormick KG, et al. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: results from the Welcome* study. Hepatology. 2014;60(4):1211–1221. [DOI] [PubMed] [Google Scholar]
- 14.Zohrer E, Alisi A, Jahnel J, et al. Efficacy of docosahexaenoic acid-choline-vitamin E in paediatric NASH: a randomized controlled clinical trial. Appl Physiol Nutr Metab. 2017;42(9):948–954. [DOI] [PubMed] [Google Scholar]
- 15.Liu XJ, Duan NN, Liu C, Niu C, Liu XP, Wu J. Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water. Lab Invest. 2018;98(9):1184–1199. [DOI] [PubMed] [Google Scholar]
- 16.Duan NN, Liu XJ, Wu J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017;176:42–53. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J, Wu J, Frizell E, et al. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117(5):1198–1204. [DOI] [PubMed] [Google Scholar]
- 18.Zhan SS, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43(3):435–443. [DOI] [PubMed] [Google Scholar]
- 19.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. [DOI] [PubMed] [Google Scholar]
- 20.Du K, Hyun J, Premont RT, et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology. 2018;154(5):1465–1479 e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest. 2019;99(6):749–763. [DOI] [PubMed] [Google Scholar]
- 22.Schofield Z, Reed MA, Newsome PN, Adams DH, Gunther UL, Lalor PF. Changes in human hepatic metabolism in steatosis and cirrhosis. World J Gastroenterol. 2017;23(15):2685–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park SY, Le CT, Sung KY, Choi DH, Cho EH. Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem Biophys Res Commun. 2018;496(2):673–678. [DOI] [PubMed] [Google Scholar]
- 24.Li YH, Woo SH, Choi DH, Cho EH. Succinate causes alpha-SMA production through GPR91 activation in hepatic stellate cells. Biochem Biophys Res Commun. 2015;463(4):853–858. [DOI] [PubMed] [Google Scholar]
- 25.Adkins Y, Fedor DM, Mackey BE, Wu J, Kelley DS. Dietary docosahexaenoic acid reverses nonalcoholic steatohepatitis and fibrosis caused by conjugated linoleic acid supplementation in mice. J Funct Foods. 2016;20:443–452. [Google Scholar]
- 26.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaikkonen JE, Wurtz P, Suomela E, et al. Metabolic profiling of fatty liver in young and middle-aged adults: Cross-sectional and prospective analyses of the Young Finns Study. Hepatology. 2017;65(2):491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Castro GS, Calder PC. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin Nutr. 2018;37(1):37–55. [DOI] [PubMed] [Google Scholar]
- 29.Nobili V, Alisi A, Musso G, Scorletti E, Calder PC, Byrne CD. Omega-3 fatty acids: Mechanisms of benefit and therapeutic effects in pediatric and adult NAFLD. Crit Rev Clin Lab Sci. 2016;53(2):106–120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.