

Abstract
Accumulation of the parkin-interacting substrate (PARIS; ZNF746), due to inactivation of parkin, contributes to Parkinson’s disease (PD) through repression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α; PPARGC1A) activity. Here, we identify farnesol as an inhibitor of PARIS. Farnesol promoted the farnesylation of PARIS, preventing its repression of PGC-1α via decreasing PARIS occupancy on the PPARGC1A promoter. Farnesol prevented dopaminergic neuronal loss and behavioral deficits via farnesylation of PARIS in PARIS transgenic mice, ventral midbrain transduction of AAV-PARIS, adult conditional parkin KO mice, and the α-synuclein preformed fibril model of sporadic PD. PARIS farnesylation is decreased in the substantia nigra of patients with PD, suggesting that reduced farnesylation of PARIS may play a role in PD. Thus, farnesol may be beneficial in the treatment of PD by enhancing the farnesylation of PARIS and restoring PGC-1α activity.
INTRODUCTION
Parkinson’s disease (PD) is a severe progressive neurodegenerative disease (1, 2). It is characterized clinically by motor dysfunction mainly due to the preferential loss of dopaminergic neurons in the substantia nigra (SN) (1–3). Several genetic causes of PD have been identified, providing an opportunity to understand the molecular mechanisms underlying the disease process (4, 5). Current treatment strategies for PD are mainly limited to the management of the motor symptoms with drugs such as levodopa (l-DOPA) or dopamine receptor agonists and deep brain stimulation (2, 6). Unfortunately, these therapies fail to prevent the progressive death of dopaminergic (DA) neurons. In addition, there are no proven therapies that delay or prevent the onset or progression of PD. Thus, there is an unmet need for new pharmacologic approaches to treat PD.
Previously, we identified the parkin-interacting substrate (PARIS; ZNF746), whose expression increases in models of parkin inactivation and in human PD brain (7–13). Through occupation of insulin response sequences (IRSs) in the peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α; PPARGC1A) promoter, PARIS transcriptionally represses the expression of PGC-1α and its target gene, nuclear respiratory factor 1 (NRF1) (12). PARIS is required for progressive loss of dopaminergic neurons in conditional knockout (KO) of parkin and knockdown of Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN)–induced putative kinase 1 (PINK1) in adult animals, and overexpression of PARIS leads to the selective DA neuronal degeneration (10, 12, 13). Accompanying the DA neuronal degeneration is PARIS-dependent declines in mitochondrial mass and respiration, suggesting that parkin loss impairs mitochondrial biogenesis, consistent with a defect in PGC-1α (9, 11–15).
PGC-1α is a master regulator of mitochondrial function through coregulating transcriptional programs important for mitochondrial biogenesis and protecting against mitochondrial oxidative stress (16–18). Emerging evidence suggests that PGC-1α plays a role in PD. PGC-1α concentration is decreased in patients with PD (12, 19). PGC-1α down-regulation in PD may be due to PGC-1α promoter methylation (20). PGC-1α KO mice are more susceptible to the degenerative effects of the PD neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (18), and overexpression of PGC-1α protects against N-methyl-4-phenylpyridinium ion toxicity, the active metabolite of MPTP (21). In addition, overexpression of PGC-1α protects against α-synuclein (α-syn), MPTP, oxidative stress, and rotenone-induced degeneration (22). The age of onset and risk of PD are associated with polymorphisms in PGC-1α, and in parkin-associated PD, PGC-1α is dysfunctional (23). PGC-1α–responsive genes are down-regulated in dopaminergic neurons from patients with PD, suggesting that it plays an important role in PD pathogenesis (24). The loss of dopamine neurons when PARIS is overexpressed is prevented by PGC-1α overexpression, suggesting that PGC-1α is the primary target linking PARIS to dopaminergic neuronal degeneration (12, 13). PARIS plays a role in the loss of DA neurons due to adult knock down of PINK1 (10), and recently, defects in PGC-1α signaling pathway via inhibition by PARIS has been implicated in the common sporadic form of PD (7, 14, 15). PARIS also plays a major role in the loss of DA neurons and mitochondrial dysfunction in Drosophila lacking parkin and PINK1 (11). Moreover, up-regulation of PARIS and down-regulation of PGC-1α drive mitochondrial alteration in human DA neurons lacking parkin (9). Thus, defects in PGC-1α signaling are emerging as important contributors to dopaminergic degeneration in PD, and the regulation of PARIS by parkin may be the underlying mechanism linking PGC-1α to PD (25).
Because the parkin-PARIS–PGC-1α pathway seems to play an important role in the death of dopaminergic neurons in PD, an unbiased screen was developed and conducted to identify compounds that maintain PGC-1α function in the presence of elevated PARIS. Here, we show that CSU-1806 (farnesol) is a potent inducer of PGC-1α via farnesylation of PARIS, which inhibits its transcriptional repression of PGC-1α. Moreover, farnesol prevents the loss of dopamine neurons in PARIS transgenic mice, adult conditional parkin KO mice, and α-syn preformed fibril model of sporadic PD. We show that PARIS farnesylation is decreased in aged mice and the SN of patients with PD compared to control, suggesting that PARIS farnesylation may play a pathophysiological role in PD.
RESULTS
Farnesol induces PGC-1α in PARIS overexpression
We first used a stable reporter cell line expressing luciferase and destabilized green fluorescent protein (GFP) under control of the 1-kb human PPARGC1A promoter (SH-PGC-1α) to screen for compounds that elevate the expression of PGC-1α in the presence of PARIS (Fig. 1A). SH-PGC-1α cells were responsive to daidzein (26), a PGC-1α inducer (fig. S1, A and B). Minimal variations within and between days and between plates were observed (fig. S1C). The Z′ factors were within the 0.5 to 1 range, and dimethyl sulfoxide (DMSO) up to 0.5% had no effect on assay sensitivity and cell viability (fig. S1, B and E, and table S1).
Fig. 1. Identification of farnesol as a PGC-1α inducer.
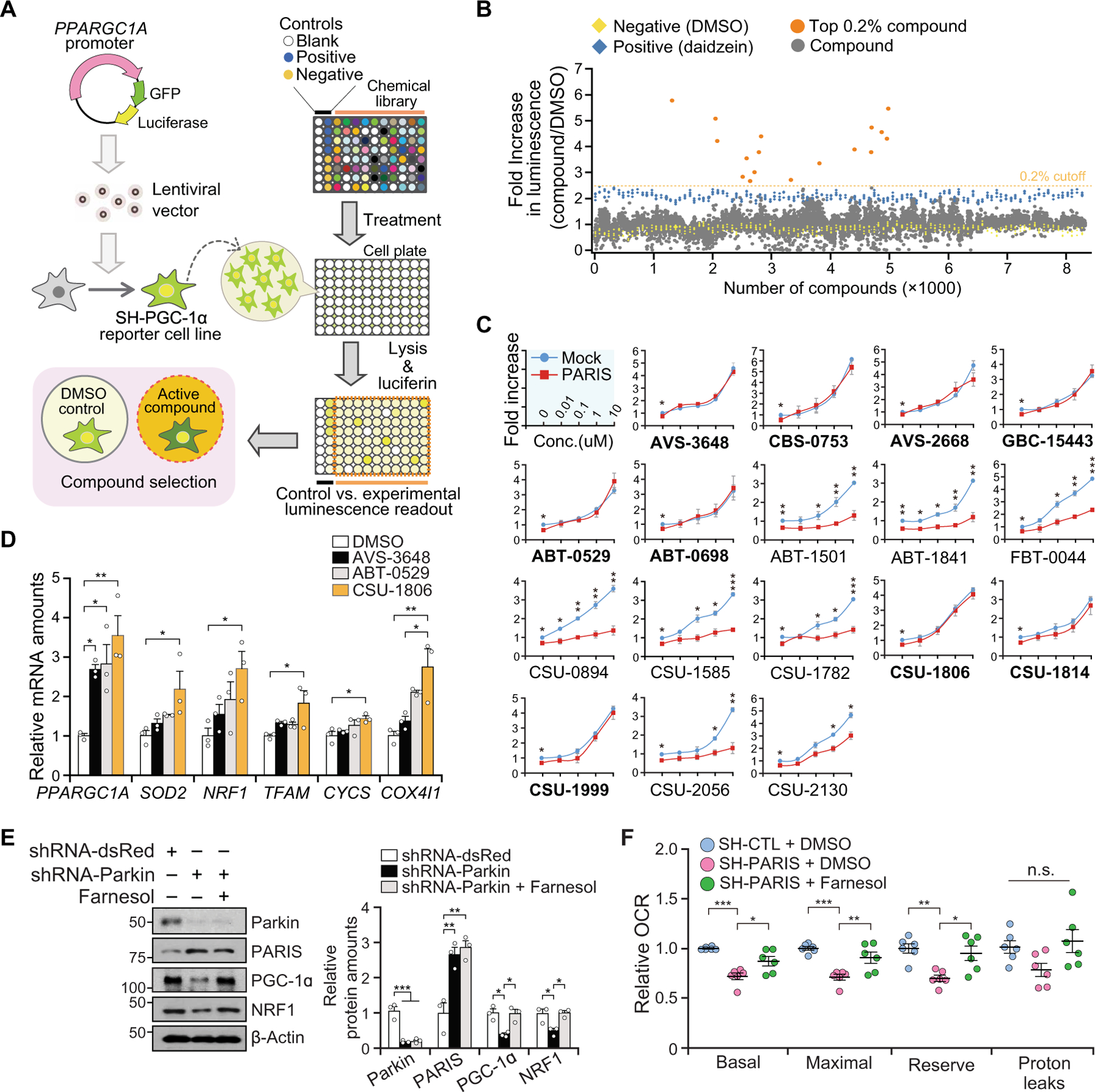
(A) Experimental illustration of compound screening. A reporter cell line (SH-PGC-1α) expressing GFP and luciferase under control of PPARGC1A promoter was used to screen for compounds that induce PPARGC1A promoter activity. (B) Promoter activity of PPARGC1A by luciferase assay. Dotted line indicates 2.5-fold activation. All readouts are displayed with color-coded shapes: yellow diamond (DMSO), blue diamond (daidzein), and gray/orange dot (experimental compounds). (C) The PPARGC1A promoter activity of the top 0.2% compounds (17 compounds) [orange dots in (B)] in the setting of overexpression of PARIS. Bold-named (nine) compounds dose-dependently increase PPARGC1A promoter activity in the presence of PARIS overexpression, n = 3 independent experiments. (D) Relative mRNA amounts of PPARGC1A and its target genes normalized to GAPDH in SH-SY5Y cells treated with DMSO, 10 µM AVS-3648, ABT-0529, or CSU-1806 (farnesol); n = 3 independent experiments. PPARGC1A, peroxisome proliferator-activated receptor γ coactivator-1α; SOD2, superoxide dismutase 2; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial transcription factor A; CYCS, cytochrome c, somatic; COX4I1, cytochrome c oxidase 4. (E) Immunoblot analysis of parkin, PARIS, PGC-1α, and NRF1 in shRNA-parkin knockdown cells ± farnesol (10 µM, 48 hours) treatment normalized to β-actin; n = 3 independent experiments. Quantitation of the immunoblots in the right panel. (F) Relative oxygen consumption rate (OCR) measured by microplate-based respirometry in stable SH-SY5Y cells overexpressing PARIS (SH-PARIS) ± farnesol (10 µM, 24 hours) treatment as compared to control SH-SY5Y cells (SH-CTL), n = 6 per group. n.s., not significant. Data are expressed as means ± SEM. Statistical significance was evaluated by applying an unpaired two-tailed Student t test (C) or using one-way ANOVA with Tukey post hoc test (D to F). Differences are considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
We assessed luciferase activity of SH-PGC-1α cells in the presence of 8320 compounds (10 µM) at 48 hours and normalized to DMSO (Fig. 1B). One hundred twenty-eight compounds increased luciferase activity 1.5-fold and were rescreened in triplicate (fig. S1D). Seventeen compounds increased luciferase activity 2.5-fold or greater without cellular toxicity at 10 µM (fig. S1, D and E, and table S2). To identify the chemicals that could overcome the suppression of PGC-1α by PARIS, we treated PARIS-overexpressing SH-SY5Y cells with these 17 compounds. Nine of the 17 compounds prevented the suppression of PGC-1α in the presence of PARIS and activated the PPARGC1A promoter in a dose-dependent manner (Fig. 1C and fig. S1F). PGC-1α protein was also increased by these nine compounds (fig. S1G). AVS-3648, ABT-0529, and CSU-1806 (farnesol, 3,7,11-trimethyl-2,6,10-dodecatriene-1-ol) showed a high likelihood of blood-brain barrier (BBB) permeability and no interaction with p-glycoprotein transporter as assessed by StarDrop (www.optibrium.com/stardrop/) (table S3).
PPARGC1A mRNA expression was increased in SH-SY5Y cell treated with AVS-3648, ABT-0529, or farnesol (CSU-1806) (Fig. 1D). Because real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) revealed that farnesol increases mRNA amounts of PGC-1α–associated genes including manganese superoxide dismutase (SOD2), nuclear respiratory factor 1 (NRF1), mitochondrial transcription factor A (TFAM), cytochrome C (CYCS), and cytochrome c oxidase (COX4I1) at a given concentration, we focused our attention on further characterization of farnesol (Fig. 1D). These same PGC-1α–associated genes involved in oxidant metabolism, mitochondrial biogenesis, and oxidative phosphorylation were identified as potential PARIS–PGC-1α pathway target genes in PD models (12).
Lentiviral short hairpin RNA (shRNA)–parkin–mediated knockdown of parkin led to a 2.7-fold increase in the amount of PARIS and a 61% decrease of PGC-1α (12, 13) (Fig. 1E). In the setting of knockdown of parkin, farnesol also restored the expression of PGC-1α without affecting the expression of parkin and PARIS. Real-time qRT-PCR revealed that farnesol rescued the mRNA reduction of PPARGC1A, NRF1, TFAM, and CYCS in the setting of parkin knockdown (fig. S1H). Farnesol treatment also restored mitochondrial deficits caused by elevated PARIS (12, 13) because a 28% reduction in basal, a 29% reduction in maximal, and a 30% reduction in reserve mitochondrial respiration as assessed by a XF-24 analyzer (Seahorse Bioscience) were restored by farnesol (Fig. 1F). To confirm that the increase of PGC-1α and NRF1 by farnesol is PARIS dependent, farnesol was administered to SH-SY5Y cells after shRNA knockdown of PARIS. Knockdown of PARIS led to a twofold increase of PGC-1α and NRF1 (12, 13), and it failed to enhance their expression by farnesol (fig. S1I). To further confirm whether PARIS down-regulation counteracts the effects of farnesol on the up-regulation of PGC-1α and related genes in the setting of parkin down-regulation, we investigated the mRNA amounts of PPARGC1A, SOD2, NRF1, TFAM, CYCS, and COX4I1 in SH-SY5Y cells transfected with small interfering RNA (siRNA)–Parkin ± siRNA-PARIS along with farnesol. Farnesol treatment restored the mRNA expression of PPARGC1A, SOD2, NRF1, TFAM, CYCS, and COX4I1 that were repressed in a PARIS-dependent manner by parkin knockdown (fig. S1J). This beneficial effect by farnesol treatment was not observed in the experimental groups with both parkin and PARIS knockdown (fig. S1J), indicating that farnesol-mediated transcriptional regulation of these genes in the absence of parkin is PARIS dependent.
To determine whether exogenous farnesol penetrates the central nervous system, we fed mice with either a control chow diet (AIN-76A diet, Research diet) or a farnesol diet [0.5% (w/w) of trans-farnesol in AIN-76A diet, Research diet] for 7 days, and we measured the concentration of farnesol in the brain and plasma by mass spectrometry (fig. S1K). Plasma concentrations of farnesol increased to 113 ng/ml in plasma from nondetectable concentrations in farnesol-fed mice versus control diet–fed mice. Farnesol concentrations in the brain increased by 37% to farnesol (155 ng/ml) in farnesol-fed mice versus control diet–fed mice. In farnesol-fed mice, PGC-1α and NRF1 amounts increased in the SN without a change of PARIS (fig. S1L).
PARIS can be farnesylated
Farnesyltransferase (FTase) uses farnesol and its intermediates for protein farnesylation via modification on cysteine residues at C termini with a CaaX motif (27). PARIS has a CaaX motif near the C terminus of human PARIS at 631CGLS634 (Fig. 2A). [3H]–Farnesyl pyrophosphate (FPP)–treated SH-SY5Y cells were transfected with Flag-tagged PARIS and either siRNA-control or siRNA to FTase α followed by Flag-tagged PARIS immunoprecipitation. Autoradiography revealed that PARIS was farnesylated, whereas knockdown of FTase α eliminated the farnesylation of PARIS (Fig. 2B). Immunoblot analysis with an α-farnesyl antibody confirmed the farnesylation of PARIS, and knockdown of FTase α reduced the farnesylation of PARIS in [3H]-FPP–treated SH-SY5Y cells (Fig. 2B). To determine the endogenous amount of PARIS farnesylation as compared to that of HRas, a well-known farnesylation substrate (28), we incubated SH-SY5Y lysate with a biotin-functionalized geranyl pyrophosphate analogue (Biotin-GPP) and glutathione S-transferase (GST)–tagged FTaseW102T/Y154T in an in vitro farnesylation (IVF) assay. Farnesylated proteins were purified by streptavidin-conjugated beads and were subjected to immunoblot analysis for monitoring relative amounts of farnesylated PARIS, and HRas normalized to their input expression, indicating that the amount of PARIS farnesylation was approximately 44% of that of HRas (fig. S2A).
Fig. 2. PARIS is farnesylated.
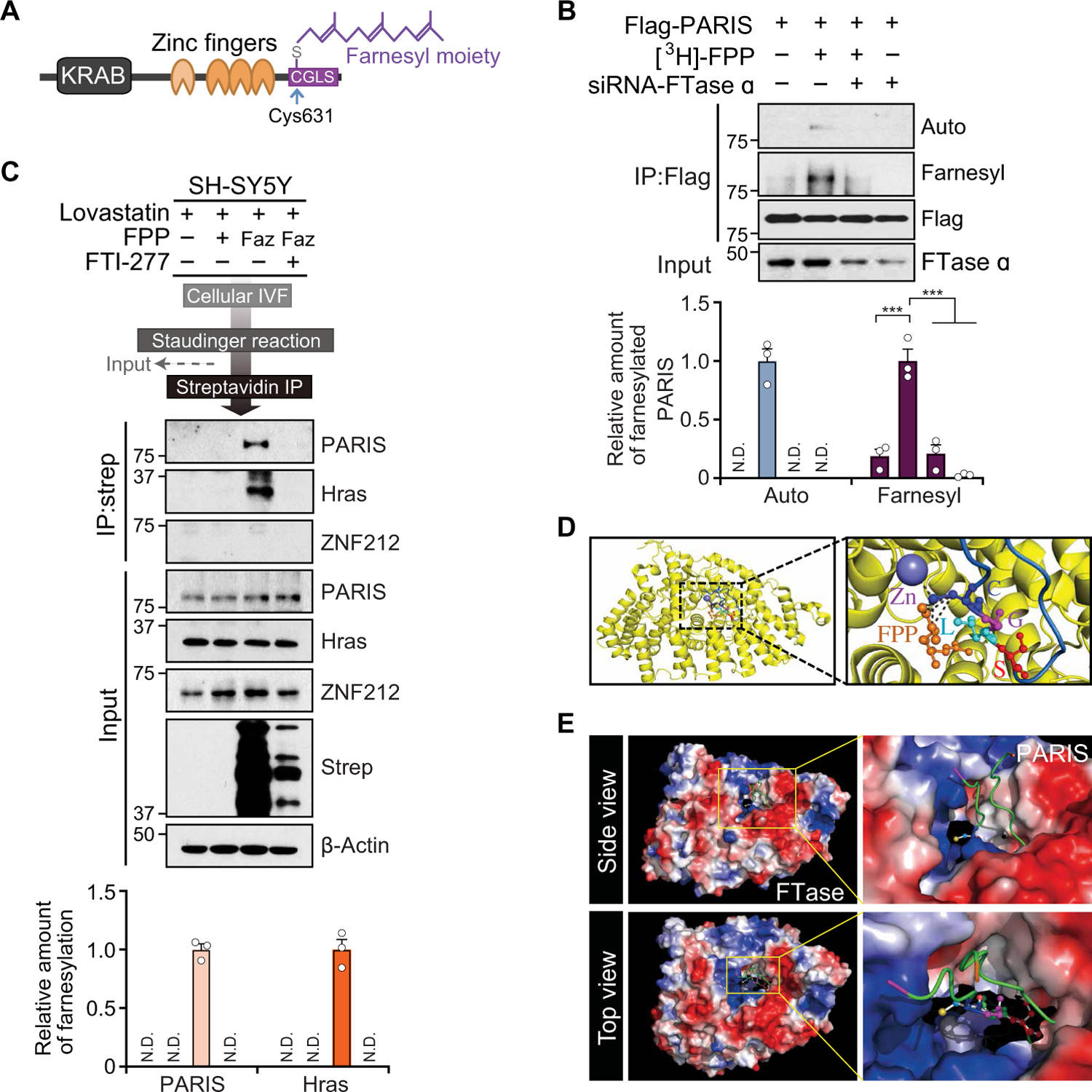
(A) Illustration showing domain and putative farnesylation site of PARIS with the farnesyl moiety on C631 of PARIS. (B) Metabolic farnesylation assay of SH-SY5Y cells expressing Flag-tagged PARIS and siRNΑ-farnesyl transferase (FTase) α incubated with [3H]-farnesyl pyrophosphate (FPP). Farnesylation of PARIS was detected by autoradiography (Auto) or via an antibody that recognizes farnesylated proteins (Farnesyl). PARIS farnesylation was normalized to the flag signal, n = 3 independent experiments. Quantitation of the immunoblots in the bottom. (C) Detection of endogenous PARIS and HRas farnesylation by tagging-via-substrate (TAS) technique. ZNF212, negative control; Faz, azido farnesyl alcohol (F-azide-OH); Strep, streptavidin. Quantitation of the immunoblots in the bottom. (D) Superposition of PARIS C terminus with the crystal structure of FTase in complex with zinc ion (Zn) and CVLS-FPP peptide (Protein Data Bank: 2H6F). Close-up view (right) of the model of FTase in complex with Zn, PARIS C-terminal peptide, and FPP substrate. The interactions between PARIS C631, FPP, and Zn are indicated by black dashed lines. (E) Solvent-accessible surface and electrostatic surface potential showing FTase active CaaX-FPP binding site. The yellow boxes highlight the cleft where CaaX-FPP binds. N.D., not detected. Data are expressed as means ± SEM. Statistical significance was evaluated via one-way ANOVA with Tukey post hoc test (B). Differences are considered significant when P < 0.05. ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
The lysates of SH-SY5Y cells transfected with Flag-tagged PARIS were incubated with Biotin-GPP and the recombinant GST-tagged FTaseW102T/Y154Y mutant, which uses Biotin-GPP as a lipid donor (29). After immunoprecipitation of Flag-tagged PARIS, we detected Biotin-GPP–conjugated PARIS by horseradish peroxidase–conjugated streptavidin, indicating that PARIS was a substrate for FTase prenylation (fig. S2B). In addition, the immunoprecipitate of Flag-tagged PARIS from SH-SY5Y cells transfected with either siRNA-control or siRNA-FTase α was mixed with 3,7-dimethyl-8-(7-nitro-benzo[1,2,5]oxadiazol-4-ylamino)-octa-2,6-diene-1-pyrophosphate (NBD-GPP), a Cy3 fluorescent analog of FPP for IVF. We observed NBD-GPP fluorescent signal in the immunoprecipitated Flag-tagged PARIS, demonstrating that endogenous FTase α coimmunoprecipitated with Flag-tagged PARIS and the immunoprecipitated FTase α successfully transferred NBD-GPP to PARIS (fig. S2C). To determine whether FTase directly farnesylates PARIS, we incubated recombinant GST-PARIS with FTase subunits (α and β) and FPP in the presence and absence of the FTase inhibitor, FTI-277 (30). GST-PARIS was farnesylated by FTase, which was abolished by the addition of FTI-277 (fig. S2D). Moreover, FTase α knockdown or FTI-277 inhibition to SH-SY5Y cells strongly prevented the farnesylation of PARIS by farnesol, leading to the inhibition of farnesol-mediated PGC-1α induction, confirming that farnesylation of PARIS requires FTase (fig. S2E). In a similar setting, we also monitored the mRNA amount of PGC-1α and its targets in SH-SY5Y cells transfected with siRNA–FTase α ± siRNA-parkin to address whether the inhibition of FTase also affects the response to farnesol in parkin knockdown cells. Farnesol treatment led to an up-regulation of PPARGC1A, SOD2, NRF1, TFAM, CYCS, and COX4I1 in siRNA control cells. Farnesol also increased these genes in the setting of parkin knockdown, whereas farnesol did not rescue their expression in the FTase α knockdown cells (fig. S2F). These findings show that the rescue of PGC-1α and PGC-1α–associated genes by farnesol in parkin knockdown requires FTase in SH-SY5Y cells. To ascertain the role of farnesylation of endogenous PARIS in vivo, we used conditional FTase β subunit KO (FNTβ-KO) mice that were generated by intranigral stereotaxic injection of adeno-associated virus (AAV)–GFP-Cre into the SN of floxed FTase β (FNTβ flox/flox) mice. We monitored the amount of PARIS farnesylation in the SN of FNTβ-KO. The farnesylation of PARIS is blocked in the SN of conditional FNTβ-KO mice, leading to PGC-1α reduction (fig.S2, G to I).
Next, we applied the tagging-via-substrate technique to assess whether PARIS can be farnesylated by endogenous farnesylation machinery (31). To produce azido-farnesylated proteins, we treated SH-SY5Y cells with lovastatin, azido farnesyl alcohol (Faz, farnesyl substrate), and FTI-277 for 24 hours. The azido-farnesyl moiety was linked with phosphine–polyethylene glycol 3–biotin (Staudinger reaction), followed by incubation with streptavidin-conjugated Dynabeads to isolate azido-farnesylated proteins. Metabolic incorporation of azido farnesyl moiety was found in endogenous PARIS and HRas (as a positive control), but not in ZNF212 (a PARIS homolog as a negative control), suggesting that PARIS is an endogenous substrate for farnesylation (Fig. 2C). To ascertain whether farnesol leads to enhanced farnesylation of PARIS, SH-SY5Y cells were transfected with Flag-tagged PARIS and subjected to farnesol treatment for 48 hours followed by immunoprecipitation (fig. S3A). Farnesol increased PARIS farnesylation in a dose-dependent manner. Accompanying the increase in PARIS farnesylation was an increase in PGC-1α. To ascertain the specificity of farnesol, we used geraniol or geranylgeraniol because they have similar structures to farnesol. Geraniol or geranylgeraniol had no effect on PGC-1α expression (fig. S3B). To determine whether farnesol increases PARIS farnesylation in vivo, PARIS was immunoprecipitated from the SN of farnesol-fed mice. Immunoblot analysis showed that endogenous PARIS was farnesylated, and farnesol diet enhanced the amount of farnesylated PARIS along with the up-regulation of PGC-1α (fig. S3C).
Superposition of FTase in the complex with PARIS C terminus, zinc ion, and FPP shows cysteine 631 (C631) of PARIS coordinated with zinc and FPP in the catalytic pocket of FTase (Fig. 2D). Solvent-accessible surface and electrostatic surface potential indicate that the CGLS motif of PARIS C terminus sits in the cleft where FTase binds to CaaX and FPP (Fig. 2E), suggesting that the PARIS C-terminal loop containing CGLS fits in the FTase binding site. Prior investigations indicate that farnesylation occurs on the C terminus of CaaX motifs (32, 33). Because farnesylation of PARIS occurs on an atypical CaaX motif, we used biochemical approaches and three-dimensional (3D) structural modeling to compare that CGLS motif of PARIS to CVLS motif of HRas, a prototypical farnesylation substrate. 3D structure prediction using the CPHmodels-3.2 Server (www.cbs.dtu.dk/services/CPHmodels/) demonstrated that the CaaX motif of PARIS (CGLS) was incorporated into the catalytic pocket of FTase similar to the CaaX motif of HRas (CVLS) (fig. S4A). Moreover, the C-terminal Flag-tagged PARIS also successfully inserted into the catalytic pocket of FTase (fig. S4B). To test that the tag does not interfere with PARIS farnesylation or activity, we assessed the amounts of PGC-1α, NRF1, and PARIS farnesylation after expression of an N-terminal or C-terminal Flag-tagged PARIS in SH-SY5Y cells (fig. S4C). Both N-terminal and C-terminal Flag-tagged PARIS wild-type (WT) were farnesylated, and both repress the expression of PGC-1α and NRF1, whereas the farnesylation of Flag-tagged PARIS under basal condition was abolished for C631S mutant (fig. S4C).
Farnesol increases PARIS farnesylation on C631
To confirm that PARIS is farnesylated and identify the farnesylation site, we applied tandem mass spectrometric (MS) analysis. First, we optimized the MS procedure using a synthetic farnesylated PARIS C-terminal peptide to improve the detectability of the hydrophobic farnesylated peptides (see Materials and Methods). MS specimens included recombinant PARIS WT and PARIS C631S protein that were subject to in vitro farnesylation and immunoprecipitation of PARIS from SH-SY5Y cells transfected with Flag-tagged PARIS and farnesol treatment. We observed that C-terminal peptides containing both the CaaX motif of nonfarnesylated PARIS WT and PARIS C631S were detected at similar retention times of ~69 min in the C18 analytical column (fig. S5A). However, we observed a shift of the retention time to around 85 min in farnesylated PARIS WT but not in PARIS C631S that was subjected to in vitro farnesylation (fig. S5, A and B). The MS1 mass was shifted as much as 204.187801 Da when it was farnesylated. Because the PARIS C-terminal peptide has a methionine at the end, the oxidized and nonoxidized forms of peptides were observed, and the mass difference between expected and observed mass was within 5 ppm, proving the accuracy of these MS experiments (fig. S5). We did not observe farnesylation of nonfarnesylated control samples nor the PARIS C631S samples (fig. S5, A and B). Furthermore, we validated the farnesylated peptides by comparing their MS2 spectra to the one acquired from the synthetic farnesylated peptide, revealing that the fragmentation pattern was almost identical. The MS2 spectra also validated that the farnesylation site was on the C631 residue (fig. S5C).
To confirm that C631 is a physiologically relevant site of PARIS farnesylation, we performed an in vitro farnesylation assay, showing that a synthesized C-terminal peptide of PARIS was robustly farnesylated (Fig. 3A). Flag-PARIS WT or Flag-PARIS C631S mutant was immunoprecipitated after incubation with or without farnesol. An increased amount of farnesylation by farnesol was found for the immunoprecipitated PARIS WT but not for C631S (Fig. 3B). The minimal farnesyl signal in the PARIS C631S mutant is due to coimmunoprecipitated endogenous PARIS WT. Accompanying the farnesylation of PARIS WT by farnesol was an up-regulation of PGC-1α, whereas farnesol had no effect on PGC-1α expression in the setting of the PARIS C631S mutant (Fig. 3B).
Fig. 3. PARIS can be farnesylated on C631 and is promoted by farnesol.
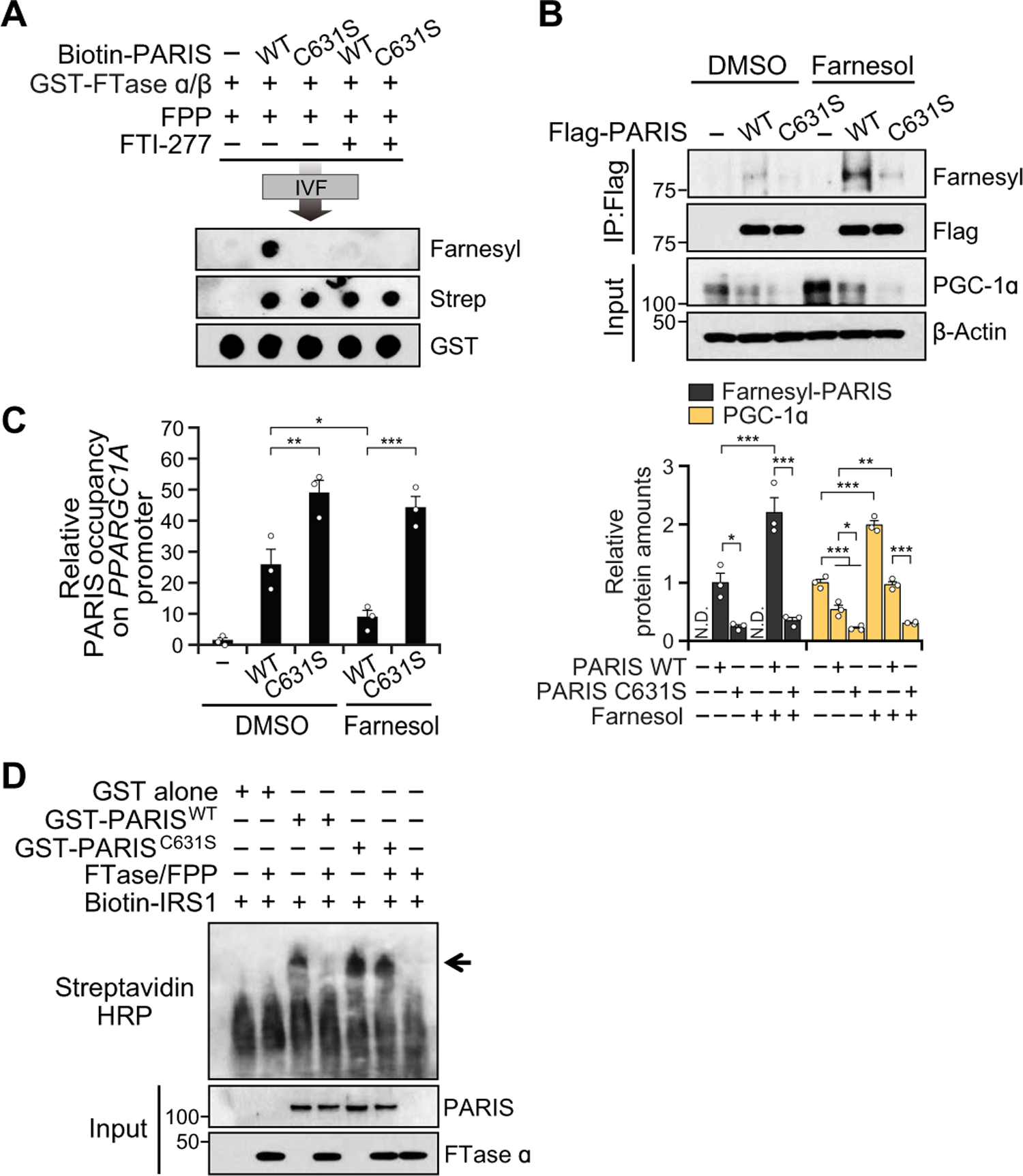
(A) In vitro farnesylation (IVF) assay with biotin-labeled PARIS WT peptide (VTDWTC631GLSVLGPTDGGDM) or C631S mutant peptide. (B) Representative immunoblot image of farnesylated PARIS and PGC-1α amounts in SH-SY5Y cells transfected with PARIS WT or C631S mutant with DMSO control or farnesol (10 µM), n = 3 independent experiments. Quantitation of farnesylated PARIS and PGC-1α amounts normalized to immunoprecipitated Flag-PARIS and β-actin, respectively (bottom). (C) Real-time qPCR of ChIP eluate normalized to input chromatin, n = 3 independent experiments. (D) Electrophoretic mobility shift assay (EMSA) of in vitro farnesylated GST, GST-PARIS WT, or C631S incubated with biotin-labeled insulin responsive sequence 1 (Biotin-IRS1) motif of PPARGC1A promoter. The arrow indicates GST-PARIS binding with IRS1. Data = means ± SEM. Statistical significance was determined via ANOVA test with Tukey post hoc (C, one-way; B, two-way). Differences were considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
A chromatin immunoprecipitation (ChIP) assay of the IRS motif of the PPARGC1A promoter from SH-SY5Y cells transfected with GFP-tagged PARIS WT or GFP-tagged PARIS C631S revealed that farnesol reduced the chromatin occupancy of PARIS WT but not PARIS C631S, on the PPARGC1A promoter (Fig. 3C and fig. S6A). We used subcellular fractionation to monitor the expression of PARIS in cytosolic, membrane, soluble nuclear, and chromatin-bound nuclear fractions (fig. S6, B to D). Consistent with the ChIP assay, farnesol abolished the expression of chromatin-bound PARIS WT but not PARIS C631S, whereas there was no change in the distribution of PARIS in the cytoplasmic and membrane fractions. Accompanying the farnesylation of PARIS WT by farnesol treatment was an increase of PPARGC1A promoter activity that was not observed with the PARIS C631S mutant (fig. S6E). An electrophoretic mobility shift assay (EMSA) showed that in vitro farnesylated GST-tagged PARIS WT failed to bind to the IRS of PPARGC1A promoter, whereas naive GST-tagged PARIS WT and C631S formed DNA-protein complexes (Fig. 3D). These results together indicate that PARIS is farnesylated, and farnesylation of PARIS eliminates its DNA binding affinity, thereby preventing its suppression of PPARGC1A. Moreover, we found that PARIS overexpression led to a decrease in the relative mitochondrial copy number, increased H2O2 concentration, and a reduction in adenosine triphosphate (ATP) concentration, which was restored by farnesol treatment (fig. S6, F to H). Farnesol had no effect on these indices in SH-SY5Y cells transfected with PARIS C631S. Thus, farnesylation of PARIS has functional consequences on PGC-1α activity.
Farnesol prevents DA neuronal loss in PARIS accumulating models
We created transgenic lines overexpressing C-terminal Flag-tagged human PARIS under the control of a tetracycline-responsive regulator (TetP-PARIS-Flag) to determine whether farnesol can protect dopaminergic neurons against increased PARIS and decreased PGC-1α expression in vivo (fig. S7A). Three high copy number male founder mice from 29 founders were crossed with the camodulin kinase IIα–tetracycline–controlled transactivator (CamKIIα-tTA) transgenic mice (fig. S7B) (34). No mice expressing both CamKIIα-tTA and TetP-PARIS-Flag were born. Thus, dams were maintained on doxycycline to suppress PARIS expression, and pups were maintained on doxycycline diet for 3 weeks after birth (fig. S7C). CamK-PARIS mice expressing both CamKIIα-tTA and TetP-PARIS-Flag were identified by PCR (fig. S7D). The doxycycline diet was withdrawn at 3 weeks of age. We detected the overexpression of PARIS at 2 weeks after withdrawal in the olfactory bulb (OB), cortex (CTX), striatum (STR), ventral midbrain (VM), cerebellum, and brain stem of CamK-PARIS mice compared to littermate controls (CamKIIα-tTA or TetP-PARIS-Flag alone) (fig. S7E). PARIS was overexpressed 7.1- to 13.7-fold in the OB, CTX, and STR in CamK-PARIS line #158 (fig. S7E). In the VM, which contains the SN, PARIS was expressed threefold, similar to the enhanced expression of PARIS in the adult conditional parkin KO mice, human postmortem brain from sporadic patients with PD (12), and adult conditional knockdown PINK1 mice (10). Overexpression of PARIS was tetracycline responsive because doxycycline administration completely blocked the up-regulation of PARIS (fig. S7, F and G). In addition, there was no alteration of endogenous PARIS in CamK-tTA or CamK-PARIS mice fed with the doxycycline diet (fig. S7G). Two independent CamK-PARIS lines (line #121 and line #158) expressed PARIS at equivalent amounts in the CTX and VM (fig. S7H). All three lines (#121, #158, and #179) exhibited clasping, seizures, and immobility and died 3 weeks after induction of unknown causes (~6 weeks of age) (fig. S7I and movie S1). The CamK-PARIS mice had reduced body weight (fig. S7J) and overexpressed PARIS in the intestine, and the mice had enlarged stomachs, suggesting impaired gastrointestinal transport (fig. S7, K and L). These abnormalities may contribute to their weight loss and premature death. No other abnormalities were identified in the CamK-PARIS mice.
Flag (PARIS) immunostaining colocalized with tyrosine hydroxylase (TH)–positive neurons throughout the SN (fig. S7M). In addition, we confirmed the activity of the CamKIIα promoter in DA neurons by crossing the CamKIIα-tTA mice with TetP-LacZ reporter mice (fig. S7N) (34). The CamKIIα promoter drove expression in the hippocampus, forebrain, VM, and SN (fig. S7, M and N). At 6 weeks of age, unbiased stereology counting of TH- and Nissl-positive neurons revealed a greater than 35% reduction of DA neurons and a 45% reduction in striatal TH immunoreactivity in CamK-PARIS line #158 (fig. S7, O and P). In CamK-PARIS line #121, there was an approximate 26% reduction of DA neurons (fig. S7Q). In the CamK-PARIS line #158, there was a reduction of striatal DA and its metabolites and increased DA catabolism as determined by high-performance liquid chromatography (HPLC) (fig. S7, R to T). Norepinephrine and epinephrine concentrations were unchanged (fig. S7U). There was a delay in time for CamK-PARIS mice to descend on the pole test, a sensitive measure of DA function at 5 to 6 weeks of age (fig. S7V and movie S2). There was no neuronal loss in the CTX of CamK-PARIS mice despite the high concentration of PARIS (fig. S7, E and W).
CamK-PARIS mice were fed a farnesol diet 3 days before doxycycline withdrawal and were maintained for the duration of the study. Separate cohorts of CamK-PARIS mice were maintained on normal mouse chow diet before doxycycline withdrawal. Control mice were subjected to the same dietary regimen. Farnesol concentrations in the brain were increased by 52 and 50% in farnesol-fed control and farnesol-fed CamK-PARIS mice versus control diet–fed mice, respectively (Fig. 4A). Doxycycline administration did not interfere with farnesol absorption and brain distribution (Fig. 4A). Farnesol treatment prevented the loss of DA neurons in the SN of CamK-PARIS mice, and there was a complete behavioral rescue as assessed by the pole test in CamK-PARIS mice (Fig. 4, B and C, and movie S2). Farnesol also delayed the premature lethality of the CamK-PARIS mice (Fig. 4D). CamK-PARIS mice on normal mouse chow exhibited an approximate 55% reduction in PGC-1α expression and a 40% reduction in NRF1 expression that was blocked by farnesol treatment (Fig. 4E). Farnesol treatment of control mice increased PGC-1α and NRF1 expression (Fig. 4E). Farnesol treatment led to enhanced farnesylation of PARIS as detected by immunoprecipitation with Flag and Western blot with an antibody to farnesyl and was accompanied by the restoration of PGC-1α and NRF1 amounts in CamK-PARIS mice (Fig. 4E). mRNA expression of Ppargc1a and Nrf1 was up-regulated in farnesol-fed mice (Fig. 4F). Reduced mRNA expression of Tfam and Cycs in the CamK-PARIS mice was restored by farnesol (Fig. 4F). Coimmunostaining of PGC-1α and TH indicated that farnesol restored PGC-1α immunoreactivity in DA neurons in CamK-PARIS mice (fig. S8, A and B). We measured the amount of protein carbonylation, a marker of oxidative stress as a potential functional readout of PGC-1α activity in vivo. There was an increase in the amount of protein carbonylation in the SN of CamK-PARIS mice (fig. S8C) that was reduced by farnesol treatment. In addition, farnesol reduced the increment of H2O2 concentration in the SN of CamK-PARIS mice (fig. S8D).
Fig. 4. Farnesol prevents DA neuron loss in CamK-PARIS transgenic mice.
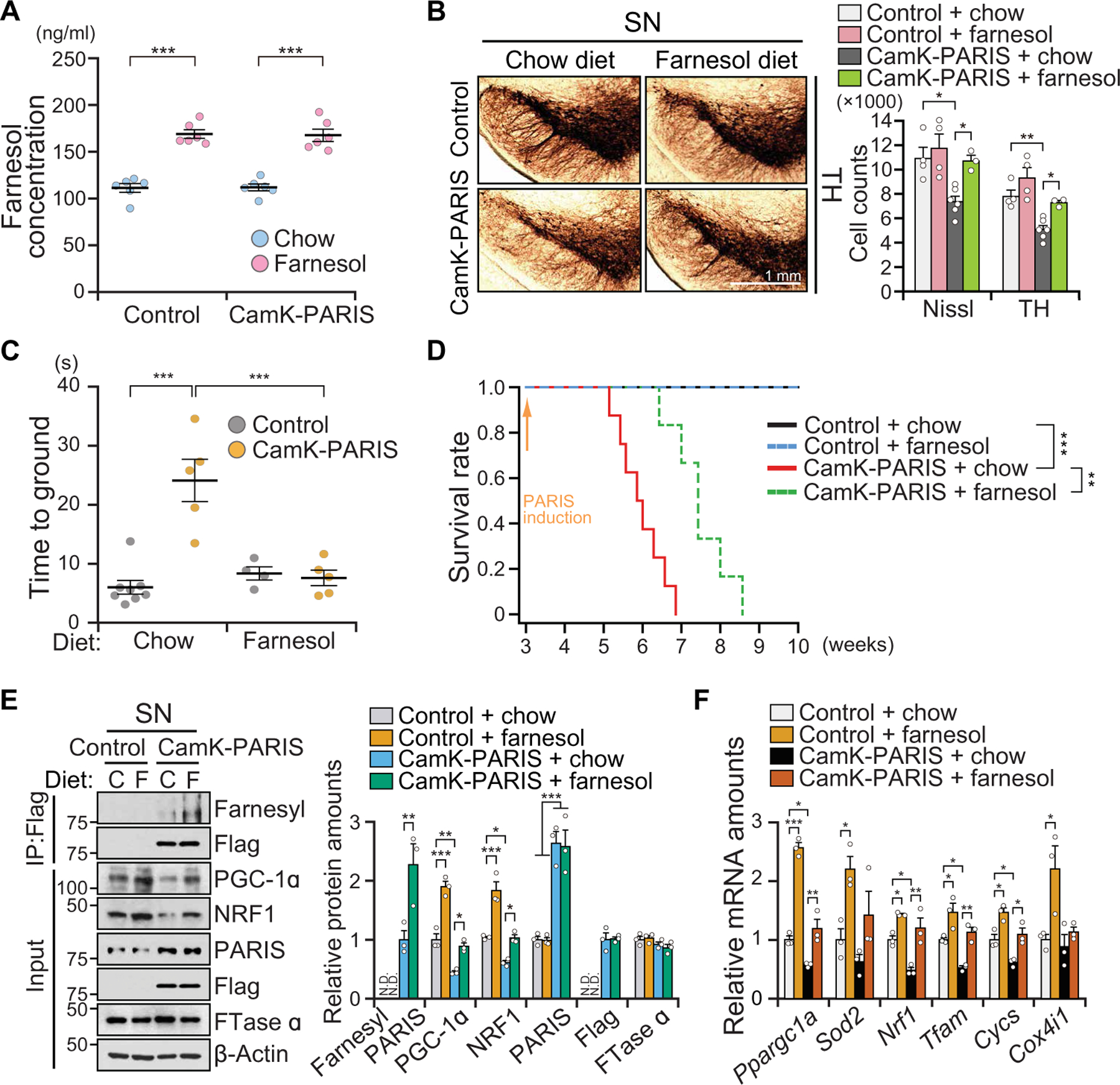
(A) Concentration of farnesol in the brain of control and CamK-PARIS ± farnesol diet (for 1 week) at 3 weeks of age, n = 6 mice per group. Chow diet (AIN-76A diet, Research diet, NJ) and farnesol diet [0.5% (w/w) of trans-farnesol in AIN-76A diet, Research diet, NJ]. (B) Representative TH immunohistochemistry of the SN of 6-week-old CamK-PARIS mice and age-matched littermate controls fed with chow or farnesol diet. PARIS is induced for 3 weeks by doxycycline withdrawal at 3 weeks of age. The farnesol diet was initiated 3 days before doxycycline withdrawal. Stereological assessment of TH or Nissl-positive neurons (right). Control ± farnesol diet, n = 4 mice per group; CamK-PARIS + chow diet, n = 6 mice; CamK-PARIS + farnesol diet, n = 3 mice. (C) Assessment of dopamine-related motor performance by pole test for CamK-PARIS and littermate control mice fed with chow or farnesol diet. PARIS was induced at 3 weeks of age, and behavior is assessed at 5 weeks of age. Control + chow diet, n = 8 mice; CamK-PARIS + chow diet, n = 5 mice; control + farnesol diet, n = 4 mice; CamK-PARIS + farnesol diet, n = 5 mice per group. (D) Survival rate of control and CamK-PARIS fed with a chow or farnesol diet. PARIS induction was started at 3 weeks of age by withdrawing Dox diet (control + chow, n = 8 mice; control + farnesol, n = 6 mice; CamK-PARIS + chow, n = 8 mice; CamK-PARIS + farnesol, n = 6 mice). (E) Representative immunoblot image of farnesylated PARIS, PGC-1α, NRF1, PARIS, and FTase α in the SN of 5- to 6-week-old CamK-PARIS mice and age-matched littermate controls (control) fed with chow (C) or farnesol (F) diet; n = 3 mice per group. Quantitation of the immunoblots in the right panel normalized to immunoprecipitated Flag or β-actin. (F) Real-time qRT-PCR analysis of Ppargc1a and its target genes in the SN of 5- to 6-week-old CamK-PARIS mice or age-matched littermate control mice fed with chow or farnesol diet normalized to GAPDH, n = 3 mice per group. Data = means ± SEM. Statistical significance was determined by two-way ANOVA test with Tukey post hoc analysis (A to C, E, and F) or log-rank (Mantel-Cox) test (D). Differences were considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
To confirm that DA neuronal death in CamK-PARIS mice was due to decrements in PGC-1α, TetP-PARIS-Flag mice were crossbred with TetP-PGC-1α and CamKIIα-tTA driver mice, which simultaneously express PGC-1α and PARIS (dCamK-PARIS/PGC-1α) (fig. S8, E and F). Overexpression of PGC-1α prevented PARIS-induced loss of DA neurons in CamK-PARIS mice (fig. S8, G and H), similar to the prevention of the loss of DA neurons afforded by farnesol treatment. These results together suggest that the DA cell loss induced by PARIS accumulation is mediated by PGC-1α repression.
To determine whether the farnesylation of PARIS is required for the protective effects of farnesol in vivo, AAV-PARIS WT, AAV-PARIS C631S, or AAV-GFP was stereotaxically injected into the SN of C57BL/6N mice (fig. S9A). Mice were either fed a normal mouse chow or a farnesol diet for 1 week before stereotaxic injection of AAV vector (fig. S9A). We confirmed equivalent expression of transduced AAV vectors (fig. S9B). Both AAV-PARIS WT and AAV-PARIS C631S led to a greater than threefold increase in the expression of PARIS, and farnesol did not affect the expression of PARIS WT or PARIS C631S (Fig. 5A). We observed a greater amount of farnesylated PARIS in AAV-PARIS WT–injected mice as compared to AAV-PARIS C631S 3 weeks after AAV injection (Fig. 5A). Detection of farnesylated PARIS in AAV-PARIS C631S–injected mice resulted from endogenous PARIS. Both AAV-PARIS WT and AAV-PARIS C631S led to loss of DA neurons as assessed by TH and Nissl stereology (Fig. 5B). Farnesol treatment completely prevented the loss of DA neurons in the AAV-PARIS WT mice similar to its protection in CamK-PARIS transgenic mice, but it had no effect on DA neuronal survival in the AAV-PARIS C631S mice (Fig. 5B). Because the AAV-PARIS injections were unilateral, we performed amphetamine-induced turning test to monitor dopaminergic-dependent behavior (35). Accompanying the dopaminergic neuronal rescue by farnesol was a restoration of amphetamine-induced rotation behavior in the AAV-PARIS WT, whereas there was no rescue in AAV-PARIS C631S mice (Fig. 5C). Thus, the behavioral and DA neuron rescue by farnesol is mediated by PARIS farnesylation on C631.
Fig. 5. Farnesol’s protection against PARIS-induced DA degeneration requires PARIS farnesylation.
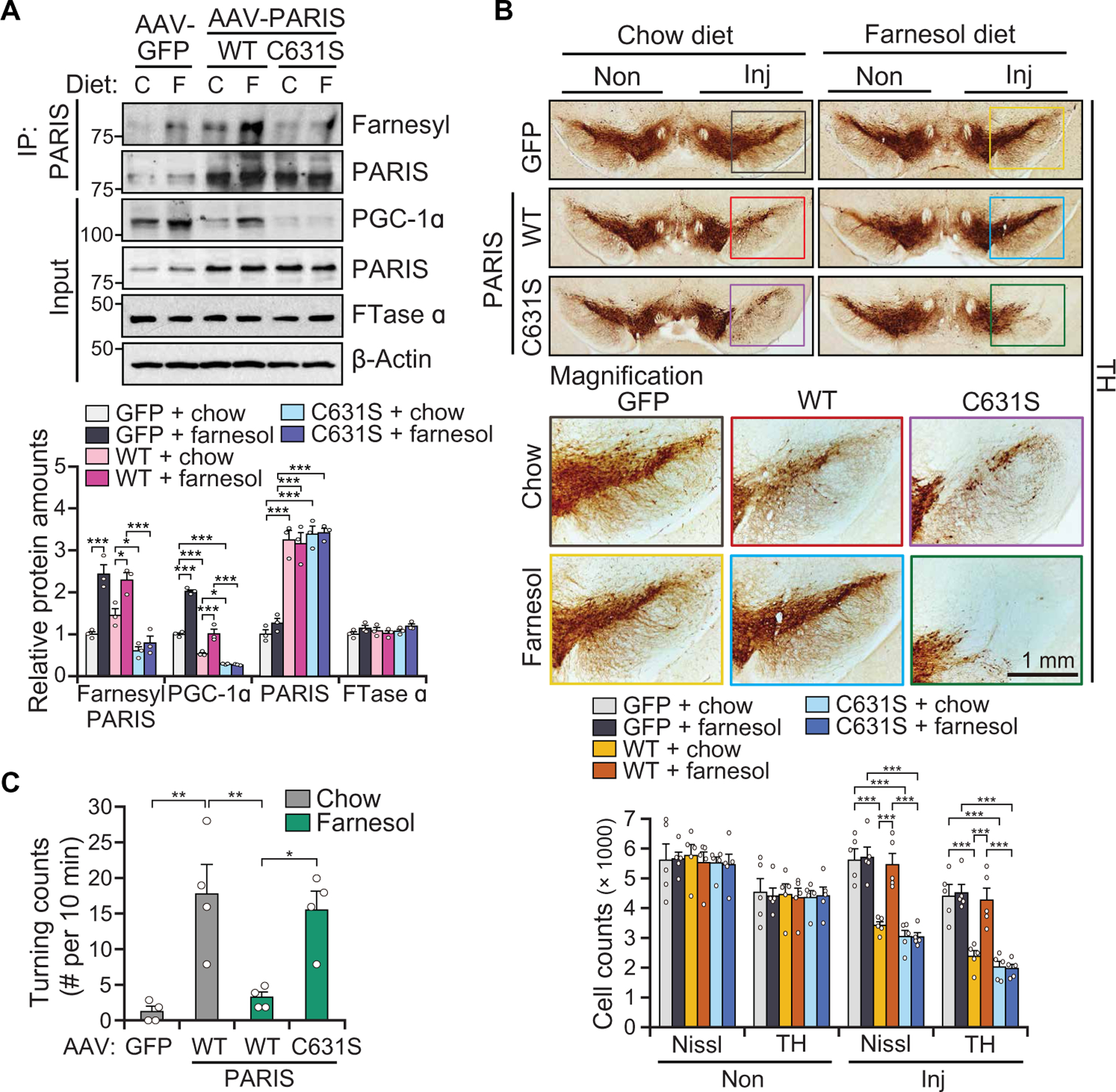
(A) Representative immunoblot image of farnesylated PARIS, PGC-1α, PARIS, and FTase α at 3 weeks postintranigral injection of AAV-GFP, AAV-PARIS WT, or C631S into the SN of 6-week-old C57BL/6N mice fed with control chow or farnesol diet, n = 3 mice per group. Quantitation of the immunoblots in the bottom normalized to immunoprecipitated PARIS or β-actin. (B) TH staining of a representative SN section of mice injected with AAV-GFP, AAV-PARIS WT, or AAV-PARIS C631S ± farnesol diet at 4 weeks after injection. Top six panels show the noninjected side (Non) and ipsilateral injected side (Inj). Colored rectangle box indicates enlarged area (bottom six panels). Stereological TH, Nissl-positive neuronal counting was indicated at the bottom; n = 5 mice per group. (C) Amphetamine-induced rotation test with mice injected with AAV-GFP, AAV-PARIS WT, or AAV-PARIS C631S ± farnesol diet at 4 weeks after injection, n = 4 mice per group. Data = means ± SEM. Statistical significance was determined by ANOVA test with Tukey post hoc analysis (A and B, two-way; C, one-way). *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
Farnesol’s prevention of DA neuronal degeneration induced by parkin loss is dependent on PARIS farnesylation at C631
To determine whether farnesol prevents the loss of DA neurons in adult conditional parkin KO (cPK-KO) mice (12, 13) and whether this potential prevention of DA neurodegeneration is dependent on PARIS farnesylation, we compared the effect on farnesol in adult cPK-KO mice with WT PARIS versus adult cPK-KO mice C638S mutant PARIS. Murine PARIS C638 is equivalent to human PARIS C631. For these experiments, we first generated PARISC638S/C638S knock-in mice (C638S KI) by CRISPR-Cas9 system (Fig. 6A). The C638S KI mice were then mated with floxed parkin mice (parkinflox/flox), and parkinflox/flox/PARISC638S/C638S were generated (Fig. 6A). AAV-Cre was stereotaxically injected in the VM of 8-week-old parkinflox/flox and parkinflox/flox/PARISC638S/C638S mice to delete parkin and create cPK-KO/WT PARIS and cPK-KO/C638S PARIS KI mice (Fig. 6B). AAV-GFP was used for control injections. These mice were fed either a farnesol diet or a normal mouse chow diet 7 days before the stereotaxic injection with AAV-Cre and were further maintained on this diet for 3 months. PARIS amounts were increased about twofold, and PGC-1α amounts were reduced in the cPK-KO/WT PARIS and cPK-KO/C638S PARIS KI mice (Fig. 6C) similar to that observed in the CamK-PARIS mice (Fig. 4E). Farnesol treatment increased PGC-1α expression in the SN of control mice, led to enhanced PARIS farnesylation, and prevented the decrements of PGC-1α in the SN of cPK-KO/WT PARIS. However, farnesol failed to increase the expression of PGC-1α and PARIS farnesylation in the SN of cPK-KO/C638S PARIS KI mice (Fig. 6C). Farnesol treatment led to an up-regulation of Ppargc1a, Sod2, Nrf1, Tfam, Cycs, and Cox4i1 in the SN of control mice. Ppargc1a and Nrf1 mRNA were down-regulated in the SN of cPK-KO/ WT PARIS mice, which were restored by the farnesol diet (Fig. 6D). Accordingly, farnesol treatment completely prevented the loss of DA neurons observed in the SN of cPK-KO WT PARIS mice (Fig. 6E). However, the neuroprotective effects of farnesol were not observed in the SN of cPK-KO/C638S PARIS KI mice (Fig. 6, D and E).
Fig. 6. Farnesol’s prevention of DA neuronal degeneration induced by parkin loss is dependent on PARIS farnesylation at C631.
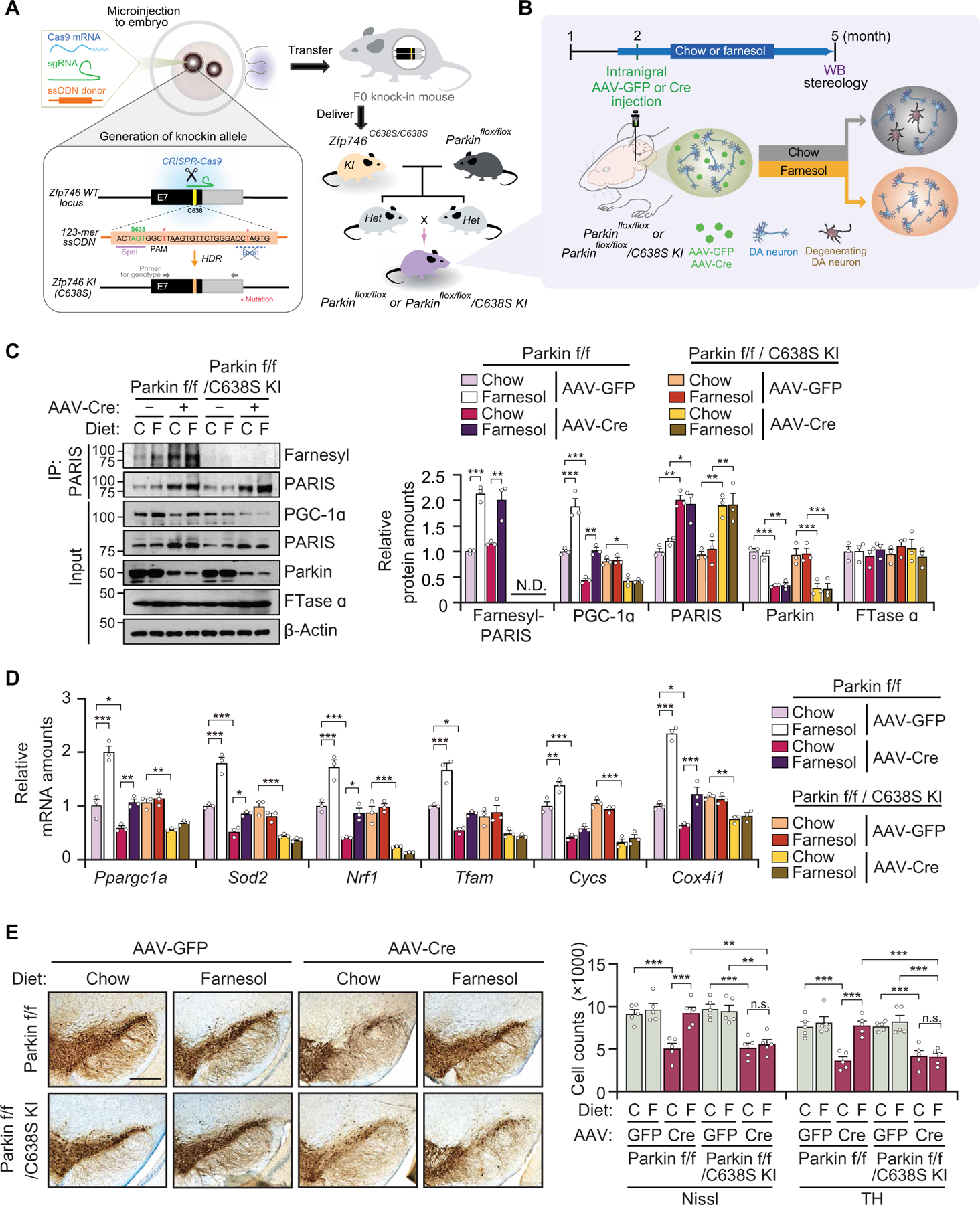
(A) Schematic illustration of generation of Zfp746(PARIS)C638S/C638S knock-in mice (C638S KI) by CRISPR/Cas9. C638S KI mice were bred with floxed parkin mice (parkinflox/flox) to generate parkinflox/flox/PARISC638S/C638S mice. (B) Experimental design to conditionally KO parkin in adult mice. AAV-Cre was injected into the SN of 8-week-old parkinflox/flox and parkinflox/flox/PARISC638S/C638S mice to generate cPK-KO and cPK-KO/C638S KI mice, respectively. Mice were fed with control chow or farnesol diet for 7 days before injection and euthanized after injection at 3 months. (C) Representative immunoblot images of farnesylated PARIS, parkin, PARIS, PGC-1α, and FTase α in the SN of cPK-KO and cPK-KO/C638S KI mice fed with control chow or the farnesol diet, n = 3 mice per group. Quantitation of the immunoblots in the right panel normalized to immunoprecipitated PARIS or β-actin. f/f, flox/flox. (D) Relative mRNA expression of Ppargc1a and its dependent genes normalized to GAPDH by real-time qRT-PCR in the SN of cPK-KO and cPK-KO/C638S KI mice fed with control chow or the farnesol diet, n = 3 mice per group. (E) TH staining of a representative section of 5-month-old cPK-KO and cPK-KO/C638S KI generated by stereotaxic injection of AAV-Cre into the SN of 8-week-old parkinflox/flox and parkinflox/flox/C638S KI mice, respectively. Scale bar, 400 µm. Stereological TH, Nissl-positive neuronal counting, n = 5 mice per group (right). Data = means ± SEM. Statistical significance was determined by two-way ANOVA test with Tukey post hoc analysis (C to E). Differences were considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
α-syn preformed fibril-mediated DA degeneration is rescued by farnesol
We used PARIS C638S KI mice to test the role of PARIS farnesylation in the α-syn preformed fibrils (α-syn PFFs) model of sporadic PD (Fig. 7A and fig. S10A) (36–38). Two-month-old PARIS C638S KI mice and littermate controls received a single intrastriatal injection of α-syn PFFs (Fig. 7A). Two weeks after α-syn PFF injection, mice were fed either a farnesol diet or a normal mouse chow diet until sacrifice at 8 months of age (Fig. 7A). Farnesylation of PARIS was decreased in α-syn PFF–injected mice, which was restored by the administration of farnesol (Fig. 7B). We observed complete loss of farnesylated PARIS in the SN of C638S KI mice, confirming that endogenous murine PARIS is farnesylated at C638 (Fig. 7B). The α-syn PFF injection led to a greater than twofold increase of PARIS in the SN of WT and C638S KI mice, as well as a reduction of PGC-1α (Fig. 7B) and a reduction of Ppargc1a and its target genes Sod2, Nrf1, and Tfam mRNA (fig. S10B). Farnesol administration reversed the α-syn PFF–induced reduction of PGC-1α and Ppargc1a and its target gene’s mRNA in WT mice, whereas farnesol had no effect in PARIS C638S KI mice (Fig. 7B and fig. S10B). The increase of PARIS and decrease of FTase α expression by α-syn PFF were not affected by farnesol administration or PARIS farnesylation in WT and C638S KI mice (Fig. 7B).
Fig. 7. Farnesol fails to rescue α-syn PFF–mediated DA neuron loss in Zfp746(PARIS)C638S/C638S knock-in mice.
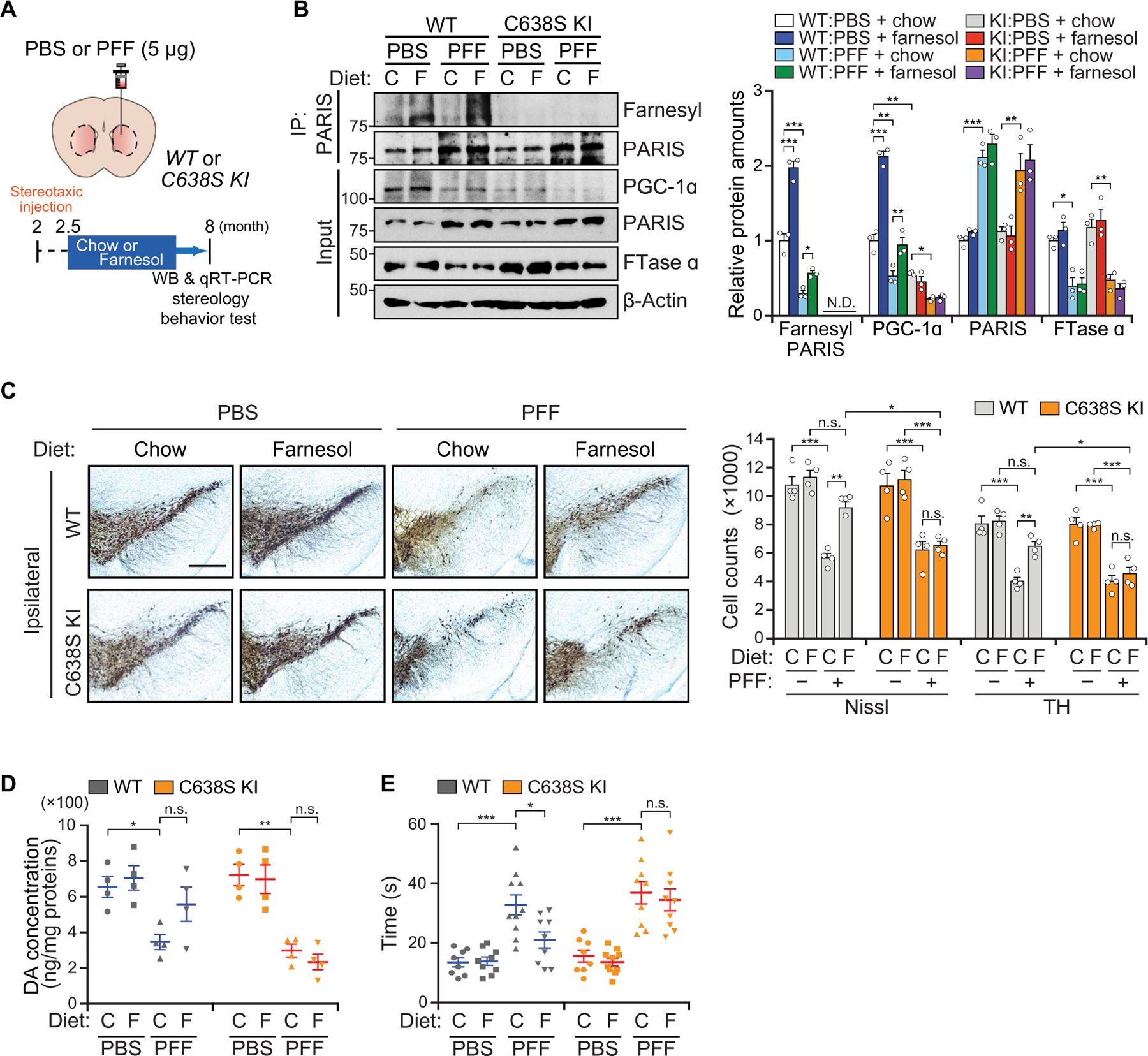
(A) Phosphate-buffered saline (PBS) or α-syn PFF was injected into the STR of 2-month-old PARIS C638S KI (C638S K) mice, and age-matched littermate controls (WT) were fed with control chow or farnesol diet, 2 weeks after injection. (B) Representative immunoblot image of farnesylated PARIS, PGC-1α, PARIS, and FTase α in the SN of C638S KI and WT mice ± α-syn PFF ± farnesol diet at 6 months after injection of α-syn PFF; n = 3 mice per group. Quantitation of the immunoblots in the bottom normalized to immunoprecipitated PARIS or β-actin. WB, western blot analysis. (C) TH staining of a representative section of 8-month-old C638S KI and WT mice ± α-syn PFF ± farnesol diet. Scale bar, 400 µm. Stereological TH, Nissl-positive neuronal counting was indicated at the bottom, n = 4 mice per group. (D) HPLC assessment of the content of dopamine (DA) normalized to protein expression in the STR of 8-month-old C638S KI and WT mice ± α-syn PFF ± farnesol diet; n = 4 mice per group. (E) Assessment of DA-related motor performance by pole test for C638S KI and WT mice ± α-syn PFF ± farnesol diet. WT, n = 8, 9, 10, and 9 mice for each group; C638S KI, n = 8, 10, 9, and 9 mice for each group. Data = means ± SEM. Statistical significance was determined by applying a two-way ANOVA test with Tukey post hoc analysis (C to F). Differences were considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
We detected pathologically aggregated and phosphorylated α-syn in the SN of α-syn PFF injected WT, whereas in the administration of farnesol, these species of α-syn were decreased in WT mice (fig. S10C). In the SN of PARIS C638S KI mice, there was no difference in pathologically aggregated α-syn between farnesol-fed and control chow–fed mice (fig. S10C). Accompanying the increase of phosphorylated α-syn in the SN was the loss of dopaminergic neurons in the ipsilateral SN (Fig. 7C) and the loss of striatal TH and DA transporter expression by immunoblot (fig. S10D). There was also the loss of striatal DA (Fig. 7D) and metabolites (fig. S10E) as measured by HPLC. Dopaminergic neurons in the contralateral SN were comparable (fig. S10F). We monitored behavioral dysfunction by the pole test (Fig. 7E) and grip strength test (fig. S10G) in α-syn PFF–injected WT and PARIS C638S KI mice. Farnesol administration prevented the loss of the dopaminergic neurons (Fig. 7, C and D, and fig. S10, D and E) and behavioral deficits (Fig. 7E, fig. S10G, and movie S3) in the α-syn PFF–injected WT mice but not in α-syn PFF–injected C638S KI mice, indicating that the neuroprotective effects of farnesol is dependent on PARIS farnesylation.
PARIS farnesylation was down-regulated in the SN of patients with PD
To determine whether PARIS farnesylation plays a role in patients with PD, the amount of PARIS farnesylation was assessed in patients with PD versus controls (Fig. 8, A and B, and table S5). As previously reported (12), PARIS was accumulated, and PGC-1α was reduced in the SN, but not in the CTX, of patients with PD compared to control (Fig. 8A). The amounts of farnesylated PARIS were decreased in the SN of patients with PD (Fig. 8A) and were unchanged in the CTX (Fig. 8B). Accompanying the decrease in PARIS farnesylation in the SN was a reduction in the expression of FTase α in the SN (Fig. 8A) with no change in FTase α in the CTX (Fig. 8B). A graphic summary shows that farnesol, a 15-carbon sesquiterpenoid molecule, increases PGC-1α expression in cellular and animal models of PD via enhancing PARIS farnesylation (fig. S11).
Fig. 8. PARIS farnesylation is decreased in the SN of sporadic patients with PD.
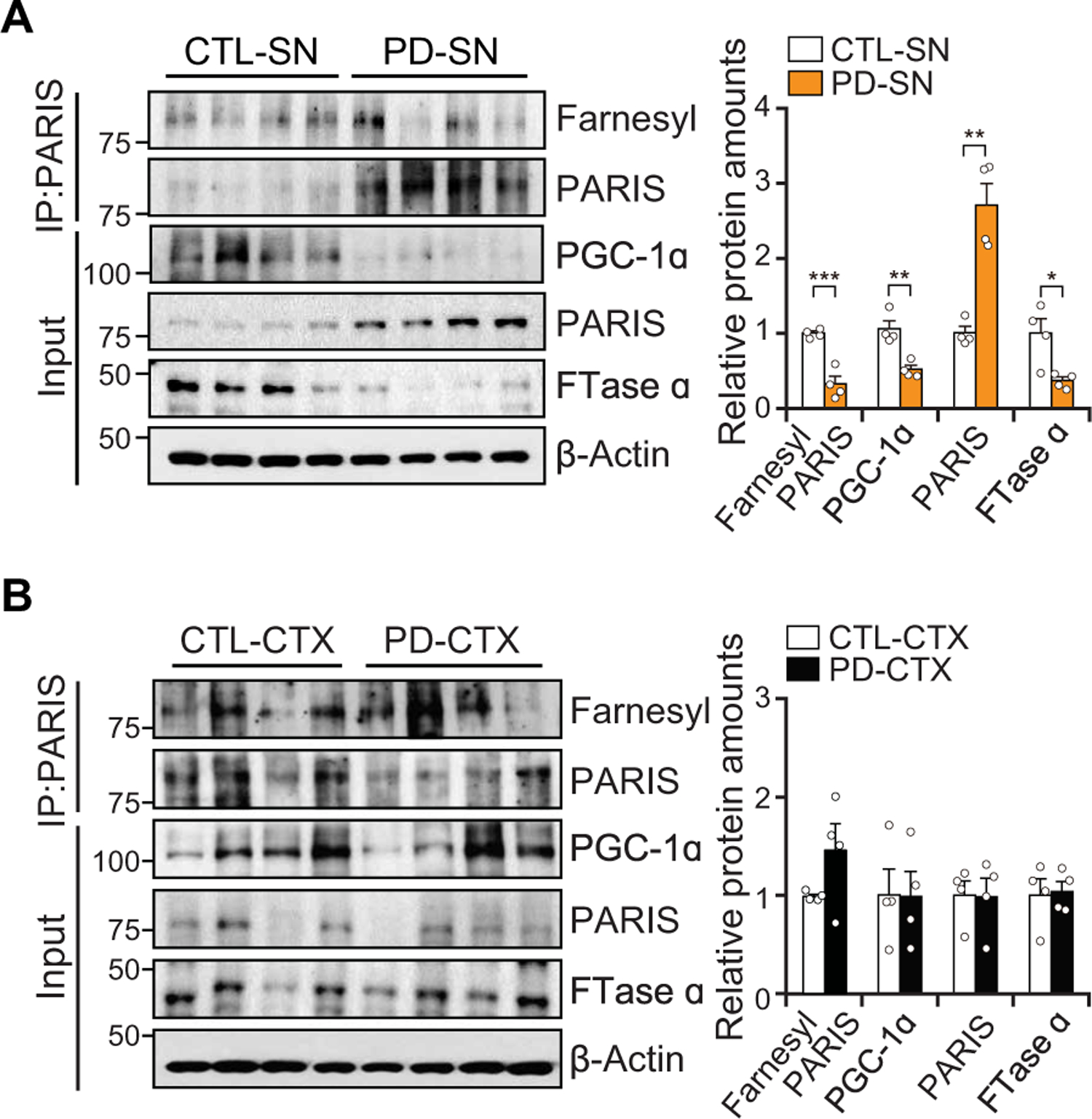
(A) PARIS farnesylation, PGC-1α, PARIS, and FTase α amounts in the SN of sporadic patients with PD brains as compared to age-matched controls; n = 4 per group. Quantification of farnesylated PARIS is normalized to immunoprecipitated PARIS (right). (B) Immunoblot analysis of PARIS farnesylation, PGC-1α, PARIS, and FTase α in the CTX from sporadic patients with PD compared to age-matched controls; n = 4 per group. Quantification of farnesylated PARIS is normalized to immunoprecipitated PARIS (right). Data = means ± SEM. Statistical significance was determined by applying an unpaired two-tailed student t test. Differences were considered significant when P < 0.05. *P < 0.05, **P < 0.01, and ***P < 0.001. Exact P values can be found in the accompanying statistical raw data.
DISCUSSION
Screening for PGC-1α inducers followed by a secondary screen of PGC-1α inducers in the setting of PARIS expression led to the identification of several compounds that prevented the repression of PGC-1α by PARIS. Farnesol was selected for further characterization because it had absorption, distribution, metabolism and excretion (ADME) properties, suggesting that it could be a viable, orally bioavailable, and brain permeable compound that could modify the expression of PGC-1α in the central nervous system. Oral administration of farnesol in mouse chow effectively permeates the BBB to enhance PGC-1α expression. Exploration of the mechanism by which farnesol enhances PGC-1α expression led to the discovery that human PARIS is farnesylated on C631. Farnesylation of PARIS prevents its ability to bind chromatin and inhibit gene transcription. The inhibition of PARIS by farnesol led to protection against loss of DA neurons induced by loss of parkin function, PARIS overexpression, and the α-syn PFF models of PD.
Farnesol prevented the loss of DA neurons and rescued the behavioral deficits due to overexpression of PARIS via a conditional transgenic approach and an AAV-mediated overexpression of PARIS. Farnesol also prevented the loss of DA neurons and rescued the behavioral deficits in the adult conditional KO of parkin and the α-syn PFF model of sporadic PD. Farnesol’s protective effects are through inhibition of transcriptional repression by PARIS through enhancing its farnesylation. Consistent with this notion are the observations that farnesol is not capable of protecting against the degeneration of DA neurons and the behavioral deficits induced by the AAV-mediated overexpression of the farnesylation-deficient human PARIS C631S mutant. In addition, farnesol fails to recover the loss of DA neurons and the behavioral deficits induced by adult conditional KO of parkin or α-syn PFFs in the farnesylation-deficient C638S KI mice.
Farnesylation and geranylgeranylation (protein prenylation) is a lipid modification involving the covalent addition of either farnesyl or geranylgeranyl isoprenoids to conserved cysteine residues at the C terminus of many proteins (33, 39). A large number of proteins are known to be prenylated, where prenylation is involved in protein targeting to membranes, protein-protein interactions and, as a consequence, changes the proteins cellular activity. To our knowledge, the regulation of transcription by protein prenylation is unprecedented. Farnesylation of the Arabidopsis MADS box transcription factor APETALA1 (AP1) has been described, but its role in the function and specificity of AP1 needs to be determined (40). Farnesylation of PARIS prevents its binding to chromatin where it is unable to influence gene transcription. This is a unique mechanism of transcriptional control. It will be important to determine whether other transcription regulators are controlled by protein prenylation.
There are some limitations of this study. Although the reduction of PGC-1α was found in the SN of C638S KI mice, we did not observe any evidence of DA loss or behavioral abnormalities in C638S KI mice. Also, C638S KI failed to exacerbate neurodegeneration observed in the adult conditional KO of parkin and the α-syn PFF model of sporadic PD. The lack of degeneration might be due to compensatory processes that occur in germline C638S KI mice. Future studies are required to address these issues. PARIS interacts with FTase α, and our molecular modeling suggests that the C terminus of PARIS containing the CaaX motif is predicted to form a loop, which, in theory, can be accommodated by the flexibility within the binding pocket of FTase α. Consistent with this notion is the observation that different amino acid residues such as Met, Gln, Cys, Ser, Thr, or Ala and inhibitors can be accommodated by the binding pocket of FTase α (41) and the mass spectroscopy confirmation of PARIS farneslylation. Future structural studies will need to be conducted to support the biology and the crystal lattice structures.
Farnesol is normally found in herbs, berries, and fruits (42), which might account, in part, for their beneficial effects on disorders of lipid metabolism and cardiovascular health (43). Farnesol can be phosphorylated in vivo to form FPP where it can enhance protein farnesylation and geranylgeranylation (44, 45). Thus, farnesol enhancement of protein prenylation seems to be a major mechanism of its beneficial effects in both the brain and heart. According to our proposed mechanistic model, PARIS farnesylation by FTase may play a role in maintaining PGC-1α expression under physiological conditions, and reduced PARIS farnesylation in PD brains may contribute to the demise of DA neurons in this disorder. In this regard, restoring protein farnesylation by farnesol could be considered as a potential disease-modifying therapeutic in PD. It will be important to determine the amount of farnesol that can be safely tolerated in humans and its pharmacokinetic properties and whether farnesol protects against the degenerative effects of PD in humans.
MATERIALS AND METHODS
Study design
This study aimed to identify potential compounds that induce PGC-1α expression and subsequently prevent PARIS-mediated DA death. High-throughput screening, promoter assay, and real-time qPCR revealed farnesol as a potent PGC-1α inducer, and we studied the molecular mechanism underlying PGC-1α induction in the presence of PARIS overexpression. To determine whether PARIS is a farnesylation substrate and farnesol enhances PARIS farnesylation, in vitro farnesylation assays, tagging-via-substrate, in vivo farnesylation assay, C631S mutant production, MS analysis, dot blot, immunoblot, and 3D structure prediction were performed. The relationship between DNA and farnesylated PARIS was explored by ChIP, EMSA, luciferase assay, and subcellular fractionation. The consequence of PARIS farnesylation was also examined by the measurement of mitochondrial DNA copy number, ATP concentration, and H2O2 concentrations. Most experiments were done in at least triplicate for statistical analysis. To evaluate the neuroprotective effect of farnesol in vivo, we used PD animal models of AAV-PARIS injection, CamK-PARIS transgenic mice, cPK-KO/C638S KI mice, and α-syn-PFF/C638S KI mice. For all animal experiments, mice groups were randomized, and the experimenters assessing the outcomes were blinded to the genotypes and intervention. For PD relevance, the expressions of farnesylated PARIS, PGC-1α, and FTase α were investigated in the brain of patients with PD. The numbers for all biological repeats are provided in the figure legends. Detailed descriptions of the materials and methods are also given in the Supplementary Materials.
Statistical analysis
For immunoblot analysis, the densitometric analysis of the bands was performed using ImageJ (National Institutes of Health, Bethesda, MO, USA; http://rsb.info.nih.gov/ij/). The intensities of protein bands normalized by those of internal controls were subjected to statistical analysis. Statistical parameters including the exact value of n, the definition of center, dispersion and precision measures (means ± SEM), and statistical significance were reported in the relevant figures and the figure legends. Statistical analyses were performed using GraphPad Prism version 7 (GraphPad Software). Data were determined to be statistically significant when P < 0.05 by applying the unpaired two-tailed Student’s t test (for comparison between two groups) or analysis of variance (ANOVA) test with Tukey’s post hoc (for comparison among three groups or more) or log-rank test (for comparison of Kaplan-Meier survival curves). In figures, asterisks denote statistical significance as calculated by Student’s t test or ANOVA test (*P < 0.05, ** P < 0.01, and ***P < 0.001) as compared to controls, unless otherwise specified by lines connecting the compared pieces of data. For data analysis of fig. S8B, data were assessed for normality using the D’Agostino and Pearson omnibus test, indicating that all datasets were sampled from normal distribution. Comparative data passing the normality test was further analyzed using two-way ANOVA test with Tukey’s post hoc test. For immunoblot and PCR analysis, sample sizes were determined as similar to those used by others in the field. No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Supplementary Material
Acknowledgments:
The chemical library used in this study was provided by Korea Chemical Bank of Korea Research Institute of Chemical Technology (www.chembank.org/). We would like to thank J.-H. Kim for help with the drug screen. We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human PD brain tissue. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901, and 1001 to the Arizona Parkinson’s Disease Consortium), and the Michael J. Fox Foundation for Parkinson’s Research.
Funding:
This work was supported by grants from the JPB Foundation, the Cure Parkinson’s Trust, and the Bachmann-Strauss Dystonia and Parkinson Foundation to T.M.D. Y.L. is supported by the Samsung Scholarship Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Programs M-1, M-2, and H-2014 to T.M.D. and V.L.D. This research was also supported by grants from the NRF (NRF-2016R1A2B4008271, NRF-2016R1A5A2945889, and NRF-2017R1E1A1A01073945 to J.-H.S; NRF-2017M3C7A1043848 to Y.L.), funded by the Korea Ministry of Science, ICT and Future Planning (MSIP), and is also supported by a Samsung Biomedical Research Institute grant (SBRI, SMX1151191, SMX1161351, SMX1161191, and SMX1170371 to J.-H.S.). This research was supported by Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2020R1A6C101A191 to J.-H.S.). This work was also supported by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute to D.A.S. and L.T.
Footnotes
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/604/eaax8891/DC1
View/request a protocol for this paper from Bio-protocol.
Competing interests: Patents related to this work include US9274128B2 entitled “Transcriptional repression leading to Parkinson’s disease” and WO2017161155A1 “Methods for preventing or treating Parkinson’s disease by the farnesylation of PARIS.” T.M.D. is a member of the Linked Clinical Trials Committee. T.M.D. and V.L.D. are founders of Valted LLC and hold an ownership equity interest in the company. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. T.M.D. is a member of Scientific Advisory Board of CurePSP, is a consultant and advisor to Sun Pharma Advanced Research Company Ltd., has received personal compensation in an editorial capacity for Journal of Clinical Investigation, is a member of American Gene Technologies International Inc., advisory board, and is a consultant for Mitokinin. T.M.D. and V.L.D. are consultants to Inhibikase Therapeutics Inc.; T.M.D. serves on the Board of Directors and is compensated for his roles as a consultant and interim Chief Scientific Officer of Valted Seq Inc.; V.L.D. serves on the Board of Directors, is a consultant, and is compensated for her role as interim Chief Executive Officer of Valted Seq Inc.; V.L.D. serves on the Scientific Advisory Board for the Burke Neurological Institute. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. The other authors declare that they have no competing interests.
Data and materials availability: The authors declare that data supporting the findings of this study are included in this manuscript. There is a material transfer agreement between Johns Hopkins University of the National Cancer Institute for the mPrP-TetP-PARIS transgenic mice.
REFERENCES AND NOTES
- 1.Mullin S, Schapira AH, Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol. Clin 33, 1–17 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Savitt JM, Dawson VL, Dawson TM, Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Invest 116, 1744–1754 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jellinger KA, Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord 27, 8–30 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Corti O, Lesage S, Brice A, What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev 91, 1161–1218 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Martin I, Dawson VL, Dawson TM, Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genomics Hum. Genet 12, 301–325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ossig C, Reichmann H, Treatment strategies in early and advanced Parkinson disease. Neurol. Clin 33, 19–37 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Brahmachari S, Lee S, Kim S, Yuan C, Karuppagounder SS, Ge P, Shi R, Kim EJ, Liu A, Kim D, Quintin S, Jiang H, Kumar M, Yun SP, Kam TI, Mao X, Lee Y, Swing DA, Tessarollo L, Ko HS, Dawson VL, Dawson TM, Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson’s disease. Brain 142, 2380–2401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS, The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep 4, 4874 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar M, Acevedo-Cintron J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, Karuppagounder SS, Brahmachari S, Chen R, Kim H, Ko HS, Dawson VL, Dawson TM, Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Rep 15, 629–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, Scarffe LA, Umanah GE, Kang H, Ham S, Kam TI, Allen K, Brahmachari S, Kim JW, Neifert S, Yun SP, Fiesel FC, Springer W, Dawson VL, Shin JH, Dawson TM, PINK1 primes parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep 18, 918–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirooznia SK, Yuan C, Khan MR, Karuppagounder SS, Wang L, Xiong Y, Kang SU, Lee Y, Dawson VL, Dawson TM, PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener 15, 17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM, PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM, Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. U.S.A 112, 11696–11701 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK, Mitochondrial quality control via the PGC1a-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by rapamycin. J. Neurosci 35, 12833–12844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siddiqui A, Rane A, Rajagopalan S, Chinta SJ, Andersen JK, Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1alpha. Neurobiol. Dis 93, 115–120 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Austin S, St-Pierre J, PGC1a and mitochondrial metabolism – Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci 125, 4963–4971 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Scarpulla RC, Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM, Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Eschbach J, von Einem B, Muller K, Bayer H, Scheffold A, Morrison BE, Rudolph KL, Thal DR, Witting A, Weydt P, Otto M, Fauler M, Liss B, McLean PJ, Spada AR, Ludolph AC, Weishaupt JH, Danzer KM, Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1a deregulation and α-synuclein oligomerization. Ann. Neurol 77, 15–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su X, Chu Y, Kordower JH, Li B, Cao H, Huang L, Nishida M, Song L, Wang D, Federoff HJ, PGC−1a promoter methylation in Parkinson’s disease. PLOS ONE 10, e0134087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye Q, Huang W, Li D, Si E, Wang J, Wang Y, Chen C, Chen X, Overexpression of PGC-1α influences mitochondrial signal transduction of dopaminergic neurons. Mol. Neurobiol 53, 3756–3770 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Wang KZ, Chu CT, After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9, 1663–1676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark J,Reddy S,Zheng K,Betensky RA,Simon DK,Associationof PGC-1αlphapolymorphisms with age of onset and risk of Parkinson’s disease. BMC Med. Genet 12, 69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grunblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wullner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, Young AB, Vance JM, Davis RL, Hedreen JC, Adler CH, Beach TG, Graeber MB, Middleton FA, Rochet JC, Scherzer CR; Global PD Gene Expression (GPEX) Consortium, PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med 2, 52ra73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scarffe LA, Stevens DA, Dawson VL, Dawson TM, Parkin and PINK1: Much more than mitophagy. Trends Neurosci 37, 315–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasbach KA, Schnellmann RG, Isoflavones promote mitochondrial biogenesis. J. Pharmacol. Exp. Ther 325, 536–543 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Hang HC, Linder ME, Exploring protein lipidation with chemical biology. Chem. Rev 111, 6341–6358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hancock JF, Magee AI, Childs JE, Marshall CJ, All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 (1989). [DOI] [PubMed] [Google Scholar]
- 29.Nguyen UT, Guo Z, Delon C, Wu Y, Deraeve C, Franzel B, Bon RS, Blankenfeldt W, Goody RS, Waldmann H, Wolters D, Alexandrov K, Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat. Chem. Biol 5, 227–235 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Lerner AEC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton D, Sebti SM, Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J. Biol. Chem 270, 26802–26806 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J, Zhao Y, A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A 101, 12479–12484 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang M, Casey PJ, Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol 17, 110–122 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Zhang FL, Casey PJ, Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem 65, 241–269 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER, Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Christie JE, Crow TJ, Turning behaviour as an index of the action of amphetamines and ephedrines on central dopamine-containing neurones. Br. J. Pharmacol 43, 658–667 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS, Rosenthal LS, Dawson TM, Dawson VL, Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 362, eaat8407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y, Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Goncalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DA, Dawson VL, Ko HS, Dawson TM, Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casey PJ, Biochemistry of protein prenylation. J. Lipid Res 33, 1731–1740 (1992). [PubMed] [Google Scholar]
- 40.Yalovsky S, Rodriguez-Concepcion M, Bracha K, Toledo-Ortiz G, Gruissem W, Prenylation of the floral transcription factor APETALA1 modulates its function. Plant Cell 12, 1257–1266 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reid TS, Terry KL, Casey PJ, Beese LS, Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J. Mol. Biol 343, 417–433 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Tatman D, Mo H, Volatile isoprenoid constituents of fruits, vegetables and herbs cumulatively suppress the proliferation of murine B16 melanoma and human HL-60 leukemia cells. Cancer Lett 175, 129–139 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Giugliano D, Ceriello A, Esposito K, Are there specific treatments for the metabolic syndrome? Am. J. Clin. Nutr 87, 8–11 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Bentinger M, Grunler J, Peterson E, Swiezewska E, Dallner G, Phosphorylation of farnesol in rat liver microsomes: Properties of farnesol kinase and farnesyl phosphate kinase. Arch. Biochem. Biophys 353, 191–198 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Szucs G, Murlasits Z, Torok S, Kocsis GF, Paloczi J, Gorbe A, Csont T, Csonka C, Ferdinandy P, Cardioprotection by farnesol: Role of the mevalonate pathway. Cardiovasc. Drugs Ther 27, 269–277 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP, Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res 94, 525–533 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D, Jiang H, Kang SU, Lee BD, Kang HC, Kim D, Tessarollo L, Dawson VL, Dawson TM, Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci 16, 1392–1400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM, Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc. Natl. Acad. Sci. U.S.A 101, 10744–10749 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A, Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 52, 642–649 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Fatokun AA, Liu JO, Dawson VL, Dawson TM, Identification through high-throughput screening of 4′-methoxyflavone and 3′,4′-dimethoxyflavone as novel neuroprotective inhibitors of parthanatos. Br. J. Pharmacol 169, 1263–1278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh RN, DeBiasio R, Hudson CC, Ramer ER, Cowan CL, Oakley RH, Quantitative cell-based high-content screening for vasopressin receptor agonists using transfluor technology. J. Biomol. Screen 10, 476–484 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Zhang JH, Chung TD, Oldenburg KR, A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Nguyen UT, Wu Y, Goodall A, Alexandrov K, Analysis of protein prenylation in vitro and in vivo using functionalized phosphoisoprenoids. Curr. Protoc. Protein Sci Chapter 14, Unit14.13 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Wu YW, Alexandrov K, Brunsveld L, Synthesis of a fluorescent analogue of geranylgeranyl pyrophosphate and its use in a high-throughput fluorometric assay for Rab geranylgeranyltransferase. Nat. Protoc 2, 2704–2711 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Teshima K, Kondo T, Analytical method for determination of allylic isoprenols in rat tissues by liquid chromatography/tandem mass spectrometry following chemical derivatization with 3-nitrophtalic anhydride. J. Pharm. Biomed. Anal 47, 560–566 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Kang H, Shin JH, Repression of rRNA transcription by PARIS contributes to Parkinson’s disease. Neurobiol. Dis 73, 220–228 (2015). [DOI] [PubMed] [Google Scholar]
- 57.During MJ, Young D, Baer K, Lawlor P, Klugmann M, Development and optimization of adeno-associated virus vector transfer into the central nervous system. Methods Mol. Med 76, 221–236 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Volpicelli-Daley LA, Luk KC, Lee VM, Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc 9, 2135–2146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karl T, Pabst R, von Horsten S, Behavioral phenotyping of mice in pharmacological and toxicological research. Exp. Toxicol. Pathol 55, 69–83 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Nielsen M, Lundegaard C, Lund O, Petersen TN, CPHmodels-3.0–Remote homology modeling using structure-guided sequence profiles. Nucleic Acids Res 38, W576–W581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.