

Abstract
N6–methyladenosine (m6A) is the most abundant mRNA modification and plays crucial roles in diverse physiological processes. Utilizing a Massively Parallel Assay for m6A (MPm6A), we discover that m6A specificity is globally regulated by “suppressors” that prevent m6A deposition in unmethylated transcriptome regions. We identify Exon Junction Complexes (EJCs) as m6A suppressors that protect exon junction-proximal RNA within coding sequences from methylation and regulate mRNA stability through m6A suppression. EJC suppression of m6A underlies multiple global characteristics of mRNA m6A specificity, with the local range of EJC protection sufficient to suppress m6A deposition in average-length internal exons, but not in long internal and terminal exons. EJC-suppressed methylation sites co-localize with EJC-suppressed splice sites, suggesting that exon architecture broadly determines local mRNA accessibility to regulatory complexes.
N6–methyladenosine (m6A), the most prevalent mRNA modification in mammals, influences wide-ranging aspects of gene expression in diverse physiological and pathophysiological processes (1–3). The METTL3-METTL14 methyltransferase complex installs m6A methylation on mRNA in a common DRACH (D = A, G, or U; R= A or G; H= A, C, or U) sequence motif, but only a fraction of DRACH sequences (~5%) in a subset of cellular transcripts are selected for methylation (4). Additionally, m6A exhibits a marked regional bias in its transcriptomic distribution, being strongly enriched in unusually long internal exons and near stop codons (5, 6). Despite the central importance of specific m6A deposition in m6A-mediated gene regulation, the mechanistic basis for m6A specificity has remained poorly understood.
In this study, we discover the existence of prevalent regulatory mechanisms that restrict m6A methylation to specific transcript regions through targeted suppression of m6A in unmethylated regions. We find that pre-mRNA splicing selectively suppresses m6A deposition in average-length internal exons, but not in longer exons. We identify Exon Junction Complexes (EJC) as major m6A suppressors that mediate this effect and control several key characteristics of global m6A specificity. EJC depletion results in pervasive aberrant methylation of mRNAs and m6A-mediated transcript destabilization. EJCs, together with interacting proteins, package and protect long stretches of proximal RNA from cellular methylation deposition, which may represent a general mechanism by which exon architecture and EJC positioning determine local mRNA accessibility to regulatory machineries.
Massively Parallel Assay for m6A
The extent to which global m6A specificity is controlled has important implications for m6A regulation but is poorly understood (4). We approached this problem by asking: is the local sequence surrounding an m6A methylated site, when uncoupled from its endogenous context, sufficient to specify methylation at that site? Conversely, is the local sequence surrounding an unmethylated DRACH site, when uncoupled from its endogenous context, sufficient to prevent methylation at that site?
To assess this systematically on an epitranscriptome-wide scale, we developed a Massively Parallel Reporter Assay (MPRA) that enables high-throughput assessment of the m6A methylation status of thousands of designed sequences, which we termed Massively Parallel assay for m6A (MPm6A) (Fig. 1A and fig. S1A). In the MPm6A workflow, thousands of endogenously methylated m6A sites and endogenously unmethylated DRACH sites, with 102 nucleotides of sequence surrounding each site, were synthesized and cloned into the 3′UTR of a plasmid-based intronless GFP transgene. For each sequence, we also designed a corresponding negative control sequence in which all DRACH motifs were mutated to prevent methylation. The sequences were expressed and then m6A methylated through transfection into cells, or, when specified, through in vitro transcription and in vitro m6A methylation. The methylation status of each individual sequence was assessed by its enrichment following m6A-immunoprecipitation (IP) of mRNA. We selected 6,897 HeLa m6A sites and 3,058 unmethylated DRACH sites to assay in HeLa cells and validated the assay’s precision and accuracy (fig. S1, B to D).
Fig. 1. MPm6A reveals suppression of thousands of m6A sites in unmethylated transcriptome regions.
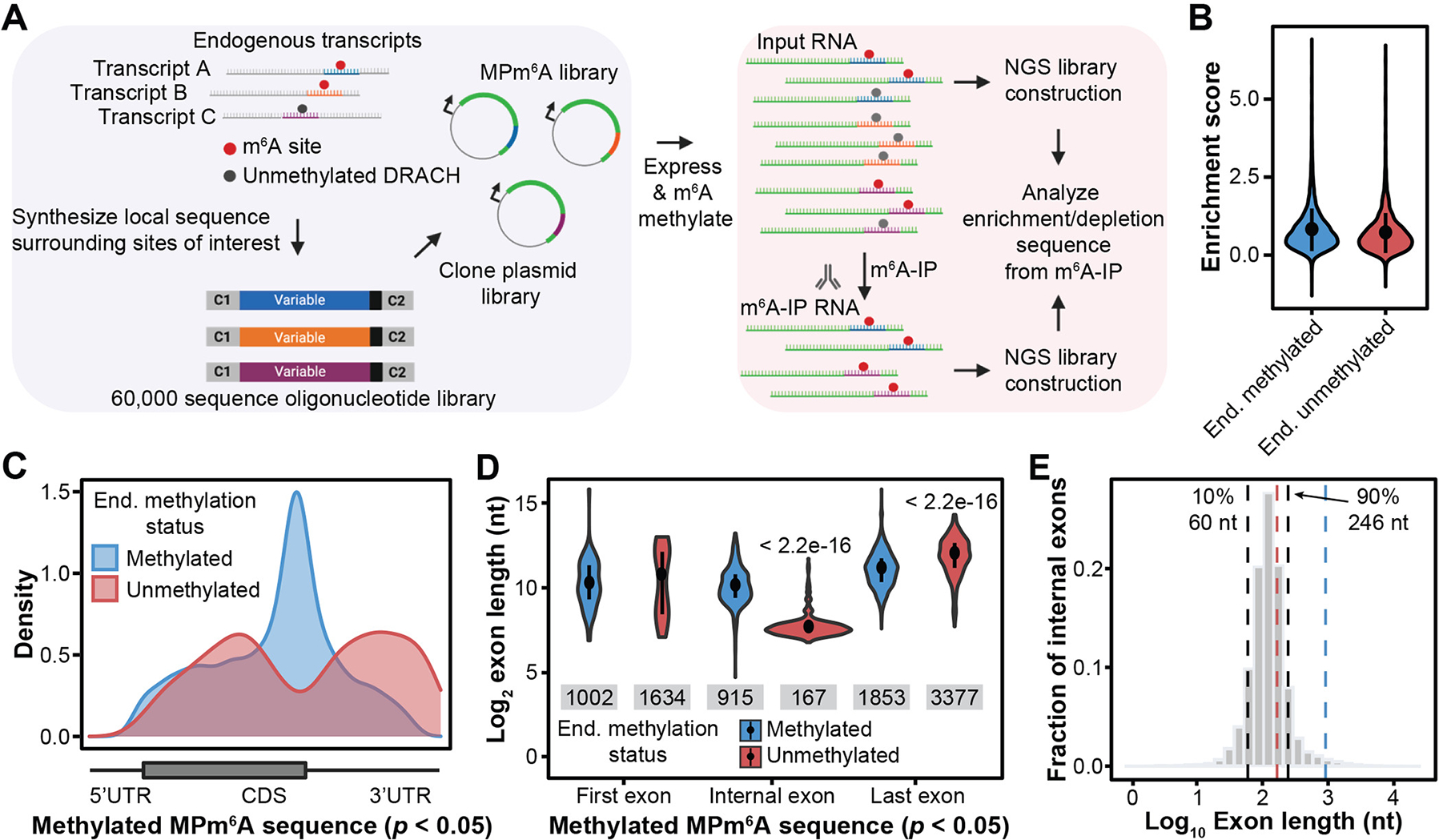
(A) Schematic of the MPm6A workflow. (B) MPm6A enrichment scores (experimental IP/input – negative control IP/input) for endogenously methylated (n = 6,095) and unmethylated (n = 2,716) sequences, mean ± SD, four biological replicates. (C) Metagenes of endogenously methylated and unmethylated sequences that are significantly methylated in MPm6A. (D) Exon lengths of endogenously methylated and unmethylated sequences that are significantly methylated in MPm6A. Median, and IQR, Wilcoxon rank sum test. Sample size for each violin plot from left to right is: n = 175, n = 22, n = 696, n = 519, n = 3,539, and n = 1,328. (E) Distribution of internal exon lengths in the human genome. Black lines indicate 10th percentile (left, 60 nt) and 90th percentile (right, 246 nt). Blue and red lines indicate median internal exon length for MPm6A endogenously methylated (915 nt) and unmethylated (167 nt) sequences, respectively.
Widespread mRNA m6A suppression controls m6A epitranscriptome specificity
When we compared the methylation levels of the endogenously methylated sequences to their negative control sequences, we found that 92.8% of the sequences exhibited significant methylation in this reporter assay (Fig. 1B and fig. S1E), indicating that most endogenously methylated sites do not strictly require their larger surrounding native context for methylation. Unexpectedly, 90.2% of endogenously unmethylated sequences also exhibited significant methylation (Fig. 1B and fig. S1E). The MPm6A enrichment scores of the endogenously unmethylated group were similar to the endogenously methylated group, despite their diverging endogenous methylation states (Fig. 1B). We observed similar results when the sequences were in vitro transcribed and methylated with recombinant METTL3-METTL14 (fig. S2). Thus, thousands of endogenously unmethylated DRACH sites became methylated when they were uncoupled from their endogenous contexts and expressed in an artificial reporter context. We term these identified sites “suppressed m6A sites”. We validated these results for three selected sequences (fig. S1F), and confirmed that methylation was not notably influenced by the CMV promoter of the MPm6A plasmid (fig. S3). We observed similar results when sequences were expressed within CDS or 5′UTR, though m6A enrichment was significantly lower for many sequences when placed in the CDS or 5′UTR versus in the 3′UTR (fig. S4, A to D). This suggests that 5′ regions are generally less conducive for m6A methylation than 3′ regions (fig. S4E). Collectively, these results reveal the existence of thousands of suppressed m6A sites that are silenced by unknown mechanisms.
We noted that suppressed m6A sites were enriched in the CDS and 3′UTR and were depleted near the stop codon, which is the reverse of endogenous m6A site enrichment (Fig. 1C and fig. S5A). Further, suppressed m6A sites in internal exons reside within much shorter exons (median = 167 nt) than endogenous m6A sites (median = 915 nt) (Fig. 1D). These observations suggest that endogenous m6A enrichment in long internal exons may be a consequence of suppression of m6A deposition in shorter internal exons, which comprise most exons (90% of internal exons are < 246 nt) (Fig. 1E). 942 genes containing suppressed m6A sites did not contain any endogenous m6A peaks on their transcripts (fig. S5B). Suppression of these sites appears to involve suppression of m6A deposition rather than active demethylation, as binding sites for RBM15, a METTL3-METTL14 methyltransferase complex accessory subunit, were highly enriched near endogenous m6A sites compared to suppressed m6A sites (fig. S6A), while FTO and ALKBH5 binding sites were not significantly enriched near suppressed sites and exhibited little binding near suppressed sites overall (fig. S6B). These results were unexpected as previous reports on m6A specificity had mainly focused on activating mechanisms (7–12). Our MPm6A assay suggests the existence of unknown m6A “suppressors” that govern global m6A specificity by suppressing m6A deposition.
Pre-mRNA splicing suppresses m6A methylation proximal to splice sites
We next examined the relative enrichment of binding sites for 120 RBPs near endogenously methylated versus suppressed m6A sites to identify candidate suppressors (13). Several spliceosome components (BUD13, SF3B4, EFTUD2) were significantly enriched near suppressed sites, suggesting that splicing may suppress m6A (fig. S6A). Because suppressed m6A sites in CDS primarily reside within average-length internal exons, we hypothesized that the splicing of average-length internal exons may suppress m6A methylation. To test this, we cloned a suppressed m6A site from an average-length internal exon in the CRY1 gene (fig. S7A) into a rabbit beta-globin minigene reporter (BG), as well as a version with the introns removed (BG Δi1,i2). First, we cloned the suppressed CRY1 site and 50/51 nt of flanking sequence into the internal exon, or in the last exon of these constructs. Notably, the spliced construct strongly suppressed methylation of the sequence placed within the internal exon, but not within the last exon (fig. S7B). Removal of either intron (BG Δi1 CRY1 102, BG Δi2 CRY1 102) resulted in partial loss of suppression, indicating that splicing of both introns contributed to methylation suppression (Fig. 2A). Consistent with this notion, deletion of all splice sites also resulted in a decrease in m6A suppression (fig. S7C). Cloning in 912 nt of the CRY1 exonic sequence surrounding the suppressed site into the internal exon (BG CRY1 912), forming a long internal exon, resulted in a loss of suppression (Fig. 2A). We hypothesized that the suppression is dependent on the proximity of the m6A site, located within the center of the exon, to splice sites. Expanding the length of the BG CRY1 102 internal exon by cloning in larger amounts of CRY1 flanking exonic sequence resulted in a progressive loss of suppression, with a > 476 nt internal exon unable to suppress m6A (Fig. 2B and fig. S7D). These results reveal a causal role for pre-mRNA splicing in m6A regulation.
Fig. 2. Pre-mRNA splicing suppresses m6A methylation in average-length exons.
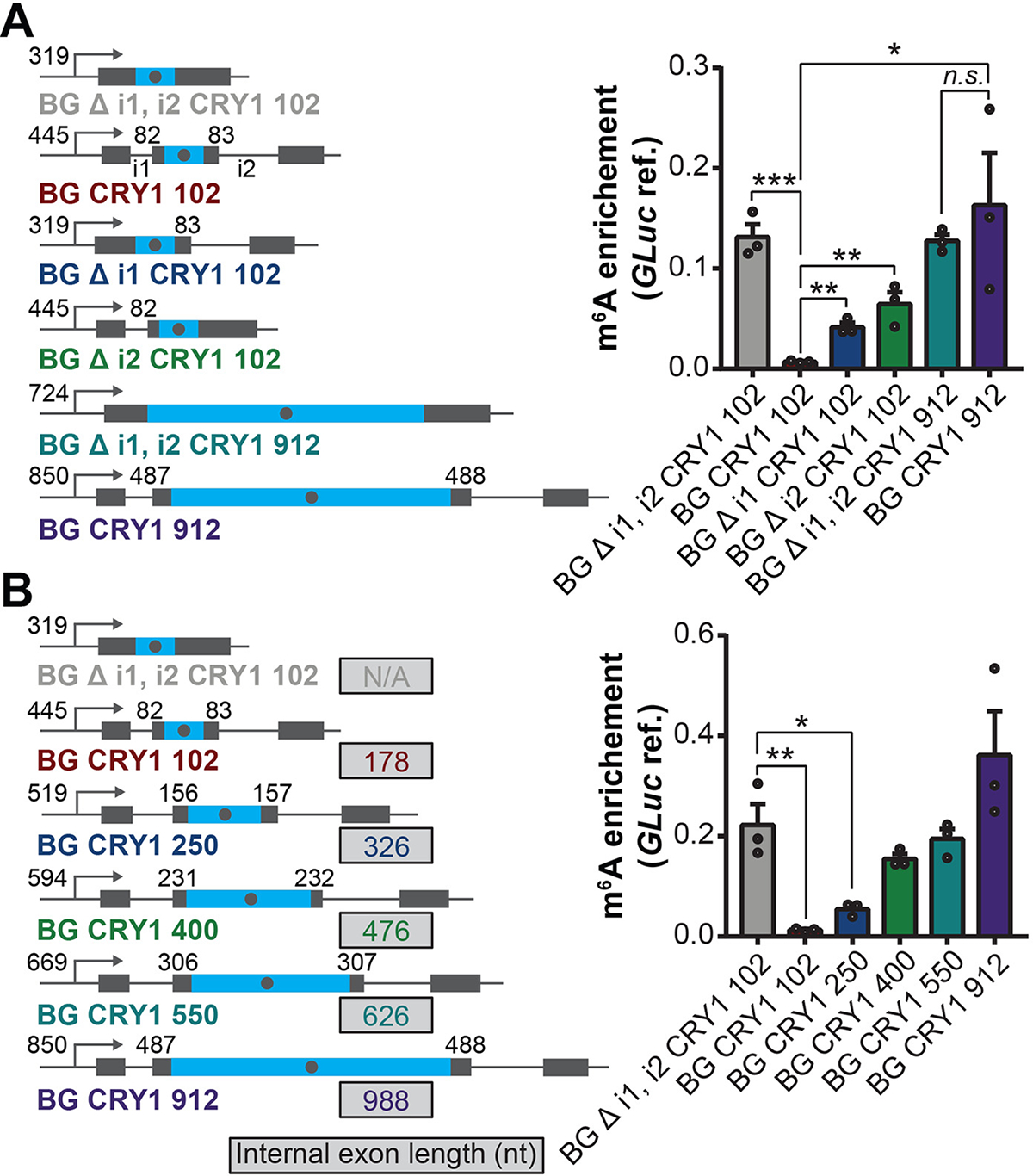
(A and B) Left: schematic of specified BG CRY1 constructs. Blue regions indicate sequences derived from the CRY1 endogenous sequence, gray regions indicate sequences derived from rabbit beta-globin (BG). Number following CRY1 refers to the number of nucleotides of exonic sequence surrounding the CRY1 suppressed m6A site in the CRY1 endogenous mRNA that was cloned into the BG construct. Grey dot in the blue region denotes the suppressed m6A site; the number at the left and right of the m6A site shows the distance (nt) between the m6A site and the 3′ and 5′ splice site, respectively; the number next to the TSS shows the distance (nt) between the m6A site and the promoter. Δ denotes deletion of the specified intron(s). Details of each construct are described in the supplementary method. Right: m6A enrichment at a CRY1 suppressed m6A site. Primers amplifying a 62 nt-fragment containing the CRY1 suppressed m6A site. m6A enrichment was calculated as IP/input normalized to m6A-marked Gaussia luciferase RNA spike-in IP/input. Mean ± SEM, two-tailed t-test, *P < 0.05; **P < 0.01, ***P < 0.001. Three biological replicates.
Exon junction complexes control m6A epitranscriptome specificity
We next sought to understand the mechanism by which splicing suppresses m6A deposition. Exon junction complexes (EJCs) are deposited by spliceosomes onto mRNA ~24 nt upstream of exon-exon junctions and plays multifaceted roles in gene expression regulation (14, 15). Notably, two recent studies reported that EJCs efficiently block splicing at proximal aberrant splice sites (16, 17). Additionally, EJCs, together with interacting serine and arginine-rich (SR) proteins, package and compact mRNA and can protect long stretches of proximal RNA from nuclease accessibility in vitro, and also block 5′ to 3′ exonuclease degradation in vivo (18, 19). We reasoned that suppressed m6A sites within average-length internal exons are within relatively close proximity to both an upstream and downstream EJC. Conversely, m6A sites within long internal exons and near stop codons (which generally reside in long last exons) can be hundreds of nucleotides away from the nearest EJC. We therefore hypothesized that EJCs could mediate the splice site-proximal suppression of m6A we observed.
We knocked down (KD) the core EJC factor EIF4A3 in HeLa cells and assessed the effect on m6A deposition transcriptome-wide using m6A-MeRIP-seq. Notably, 24,350 regions were significantly hypermethylated upon EIF4A3 KD, while 3,140 regions were hypomethylated (Fig. 3A). 39% of these hypermethylated regions exhibited a greater than 8-fold increase in m6A enrichment compared to the non-targeting siRNA control. We knocked down RBM8A, another core EJC factor (20), and observed similar, though relatively milder, transcriptome-wide m6A changes, with 14,034 significantly hypermethylated regions observed, of which 57% overlapped with hypermethylated regions observed in EIF4A3 KD (Fig. 3A and fig. S8, A and B). The relatively milder m6A changes upon RBM8A KD may result from relatively lower KD efficiency (table S1) or may indicate a stronger requirement of EIF4A3 for suppression. Concordant with these transcriptome-wide m6A changes, using UHPLC-QQQ-MS/MS, we found that EIF4A3 KD increased global levels of m6A in polyadenylated RNA by two-fold, while RBM8A KD resulted in a ~25% increase (fig. S8C).
Fig. 3. EJCs protect exon junction-proximal RNA in average-length exons within CDS regions from m6A methylation.
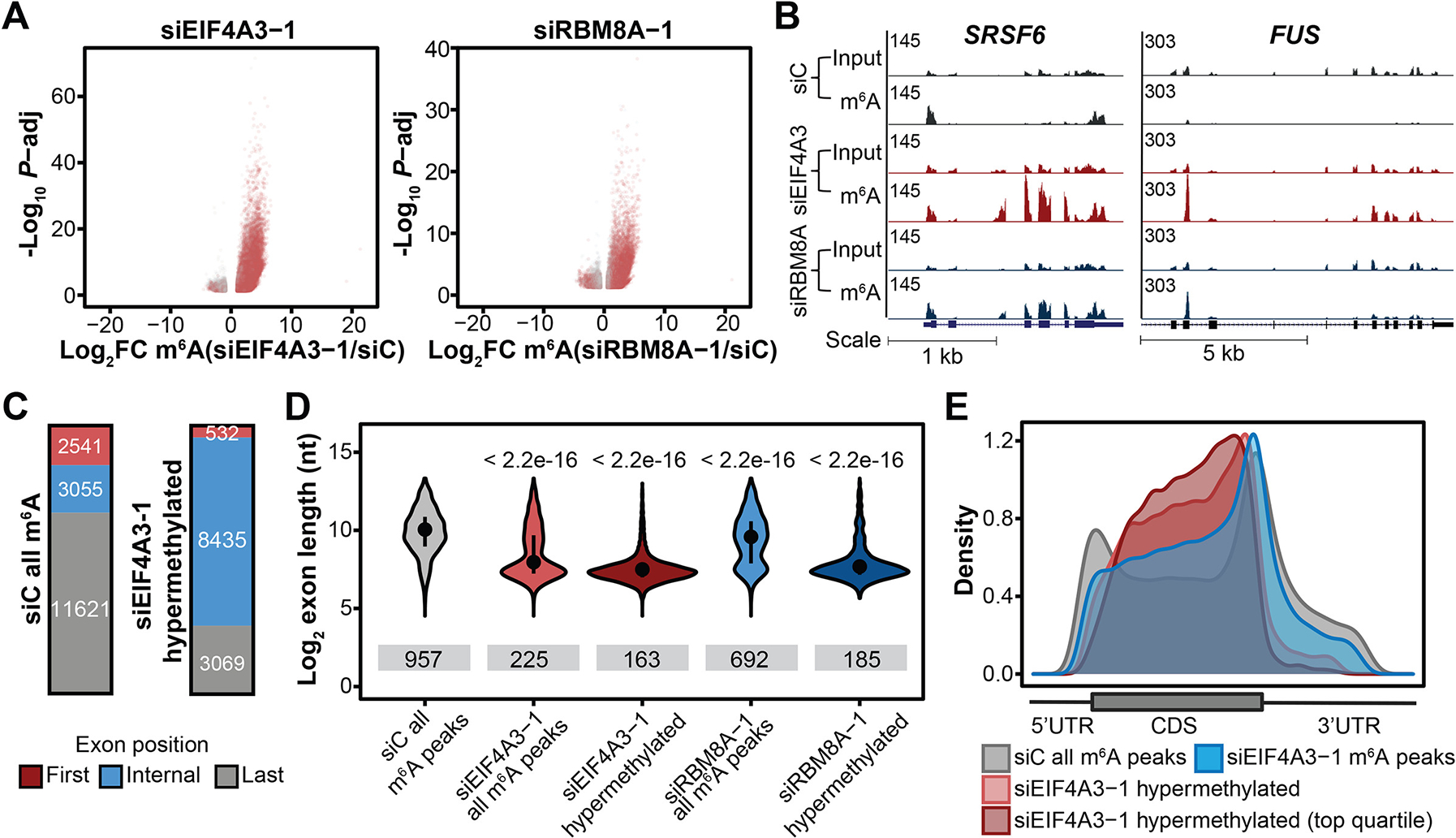
(A) Differentially methylated regions upon EIF4A3 KD (left) and RBM8A KD (right) in HeLa cells (FDR<.1, |log2FC|>1). Three biological replicates. Gray and red dots indicate differentially methylated regions that overlap and do not overlap m6A peaks in the control cells, respectively. (B) Input and m6A-IP read coverage at FUS and SRSF6 in EIF4A3 KD, RBM8A KD, and control HeLa cells. (C) Numbers of EIF4A3 KD hypermethylated regions (left) and m6A peaks in control cells (right) that reside within first, internal or last exons (D) Exon lengths for m6A peaks residing within internal exons in control KD, EIF4A3 KD and RBM8A KD cells, and exon lengths of hypermethylated regions residing within internal exons in EIF4A3 and RBM8A KD cells. Dot and bar represent median and interquartile range, Wilcoxon rank sum test of indicated group vs. siC all m6A peaks. Sample size for each violin plot from left to right is: n = 3166, n = 6659, n = 8438, n = 3817, and n = 3827. (E) Metagenes of m6A peaks and significantly hypermethylated m6A regions (and top quartile) in EIF4A3 KD HeLa cells, and m6A peaks in control cells.
94% of hypermethylated regions from EIF4A3 KD and 82% of hypermethylated regions from RBM8A KD did not overlap with m6A peaks identified under the non-targeting siRNA control conditions, suggesting that these regions contain newly methylated suppressed m6A sites (Fig. 3, A and B, and fig. S8D). Indeed, out of 1,024 CDS sequences identified by MPm6A to contain suppressed m6A sites, 46% become methylated upon EIF4A3 and/or RBM8A KD, including the CRY1 suppressed site (fig. S8E), with three selected suppressed sites validated (fig. S8, F and G) (21). Furthermore, EIF4A3 KD substantially alleviated the previously observed m6A suppression within the internal exon of BG CRY1 102 (fig. S8H).
Consistent with our model, newly methylated and hypermethylated regions were highly enriched in average-length internal exons within CDSs (Fig. 3, C to E, and fig. S8, I and J), with transcriptome-wide increases in m6A enrichment in exon junction-proximal regions observed (fig.S9, A and B) upon EIF4A3 or RBM8A KD. EIF4A3 KD disrupted m6A epitranscriptome specificity globally, resulting in substantial loss of enrichment of m6A peaks in long internal exons and increased density of m6A in the CDS relative to the stop codon (Fig. 3, D and E). It was previously reported that the peak of m6A density near stop codons on metagene plots can be more precisely visualized as an increased enrichment 150 nt past the start of last exons (6). EIF4A3 KD resulted in a global increase in m6A enrichment < 150 nt past the start of last exons (fig. S9, A to C), indicating that EJC suppression of methylation proximal to last exon-exon junctions is responsible for the characteristic m6A peak density near stop codons. While most transcripts exhibited hypermethylation and contained one or more endogenous m6A peaks upon EIF4A3 KD, over a thousand transcripts that ordinarily lack endogenous m6A peaks also gained aberrant m6A methylation upon EIF4A3 KD, revealing a major role for EJCs in suppressing m6A deposition on the subset of transcripts that ordinarily are not subject to m6A regulation (fig. S9, D to F).
The widespread suppression of m6A by the EJCs also implies that many m6A are deposited following splicing, which we confirmed using pulse-chase metabolic labeling experiments and UHPLC-QQQ-MS/MS (supplementary text and fig. S10). Two genes used in gene therapies for mucopolysaccharidosis type II and spinal muscular atrophy, IDS and SMN, contain EJC-suppressed m6A sites in their mRNAs, respectively. As expected, when these mRNAs were expressed from cDNA constructs, and thus not bound by EJCs, they were significantly hypermethylated relative to the corresponding endogenous mRNAs (fig. S11). Further, lncRNAs that contain three or more exons globally exhibit EJC suppression of m6A in internal exons, while those with two or less do not (supplementary text and fig. S12). We depleted EIF4A3 with a different siRNA in HeLa cells, and knocked down EIF4A3 in HEK293T cells as well as in a glioblastoma cancer cell line (U87) that is sensitive to EIF4A3 perturbation (22), and observed similar transcriptome-wide m6A changes in each case (figs. S13 to S15). Altogether, our results indicate that spliceosomes widely suppress m6A methylation via deposition of EJCs that protect proximal RNA from methylation.
EJCs regulate mRNA stability by suppressing m6A methylation
m6A is known to mainly accelerate mRNA degradation via the reader protein YTHDF2 (23, 24). Accordingly, we observed globally reduced mRNA half-life of hypermethylated transcripts (~90%) upon EIF4A3 KD (Fig. 4, A and B). Consistently, we observed generally increased YTHDF2 binding on hypermethylated mRNAs, accompanied with the decreased mRNA half-life (Fig. 4C). YTHDF2 KD could rescue accelerated degradation of YTHDF2 target transcripts upon EIF4A3 KD (fig. S16). Further, the density of EJC-loading on transcripts (estimated by the number of exons within CDS regions per 1 kb) correlated with transcriptome-wide mRNA stability (fig. S17). Higher EJC density on transcripts tended to correlate with reduced m6A methylation and higher mRNA stability, and the strength of this correlation was diminished by Mettl3 KO (fig. S17, A and B).
Fig. 4. mRNA m6A hypermethylation upon EJC depletion destabilizes mRNAs.
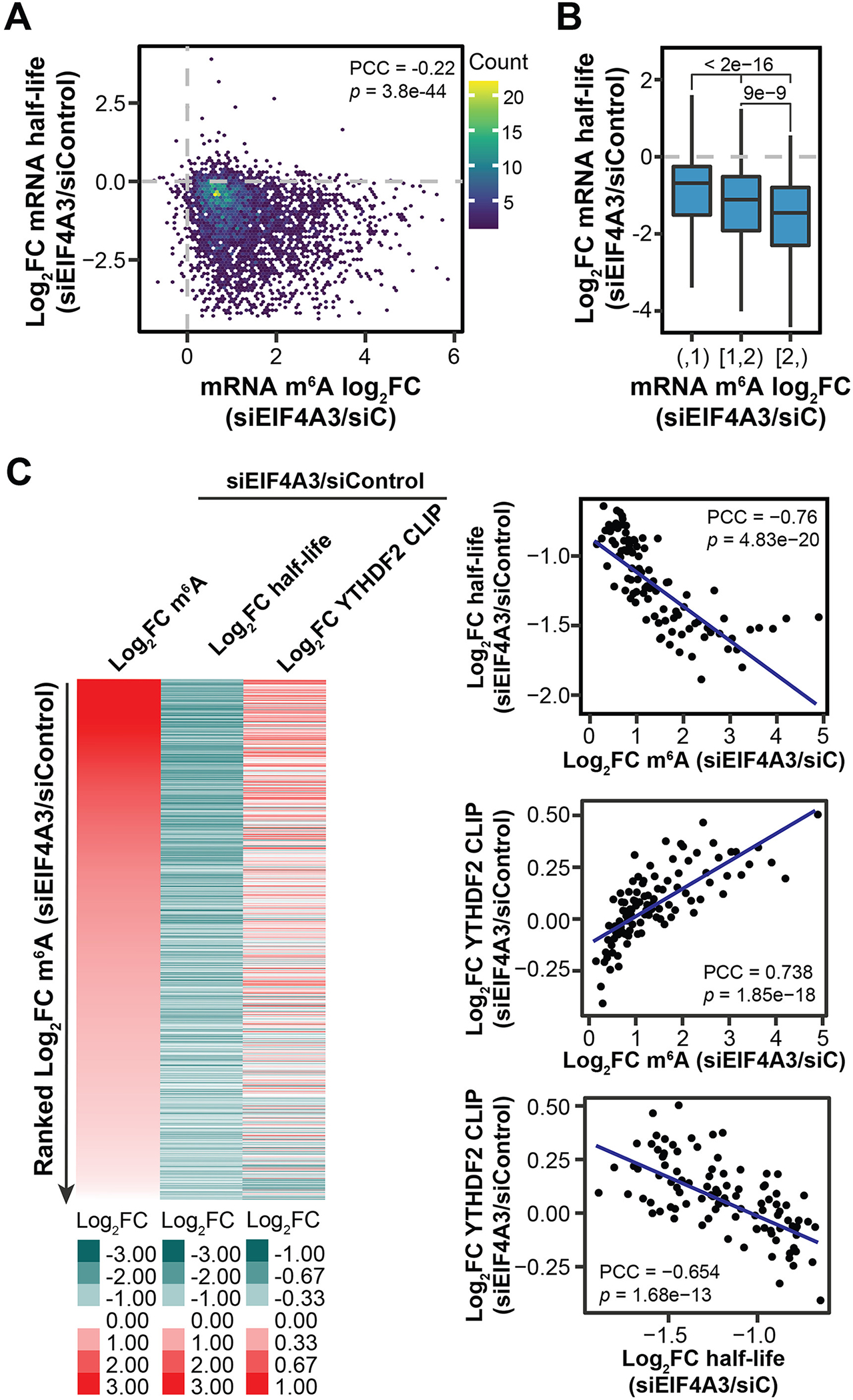
(A) Correlation between fold changes in mRNA half-life and m6A level upon EIF4A3 KD in HeLa cells (n = 3840). (B) Boxplots showing half-life fold changes of hypermethylated mRNAs upon EIF4A3 KD in HeLa cells. mRNAs were categorized into three groups according to their methylation changes upon EIF4A3 KD in HeLa cells. P values from Wilcoxon rank sum test. Sample size for each boxplot plot from left to right is: n = 1887, n = 1201, and n = 752. (C) Left: heatmap showing fold changes in m6A level, mRNA half-life, and YTHDF2 binding upon EIF4A3 KD in HeLa cells. Right: scatter plots showing the correlation among fold changes in m6A level, mRNA half-life, and YTHDF2 binding upon EIF4A3 KD in HeLa cells. The hypermethylated mRNAs (m6A log2FC > 0; n = 3424) were categorized into 100 bins based on ranked fold change of m6A level upon EIF4A3 KD. For (A) and (C), PCC and P values are shown.
We also found that METTL3 depletion could generally reduce the expression level changes of hypermethylated genes upon EJC depletion in HeLa cells (supplementary text and fig. S18), indicating that these EJC-dependent gene expression changes are at least in part mediated by m6A methylation.
While the vast majority of hypermethylated transcripts were destabilized by EIF4A3 KD, a small subset of hypermethylated transcripts were stabilized (Fig. 4A). One example is p53, which mediates neurodevelopmental defects in mouse models of EJC haploinsufficiency (25). The TP53 transcript was hypermethylated but also up-regulated upon EIF4A3 KD. Mechanistically, we observed increased binding to TP53 mRNA by IGF2BP proteins, which are known to stabilize methylated transcripts (supplementary text and fig. S19). In summary, while the predominant effect of EJC-mediated m6A suppression is to stabilize mRNAs by preventing the YTHDF2-mediated decay, in a minority of instances hypermethylated transcripts can be stabilized by other mechanisms, such as binding by IGF2BPs (26).
Consistent with a general role for m6A in promoting translation (12, 27), EIF4A3 KD led to slightly increased translation efficiency of hypermethylated transcripts, with more highly hypermethylated transcripts exhibiting greater increases in translation efficiency (fig. S20), although the impact was modest relative to the effects observed on mRNA stability.
Differential m6A methylation across tissues and species through EJC suppression
Our model suggests that the cellular EJC levels may impact global mRNA m6A deposition in different tissues. Indeed, we observed a negative correlation between EIF4A3 expression level and global mRNA m6A modification level in 25 different human tissues with available transcriptome-wide m6A profiles (fig. S21A) (28). We examined the top 10% of genes with the strongest correlations and found that the majority (> 70%) exhibited a negative correlation between m6A and EIF4A3 levels in different tissues. Further, m6A levels of these genes also negatively correlated with their transcript abundances (fig. S21B). Similar trends were also observed in mouse tissues (fig. S21C). These results further support m6A suppression by EJCs and subsequently mRNA stability regulation in mammalian tissues.
Notably, we observed the lowest EIF4A3 expressions in brain tissues, which exhibited the highest overall mRNA m6A levels (fig. S21A). We further compared the methylome of the human cerebellum (lowest EIF4A3 level and highest overall mRNA m6A) with that of the heart (higher EIF4A3 level and lower overall mRNA m6A). Regions that are hypermethylated in the cerebellum (compared to heart) reside within short internal exons (fig. S21D), suggesting reduced m6A suppression due to low EIF4A3 expression in cerebellum. This association between high m6A level and low EIF4A3 expression in cerebellum was attenuated upon depletion of METTL3 (fig. S21C). These observations further indicate the widespread suppression by EJCs contributes to tissue-specific m6A deposition. We also found that a subset of EJC-suppressed m6A sites physiologically escape suppression in certain tissues via methylation of alternative transcript isoforms. These isoforms contain longer exons and thus altered EJC positioning; methylation of these isoforms generates tissue-specific m6A patterns (supplementary text and fig. S22).
Lastly, the effect of exon-intron architecture on mRNA stability may have co-evolved with YTHDF2 in vertebrates. The strong correlation between EJC loading, represented by the number of exons, and mRNA level across tissues is maintained across humans, mice, and zebrafish, but not fly and worm, which lack YTHDF2 orthologs (supplementary text and fig. S23).
EJCs and peripheral EJC factor RNPS1 protect exon junction-proximal RNA regions from aberrant mRNA processing
We did not observe interactions between the methyltransferase complex and EJC complexes (fig. S24), suggesting that steric hindrance from EJCs, rather than a specific inhibitory interaction, accounts for methylation suppression. Nuclear EJCs bound with the peripheral EJC factor RNPS1 multimerize and associate with wide variety of SR and SR-like proteins to package and compact mRNA into higher-order, megadalton-scale mRNPs that ensheathe proximal RNA well beyond the canonical EJC deposition sites (18, 29, 30). Tens to hundreds of nucleotides of proximal RNA could be protected by this mega-complex from nuclease digestion due to this packaging (18, 31). To examine whether the mRNA packaging function of the EJC-mediates suppression of proximal methylation, we isolated EJCs/EJC-bound RNA from cellular extracts, digested away physically accessible RNA with in vitro nuclease treatment, and then measured m6A levels on the EJC-protected RNA footprints (fig. S25, A and B). EJC-protected footprints were strongly depleted of m6A, indicating that these inaccessible RNA regions are largely protected from m6A deposition within cells (fig. S25C). EJCs also protected these footprints from in vitro methylation by recombinant METTL3-METTL14 (fig. S25D). This was not due to general inhibition of methyltransferase activity or lack of methylatable sites on the EJC footprints, as free, unmethylated RNA spiked into the methylation reaction as well as deproteinized footprints were both robustly methylated (fig. S25, D and E). Therefore, EJCs suppress local m6A deposition by packaging proximal RNA.
We next asked whether the peripheral EJC factor RNPS1, which associates with high molecular weight EJCs in these highly packaged mRNP structures (29), plays a role. RNPS1 knockdown led to substantial transcript m6A hypermethylation within average-length internal exons and CDS regions (Fig. 5, A to C, and fig. S26, A to C). We detected fewer hypermethylated regions overall compared to depletion of the core EJC factors; however, we did observe high overlap (45%) between siRNPS1 hypermethylated regions and siEIF4A3/siRBM8A hypermethylated regions (Fig. 5C and fig. S26C). In contrast, depletion of UPF1, a central NMD factor that interacts with the EJC in the cytoplasm, did not result in m6A changes similar to those of the core EJC (fig. S27).
Fig. 5. EJCs and RNPS1 protect proximal RNA regions from aberrant mRNA processing.
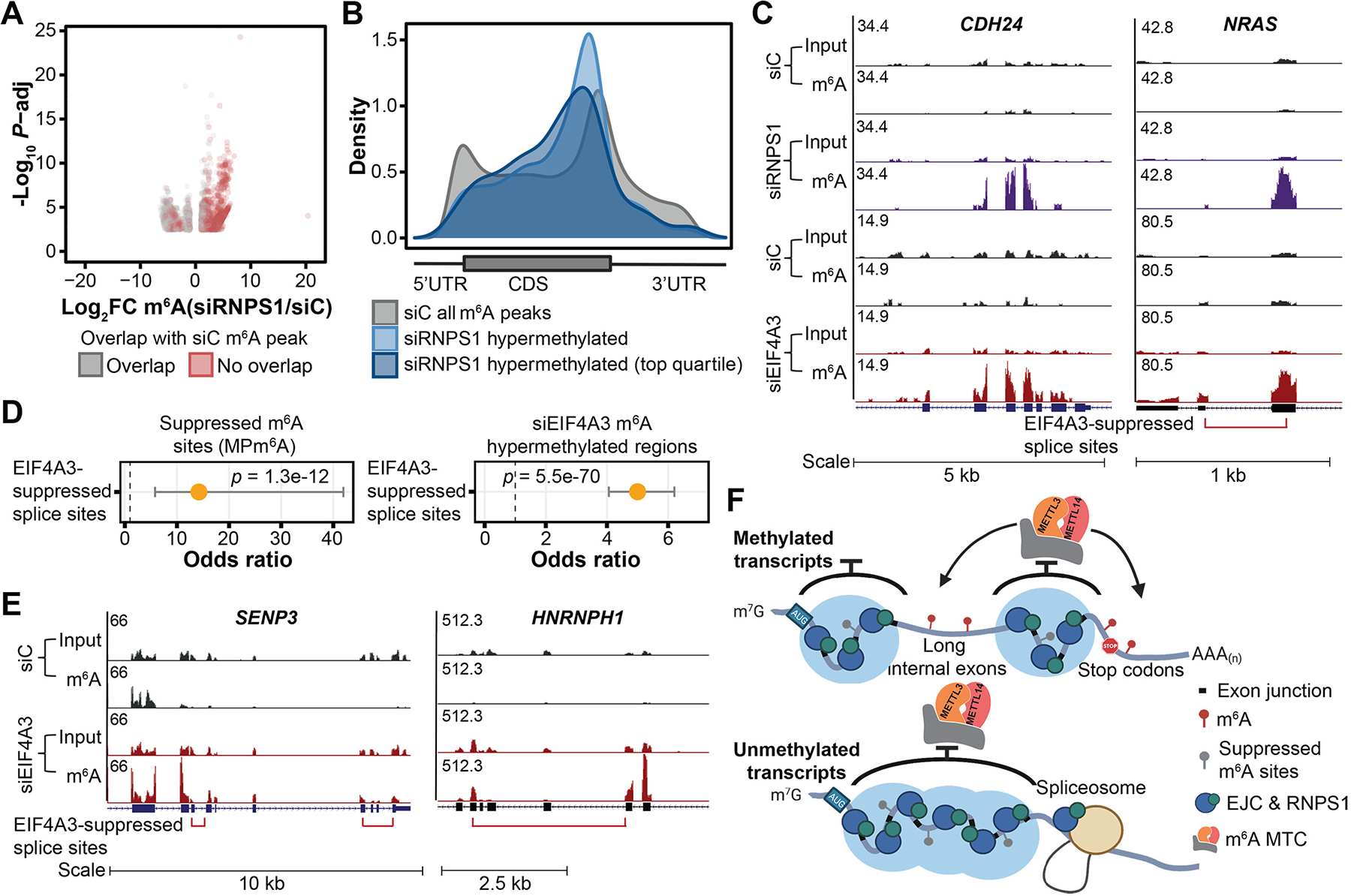
(A) Differentially methylated regions upon RNPS1 KD in HeLa cells (FDR < 0.1, |log2FC| > 1), three biological replicates. Gray and red dots indicate differentially methylated regions that overlap and do not overlap with m6A peaks in the control KD cells, respectively. (B) Metagenes of significantly m6A hypermethylated regions (and top quartile) upon RNPS1 KD in HeLa cells in comparison with that of all m6A peaks in control cells. (C) Input and m6A-IP read coverage at CDH24 and NRAS upon RNPS1 KD and EIF4A3 KD, respectively, as well as corresponding controls in HeLa cells. (D) Enrichment of suppressed m6A sites (identified from MPm6A) at EIF4A3-suppressed splice sites (left) and enrichment of EIF4A3 KD hypermethylated regions at EIF4A3-suppressed splice sites (right). Fisher’s exact test, dot and bar represent odds ratio and 95% confidence interval. (E) Input and m6A-IP read coverage at SENP3 and HNRNPH1 in EIF4A3 KD and control HeLa cells. (F) Schematic model depicting that EJCs and RNPS1 (and potentially other EJC-associated proteins) protect exon junction-proximal RNA from m6A deposition through local mRNA packaging. For (C) and (E), red bracket indicates EIF4A3-suppressed splice variant, with ends of bracket indicating the suppressed splice junctions.
The ability of EJCs to protect proximal RNA regions from methylation resembles the recently characterized EJC- and RNPS1-mediated suppression of proximal aberrant splice sites and recursive splicing (16, 17). Transcriptome-wide, EJC-suppressed splice sites significantly colocalize with EJC-suppressed m6A sites (supplementary text; Fig. 4, C to E; fig. S26, D to F; and table S2). Altogether, these results suggest that RNPS1-associated EJCs suppress both local cellular m6A methylation and splicing through packaging of proximal RNA and point to exon architecture as an important determinant of local RNA accessibility to regulatory machineries. Additionally, beyond components of the m6A methyltransferase complex, a number of other RBPs also exhibit preferential binding at long internal exons, suggesting that EJCs may regulate mRNA accessibility to a broader range of mRNA regulators in addition to the splicing and m6A methylation machineries through their mRNA packaging function (supplementary text and fig. S28).
Discussion
Previously identified m6A effector proteins fall broadly into three categories according to their activities: “writers”, which catalyze m6A methylation, “readers”, which preferentially bind m6A, and “erasers”, which reverse m6A methylation. Here we establish the EJCs as a member of a new class of m6A regulators: “suppressors”, which broadly suppress the deposition of m6A (fig. S28). EJCs appear to be a major regulator of m6A deposition that mediate multiple key aspects of global m6A epitranscriptome specificity, including enrichment of m6A in long internal exons, depletion of m6A in CDSs and enrichment of m6A in last exons near stop codons, and methylation selectivity for transcripts possessing long internal exons. This mechanism may also explain the high abundance of m6A on certain non-coding RNAs, such as LINE-1 elements that are generally unspliced and thus not bound by the EJCs (32, 33). Further, our systematic analysis of m6A determinants using MPm6A may suggest the existence of additional m6A suppressing pathways, including m6A suppression within the CDS, as EIF4A3 KD does not appear to completely restore methylation to unspliced levels (supplementary text, fig. S8H, and fig. S29).
Our results point to exon length within transcripts as a functionally relevant element for post-transcriptional gene expression regulation. Mammalian EJCs stably bind the vast majority of pre-translational mRNAs in the transcriptome at closely spaced intervals. Long internal exons and terminal exons, which usually encode UTRs, are notably free of EJCs. This widespread binding, in conjunction with their mRNA packaging function, appears to uniquely position EJCs to broadly determine mRNA accessibility to regulatory machineries, such as the m6A methylation and splicing machineries (fig. S30). Our work has relevance for the use of cDNA expression constructs in research studies and gene therapies, as loss of endogenous mRNA exon architecture and EJC protection results in m6A hypermethylation (fig. S11), which could modulate gene expression outcome. Finally, our study also suggests that exon length and architecture co-evolved with mRNA processing steps as an additional regulatory layer of gene expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tao Pan, Erin Adams, Marcus Clark, Marcelo Nobrega, Jonathan Staley, and Amelia Joslin for comments and suggestions. We thank the Genomics Facility of the University of Chicago and the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility for assistance with sequencing.
Funding:
National Institutes of Health HG008935 (C.H.); National Institutes of Health grant T32 HD007009 (P.C.H); National Institutes of Health grant F32 CA221007 (B.T.H); C.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc., Aferna Bio, Inc., and AccuraDX Inc. The other authors declare no competing interests.
SUPPLEMENTARY MATERIALS
References (35–79)
Data and materials availability:
Raw and processed data can be found at NCBI GEO accession GSE162199. Custom scripts available on Zenodo (34). All other data are available in the manuscript or the supplementary materials.
REFERENCES AND NOTES
- 1.Frye M, Harada BT, Behm M, He C, RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018).doi: 10.1126/science.aau1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert WV, Bell TA, Schaening C, Messenger RNA modifications: Form, distribution, and function. Science 352, 1408–1412 (2016).doi: 10.1126/science.aad8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roundtree IA, Evans ME, Pan T, He C, Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).doi: 10.1016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He PC, He C, m6A RNA methylation: From mechanisms to therapeutic potential. EMBO J. 40, e105977 (2021).doi: 10.15252/embj.2020105977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G, Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).doi: 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- 6.Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, Vågbø CB, Kusśnierczyk A, Klungland A, Darnell JE Jr., R. B. Darnell, A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 29, 2037–2053 (2015).doi: 10.1101/gad.269415.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, De Braekeleer E, Ponstingl H, Hendrick A, Vakoc CR, Vassiliou GS, Kouzarides T, Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131 (2017).doi: 10.1038/nature24678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, Sadée C, Lenaerts A-S, Nakanoh S, Grandy R, Farnell E, Ule J, Stunnenberg HG, Mendjan S, Vallier L, The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 555, 256–259 (2018).doi: 10.1038/nature25784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fish L, Navickas A, Culbertson B, Xu Y, Nguyen HCB, Zhang S, Hochman M, Okimoto R, Dill BD, Molina H, Najafabadi HS, Alarcón C, Ruggero D, Goodarzi H, Nuclear TARBP2 drives oncogenic dysregulation of RNA splicing and decay. Mol. Cell 75, 967–981.e9 (2019).doi: 10.1016/j.molcel.2019.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slobodin B, Han R, Calderone V, Vrielink JAFO, Loayza-Puch F, Elkon R, Agami R, Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 169, 326–337.e12 (2017).doi: 10.1016/j.cell.2017.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, Klemm L, Wu H, Zuo Z, Qin X, Dong Y, Zhou Y, Qin H, Tao S, Du J, Liu J, Lu Z, Yin H, Mesquita A, Yuan CL, Hu Y-C, Sun W, Su R, Dong L, Shen C, Li C, Qing Y, Jiang X, Wu X, Sun M, Guan J-L, Qu L, Wei M, Müschen M, Huang G, He C, Yang J, Chen J, Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567, 414–419 (2019).doi: 10.1038/s41586-019-1016-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Luo K, Zou Z, Qiu M, Tian J, Sieh L, Shi H, Zou Y, Wang G, Morrison J, Zhu AC, Qiao M, Li Z, Stephens M, He X, He C, Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat. Genet. 52, 939–949 (2020).doi: 10.1038/s41588-020-0644-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NAL, Dominguez D, Olson S, Sundararaman B, Zhan L, Bazile C, Bouvrette LPB, Bergalet J, Duff MO, Garcia KE, Gelboin-Burkhart C, Hochman M, Lambert NJ, Li H, McGurk MP, Nguyen TB, Palden T, Rabano I, Sathe S, Stanton R, Su A, Wang R, Yee BA, Zhou B, Louie AL, Aigner S, Fu X-D, Lécuyer E, Burge CB, Graveley BR, Yeo GW, A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020).doi: 10.1038/s41586-020-2077-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Hir H, Saulière J, Wang Z, The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 17, 41–54 (2016).doi: 10.1038/nrm.2015.7 [DOI] [PubMed] [Google Scholar]
- 15.Boehm V, Gehring NH, Exon junction complexes: Supervising the gene expression assembly line. Trends Genet. 32, 724–735 (2016).doi: 10.1016/j.tig.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 16.Boehm V, Britto-Borges T, Steckelberg A-L, Singh KK, Gerbracht JV, Gueney E, Blazquez L, Altmüller J, Dieterich C, Gehring NH, Exon junction complexes suppress spurious splice sites to safeguard transcriptome integrity. Mol. Cell 72, 482–495.e7 (2018).doi: 10.1016/j.molcel.2018.08.030 [DOI] [PubMed] [Google Scholar]
- 17.Blazquez L, Emmett W, Faraway R, Pineda JMB, Bajew S, Gohr A, Haberman N, Sibley CR, Bradley RK, Irimia M, Ule J, Exon junction complex shapes the transcriptome by repressing recursive splicing. Mol. Cell 72, 496–509.e9 (2018).doi: 10.1016/j.molcel.2018.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ, The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151, 750–764 (2012).doi: 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee W-C, Hou B-H, Hou C-Y, Tsao S-M, Kao P, Chen H-M, Widespread exon junction complex footprints in the RNA degradome mark mRNA degradation before steady state translation. Plant Cell 32, 904–922 (2020).doi: 10.1105/tpc.19.00666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tange TØ, Shibuya T, Jurica MS, Moore MJ, Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA 11, 1869–1883 (2005).doi: 10.1261/rna.2155905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Y, Wang Y, Tang Q, Wei L, Zhang X, Jia G, An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N6-methyladenosine modification. Angew. Chem. Int. Ed. 57, 15995–16000 (2018).doi: 10.1002/anie.201807942 [DOI] [PubMed] [Google Scholar]
- 22.Tang W, Wang D, Shao L, Liu X, Zheng J, Xue Y, Ruan X, Yang C, Liu L, Ma J, Li Z, Liu Y, LINC00680 and TTN-AS1 stabilized by EIF4A3 promoted malignant biological behaviors of glioblastoma cells. Mol. Ther. Nucleic Acids 19, 905–921 (2020).doi: 10.1016/j.omtn.2019.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, He C, N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).doi: 10.1038/nature12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L, YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016).doi: 10.1038/ncomms12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao H, McMahon JJ, Tsai Y-H, Wang Z, Silver DL, Haploinsufficiency for Core Exon Junction Complex Components Disrupts Embryonic Neurogenesis and Causes p53-Mediated Microcephaly. PLOS Genet. 12, e1006282 (2016).doi: 10.1371/journal.pgen.1006282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu Y-C, Hüttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan J-L, He C, Yang J, Chen J, Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).doi: 10.1038/s41556-018-0045-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C, N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).doi: 10.1016/j.cell.2015.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Li K, Cai J, Zhang M, Zhang X, Xiong X, Meng H, Xu X, Huang Z, Peng J, Fan J, Yi C, Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Mol. Cell 77, 426–440.e6 (2020).doi: 10.1016/j.molcel.2019.09.032 [DOI] [PubMed] [Google Scholar]
- 29.Mabin JW, Woodward LA, Patton RD, Yi Z, Jia M, Wysocki VH, Bundschuh R, Singh G, The exon junction complex undergoes a compositional switch that alters mRNP structure and nonsense-mediated mRNA decay activity. Cell Rep. 25, 2431–2446.e7 (2018).doi: 10.1016/j.celrep.2018.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metkar M, Ozadam H, Lajoie BR, Imakaev M, Mirny LA, Dekker J, Moore MJ, Higher-order organization principles of pre-translational mRNPs. Mol. Cell 72, 715–726.e3 (2018).doi: 10.1016/j.molcel.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Hir H, Izaurralde E, Maquat LE, Moore MJ, The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 19, 6860–6869 (2000).doi: 10.1093/emboj/19.24.6860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D, He C, N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020).doi: 10.1126/science.aay6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Yu X, Yang L, Liu X, Gao B, Huang B, Dou X, Liu J, Zou Z, Cui X-L, Zhang L-S, Zhao X, Liu Q, He PC, Sepich-Poore C, Zhong N, Liu W, Li Y, Kou X, Zhao Y, Wu Y, Cheng X, Chen C, An Y, Dong X, Wang H, Shu Q, Hao Z, Duan T, He Y-Y, Li X, Gao S, Gao Y, He C, FTO mediates LINE1 m6A demethylation and chromatin regulation in mESCs and mouse development. Science 376, 968–973 (2022).doi: 10.1126/science.abe9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He PC, Dou X, Custom scripts associated with “Exon architecture controls mRNA m6A suppression and gene expression”, Zenodo (2023); 10.5281/zenodo.7541415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uphoff CC, Drexler HG, in Cancer Cell Culture: Methods and Protocols, Langdon SP, Ed., vol. 88 of Methods in Molecular Medicine (Humana Press, 2004), pp. 319–326.14634244 [Google Scholar]
- 36.Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, Wang F, Wang X, Shen B, Wang Y, Feng X, He C, Liu J, VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10 (2018).doi: 10.1038/s41421-018-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Zeng P, Li Y-H, Zhang Z, Cui Q, SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 44, e91 (2016).doi: 10.1093/nar/gkw104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE, Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011).doi: 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D, Langmead B, Salzberg SL, HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).doi: 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Zhan Q, Eckert M, Zhu A, Chryplewicz A, De Jesus DF, Ren D, Kulkarni RN, Lengyel E, He C, Chen M, RADAR: Differential analysis of MeRIP-seq data with a random effect model. Genome Biol. 20, 294 (2019).doi: 10.1186/s13059-019-1915-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G, Wang L-G, Han Y, He Q-Y, clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D, The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).doi: 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolger AM, Lohse M, Usadel B, Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S, Pyl PT, Huber W, HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 48.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 50.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS, Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).doi: 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buschmann T, Bystrykh LV, Levenshtein error-correcting barcodes for multiplexed DNA sequencing. BMC Bioinformatics 14, 272 (2013).doi: 10.1186/1471-2105-14-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agarwal V, Bell GW, Nam J-W, Bartel DP, Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005 (2015).doi: 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE Jr., R. B. Darnell, m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 (2017).doi: 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, He C, Litzow MR, Liu S, A dynamic N6-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 28, 1062–1076 (2018).doi: 10.1038/s41422-018-0097-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ-L, Bomane A, Cosson B, Eyras E, Rasko JEJ, Ritchie W, IRFinder: Assessing the impact of intron retention on mammalian gene expression. Genome Biol. 18, 51 (2017).doi: 10.1186/s13059-017-1184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann CJ, Schmidt R, Kanitz A, Artimo P, Gruber AJ, Zavolan M, PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 48, D174–D179 (2020).doi: 10.1093/nar/gkz918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M, The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46, 674–690 (2012).doi: 10.1016/j.molcel.2012.05.021 [DOI] [PubMed] [Google Scholar]
- 58.Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S, N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res. 45, 11356–11370 (2017).doi: 10.1093/nar/gkx778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Busch A, Hertel KJ, HEXEvent: A database of Human EXon splicing Events. Nucleic Acids Res. 41, D118–D124 (2013).doi: 10.1093/nar/gks969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).doi: 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- 61.Louloupi A, Ntini E, Conrad T, Ørom UAV, Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep. 23, 3429–3437 (2018).doi: 10.1016/j.celrep.2018.05.077 [DOI] [PubMed] [Google Scholar]
- 62.Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Ż, Pan JN, He C, Parisien M, Pan T, Regulation of co-transcriptional pre-mRNA splicing by m6A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81.e9 (2019).doi: 10.1016/j.molcel.2019.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, Sun H-Y, Li A, Ping X-L, Lai W-Y, Wang X, Ma H-L, Huang C-M, Yang Y, Huang N, Jiang G-B, Wang H-L, Zhou Q, Wang X-J, Zhao Y-L, Yang Y-G, Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016).doi: 10.1016/j.molcel.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 64.Viphakone N, Sudbery I, Griffith L, Heath CG, Sims D, Wilson SA, Co-transcriptional loading of RNA export factors shapes the human transcriptome. Mol. Cell 75, 310–323.e8 (2019).doi: 10.1016/j.molcel.2019.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neugebauer KM, Nascent RNA and the Coordination of Splicing with Transcription. Cold Spring Harb. Perspect. Biol. 11, a032227 (2019).doi: 10.1101/cshperspect.a032227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akhtar J, Kreim N, Marini F, Mohana G, Brüne D, Binder H, Roignant J-Y, Promoter-proximal pausing mediated by the exon junction complex regulates splicing. Nat. Commun. 10, 521 (2019).doi: 10.1038/s41467-019-08381-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silver DL, Watkins-Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, Liaw H-J, Myung K, Walsh CA, Gaiano N, Pavan WJ, The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nat. Neurosci. 13, 551–558 (2010).doi: 10.1038/nn.2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Murigneux V, Le Hir H, Transcriptome-wide modulation of splicing by the exon junction complex. Genome Biol. 15, 551 (2014).doi: 10.1186/s13059-014-0551-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu C-C, Lee C-C, Tseng C-T, Tarn W-Y, Y14 governs p53 expression and modulates DNA damage sensitivity. Sci. Rep. 7, 45558 (2017).doi: 10.1038/srep45558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sendinc E, Valle-Garcia D, Jiao A, Shi Y, Analysis of m6A RNA methylation in Caenorhabditis elegans. Cell Discov. 6, 47 (2020).doi: 10.1038/s41421-020-00186-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kan L, Ott S, Joseph B, Park ES, Dai W, Kleiner RE, Claridge-Chang A, Lai EC, A neural m6A/Ythdf pathway is required for learning and memory in Drosophila. Nat. Commun. 12, 1458 (2021).doi: 10.1038/s41467-021-21537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, He C, N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).doi: 10.1038/nchembio.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, Shi H, Cui X, Su R, Klungland A, Jia G, Chen J, He C, Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 71, 973–985.e5 (2018).doi: 10.1016/j.molcel.2018.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, Yu M, Skibbe J, Dai Q, Zou D, Wu T, Yu K, Weng H, Huang H, Ferchen K, Qin X, Zhang B, Qi J, Sasaki AT, Plas DR, Bradner JE, Wei M, Marcucci G, Jiang X, Mulloy JC, Jin J, He C, Chen J, R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell 172, 90–105.e23 (2018).doi: 10.1016/j.cell.2017.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, Lu Z, Bosmans RPG, Dai Q, Hao Y-J, Yang X, Zhao W-M, Tong W-M, Wang X-J, Bogdan F, Furu K, Fu Y, Jia G, Zhao X, Liu J, Krokan HE, Klungland A, Yang Y-G, He C, ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).doi: 10.1016/j.molcel.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, Majumder S, He C, Huang S, m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 31, 591–606.e6 (2017).doi: 10.1016/j.ccell.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Molinie B, Wang J, Lim KS, Hillebrand R, Lu Z-X, Van Wittenberghe N, Howard BD, Daneshvar K, Mullen AC, Dedon P, Xing Y, Giallourakis CC, m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 13, 692–698 (2016).doi: 10.1038/nmeth.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu L, Liu S, Peng Y, Ge R, Su R, Senevirathne C, Harada BT, Dai Q, Wei J, Zhang L, Hao Z, Luo L, Wang H, Wang Y, Luo M, Chen M, Chen J, He C, m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol. 40, 1210–1219 (2022).doi: 10.1038/s41587-022-01243-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ha KCH, Blencowe BJ, Morris Q, QAPA: A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 19, 45 (2018). doi: 10.1186/s13059-018-1414-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data can be found at NCBI GEO accession GSE162199. Custom scripts available on Zenodo (34). All other data are available in the manuscript or the supplementary materials.