

Summary
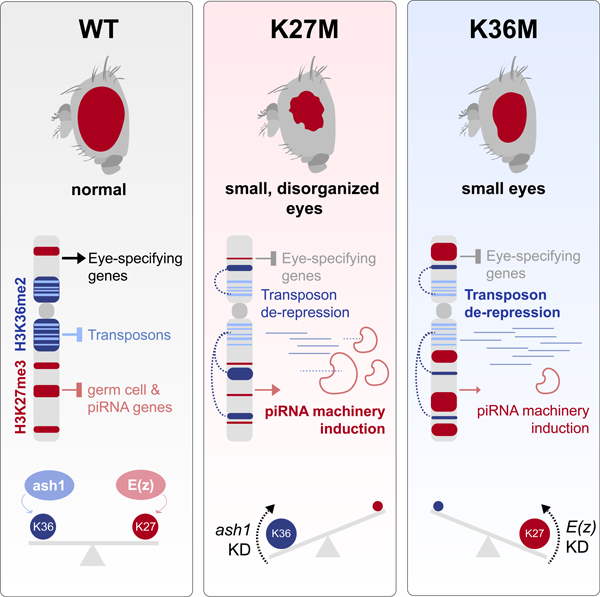
Histone H3.3 Lysine-to-Methionine substitutions K27M and K36M impair the deposition of opposing chromatin marks, H3K27me3/me2 and H3K36me3/me2. We show that these mutations induce hypotrophic and disorganized eyes in Drosophila eye primordia. Restriction of H3K27me3 spread in H3.3K27M and its redistribution in H3.3K36M result in transcriptional deregulation of PRC2-targeted eye development and of piRNA biogenesis genes including krimp. Notably, both mutants promote redistribution of H3K36me2 away from repetitive regions into active genes, which associates with retrotransposon de-repression in eye discs. Aberrant expression of krimp represses LINE retrotransposons but does not contribute to the eye phenotype. Depletion of H3K36me2 methyltransferase ash1 in H3.3K27M, and of PRC2 component E(z) in H3.3K36M, restores the expression of eye developmental genes and normal eye growth, showing that redistribution of antagonistic marks contributes to K-to-M pathogenesis. Our results implicate a novel function for H3K36me2 while they showcase convergent downstream effects of oncohistones that target opposing epigenetic marks.
Graphical Abstract
Introduction
Post-translational modifications (PTMs) of histones, the core components of chromatin in eukaryotes, regulate virtually all DNA-mediated processes including transcription (Jenuwein and Allis, 2001). Methylation of two histone H3 tail residues, H3K27 and H3K36, generates two opposing epigenetic marks that regulate cell fate and memory during metazoan development (Weinberg et al., 2017). In mammals and flies, Polycomb Repressive Complex 2 (PRC2) catalyzes di- and tri-methylation of H3K27 (H3K27me2/me3), two transcriptionally repressive marks that subsequently lead to Polycomb Repressive Complex 1 (PRC1) recruitment and chromatin compaction (Margueron and Reinberg, 2011). Conversely, H3K36 methylation is mediated by several enzymes, and this methylation has been implicated in regulating transcriptional elongation, RNA processing, and DNA damage sensing (Guo et al., 2014; Kuo et al., 2011; Wen et al., 2014). In mammalian cells, SETD2 (Set2 in Drosophila) is the only H3K36 tri-methyltransferase, while multiple enzymes catalyze formation of H3K36me1/2, including NSD1/2/3 (Nsd in Drosophila), and ASH1L (Ash1 in Drosophila) (Wagner and Carpenter, 2012). H3K36 di-and tri-methylation (H3K36me2/3) have been previously shown to act as a euchromatin barrier to restrict H3K27me3 deposition (Streubel et al., 2018). Accordingly, H3K27me3 and H3K36me3 rarely co-exist in nucleosome pools and genomic distributions (Mao et al., 2015; Xiao et al., 2012; Yuan et al., 2011), and pre-methylated H3K27 histones are refractory to the catalytic activity of H3K36 methyltransferases (KMTs) (Jani et al., 2019).
Somatic mutations leading to lysine to methionine (K-to-M) substitutions at K27 and K36 in H3 variants (H3K27M and H3K36M) are recurrent in several cancers (Behjati et al., 2013; Boileau et al., 2019; Downing et al., 2012; Lu et al., 2016; Papillon-Cavanagh et al., 2017; Schwartzentruber et al., 2012; Wu et al., 2012). K-to-M mutations on H3.3 are dominant negative, effectively inhibiting the enzymatic activity of the relevant SET methyltransferases targeting the lysine on which they occur (Lewis et al., 2013). Consequently, there is a drastic decrease in the global levels of the methylation marks, despite mutant histones comprising less than 25% of the total pool of H3 (Lewis et al., 2013; Lu et al., 2016). The effects of H3K27M, however, do not simply mimic loss-of-function of PRC2. Rather, PRC2 retains its recruitment to nucleation sites at CpG islands (Harutyunyan et al., 2019; Mohammad et al., 2017) but is unable to propagate H3K27me3/me2 marks in the presence of H3K27M, thus possibly preventing differentiation-coupled epigenomic reprogramming (Harutyunyan et al., 2019). Conversely, in H3K36M the reduction of H3K36me2 in intergenic loci relieves repression of PRC2, leading to intergenic spread of H3K27me3 and subsequent recruitment of PRC1 away from its target genes, alleviating transcriptional silencing (Lu et al., 2016). The restricted deposition of H3K36me2/me3 marks was associated with impaired differentiation in the context of H3K36M (Lu et al., 2016), indicative that K-to-M H3 mutations may converge on impaired development through perturbation of K27/K36 methylation.
The Drosophila model has been instrumental in the identification and functional characterization of Polycomb group (PcG) components. In Drosophila, substitution of K27 by an arginine on H3 induces transcriptional de-repression and homeotic transformations mirroring loss of PRC2 and underscoring the crucial link between H3K27 methylation and PRC2 activity (Pengelly et al., 2013). In this study we expressed H3.3K27M and H3.3K36M in Drosophila tissues to investigate resulting effects on H3 PTMs, gene expression, and development.
Results
Overexpression of H3.3K27M and H3.3K36M in eye imaginal discs produces deleterious phenotypes in adult eyes and disrupts ommatidial architecture
To model the effect of K-to-M histone mutants on animal development, we overexpressed H3.3K27M, H3.3K36M and H3.3 wild-type (WT) control with a variety of tissue-specific promoters. Expressing either H3.3K27M or H3.3K36M under the control of 17 different promoters using the Gal4-UAS system produced larval or pupal lethality, and other deleterious phenotypes, while no detectable effects were observed from overexpression of control H3.3WT (Fig. S1A).
Expressing H3.3K27M under control of the eyeless (ey) driver resulted in reduced eye size, disorganization of the remaining ommatidia and loss of photoreceptor neurons, a finding also seen, albeit to a variable extent, when overexpressing H3.3K36M (Fig. 1A, Fig. S1B). Nearly 40% of ey>H3.3K27M flies and ~2% of ey>H3.3K36M flies exhibited disorganized tissue growth. This indicates that expression of H3.3K27M in this model system produces overgrowth on its own, an effect worsened in flies with a well-established sensitized retina tumor model that involves overexpression of Delta, a Notch-activating ligand (Bossuyt et al., 2009; Dominguez and Casares, 2005). In this genetic background, expression of H3.3K27M produced a stronger phenotype, with tumorous eyes seen in ~80% of flies and a high rate of ectopic growth (63%), directly adjacent to the eye area or distal in the head region (Fig. 1B, Fig. S1C). A smaller number of H3.3K36M-expressing eyes showed overgrowth (6%) and ectopic growth (13%), or complete loss of eye tissue (~1%) in the UAS-Dl background.
Fig. 1. Overexpression of H3.3K27M and H3.3K36M in eye imaginal discs using ey-Gal4 produces hypotrophic and tumorous eyes.
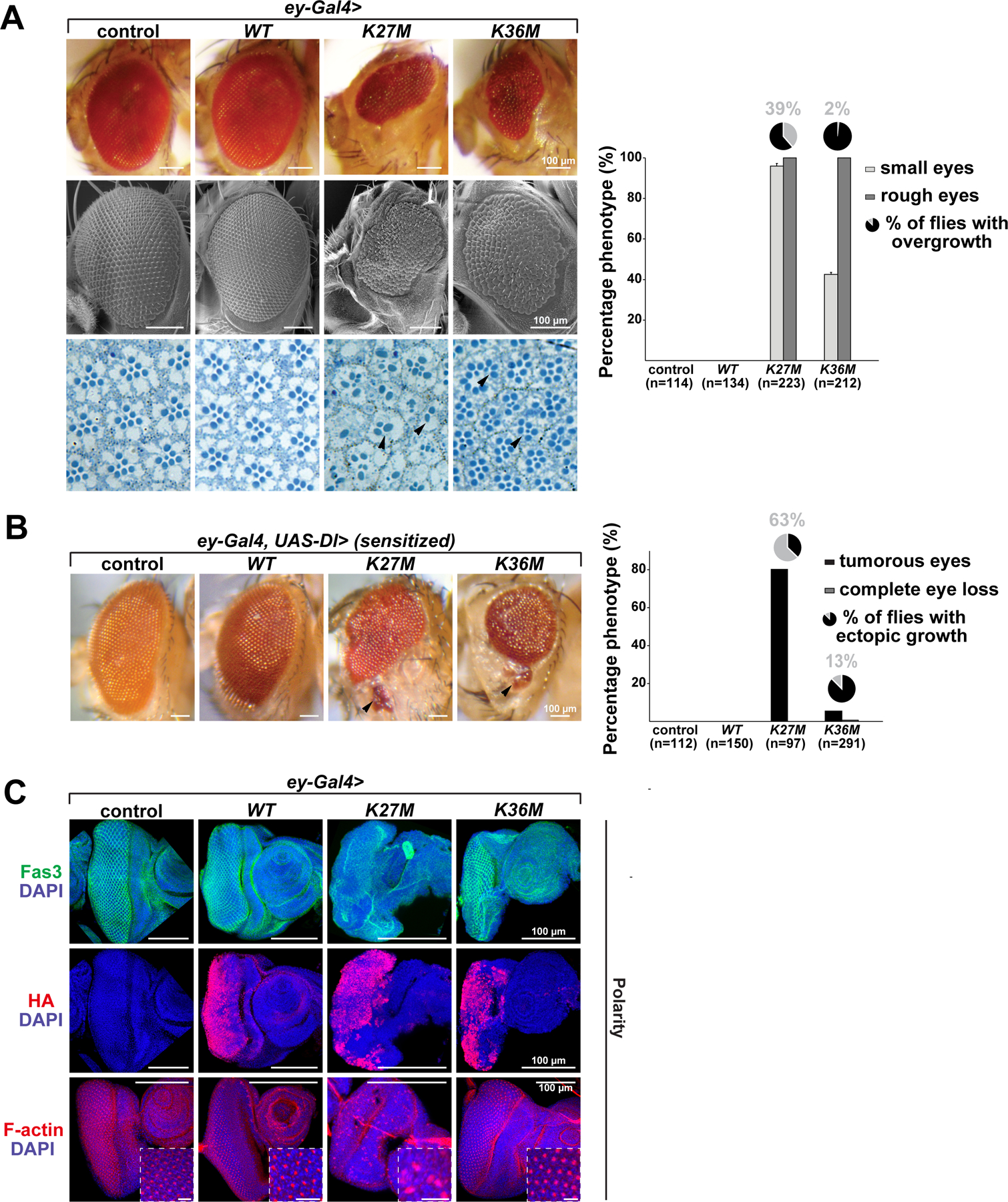
(A) Expression of H3.3K27M and H3.3K36M induces small eyes (upper panel), disorganization of ommatidia (rough eye phenotype), in some cases overgrowth, and decreased photoreceptor neurons (lower panel, black arrowheads) while expression of H3.3 wild-type (WT), and a Gal4 control have no effect on eye size and architecture; quantifications for the eye phenotypes are depicted in the right panel. Light grey bars represent percentage of small eyes, dark grey bars represent percentage of rough eyes, and pie charts percentage of flies with overgrowth phenotype; (n) represents total number of flies counted. (B) Sensitized flies overexpressing the Notch ligand Delta (Dl) in the developing eye display increased eye size. Tumorous eyes with metastasis of eye tissue (black arrowheads) are obtained upon expression of H3.3K27M and to much lower extent of H3.3K36M. Black bars represent percentage of tumorous eyes, grey bars represent percentage of flies with complete eye loss, and pie charts metastasis incidence within the tumorous eye phenotypes; (n) represents total number of flies counted. (C) Immunofluorescence staining for Fasciclin III, HA and the polarity marker F-actin in Gal4 control, H3.3 WT, H3.3K27M and H3.3K36M overexpressing eye imaginal discs. Transgene expression is visualized by HA staining. Areas in squares are shown at higher magnification. Scale bar 10µm. See also Fig. S1.
We next evaluated the phenotypes of ey>H3.3K27M flies and ey>H3.3K36M in third-instar larval eye-antennal discs. While only modest effects were observed when expressing H3.3K36M, expression of H3.3K27M in eye imaginal discs severely affected cell polarity and disc morphology. Immunostaining for the cone cell marker Fasciclin III showed severe disruption of the stereotypic repeated pattern of cone cells within the ey-GAL4 expression domain (Fig. 1C). The distribution of F-actin, a marker of cell polarity, was also abnormal in H3.3K27M-expressing eye imaginal discs. As well, ectopic expression of wingless (wg) was seen in 38% of H3.3K27M-expressing eye imaginal discs indicating transformations from eye disc to wing disc in dorsal regions neighbouring the ey-GAL4 expression domain (Fig. S1D). Deregulated expression of classical PcG target genes regulated by H3K27me3 deposition including Hox family members has been previously observed in an engrailed (en)-GAL4 H3.3K27M model (Herz et al., 2014). We thus assessed the expression of several Hox genes in our model system. Expression of Ultrabithorax (Ubx), previously shown to be derepressed in the wing imaginal disc upon H3.3K27M expression (Herz et al., 2014), and of Abdominal-B (Abd-B) were not altered in eyes expressing either H3.3 mutant (Fig. S1E). However, we observed ectopic expression of Antennapedia (Antp), which is normally expressed in thoracic imaginal discs, in cells anterior to the ey-GAL4 expression domain of H3.3K27M, but not in H3.3K36M mutants. We also observed homeotic transformations in the head regions near the eyes of H3.3K27M expressing adult flies, including extra wings, bristles or leg like structures (Fig. S1F). These non-cell autonomous phenotypes probably result from trans-determinative effects whereby H3.3K27M expressing cells perturb the developmental program of the neighbouring tissues. In all, these results indicate that H3.3K27M and H3.3K36M expression in imaginal eye discs affects eye growth and cell polarity, in a manner reminiscent of PRC2 and PRC1 mutant eye discs (Loubiere et al., 2016).
Overexpression of H3.3K27M and H3.3K36M in eye imaginal discs causes aberrant distribution of antagonistic methylation marks
Given that K-to-M mutations inhibit H3 lysine methylation, we investigated post-translational modification of H3K27 and H3K36 in the context of these mutations. As expected, expression of H3.3K27M in eye imaginal discs markedly reduced H3K27me2 and H3K27me3 levels, while H3.3K36M expression led to decreased H3K36me2 and H3K36me3 levels (Fig. 2A–B, Fig. S2A–B). Notably, we observed a strong increase of H3K36me2 in H3.3K27M expressing eyes, while the level of the much less abundant H3K36me3 was harder to quantitate. Reciprocal effects were observed in H3.3K36M-expressing eyes where higher levels of the antagonistic H3K27me2 and H3K27me3 marks were observed. Taken together, these data suggest that expression of H3.3 K27M and K36M in the eye disc depletes the cognate lysine methylation modification, and results in inappropriate accumulation of the respective antagonistic H3K27me3/H3K36me2 marks.
Fig. 2. Overexpression of H3.3K27M and H3.3K36M in eye imaginal discs using ey-Gal4 produces aberrant methylation of antagonistic methylation marks.
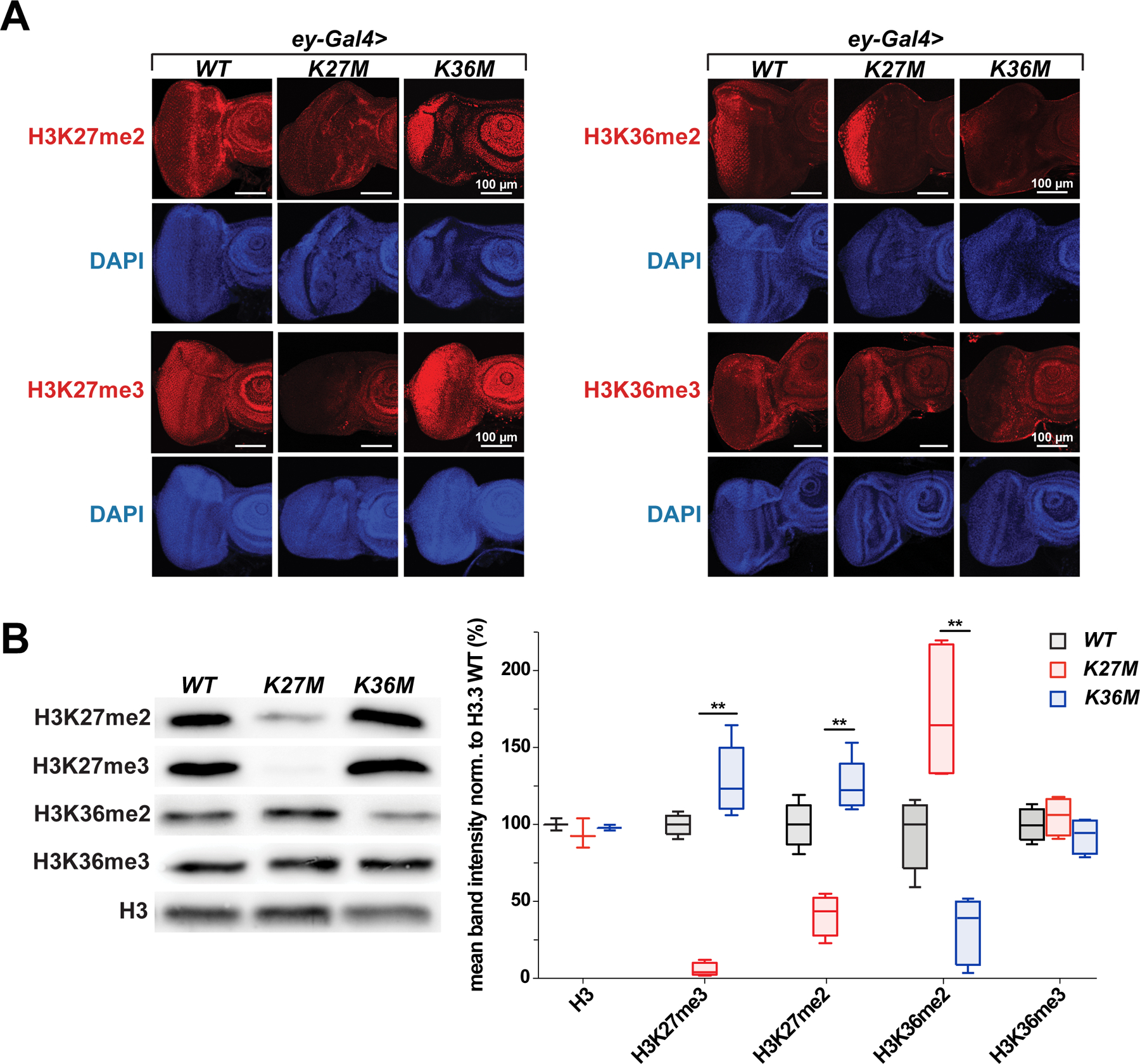
(A) Immunofluorescence staining for H3K27me2/3 (left) and H3K36me2/3 (right) in H3.3K36M, H3.3K27M and H3.3 WT overexpressing eye discs. (B) Quantification of H3K36 di-/ trimethylation, H3K27 di-/ trimethylation and H3 levels in histone extracts from adult eyes by western blot. Histone H3 was used as a control and β-Actin was used as a loading control. Band intensity of western blot was quantitated in graph to the right. See also Fig. S2.
H3.3K27M or H3.3K36M expressing eyes down-regulate eye development genes, and up-regulate piwi-interacting RNA (piRNA) biogenesis
To investigate molecular mechanism underlying the developmental defects following expression of K-to-M mutations, we carried out transcriptome profiling on Drosophila eye-antennal imaginal discs expressing H3.3WT, H3.3K27M, or H3.3K36M, along with yw controls (Fig. 3A, Table S1). We observed dysregulation of well-defined, functionally distinct pathways in both H3.3K27M and H3.3K36M mutants relative to (yw) and H3.3WT controls. Consistent with its more severe phenotype, a larger number of differentially expressed transcripts were identified for H3.3K27M (882 genes compared to 488 genes in H3.3K36M) (Fig. 3B). Despite these differences, we observed a convergence of transcriptional profiles, with an important overlap of significantly dysregulated genes in both mutants. In particular, mutually downregulated genes were found in pathways involved or related to eye development and photoreceptor cell differentiation, consistent with the phenotypes observed (Fig. 3C, Table S2). The significant changes we observed in H3K27M in higher level pathways such as ‘Generation of neurons’ and ‘Neuron differentiation’, were also driven by genes related to eye development, as the function of 26 out of 50 genes in these pathways is directly related to eye development. Examples of genes downregulated in both mutants include sevenless (sev), a gene expressed in photoreceptors and cone cells and required for R7 photoreceptor cell specification (Raabe, 2000), bride of sevenless (boss), the receptor for Sev, and lozenge (lz), which encodes a transcription factor involved in pre-patterning photoreceptor precursors in the developing eye.
Fig. 3. H3.3K27M or H3.3K36M expression results in downregulation of genes involved in eye development and upregulation of the piwi-interacting RNA (piRNA) pathway.
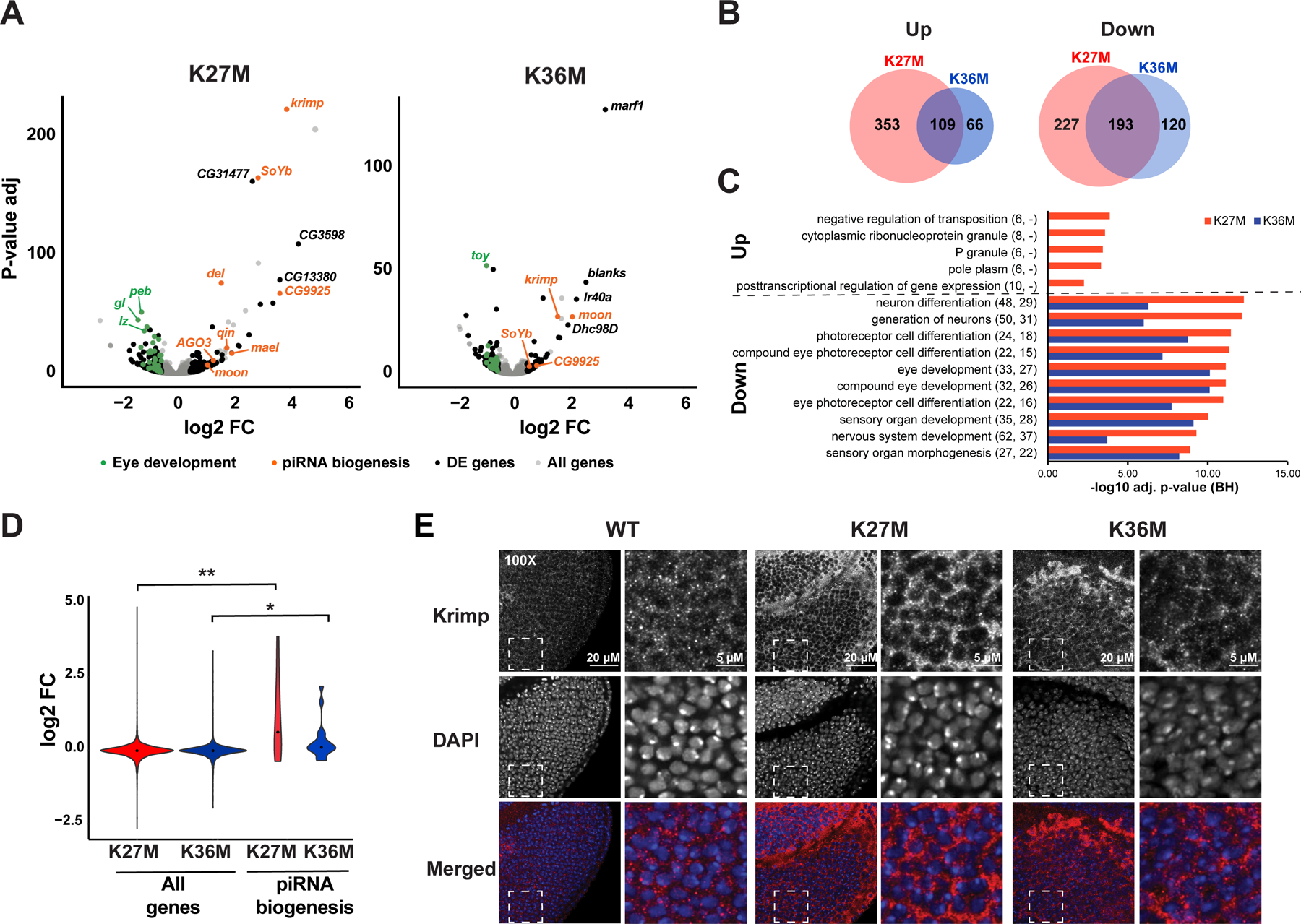
(A) Differential gene expression analysis of H3.3K27M (left) or H3.3K36M (right) compared to yw baseline control. Grey: all genes. Black: significantly deregulated genes (adjusted p-value < 0.05, absolute log2 fold change > 0.5, mean expression > 100) not affected when overexpressing H3.3 WT. Orange: piRNA pathway genes. Green: genes involved in eye development. (B) Overlap of statistically significant genes associated with expression of each H3.3 mutant. (C) Gene ontology analysis of top enriched biological processes in differentially expressed genes, as in (A). Numbers in parentheses indicate number of differentially expressed genes observed under each category in H3.3K27M and H3.3K36M mutant respectively. (D) piRNA biogenesis genes show significant increase in expression upon H3.3K27M expression. (E) Right: immunofluorescent staining (red in merged) for Krimp in ey>H3.3 WT, ey>H3.3K27M, and ey>H3.3K36M eye discs. The DNA stain DAPI (blue in merged) is used as an exposure control. White dashed square indicates field sampled for inset. Left: inset images showing cytoplasmic distribution of Krimp. See also Fig. S3, Table S1–S4.
Notably, expression of genes encoding proteins involved in biogenesis of piwi-interacting RNAs (piRNAs), which are normally associated with suppression of transposons in the germline (Klattenhoff and Theurkauf, 2008) showed striking changes in H3.3K27M, with krimper (krimp) the most highly up-regulated. The set of genes involved in piRNA biogenesis was upregulated in both H3.3K27M and H3.3K36M (Fig. 3D, Table S3), which we further confirmed by quantitative RT-PCR (Fig. S3A). This effect was more pronounced in H3.3K27M than in H3.3K36M, and we also observed increased expression of other germ cell developmental genes including fate-specification factor bam (McKearin and Spradling, 1990) and meiosis regulator Marf1 (Kawaguchi et al., 2020). Consistent with transcriptomic data, immunostaining showed increased Krimp expression in H3.3K27M, and to a lesser extent in H3.3K36M expressing eyes (Fig. 3E). Furthermore, RNA-seq of small RNAs confirmed expression of piRNA clusters, that are normally expressed in the germline, in H3.3K27M and H3.3K36M expressing eye imaginal discs (Fig. S3B, Fig. S3C). Within piRNA clusters, we observed differences in expression patterns of individual transcripts between the mutants (Table S4). Globally, the trend was towards up-regulation of piRNA clusters, with most clusters displaying greater positive change in the mutants compared to H3.3 WT, and with the upregulated clusters displaying more significant p-values and a larger size effect. Principal component analysis based on expression of piRNA clusters segregated both K-to-M mutants, while no correlation with genotype was observed for other small RNAs (Fig. S3D), suggesting that the effect of the H3.3 mutants on expression is stronger for piRNAs. Taken together, K27M and K36M disrupt expression of canonical eye developmental genes and promote aberrant expression of germ cell markers, including those involved in the piRNA biogenesis pathway.
H3.3 K27M and K36M disrupt H3K27me3 deposition on chromatin
To determine whether the transcriptome dysregulation relates to epigenetic perturbation, we performed H3K27me3 and H3K36me2 chromatin immunoprecipitation sequencing (ChIP-seq) on imaginal discs expressing H3.3WT, H3.3K27M, or H3.3K36M. Globally, we observed decreased deposition of the H3K27me3 mark in H3.3K27M, and global gain of H3K27me3 in H3.3K36M flies (Fig. S4A), in keeping with immunofluorescence and western blot data. As expected, H3K36me3 was depleted from gene bodies (Fig. S4B), while the globally distributed H3K36me2 mark was modestly reduced in H3.3K36M-expressing flies. Interestingly, H3.3K27M flies showed bimodal H3K36me2 change, with distinct genomic regions showing gains and losses (Fig. S4A).
On the chromosomal level, H3K27me3 and H3K36me2 occupied distinct regions of the genome and were mutually exclusive (Fig. 4A, Fig. S4C), as expected from the known antagonism of the two marks. In H3.3WT control, H3K27me3 formed discrete, punctate peaks over repressed genes along the chromosomal arm (Fig. S4D). Upon expression of H3.3K27M, H3K27me3 was retained and heavily enriched at strong peaks also observed in H3.3WT, but showed decreased deposition in their immediate flanking regions, consistent with H3.3K27M concentrating H3K27me3 at canonical sites and inhibiting spreading of this mark beyond PRC2 nucleation sites (Harutyunyan et al., 2019). In contrast, H3.3K36M showed depletion of H3K27me3 at highly enriched sites in H3.3WT, and gains of this mark in their flanking regions, suggesting the mutation promotes undue spreading of H3K27me3, diluting the mark from canonical sites observed in H3.3WT eye discs.
Fig. 4. K27M and K36M redistribute H3K36me2 away from repetitive sequences to actively transcribed genes.
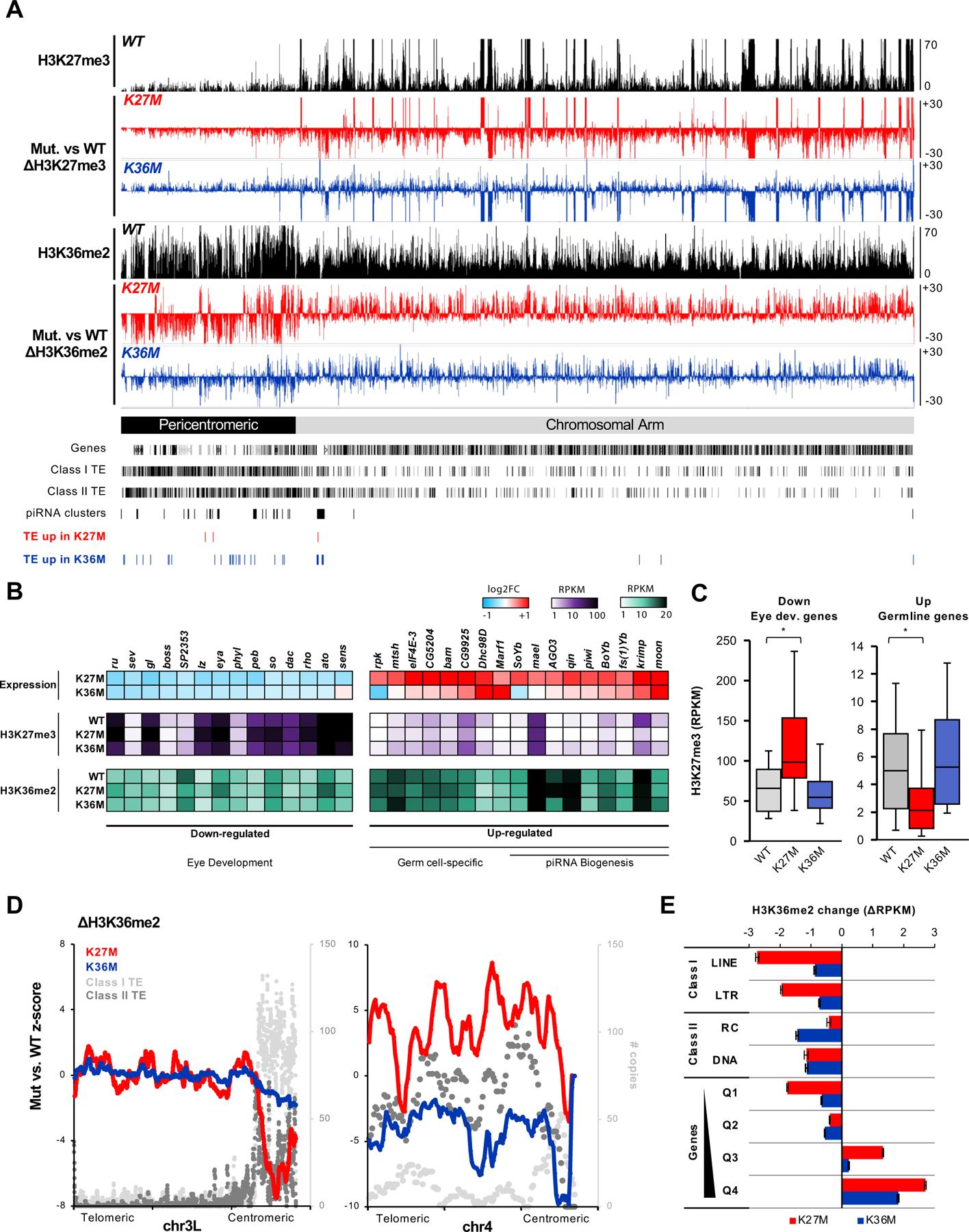
(A) Genome browser snapshot depicting H3K27me3 and H3K36me2 changes in H3.3K27M and H3.3K36M flies on chr2R, relative to H3.3 WT flies. Gain is represented as bars above zero, whereas loss is represented bars below zero. (B) Significantly differentially expressed eye developmental and germline-specific genes identified in H3.3K27M and their expression in H3.3K36M. Below, RPKM enrichment of H3K27me3 and H3K36me2 for each gene is depicted for H3.3 WT, H3.3K27M, and H3.3K36M flies. (C) H3K27me3 levels at eye developmental genes and germline specific genes, as in (B). * denote significant change in H3K27me3 (p < 0.001, two-tailed paired t-test). (D) Smoothed line plot depicting H3.3K27M- or H3.3K36M-mediated H3K36me2 change in relation to transposable element (TE) copy numbers over chr3L (left) and chr4 (right). (E) Bar plot depicting H3K36me2 change at LTR repeats and genic regions in H3.3K27M (red) or H3.3K36M (blue) mutants relative to H3-WT. Genes were stratified into 4 quartile bins based on expression in WT, with Q1 being silenced to Q4 being the highest expressed genes. See also Fig. S4–5.
Intersection of ChIP-seq data with differentially expressed genes, revealed that, compared to H3.3WT, H3K27me3 deposition is significantly increased at eye developmental genes in H3.3K27M mutants, likely leading to their continued transcriptional repression (Fig. 4B–C). In contrast, germ cell-specific genes including piRNA biogenesis components are marked with more intermediate levels of H3K27me3 in H3.3WT flies, and are significantly depleted of H3K27me3 in H3.3K27M mutants, concordant with their increased expression. In H3.3K36M, a subset of germline genes (e.g. krimp, CG9925) also showed modest loss of H3K27me3 (Fig. S4E). We further confirmed that a majority of the dysregulated genes are direct targets of PRC2, by intersecting the H3.3K27M/H3.3K36M transcriptomes with published transcriptomes of PRC1-or PRC2-deficient Drosophila eyes (Loubiere et al., 2016) (Fig. S4F). Altogether, the transcriptomic and epigenomic analysis suggest that H3.3K27M and H3.3K36M perturb PRC2-mediated H3K27me3 deposition resulting in down-regulation of eye development genes and aberrant expression of piRNA biogenesis genes.
H3.3K27M and H3.3K36M re-distribute H3K36me2 away from pericentromeric transposon-rich regions
H3K36me2 is widely distributed in eye-antennal disc tissue, where it marked close to ~60% of the genome, at euchromatic gene bodies as previously described (Bell et al., 2007), with levels of enrichment correlating with transcription activity (Fig. S4D). Importantly, we observed striking H3K36me2 domains that are over hundreds of kilobases long at pericentromeric regions in H3.3WT control eye discs (Fig. 4A). In the Drosophila genome, these pericentromeric regions are relatively gene-poor and are densely populated with transposable elements (TEs), with some degenerate TE copies transcribed as piRNA clusters (Brennecke et al., 2007). In H3.3K27M-expressing eye-antennal discs, H3K36me2 is drastically depleted at these pericentromeric regions, with boundaries of loss aligning precisely with TE density (Fig. 4D, Fig. S5A). This mark is redistributed from these areas and is instead gained along the chromosomal arm of H3.3K27M flies, mostly at highly expressed gene bodies (Fig. S4D). H3.3K36M-expressing flies showed a similar H3K36me2 redistribution pattern on most chromosomes, albeit to a lesser degree compared to H3.3K27M. Chromosome 4, the smallest and most TE enriched in Drosophila, was the notable exception, showing severe loss of H3K36me2 in H3.3K36M flies but not in H3.3K27M (Fig. 4D). Regardless, H3.3K27M and H3.3K36M both showed genome-wide redistribution of H3K36me2 away from TE-rich regions into highly transcribing gene bodies (Fig. 4E). Indeed, regions concurrently losing H3K27me3 and reciprocally gaining H3K36me2 in H3.3K27M were enriched for genes relative to randomly sampled bins; similarly, genic enrichment was also observed for regions losing H3K36me2 and gaining H3K27me3 in H3.3K36M (Fig. S5B). Altogether, these data suggest H3.3K27M and H3.3K36M collectively perturb H3K27me3/H3K36me2 crosstalk at gene-dense euchromatin, causing redistribution of H3K36me2 away from transposon-rich regions into gene bodies.
De-repression of transposable elements in H3.3 K27M and K36M flies
Given that H3K36me2 is severely depleted at repetitive regions, we then examined whether TEs become transcriptionally de-repressed in the K-to-M mutants. Using uniquely aligned RNA-seq reads at annotated repetitive elements, we observed significant induction of individual Long Terminal Repeat endogenous retroviral elements (LTR-ERVs) in both H3.3K27M and H3.3K36M flies compared to other classes of repeats (Fig. 5A), and induction of Long Interspersed Nuclear Elements (LINEs) specifically in H3.3K36M mutants. Significantly up-regulated TEs are mostly embedded in pericentromeric regions heavily enriched for H3K36me2, and generally do not overlap with the piRNA clusters (Fig. S5C). Notably, H3.3K36M mutants showed greater induction of transposon expression, and de-repressed a wider spectrum of repeat families than H3.3K27M (Fig. 5B). Nevertheless, both K-to-M mutants likely perturbed H3K36me2-mediated repression of transposable elements in Drosophila, as they both impacted the deposition of this mark on repeat elements. Specifically, full length copies of gypsy LTR-ERVs are generally enriched for H3K36me2 in WT eye discs, relative to H3K27me3 or H3K36me3 (Fig. S6A, Table S5). In both H3.3K27M and H3.3K36M mutants, gypsy LTRs and internal coding regions were depleted of H3K36me2 and constituted the majority of up-regulated TEs (Fig. 5C, Fig. S6A).
Fig. 5. H3.3K27M and K36M de-repress transposable elements in flies.
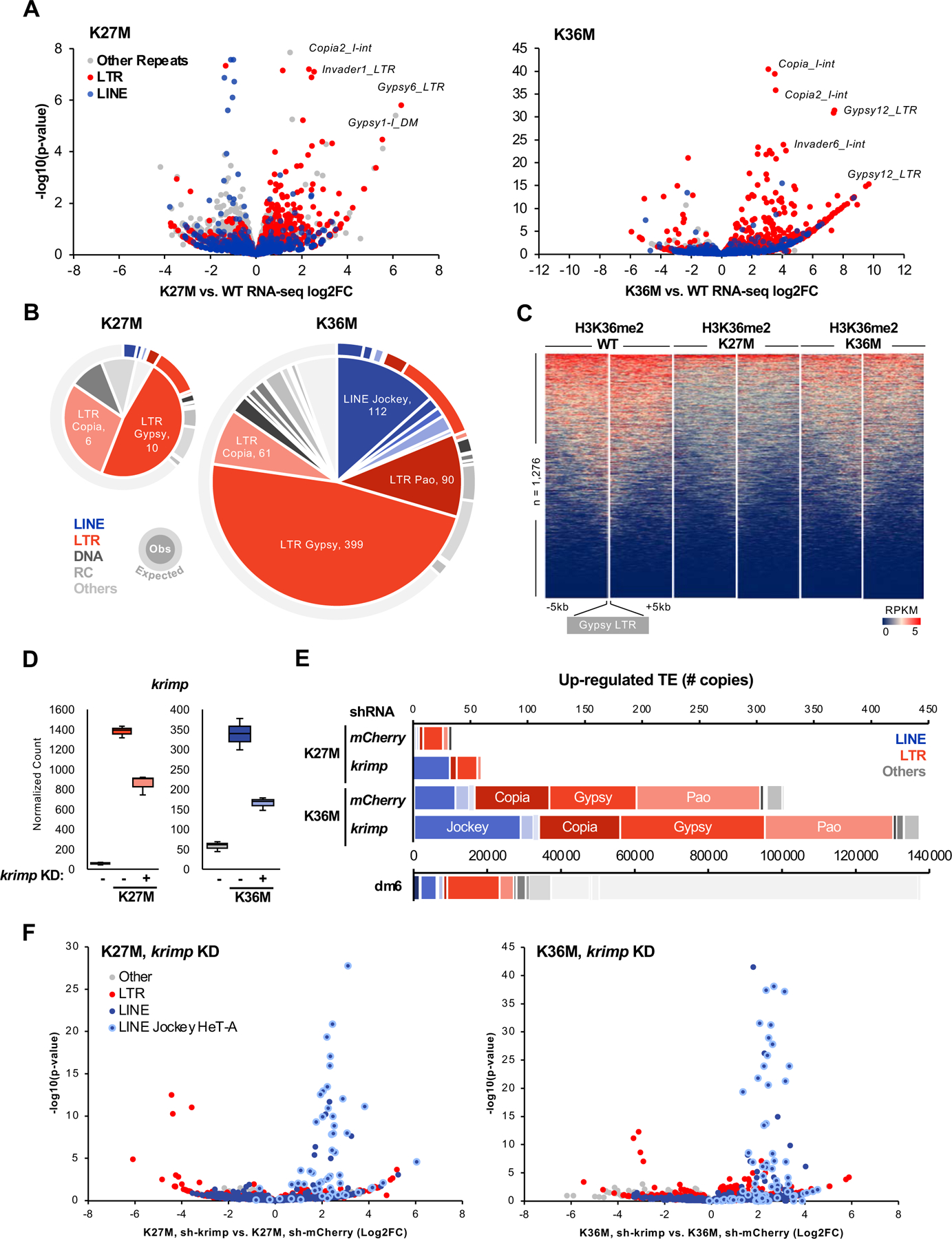
(A) Volcano plot depicting K27M- (left) and K36M-mediated transcrional dysregulation in repetitive sequences. Each dot represents an individual copy of repeat, red dots represent individual LTR elements while blue dots depict LINEs, grey dots represent all other repetitive classes. Note that K36M de-represses LTR repeats to a greater extent than K27M. (B) Pie chart representing the number of significantly up-regulated (log2FC > 2, FDR < 0.05) elements in each repeat class in H3.3K27M and H3.3K36M eye discs. Outer ring depicts copy number of each repeat class in Drosophila melanogaster genome dm6. (C) Stacked heatmap depicting H3K36me2 enrichment at all gypsy LTR (n = 1,276) in H3.3 WT, H3.3K27M, and H3.3K36M flies. (D) RNA-seq normalized count for krimp knockdown validation in H3.3K27M and H3.3K36M flies. Negative sign (-) indicate mCherry RNAi. (E) Number of transposon elements significantly up-regulated (log2FC > 0, FDR < 0.05) in krimp KD K27M and K36M, compared to mCherry RNAi negative control. Below, copy number of each repeat class in dm6. (F) Volcano plot depicting repeat sequence dysregulation upon krimp KD, compared to mCherry KD control, in H3.3K27M and H3.3K36M flies. Each dot represents an individual copy of repeat, red dots represent individual LTR elements, blue dots depict LINEs (light blue circle: Jockey HeT-A family), grey dots represent all other repetitive classes. See also Fig. S6, Table S5.
We had previously reported increased transcription of ERVs in human H3.3K27M glioma (Krug et al., 2019). To determine whether K36M similarly disrupts TE repression in mammals, we analyzed published RNA-seq (Lu et al., 2016) from a panel of K36-mutant murine mesenchymal stem cell (MSCs) and human K36M chondroblastoma tumors (Fig. S6B). Consistent with results in Drosophila, we observed de-repression of both LTR and LINE1 families in K36M/I, but not K36R-expressing murine MSCs (Fig. S6C–D). Finally, we also confirmed significant induction of individual LINE and LTR transposons, predominantly from the L1 and L2 families, in human K36M chondroblastoma compared to H3 WT tumors (Fig. S6B–D). Collectively, our data suggest K36M compromises retrotransposon silencing across animal species.
krimp modulates TE expression in H3.3K27M/H3.3K36M
In Drosophila germ cells, the piRNA component Krimp recruits Aubergine and AGO3 proteins to perinuclear nuage and coordinates the assembly of ping-pong piRNA-processing complex (Sato et al., 2015; Webster et al., 2015) to mediate LINE silencing (Lim & Kai, 2007). To investigate whether ectopic piRNA gene expression in the mutants contributes to TE silencing, we expressed short-hairpin RNA targeting krimp in ey>H3.3K27M and ey>H3.3K36M flies and performed RNA-seq on larval eye discs. As expected, H3.3K27M and H3.3-K36M expression led to robust up-regulation of krimp, inducing 24- and 6-fold higher expression respectively compared to mCherry KD negative control (Fig. 5D). In both H3.3 mutants, down-regulation of krimp had modest effects on the abnormal eye phenotype (Fig. S6E) and did not alter the levels of eye developmental genes or the expression of other piRNA biogenesis genes (Fig. S6F). In contrast, decreased krimp expression led to specific de-repression of Jockey HeT-A elements in H3.3K27M and H3.3K36M eye discs (Fig. 5F), in keeping with the known regulation of this LINE family elements by this piRNA biogenesis member (Lim & Kai, 2007). Thus, high krimp expression likely accounts for the silencing of LINE family elements in H3.3K27M-expressing eyes (Fig. 5B, 5E), as its expression is much higher in these mutants compared to K36M (Fig. 5D, Fig. 3E). Taken together, we demonstrate that loss of H3K27me3 in H3.3K27M and its redistribution in H3.3K36M lead to aberrant expression of the piRNA biogenesis pathway, which in turn dampens the extent of TE de-repression especially in H3.3K27M mutant flies.
Reduction of ash1 and E(z) rescues H3.3K27M and H3.3K36M phenotypes respectively
To assess whether redistribution of antagonizing H3K36 and H3K27 methylation marks is causal to the observed phenotypes in Drosophila, we performed modifier screens selectively focusing on PRC1 and PRC2 components and H3K36 histone methyltransferases. We used the UAS-Gal4 system to express short hairpin RNA (shRNA) against 18 selected genes in ey>H3.3K27M and ey>H3.3K36M flies (Table S6). In both H3.3K27M and H3.3K36M expressing eyes, knock-down of members of the PRC1 complex Suppressor of zeste-2 (Su(z)2), Sex combs extra (Sce), Polycomb (Pc), or Sex comb on midleg (Scm) or PRC2 components jing and Chromatin assembly factor 1, p55 subunit (Caf1–55), also a component of the ASH1 complex (Huang et al., 2017), worsened the phenotype (Fig. 6A, Table S7–8). Remarkably, expression of shRNA targeting Enhancer of zeste (E(z)), a component of PRC2, or discs absent, small, or homeotic-1 (ash1) that encodes a trithorax-group H3K36 methyltransferase (Dorighi and Tamkun, 2013), restored normal growth and ommatidial organization in H3.3K36M and H3.3K27M, respectively (Fig. 6B, Fig. S7A–C). Specifically, the expression of two different shRNAs targeting E(z) in H3.3K36M expressing eyes, rescued the small-eye phenotype in 70%-86% of flies, while it worsened the phenotype and caused lethality in H3.3K27M expressing flies (Fig. 6B, Fig. S7B–C, Table S7–8). Conversely, expression of four different shRNAs targeting ash1 produced striking modifier effects resulting in the rescue of the H3.3K27M small-eye phenotype in 56%-94% of flies (Fig. 6B, Fig. S7A, Table S9), and dramatically exacerbated the H3.3K36M phenotype (Fig. 6B). This effect was not unique to eye discs, as ash1 knock-down also rescued H3.3K27M-induced wing and leg defects (Table S7).
Fig. 6. shRNA-mediated knockdown of E(z) and ash1 restores normal eyes development in respectively H3.3K36M and H3.3K27M mutant flies.
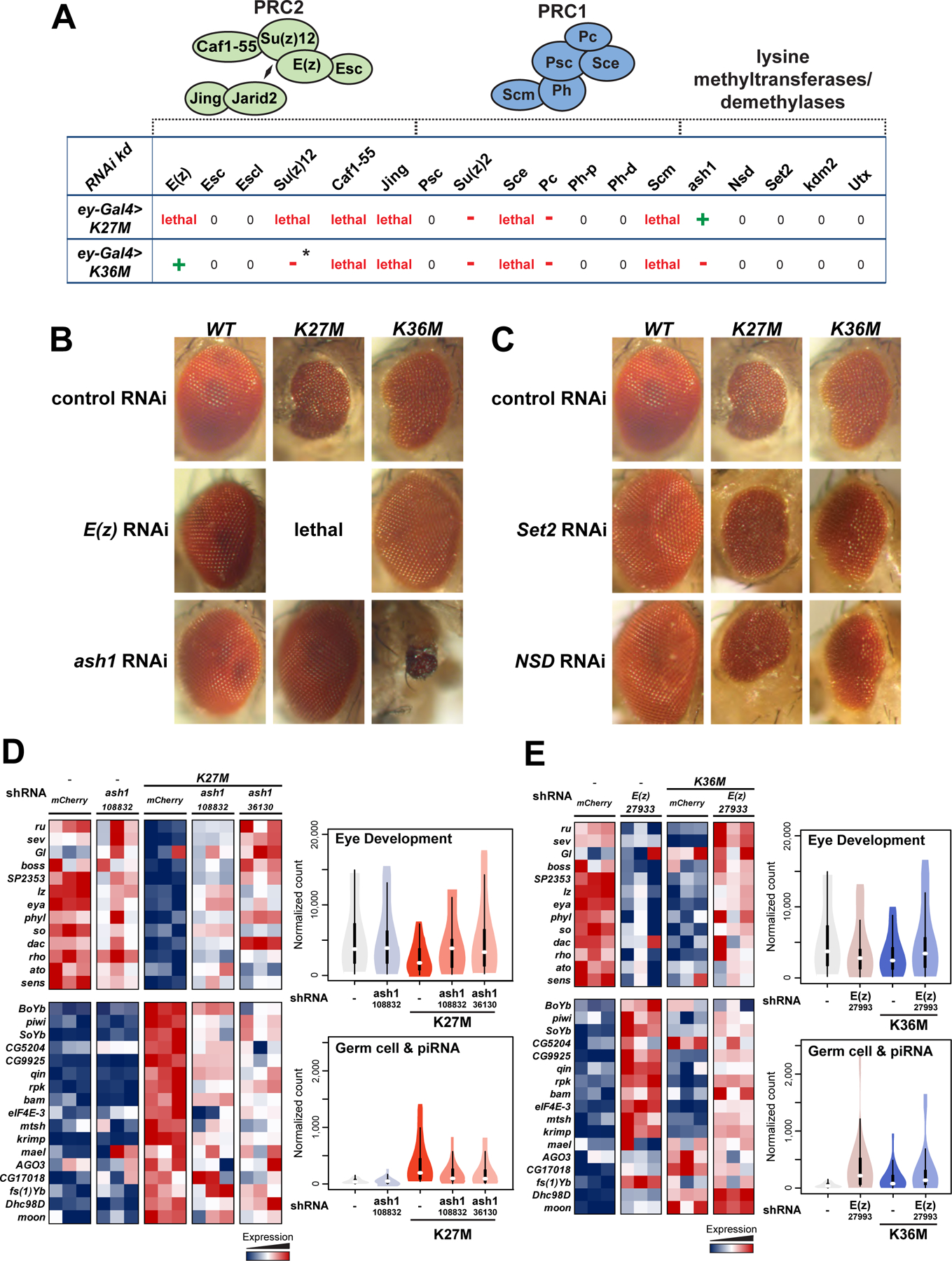
(A) Eye disc-specific H3.3K27M and H3.3K36M expressing flies were crossed with 18 selected strains expressing specific short hairpin (sh)RNAs targeting PRC2/PRC1 members as well as lysine-methyltransferases or demethylases. Green crosses indicate suppression of phenotype, red dashes indicate worsening of phenotype, while zeroes indicate no effect. Asterisk indicates that the effect was only observed in females, not in males. (B) Micrographs of adult eyes illustrating modifier effects of E(z) (BDSC: 27933) and ash1 (VDRC:108832) knockdown on ey>H3.3 WT, ey>H3.3K27M, and ey>H3.3K36M flies. (C) Micrographs of adult eyes illustrating the lack of a modifier effect from Set2 or NSD knockdown on ey>H3.3 WT, ey>H3.3K27M, and ey>H3.3K36M flies. (D) Left: heatmap depicting relative expression of eye developmental genes (upper panel) and germ cell genes (lower panel) in ash1-KD rescued H3.3K27M, relative to ash1-KD alone and mCherry KD controls. Right: violin plot representing expression quantification of gene sets depicted in the heatmap. (E) Left: heatmap depicting relative expression of eye developmental genes (upper panel) and germ cell genes (lower panel) in E(z)-KD rescued H3.3K36M, relative to E(z)-KD alone and mCherry KD controls. Right: violin plot representing expression quantification of gene sets depicted in the heatmap. See also Fig. S7, Table S6–9.
Targeting other H3K36 methyltransferases, namely NSD and Set2, had no effect on the small eye phenotype or leg defects in Drosophila despite the use of multiple shRNAs and validation of knock-down (Fig. 6C, Table S7–8). Moreover, we observed no effect on eye development and organization upon NSD and Set2 knock-down in a H3.3 wild-type genetic background, while as expected, knock-down of ash1 and E(z) alone induced eye hypotrophy with variable penetrance (Loubiere et al., 2016; Shearn and Garen, 1974) (Fig. 6C, Fig. S7A–B). Notably, rescue of the eye phenotype was dose-dependent, as it was inversely proportional to the construct’s ability to induce eye defects in H3 WT flies, and by extension to residual ash1 or E(z) levels following knock-down (Fig. S7A–B). Our data show that dosage reduction of the antagonizing lysine methyltransferases restores normal eye, leg, and wing development, suggesting that aberrant accumulation of H3K36me2 in H3.3K27M, and of H3K27me3 in H3.3K36M participate in generating the phenotype induced by these mutations.
ash1- and E(z)-depletion restores eye developmental gene expression in K27M and K36M
To delineate whether genic or TE dysregulation underlie the developmental eye defect, we performed RNA-seq on K27M-ash1 and K36M-E(z) rescued flies and on ash1- and E(z)-alone RNAi lines, as they also exhibited hypotrophic eyes. In K27M, ash1-KD significantly increased the expression of eye developmental genes, with ash1 RNAi line 36130 showing near complete complementation relative to control consistent with its higher rescue efficiency (Fig. S7A). In contrast, germ cell genes and LTR-ERVs remained derepressed in the two ash1-rescued lines with respect to negative control (Fig. S7D). Depletion of ash1-alone caused significant down-regulation of a subset of eye developmental genes (e.g. lz, eya, and so), consistent with the phenotype it induced, but had only modest effects on germ cell/piRNA biogenesis genes and TE expression (Fig. 6D, Fig. S7D). In E(z)-rescued K36M flies, expression of eye developmental genes was similarly restored to normal levels. In contrast, germ cell and piRNA biogenesis genes became further up-regulated upon E(z)-KD, and this increase was concurrent with dampened TE de-repression in rescued K36M-mutant eyes (Fig. S7D). E(z) KD in the eye of normal flies recapitulated dysregulation of both eye developmental and germ cell genes, phenocopying in this aspect K27M mutants (Fig. 6E). Collectively, these results suggest that the developmental defects in H3.3K27M and H3.3K36M can be reverted by re-expression of canonical eye specification genes, and are likely independent of aberrant expression of piRNA genes and TEs.
Discussion
We show that expression of H3.3K27M and H3.3K36M in Drosophila eye imaginal discs leads to hypotrophy of the eye and disorganized ommatidial structures. With respect to these phenotypes H3.3K27M seems to recapitulate aspects of both PRC2 and PRC1 knock-down (Herz et al., 2014), with hypotrophy resembling PRC2 mutants (Loubiere et al., 2016; Pengelly et al., 2013), while the overgrowth and changes in cell identity may reflect what has been observed in PRC1 mutants (Loubiere et al., 2016). Indeed, PRC2/1-target genes are transcriptionally dysregulated in both H3.3K27M- and H3.3K36M-expressing eye imaginal discs, confirming these mutations interfere with their transcriptional silencing.
Our work reinforces the notion that disease phenotypes observed in K-to-M mutants result not only from direct loss of the cognate lysine methylation, but also from secondary redistribution of antagonistic chromatin modifications leading to further transcriptional dysregulation. We show here in Drosophila eye discs that H3.3K27M and H3.3K36M mutations directly deplete H3K27me3 and H3K36me2, respectively, and inappropriately re-distribute the reciprocal mark away from canonical sites, compromising silencing of germline specification genes and transposable elements (Fig. 7). In the normal eye disc, H3K27me3 repress developmentally regulated genes and germ cell markers (including piRNA genes), whereas H3K36me2 predominantly mark transposon-rich pericentromeric regions and transcribing gene bodies. Upon expression of H3.3K27M, H3K27me3 domains contract, causing this mark to be unduly retained at eye developmental genes and to be lost at intermediate sites, leading to aberrant expression of germ cell-specific genes including the piRNA biogenesis machinery. In the absence of H3K27me3 in H3.3K27M, H3K36me2 is titrated away from repetitive regions and accumulates at gene-dense regions. This redistribution is associated with only modest transposon activation as expression of these elements is probably further regulated by piRNA-mediated surveillance in the H3.3K27M eye discs, as suggested by the modulation of specific TEs by krimp in our model system. As the majority of piRNA biogenesis machinery (e.g. ago3, piwi, krimp) is aberrantly expressed in H3.3K27M flies, this may play a compensatory role in processing and degradation of TE transcripts. Conversely, H3.3K36M expression causes direct loss of H3K36me2 at transposon-rich regions, leading to rampant TE induction. Loss of H3K36me2 promotes redistribution of H3K27me3, left unchecked in the H3.3K36M mutants, which propagates into flanking regions at the expense of canonical PRC2 targets, relaxing repression at a smaller subset of germline genes.
Figure 7. Model depicting K27M and K36M-mediated perturbation of H3K27me3 and H3K36me2 at genes and transposons.
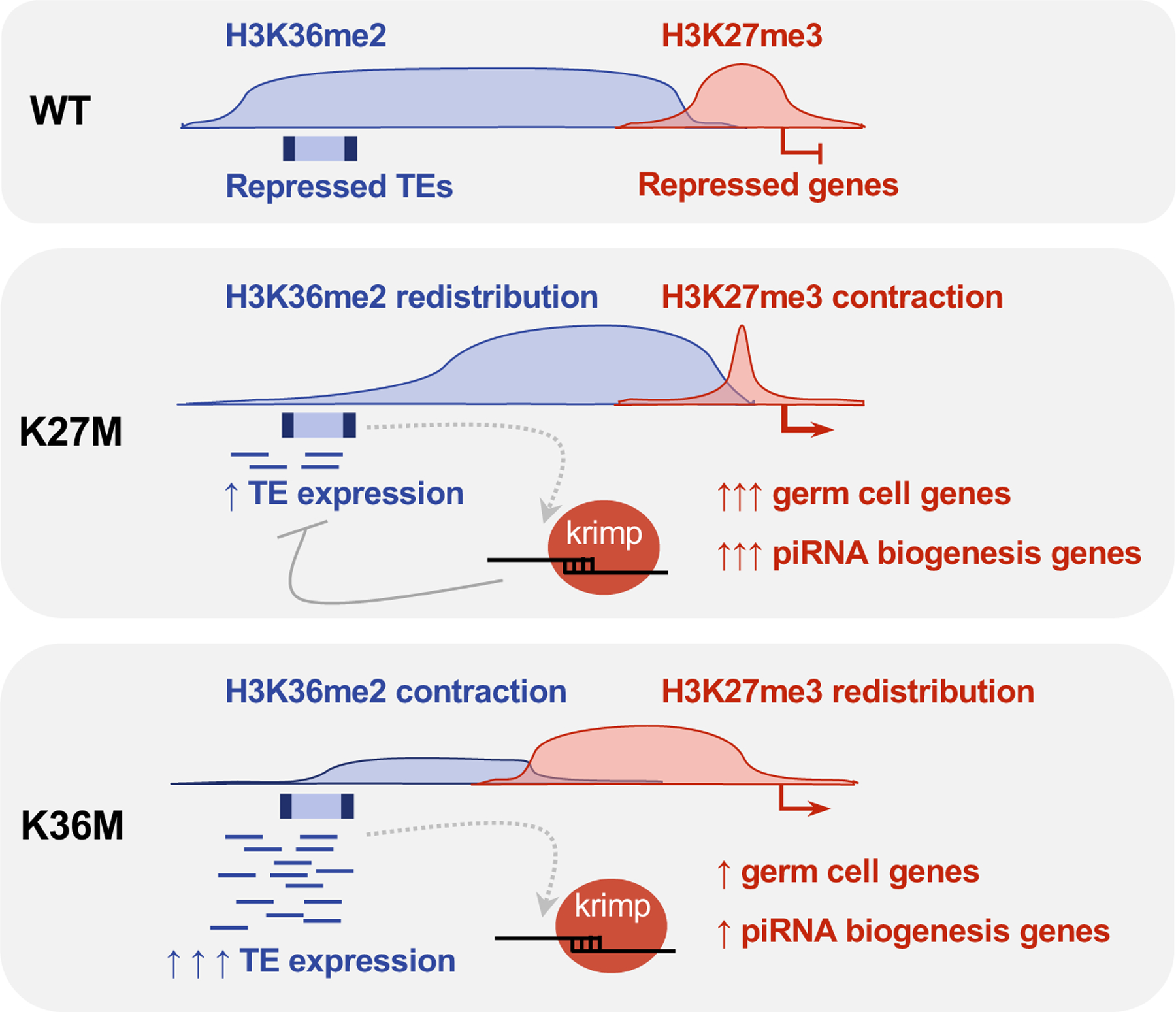
In WT eye discs, H3K27me3 and H3K36me2 mark and transcriptionally repress distinct regions of the genome, with H3K27me3 repressing developmentally-regulated genes and H3K36me2 repressing transposons. In the presence of K27M, the repressive H3K27me3 chromatin contracts and causes rampant activation of germ cell-specific genes including piRNA biogenesis components. In the absence of H3K27me3, H3K36me2 is redistributed into genic regions and lost from transposon-rich pericentromeric heterochromatin resulting in transposon de-repression. The aberrantly expressed piRNA components is then be utilized as a surveillance mechanism to degrade these TE transcripts primarily in K27M. In K36M-expressing eye discs, H3K36me2 is lost from pericentromeric regions and directly causes de-repression of transposable elements. In the absence of H3K36me2, H3K27me3 aberrantly spreads into flanking sites and is diluted from its canonical targets, resulting in mild up-regulation of germ cell and a subset of piRNA biogenesis genes.
The upregulation of TE in H3.3K27M and K36M mutants in Drosophila is reminiscent of what we previously observed in human H3.3K27M glioma (Krug et al., 2019). When we investigated K36M mammalian models, we observed similar effects in mouse K36M/I MSCs, and in H3.3K6M-mutant human chondroblastoma cell lines, supporting a role for H3K36me2 in the containment of TE in mammalian cells. In somatic mammalian cells, DNA cytosine methylation plays a critical role in maintaining retrotransposon silencing (Walsh et al., 1998), and intergenic DNA methylation is mediated, in part, by H3K36me2. Indeed, recent work in mammalian cells shows that H3K36me2 targets the recruitment of the de novo DNA methyltransferase DNMT3A to intergenic regions (Weinberg et al., 2019). As H3K36me2 is enriched at repeat elements, and its decrease in K36M-expressing eyes or its redistribution away from these elements in K27M-expressing eyes correspond to de-repression, this raises the intriguing possibility that the H3K36me2 mark by itself could mediate transcriptional repression in Drosophila, which lacks DNA methyltransferases. Whether H3K36me2 confers this repression directly in Drosophila, or indirectly via crosstalk with other heterochromatin machinery such as eggless/SETDB1-mediated H3K9me3, requires further investigation. This undue de-repression of repetitive elements by K-to-M mutants may promote antiviral host response such as interferon response in K27M glioma (Krug et al., 2019) and, as we show here, piRNA-mediated surveillance in flies.
Importantly, the developmental defects associated with K27M and K36M-expression can be reverted by knockdown of the antagonistic lysine methyltransferase: in H3K36M by decreasing E(z), and in K27M by decreasing ash1. Notably, individual K36 methyltransferases in Drosophila - Set2 (SETD2), NSD (NSD1/2) and Ash1 (ASH1L) - are non-redundant in their capacity to revert the K27M phenotype. NSD and Set2 have highest expression levels during early Drosophila development and are responsible for most H3K36 methylation in larval brains (Dorafshan et al., 2019). In contrast, within somatic tissues, ash1 expression is highest in imaginal discs, in keeping with a preponderant role for this methyltransferase in eye and limb development (Dorafshan et al., 2019). This further argues for a boundary effect mediated by antagonistic chromatin marks where deposition of H3K36me2 by Ash1 may oppose PRC2 recruitment or spread, preventing H3K27me3 deposition to maintain expression of key developmental transcription factors. This is similar to the recently uncovered differential role of K36 methyltransferases in Neurospora fungi (Bicocca et al., 2018), where Ash1-mediated H3K36me functions to maintain repression at H3K27me3-competent chromatin, whereas Set2 deposits H3K36me at active genes. Better understanding of ash1/ASH1L would therefore be critical in evaluating its potential as a suppressor of K27M oncohistone in human cancers.
Our data suggest the developmental defect associated with K27M and K36M in Drosophila stems from insufficient expression of canonical eye specification genes, and is likely independent from aberrant expression of germ cell genes and transposons in these mutations. Indeed, piRNA genes and transposons remain transcriptionally de-repressed in ash1-rescued K27M and E(z)-rescued K36M animals, and depletion of krimp did not notably shift the hypotrophic eye phenotype in K27M and K36M mutants. We also show that these eye developmental genes are canonical targets of PRC2 and abnormally retain H3K27me3 in K27M flies. ash1 depletion in K27M may therefore allow PRC2 to spread locally to alleviate this repression, whereas E(z) depletion would presumably relax repression similarly in K36M flies. Whether protracted PRC2-silencing causes similar developmental stalling in K27M- and K36M-associated cancers remains to be investigated.
In summary, we show that H3.3K27M and H3.3K36M oncohistones exert dominant negative effect on lysine methyltransferases inducing profound secondary effects on the epigenetic landscape, which contribute to the phenotype in Drosophila and likely play similar roles in human tumorigenesis. The study of these oncohistones reveals a previously unappreciated role of the H3K36me2 mark in possibly containing the silent genome, and further highlight the non-redundancies of the individual H3K36 lysine methyltransferases in their specificity on chromatin. Importantly, this work further implicates the therapeutic potential of PRC2 inhibition in H3.3 K27M and K36M oncohistone-associated cancers and identifies H3K36 methylation as a potential target for deadly H3.3K27M-associated tumors.
Limitations of the Study
Many questions remain to be addressed regarding the role of opposing chromatin marks in K-to-M mutagenesis, and the exact function of H3K36me2 in Drosophila and mammalian systems. H3K36me2 exist as broad domains over transcriptionally silenced region in mammals (Lu et al., 2016; Shirane et al., 2020), and as we show here, flies; however, how this intergenic H3K36me2 is deposited and maintained, and its functions are poorly understood. H3K36me2 is widely redistributed in the context of both K27M and K36M mutations in the fly model and specifically depleted from TE-rich regions, concurrent with their de-repression. While this indicates H3K36me2 may have a repressive role on transcription, our results are correlative and we cannot exclude secondary epigenetic dysregulation resulting in TE induction. The small amounts of chromatin from larvae eye discs limited a more comprehensive exploration of additional epigenetic modifications, and the functional effects of TE de-repression. Future work deciphering the differential recruitment mechanisms of H3K36 di-methyltransferases, the role of H3K36me2 and the interplay with other epigenetic marks in the fly and in more elaborate mammalian systems are warranted to further validate its role as a repressive mark.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nada Jabado (nada.jabado@mcgill.ca).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
ChIP-seq, RNA-seq, smRNA-seq datasets for Drosophila samples have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE140979. H3.3 K36M murine and human RNA-seq datasets have been previously deposited in GEO under dataset accession number GSE69291.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila strains
The following fly strains were used throughout the study: Act5C-Gal4 (BDSC #3954, Christian Klämbt), ey-Gal4 (BDSC #5535), ey-Gal4, UAS-Dl/CyO flies (a gift from B. Hassan), robo2-Gal4 (BDSC #48074), en-Gal4 (BDSC #6356), ptc-Gal4 (BDSC #52212), C179-Gal4 (BDSC #6450), mef2-Gal4 (BDSC #27390), CG25C-Gal4 (BDSC #7011), elav-Gal4 (BDSC #458), slit-Gal4 (BDSC #9580, Christian Klämbt), repo-Gal4 (BDSC #7415, Christian Klämbt), gcm-Gal4 (BDSC #35541), crol-Gal4 (VDRC #200123), ama-Gal4 (#205487), appl-Gal4 (a gift from Doris Kretzschmar, Münster) and tkk-Gal4 (#45606). All RNAi strains used are listed in (Table S6).
Generation of H3.3 WT and mutant transgenic flies
To generate transgenic flies PCR8/GW/TOPO vectors containing H3.3 WT and H3.3K27M mutant cDNAs with 1xHA tag were subcloned into the pUASg.attB gateway vector for use in the φC31-based integration system. For H3.3K36M, pUC57 vector containing the appropriately mutated mouse H3.3 sequence was used as a template for amplification and subsequent cloning into the pENTR/SD/D-TOPO vector. The insert from the pENTR vector was then subcloned into the pUASg-HA.attB vector, which contains 3x HA tags. All vectors were then injected into embryos of the attP-86Fb acceptor fly line for integration at the same genomic location. Flies were raised and crossed at 25°C; standard Drosophila husbandry procedures were followed. Flies were crossed against cell type-specific Gal4 driver lines (Bloomington Drosophila Stock Center, Bloomington, USA and Vienna Drosophila Resource Center, Vienna, Austria) to activate expression of UAS constructs.
Genetic modifier screen
Cell-type-specific screening strains were generated by balancing the second and third chromosomes carrying the GAL4 or UAS construct, respectively. Tissue specific expression of transgenic H3.3K36M and H3.3K27M was achieved using eyeless-Gal4 (ey-GAL4) or patched-Gal4 (ptc-GAL4) driver strains.
Genetic modifier screens were conducted by crossing screening strains to RNAi lines (GD or KK stocks obtained from Vienna Drosophila Resource Center (Vienna, Austria) and Bloomington Drosophila Stock Center (Bloomington, IN) to test for a modification of phenotypes by mutant H3.3 expression. To control for GAL4 dosage effects, screening strains were crossed to UAS-mCherry-RNAi. Positive and negative shifts were verified in triplicates. ey-GAL4 modifier screen was evaluated regarding a rescue of the eye phenotype by scoring the number of small eyes, rough eyes and eyes with overgrowth under control conditions as well as following RNAi expression in triplicates for each tested gene. ptc-GAL4 modifier screen was evaluated regarding a rescue of wing defects (wing unfolded) and leg defects (deformed legs).
METHOD DETAILS
Scanning electron microscopy (SEM)
Adult flies were fixed in 2% glutaraldehyde + 2% formaldehyde solution in PBS (1X). Flies were then washed and dehydrated in increasing concentrations of ethanol (30%-100%) as described in42. Critical point drying was performed using a CPD machine (K850, Quorum Technology). Adult flies were mounted on carbon on aluminum stubs and coated with gold or platinum at 15 mA for 60 seconds (20nm). Scanning electron microscopy (SEM) was performed using a Tescan, Mira III LMU, FEG SEM (Field emission gun), and a secondary electron detector was used for capturing the electron microscopy images.
Semi-thin sections of Drosophila compound eyes
Adult flies were decapitated and heads were fixed in 2.5% glutaraldehyde in PBS for 96 hours. Drosophila heads were postfixed in 1% osmium tetroxide, dehydrated in increasing concentrations of alcohol, passed through propylene oxide and embedded in Araldite. Araldite blocks were cut in 1 µm sections and stained with Richardson’s stain (1% methylene blue in 1% borax solution and 1% azur II solution) for examination by light microscopy.
Immunofluorescence
Eye-antennal imaginal discs of third instar larvae were dissected, fixed in 3.7% formalin and subsequently stained using primary antibodies [rabbit anti-Tri-Methyl-Histone H3 (Lys27, 1:500 or 1:100, C36B11, Cell Signaling, Cambridge, UK), rabbit anti-Di-Methyl-Histone H3 (Lys27 1:200, D18C8, Cell Signaling, Cambridge, UK), rabbit anti-Tri-Methyl-Histone H3 (Lys36, 1:50, #9763, Cell Signaling), rabbit anti-di-Methyl-Histone H3 (Lys36, 1:100 or 1:25, C75H12, Cell Signaling, Cambridge, UK), mouse anti-Antp (1:200, 8C11, DSHB), mouse anti-Abd-B (1:10, 1A2E9, DSHB), mouse anti-Ubx (1:10, FP3.38, DSHB), mouse anti-Fas III (1:20, 7G10, DSHB), mouse anti-wingless (1:30, 4D4, DSHB), rhodamine phalloidin (1:40, Life Technologies), rabbit anti-Hemagglutinin (1:500, ab9110, Abcam), rat anti-Hemagglutinin (1:100, 3F10, Sigma Aldrich, Munich) and guinea pig anti-Krimper (1:2000, gift from Toshie Kai, Osaka, Japan). As secondary antibodies [goat Alexa Fluor ® 488 anti-rabbit, goat Alexa Fluor ® 568 anti-rabbit, goat Alexa Fluor ® 488 anti-mouse, goat Alexa Fluor ® 647 anti-rat (1:1000, Thermo Fisher Scientific, Waltham, MA)], goat AlexaFluor ® 488 anti-mouse (1:500, Invitrogen), goat AlexaFluor ® 594 anti-rabbit (1:500, Abcam), goat AlexaFluor ® 635 anti-rabbit (1:500, Life Technologies) were used. As a nuclear counterstain DNA stain DAPI (1:5000, 10−3 mg/mL, Molecular Probes) was used. Eye-antennal imaginal discs were mounted in Roti ®-Mount FluorCare (Carl Roth, Karlsruhe, Germany) or gold anti-fade solution (Invitrogen). The ey-Gal4 screening lines were used to stain for actin, Fas3, Wg and H3K27me3 and H3K36me2 marks in rescued eye-antennal discs (Fig. 6). All other stains were performed on eye-Gal4 crossed to control, H3.3 WT or mutant H3.3 lines.
Confocal microscopy and image analysis
For image acquisition of eye-antennal imaginal discs an Axio Imager M2 with an LSM 700 confocal unit (Carl Zeiss Jena, Germany) (lenses: 10x Plan Apo, NA = 0, 45; 20x Plan Apo, NA = 0,8; 40x Plan Apo, NA = 1,4), Leica SP8 confocal laser-scanning microscope (McGill University Life Sciences Complex Advanced BioImaging Facility (ABIF) or Zeiss LSM 710 laser scanning confocal microscope (Biological Imaging core facility, American University of Beirut). Zen 2009 software (Zeiss), ImageJ software (available at http://imagej.nih.gov/ij/), Fiji(Schindelin et al., 2012) were used for image processing. For quantification of pS10-H3-positive cells the ImageJ plugin Image-based Tool for Counting Nuclei (ITCN) was utilized.
Western blot
Adult eyes were dissected, subsequently transferred in 200 µl cold TEB buffer (PBS containing 0.5% Triton X 100 (v/v), 2 mM phenylmethylsulfonyl fluoride, 0.02% (w/v) NaN3, 1:1000 proteinase-inhibitor) and grounded with pestles. Samples were centrifuged for 10 min at 6.500 g and 4°C. The supernatant was discarded, the pellet washed in 100µl TEB buffer and centrifuged as before. The supernatant was transferred into a fresh reaction tube and histones were acid extracted over night at 4°C in 0.2 N HCl. Histone extracts were heated for 5 min at 95°C in sample buffer and run on a 12.5% SDS-PAGE gel. Separated proteins were blotted using nitrocellulose and incubated with primary antibodies [rabbit anti Histone H3.3 (1:1000, 09–838, Merck Millipore, Darmstadt, GER), rabbit anti-Tri-Methyl-Histone H3 (Lys27, 1:1000, C36B11, Cell Signaling, Cambridge, UK), rabbit anti-Di-Methyl-Histone H3 (Lys27, 1:1000, D18C8, Cell Signaling, Cambridge, UK and rabbit anti-Di-Methyl-Histone H3 (Lys36, 1:1000, C75H12, Cell Signaling, Cambridge, UK)]. Secondary antibodies [goat Alexa Fluor ® 488 anti-rabbit (Thermo Fisher Scientific, Waltham, MA)] were used at dilution of 1:3000. Stained protein bands were visualized using ChemiDocTM MP imaging system (Bio-rad, Hercules, CA).
Negative geotaxis assay
Flies were crossed and separated immediately after hatching into groups of 10 animals and kept on Drosophila standard food 25°C in an incubator. Climbing assay was conducted weekly at the same time of the day (10.00 am). Before climbing assay, flies were transferred into a 15 ml falcon tube without using anesthesia. After one minute of habituation, flies were gently tapped down to the bottom of the tube and the number of animals attaining a threshold at height of 9 cm was counted. The procedure was repeated 10 times to obtain mean values for each single group. To exclude an effect of lighting conditions, the assay was carried out under red light.
Reverse transcriptase quantitative PCR (RT-qPCR)
500ng of RNA was used to synthesize cDNA using the Maxima H Minus First Strand cDNA Synthesis Kit with random hexamer primers (ThermoScientific). Gene expression was measured using the DyNAmo Flash SYBR Green qPCR Kit (ThermoScientific) with the Bio-Rad CFX 96 Real-Time System and C1000 Thermal Cycler. For all RT-qPCR experiments, the data was normalized using the housekeeping gene rp49 and to the control (yw). The following primers were used:
rp49 forward: TACAGGCCCAAGATCGTGAAG
rp49 reverse: GACGCACTCTGTTGTCGATACC
qin forward: CCTCAGTTCCGCCGACTATC
qin reverse: ACGGAAGTCCGAACAACGAA
AGO3 forward: TCCAGCTTGTTGTATGTACGCT
AGO3 reverse: TGCTTCAGCCACATCTCGTT
moon forward: TATCGGCGTAAGCAGCGTAG
moon reverse: CGCGGAATAGTCCCTCCATC
krimp forward: TTCGATAGCAACATGCACAAGC
krimp reverse: CAGCGGAATGAGACTCCTGTA
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA extraction and RNA-seq
20–30 pairs of eye-antennal imaginal discs were dissected from wandering third instar larvae in PBS for each biological replicate, and flash frozen in liquid nitrogen prior to RNA extraction. 3 biological replicates were collected for each genotype profiled. RNA was extracted using the RNeasy mini kit (Qiagen, 74104) according to instructions from the manufacturers. To ensure elimination of genomic DNA contamination, RNA was treated with on-column RNAse-free DNase I (Qiagen, 79254). Library preparation was performed with NEBNext directional poly-A mRNA-seq kit according to manufacturer’s instructions. 100bp paired-end sequencing was performed at the McGill University and Génome Québec Innovation Centre on Illumina HiSeq 4000 and NovaSeq 6000.
Bioinformatics – RNA-seq
Raw sequencing reads were trimmed using Trimmomatic v0.32 (Bolger et al., 2014) to remove low-quality bases at the ends of reads (average quality phred33 < 30 in sliding windows of 4 nucleotides), clip the first four bases, and remove Illumina adaptor sequences using palindrome mode. Reads shorter than 30 nucleotides after trimming were discarded. The resulting high-quality reads were aligned to the Drosophila melanogaster reference genome (Illumina iGenome UCSC dm6) using STAR v2.30e (Dobin et al., 2013) with default settings. Mapped reads were quantified with featureCounts v1.4.4 (Liao et al., 2014) using dm6 ensGene Ensembl genes. For repetitive element analysis, uniquely aligned reads (mapQ > 0) were quantified with SeqMonk v1.47 using dm6 RepeatMasker annotations. Normalization and differential expression analysis for genes and repeats were performed using DESeq2 v1.14.1 (Love et al., 2014). Differentially expressed genes were defined as expressed genes (average normalized expression > 100) presenting large expression changes (absolute log2 fold change > 0.5) and statistically significant (adjusted p-value < 0.05). To control for changes that may be induced by expression of the human construct, genes detected as differentially expressed in the comparison between Control and H3.3 WT samples genotypes were filtered out. This analysis produces a very conservative list of differentially expressed genes, removing several genes where expression of H3.3 WT resulted in an intermediate level of expression, that would be statistically significant in a H3.3 WT vs H3.3K-to-M comparison. To perform gene ontology analysis, EnrichGO function from clusterProfiler v3.2.14 (Yu et al., 2012) was used to find enriched pathways from biological process, molecular function and cellular component. The background gene list was defined as all genes with expression levels greater than 0 in at least one sample in the complete dataset. The Bioconductor org.Dm.eg.db v3.4.0 database was used and a Benjamini and Hochberg adjusted p-value of 0.05 was used as threshold. Pathway analysis was complemented with Panther (Thomas et al., 2003) and DAVID (Huang da et al., 2009). Differentially expressed repeats were defined as elements with large expression changes (absolute log2 fold change > 2) and statistically significant (adjusted p-value < 0.05).
Murine and human RNA-seq datasets were obtained from Gene Expression Omnibus accession # GSE69291. Raw reads were processed using GenPipes (Bourgey et al., 2019a) v.3.1.2 RNA-seq module under default parameters. Briefly, reads were trimmed for Illumina adapter sequence using Trimmomatic v.0.36 using quality cut-off of phred < 33. PCR duplicates were identified and filtered using Picard v.2.9.0. Resulting unique reads were then aligned to Mus musculus reference genome (mm10) and Homo sapiens reference genome (hg19) using STAR v.2.5.3a. For repetitive element analysis, uniquely aligned reads (mapQ > 0) were quantified with SeqMonk v1.47 using mm10 or hg19 RepeatMasker LTR and LINE annotations. Normalization and differential expression analysis for repeats were performed using DESeq2 v1.14.1 (Love et al., 2014). Differentially expressed repeats were defined as elements with large expression changes (absolute log2 fold change > 2) and statistically significant (adjusted p-value < 0.05).
Small RNA-seq (smRNA-seq)
20–30 pairs of eye-antennal imaginal discs were dissected from wandering third instar larvae in PBS for each biological replicate. PBS was removed and samples kept in QIAzol reagent, RNA was extracted using the miRNeasy micro kit (Qiagen, 217084) according to manufacturer’s instructions. To ensure elimination of genomic DNA contamination, RNA was treated with on-column RNAse-free DNase I (Qiagen, 79254). 3 technical replicates were collected for each genotype profiled. smRNA-seq libraries were then prepared using NEBNext Small RNA library kit according to manufacturer’s instructions. smRNA libraries were then sequenced (single-end, 50bp) at the McGill University and Génome Québec Innovation Centre on Illumina HiSeq 4000.
Bioinformatics – smRNA-seq
Sequencing reads were processed as described for RNA-seq reads, with the following modifications to the workflow: the first four bases were not clipped; trimmed reads shorter than 15 bases (as opposed to 30) were discarded. The resulting clean set of reads were then aligned to the drosophila reference genome (dm6) using novoalign (v3.02.12) with a score difference of 0 for calling multiple mapping (-R 0), reporting a maximum of 100 alignments per read (-r All 100), setting miRNA mode (-m 100) and using −l 14 and -t 10,5.5 as alignment scoring parameters.
Gene expression levels were estimated by quantifying uniquely-aligned and multi-mapped (at primary alignments only) to the Ensembl genes, miRNAs (miRBase), tRNAs (GtRNAdb), piRNA clusters (Brennecke et al., 2007) and the piRNA database (Sai Lakshmi and Agrawal, 2008) annotation sets using featureCounts (v1.4.4) (Liao et al., 2014) piRNA clusters and piRNA database annotations were converted to dm6 genome build using the UCSC liftOver tool with dm3toDm6 chain. Normalization (mean of ratios) and variance-stabilized transformation of the data were performed using DESeq2 (v1.14.1) (Love et al., 2014) using a concatenation of the miRNAs, tRNA and piRNA database genes. Multiple control metrics were obtained using FASTQC (v0.11.2), samtools (v0.1.19)(Li et al., 2009) BEDtools (v2.17.0) (Quinlan and Hall, 2010) and custom scripts. For visualization, normalized Bigwig tracks of all reads (MapQ > 0) and uniquely aligned reads (MapQ > 1) were generated using strand-specific quantification pipeline from SeqMonk. Integrative Genomic Viewer (Thorvaldsdottir et al., 2013) was used for data visualization.
Chromatin immunoprecipitation sequencing (ChIP-seq)
Native ChIP-seq for H3K27me3, H3K36me2 and H3K36me3 was performed as described previous(Karimi et al., 2011) with minor adjustments. Briefly ~80–200 pairs of eye disc tissue were dissected from H3.3 WT, K27M, K36M, and negative control yw flies and flash frozen. Tissue was disassociated passing through a syringe (25 5/8 gauge) 20x in douncing buffer (10mM Tris Cl pH7.5, 4mM MgCl2, 1mM CaCl2, and protease inhibitor cocktail (Roche)). Disassociated cells were then digested with 30U of micrococcal nuclease (Worthington) at 37°C for 7 mins, yielding predominantly mono- and di-nucleosomes. Cells were then lysed in ice-cold hypotonic lysis buffer (0.2 mM EDTA, 0.1 mM benzamidine, 0.1 mM PMSF, 1.5 mM DTT, and protease inhibitor cocktail (Roche)) to yield chromatin. Chromatin was then incubated with pre-conjugated antibody:bead complex containing H3K27me3 (CST 9733, 3 μL / IP), H3K36me2 (Active Motif 39255, 3 μL / IP), H3K36me3 (Active Motif 61022, 3 μL / IP) complexed to Protein A Dynabeads (Invitrogen, 20 μL / IP) or anti-mouse IgG Dynabeads (Invitrogen, 20 μL / IP) rotating at 4°C overnight. Beads were then washed 2x in ChIP Wash Buffer (20 mM Tris-HCl pH 8, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and protease inhibitor cocktail) and 1x in Final Wash Buffer (20 mM Tris-HCl pH 8, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, and protease inhibitor cocktail). DNA was then eluted in 100mM NaHCO3 and 1% SDS at 65°C for 2h, and purified using phenol:chloroform extraction followed by ethanol precipitation.
Library preparation was carried out using KAPA HTP Illumina library preparation reagents, following manufacturer’s instructions. Briefly, 50 µl of ChIP sample was incubated with 45 µl end repair mix at 20 °C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50 µl buffer enzyme mix at 30 °C for 30 min, followed by PEG/NaCl purification. Adaptor ligation: bead-bound sample was incubated with 45 µl buffer enzyme mix and 5 µl of TruSeq DNA adapters (Illumina), for 20 °C 15 min, followed by PEG/NaCl purification (twice). Library enrichment: 14 cycles of PCR amplification. Size selection was performed after PCR using gel excision to collect 150–450 bp fragments. ChIP libraries were sequenced on Illumina NovaSeq 6000 platforms at 50 bp paired-reads.
Bioinformatics - ChIP-seq
ChIP-seq datasets were processed using the ChIP-seq module of GenPipes (Bourgey et al., 2019b). Briefly, raw reads were trimmed using Trimmomatic (Bolger et al., 2014) v0.32 to remove adaptor and sequencing-primer associated reads, then aligned to dm6 using bwa-mem (Li and Durbin, 2009) (v0.7.12) with default parameters. PCR duplicate reads as defined by reads with identical mapping coordinates were then collapsed by Picard v2.0.1 to produce uniquely aligned reads. Uniquely aligned reads with mapping quality of >5 were then used to quantify enrichment. Reads per kilobase mapped (RPKM) was calculated using SeqMonk v1.47 at annotated promoters (defined as 5 kilobase centred on transcription start site) using dm6 ensGene Ensembl gene annotation and at genomic tiled bins. Differential enrichment between the mutant and WT control were then calculated as Z-scores, with |Z-score| above 0.5 is then considered as change in histone modification.
Alignment to Drosophila gypsy LTR (Dfam accession DF0001639) and internal (Dfam accession DF0001638) consensus sequence was performed using bwa-mem (Li and Durbin, 2009) (v0.7.17) with default parameters. PCR duplicate reads as defined by reads with identical mapping coordinates were then collapsed by Picard v2.0.1 to produce uniquely aligned reads. Read count and normalization was performed using SeqMonk v1.47. To normalize for sequencing depth, read count at transposon consensus was divided by total number of mapped reads to dm6 to derive Reads Per Million mapped (RPM).
Supplementary Material
Fig. S1. Detailed analysis of eye phenotypes in mutant histone H3.3 expressing flies.
(A) Phenotypes associated with targeted expression of H3.3K27M, H3.3K36M and H3.3 WT. (B) Expression of H3.3K27M and H3.3K36M in eye imaginal discs resulted in a small/rough eye phenotype with a variation regarding size of compound eyes and tumorous growth of additional head and eye tissue (black arrowheads). The eye phenotype upon H3.3K27M expression was notably more severe as compared to H3.3K36M expression. (C) Range of eye phenotypes upon expression of H3.3K27M or H3.3K36M in a sensitized ey-Gal4, UAS-Dl background. Expression of H3.3K27M or H3.3K36M resulted in tumorous eyes with variable severity including overgrowth, hyperplastic tissue with folds, metastasis (black arrowheads) or complete loss of eye tissue. (D) Immunofluorescence staining for Wg, a ligand of the Wnt/Wg signaling pathway. In approximately one third of eye discs expressing H3.3K27M, expression of Wg shifts to a wing-like pattern in the dorsal part of the eye disc as marked by the white outline. Scale bar 100µm. (E) Immunofluorescence staining for the Hox proteins Antp, Abd-B, and Ubx in H3.3K36M, H3.3K27M and H3.3 WT overexpressing eye discs. Transgene expression is visualized by HA staining. There is no de-repression of Abd-B or Ubx in histone H3 mutant expressing discs visible. (F) Micrographs of adult eyes illustrating ectopic tissues observed in the eye and head region of H3.3K27M expressing animals (ey-Gal4) including leg-like structures, thoracic bristles and an extra pair of wings (white arrows).
Fig. S2. Quantification of global changes in histone marks in mutant and wild-type histone H3.3 expressing flies.
(A) Immunofluorescence staining for H3K27me3 or (B) H3K36me2 combined with histone transgene expression (HA staining) in Gal4 control, H3.3 WT, H3.3K27M and H3.3K36M overexpressing eye discs.
Fig. S3. K-to-M mutations result in induction of piRNA genes.
(A) Dot plots showing qPCR results from three independent experiments validating that krimp, moon, AGO3, and qin RNA levels are elevated in H3.3K27M eye discs, and that krimp and moon RNA levels are elevated in H3.3K36M eye discs. The y-axis shows fold increase relative to yw controls. (B) Differential expression analysis of piRNA clusters following small RNAseq between H3.3K27M or H3.3K36M compared to baseline control. (C) Genome browser snapshot depicting piRNA expression at cluster 1 (left) and flamenco/cluster 8 (right) in H3.3WT (black), H3.3K27M (red), and H3.3K36M mutants (blue). Upper panel: all aligned reads in the sense (positive, darker) and anti-sense (negative, lighter) orientation. Lower panel: uniquely aligned (mapQ > 1) reads. (D) Principal component analysis of the smRNA-seq data, using as features expression of piRNA clusters (left), microRNA genes (middle), or tRNAs (right).
Fig. S4. H3.3K27M and H3.3K36M perturb global H3K27me3/H3K36me2 landscape.
(A) Histogram of genome-wide H3K27me3 (left) and H3K36me2 (right) changes in H3.3K27M and H3.3K36M mutant flies. Positive z-score indicate gain of the mark in mutant vs. H3.3 WT flies. (B) Left: browser snapshot of H3K36me3 and H3K36me2 loss in H3.3K36M eye disc compared to H3.3WT. Right: genome-wide 5kb quantification of H3K36me3 in H3.3 WT vs. H3.3K36M. (C) Scatterplot depicting genome-wide anti-correlation between H3K27me3 and H3K36me2 in H3.3 WT eye disc. (D) Metagene plot depicting H3K27me3 and H3K36me2 level in relation to transcriptional activity in H3.3 WT, H3.3K27M, and H3.3K36M. Genes were stratified into 4 quartiles based on expression in H3.3 WT (Q1 being silenced and Q4 being highly transcribed), and H3K27me3/H3K36me2 enrichment (RPKM) were plotted in 5 kilobase window centered on the transcription start site. (E) Scatterplot depicting the relationship of gene expression change vs. H3K27me3 at all annotated promoters. (F) Overlap of differentially expressed genes with genes deregulated upon loss-of-function of PRC1 or PRC2 in the Drosophila eye (Loubiere et al., 2016).
Fig. S5. H3.3K27M and H3.3K36M redistribute H3K36me2 away from repetitive regions.
(A) Smoothed line plot depicting H3.3K27M- or H3.3K36M-mediated H3K36me2 change in relation to transposable element (TE) copy numbers on autosomes and chromosome X. (B) Venn diagram of genomic bins showing concurrent H3K27me3-loss/H3K36me2-gain in H3.3K27M (left), and reciprocally H3K36me2-loss/H3K27me3-gain in H3.3K36M (right). Below, the percentage of bins falling into genic vs. intergenic region in each category. Note enrichment of genic bins in the intersection of H3K27me3/H3K36me2 change in both mutants. (C) Genome browser snapshot of pericentromeric region of chr2R, showing localization of up-regulated TE observed in H3.3K27M and H3.3K36M flies. Black boxes: WT H3K27me3 and WT H3K36me2 called peaks.
Fig. S6. H3.3K27M and K36M de-repress transposable elements in flies.
(A) Alignment of H3K27me3 and H3K36me2/3 at Gypsy LTR (upper panels) and internal (lower panels) consensus sequence in H3.3 WT (black), K27M (red), and K36M (blue). (B) Volcano plot depicting K36M-mediated transcriptional dysregulation in repetitive sequences in murine mesenchymal stem cells, or MSCs (left), and human primary chondroblastoma (right). Each dot represents an individual copy of repeat, and red dots represent individual LTR elements while blue dots depict LINEs. (C) Heatmap depicting TE de-repression in panel of K36 mutants relative to H3.3WT-expressing MSCs. (D) Pie chart depicting number and diversity of repeat copies showing significant up-regulation (log2FC > 2, FDR < 0.05) in murine MSCs and human chondroblastoma. Outer ring: copy number of each repeat class in the respective mouse (mm10) and human (hg19) genomes. (E) The krimp shRNA strain BDSC#35230 was tested regarding its potential to rescue the eye phenotype compared to H3.3K27M or H3.3K36M expression combined with control RNAi (mCherry BDSC#35785) knockdown. Micrographs (lower panel) show representative images of H3.3K27M/krimp RNAi (small, rough with overgrowth) and H3.3K36M/krimp RNAi (small, not rough) expressing flies compared to control flies. Eye phenotypes of H3.3K27M and H3.3K36M expressing flies are scored according to the following criteria: small eyes, rough eyes and overgrowth. Note that there was no major rescue detected upon krimp knockdown. (F) Violin plot representing normalized expression score for eye developmental genes and germ cell/piRNA genes in krimp-KD H3.3K27M and H3.3K36M flies, relative to mCherry KD controls.
Fig. S7. shRNA-mediated knockdown of the H3K27 methyltransferase E(z) and the H3K36 methyltransferase ash1 restores global histone methylation marks in mutant H3.3 expressing eye discs.
(A) Three independent ash1 shRNA strains were tested regarding their potential to rescue the eye phenotype observed upon ey>H3.3K27M expression (BDSC ID 36130: 94.39% rescue, sem: 1.56, n=95; VDRC ID 108832: 56.54% rescue, sem: 5.30, n=104; BDSC ID 36803: 66.70% rescue, sem: 6.67, n=73). Micrographs (right panel) show H3.3K27M/ash1 RNAi expressing flies which were not completely rescued, as well as H3.3 WT/ash1 RNAi expressing flies exhibiting an eye phenotype (BDSC ID 36130: 1.79% eye phenotype, sem: 1.03, n=115; VDRC ID 108832: 17.44% eye phenotype, sem: 0.59, n=93; BDSC ID 36803: 0% eye phenotype, n=87). (B) Two independent E(z) shRNA strains were tested regarding their potential to rescue the eye phenotype observed upon eyeless-specific H3.3K36M expression (BDSC ID 27993: 86.62% rescue, sem: 5.47, n=134; VDRC ID 107072: 70.29% rescue, sem:3.29, n=144). Micrographs (right panel) show H3.3K36M/E(z) RNAi expressing flies which were not completely rescued, as well as H3.3 WT/E(z) RNAi expressing flies exhibiting an eye phenotype (BDSC ID 27993: 3.48% eye phenotype, sem: 0.54, n=112; VDRC ID 107072: 0% eye phenotype, n=101). (C) SEM micrographs of adult eyes illustrating modifier effects of E(z) and ash1 knockdown on eyeless>H3.3K27M and ey>H3.3K36M flies. Expression of either E(z) or ash1 shRNA alone has no significant phenotype. (D) Number of transposon elements significantly up-regulated (log2FC > 0, FDR < 0.05) in ash1-rescued K27M and E(z)-rescued K36M, compared to ash1-KD alone, E(z)-KD alone, and mCherry RNAi negative control. Below, copy number of each repeat class in dm6.
Table S3: Differential expression analysis of piRNA biogenesis genes in eye imaginal discs expressing H3.3K27M compared to y w controls.
Table S6: RNAi strains for genetic modifier screen including PRC1, PRC2 members, lysine methyltransferases and candidates from the piwi-interacting RNA (piRNA) pathway.
Table S7: Patched-specific modifier screen.
Table S8: Modifier screen including PRC1, PRC2 members and lysine methyltransferases.
Table S1. RNA-seq differential expression analysis of H3.3K27M, H3.3K36M and wild-type H3.3 samples against yw controls.
Table S2. Statistically significant (BH padj < 0.05) gene ontology pathways for genes that are: 1) downregulated in K27M, 2) downregulated in K36M and 3) upregulated in K27M.
Table S4. smRNA-seq differential expression analysis of piRNA clusters in H3.3K27M and H3.3K36M against yw controls and wild-type H3.3 samples.
Table S5. H3K27me3 and H3K36me2 signal over individual TE in H3.3K27M, H3.3K36M and wild-type H3.3 samples.
Acknowledgments
We acknowledge the Bloomington Drosophila Stock Center (NIH P40OD018537) and Vienna Drosophila RNAi Center for providing fly stocks. Funding: This work was supported by a Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l’Économie, de la Science et de l’Innovation du Québec, with the support of the Ontario Research Fund through funding provided by the Government of Ontario to N.J. and C.L.K; the Canadian Institutes for Health Research (CIHR grant PJT-156086 to C.L.K, MOP-44050 and IOP-107945 to P.L., and MOP-286756 and FDN-154307 to N.J.); the National Sciences and Engineering Research Council (NSERC RGPIN-2016-04911 to C.L.K.); the Medical Faculty Münster (Technology platform “Drosophila”). CCLC is supported by fellowship from RI-MUHC sponsored by Toronto Dominion Bank and Alex’s Lemonade Stand Foundation. M.H. is supported by IZKF Münster (Ha3/019/15) and (together with AJ) by Deutsche Forschungsgemeinschaft (DFG HA 3060/10-1; JE 610/4-1); C.L.K. is supported by a salary award from the FRQS; M.S. is supported by the American University of Beirut Medical Practice Plan. P.L. is supported by a Visiting Professorship from Radboud University during a sabbatical year visit and by a James McGill Distinguished Professor award. N.J. is a member of the Penny Cole Laboratory and holds a Canada Research Chair Tier 1 in Pediatric Oncology from CIHR. This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium. We are especially grateful for the generous philanthropic donation of Charles Bruneau Foundation, the WeLoveYouConnie Foundation, and the Cedars/Sarah Cook funds.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45, 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Wirbelauer C, Hild M, Scharf AN, Schwaiger M, MacAlpine DM, Zilbermann F, van Leeuwen F, Bell SP, Imhof A, et al. (2007). Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J 26, 4974–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicocca VT, Ormsby T, Adhvaryu KK, Honda S, and Selker EU (2018). ASH1-catalyzed H3K36 methylation drives gene repression and marks H3K27me2/3-competent chromatin. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau M, Shirinian M, Gayden T, Harutyunyan AS, Chen CCL, Mikael LG, Duncan HM, Neumann AL, Arreba-Tutusaus P, De Jay N, et al. (2019). Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat Commun 10, 2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt W, De Geest N, Aerts S, Leenaerts I, Marynen P, and Hassan BA (2009). The atonal proneural transcription factor links differentiation and tumor formation in Drosophila. PLoS Biol 7, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgey M, Dali R, Eveleigh R, Chen KC, Letourneau L, Fillon J, Michaud M, Caron M, Sandoval J, Lefebvre F, et al. (2019a). GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgey M, Dali R, Eveleigh R, Chen KC, Letourneau L, Fillon J, Michaud M, Caron M, Sandoval J, Lefebvre F, et al. (2019b). GenPipes: an open-source framework for distributed and scalable genomic analyses. GigaScience 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, and Hannon GJ (2007). Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez M, and Casares F (2005). Organ specification-growth control connection: new in-sights from the Drosophila eye-antennal disc. Dev Dyn 232, 673–684. [DOI] [PubMed] [Google Scholar]
- Dorafshan E, Kahn TG, Glotov A, Savitsky M, Walther M, Reuter G, and Schwartz YB (2019). Ash1 counteracts Polycomb repression independent of histone H3 lysine 36 methylation. EMBO Rep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, and Tamkun JW (2013). The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila. Development 140, 4182–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, and Evans WE (2012). The Pediatric Cancer Genome Project. Nat Genet 44, 619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al. (2014). BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell 56, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, et al. (2019). H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nature Communications 10, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, and Shilatifard A (2014). Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Yang F, Zhang Z, Zhang J, Cai G, Li L, Zheng Y, Chen S, Xi R, and Zhu B (2017). Mrg15 stimulates Ash1 H3K36 methyltransferase activity and facilitates Ash1 Trithorax group protein function in Drosophila. Nature Communications 8, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jani KS, Jain SU, Ge EJ, Diehl KL, Lundgren SM, Muller MM, Lewis PW, and Muir TW (2019). Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc Natl Acad Sci U S A 116, 8295–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. (2011). DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Ueki M, and Kai T (2020). Drosophila MARF1 ensures proper oocyte maturation by regulating nanos expression. PLoS One 15, e0231114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, and Theurkauf W (2008). Biogenesis and germline functions of piRNAs. Development 135, 3–9. [DOI] [PubMed] [Google Scholar]
- Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, Bertrand KC, Mikael LG, McConechy MK, Chen CCL, et al. (2019). Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 35, 782–797 e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. (2011). NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Loubiere V, Delest A, Thomas A, Bonev B, Schuettengruber B, Sati S, Martinez AM, and Cavalli G (2016). Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat Genet 48, 1436–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science (New York, N.Y.) 352, 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Han G, Xu L, Zhu D, Lin H, Cao X, Yu Y, and Chen CD (2015). Cis-existence of H3K27me3 and H3K36me2 in mouse embryonic stem cells revealed by specific ions of isobaric modification chromatogram. Stem Cell Res Ther 6, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKearin DM, and Spradling AC (1990). bag-of-marbles: a Drosophila gene required to initiate both male and female gametogenesis. Genes Dev 4, 2242–2251. [DOI] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA, et al. (2017). Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet 49, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Copur O, Jackle H, Herzig A, and Muller J (2013). A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339, 698–699. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe T (2000). The sevenless signaling pathway: variations of a common theme. Biochim Biophys Acta 1496, 151–163. [DOI] [PubMed] [Google Scholar]
- Sai Lakshmi S, and Agrawal S (2008). piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res 36, D173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Shearn A, and Garen A (1974). Genetic control of imaginal disc development in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 71, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane K, Miura F, Ito T, and Lorincz MC (2020). NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat Genet 52, 1088–1098. [DOI] [PubMed] [Google Scholar]
- Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, et al. (2018). The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell 70, 371–379.e375. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, and Narechania A (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, and Carpenter PB (2012). Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, and Bestor TH (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20, 116–117. [DOI] [PubMed] [Google Scholar]
- Weinberg DN, Allis CD, and Lu C (2017). Oncogenic Mechanisms of Histone H3 Mutations. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. (2014). ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Xie D, Cao X, Yu P, Xing X, Chen CC, Musselman M, Xie M, West FD, Lewin HA, et al. (2012). Comparative epigenomic annotation of regulatory DNA. Cell 149, 1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, and Zhu B (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 286, 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Detailed analysis of eye phenotypes in mutant histone H3.3 expressing flies.
(A) Phenotypes associated with targeted expression of H3.3K27M, H3.3K36M and H3.3 WT. (B) Expression of H3.3K27M and H3.3K36M in eye imaginal discs resulted in a small/rough eye phenotype with a variation regarding size of compound eyes and tumorous growth of additional head and eye tissue (black arrowheads). The eye phenotype upon H3.3K27M expression was notably more severe as compared to H3.3K36M expression. (C) Range of eye phenotypes upon expression of H3.3K27M or H3.3K36M in a sensitized ey-Gal4, UAS-Dl background. Expression of H3.3K27M or H3.3K36M resulted in tumorous eyes with variable severity including overgrowth, hyperplastic tissue with folds, metastasis (black arrowheads) or complete loss of eye tissue. (D) Immunofluorescence staining for Wg, a ligand of the Wnt/Wg signaling pathway. In approximately one third of eye discs expressing H3.3K27M, expression of Wg shifts to a wing-like pattern in the dorsal part of the eye disc as marked by the white outline. Scale bar 100µm. (E) Immunofluorescence staining for the Hox proteins Antp, Abd-B, and Ubx in H3.3K36M, H3.3K27M and H3.3 WT overexpressing eye discs. Transgene expression is visualized by HA staining. There is no de-repression of Abd-B or Ubx in histone H3 mutant expressing discs visible. (F) Micrographs of adult eyes illustrating ectopic tissues observed in the eye and head region of H3.3K27M expressing animals (ey-Gal4) including leg-like structures, thoracic bristles and an extra pair of wings (white arrows).
Fig. S2. Quantification of global changes in histone marks in mutant and wild-type histone H3.3 expressing flies.
(A) Immunofluorescence staining for H3K27me3 or (B) H3K36me2 combined with histone transgene expression (HA staining) in Gal4 control, H3.3 WT, H3.3K27M and H3.3K36M overexpressing eye discs.
Fig. S3. K-to-M mutations result in induction of piRNA genes.
(A) Dot plots showing qPCR results from three independent experiments validating that krimp, moon, AGO3, and qin RNA levels are elevated in H3.3K27M eye discs, and that krimp and moon RNA levels are elevated in H3.3K36M eye discs. The y-axis shows fold increase relative to yw controls. (B) Differential expression analysis of piRNA clusters following small RNAseq between H3.3K27M or H3.3K36M compared to baseline control. (C) Genome browser snapshot depicting piRNA expression at cluster 1 (left) and flamenco/cluster 8 (right) in H3.3WT (black), H3.3K27M (red), and H3.3K36M mutants (blue). Upper panel: all aligned reads in the sense (positive, darker) and anti-sense (negative, lighter) orientation. Lower panel: uniquely aligned (mapQ > 1) reads. (D) Principal component analysis of the smRNA-seq data, using as features expression of piRNA clusters (left), microRNA genes (middle), or tRNAs (right).
Fig. S4. H3.3K27M and H3.3K36M perturb global H3K27me3/H3K36me2 landscape.
(A) Histogram of genome-wide H3K27me3 (left) and H3K36me2 (right) changes in H3.3K27M and H3.3K36M mutant flies. Positive z-score indicate gain of the mark in mutant vs. H3.3 WT flies. (B) Left: browser snapshot of H3K36me3 and H3K36me2 loss in H3.3K36M eye disc compared to H3.3WT. Right: genome-wide 5kb quantification of H3K36me3 in H3.3 WT vs. H3.3K36M. (C) Scatterplot depicting genome-wide anti-correlation between H3K27me3 and H3K36me2 in H3.3 WT eye disc. (D) Metagene plot depicting H3K27me3 and H3K36me2 level in relation to transcriptional activity in H3.3 WT, H3.3K27M, and H3.3K36M. Genes were stratified into 4 quartiles based on expression in H3.3 WT (Q1 being silenced and Q4 being highly transcribed), and H3K27me3/H3K36me2 enrichment (RPKM) were plotted in 5 kilobase window centered on the transcription start site. (E) Scatterplot depicting the relationship of gene expression change vs. H3K27me3 at all annotated promoters. (F) Overlap of differentially expressed genes with genes deregulated upon loss-of-function of PRC1 or PRC2 in the Drosophila eye (Loubiere et al., 2016).
Fig. S5. H3.3K27M and H3.3K36M redistribute H3K36me2 away from repetitive regions.
(A) Smoothed line plot depicting H3.3K27M- or H3.3K36M-mediated H3K36me2 change in relation to transposable element (TE) copy numbers on autosomes and chromosome X. (B) Venn diagram of genomic bins showing concurrent H3K27me3-loss/H3K36me2-gain in H3.3K27M (left), and reciprocally H3K36me2-loss/H3K27me3-gain in H3.3K36M (right). Below, the percentage of bins falling into genic vs. intergenic region in each category. Note enrichment of genic bins in the intersection of H3K27me3/H3K36me2 change in both mutants. (C) Genome browser snapshot of pericentromeric region of chr2R, showing localization of up-regulated TE observed in H3.3K27M and H3.3K36M flies. Black boxes: WT H3K27me3 and WT H3K36me2 called peaks.
Fig. S6. H3.3K27M and K36M de-repress transposable elements in flies.
(A) Alignment of H3K27me3 and H3K36me2/3 at Gypsy LTR (upper panels) and internal (lower panels) consensus sequence in H3.3 WT (black), K27M (red), and K36M (blue). (B) Volcano plot depicting K36M-mediated transcriptional dysregulation in repetitive sequences in murine mesenchymal stem cells, or MSCs (left), and human primary chondroblastoma (right). Each dot represents an individual copy of repeat, and red dots represent individual LTR elements while blue dots depict LINEs. (C) Heatmap depicting TE de-repression in panel of K36 mutants relative to H3.3WT-expressing MSCs. (D) Pie chart depicting number and diversity of repeat copies showing significant up-regulation (log2FC > 2, FDR < 0.05) in murine MSCs and human chondroblastoma. Outer ring: copy number of each repeat class in the respective mouse (mm10) and human (hg19) genomes. (E) The krimp shRNA strain BDSC#35230 was tested regarding its potential to rescue the eye phenotype compared to H3.3K27M or H3.3K36M expression combined with control RNAi (mCherry BDSC#35785) knockdown. Micrographs (lower panel) show representative images of H3.3K27M/krimp RNAi (small, rough with overgrowth) and H3.3K36M/krimp RNAi (small, not rough) expressing flies compared to control flies. Eye phenotypes of H3.3K27M and H3.3K36M expressing flies are scored according to the following criteria: small eyes, rough eyes and overgrowth. Note that there was no major rescue detected upon krimp knockdown. (F) Violin plot representing normalized expression score for eye developmental genes and germ cell/piRNA genes in krimp-KD H3.3K27M and H3.3K36M flies, relative to mCherry KD controls.
Fig. S7. shRNA-mediated knockdown of the H3K27 methyltransferase E(z) and the H3K36 methyltransferase ash1 restores global histone methylation marks in mutant H3.3 expressing eye discs.
(A) Three independent ash1 shRNA strains were tested regarding their potential to rescue the eye phenotype observed upon ey>H3.3K27M expression (BDSC ID 36130: 94.39% rescue, sem: 1.56, n=95; VDRC ID 108832: 56.54% rescue, sem: 5.30, n=104; BDSC ID 36803: 66.70% rescue, sem: 6.67, n=73). Micrographs (right panel) show H3.3K27M/ash1 RNAi expressing flies which were not completely rescued, as well as H3.3 WT/ash1 RNAi expressing flies exhibiting an eye phenotype (BDSC ID 36130: 1.79% eye phenotype, sem: 1.03, n=115; VDRC ID 108832: 17.44% eye phenotype, sem: 0.59, n=93; BDSC ID 36803: 0% eye phenotype, n=87). (B) Two independent E(z) shRNA strains were tested regarding their potential to rescue the eye phenotype observed upon eyeless-specific H3.3K36M expression (BDSC ID 27993: 86.62% rescue, sem: 5.47, n=134; VDRC ID 107072: 70.29% rescue, sem:3.29, n=144). Micrographs (right panel) show H3.3K36M/E(z) RNAi expressing flies which were not completely rescued, as well as H3.3 WT/E(z) RNAi expressing flies exhibiting an eye phenotype (BDSC ID 27993: 3.48% eye phenotype, sem: 0.54, n=112; VDRC ID 107072: 0% eye phenotype, n=101). (C) SEM micrographs of adult eyes illustrating modifier effects of E(z) and ash1 knockdown on eyeless>H3.3K27M and ey>H3.3K36M flies. Expression of either E(z) or ash1 shRNA alone has no significant phenotype. (D) Number of transposon elements significantly up-regulated (log2FC > 0, FDR < 0.05) in ash1-rescued K27M and E(z)-rescued K36M, compared to ash1-KD alone, E(z)-KD alone, and mCherry RNAi negative control. Below, copy number of each repeat class in dm6.
Table S3: Differential expression analysis of piRNA biogenesis genes in eye imaginal discs expressing H3.3K27M compared to y w controls.
Table S6: RNAi strains for genetic modifier screen including PRC1, PRC2 members, lysine methyltransferases and candidates from the piwi-interacting RNA (piRNA) pathway.
Table S7: Patched-specific modifier screen.
Table S8: Modifier screen including PRC1, PRC2 members and lysine methyltransferases.
Table S1. RNA-seq differential expression analysis of H3.3K27M, H3.3K36M and wild-type H3.3 samples against yw controls.
Table S2. Statistically significant (BH padj < 0.05) gene ontology pathways for genes that are: 1) downregulated in K27M, 2) downregulated in K36M and 3) upregulated in K27M.
Table S4. smRNA-seq differential expression analysis of piRNA clusters in H3.3K27M and H3.3K36M against yw controls and wild-type H3.3 samples.
Table S5. H3K27me3 and H3K36me2 signal over individual TE in H3.3K27M, H3.3K36M and wild-type H3.3 samples.
Data Availability Statement
ChIP-seq, RNA-seq, smRNA-seq datasets for Drosophila samples have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE140979. H3.3 K36M murine and human RNA-seq datasets have been previously deposited in GEO under dataset accession number GSE69291.