

Abstract
Background and Objective
Neuromyelitis optica spectrum disorder (NMOSD) is a rare autoimmune condition, which can lead to significant disability, and up to 3%–5% of the cases have a pediatric onset. There are limited studies to guide physicians in disease-modifying treatment (DMT) choices for children with NMOSD.
Methods
This retrospective cohort study evaluated children with NMOSD cases followed at 12 clinics in the US Network of Pediatric MS Centers. Cases were classified as aquaporin-4 antibody positive (AQP4+) and double seronegative (DS) when negative for AQP4+ and for myelin oligodendrocyte glycoprotein (MOG) antibody. The effect of initial DMTs including rituximab, mycophenolate, azathioprine, and IV immunoglobulin (IVIg) on the annualized relapse rate (ARR) was assessed by negative binomial regression. Time to disability progression (EDSS score increase ≥1.0 point) was modeled with a Cox proportional-hazards model.
Results
A total of 91 children with NMOSD were identified: 77 AQP4+ and 14 DS (85.7% females; 43.2% White and 46.6% African American). Eighty-one patients were started on a DMT, and 10 were treatment naive at the time of the analysis. The ARR calculated in all serogroups was 0.25 (95% CI 0.13–0.49) for rituximab, 0.33 (95% CI 0.19–0.58) for mycophenolate, 0.40 (95% CI 0.13–1.24) for azathioprine, and 0.54 (95% CI 0.28–1.04) for IVIg. The ARR in the AQP4+ subgroup was 0.28 (95% CI 0.14–0.55) for rituximab, 0.39 (95% CI 0.21–0.70) for mycophenolate, 0.41 (95% CI 0.13–1.29) for azathioprine, and 0.54 (95% CI 0.23–1.26) for IVIg. The ARR in the treatment-naive group was 0.97 (95% CI 0.58–1.60) in all serogroups and 0.91 (95% CI 0.53–1.56) in the AQP4+ subgroup. None of the initial DMT had a statistically significant effect on EDSS progression.
Discussion
The use of DMTs, particularly rituximab, is associated with a lowered annualized relapse rate in children with NMOSD AQP4+.
Classification of Evidence
This study provides Class IV evidence that use of disease-modifying treatments is associated with a lowered annualized relapse rate in children with NMOSD AQP4+.
Neuromyelitis optica spectrum disorder (NMOSD) is a rare autoimmune astrocytopathy1 that affects adults and children. Initially interpreted as a clinical variant of multiple sclerosis (MS), it was later identified as a distinct disorder given specific features such as longitudinally extensive transverse myelitis (LETM), recurrent optic neuritis (ON), and typical brain lesions.2 About 3%–5% of cases have a pediatric onset,3 with a typical age at onset between 10-12 years4 and female predominance.5 Approximately 31% of pediatric patients with NMOSD have detectable serum anti–AQP4-IgG, and 57% have serum MOG-IgG, and roughly 12% are double seronegative (DS).6,7 Children with MOG antibodies present with a variety of clinical syndromes that include NMOSD; in addition, recent data support the consideration of this subgroup of patients as a separate entity with distinct pathophysiology targeting oligodendrocytes6,8 and were therefore not included in this study. Compared with MS, children with NMOSD have a higher attack rate and an EDSS score within 2 years of disease onset.4 Over the past decade, diagnostic NMOSD criteria have been modified to increase diagnostic accuracy. The 2015 International Consensus Diagnostic Criteria9 considered ON, LETM, and area postrema syndrome as cardinal manifestations and stratified patients as AQP4-IgG+ and AQP4-IgG−. These criteria seem to apply as well to the pediatric population allowing early differentiation of NMOSD from other demyelinating disorders.4 This is of critical importance given the high level of disability associated with NMOSD, allowing for timely initiation of treatment. There is limited Class I evidence for the treatment of NMOSD, and 3 new drugs were recently approved by the FDA for adult NMOSD (eculizumab, satralizumab, and inebilizumab).10 Little data are available about the treatment of this disease in the pediatric population.6 We aimed to evaluate the usage patterns and real-world effectiveness on the relapse rate and disability progression of initial disease-modifying treatments (DMTs) in children with NMOSD, providing evidence that DMTs are associated with a lowered annualized relapse rate in this pediatric population.
Methods
Study Design
This is a multicenter retrospective cohort study that used prospectively collected data and that includes patients from 12 regional pediatric MS/neuroimmunology referral centers from across the United States participating in the US Network of Pediatric MS Centers.11 The sites include Boston Children's Hospital, Massachusetts General Hospital, Mayo Clinic, Cleveland Clinic, New York University Langone Medical Center, State University of New York at Buffalo, Children's Hospital Colorado, Loma Linda University, University of Utah, Texas Children's Hospital, University of Alabama at Birmingham, Washington University in Saint Louis, University of California San Diego, and University of California San Francisco. This is a descriptive study aimed to analyze the pattern of DMT use, treatment response, relapse rate, and effect on disability progression in children with NMOSD within the United States. Clinical data including demographics, attack details, type of initial DMT, and neurologic examination data were prospectively collected and stored in a central database from May 2011 through January 1, 2020. We evaluated treatment response in the 2 serostatus subgroups on the annualized relapse rate (ARR) and disability progression. We also gauged the ARR in a group of treatment-naive patients. Data from prior to 2011 were retrospectively entered from medical records. The data are stored and managed by the Data Coordinating and Analysis Center (DCAC) at the University of Utah, which also performs quality control.
Study Population
Pediatric patients were diagnosed with NMOSD at the most recent visit before 18 years of age by expert neurologists from the US Network of Pediatric MS Centers based on the 2015 International Consensus Diagnostic Criteria. The patients were identified from the database with complete data. Included patients were either AQP4 antibody seropositive or double seronegative with negative testing for both AQP4 and MOG antibodies. Patients enrolled in clinical trials were excluded because of unknown treatment allocation.
Measurements
Baseline characteristics were considered at the time of starting the first DMT, including age, sex, race, ethnicity, network site, height, weight, diagnosis, and disease duration. Disease serostatus was also recorded, which allowed the subclassification in AQP4 and DS. Therapies of interest included rituximab, mycophenolate mofetil, azathioprine, and IVIg. We considered IVIg as DMT only when their use was consistent over time (i.e., the treatment lasted at least 60 days) and not for the symptomatic treatment of an acute relapse. Data on the type of the first DMT initiated, year the DMT was initiated, and duration of use were recorded. The Expanded Disability Status Scale (EDSS) score at the first visit and during the subsequent visits while on the first DMT was also collected.
The primary outcome was the relapse rate from baseline (i.e., time of initiation of the first DMT for those on DMT; time at disease onset for the treatment-naive patients) to the time of discontinuation of the initial DMT. Relapses were identified by the primary neurologist and defined as new or worsening neurologic symptoms for at least 24 hours in the absence of infection or fever separated by 30 days at least from prior relapse. The secondary outcome was EDSS progression, which was defined as having 1.0 point or greater EDSS score increase, not within 30 days of a relapse or pulse steroid treatment. In this analysis, only those with a baseline EDSS score in the 6 months before initial DMT and at least 1 EDSS score during treatment were included. The time to the first EDSS progression was found; those without EDSS progression were censored at their last visit. The timeline for treatment-naive patients started at disease onset, and the timeline for those on a first-line DMT was at DMT start date.
Statistical Analysis
Characteristics of the analysis population, including age at first event, sex, race, ethnicity, first DMT, disease duration at the time of the first DMT, years on the first DMT, number of events and EDSS score in the 6 months before the first DMT, number of events while on the first DMT, and time to EDSS progression, were calculated by serostatus groups. Continuous variables were summarized using means and SDs and were compared with a Kruskal-Wallis test. Categorical variables were summarized using frequencies and percentages and were compared with a Fisher exact test (with and without Monte Carlo approximation).
The ARR was modeled as the number of events over the DMT duration in years using a negative binomial regression model. This was analyzed for azathioprine, mycophenolate, rituximab, and IVIg. Treatment-naive patients were modeled as the number of events over the follow-up time in the registry. This was also reported for the AQP4+ subset. We used Cox proportional-hazards regression to model time to the first event for azathioprine, IVIg, mycophenolate, and rituximab, and DMTs were compared using a logrank (score) test. We assessed the proportional-hazards assumption and did not find compelling evidence of an egregious violation. Those without an event occurring while on the DMT were censored at DMT stop date (or last follow-up). Treatment-naive patients were modeled as the time to the first event since disease onset. The time to EDSS progression was also modeled for the DMTs in a similar fashion. Analyses were conducted using SAS software, version 9.4 (SAS Institute Inc., Cary, NC). A p value of 0.05 or less was considered statistically significant.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by ethics committees of participating institutions: The University of Alabama at Birmingham IRB (IRB-030409007); Boston Children's Hospital IRB (IRB-P00000493); Cleveland Clinic IRB (15-1026); University of Colorado, Colorado Multiple IRB (PAM010-1); Loma Linda University Hospital IRB (5110192); Mayo Clinic IRB (PR06-005020-15); Mass General Brigham Human Research Committee IRB (2008P002065); NYU Langone Health IRB (i15-01261_CR6); Baylor College of Medicine IRB (H-29147); University of California San Francisco and University of California San Diego HRPP/IRB (11-05873); Washington University in St. Louis HRPO (IRB ID# 201508048); The University of Utah IRB (IRB_00063428); and The State University of New York, University at Buffalo IRB (MODCR00005553). Parents and participants signed consent forms and assent forms before enrollment as required by each site's institutional review board.
Data Availability
Qualified investigators can request the data for purposes of replicating procedures or results by contacting the corresponding author.
Role of the Funding Source
The funding sources were not involved in study design, collection, analysis, and interpretation of data or in writing the report or the decision to submit for publication. All authors had full access to data in the study, and the corresponding author had the final decision to submit for publication.
Results
Patient Characteristics
Our database included 91 children with NMOSD, and data were collected from May 2011 through January 1, 2020. Cases met the NMOSD International Consensus Diagnostic Criteria9 and were classified based on their serostatus as AQP4+ (n = 77) or DS (n = 14). Ten patients with MOG+ antibody and those with unknown serostatus were excluded (eFigure 1, links.lww.com/WNL/C517). NMO antibody testing was performed in all patients with a median of 2 tests (IQR 1–4), whereas MOG antibody testing was performed in 27 patients with a median of 1 test (IQR 1-1). In the AQP4+ group, 13 patients were all tested once for MOG antibody. All DS patients were tested for AQP4 and MOG antibodies, with medians of 3 NMO antibody tests (IQR 2–4) and 1 MOG antibody test (IQR 1–2). Eighty-one patients were started on an initial DMT, of which 68 were AQP4+ and 13 DS. There were 10 patients (9 AQP4+ and 1 DS) treatment naive. In addition, 3 patients were either started on treatments not usually used for NMOSD or excluded from the analyses because of the small number of partial data available (i.e., 2 patients were on plasmapheresis; 1 patient was on ocrelizumab), and 2 patients were excluded because they were enrolled in clinical trials. A total of 78 patients were females; 38 patients were White, 41 patients were African American, and 18 patients were Hispanic (Table 1). No statistically significant differences were seen between groups in terms of sex, race, ethnicity, or BMI (Table 1). The mean age at the time of the first event did not differ between the serogroups.
Table 1.
Characteristics of the Analysis Population

Pattern of DMT Use and Duration
In total, 12.3% of patients remained treatment naive (9 AQP4+ and 1 DS). In the other 87.7% of children with NMOSD, the most frequently used treatments were rituximab (n = 38), followed by mycophenolate (n = 16), azathioprine (n = 15), and IVIg (n = 9). There were no patients receiving 2 DMTs as first-line treatment. First-line DMTs varied by serostatus, but the difference was not statistically significant (Table 1). Patients on rituximab, mycophenolate, and azathioprine tended to be on these treatments for a longer period of time when compared with IVIg (Table 2). When evaluating the first DMT duration for each serogroup, there was no statistical difference between them (p = 0.16) (Table 1).
Table 2.
DMT Duration (Years) for First-Line Treatment and Disease Duration at the Start of First-Line Treatment

Pattern of Disease Duration
Disease duration at the time of initiation of the first DMT was 0.9 ± 1.4 years for the AQP4+ group and 1.2 ± 1.8 years for the DS group (p = 0.59; Table 1). We observed an average longer disease duration at the start of first-line treatment in the rituximab group (1.2 ± 1.2 years), followed by mycophenolate (1.1 ± 1.7), azathioprine (1.0 ± 1.8), and IVIg (0.7 ± 0.7), as detailed in Table 2.
Treatment Response by Serostatus
There were no statistically significant differences in the number of relapses 6 months before the first DMTs among the serogroups (p = 0.79). Similarly, the number of relapses on the first DMTs did not differ significantly among serogroups (p = 0.11; Table 1). Relapse data stratified by serostatus are summarized in eTable 1, links.lww.com/WNL/C517. Among the patients on DMTs, the ARR in all patients was 0.25 (95% CI 0.13–0.49) for rituximab, 0.33 (95% CI 0.19–0.58) for mycophenolate, 0.40 (95% CI 0.13–1.24) for azathioprine, and 0.54 (95% CI 0.28–1.04) for IVIg. In the AQP4+ subgroup, the patients started on rituximab had an adjusted ARR of 0.28 (95% CI 0.14–0.55), those on mycophenolate 0.39 (95% CI 0.21–0.70), those on azathioprine 0.41 (95% CI 0.13–1.29), and those on IVIg 0.54 (95% CI 0.23–1.26). When comparing the ARR by race, we observed the data reported in Table 3; when adjusting the ARR model for race, DMT, and a race by DMT interactions, none of those factors resulted as significant. Kaplan-Meier (KM) survival curves were generated for the 4 groups (Figure 1). As shown in Figure 1, patients who used rituximab as a first DMT experienced longer event-free time intervals, followed by azathioprine (HR 1.19, 95% CI 0.41–3.42), mycophenolate (HR 2.20, 95% CI 0.92–5.28), and IVIg (HR 3.22, 95% CI 1.01–10.24), although the difference was not statistically significant (p = 0.11).
Table 3.
Annualized Relapse Rate by First-Line Treatment, Race Comparison
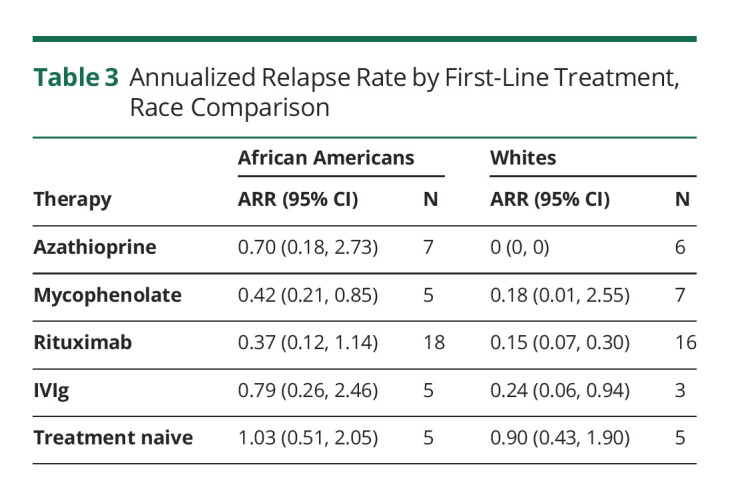
Figure 1. Time to First Relapse of DMT for First-Line Treatment and Treatment-Naive Patients With Neuromyelitis Optica Spectrum Disorder.

For DMT groups, time is measured from the initiation of the first DMT. For the treatment-naive group, time is measured from disease onset. aKaplan-Meier method; bCox model; and clogrank test. DMT = disease-modifying therapies.
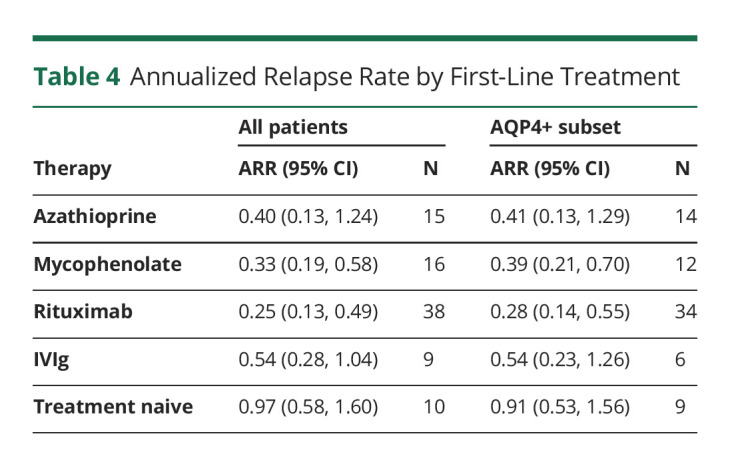
In the treatment-naive group, the average number of events was 1.5 ± 1.4, and the ARR was 0.97 (95% CI 0.58–1.60) in all patients, whereas the treatment-naive AQP4+ subgroup had an ARR of 0.91 (95% CI 0.53–1.56) (Table 4).
Table 4.
Annualized Relapse Rate by First-Line Treatment
This study provides Class IV evidence that use of disease-modifying treatments is associated with a lowered annualized relapse rate in children with NMOSD.
Disability Progression
The baseline EDSS score was available for 46 patients at the time of the first DMT start (±6 months) and for 5 patients in the treatment-naive group. The baseline median EDSS score (years) was 2.0 in the AQP4+ group, 3.0 in the DS subgroup, and 3.0 in the treatment-naive group (eTable 2, links.lww.com/WNL/C517). KM survival curves show that there was no statistically significant difference in EDSS progression among the DMT groups (Figure 2). Rituximab and mycophenolate appear to be the DMTs with the lowest number of EDSS progression events, followed by azathioprine. Given the small number of EDSS data in the IVIg group, the EDSS progression in this group has been omitted in Figure 2. Where the AQP4+ group is concerned, we report in Figure 3 the KM curves of the EDSS progression on the first DMT; as above, these differences were not statistically significant (p = 0.13).
Figure 2. Time to Expanded Disability Status Scale Progression on First-Line Treatment.
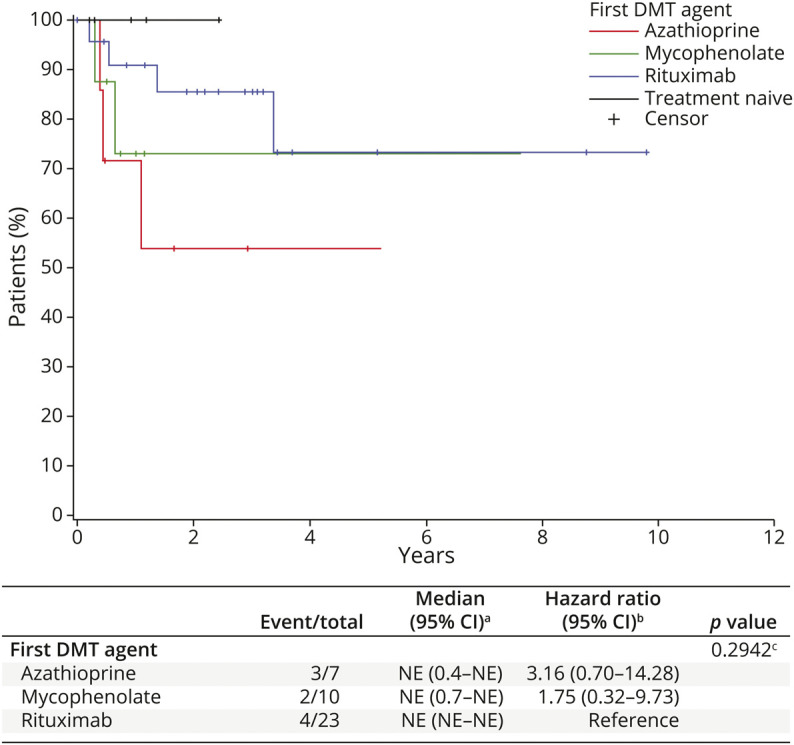
For DMT groups, time is measured from the initiation of the first DMT. For the treatment-naive group, time is measured from disease onset. aKaplan-Meier method; bCox model; and clogrank test. DMT = disease-modifying therapies.
Figure 3. Time to Expanded Disability Status Scale Progression on the First DMT for AQP4+ Patients.

For DMT groups, time is measured from the initiation of the first DMT. For the treatment-naive group, time is measured from disease onset. aCox model; blogrank test. DMT = disease-modifying therapies.
For completeness, we show the KM curves for all data available together with the number of events and the hazard ratios for the 4 DMTs considered in these analyses (Figures 2 and 3).
We have also calculated the progression independent of relapse activity (PIRA), which was defined as an increase in the EDSS score (≥1.5 points for patients with a baseline EDSS score of zero, ≥1.0 point for patients with a baseline EDSS score of 1–5, and 0.5 points for patients with a baseline EDSS score of ≥5.5), with the EDSS score obtained at least 90 days from the last relapse. No relapse occurred within 30 days before or after the EDSS confirmation. Only 21 of 91 patients had PIRA (14 AQP4+ and 7 double negative). We also calculated PIRA by the first DMT agent and the treatment-naive group (eTable 3, links.lww.com/WNL/C517). PIRA was not statistically significant with a p value of 0.34.
Discussion
As there have been no investigational or randomized controlled clinical trials of any immunotherapy in pediatric NMOSD, treatments used for these conditions are not FDA approved. Despite the lack of consensus on the best initial therapy, and the lack of randomized clinical trials, observational data suggest that rituximab, azathioprine, mycophenolate mofetil, IVIg, and plasmapheresis reduce the relapse rates in adult and pediatric forms of NMOSD12,13; however, significant gaps in knowledge remain. Prompt and effective treatment of children with NMOSD is fundamental, given the higher disability accumulation associated with relapses.4,14 Our retrospective study provides Class IV evidence that first-line rituximab, mycophenolate, azathioprine, and IVIg are associated with reduced ARRs in pediatric NMOSD AQP4+ when compared with treatment-naive cases. Moreover, rituximab is associated with (1) a longer event-free period when used as a first DMT and (2) a lowered annualized relapse rate in the AQP4+ subgroup. The latter is consistent with a recent large study evaluating the effectiveness of DMTs in children15 as well as prior studies in smaller cohorts of pediatric patients with NMOSD.16–19 A prior study of 180 children with NMOSD who received rituximab during their first hospitalization showed that the therapy was not associated with a reduced risk of rehospitalization, although the duration of rehospitalization was shorter.20
Overall, the current literature and our results seem to support the use of rituximab, a B-cell–depleting drug, as a first-line agent showing good tolerability and effectiveness in children, particularly in NMOSD AQP4+ patients. Given that B-cell repopulation may occur earlier than 6 months in pediatric patients, from the time of rituximab infusion, B-cell monitoring and redosing might help preventing relapses.16 Moreover, patients need to be closely monitored because of the risk of developing hypogammaglobulinemia and infections, which have been associated with the use of rituximab in both adult and pediatric populations.21,22
Patients on first-line mycophenolate had a similar ARR as rituximab, with a slightly higher ARR than rituximab in the AQP4+ subgroup, followed by azathioprine and IVIg (Table 4). This is in line with findings from other studies. For example, a study from 2016 showed good efficacy and tolerability of mycophenolate used as first-line therapy in adult patients with NMOSD, independent of AQP4+ serostatus.23 Another multicenter study including 59 adult NMOSD cases showed that mycophenolate was more effective than azathioprine with a reduction of relapse rates in 60% of the patients and stabilization or improvement of disability scores in 91% of the cases, with good overall tolerability.24 A retrospective multicenter analysis of relapses in 90 adult and pediatric patients with NMOSD (48 of whom were AQP4+)25 showed that mycophenolate and rituximab were more effective than azathioprine with a reduction of relapse rates of 88.2% and 87.4%, respectively, vs 72.1% in the azathioprine group. Furthermore, two other studies demonstrated the efficacy of mycophenolate in children with NMOSD.15,26
In our study, patients treated with azathioprine had a high ARR both in all the patients and in the AQP4+ serogroup. This is in line with the findings from Mealy et al.,25 which included adult and pediatric cases treated with azathioprine and concomitant prednisone.25 Little data are available about the use of this drug in pediatric cases, showing again modest efficacy.15,27,28
The use of IVIg as a preventive agent is supported by case series, mainly adult patients.13,29,30 IVIg is usually used as an alternative agent in patients with contraindication to one of the other treatments. In our study, we also aimed to investigate the use of IVIg as DMT in children with NMOSD. Only 9 of 91 patients (6 AQP4+) were treated with IVIg as first-line monotherapy. Despite the low number of patients, IVIg seemed to have a possible effect in preventing NMOSD relapses; however, a larger study is needed.
Current data in the literature showed that NMOSD may be more aggressive in African American than in White patients, and therefore, they have a greater risk of severe disability.31 Our study included an equal number of White (n = 38) and African American (n = 41) children, and no difference in the ARR by race and DMT interactions was noted in these 2 subgroups when compared with the treatment-naive group, supporting the use of these treatments independently from the race. Therefore, despite a potentially more aggressive course, African American patients benefit from immunomodulating treatments similarly to White patients without the need of more aggressive treatments.
Another interesting finding of our study is the fewer EDSS progression events in the group of patients on rituximab and mycophenolate, followed by azathioprine, although the difference is not statistically significant. Similarly, PIRA by first DMT or by serostatus was not significant. More data and longer follow-up duration are needed in establishing the effect of DMT on disability progression in pediatric patients with NMOSD.
Limitations of our study include the lack of information about short-term safety, dose, tolerability and side effects of the different DMTs, reason for discontinuation of the first DMT, and missed longitudinal EDSS data between groups, as well as residual confounding given the observational design. A comparison between the different DMTs cannot be performed reliably because there was no initial randomization, and the therapies were chosen at the discretion of the treating physicians. To avoid selection bias, we included all patients started on initial DMT with follow-up from DMT start date. Furthermore, we could not systematically assess infection rates in our cohort because these data were not prospectively and uniformly collected. Measurement bias is unlikely to explain the effect of DMTs on outcomes because data were prospectively collected. No data on the new FDA-approved monoclonal antibodies (eculizumab, satralizumab, and inebilizumab) were available.
Strengths of this study include the large cohort of pediatric patients with NMOSD from different centers in the United States, with uniform data prospectively collected over the past 10 years, and a diverse cohort with White and African American patients present in almost equal proportions. Our study includes a large cohort of AQP4+ patients and a small proportion of DS patients. Given the low number of MOG+ patients, these were excluded from the analysis. A recent large study analyzed 67 children with NMOSD AQP4+ only from a European and Brazilian cohort of pediatric patients showing the median cumulative ARR of rituximab, mycophenolate mofetil, and azathioprine as first-, second-, third-, and even fourth-line treatments in different subcohorts, with overlapping use of steroids at times, for a median follow-up of 4 years.15 No treatment-naive patients were included in the study for comparison. The study showed that these treatments were associated with a reduction of ARRs and no relapses seen in patients (n = 14) treated with rituximab as a first-line therapy during the time of the follow-up. Our study analyzed a larger cohort of children with different serotypes of NMOSD treated in the United States. We specifically analyzed the ARR of the most used DMTs in the prevention of relapses in NMOSD including a high number of AQP4+ patients receiving rituximab (n=34). The focus of our study is on first line treatment; this choice was made with the objective of minimizing the risk of confounding biases such as the remaining effect of the preceding treatments or combination therapies such with steroids.
Randomized controlled trials are the gold standard for the analysis of treatment efficacy. Despite that, their implementation in rare diseases, particularly in children with NMOSD, is challenging. This study provides real-world efficacy data on different first-line DMTs suggesting a role in reducing the relapses and disability progression in this patient population, aiming at guiding physicians in their treatment choices for children with NMOSD AQP4+. Further data and analyses on long-term efficacy, imaging data, safety, and tolerability are necessary, as well as comparative studies that should include the newly FDA-approved therapies.
Acknowledgment
The authors thank the dedicated research coordinators and the patients and their families who participated in the study.
Glossary
- ARR
annualized relapse rate
- DCAC
Data Coordinating and Analysis Center
- DMTs
disease-modifying treatments
- EDSS
Expanded Disability Status Scale
- LETM
longitudinally extensive transverse myelitis
- MS
multiple sclerosis
- NMOSD
neuromyelitis optica spectrum disorder
Appendix. Authors
Footnotes
Class of Evidence: NPub.org/coe
Study Funding
Sumaira Foundation Unicorn Grant (TSF.NMO19-02) to T.C. The US NPMSC is sponsored by the National MS Society (SI-1808-32326).
Disclosure
Dr. R. Pizzolato Umeton received research support from the National MS Society. Dr. T. Chitnis is an advisory board member for Biogen, Novartis, and Sanofi-Genzyme; has received research support from the National MS Society, Department of Defense, Guthy-Jackson Charitable Foundation, Biogen, Novartis, Octave, Serono, and Verily; and has participated in clinical trials sponsored by Sanofi-Genzyme and Novartis. Dr. J. Graves has received recent grant and clinical trial support from the National MS Society, Race to Erase MS, UCSF CTSI RAP program, Biogen, and Genentech. She has received honoraria from Biogen and Genzyme for nonpromotional trainee education events. She has received personal fees from Novartis and Celgene. Dr. M. Rensel has served as a consultant/speaker for Biogen, Teva, Genzyme, and Novartis and received research support from MedImmune. Dr. B. Weinstock-Guttman has served as a consultant/speaker for Biogen, Teva, Novartis, Genzyme, Genentech, and EMD Serono and received research support from Biogen, Teva, Novartis, Genentech, and EMD Serono. Dr. G. Aaen has participated in clinical trials funded by Biogen and Roche. Dr. L. Benson has received funding for research unrelated to this work for a Biogen-sponsored clinical trial and Boston Children's Hospital Office of Faculty Development Grant. She has also acted as a paid consultant to the National Vaccine Injury Compensation Program. Dr. M. Gorman has participated in clinical trials funded by Novartis and Biogen and received research funding from Pfizer. Dr. M. Goyal has received fees for providing consultations on medicolegal cases related to neuroradiology and has stock in IBM, Moderna, and BioNTech. Dr. Y. Harris reports no disclosures relevant to the manuscript. Dr. L. Krupp received payments as a consultant for Biogen, Novartis, Everyday Health, Genentech, Gerson Lehrman, and Sanofi; served as an uncompensated consultant for Celgene; and received licensing payments from biotechnology and pharmaceutical companies for the Fatigue Severity Scale. Dr. T. Lotze has served as a consultant/speaker for Biogen. Dr. N. Shukla, Dr. S. Mar, Dr. J. Ness, and Dr. M. Rodriguez report no disclosures relevant to the manuscript. Dr. J. Rose has received research support from the NMSS, NIH, Guthy-Jackson Charitable Foundation, PCORI, Teva Neuroscience, Biogen, and VA. Dr. T. Schreiner has participated in trials funded by Biogen and MSDx. Dr. J.-M. Tillema, Mr. M. Waltz, and Dr. T.C. Casper report no disclosures relevant to the manuscript. Dr. E. Waubant has participated in multicenter clinical trials funded by Genentech and Biogen. She has current support from the NIH, NMSS, PCORI, and Race to Erase MS. Go to Neurology.org/N for full disclosures.
References
- 1.Lucchinetti CF, Guo Y, BFGh Popescu, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol 2014;24(1):83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder. Neurology 2015;84(11):1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quek AML, McKeon A, Lennon VA, et al. Effects of age and sex on aquaporin-4 autoimmunity. Arch Neurol-chicago 2012;69(8):1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chitnis T, Ness J, Krupp L, et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology 2015;86(3):245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenembaum S, Chitnis T, Nakashima I, et al. Neuromyelitis optica spectrum disorders in children and adolescents. Neurology 2016;87(9 Supplement 2):S59–S66. [DOI] [PubMed] [Google Scholar]
- 6.Chitnis T. Pediatric central nervous system demyelinating diseases. Continuum Lifelong Learn Neurol 2019;25(3):793–814. [DOI] [PubMed] [Google Scholar]
- 7.Hacohen Y, Mankad K, Chong WK, et al. Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology 2017;89(3):269–278. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan EP. Neuromyelitis optica spectrum disorder and other non–multiple sclerosis central nervous system inflammatory diseases. Continuum Lifelong Learn Neurol 2019;25(3):815–844. [DOI] [PubMed] [Google Scholar]
- 9.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85(2):177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallach AI, Tremblay M, Kister I. Advances in the treatment of neuromyelitis optica spectrum disorder. Neurol Clin. 2020;39(1):35–49. [DOI] [PubMed] [Google Scholar]
- 11.Casper TC, Rose JW, Roalstad S, et al. The US Network of Pediatric Multiple Sclerosis Centers. J Child Neurol 2014;30(10):1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baghbanian SM, Asgari N, Sahraian MA, Moghadasi AN. A comparison of pediatric and adult neuromyelitis optica spectrum disorders: a review of clinical manifestation, diagnosis, and treatment. J Neurol Sci. 2018;388(Neurology 85 2 2015 Jul 14):222-231. [DOI] [PubMed] [Google Scholar]
- 13.NEMOS, NOSG, Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the neuromyelitis optica study group (NEMOS). J Neurol 2014;261(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fragoso YD, Ferreira MLB, Oliveira EML, et al. Neuromyelitis optica with onset in childhood and adolescence. Pediatr Neurol 2014;50(1):66–68. [DOI] [PubMed] [Google Scholar]
- 15.Paolilo RB, Hacohen Y, Yazbeck E, et al. Treatment and outcome of aquaporin-4 antibody–positive NMOSD: a multinational pediatric study. Neurol - Neuroimmunol Neuroinflammation 2020;7(5):e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nosadini M, Alper G, Riney CJ, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflammation 2016;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longoni G, Banwell B, Filippi M, Yeh EA. Rituximab as a first-line preventive treatment in pediatric NMOSDs Preliminary results in 5 children. Neurol - Neuroimmunol Neuroinflammation 2014;1(4):e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivieri G, Nociti V, Iorio R, et al. Rituximab as a first-line treatment in pediatric neuromyelitis optica spectrum disorder. Neurol Sci. 2015;36(12):2301–2302. [DOI] [PubMed] [Google Scholar]
- 19.Mahmood NA, Silver K, Onel K, Ko M, Javed A. Efficacy and safety of rituximab in pediatric neuromyelitis optica. J Child Neurol. 2011;26(2):244–247. [DOI] [PubMed] [Google Scholar]
- 20.Gmuca S, Xiao R, Weiss PF, Waldman AT, Gerber JS. Use of rituximab and risk of re-hospitalization for children with neuromyelitis optica spectrum disorder. Mult Scler Demyelinating Disord 2018;3(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghezzi A, Banwell B, Bar-Or A, et al. Rituximab in patients with pediatric multiple sclerosis and other demyelinating disorders of the CNS: practical considerations. Mult Scler J. 2020:135245852093279. [DOI] [PubMed] [Google Scholar]
- 22.Khojah AM, Miller ML, Klein-Gitelman MS, et al. Rituximab-associated Hypogammaglobulinemia in pediatric patients with autoimmune diseases. Pediatr Rheumatol. 2019;17(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montcuquet A, Collongues N, Papeix C, et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler J 2016;23(10):1377–1384. [DOI] [PubMed] [Google Scholar]
- 24.Huh S-Y, Kim S-H, Hyun J-W, et al. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. Jama Neurol 2014;71(11):1372–1378. [DOI] [PubMed] [Google Scholar]
- 25.Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. Jama Neurol 2014;71(3):324–330. [DOI] [PubMed] [Google Scholar]
- 26.Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol-chicago 2009;66(9):1128–1133. [DOI] [PubMed] [Google Scholar]
- 27.Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody–associated disease. Jama Neurol 2018;75(4):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Huang Q, Lu T, et al. Azathioprine therapy in a case of pediatric multiple sclerosis that was seropositive for MOG-IgG. J Clin Neurosci 2017;38:71–73. [DOI] [PubMed] [Google Scholar]
- 29.Elsone L, Mutch K, Jacob A. IVIG in NMO refractory/intolerant to rituximab. J Neurol Neurosurg Psychiatry 2014;85(10):e4.65–e4. [Google Scholar]
- 30.Magraner MJ, Coret F, Casanova B. Estudio del efecto del tratamiento con inmunoglobulinas por vía intravenosa en la neuromielitis óptica. Neurología 2013;28(2):65–72. [DOI] [PubMed] [Google Scholar]
- 31.Kim S-H, Mealy MA, Levy M, et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018;91(22):e2089–e2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Qualified investigators can request the data for purposes of replicating procedures or results by contacting the corresponding author.