

Abstract

The Ir-MaxPHOX-type catalysts demonstrated high catalytic performance in the hydrogenation of a wide range of nonchelating olefins with different geometries, substitution patterns, and degrees of functionalization. These air-stable and readily available catalysts have been successfully applied in the asymmetric hydrogenation of di-, tri-, and tetrasubstituted olefins (ee′s up to 99%). The combination of theoretical calculations and deuterium labeling experiments led to the uncovering of the factors responsible for the enantioselectivity observed in the reaction, allowing the rationalization of the most suitable substrates for these Ir-catalysts.
Keywords: asymmetric hydrogenation; iridium; P−N ligands, DFT calculations, olefins
Introduction
Advances in the synthesis of chiral molecules, whether creating new compounds or improving existing synthetic procedures, are made possible by the continuous innovations in asymmetric catalysis.1 Among the asymmetric catalytic reactions that lead to enantiomerically pure products, the hydrogenation of olefins is one of the most powerful.1,2 This 100% atom economy process has a large record of successful examples in the production of single enantiomer intermediates, especially in the pharmaceutical industry, using substrates ranging from olefins with coordinating functional groups to nonfunctionalized counterparts, passing through olefins with intermediate coordinating properties.3 As the number of substrates continues to increase to reach more complex molecules, finding a catalyst that performs well with many of them regardless of geometry, substitution patterns, and functionalization remains a challenge. While Rh- and Ru-catalysts (mainly with diphosphine ligands) have been shown to be optimal for the reduction of olefins with strong coordinating functional groups,4 the Ir–P,X-catalysts (X = N, S, and O; mainly with phosphine/phosphinite/phosphite–oxazoline ligands) gave the best results for the hydrogenation of nonchelating alkenes.5 Particularly, the reduction of nonchelating olefins is the most difficult and less explored field since they do not have a coordinating group to help transfer the chiral information to the product. Currently, Ir-catalysts only perform well for specific types of olefins. The most common substitution patterns are E-trisubstituted alkenes and, to a lesser extent, Z-trisubstituted and 1,1-disubstituted alkenes. The hydrogenation of tetrasubstituted olefins is the least developed category.5 Even for the most studied trisubstituted olefins, there is still room for improvement. For example, the reduction of the so-called purely alkyl-trisubstituted olefins, those without functional groups or aryl substituents, has been achieved in very few cases6 and the effectiveness for exocyclic substrates needs to be improved.7 For tetrasubstituted olefins, only a few specific Ir-catalysts have provided high performance for certain substrates, with variable enantioselectivity and low functional group tolerance. Most of the substrates studied were restricted to cyclic olefins and only a few were acyclic, mainly trimethyl styrene derivatives,7b,8 until recently when Gosselin′s group in collaboration with Bigler, Pfaltz, and Denmark9 presented the reduction of a wide range of acyclic olefins with two or more aryl substituents. In addition, there are fewer reports of tetrasubstituted olefins with poorly coordinative groups that are useful for further synthesis, and, in most cases, the same catalyst was unsuccessful for tetrasubstituted olefins without a poorly coordinative group.10 The finding of a catalyst that could work on all of them is highly desirable to limit time-consuming catalyst design and avoid a variety of preparation methods.
The bottleneck in finding the best catalysts is the identification of the right ligands with a broad substrate scope.11 To overcome the substrate scope limitation in the asymmetric hydrogenation of nonchelating olefins, we recently reported on the first P,N-ligand library that could reduce different types of nonchelating olefins.7b From a common backbone, the selection of the phosphite or phosphinite group led to ligands that were suitable for 56 examples of di-, tri-, and tetrasubstituted olefins. However, only 11 examples of tetrasubstituted olefins could be reduced, mainly indene derivatives and some acyclic olefins, to the detriment of tetrasubstituted acyclic alkenes with relevant poorly coordinative groups. Even for trisubstituted olefins, only one example of Z-olefin was successfully reduced and none of purely alkyl-substituted. Later on, we reported the successful application of a family of P-stereogenic aminophosphine-oxazoline (MaxPHOX) ligands in the Ir-catalyzed hydrogenation of the aforementioned unfunctionalized tetrasubstituted olefins and also in the reduction of several tetrasubstituted substrates with poorly coordinative groups, such as acyclic-tetrasubstituted vinyl fluorides with ester functionalities.8c
To advance the search for a ligand library capable of hydrogenating a larger range of substituted nonchelating olefins, here we report an extension of the scope of olefins that Ir-MaxPHOX-type catalysts can successfully reduce. With the Ir-MaxPHOX 1-4a-c family of catalysts (Figure 1), we have been able to hydrogenate, with high catalytic performance, a wide range of di- and trisubstituted olefins and we have also increased the number of tetrasubstituted olefins containing neighboring poorly coordinative polar groups that could be used successfully. These catalysts have the advantage that they are prepared in four steps from available starting materials12 and allow us to easily study the effect of varying some ligand properties, such as the bulkiness of the oxazoline and its configuration and the configuration of the stereogenic center at the alkyl backbone chain. Together with mechanistic studies based on density functional theory (DFT) calculations and deuterogenation experiments, we were able to explain the origin of enantioselectivity, identify the preferred pathway, and predict enantioselectivities with good accuracy.
Figure 1.

Family of aminophosphine-oxazoline iridium(I) catalysts (Ir-MaxPHOX) 1-4a-c.
Results and Discussion
Initial Catalytic Screening
As mentioned in the introduction, the hydrogenation of nonchelating olefins depends largely on the substitution pattern of the substrate. The most successful examples have been reported for E-trisubstituted, while 1,1′-disubstituted olefins are usually hydrogenated less enantioselectively and tetrasubstituted olefins are still underdeveloped.5 To explore the scope of the Ir-MaxPHOX catalysts (1-4a-c), we initially applied them in the asymmetric hydrogenation of the nonfunctionalized disubstituted olefin S1 and the widely used benchmark trisubstituted substrate S2 (Table 1). The initial test conditions were the optimal conditions reported in previous studies with other P,N ligands.5 Therefore, the reactions were carried out at room temperature using 1 mol % of the catalyst in dichloromethane under 1 bar of H2 for the disubstituted substrate S1 and 50 bar of H2 for the trisubstituted olefin S2. The previous results for the model acyclic-tetrasubstituted substrate S3 were also included in Table 1 for comparison.8c
Table 1. Asymmetric Hydrogenation of Substrates S1, S2, and S3(8c) with Ir-Catalysts 1-4a-ca.

| entry | Ir complex | % convb | % eec | % convb | % eec | % convb | % eec |
|---|---|---|---|---|---|---|---|
| 1 | 1a | 100 | 74 (S) | 100 | 67 (R) | 100 | 75 (R) |
| 2 | 1b | 100 | 66 (S) | 100 | 75 (R) | 100 | 85 (S)d |
| 3 | 1c | 100 | 81 (S) | 100 | 77 (R) | 85 | 44 (R) |
| 4 | 2b | 100 | 15 (S) | 100 | 15 (S) | 85 | 33 (S) |
| 5 | 3b | 100 | 80 (R) | 100 | 23 (S) | 100 | 44 (R) |
| 6 | 4a | 100 | 83 (R) | 100 | 82 (S) | 100 | 28 (R) |
| 7 | 4b | 100 | 88 (R) | 100 | 85 (S) | 100 | 25 (R) |
| 8 | 4c | 100 | 91 (R) | 100 | 88 (S) | 100 | 31 (R) |
| 9e | 4c | 100 | 91 (R) | 100 | 89 (S) | ||
| 10e | 1b | 100 | 98 (S)f |
Reaction conditions: catalyst (1 mol %), CH2Cl2, 1 bar of H2 (S1), 50 bar of H2 (S2), or 75 bar of H2 (S3), rt, 4 h (S1 and S2) or 24 h (S3).
Conversions were measured by 1H NMR spectroscopy after 4 h (S1 and S2) or 24 h (S3).
Enantiomeric excess determined by GC.
Using 2 bar of H2—98% (S) ee.
Reactions carried out in PC instead of CH2Cl2 after 6 h (S1 and S2) and 30 h (S3).
Using 2 bar of H2.
For substrates S1 and S2, the best enantioselectivities were obtained with Ir-catalyst 4c (ee′s up to 91%, entry 8) regardless of the substitution pattern of the substrate. The results showed that both the oxazoline substituent and the diastereoisomeric backbone of the ligand had a noticeable effect on the stereochemical outcome. This effect also occurred in the hydrogenation of the tetrasubstituted olefin S3. However, while for the di- and trisubstituted substrates (S1 and S2) the best results were obtained with the bulkier tBu group in the oxazoline (e.g., see entry 8 vs. 6–7), the best results for the tetrasubstituted substrate S3 were obtained with the less bulky iPr group, in accordance with the higher steric hindrance of S3 (entry 2). Similarly, the effect of the diastereoisomeric backbone differed between the di/trisubstituted alkenes S1 and S2 and the tetrasubstituted olefin S3. While backbone 4 (Figure 1) was best for S1 and S2 (ee′s up to 91%), the best backbone for S3 was 1 (ee′s up to 98% at 2 bars of H2, entry 2). In summary, optimizing the ligand structure led us to identify 1b and 4c as the best catalysts of the family for the hydrogenation of olefins with different substitution patterns.13
To make the process more sustainable, the reaction was carried out in 1,2-propylene carbonate (PC),14 an eco-friendly alternative to standard organic solvents due to its high boiling point, low toxicity, and green synthesis (Table 1, entries 9 and 10). Advantageously, enantioselectivities remained as high as those obtained with dichloromethane (ee’s up to 98%). In addition, the catalyst could be recycled up to five times with a simple two-phase extraction with hexane with a minimal decrease in enantioselectivity (see the Supporting Information).
Mechanistic Studies: The Origin of Enantioselectivity
To understand why the best ligand for tetrasubstituted olefins is different from that of di- and trisubstituted analogues, we performed a density functional theory (DFT) study. The transition states (TSs) involved in the enantiodetermining step of the reaction for the tri- and tetrasubstituted olefins, S2 and S3, with catalyst 4c (for S2) and catalysts 1b and 4c (for S3) were searched using the B3LYP15 functional with the Grimme Dispersion correction, GD3.16 Mechanistically it is well known that Ir-catalyzed hydrogenation of nonfunctionalized alkenes proceeds through an Ir(III)/Ir(V) tetrahydride intermediate17 and enantioselectivity is determined in the first hydrogen transfer from the metal to the coordinated olefin. Our calculations also support this mechanism; the free energy reaction profile is presented in the Supporting Information. Consequently, enantioselectivity can be reliably estimated from the relative energies of the TSs of this step. Nevertheless, two different mechanisms can be considered for this process: (i) an Ir(III)/Ir(V) migratory-insertion step (mechanism 3/5-MI, Scheme 1) and (ii) an Ir(III)/Ir(V) σ-bond metathesis (mechanism 3/5-Meta, Scheme 1). While (i) is usually the most favorable mechanism, (ii) is also energetically feasible and cannot be immediately discarded. We, therefore, computed the TSs for both pathways (see the Supporting Information for the full set of calculated TSs). A data set collection of computational results is available in the ioChem-BD repository.18
Scheme 1. Proposed Catalytic Cycles 3/5-MI and 3/5-Meta for the Asymmetric Hydrogenation of Nonchelating Olefins.

The calculated relative energies for the most stable isomers of the TSs for both pathways (TSMI and TSMeta) are shown in Table 2. These key isomers are the result of the relative arrangement of the hydride (up or down), the coordination of the olefin through the Re- or Si-face, and the attack of the hydride through the two olefinic carbons (C1 or C2). In addition, in these calculations, we also considered the rotamers of the isopropyl group. As in other reported studies, the results show that in all cases, the migratory insertion is the preferred reaction pathway.
Table 2. Calculated Relative Energies (kJ/mol) for the Transition States TSMI and TSMeta with Substrates S2 and S3 Using Ir-Catalyst 4c (for S2) and Ir-Catalysts 1b and 4c (for S3)a,b.

Values in blue and bold indicate the lowest Re and Si energy TSs for each combination of substrate and catalyst.
Relative Gibbs free energies (kJ/mol) in solution (B3LYP-D3/6-31G(d,p)&LANL2DZ) with respect to the corresponding lowest energy transition state; for S2 Ar = 4-CH3O-C6H4 and R = H and for S3 Ar = C6H5 and R = CH3; C1 is the least electronegative olefinic carbon atom and C2 is the most electronegative one. In all TSs, the most stable rotamer was selected.
Positively, the calculations for the trisubstituted substrate S2 with the Ir-catalyst 4c reproduce the experimental outcome. The favored pathway TSL (Table 2) proceeds through the Re-face, which leads to the formation of the (S)-product, and the energy difference between the two most stable TSs (TSL and TSO, Table 2), which lead to opposite enantiomers, is 5.3 kJ/mol (eecalc = 79% (S)) in agreement with the experimental enantioselectivity (88% (S)). Single-point calculations on the most stable TSs with larger basis sets, B97D3/cc-pVTZ & cc-pVTZ-PP, improve the agreement eecalc = 85% (S) (see the Supporting Information for further details). Thus, the factors responsible for enantioselectivity can be deduced by analyzing the structures of both TSs via quantitative quadrant diagram representations using MolQuO19 software (Figure 2).
Figure 2.

Models of the most favored TSs for the asymmetric hydrogenation of S2 and S3 with 4c; (a) schematic quadrant model for 4c (the olefin coordinates above the plane of the paper), (b) the most favorable coordination of S2 giving the major (S)-product, (c) the most favorable coordination of S2 giving the minor (R)-product, (d) the most favorable coordination of S3 giving the major (R)-product, and (e) the most favorable coordination of S3 giving the minor (S)-product.
Figure 2a shows the quadrant diagram obtained by analyzing the two most stable TSs for the hydrogenation of S2 (TSL and TSO, Table 2).20 In this diagram, the oxazoline substituent (tBu) blocks the lower-left quadrant Q3 (quadrant occupancy = 3.8), while the methylenic carbon of the oxazoline partly occupies the upper-left quadrant Q1 (quadrant occupancy = 1.6) making it semihindered (Figure 2a). The other two quadrants, Q2 and Q4, free from bulky groups, are empty (quadrant occupancy = 0). According to this model, the coordination of the trisubstituted olefin S2 through the Re-face is favored because the smallest substituent, the olefinic hydrogen, is located in the most hindered quadrant Q3 and the aryl substituent (4-OMe-C6H5) is located in the semihindered quadrant Q1 (Figure 2b). In contrast, when the olefin coordinates through the Si-face, which leads to the opposite enantiomer ((R)-enantiomer, TSO, Table 2), the aryl group is located at the most hindered quadrant resulting in a less favorable TS (Figure 2c). The occupancy value for this quadrant (3.1) is slightly lower than that obtained for the TS leading to the major product, indicating that the ligand adapts its chiral pocket to suit the olefin in this coordination manner. It is noteworthy that all TSs with the methyl group located in Q3 are less stable, at least 26.3 kJ/mol higher in energy than the most stable one. Note that despite the small size of a methyl group, the flat 4-MeO-C6H5 group fits better into the cavity in Q3. In summary, the model indicates that the stereochemical outcome with trisubstituted olefin S2 depends on steric factors. Following this observation, it can be hypothesized that the catalyst may also work for other aryl-containing trisubstituted olefins, including the less studied triaryl trisubstituted and Z-olefins (see Table 3 below), where the TS with olefinic hydrogen located in the most hindered quadrant Q3 will continue to be more stable than a TS with the aryl substituent (for triaryl olefins) or the methyl substituent (for Z-olefins) in Q3. In addition, this model suggests that if the olefinic aryl group is replaced by a bulkier substituent (e.g., purely alkyl-substituted olefins), then a higher destabilization of the TSO could be expected, resulting in a higher energy gap between the TSs and high enantioselectivity (see results for S20 and S21, Table 3 below).
Table 3. Asymmetric Hydrogenation of Nonfunctionalized Trisubstituted Olefins with Only Aryl and/or Alkyl Substituents S5–S30.


Reaction conditions: 4c (1 mol %), CH2Cl2, 23 °C, 4 h, using 1 bar of H2 for S5–S12 or 50 bar of H2 for S13–S30.
Reaction carried out using propylene carbonate (PC) as a solvent for 6 h.
In contrast, the most favorable TS with the same Ir-catalyst 4c system but with the tetrasubstituted olefin S3 was TSO (Table 2), where the olefin coordinates through the Si-face and the (R)-enantiomer would be obtained as observed experimentally. The quadrant diagrams of the two most stable TSs (TSO and TSP, Table 2) with the tetrasubstituted olefin S3 and 4c were analyzed (Figure 2d,e). The diagrams show that the preferred coordination of S3 is through the Si-face with the olefinic phenyl substituent occupying the most hindered quadrant (Q3, Figure 2d), which explains why the enantioselectivity is opposite to that of S2. Again, the planarity of the phenyl substituent makes the TS less crowded in Q3 than with a methyl group. This is reflected in the fact that the distance between the hydrogen of the C4 of the oxazoline and the olefinic phenyl substituent (TSO) is greater than the distance between the hydrogen of the C4 of the oxazoline and the methyl substituent in the TSP (Figure 3).
Figure 3.

Representation of the two most stable TSs (TSO and TSP) for 4c and substrate S3. Relative Gibbs free energies in solution (kJ/mol) with respect to the corresponding lowest TS.
When Ir-catalyst 1b was used in the hydrogenation of tetrasubstituted olefin S3, reverse enantioselectivity was obtained compared to Ir-catalyst 4c. This can be rationalized by analyzing the quadrant model of the most stable transition state, TSN (Table 2), for the hydrogenation of S3 with 1b (Figure 4). Ir-catalyst 1b has the opposite configuration in the oxazoline substituent compared to 4c, making the upper-left quadrant Q1 the most hindered (Figure 4a). Therefore, the preferred coordination of S3 is through the Re-face (the opposite of 4c) with the olefinic phenyl located in the most hindered quadrant (Q1) (Figure 4b).
Figure 4.

Model of the most favored TS for the asymmetric induction of S3 with 1b; (a) schematic quadrant model for 1b (the olefin coordinates above the plane of the paper) and (b) the most favorable coordination of S3 giving the major (S)-product.
Although the sense of enantioselectivity for S3 was well predicted for both Ir-catalysts 4c and 1b, the enantioselectivity value was greatly overestimated with 4c (82% (R) B3LYP-D3/6-31G(d,p)&LANL2DZ and 85% (R) B97D3/cc-pVTZ&cc-pVTZ-PP//B3LYP-D3/6-31G(d,p)&LANL2DZ predicted ee vs. 31% (R) observed ee). To explain this disagreement, we conducted deuterium labeling experiments with 1b and 4c (Scheme 2) in which the related tetrasubstituted olefin S4 was reduced with deuterium. Note that in these deuterogenation experiments, we used substrate S4, which differs from the tetrasubstituted olefin S3 in a methoxy group in the aryl group, which was introduced to facilitate product analysis. Both substrates performed in the same way. As expected, no deuteration at the methyl groups was observed using 1b. However, in the case of 4c, a substantial deuteration was found at the allylic position, indicating the existence of a competing isomerization process. This isomerization would explain the lower enantioselectivity observed when using 4c in the hydrogenation of tetrasubstituted alkenes such as S3 or S4 (Table 1, entry 2 vs. 7).
Scheme 2. Deuterium Labeling Experiments of the Tetrasubstituted Substrate (S4).

Percentage of deuterium incorporation is shown in brackets.
Substrate Scope
We first evaluated the Ir-precatalysts 1-4a-c in the reduction of a wide range of di- and trisubstituted substrates with E- and Z-geometries and different neighboring polar groups.
We first focused on the hydrogenation of nonfunctionalized olefins with aryl and/or alkyl substituents only (Table 3). According to the previous screening, Ir-catalyst 4c was selected for the hydrogenation of a wide range of 1,1′-disubstituted olefins. As expected, this catalyst provided high enantioselectivities (up to 94% ee) for other α-tert-butylstyrenes (substrates S5–S11) with a range of electronic and steric properties at the aryl group. These are significant results because disubstituted substrates suffer more face-selectivity indetermination than the trisubstituted equivalents and therefore there are fewer catalysts21 that can provide those high ee′s. Nevertheless, the hydrogenation of α-alkylstyrene S12, which has a less bulky ethyl group, proceeded with lower enantioselectivity (ee′ up to 80%) than α-tert-butylstyrenes. Although this is still a remarkable result for this challenging substrate, the lower ee was due to the isomerization of S12 (as observed in deuteration experiments; see the Supporting Information). Thus, like the most successful cases reported in the literature,22 the competition between direct hydrogenation and isomerization is responsible for the observed decrease in enantioselectivity. Börner et al. found that the use of 1,2-propylene carbonate (PC) as a solvent reduces the isomerization rate.14a We, therefore, performed the reaction of S12 in PC and we were glad to see that the enantioselectivity increased to 90% ee (entry 9).
As far as the hydrogenation of aryl trisubstituted olefins is concerned (S13–S19; Table 3, entries 10–16), the catalyst 4c also worked well for those with an E-geometry, S13 and S14 (ee′s up to 94%), which differ from S2 in the substituent of the aryl ring and the substituent trans to the aryl group as well as for the more challenging Z-geometry alkenes S15–S17 (ee′s up to 91%). In addition, the substrate scope was extended to the triaryl trisubstituted substrates S18 and S19 (ee′s up to 99%), whose reduction has been less studied despite the fact that they are an easy entry point to obtain diaryl methine chiral centers present in natural products and medicines.23 These catalytic results are completely consistent with the calculated TSs (vide supra). The analysis of the TSs indicated that the stereochemical outcome for the E-olefins mainly depends on steric factors. This finding suggested that enantioselectivities could also be high for substrates such as S2 that have a bulkier group in the position of the phenyl moiety. This hypothesis was confirmed by the high enantioselectivities (ee′s > 98%) found in the hydrogenation of substrates S20 and S21, which contain a bulky isopropyl and cyclohexyl group, respectively (Table 3, entries 17 and 18).24 These are valuable results because the highly enantioselective hydrogenation of purely alkyl substrates is rare,6 and indicate that the chiral pocket of the catalyst 4c is suitable for achieving the hydrogenation of these elusive substrates with excellent enantiocontrol.
The results up to this point led us to test the reduction of exocyclic trisubstituted olefins (S22–S30, Table 3). The hydrogenation of these substrates is of interest because the chiral benzofused ring motif is present in pharmaceuticals, natural products, and intermediates of relevant bioactive drugs.25 Despite the similarities with the acyclic olefins discussed above, the asymmetric hydrogenation of exocyclic olefins has hardly been explored and has yet to be resolved. The main challenge with exocyclic olefins is that the stereochemical outcome is highly influenced by ring size, and until recently, only a few examples had been able to provide high enantiocontrol, particularly for exocyclic olefins with a benzofused 5-membered ring7a,7b,26 although enantioselectivity decreased when an ortho-substituent was present and required an additive to work.27 Positively, the stereochemical outcome using Ir-catalyst 4c was barely affected by the size of the ring of the substrate, being able to hydrogenate five- and six-membered ring benzofused olefins with high enantioselectivities (up to 86% ee, Table 3) at room temperature without additives. In addition, 4c tolerates well the presence of several substituents that decorate the aryl group, even an ortho group. Note also that, surpassing the previously reported results, the more challenging benzofused olefin with a four-membered ring S30 could also be hydrogenated with a significant enantioselectivity of 74% ee.
We then moved on to asymmetric hydrogenation of key acyclic olefins with neighboring polar groups. In this context, a set of α,β-unsaturated trisubstituted acyclic enones S31–S36 (Scheme 3) could be hydrogenated with enantioselectivities comparable to the best ones reported but, in contrast to the asymmetric hydrogenation of di- and trisubstituted alkenes mentioned above, this was done with the catalytic system 4a.7d−7f,28 The reduction of these olefins opens a direct, atom-efficient path to prepare optically pure ketones, the synthesis of which until now has been mainly based on noncatalytic methods with a limited substrate scope. The attained enantioselectivities, between 95 and 98% ee, were quite independent of the nature of the substituents, which also allowed the successful hydrogenation of the highly appealing α-fluoride substituted enone S36.29 It has been reported that the stereochemical outcome in the hydrogenation of acyclic enones is greatly influenced by the enone substitution pattern and, therefore, only a few catalysts have been able to hydrogenate both α,β- and β,β-unsaturated trisubstituted enones with high enantioselectivities.28c,28d Gratifyingly, the catalytic system 4a also proved to be very efficient in the hydrogenation of β,β-unsaturated enones S37 and S38 (Scheme 3).
Scheme 3. Asymmetric Hydrogenation of α,β- and β,β-Unsaturated Trisubstituted Enones.

Full conversions were achieved in all cases.
We then tested whether the high enantioselectivities were maintained for acyclic olefins containing other relevant neighboring polar groups (see Scheme 4, substrates S39–S48). High enantioselectivities up to 98% in alkenyl boronic esters and enol phosphinates were obtained. Among these results, one can highlight the effective hydrogenation of the pure alkyl-trisubstituted enol phosphinates S44 and S46, a good alternative to the hydrogenation of dialkyl ketones to alcohols whose hydrogenation is still elusive. While for the reduction of vinyl boronate, the best enantioselectivity was achieved with 4b (95% ee); for enol phosphinates, the highest enantioselectivities (up to 98% ee) were with 4a. Both types of substrates are of interest because their reduction opens up straightforward routes for preparing enantiomerically pure organoboron and organophosphorus compounds, which can be easily transformed into high-value compounds.30 The excellent enantioselectivities obtained in the hydrogenation of the trisubstituted alkenyl boronic ester and enol phosphinates were also reached in the even more challenging disubstituted analogues (S40, S41 and S47, S48; up to 92% ee), including the hydrogenation of nonaromatic disubstituted olefins S41 and S47.
Scheme 4. Asymmetric Hydrogenation of Vinyl Boronates S39–S41 and Enol Phosphinates S42–S48.

Full conversions were achieved in all cases.
Reactions carried out using 4b.
Reactions carried out with 4a.
Subsequently, we focused on the asymmetric hydrogenation of exocyclic olefins containing a neighboring polar group (Scheme 5, S49–S68). In particular, we considered the hydrogenation of α,α-unsaturated exocyclic enones and α,α-unsaturated lactones and lactams, since the reduced products of these olefins are encountered in natural products and drugs.31 These substrates suffer from the same ring size limitation that was discussed for exocyclic olefins without a neighboring polar group.7 In our case, however, the hydrogenation of the exocyclic enones S49 and S50 using 4a proceeded with high enantioselectivities (up to 97%), comparable to the best ones, regardless of the size of the ring. In addition, hydrogenation of α,α-unsaturated lactones (S51–S59) also proceeded with excellent levels of enantioselectivity (ee′s up to 99%) regardless of the size of the lactone ring. In addition, ee′s were found to be quite independent of the electronic and steric nature of the olefinic substituent. Chiral α-substituted-δ-valerolactones and γ-butyrolactones were therefore attained with ee′s up to 99%. The hydrogenation of α,α-unsaturated lactams (S60–S68) followed the same trend as related lactones, with ee′s up to >99%. Note that the Ir-catalyst 4a also allows the presence of different protecting groups, such as Bn, Ac, and Boc, albeit in the latter case, the Boc group can also be partially cleaved under the reaction conditions.
Scheme 5. Asymmetric Hydrogenation of Exocyclic α,α-Unsaturated Enones, Lactones, and Lactams (S49–S68).
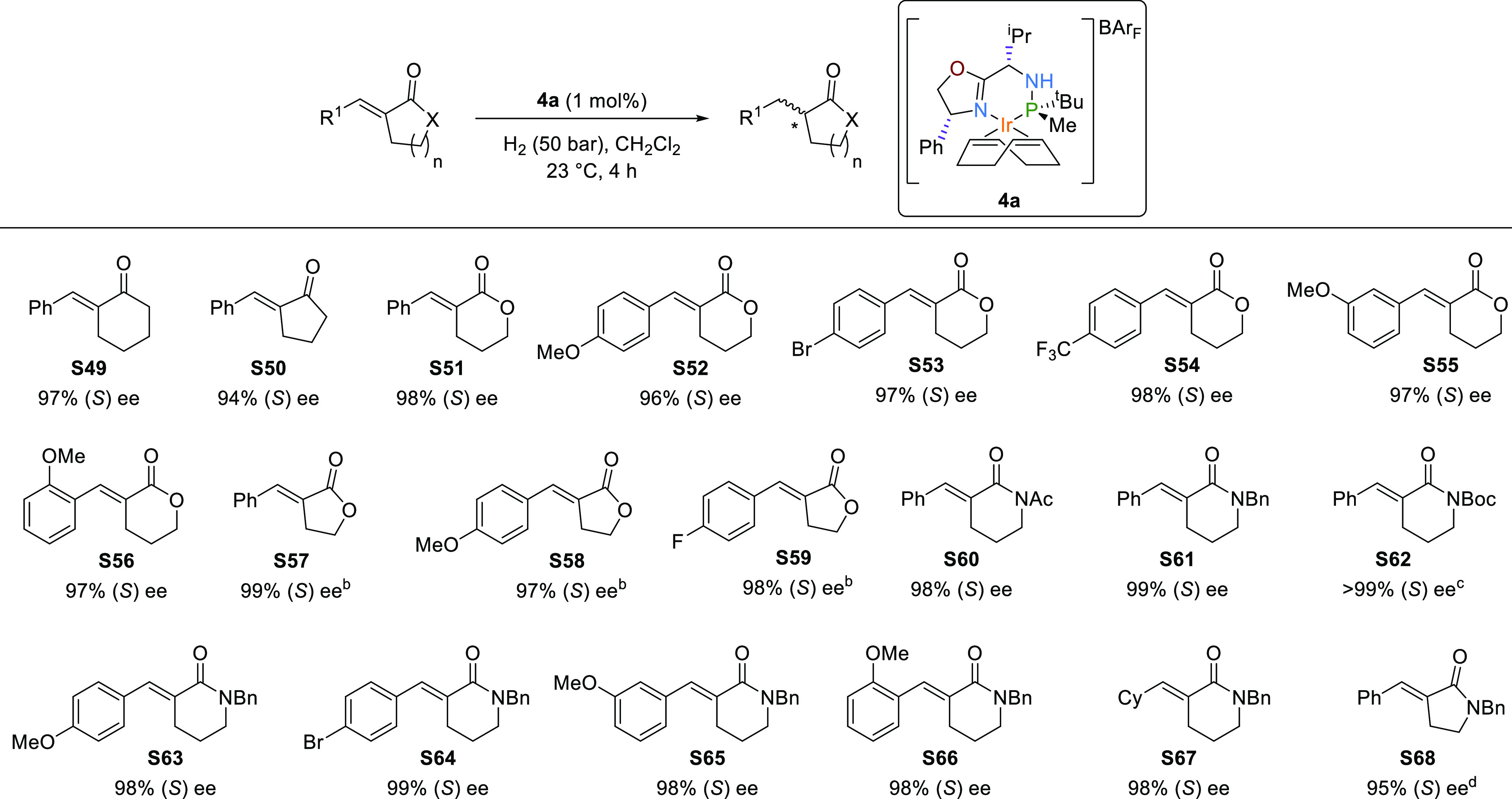
Full conversions were attained in all cases otherwise noted.
Reactions carried out using 2 mol% of catalysts.
28% of deprotected lactam was also obtained.
76% conversion was attained.
Finally, we studied how using Ir-catalysts 1-4a-c, we can extend the asymmetric hydrogenation domain to new types of tetrasubstituted olefins. Tetrasubstituted acyclic olefins are considered to be some of the most challenging substrates to be hydrogenated due to the difficulty in differentiating the prochiral faces and the slow activities that result from their steric hindrance. Compared to the progress made with functionalized tetrasubstituted olefins, the reduction of nonchelating tetrasubstituted acyclic olefins remains an open challenge. Furthermore, there are only a few reports on the hydrogenation of tetrasubstituted olefins with poorly coordinative groups that can create intermediates useful for subsequent synthesis.10 As mentioned in the Introduction section, the Ir-catalysts 1-4a-c were successfully applied in reducing a range of nonchelating tetrasubstituted substrates, most of them without poorly coordinative groups. However, high enantioselectivities were attained in the reduction of several acyclic-tetrasubstituted vinyl fluorides containing an ester functionality such as substrates S69 type (Scheme 6).8c The challenge of these substrates is that the catalysts must not only control enantioselectivity but also diastereoselectivity (two vicinal stereogenic centers are created) and the defluorination side reaction. We first studied whether we could further expand the previous olefin scope to the reduction of the elusive vinyl fluoride S70 with an ester functionality and also a CF3-functional group instead of the methyl group of S69.32 Improving on previous results reported in the literature (67% ee)10c the reduction proceeded for the first time with high enantioselectivity (87% ee; Scheme 6), excellent diastereoselectivity without any defluorination with 4c. The result is in line with the quadrant model developed for 4c (vide supra, Figure 2a). The smallest substituent of the olefin (F) is placed in the most hindered quadrant (Q3) and the aryl substituent is in the semihindered quadrant Q1. According to this model, the predicted absolute configuration of the reduced product would be 2S, 3R, in agreement with the experimental result. Positively, the high enantioselectivity was extended for the first time to substrates with different aryl substituents S71–S73 (Scheme 6).
Scheme 6. Asymmetric Hydrogenation of Tetrasubstituted Olefins S69–S82.
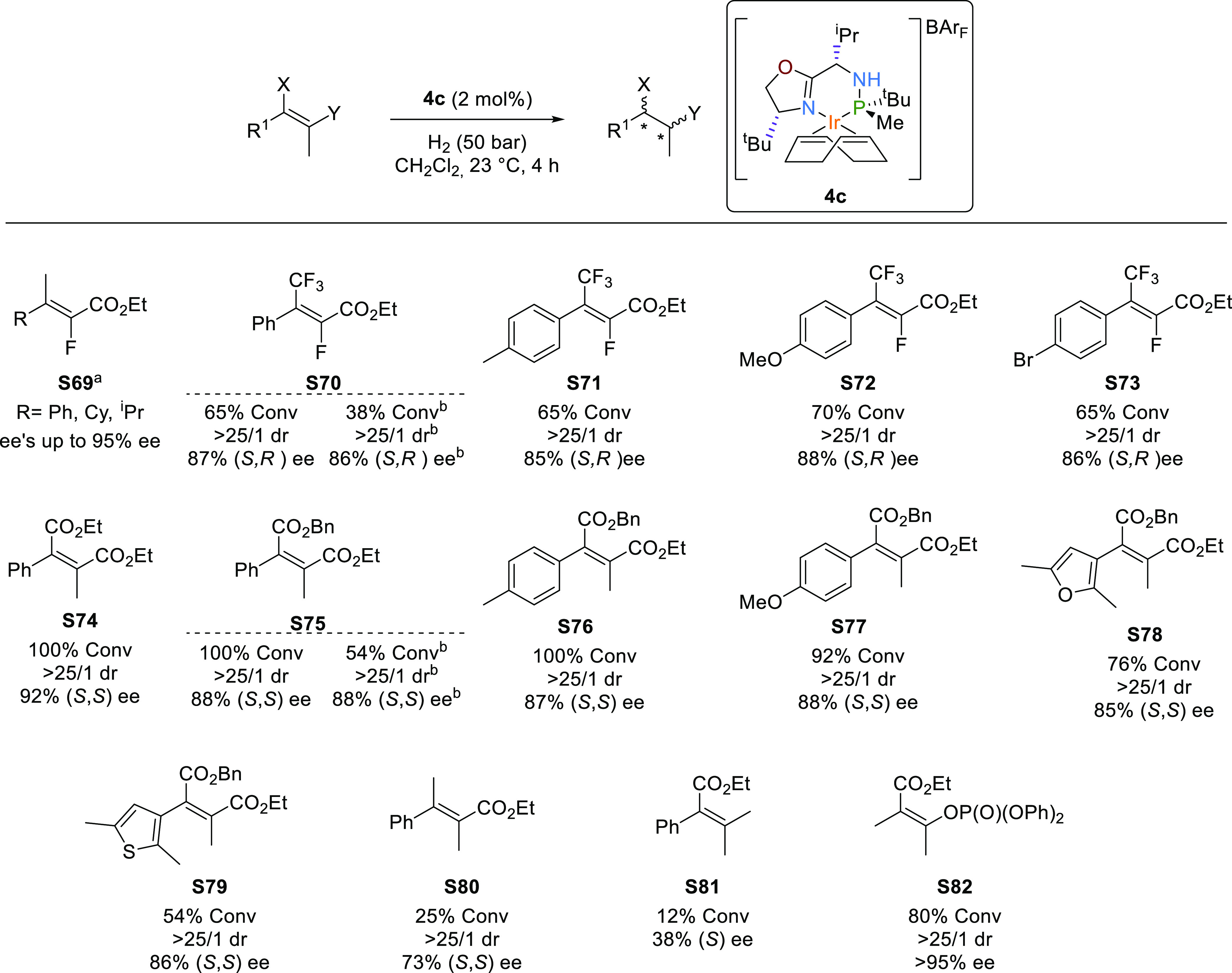
Data from ref (8).
Reaction carried out at 1 mol % of catalysts.
Encouraged by these results, we then studied other functionalized tetrasubstituted olefins lacking a strong coordinative group. Due to the importance of succinic acid derivatives,33 we focused on the asymmetric hydrogenation of tetrasubstituted maleates, with two vicinal ester groups (substrates S74–S79; Scheme 6), as an atom-efficient method for their preparation. The reactions with 4c proceeded smoothly, providing the hydrogenated products with excellent diastereoselectivity (>25/1 dr) and high enantioselectivities (up to 92%). Moreover, the enantioselectivity was almost unaffected by the electronic nature of the aromatic group (S75–S77) or the presence of heteroaromatic cyclic substituents (S78 and S79).
Next, we studied whether these results could be reproduced by replacing one of the ester groups with other substituents (Scheme 6). While the exchange of any of the esters by a methyl group (S80 and S81) led to a decrease in activity and enantioselectivity (ee′s up to 73%), positively the reduction of S82, with a phosphate instead of one of the ester groups, proceeded with high enantioselectivity (>95% ee) and diastereoselectivity (>25/1 dr), being the first time that this substrate class was hydrogenated.
Based on the recent findings by Gosslein and collaborators of an Ir–P,N catalyst applicable to a wide range of unfunctionalized tetrasubstituted acyclic olefins containing two or three aryl substituents,9 the scope of our iridium catalysts 1–4 was also studied in the reduction of some of these unfunctionalized olefins (Scheme 7 and the Supporting Information for pressure and catalyst loading effects). Initially, we studied the hydrogenation of substrate S83, having two phenyl groups in a trans disposition. In agreement with our quadrant model, high diastereo- and enantioselectivities were attained (>25/1 dr and 99% ee). Calculations performed for substrate S83 with the catalyst 4c reproduce the enantiomeric excess (computed 99% ee (R,R)) and support our quadrant model; see the Supporting Information for further details. We then proceed to study several E-1,2-dialkyl-1,2-diaryl olefins (S84–S86). Overcoming the limitations of Gosselin′s system,9 our catalyst was able to differentiate the Re- and Si-faces in substrates differentiated only in the length of an alkyl substituent S84 and S85 and in the electronic properties of the aromatic substituents S86. Thus, enantioselectivities >95% ee were achieved for these elusive substrate types.
Scheme 7. Asymmetric Hydrogenation of Tetrasubstituted Olefins S83–S86.

Finally, to show the potential utility of our catalysts, we also carried out the reaction of some representative di-, tri-, and tetrasubstituted substrates (S1, S31, and S83) on a 7.5 mmol scale (Scheme 8).
Scheme 8. Practical Hydrogenation of S1, S31, and S83.

Conclusions
In summary, we have shown that Ir-MaxPHOX catalysts (1-4a-c) that had been previously found to be successful in the asymmetric hydrogenation of nonfunctionalized cyclic and few acyclic-tetrasubstituted olefins are also good performers in the hydrogenation of a new set of 84 olefins which included di- and trisubstituted olefins, some with key poorly coordinative groups (such as lactams, lactones, enol phosphinates, ···) and some new examples of challenging tetrasubstituted alkenes. This family of Ir-MaxPHOX-type catalysts allowed the hydrogenation of exocyclic olefins, Z-olefins, pure alkyl-substituted olefins, and a broad range of tetrasubstituted olefins, thus improving over a previous family7b also based on P,N ligands, that was so far the only one able to hydrogenate di-, tri-, and tetrasubstituted olefins. DFT calculations and deuterium labeling experiments allowed the rationalization of the stereochemical outcomes of the reactions and helped in the selection of suitable substrates for these Ir-MaxPHOX-type catalysts. The analysis of the TSs indicated that the high catalytic performance of these catalysts is due to their ability to adapt to the demands of each substrate. This ability also explains its excellent performance in the hydrogenation of functionalized olefins such as allyl amines and phthalimides,34 cyclic α- and β-enamides,12 and imines.35 These results open a new perspective for the growth of ligand libraries for the asymmetric hydrogenation of nonchelating olefins, where the Ir/P-stereogenic aminophosphine-oxazoline catalysts could be a good choice for further development.
Experimental Section
General Considerations
All reactions were carried out using standard Schlenk techniques under an atmosphere of argon. Solvents were purified and dried by standard procedures. All reagents were used as received. Ir-catalyst precursors 1-4a-c were prepared as previously reported.121H and 13C{1H} were recorded using a 400 MHz spectrometer. Chemical shifts are relative to that of SiMe4 (1H and 13C). 1H and 13C assignments were made based on 1H–1H gCOSY and 1H–13C gHSQC.
Typical Procedure for the Hydrogenation of Olefins
The alkene (0.5 mmol) and Ir complex (1 or 2 mol %) were dissolved in CH2Cl2 (2 mL) in a high-pressure autoclave, which was purged four times with hydrogen. The apparatus was pressurized to the desired pressure, and after the required reaction time, the autoclave was depressurized and the solvent evaporated. The residue was dissolved in Et2O (1.5 mL) and filtered through a short Celite plug.
Computational Details
All species were optimized using the B3LYP15-D316 functional as implemented in Gaussian 09.36 The LANL2DZ37 basis set together with the associated pseudopotential was used for iridium, and the 6-31G**38 basis set was used for all other atoms. Implicit solvation using the PCM39 model with the parameters for dichloromethane was included in geometry optimizations. The reported energies are Gibbs free energies in solution within the quasi-harmonic approximation to the Rigid Rotor Harmonic Oscillator Model proposed by Cramer and Truhlar;40 corrections were done using the GoodVibes program.41 Single-point calculations were done using Grimme’s standalone pure functional B97D342 and the larger basis sets cc-pVTZ43 for all atoms except for Ir, for which the cc-pVTZ-PP44,45 basis sets were used instead (see the Supporting Information).
Quadrant analysis was done by means of MolQuO (Quantitative Quadrant Diagram Representation of Molecular Systems).19 Note that this analysis was done taking the geometry of the whole TS, as shown in the figure, but removing the atoms of the olefin in the MolQuO calculation.
Acknowledgments
This work was supported by grants from the FEDER/Ministerio de Ciencia e Innovación (MICINN)/AEI (PID2019-104904GB-I00, PID2021-128128NB-100, PID2020-115074GB-I00, PID2020-112825RB-I00, and CEX2019-000925-S). Grant 2017SGR1472, funded by the Catalan Government, is also gratefully acknowledged. ICIQ and IRB Barcelona are recipients of institutional funding from MICINN through the Centres of Excellence Severo Ochoa award and from the CERCA Program of the Catalan Government.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c05579.
Calculated energies and computed cartesian coordinates for all TSs; synthesis of substrates; characterization details and enantiomeric excess determination of hydrogenated products; copies of NMR spectra and GC or HPLC traces for ee determination of hydrogenated products, and hydrogenation experiments carried out in PC and deuterogenation experiments (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Noyori R.Asymmetric Catalysis in Organic Synthesis; Wiley: New York, 1994. [Google Scholar]; b Comprehensive Asymmetric Catalysis; Jacobsen E. N.; Pfaltz A.; Yamamoto H., Eds.; Springer-Verlag: Berlin, 1999. [Google Scholar]; c Asymmetric Catalysis in Industrial Scale: Challenges, Approaches and Solutions, 2nd ed.; Blaser H.-U.; Federsel H.-J., Eds.; Wiley: Weinheim, 2010. [Google Scholar]; d Catalytic Asymmetric Synthesis, 4th ed.; Akiyama T.; Ojima I., Eds.; John Wiley & Sons, Inc: Hoboken, 2022. [Google Scholar]
- a Busacca C. A.; Fandrick D. R.; Song J. J.; Senanayake C. H. The Growing Impact of Catalysis in the Pharmaceutical Industry. Adv. Synth. Catal. 2011, 353, 1825–1864. 10.1002/adsc.201100488. [DOI] [Google Scholar]; b Ager D. J.; de Vries A. H. M.; de Vries J. G. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 2012, 41, 3340–3380. 10.1039/c2cs15312b. [DOI] [PubMed] [Google Scholar]; c Metal-Catalyzed Asymmetric Hydrogenation. Evolution and Prospect. In Advance Catalysis; Diéguez M.; Pizzano A., Eds.; Elsevier: Oxford, 2021; Vol. 68. [Google Scholar]
- Biosca M.; Diéguez M.; Zanotti-Gerosa A.. Asymmetric Hydrogenation in Industry. In Advances in Catalysis; Elsevier, 2021; Vol. 68, pp 341–384. [Google Scholar]
- See for example:; a Genêt J. P.Modern Reduction Methods; Andersson P. G.; Munslow I. J., Eds.; Wiley-VCH: Weinheim, 2008; pp 3–38. [Google Scholar]; b Tang W.; Zhang X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3069. 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]; c Chi Y.; Tang W.; Zhang X.. Modern Rhodium-Catalyzed Organic Reactions; Evans P. A., Ed.; Wiley-VCH: Weinheim, 2005; pp 1–31. [Google Scholar]; d Kitamura M.; Noyori R.. Ruthenium in Organic Synthesis; Murahashi S.-I., Ed.; Wiley-VCH: Weinheim, 2004; pp 3–52. [Google Scholar]; e Weiner B.; Szymanski W.; Janssen D. B.; Minnaard A. J.; Feringa B. L. Recent Advances in the Catalytic Asymmetric Synthesis of Beta-amino Acids. Chem. Soc. Rev. 2010, 39, 1656–1691. [DOI] [PubMed] [Google Scholar]; f Xie J.-H.; Zhu S.-F.; Zhou Q.-L. Transition Metal-Catalyzed Enantioselective Hydrogenation of Enamines and Imines. Chem. Rev. 2011, 111, 1713–1760. 10.1021/cr100218m. [DOI] [PubMed] [Google Scholar]; g Etayo P.; Vidal-Ferran A. Rhodium-catalysed asymmetric hydrogenation as a valuable synthetic tool for the preparation of chiral drugs. Chem. Soc. Rev. 2013, 42, 728–754. 10.1039/C2CS35410A. [DOI] [PubMed] [Google Scholar]; h Pizzano A.Asymmetric Hydrogenation of Functionalized Olefins. In Advances in Catalysis; Elsevier, 2021; Vol. 68, pp 1–134. [Google Scholar]; i Kim A. N.; Stoltz B. M. Recent advances in homogeneous catalysts for the asymmetric hydrogenation of heteroarenes. ACS Catal. 2020, 10, 13834–13851. 10.1021/acscatal.0c03958. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Cabré A.; Verdaguer X.; Riera A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 2022, 122, 269–339. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Zhang Z.; Butt N. A.; Zhang W. Asymmetric Hydrogenation of Nonaromatic Cyclic Substrates. Chem. Rev. 2016, 116, 14769–14827. 10.1021/acs.chemrev.6b00564. [DOI] [PubMed] [Google Scholar]
- a Cui X.; Burgess K. Catalytic Homogeneous Asymmetric Hydrogenations of Largely Unfunctionalized Alkenes. Chem. Rev. 2005, 105, 3272–3296. 10.1021/cr0500131. [DOI] [PubMed] [Google Scholar]; b Roseblade S. J.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Olefins. Acc. Chem. Res. 2007, 40, 1402–1411. 10.1021/ar700113g. [DOI] [PubMed] [Google Scholar]; c Woodmansee D. H.; Pfaltz A. Asymmetric Hydrogenation of Alkenes Lacking Coordinating Groups. Chem. Commun. 2011, 47, 7912–7916. 10.1039/c1cc11430a. [DOI] [PubMed] [Google Scholar]; d Zhu Y.; Burgess K. Filling Gaps in Asymmetric Hydrogenation Methods for Acyclic Stereocontrol: Application to Chirons for Polyketide-Derived Natural Products. Acc. Chem. Res. 2012, 45, 1623–1636. 10.1021/ar200145q. [DOI] [PubMed] [Google Scholar]; e Verendel J. J.; Pàmies O.; Diéguez M.; Andersson P. G. Asymmetric Hydrogenation of Olefins Using Chiral Crabtree-type Catalysts: Scope and Limitations. Chem. Rev. 2014, 114, 2130–2169. 10.1021/cr400037u. [DOI] [PubMed] [Google Scholar]; f Margarita C.; Andersson P. G. Evolution and Prospects of the Asymmetric Hydrogenation of Unfunctionalized Olefins. J. Am. Chem. Soc. 2017, 139, 1346–1356. 10.1021/jacs.6b10690. [DOI] [PubMed] [Google Scholar]; g Pàmies O.; Zheng J.; Faiges J.; Andersson P. G.. Asymmetric Hydrogenation of Unfunctionalized Olefins or with Poorly Coordinative Groups. In Advances in Catalysis; Elsevier, 2021; Vol. 68, pp 135–203. [Google Scholar]; For specific examples of application of P,O and P,S-ligands in the Ir-catalyzed asymmetric hydrogenation see:; h Margalef J.; Pàmies O.; Pericas M. A.; Diéguez M. )Evolution of phosphorus-thioether ligands for asymmetric catalysis. Chem. Commun. 2020, 56, 10795–10808. 10.1039/D0CC04145A. [DOI] [PubMed] [Google Scholar]; i Margalef J.; Pericàs M. A.. Chiral Bidentate Heterodonor P-S/O Ligands. In Chiral Ligands; Diéguez M., Ed.; CRC Press, 2021; pp 81–108. [Google Scholar]; j Rageot D.; Woodmansee D. H.; Pugin B.; Pfaltz A. Proline-Based P,O Ligand/Iridium Complexes as Highly Selective Catalysts: Asymmetric Hydrogenation of Trisubstituted Alkenes. Angew. Chem., Int. Ed. 2011, 50, 9598–9601. 10.1002/anie.201104105. [DOI] [PubMed] [Google Scholar]
- a Bell S.; Wüstenberg B.; Kaiser S.; Menges F.; Netscher T.; Pfaltz A. Asymmetric Hydrogenation of Unfunctionalized, Purely Alkyl-Substituted Olefins. Science 2006, 311, 642–644. 10.1126/science.1121977. [DOI] [PubMed] [Google Scholar]; b Wang A.; Fraga R. P. A.; Hörmann E.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized, Trialkyl-Substituted Olefins. Chem. Asian J. 2011, 6, 599–606. 10.1002/asia.201000595. [DOI] [PubMed] [Google Scholar]
- The hydrogenation of such substrates is highly influenced by the size of substrate ring. So, for instance, a catalyst that provides high enantioselectivities in the hydrogenation of exocyclic olefins attached to a 5-membered ring motif is not suitable for the reduction of the 6-membered ring counterparts or vice versa. For non-functionalized olefins, see for instance:; a Xia J.; Yang G.; Zhuge R.; Liu Y.; Zhang W. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized Exocyclic C=C Bonds. Chem. - Eur. J. 2016, 22, 18354–18357. 10.1002/chem.201604298. [DOI] [PubMed] [Google Scholar]; (up to 97% for benzofused 5-membered ring olefins and 75% ee for 6-membered ring counterparts); b Biosca M.; Magre M.; Pàmies O.; Diéguez M. Asymmetric Hydrogenation of Disubstituted, Trisubstituted, and Tetrasubstituted Minimally Functionalized Olefins and Cyclic β-Enamides with Easily Accessible Ir–P,Oxazoline Catalysts. ACS Catal. 2018, 8, 10316–10320. 10.1021/acscatal.8b03170. [DOI] [Google Scholar]; (up to 93% for benzofused 5-membered ring olefins and 30% ee for 6-membered ring counterparts); c Biosca M.; de la Cruz-Sánchez P.; Tarr D.; Llanes P.; Karlsson E. A.; Margalef J.; Pàmies O.; Pericàs M. A.; Diéguez M. Filling the gaps in the challenging asymmetric hydrogenation of exocyclic benzofused-based alkenes with Ir-P,N catalysts. Adv. Synth. Catal. 2023, 365, 167–177. 10.1002/adsc.202200870. [DOI] [Google Scholar]; (up to 96% and 99% for benzofused 5- and 6-membered ring olefins, respectively, and 40% ee for the 4-memberd ring counterpart). For olefins with poorly coordinative groups, see for instance:; d Tian F.; Yao D.; Liu Y.; Xie F.; Zhang W. Iridium-Catalyzed Highly Enantioselective Hydrogenation of Exocyclic α,β-Unsaturated Carbonyl Compounds. Adv. Synth. Catal. 2010, 352, 1841–1845. 10.1002/adsc.201000185. [DOI] [Google Scholar]; (up to 99% ee for 5-membered ring cyclic enones, lactones and lactams and up to 75% ee for 6-membered ring counterparts); e Liu X.; Han Z.; Wang Z.; Ding K. SpinPhox/Iridium(I)-Catalyzed Asymmetric Hydrogenation of Cyclic α-Alkylidene Carbonyl Compounds. Angew. Chem., Int. Ed. 2014, 53, 1978–1982. 10.1002/anie.201309521. [DOI] [PubMed] [Google Scholar]; (up to 98% ee for 6-membered ring cyclic enones, lactones and lactams and up to 83% ee for 5-membered ring counterparts); f Xia J.; Nie Y.; Yang G.; Liu Y.; Gridnev I. D.; Zhang W. Ir-Catalyzed Asymmetric Hydrogenation of α-Alkylidene β-Lactams and Cyclobutanones. Chin. J. Chem. 2018, 36, 612–618. 10.1002/cjoc.201800088. [DOI] [Google Scholar]; (catalysts specially designed for 4-membered ring cyclic enones and lactams; ee′s up to 98%); g Margalef J.; Biosca M.; de la Cruz-Sánchez P.; Caldentey X.; Rodríguez-Escrich C.; Pàmies O.; Pericàs M. A.; Diéguez M. Indene Derived Phosphorus-Thioether Ligands for the Ir-Catalyzed Asymmetric Hydrogenation of Olefins with Diverse Substitution Patterns and Different Functional Groups. Adv. Synth. Catal. 2021, 363, 4561–4574. 10.1002/adsc.202100069. [DOI] [Google Scholar]; (catalysts designed for 6-membered ring cyclic enones, lactones and lactams; ee′s up to 99%)
- a Schrems M. G.; Neumann E.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized Tetrasubstituted Olefins. Angew. Chem., Int. Ed. 2007, 46, 8274–8276. 10.1002/anie.200702555. [DOI] [PubMed] [Google Scholar]; b Busacca C. A.; Qu B.; Grět N.; Fandrick K. R.; Saha A. K.; Marsini M.; Reeves D.; Haddad N.; Eriksson M.; Wu J. P.; Grinberg N.; Lee H.; Li Z.; Lu B.; Chen D.; Hong Y.; Ma S.; Senanayake C. H. Tuning the Peri Effect for Enantioselectivity: Asymmetric Hydrogenation of Unfunctionalized Olefins with the BIPI Ligands. Adv. Synth. Catal. 2013, 355, 1455–1463. 10.1002/adsc.201201104. [DOI] [Google Scholar]; c Biosca M.; Salomó E.; de la Cruz-Sánchez P.; Riera A.; Verdaguer X.; Pàmies O.; Diéguez M. Extending the Substrate Scope in the Hydrogenation of Unfunctionalized Tetrasubstituted Olefins with Ir-P Stereogenic Aminophosphine–Oxazoline Catalysts. Org. Lett. 2019, 21, 807–811. 10.1021/acs.orglett.8b04084. [DOI] [PubMed] [Google Scholar]
- Bigler R.; Mack K. A.; Shen J.; Tosatti P.; Han C.; Bachmann S.; Zhang H.; Scalone M.; Pfaltz A.; Denmark S. E.; Hildbrand S.; Gosselin F. Asymmetric Hydrogenation of Unfunctionalized Tetrasubstituted Acyclic Olefins. Angew. Chem., Int. Ed. 2020, 59, 2844–2849. 10.1002/anie.201912640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kerdphon S.; Ponra S.; Yang J.; Wu H.; Eriksson L.; Andersson P. G. Diastereo- and Enantioselective Synthesis of Structurally Diverse Succinate, Butyrolactone, and Trifluoromethyl Derivatives by Iridium-Catalyzed Hydrogenation of Tetrasubstituted Olefins. ACS Catal. 2019, 9, 6169–6176. 10.1021/acscatal.9b01508. [DOI] [Google Scholar]; b Zhao Q.-K.; Wu X.; Li L.-P.; Yang F.; Xie J.-H.; Zhou Q.-L. Asymmetric Hydrogenation of β-Aryl Alkylidene Malonate Esters: Installing an Ester Group Significantly Increases the Efficiency. Org. Lett. 2021, 23, 1675–1680. 10.1021/acs.orglett.1c00093. [DOI] [PubMed] [Google Scholar]; c Ponra S.; Rabten W.; Yang J.; Wu H.; Kerdphon S.; Andersson P. G. Diastereo-and enantioselective synthesis of fluorine motifs with two contiguous stereogenic centers. J. Am. Chem. Soc. 2018, 140, 13878–13883. 10.1021/jacs.8b08778. [DOI] [PubMed] [Google Scholar]
- a Pfaltz A.; Drury W. J. III Design of chiral ligands for asymmetric catalysis: From C2-symmetric P,P- and N,N-ligands to sterically and electronically nonsymmetrical P,N-ligands. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5723–5726. 10.1073/pnas.0307152101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoon T. P.; Jacobsen E. N. Privileged chiral catalysts.. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]; c Sommer W.; Weibel D. Asymmetric Catalysis, Privileged Ligands and Complexes. Sigma Aldrich′s ChemFiles 2008, 2, 1–91. [Google Scholar]; d Privileged Chiral Ligands and Catalysts; Zhou Q., Ed.; John Wiley& Sons Inc.: New York, 2011. [Google Scholar]; e Chiral Ligands. Evolution of Ligand Libraries for Asymmetric Catalysis; Diéguez M., Ed.; CRC Press: Boca Raton, 2021. [Google Scholar]
- a Salomó E.; Orgué S.; Riera A.; Verdaguer X. Highly Enantioselective Iridium-Catalyzed Hydrogenation of Cyclic Enamides. Angew. Chem., Int. Ed. 2016, 55, 7988–7992. 10.1002/anie.201602219. [DOI] [PMC free article] [PubMed] [Google Scholar]; The Ir-MaxPHOX library has also been applied in the asymmetric hydrogenation of functionalized olefins and imines as well as in the isomerization of alkenes, see:; b Cabré A.; Riera T.; Verdaguer X. P-Stereogenic Amino-Phosphines as Chiral Ligands: From Privileged Intermediates to Asymmetric Catalysis. Acc. Chem. Res. 2020, 53, 676–689. 10.1021/acs.accounts.9b00633. [DOI] [PubMed] [Google Scholar]
- To the best of our knowledge so far only one family of Ir-catalysts has been successfully applied to di-, tri- and tetrasubstituted olefins, see ref 7b
- a Bayardon J.; Holz J.; Schäffner B.; Andrushko V.; Verevkin S. P.; Preetz A.; Börner A. Propylene Carbonate as a Solvent for Asymmetric Hydrogenations. Angew. Chem., Int. Ed. 2007, 46, 5971–5974. 10.1002/anie.200700990. [DOI] [PubMed] [Google Scholar]; b Schäffner B.; Holz J.; Verevkin S. P.; Börner A. Organic carbonates as alternative solvents for palladium-catalyzed substitution reactions. ChemSusChem 2008, 1, 249–253. 10.1002/cssc.200700142. [DOI] [PubMed] [Google Scholar]; c Schäffner B.; Schäffner B.; Verevkin S. P.; Börner A. Organic carbonates as solvents in synthesis and catalysis. Chem. Rev. 2010, 110, 4554–4581. 10.1002/cssc.200700142. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. Density-functional thermochemistry. III. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. A 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- a Brandt P.; Hedberg C.; Andersson P. G. New Mechanistic Insights into the Iridium–Phosphanooxazoline-Catalyzed Hydrogenation of Unfunctionalized Olefins: A DFT and Kinetic Study. Chem. - Eur. J. 2003, 9, 339–347. 10.1002/chem.200390029. [DOI] [PubMed] [Google Scholar]; b Fan Y.; Cui X.; Burgess K.; Hall M. B. Electronic effects steer the mechanism of asymmetric hydrogenations of unfunctionalized aryl-substituted alkenes. J. Am. Chem. Soc. 2004, 126, 16688–16689. 10.1021/ja044240g. [DOI] [PubMed] [Google Scholar]; c Cui X.; Fan Y.; Hall M. B.; Burgess K. Mechanistic Insights into Iridium-Catalyzed Asymmetric Hydrogenation of Dienes. Chem. - Eur. J. 2005, 11, 6859–6868. 10.1002/chem.200500762. [DOI] [PubMed] [Google Scholar]; d Church T. L.; Rasmussen T.; Andersson P. G. Enantioselectivity in the Iridium-Catalyzed Hydrogenation of Unfunctionalized Olefins. Organometallics 2010, 29, 6769–6781. 10.1021/om100899u. [DOI] [Google Scholar]; e Hopmann K. H.; Bayer A. On the mechanism of iridium-catalyzed asymmetric hydrogenation of imines and alkenes: A theoretical study. Organometallics 2011, 30, 2483–2497. 10.1021/om1009507. [DOI] [Google Scholar]; f Mazuela J.; Norrby P.-O.; Andersson P. G.; Pam̀ies O.; Diéguez M. Pyranoside Phosphite–Oxazoline Ligands for the Highly Versatile and Enantioselective Ir-Catalyzed Hydrogenation of Minimally Functionalized Olefins. A Combined Theoretical and Experimental Study. J. Am. Chem. Soc. 2011, 133, 13634–13645. 10.1021/ja204948k. [DOI] [PubMed] [Google Scholar]; g Gruber S.; Pfaltz A. Asymmetric hydrogenation with iridium C, N and N, P ligand complexes: characterization of dihydride intermediates with a coordinated alkene. Angew. Chem., Int. Ed. 2014, 53, 1896–1900. 10.1002/anie.201309515. [DOI] [PubMed] [Google Scholar]
- Àlvarez-Moreno M.; de Graaf C.; Lopez N.; Maseras F.; Poblet J. M.; Bo C. Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform. J. Chem. Inf. Model. 2015, 55, 95–103. 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
- a Aguado-Ullate S.; Saureu S.; Guasch L.; Carbo J. J. Theoretical Studies of Asymmetric Hydroformylation Using the Rh–(R,S)-BINAPHOS Catalyst – Origin of Coordination Preferences and Stereoinduction. Chem. - Eur. J. 2012, 18, 995–1005. 10.1002/chem.201101230. [DOI] [PubMed] [Google Scholar]; b Aguado- Ullate S.; Urbano-Cuadrado M.; Villalba I.; Pires E.; García J. I.; Bo C.; Carbó J. J. Predicting the Enantioselectivity of the Copper-Catalysed Cyclopropanation of Alkenes by Using Quantitative Quadrant-Diagram Representations of the Catalysts. Chem. - Eur. J. 2012, 18, 14026–14036. 10.1002/chem.201201135. [DOI] [PubMed] [Google Scholar]
- Note, that this analysis was performed by taking the geometry of the whole TS, as shown in the figure, but removing the olefin atoms in the MolQuO calculation
- a Mazuela J.; Verendel J. J.; Coll M.; Schäffner B.; Börner A.; Andersson P. G.; Pàmies O.; Diéguez M. Iridium Phosphite-Oxazoline Catalysts for the Highly Enantioselective Hydrogenation of Terminal Alkenes. J. Am. Chem. Soc. 2009, 131, 12344–12353. 10.1021/ja904152r. [DOI] [PubMed] [Google Scholar]; b Pàmies O.; Andersson P. G.; Diéguez M. Asymmetric Hydrogenation of Minimally Functionalised Terminal Olefins: An Alternative Sustainable and Direct Strategy for Preparing Enantioenriched Hydrocarbons. Chem. - Eur. J. 2010, 16, 14232–14240. 10.1002/chem.201001909. [DOI] [PubMed] [Google Scholar]
- a Blankenstein J.; Pfaltz A. A New Class of Modular Phosphinite–Oxazoline Ligands: Ir-Catalyzed Enantioselective Hydrogenation of Alkenes. Angew. Chem., Int. Ed. 2001, 40, 4445–4447. [DOI] [PubMed] [Google Scholar]; b McIntyre S.; Hörmann E.; Menges F.; Smidt S. P.; Pfaltz A. Iridium-Catalyzed Enantioselective Hydrogenation of Terminal Alkenes. Adv. Synth. Catal. 2005, 347, 282–288. 10.1002/adsc.200404256. [DOI] [Google Scholar]; c Biosca M.; Paptchikhine A.; Pam̀ies O.; Andersson P. G.; Dieǵuez M. Extending the Substrate Scope of Bicyclic P-Oxazoline/Thiazole Ligands for Ir-Catalyzed Hydrogenation of Unfunctionalized Olefins by Introducing a Biaryl Phosphoroamidite Group. Chem. - Eur. J. 2015, 21, 3455–3464. 10.1002/chem.201405361. [DOI] [PubMed] [Google Scholar]; d Krajangsri S.; Wu H.; Liu J.; Rabten W.; Singhband T.; Andersson P. G. Tandem Peterson olefination and chemoselective asymmetric hydrogenation of β-hydroxy silanes. Chem. Sci. 2019, 10, 3649–3653. 10.1039/c8sc05261a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fessard T. C.; Andrews S. P.; Motoyohsi H.; Carreira E. Enantioselective Preparation of 1, 1-Diarylethanes: Aldehydes as Removable Steering Groups for Asymmetric Synthesis. Angew. Chem., Int. Ed. 2007, 46, 9331–9334. 10.1002/anie.200702995. [DOI] [PubMed] [Google Scholar]; b Prat L.; Dupas G.; Duflos J.; Quéguiner G.; Bourguignon J.; Levacher V. Deracemization of alkyl diarylmethanes using (−)-sparteine or a chiral proton source. Tetrahedron Lett. 2001, 42, 4515–4518. 10.1016/s0040-4039(01)00784-5. [DOI] [Google Scholar]; c Wilkinson J. A.; Rossington S. B.; Ducki S.; Leonard J.; Hussain N. Asymmetric alkylation of diarylmethane derivatives. Tetrahedron 2006, 62, 1833–1844. 10.1016/j.tet.2005.11.044. [DOI] [Google Scholar]
- Pure alkyls are difficult to study because the enantiomers of pure hydrocarbons are difficult to separate. This was also the case of the substrate (E)-3,4,4-trimethylpent-2-ene (with a tBu group), whose measurement of ee failed despite we obtained 100% conversion
- a Donde Y.; Nguyen J. H.. WO2015048553A12015.; b Pohlski F.; Lange U.; Ochse M.; Behi B.; Hutchins C. W.. US2012040948A12012.; c Lansbury P. T.; Justman C. J.. WO2009036275A12009.; d Pontillo J.; Gao Y.; Wade W. S.; Wu D.; Eccles W. K. U.S. Patent US2006276454A12006.; e Kolanos R.; Siripurapu U.; Pullagurla M.; Riaz M.; Setola V.; Roth B. L.; Dukat M.; Glennon R. A. Binding of isotryptamines and indenes at h5-HT6 serotonin receptors. Bioorg. Med. Chem. Lett. 2005, 15, 1987–1991. 10.1016/j.bmcl.2005.02.070. [DOI] [PubMed] [Google Scholar]; f Horwell D. C.; Howson W.; Nolan W. P.; Ratcliffe G. S.; Rees D. C.; Willems H. M. G. The design of dipeptide helical mimetics, Part I: the synthesis of 1,6-disubstituted indanes. Tetrahedron 1995, 51, 203–211. 10.1016/0040-4020(94)00887-Z. [DOI] [Google Scholar]; g Plummer E. L.; Tonawanda N. U.S. Patent US41362744A11982.; h Ardalani H.; Avan A.; Ghayour-Mobarhan M. Podophyllotoxin: a novel potential natural anticancer agent. Avicenna J. Phytomed. 2017, 7, 285–294. [PMC free article] [PubMed] [Google Scholar]; i Cervo L.; Samanin R. Potential antidepressant properties of 8-hydroxy-2-(di-n-propylamino) tetralin, a selective serotonin IA receptor agonist. Eur. J. Pharm. 1987, 144, 223–229. 10.1016/0014-2999(87)90523-1. [DOI] [PubMed] [Google Scholar]
- Biosca M.; Magre M.; Coll M.; Pàmies O.; Diéguez M. Alternatives to Phosphinooxazoline (t-BuPHOX) Ligands in the Metal-Catalyzed Hydrogenation of Minimally Functionalized Olefins and Cyclic β-Enamides. Adv. Synth. Catal. 2017, 359, 2801–2814. 10.1002/adsc.201700573. [DOI] [Google Scholar]
- Much recently it has been disclosed that the high enantiocontrol for these substrates can be further expanded to both 5- and 6-membered ring counterparts by introducing a triazole in the ligand design, see ref. 7c
- a Rageot D.; Woodmansee D. H.; Pugin B.; Pfaltz A. Proline- Based P,O Ligand/Iridium Complexes as Highly Selective Catalysts: Asymmetric Hydrogenation of Trisubstituted Alkenes. Angew. Chem., Int. Ed. 2011, 50, 9598–9601. 10.1002/anie.201104105. [DOI] [PubMed] [Google Scholar]; b Xi J. Q.; Quan X.; Andersson P. G. Highly Enantioselective Iridium-Catalyzed Hydrogenation of α,β- Unsaturated Esters. Chem. - Eur. J. 2012, 18, 10609–10616. 10.1002/chem.201200907. [DOI] [PubMed] [Google Scholar]; c Woodmansee D. H.; Müller M. A.; Tröndlin L.; Hörmann E.; Pfaltz A. Asymmetric Hydrogenation of α,β-Unsaturated Carboxylic Esters with Chiral Iridium N, P Ligand Complexes. Chem. - Eur. J. 2012, 18, 13780–13786. 10.1002/chem.201202397. [DOI] [PubMed] [Google Scholar]; d Lu S. M.; Bolm C. Highly Enantioselective Synthesis of Optically Active Ketones by Iridium-Catalyzed Asymmetric Hydrogenation. Angew. Chem., Int. Ed. 2008, 47, 8920–8923. 10.1002/anie.200803709. [DOI] [PubMed] [Google Scholar]; e Lu W.-J.; Chen Y.-W.; Hou X.-L. Iridium- Catalyzed Highly Enantioselective Hydrogenation of the C–C Bond of α,β-Unsaturated Ketones. Angew. Chem., Int. Ed. 2008, 47, 10133–10136. 10.1002/anie.200803872. [DOI] [PubMed] [Google Scholar]; f Shang J.; Han Z.; Li Y.; Wang Z.; Ding K. Highly enantioselective asymmetric hydrogenation of (E)-β,β-disubstituted α,β-unsaturated Weinreb amides catalyzed by Ir (I) complexes of SpinPhox ligands. Chem. Commun. 2012, 48, 5172–5174. 10.1039/c2cc30812f. [DOI] [PubMed] [Google Scholar]; g Biosca M.; Pam̀ies O.; Diéguez M. Giving a Second Chance to Ir/ Sulfoximine-Based Catalysts for the Asymmetric Hydrogenation of Olefins Containing Poorly Coordinative Groups. J. Org. Chem. 2019, 84, 8259–8266. 10.1021/acs.joc.9b00829. [DOI] [PubMed] [Google Scholar]
- Chiral organofluorines are important in agrochemicals and drug synthesis among other applications due to its unique physical properties. For a recent hydrogenation of this substrate class, see:; Ponra S.; Yang J.; Kerdphon S.; Andersson P. G. Asymmetric Synthesis of Alkyl Fluorides: Hydrogenation of Fluorinated Olefins. Angew. Chem., Int. Ed. 2019, 58, 9282–9287. 10.1002/anie.201903954. [DOI] [PubMed] [Google Scholar]
- a Cheruku P.; Gohil S.; Andersson P. G. Asymmetric hydrogenation of enol phosphinates by iridium catalysts having N,P ligands. Org. Lett. 2007, 9, 1659–1661. 10.1021/ol070325l. [DOI] [PubMed] [Google Scholar]; b Cheruku P.; Diesen J.; Andersson P. G. Asymmetric Hydrogenation of Di and Trisubstituted Enol Phosphinates with N,P- Ligated Iridium Complexes. J. Am. Chem. Soc. 2008, 130, 5595–5599. 10.1021/ja711372c. [DOI] [PubMed] [Google Scholar]; c Paptchikhine A.; Cheruku P.; Engman M.; Andersson P. G. Iridium-catalyzed enantioselective hydrogenation of vinyl boronates. Chem. Commun. 2009, 5996–5998. 10.1039/b912590f. [DOI] [PubMed] [Google Scholar]; d Ganic A.; Pfaltz A. Iridium-Catalyzed Enantioselective Hydrogenation of Alkenylboronic Esters. Chem. - Eur. J. 2012, 18, 6724–6728. 10.1002/chem.201200246. [DOI] [PubMed] [Google Scholar]
- See for instance:; a Theodore L. J.; Nelson W. L. Stereospecific synthesis of the enantiomers of verapamil and gallopamil. J. Org. Chem. 1987, 52, 1309–1315. 10.1021/jo00383a026. [DOI] [Google Scholar]; b Procopiou P. A.; Biggadike K.; English A. F.; Farrell R. M.; Hagger G. N.; Hancock A. P.; Haase M. V.; Irving W. R.; Snowden M. A.; Solanke Y. E.; Tralau-Stewart C. J.; Walton S. E.; Wood J. A. Novel Glucocorticoid Antedrugs Possessing a 17β-(γ-Lactone) Ring. J. Med. Chem. 2001, 44, 602–612. 10.1021/jm001035c. [DOI] [PubMed] [Google Scholar]; c Adlington R. M.; Baldwin J. E.; Becker G. W.; Chen B.; Cheng L.; Cooper S. L.; Hermann R. B.; Howe T. J.; McCoull W.; McNulty A. M.; Neubauer B. L.; Pritchard G. J. Design, synthesis, and proposed active site binding analysis of monocyclic 2-azetidinone inhibitors of prostate specific antigen. J. Med. Chem. 2001, 44, 1491–1508. 10.1021/jm000145g. [DOI] [PubMed] [Google Scholar]; d Aoyama Y.; Uenaka M.; Kii M.; Tanaka M.; Konoike T.; Hayasaki-Kajiwara Y.; Naya N.; Nakajima M. Design, synthesis and pharmacological evaluation of 3-benzylazetidine-2-one-based human chymase inhibitors. Bioorg. Med. Chem. 2001, 9, 3065–3075. 10.1016/S0968-0896(01)00209-7. [DOI] [PubMed] [Google Scholar]; e Kottirsch G.; Koch G.; Feifel R.; Neumann U. β-Aryl-Succinic Acid Hydroxamates as Dual Inhibitors of Matrix Metalloproteinases and Tumor Necrosis Factor Alpha Converting Enzyme. J. Med. Chem. 2002, 45, 2289–2293. 10.1021/jm0110993. [DOI] [PubMed] [Google Scholar]; f Higashi T.; Isobe Y.; Ouchi H.; Suzuki H.; Okazaki Y.; Asakawa T.; Furuta T.; Wakimoto T.; Kan T. Stereocontrolled Synthesis of (+)-Methoxyphenylkainic Acid and (+)-Phenylkainic Acid. Org. Lett. 2011, 13, 1089–1091. 10.1021/ol103131p. [DOI] [PubMed] [Google Scholar]; g Shan W.; Balog A.; Quesnelle C.; Gill P.; Han W.-C.; Norris D.; Mandal S.; Thiruvenkadam R.; Gona K. B.; Thiyagarajan K.; Kandeula S.; McGlinchey K.; Menard K.; Wen M.-L.; Rose A.; White R.; Guarino V.; Shen D. R.; Cvijic M. E.; Ranasinghe A.; Dai J.; Zhang Y.; Wu D.-R.; Mathur A.; Rampulla R.; Trainor G.; Hunt J. T.; Vite G. D.; Westhouse R.; Lee F. Y.; Gavai A. V. BMS-871: A novel orally active pan-Notch inhibitor as an anticancer agent. Bioorg. Med. Chem. Lett. 2015, 25, 1905–1909. 10.1016/j.bmcl.2015.03.038. [DOI] [PubMed] [Google Scholar]; h Lin X.; Yuen P.-W.; Mendonca R.; Parr B.; Pastor R.; Pei Z.; Gaz-zard L.; Jaipuri F.; Kumar S.; Li X.; Pavana R.; Potturi H.; Velvadapu V.; Waldo J.; Zhang Z.; Wu G.. WO2017107979A12017.; i Kaieda A.; Toyofuku M.; Daini M.; Nara H.; Yoshikawa M.; Ishii N.; Hidaka K. U.S. Patent. US20170015655A12017.; j Huang X.; Brubaker J.; Peterson S. L.; Butcher J. W.; Close J. T.; Martinez M.; Maccoss R. N.; Jung J. O.; Siliphaivanh P.; Zhang H.; Aslanian R. G.; Biju P. J.; Dong L.; Huang Y.; Mccormick K. D.; Palani A.; Shao N.; Zhou W.. WO2012174176A12017.
- Chiral CF3-containing molecules are of interest because the trifluoromethyl motif often occurs in pharmaceuticals and agrochemical products, see for instance; a Jeschke P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; b Zhou Y.; Wang J.; Gu Z.; Wang S.; Zhu W.; Aceña J. L.; Soloshonok V. A.; Izawa K.; Liu H. Next Generation of Fluorine- Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]; c Yang J.; Ponra S.; Li X.; Peters B. B. C.; Massaro L.; Zhou T.; Andersson P. G. Catalytic enantioselective synthesis of fluoromethylated stereocenters by asymmetric hydrogenation. Chem. Sci. 2022, 13, 8590–8596. 10.1039/D2SC02685F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See for example; a Kammermeier B.; Beck G.; Holla W.; Jacobi D.; Napierski B.; Jendralla H. Vanadium(II)- and Niobium(III)-Induced, Diastereoselective Pinacol Coupling of Peptide Aldehydes to Give a C2-Symmetrical HIV Protease Inhibitor. Chem. - Eur. J. 1996, 2, 307–315. 10.1002/chem.19960020312. [DOI] [Google Scholar]; b Fabre B.; Ramos A.; Pascual-Teresa B. Targeting Matrix Metalloproteinases: Exploring the Dynamics of the S1′ Pocket in the Design of Selective, Small Molecule Inhibitors. J. Med. Chem. 2014, 57, 10205–10219. 10.1021/jm500505f. [DOI] [PubMed] [Google Scholar]; c Vandenbroucke R. E.; Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition?. Nat. Rev. Drug Discovery 2014, 13, 904–927. 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]; d Stuart A.; McCallum M. M.; Fan D.; LeCaptain D. J.; Lee C. Y.; Mohanty D. K. Poly(vinyl chloride) plasticized with succinate esters: synthesis and characterization. Polym. Bull. 2010, 65, 589–598. 10.1007/s00289-010-0271-4. [DOI] [Google Scholar]
- a Cabré A.; Romagnoli E.; Martínez-Balart P.; Verdaguer X.; Riera A. Highly Enantioselective Iridium-Catalyzed Hydrogenation of 2-Aryl Allyl Phthalimides. Org. Lett. 2019, 21, 9709–9713. [DOI] [PubMed] [Google Scholar]; b Rojo P.; Molinari M.; Cabré A.; García-Mateos C.; Riera A.; Verdaguer X. Iridium-Catalyzed Asymmetric Hydrogenation of 2,3-Diarylallyl Amines with a Threonine-Derived P-Stereogenic Ligand for the Synthesis of Tetrahydroquinolines and Tetrahydroisoquinolines. Angew. Chem., Int. Ed. 2022, 61, e202204300 10.1002/anie.202204300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Salomó E.; Rojo P.; Hernández-Lladó P.; Riera A.; Verdaguer X. P-Stereogenic and Non-P-Stereogenic Ir-MaxPHOX in the Asymmetric Hydrogenation of N-Aryl Imines. Isolation and X-Ray Analysis of Imine Iridacycles. J. Org. Chem. 2018, 83, 4618–4627. 10.1021/acs.joc.8b00361. [DOI] [PubMed] [Google Scholar]; b Salomó E.; Gallen A.; Sciortino G.; Ujaque G.; Grabulosa A.; Lledós A.; Riera A.; Verdaguer X. Direct Asymmetric Hydrogenation of N-Methyl and N-Alkyl Imines with an Ir(III)H Catalyst. J. Am. Chem. Soc. 2018, 140, 16967–16970. 10.1021/jacs.8b11547. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 09, Revision A.02; Gaussian, Inc., 2016.
- a Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]; b Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. 10.1063/1.448975. [DOI] [Google Scholar]
- Petersson G. A.; Bennett A.; Tensfeldt T. G.; Al-Laham M. A.; Shirley W. A.; Mantzaris J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row atoms. J. Chem. Phys. 1988, 89, 2193–2218. 10.1063/1.455064. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Luchini G.; Alegre-Requena J. V.; Funes-Ardoiz I.; Paton R. S. GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data. F1000Research 2020, 9, 291 10.12688/f1000research.22758.1. [DOI] [Google Scholar]
- a Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]; b Grimme S.; Ehrlich S.; Goerigk F. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- a Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]; b Woon D. E.; Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. 10.1063/1.464303. [DOI] [Google Scholar]
- Figgen D.; Peterson K. A.; Dolg M.; Stoll H. Energy-consistent pseudopotentials and correlation consistent basis sets for the 5d elements Hf-Pt. J. Chem. Phys. 2009, 130, 164108 10.1063/1.3119665. [DOI] [PubMed] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.