

Abstract
Objective
To describe demographic, clinical and laboratory features of systemic sclerosis sine scleroderma (ssSSc) in a large multicentre systemic sclerosis (SSc) cohort.
Methods
Data involving 1808 SSc patients from Italian Systemic sclerosis PRogression INvestiGation registry were collected. The ssSSc was defined by the absence of any cutaneous sclerosis and/or puffy fingers. Clinical and serological features of ssSSc were compared with limited cutaneous (lcSSc) and diffuse cutaneous (dcSSc) subsets.
Results
Among patients with SSc, only 61 (3.4%) were classified as having ssSSc (F/M=19/1). Time from Raynaud’s phenomenon (RP) onset to diagnosis was longer in ssSSc (3 years, IQR 1–16.5) than lcSSc (2 years, IQR 0–7), and dcSSc (1 year, IQR 0–3) (p<0.001). Clinical ssSSc phenotype was comparable to lcSSc, except for digital pitting scars (DPS) (19.7% vs 42%, p=0.01), but significantly milder than dcSSc, particularly for digital ulcers (DU) (6.6% vs 35.7%, p<0.001), oesophagus (46.2% vs 63.5%, p=0.009), lung (mean diffusion capacity for carbon monoxide 72.2±19.6 vs 62.4±22.8, p=0.009; mean forced vital capacity 105.6±21.7 vs 89.2±20.9, p<0.001) and major videocapillaroscopic alterations (late pattern 8.6% vs 47.6%, p<0.001). Moreover, in ssSSc the percentages of anticentromere and antitopoisomerase were comparable to lcSSc (40% and 18.3% vs 36.7% and 26.6%), but divergent respect to dcSSc (8.6% and 67.4%, p<0.001).
Conclusion
The ssSSc is a quite rare disease variant characterised by clinico-serological features comparable to lcSSc, but significantly different from dcSSc. Overall, longer RP duration, low percentages of DPS and peripheral microvascular abnormalities, and increased anti-centromere seropositivity distinguish ssSSc. Further investigations based on national registries might provide useful insights on the actual relevance of the ssSSc within the scleroderma spectrum.
Keywords: Scleroderma, Systemic; Autoimmunity; Autoimmune Diseases; Epidemiology
WHAT IS ALREADY KNOWN ON THIS TOPIC
Currently, the literature is conflicting concerning demographics and clinico-laboratory hallmark of systemic sclerosis (SSc) sine scleroderma (ssSSc), a quite rare SSc subset without distinctive cutaneous signs, generating diagnostic uncertainties.
WHAT THIS STUDY ADDS
The analysis of the large SSc population from Systemic sclerosis PRogression INvestiGation Italian national registry allowed for an updated description of the ssSSc phenotype, mainly characterised by a longer Raynaud’s phenomenon duration at diagnosis, reduced frequencies of peripheral vascular involvement, less microcirculatory abnormalities and anticentromere positivity. The comparative analysis with other subsets revealed that ssSSc visceral involvement was nearly similar to limited cutaneous SSc and significantly milder than diffuse cutaneous SSc.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our findings may provide some important suggestions for future investigations on the biological bases of the variable distribution of both skin/visceral fibrosis and microangiopathy through the whole scleroderma spectrum, as well as on the complex etiopathogenesis of the SSc, which may lead to a novel disease subsetting.
Introduction
Systemic sclerosis (SSc) is a connective tissue disease affecting skin and internal organs, characterised by autoimmunity, microvascular injury and collagen deposition.1 2 In SSc, widespread skin and visceral fibrosis are associated with a reduction of quality of life, poor patients’ outcomes and increased mortality.1 3 The hallmark of the disease is the remarkable heterogeneity of clinical manifestations.3 The disease is clinically classified according to the extension of skin involvement in two main subsets, limited cutaneous SSc (lcSSc) and diffuse cutaneous (dcSSc), that also include SSc-specific autoantibodies, nailfold capillaroscopic patterns and fibrosis of internal organ.2 3 The two subsets have well-recognised differences with respect to disease severity and prognosis.2 4 Furthermore, SSc sine scleroderma (ssSSc) is considered as a separate subset first described in detail by Rodnan and Fennell.5 Its clinical presentation can be misleading, generating diagnostic uncertainties because of the lack of skin involvement, although it may have the involvement of lung, heart and gastrointestinal (GI) system.6 7 Currently, the literature is conflicting concerning the real prevalence of ssSSc, the female/male ratio, and the presence/severity of both visceral organ and peripheral vascular involvement, mostly depending on the characteristics of the studied population.8–14
In 2014, the Italian Society for Rheumatology promoted the development of the national SPRING (Systemic sclerosis PRogression INvestiGation) registry, which includes the clinical conditions preceding the onset of definite SSc and the main disease subsets.15 The overall baseline data have already been published15 16 while the assessment of more than 2400 consecutive patients is still in progress. The aim of the present work was to analyse the main demographic, clinical and laboratory features of patients with ssSSc in comparison with lcSSc and dcSSc subsets within the SPRING registry. Moreover, the observed findings were compared with other similar studies present in the literature.
Patients and methods
The non-profit national multicentre SPRING registry, involving 37 tertiary referral centres, collects more than 150 disease variables, such as demographic, clinical and imaging investigations, as well as ongoing treatments.
Data were collected and handled using the tool REDCap (Research Electronic Data Capture), a web-based application for assistance in data collection. Since multicentre registries are greatly heterogeneous in collecting and entering data, we minimised this issue by introducing clear-cut definitions of all registry variables; moreover, periodic quality checks were performed by the coordinating centre.15
Definitions
For the current study, data concerning patients with definite SSc aged >18, enrolled up to June 2022, were taken into account. The SPRING database has been previously described,15 16 consisting of patients classified into four different cohorts: (1) primary Raynaud’s phenomenon (RP); (2) suspected secondary RP; (3) Very Early Diagnosis of Systemic Sclerosis; (4) definite SSc according to ACR/EULAR 2013 classification criteria.17 A thorough medical chart review for all consecutive patients with definite SSc was made and cutaneous subsets were classified as dcSSc, lcSSc and ssSSc. In particular, the ssSSc was classified based on the absence of puffy fingers and skin thickening, in any skin areas, including fingers (sclerodactyly), hands, limbs and trunk. All ssSSc patients had a modified Rodnan skin score=0.2 4 7
Information collected at registration included age of disease onset, that is, that of the first non-RP sign(s)/ symptom(s), time from SSc onset to diagnosis, time from RP onset to SSc diagnosis, as well the following clinical variables: oesophageal dysfunction symptoms (dysphagia, reflux), cardiopulmonary signs and symptoms (dyspnoea, arrhythmias, heart failure), sicca syndrome (dry eyes/mouth), renal crisis (sudden onset of severe arterial hypertension with acute renal failure), skin signs (sclerodactyly, puffy fingers, calcinosis, telangiectasia), peripheral vascular signs (fingertip pitting scars (DPS), digital ulcers (DUs), gangrene) and musculoskeletal (tenosynovitis, arthritis defined as inflammatory changes observed in more than two joints, joint contractures, tendon friction rubs, osteomyelitis, carpal tunnel syndrome, myositis). Capillaroscopic patterns at nailfold videocapillaroscopy (NVC) were classified according to the current guidelines as normal (N), early (E), active (A) and late (L).18 Laboratory findings included antinuclear antibodies (ANA), antiextractable nuclear antigens, particularly the SSc-related antibodies (anticentromere/CENP-B, antitopoisomerase I/Scl-70 and anti-RNA polymerase III), as earlier described.15 16 Non-invasive cardiac diagnostic testing was performed by trans-thoracic Doppler echocardiography, collecting the following data: systolic pulmonary arterial pressure (sPAP), left ventricular ejection fraction (LVEF), anomalous diastolic function, pericardial effusion. The current algorithm was used to screen SSc patients and identify those with a high-risk of pulmonary arterial hypertension (PAH). Those with a high PAH probability underwent right hearth catheterisation (RHC).19
Investigations for lung involvement consisted of pulmonary function tests (predicted value of total lung capacity (TLC), forced vital capacity (FVC)), diffusion capacity for carbon monoxide (DLCO) and high-resolution CT (HRCT) (ground glass fibrosis, reticulation, honeycombing). Finally, information about previous/current treatments included both vasoactive/vasodilating drugs (bosentan, sildenafil, vardenafil, tadalafil, iloprost, PGE1, inhaled-INN iloprost, epoprostenol, riociguat, nifedipine, nicardipine, amlodipine, felodipine, diltiazem) and immunosuppressants (cyclophosphamide, methotrexate, leflunomide, aziatoprine, micophenolic acid, cyclosporine, rituximab, imatinib, anti-TNF-alpha, tocilizumab, abatacept) was collected.
Statistical analysis
Descriptive analyses were reported as absolute and relative frequencies for categorical variables, mean and SD for continuous ones. Median (IQR) has been provided in place of mean (SD) when significant asymmetry of distributions was present. To evaluate the differences among groups either the Pearson’s χ2 test or the Fisher’s exact test were employed, while quantitative variables were examined using the non-parametric Mann-Whitney test or the t-test, as appropriate. To avoid family-wise error rate the Simes-Benjamini-Hochberg correction was applied. Multivariable statistical analysis was also performed by using a logistic regression model. P values <0.05 were considered statistically significant.
All analyses were carried out using R statistical software (Foundation for Statistical Computing, V.4.2).
Results
Demographic, clinical and laboratory findings of the whole SSc series and cutaneous subsets are provided in table 1, whereas data regarding internal organ involvement, peripheral microcirculation abnormalities and previous/current treatments are given in table 2. Moreover, figure 1 gives a comprehensive depiction of the similarities and difference between the three cutaneous subsets. Finally, table 3 summarises the main cohort and multicentre studies on ssSSc available in the world literature.
Table 1.
Demographic, clinical characteristics and laboratory characteristics of SSc patients by cutaneous subtype classification
| All (N=1808) | ssSSc N=61 (3.4) |
lcSSc N=1377 (76.2) |
dcSSc N=370 (20.4) |
P value | |||
| ssSSc vs lcSSc | ssSSc vs dcSSc | lcSSc vs dcSSc | |||||
| Demographics | |||||||
| Female sex, n/N (%) | 1594/1803 (88.4) | 58/61 (95.1) | 1229/1372 (89.6) | 307/370 (83) | 0.71 | 0.06 | 0.001 |
| Age of onset, years, mean (SD) | 49.6 (14) | 52.8 (14.7) | 50.6 (13.9) | 45.4 (13.4) | 0.70 | 0.003 | <0.001 |
| SSc onset to diagnosis, years, median (IQR) | 0 (0–1) | 0 (0–1.5) | 0 (0–1) | 0 (0–1) | 0.99 | 0.9 | 0.22 |
| RP onset to diagnosis, years, median (IQR) | 2 (0–6) | 3 (1–16.5) | 2 (0–7) | 1 (0–3) | 0.27 | <0.001 | <0.001 |
| Clinical | |||||||
| Skin | |||||||
| Digital pitting scars, n/N (%) | 855/1798 (47.6) | 12/61 (19.7) | 574/1367 (42) | 369/370 (72.7) | 0.01 | <0.001 | <0.001 |
| Digital ulcers, n/N (%) | 403/1802 (22.4) | 4/61 (6.6) | 267/1371 (19.5) | 132/370 (35.7) | 0.18 | <0.001 | <0.001 |
| Gangrene, n/N (%) | 18/1796 (1) | 0/61 (0) | 10/1365 (0.7) | 8/370 (2.2) | 0.99 | 0.79 | 0.05 |
| Telangiectasia, n/N (%) | 1072/1801 (59.5) | 39/61 (63.9) | 783/1370 (57.2) | 250/370 (67.6) | 0.77 | 0.83 | 0.001 |
| Calcinosis, n/N (%) | 219/1795 (12.2) | 2/61 (3.3) | 153/1364 (11.2) | 64/370 (17.3) | 0.44 | 0.02 | 0.005 |
| Musculoskeletal | |||||||
| Tenosynovitis, n/N (%) | 110/1798 (6.1) | 3/61 (4.9) | 76/1367 (5.6) | 31/370 (8.4) | 0.99 | 0.66 | 0.09 |
| Arthritis, n/N (%) | 207/1790 (11.6) | 7/61 (11.5) | 138/1359 (10.2) | 62/370 (16.8) | 0.99 | 0.60 | 0.001 |
| Osteomyelitis, n/N (%) | 12/1795 (0.7) | 0/61 | 7/1365 (0.5) | 5/369 (1.4) | 0.99 | 0.99 | 0.20 |
| Carpal tunnel syndrome, n/N (%) | 83/1794 (4.6) | 0/61 | 61/1364 (4.5) | 22/369 (6) | 0.47 | 0.12 | 0.35 |
| Myositis, n/N (%) | 265/1796 (14.8) | 7/61 (11.5) | 177/1365 (13) | 81/370 (21.9) | 0.99 | 0.18 | <0.001 |
| Oesophageal symptoms, n/N (%) | 894/1801 (49.6) | 26/61 (42.6) | 633/1370 (46.2) | 235/370 (63.5) | 0.99 | 0.009 | <0.001 |
| Sicca syndrome, n/N (%) | 499/1797 (27.8) | 27/61 (44.3) | 380/1366 (27.8) | 92/370 (24.9) | 0.11 | 0.009 | 0.35 |
| Renal crisis, n/N (%) | 21/1796 (1.2) | 1/61 (1.6) | 11/1366 (0.8) | 9/369 (2.4) | 0.83 | 0.99 | 0.03 |
| Autoantibodies | |||||||
| ANA, n/N (%) | 1725/1783 (96.7) | 60/60 (100) | 1308/1358 (96.3) | 357/365 (97.8) | 0.73 | 0.82 | 0.26 |
| Antitopoisomerase I, n/N (%) | 616/1781 (34.6) | 11/60 (18.3) | 61/1359 (26.6) | 244/362 (67.4) | 0.76 | <0.001 | <0.001 |
| Anti-RNA polymerase III, n/N (%) | 27/1437 (1.9) | 1/39 (2.6) | 14/1094 (1.3) | 12/304 (3.9) | 0.81 | 0.99 | 0.01 |
| Anti-centromere, n/N (%) | 515/1655 (31.1) | 22/55 (40) | 464/1264 (36.7) | 29/336 (8.6) | 0.99 | <0.001 | <0.001 |
Significant p-values are outlined in bold
ANA, antinuclear antibodies; dcSSc, diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; RP, Raynaud’s phenomenon; SSc, systemic sclerosis; ssSSc, SSc sine scleroderma.
Table 2.
Results of diagnostic tests and treatments in SSc patients by cutaneous subtype classification
| All (N=1808) | ssSSc N=61 (3.4) |
lcSSc N=1377 (76.2) |
dcSSc N=370 (20.4) |
P value | |||
| ssSSc vs lcSSc | ssSSc vs dcSSc | lcSSc vs dcSSc | |||||
| Hearth | |||||||
| PAH, n/N (%) | 29/1494 (1.9) | 3/51 (5.9) | 17/1126 (1.5) | 9/317 (2.8) | 0.30 | 0.40 | 0.24 |
| sPAP, mm Hg, mean (SD) | 23.3 (16.4) | 25.8 (17) | 23 (16.1) | 24 (17.2) | 0.73 | 0.83 | 0.22 |
| LVEF, mean (SD) | 61.1 (5.8) | 61.7 (4.1) | 61.2 (5.7) | 61 (6.2) | 0.99 | 0.92 | 0.83 |
| Diastolic dysfunction, n/N (%) | 20/1452 (22) | 11/50 (22) | 240/1078 (22.3) | 69/324 (21.3) | 0.99 | 0.99 | 0.81 |
| Pericardial effusion, n/N (%) | 109/1474 (7.4) | 3/51 (5.9) | 72/1099 (6.6) | 34/324 (10.5) | 0.99 | 0.66 | 0.04 |
| Lung | |||||||
| ILD, n/N (%) | |||||||
| Ground glass | 371/1808 (20.5) | 11/61 (18) | 244/1377/17.7) | 116/370 (31.4) | 0.99 | 0.11 | <0.001 |
| Reticulation | 294/1808 (16.3) | 11/61 (18) | 208/1377 (15.1) | 75/370 (20.3) | 0.99 | 0.95 | 0.03 |
| Honeycomb | 97/1808 (5.4) | 1/61 (1.6) | 55/1377 (4) | 41/370 (11.1) | 0.95 | 0.09 | <0.001 |
| DLCO % predicted, mean (SD) | 68.6 (20.6) | 72.2 (19.6) | 69.8 (19.7) | 63.4 (22.8) | 0.73 | 0.009 | <0.001 |
| FVC % predicted, mean (SD) | 101 (22.8) | 105.6 (21.7) | 104.1 (22.3) | 89.2 (20.9) | 0.99 | <0.001 | <0.001 |
| TLC % predicted, mean SD | 96.6 (20.4) | 103.9 (19.1) | 98.5 (20.1) | 87.6 (19.5) | 0.44 | <0.001 | <0.001 |
| Nailfold videocapillaroscopy | |||||||
| Scleroderma pattern, n/N (%) | |||||||
| Normal (N) | 105/1609 (6.5) | 7/58 (12.1) | 92/1215 (7.6) | 6/336 (1.8) | 0.73 | 0.003 | <0.001 |
| Early (E) | 336/1609 (20.9) | 21/58 (36.2) | 265/1215 (21.8) | 50/336 (14.9) | 0.19 | 0.001 | 0.013 |
| Active (A) | 746/1609 (46.4) | 25/58 (43.1) | 601/1215 (49.5) | 120/336 (35.7) | 0.79 | 0.58 | <0.001 |
| Late (L) | 422/1609 (26.2) | 5/58 (8.6) | 257/1215 (21.2) | 160/336 (47.6) | 0.25 | <0.001 | <0.001 |
| Treatments | |||||||
| Vasoactive drugs, n/N (%) | 1391/1808 (76.9) | 38/61 (62.3) | 1036/1377 (75.2) | 317/370 (85.7) | 0.23 | <0.001 | <0.001 |
| Immunosuppressants, n/N (%) | 500/1808 (27.7) | 15/61 (24.6) | 299/1377 (21.7) | 186/370 (50.3) | 0.99 | 0.001 | <0.001 |
Significant p-values are outlined in bold
dcSSc, diffuse cutaneous SSc; DLCO, diffusing lung capacity for carbon monoxide; FVC, forced vital capacity; ILD, interstitial lung disease; lcSSc, limited cutaneous SSc; lcSSc, limited cutaneous systemic sclerosis; lcSSc, diffuse cutaneous SSc; LVEF, left ventricular ejection fraction; NVC, nailfold videocapillaroscopy; PAH, pulmonary arterial hypertension by hearth catheterisation; sPAP, systolic pulmonary arterial pressure; SSc, systemic sclerosis; ssSSc, SSc sine scleroderma; TLC, total lung capacity.
Figure 1.
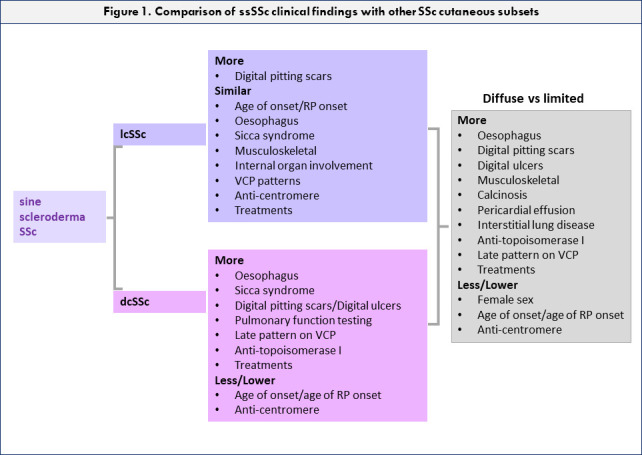
Comparison of SSc sine scleroderma clinical findings with other SSc cutaneous subsets. VCP, videocapillaroscopy; dcSSc, diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; RP, Raynaud’s phenomenon; SSc, systemic sclerosis.
Table 3.
Summary of main registry studies on systemic sclerosis sine scleroderma (ssSSc) published in the world literature
| Author, country, year (ref) | Data source | n/N (%) | F (%) | DPS-DU (%) | ILD (%) | Hearth (%) | PAH (%) | SRC (%) | OES (%) | MS (%) | AA (%) | Entry criteria |
| Poormoghim et al, Massachusetts, 20008 | Monocentric registry | 48/556 (8.6)* | 85.4 | 60.4† | 38.6 | 8.8 | 22.9 | 0 | – | 47.9 | Scl-70: 6.25 ACA: 31.2 |
No skin thickening on physical examination |
| Hunzelmann et al, Germany, 20089 | Multicentric inter-disciplinary registry | 22/1483 ( 1.4 ) | 90.9 | 33.3‡ | 59.1 | 13.6 | 13.6 | 22.7 | 72.7 | 45.5 | Scl-70: 40
ACA: 35 |
No skin alterations |
| Simeón-Aznar et al, Spain, 201210 | Multicentric registry | 67/916 (7.3) | 92.5 | 14.5‡ | 39.1 | 49.3 | 24.6 | 1.4 | 44.9 | 52.2 | Scl-70: 9.5 ACA: 41.5 |
1980 ACR 2001 LeRoy-Medsger |
| Marangoni et al, Brazil, 201311 | Retrospective bi-centric database | 79/947 (8.3) | 96.2 | 24.1‡ | 56.9 | – | 22.8 | 2.5 | 83.1 | 46.9 | Scl-70: 7.8 ACA: 41.7 |
No skin thickening on physical examination |
| Diab et al, Canada, 201412 | Multicentre registry | 27/1417 (1.9) | 88.9 | 29.6† | 25.9 | – | 11.5 | 3.7 | 51.8 | 14.8 | Scl-70: 16.6 ACA: 60 |
Physician diagnosis |
| Tolosa-Vilella et al, Spain, 201613 | Multicentric registry | 118/1326 (8.9) | 91.5 | 16.1‡ | 27.9 | 29.6 | 25.8 | 1.4 | 19.1 | 21.8 | Scl-70: 12.7 ACA: 46.6 |
1980 ACR 2001 LeRoy-Medsger 2013 ACR-EULAR |
| De Almeida Chaves et al, France, 202114 | Retrospective bi-centric database | 33/375 (8.8) | 84.8 | 9.1‡ | 9.1 | 15.1 | 9.1 | 3 | – | 42.4 | Scl-70: 12.1 ACA: 78.7 |
2001 LeRoy-Medsger 2013 ACR-EULAR |
| Present study, 2022 | Multicentric registry | 61/1808 (3.3) | 95.1 | 26.2† | 37.7 | 27.4 | 5.9 | 1.6 | 42.6 | 32.7 | Scl-70: 18.3 ACA: 40 |
2013 ACR- EULAR |
Lowest and highest values for each item are highlighted in bold
*lcSSc+ssSSc.
†DPS+DU.
‡DU.
AA, autoantibodies; ACA, anticentromere; DPS, digital pitting scars; DU, digital ulcer; ILD, interstitial lung disease; lcSSc, limited cutaneous systemic sclerosis; MS, musculoskeletal; OES, oesophageal involvement; PAH, pulmonary hypertension; SRC, scleroderma renal crisis.
Whole SSc series: demographic features and subsetting
Up to 30 June 2022, among the whole 1808 patients’ series with definite SSc included in the study, 61 (3.4%) were classified as ssSSc (table 1) that were characterised by the absence of cutaneous involvement but fulfilling the classification criteria of SSc.17 All patients with ssSSc reached the cut-off of ≥9 that satisfied the subitem scores, excluding scleroderma skin involvement, in accordance with the point score system of ACR/EULAR 2013 criteria.17
In particular, 6 (9.8%) had a total score of 9, and 33 (54.1%) a total score of 10. Next, patients (8.2%) reached the total score of 11 and 10 (16.4%) the total score of 12. Finally, seven patients (11.4%) had a score ≥13.17
These ssSSc patients had a mean age at disease onset of 52.8±14.7 years, with a 95.1% (F/M ratio 19/1) of females. The lcSSc subset consisted of 1377 patients, accounting for 76.2% of the whole cohort (F/M ratio 8.5:1), while 370 patients (20.4%) had dcSSc variant (F/M ratio 4.9:1) (table 1).
ssSSc: clinical variables and autoantibodies
ssSSc patients showed a variable percentage of other SSc signs/symptoms. Namely, telangiectasias (63.9%), oesophageal involvement (42.6%) and sicca syndrome (44.3%) were common, while DPS and DUs were less represented (19.7% and 6.6%, respectively). Musculoskeletal involvement was globally present in around one-third of patients (tenosynovitis 4.9%, arthritis 11.9% and myositis 11.9%). Joint contractures and tendon friction rubs were only anecdotally reported (only two and one case). In all ssSSc patients, serum ANA were present. Among SSc-specific autoantibodies, anticentromere were detected in 40%, followed by antitopoisomerase I in 18.3% and anti-RNA polymerase III in 2.6% of ssSSc (table 1).
ssSSc: internal organ and microcirculation abnormalities
Hearth involvement was observed in 13 out of total ssSSc patients (21.3%). Doppler echocardiography examination revealed diastolic dysfunction (22%), pericardial effusion (5.9%), mean sPAP of 25.8±17 mm Hg, and mean LVEF % of 61.7±4, while PAH at RHC was found in 5.9% of assessed individuals (table 2). More than one-third of ssSSc patients (37.7%) had ILD at HRCT. The mean values of % predicted DLCO, FVC and TLC were 72.2±19.6, 105.6±21.7 and 103.9±19.1, respectively. Among capillaroscopic findings, a normal or early pattern was more frequent and was found in almost 50% of ssSSc patients (12.1% and 36.2%, respectively), whereas late pattern was uncommon (8.6%). In ssSSc patients, vasoactive/vasodilating treatments were frequently used (62.3%), while immunosuppressants in around 24.6% of patients (table 2).
ssSSc versus limited and diffuse cutaneous subsets
The results of comparative analysis among the three SSc subsets are shown in tables 1 and 2 and figure 1.
The ssSSc and lcSSc exhibit several similarities as regards both demographic and clinical parameters, except for DPS (ssSSc 19.7% vs lcSSc 42 %, p=0.01).
Conversely, the ssSSc and the dcSSc subsets markedly differ for the rate of female sex (95.1% vs 83%, p=0.001), the age of disease onset (52.8±14.7 vs 45.4±13.4 years; p=0.003), as well as time interval (years) from RP onset to SSc diagnosis (median, IQR=3, 1–16.5 vs median, IQR=1, 0–3; p<0.001) (table 1).
The oesophageal involvement and sicca syndrome were significantly lower in ssSSc than dcSSc (p=0.009, for both), as well as DPS (p<0.001), DU (p<0.001) and calcinosis (p=0.02).
Among SSc-specific autoantibodies, anticentromere were more frequently detected in ssSSc (40%) compared with dcSSc (8.6%, p<0.001), while an opposite distribution was observed for antitopoisomerase I antibodies (18.3% vs 67.4%, p<0.001).
The frequency of ILD was similar in ssSSc and lcSSc (37.7% and 36.8%, respectively), but higher in dcSSc (62.7%). ssSSc and dcSSc differed for DLCO (mean DLCO 72.2±19.6 vs 62.4±22.8, p=0.009) and other functional tests (mean FVC predicted 105.6±21.7 vs 89.2±20.9, TLC 103.9±19.1 vs 87.6±19.5, p<0.0001 for both) (table 2).
The proportion of normal and early patterns were more frequent in ssSSc (12.1% and 36.2%) compared with both lcSSc (7.6% and 21.8%) and dcSSc (1.8% and 14.9%) (p=0.003 and 0.001, respectively), whereas the late pattern was uncommon (8.6%) in ssSSc, with an increasing prevalence from lcSSc (21.2%) to dcSSc (47.6%, p<0.001). Finally, both vasoactive/vasodilating and immunosuppressive therapies were more frequently used in dcSSc (p=0.001, table 2).
Multivariable logistic regression analysis, after adjustment for sex and age at onset, indicates that longer time from RP onset to diagnosis (OR 1.031; 95% CI 1.004 to 1.057; p=0.016) and higher prevalence of DPS (OR 0.394; 95% CI 0.188 to 0.767; p=0.009) may distinguish ssSSc from lcSSc patients, whereas longer time from RP onset to diagnosis (OR 1.062; 95% CI 1.024 to 1.105; p=0.002), higher DPS (OR 0.158; 95% CI 0.067 to 0.346, p<0.001), anticentromere positivity (OR 2.486; 95% CI 1.038 to 5.972; p=0.04) and antitopoisomerase 1 negativity (OR 0.219; 95% CI 0.086 to 0.515; p=0.001) may distinguish ssSSc from dcSSc patients.
As expected, lcSSc and dcSSc significantly differed in several clinical and laboratory findings (oesophageal involvement, renal crisis, DPS and DU, telangiectasias, calcinosis, arthritis, and myositis), including cardio-pulmonary involvement (pericardial effusion and ILD in all items), with a relevant higher frequency of antitopoisomerase I and late capillaroscopic pattern in dcSSc. Taken together these differences showed a significant higher prevalence of worse clinical-prognostic parameters in the dcSSc compared with lcSSc (figure 1).
Discussion
This cross-sectional study indicates that ssSSc subset accounts for approximately 3% of the SSc patients’ population recorded in the Italian SPRING registry. Except for cutaneous involvement, this subset fulfils the current classification criteria of SSc by exhibiting the typical disease manifestations, including the main visceral organ damages. Our national registry study, focusing on the largest SSc population so far investigated, allows for valuable comparative analysis between the three skin subgroups.
In particular, the data show that the clinical features and the autoantibodies of the ssSSc subset overlap with those of the lcSSc subset, while both differ significantly from the dcSSc subset, which is characterised by more severe microvascular and fibrotic organ involvement and increased antitopoisomerase I and anti-RNA polymerase rates. Of note, a significantly longer time interval from RP onset to SSc diagnosis was observed either in ssSSc and lcSSc compared with dcSSc, as well as an increasing trend in DU rates through the three subsets (ssSSc<lcSSc<dcSSc). Overall, a longer RP duration at diagnosis, reduced DPS frequency, less microcirculatory abnormalities and anticentromere positivity were the main features of the ssSSc subset.
Demographic and clinical hallmarks of the present ssSSc series and those previously published are summarised in table 3. The papers are characterised by a significant heterogeneity for what concerns the number, the modalities of patients’ recruitment (mono/multicentre), and the classification criteria adopted.8–14 This condition may account for the variability in the ssSSc prevalence (from 1.4% to 8.9%), as well as in the clinical phenotype, namely peripheral vascular, heart, lung, renal, oesophageal and musculoskeletal involvement. Whereas, similar data are reported concerning the higher occurrence of anticentromere antibodies, which exceeds that of antitopoisomerase in the majority of the ssSSc series. Given the large time interval of more than 20 years from the first to the last study, it may be noted that only the last three reports, including ours, used the 2013 ACR/EULAR classification criteria (table 3). Overall, the findings reported in the world literature suggested a few considerations that are addressed and developed in the discussion.
The occurrence of ssSSc might be underestimated in clinical practice, being its identification and diagnosis difficult in some cases, due to the absence of any skin involvement paralleled by mild disease manifestations. Moreover, the recognising of SSc, particularly in the early stage, is based on some cardinal signs, namely Raynaud’s phenomenon, DPS, puffy fingers, cutaneous sclerosis, sclerodactyly and/or capillaroscopic/autoantibody alterations.2 4
The ACR/EULAR 2013 classification criteria for SSc have improved the sensitivity and specificity of previous 1980 ACR criteria,20 even in the absence of oedematous/fibrotic skin involvement. However, the variable prevalence of ssSSc among the main studies of the literature might be explained by the use of different classification criteria.8–14 It is supposable that these differences are real and may reflect the variable contribution of genetic and/or geographical/environmental factors among SSc populations from different ethnic groups or geographical areas.21 22 Furthermore, the low rate of ssSSc observed in some reports,9 12 including the present study, could also be related to an inadequate network of specialised tertiary referral centres of some geographic areas where a number of ssSSc may be diagnosed very late or completely overlooked.22
Our ssSSc subgroup showed a female/male ratio comparable to that of the Brazilian study11 and of our earlier reports,15 16 but significantly higher than that observed in other studies.8 12 14 According to previous observation for the whole SSc population in Italy,1 15 the longer time from RP onset to diagnosis seems to characterise the ssSSc subset, a finding also reported by other authors in national registries.10 14 It may represent a useful prognostic factor at SSc diagnosis in individual patient, suggesting a rather slow progression of the microangiopathic dysfunction that characterises the SSc pathogenesis.23 24
The present findings confirmed the lower rate of peripheral vascular complications in ssSSc, namely the DPS and/or DU, compared with other subsets.10 11 13 14 In particular, our ssSSc patients showed the lowest rate of DU among the three subsets, and a significantly lower percentage of DPS than the lcSSc subset. In this respect, a recent study found that DPS were associated with a severe disease course and worse outcomes.25 In ssSSc, our data indicate a milder peripheral small vessel vasculopathy as shown by the rarity of major capillaroscopic modifications and the low rate use of vasoactive/vasodilating drugs.
In SSc, lung involvement includes ILD and PAH.26 The recognition of more than 37% of ssSSc patients presenting ILD on HRCT (with a DLCO close to 70%), with a percentage comparable to lcSSc, is in agreement with previous studies,8 10 but still higher than others.12–14 Noteworthy, in our ssSSc cohort, the percentage of honeycombing, which correspond to the most advanced and severe sign of lung injury,26 was very low. However, our findings demonstrate that, also in ssSSc, ILD should be a concern that should not be neglected.27 In this respect, additional data from national registries are needed to verify the real occurrence and severity of lung involvement in ssSSc, as well as of other organs manifestations (ie, cardiac, GI and musculoskeletal) that showed a wide range of variability among previously published studies (table 3).
In our ssSSc patients, the autoantibody profile was similar to the data previously reported8 10 11 13; namely, anticentromere were detected in 40% or more, with antitopoisomerase I usually belonging 20% or less of patients.10–14 This specific autoantibody dichotomy seems to be the distinctive immunological marker of the ssSSc subset.
The comparison between ssSSc and lcSSc revealed several similarities concerning demographic, clinical and immunological features. On the contrary, clear-cut differences were found in both ssSSc and lcSSc when compared with dcSSc, being the latter characterised by higher proportion of oesophageal, peripheral vascular (DPS, DU, calcinosis), pulmonary (functional alterations), worse NVC microvascular involvement and higher serum antitopoisomerase I autoantibodies. These findings were consistent with data formerly described.10 11 13
A thorough examination of the literature shows a substantial disagreement about including ssSSc within the scleroderma spectrum. Some authors recommend that ssSSc should be a separate condition to avoid misdiagnosis,4 28 while others consider ssSSc as a mild subvariant of lcSSc.8 9 12 In this scenario, several data suggested that the investigation of SSc subsets might help to better understand the disease aetiopathogenesis, and to shape the prognosis predicting the severity of organ complications.1–4 The fact that ssSSc and lcSSc share a similar clinical picture, the autoantibody profile and the peripheral microangiopathy may suggest that these subsets are strongly related, although with a different skin phenotype. The heterogeneity of skin involvement remains a matter of debate in SSc, as well as the variable combination and the severity of microangiopathy/fibrosis-related manifestations in internal organs.21 29–32
The strengths and limitations of SSc registry-based multicentric studies have been previously addressed.12 15 16 Although our SSc population is the largest reported among national registries, the present data are not conclusive. Possibly, long-term follow-up studies may verify the natural course and outcome of ssSSc patients in comparison to the other cutaneous subsets.14 33
The aim of our study was to provide an overall assessment of the ssSSc subset recruited at tertiary referral centres in our Italian SPRING registry. First, a relatively low proportion of ssSSc was observed within a large population of definite SSc, a finding that greatly varied among the few reports of the existing literature. Apart from skin involvement, the signs and symptoms of the ssSSc subset were mostly comparable with that of lcSSc. Both subsets were characterised by less frequent and less severe organ involvement, scarce NVC alterations, absence of anti-topoisomerase I positivity and a significantly different clinical pattern respect to dcSSc.
A number of issues still remain unclear: the absence of cutaneous sclerosis, and the clinical overlap between the ssSSc and lcSSc raise the question whether ssSSc represents a distinct SSc subset or a simple phenotypic variant of lcSSc. Overall, future investigations on the biological origin of the different distribution of skin fibrosis among SSc patients29–32 may provide useful insights on the complex etiopathogenesis of the disease, likewise a novel disease sub-setting.2 4 33
Acknowledgments
This study was supported by the Italian Society for Rheumatology (SIR).
Footnotes
Twitter: @Dott.Simone Parisi
Correction notice: This article has been corrected since it was first published online. The author Greta Pellagrino has been updated to Greta Pellegrino.
Collaborators: Convenors: Clodoveo Ferri (University of Modena & Reggio Emilia, Italy; clodoveo.ferri@unimore.it); Marco Matucci-Cerinic (University of Florence, Italy; marco.matuccicerinic@unifi.it). Investigators (in alphabetical order): Abignano Giuseppina (AOR San Carlo di Potenza; g.abignano@hotmail.com); Agnes Cecilia (Ospedale San Lorenzo, Carmagnola (TO), ASL-TO5; ceciliaagnes909@gmail.com); Amato Giorgio (AOU Policlinico – Vittorio Emanuele, Catania; giorgioamato@hotmail.it); Ariani Alarico (AOU Parma; dott.alaricoariani@libero.it); Bagnato Gianluca (Università degli Studi di Messina; gianbagnato@gmail.com); Bajoicchi Gianluigi (Arcispedale S. Maria Nuova, Reggio Emilia; gianluigi.bajocchi@asmn.re.it); Barsotti Simone (AOU Santa Chiara, Pisa; simone.barsotti@outlook.com); Bellando-Randone Silvia (University of Florence; s.bellandorandone@gmail.com); Benenati Alessia (AOU ‘Policlinico - Vittorio Emanuele, Catania; alessia.benenati@libero.it); Beretta Lorenzo (Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano; lorberimm@hotmail.com); Bianchi Gerolamo (ASL3 Genova; gerolamo.bianchi@asl3.liguria.it); Bosello Silvia (Policlinico “A. Gemelli” –IRCCS – UOC di Reumatologia; Roma; silvia.bosello@libero.it); Cacciapaglia Fabio (UO Reumatologia – DETO, Università di Bari; fabio.cacciapaglia79@gmail.com); Calabrese Francesca (SSD Reumatologia, Reggio Calabria; francescacalabrese81@virgilio.it); Caminiti Maurizio (Ospedale Bianchi-Melacrino-Morelli, SSD Reumatologia, Reggio Calabria; mauriziocaminiti@tin.it); Campochiaro Corrado (Ospedale S. Raffaele, Milano; corradocampochiaro@gmail.com); Carignola Renato (AOU San Luigi Gonzaga, Orbassano (TO); renatocarigno@gmail.com Cavazzana Ilaria, Spedali Civili di Brescia; ilariacava@virgilio.it); Ciano Giovanni (Ospedale Ariano Irpino, ASL Avellino; giovanni.ciano55@gmail.com); Cipolletta Edoardo (Clinica Reumatologica, Università Politecnica delle Marche, Ancona; edo.cipo@hotmail.it); Codullo Veronica (Policlinico San Matteo, Pavia; veronicacodullo@yahoo.it); Cozzi Franco (Villa Salus, Mestre; franco.cozzi@unipd.it); Cuomo Giovanna (Università degli Studi della Campania - Luigi Vanvitelli, Napoli; giovanna.cuomo@unicampania.it); D’Angelo Salvatore (AOR San Carlo di Potenza; saldangelo@katamail.com); Dagna Lorenzo (Ospedale S. Raffaele, Milano; dagna.lorenzo@hsr.it); Dall’Ara Francesca (UO Medicina Interna-Ambulatorio Reumatologia, Ospedale di Lodi; francesca.dallara@gmail.com); De Andres Ilenia (AO ARNAS Garibaldi, Catania; ilenia.deandres@gmail.com); De Angelis Rossella (Clinica Reumatologica, Università Politecnica delle Marche, Ancona; r.deangelis@staff.univpm.it); De Cata Angelo (Ospedale Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG); a.decata@operapadrepio.it); De Luca Giacomo (Ospedale S. Raffaele, Milano; deluca.giacomo@hsr.it); De Santis Maria (Istituto Clinico Humanitas, Rozzano, Milano; maria.desantis@humanitas.it); Della Rossa Alessandra (AOU Santa Chiara, Pisa; a.dellarossa69@gmail.com); Di Vico Claudio (Università degli Studi della Campania “Luigi Vanvitelli”; claudio.divico@unicampania.it); Doria Andrea (Università degli Studi di Padova; adoria@unipd.it); Doveri Marica (ASL3 Genova; marica.doveri@asl3.liguria.it); Foti Rosario (AOU Policlinico San Marco, Catania; rosfoti5@gmail.com); Furini Federica (Department of Medical Sciences, University of Ferrara; fefe.furini@gmail.com); Fusaro Enrico (AOU Città della Salute e della Scienza di Torino; fusaro.reumatorino@gmail.com); Generali Elena (Istituto Clinico Humanitas, Rozzano, Milano; e.generali@gmail.com); Gigante Antonietta (Università degli Studi “La Sapienza”, Roma; antonietta.gigante@uniroma1.it); Giollo Alessandro (AOUI Verona; alessandro.giollo@univr.it); Girelli Francesco (Ospedale GB Morgagni, Forlì; francesco.girelli@auslromagna.it); Giuggioli Dilia (University of Modena/Reggio Emilia; dilia.giuggioli@unimore.it); Govoni Marcello (AOU S. Anna, Ferrara; gvl@unife.it); Guiducci Serena (University of Florence; s.guiducci@hotmail.com); Iannone Florenzo (UO Reumatologia– DETO, Università di Bari; florenzo.iannone@uniba.it); Ingegnoli Francesca (Università degli Studi di Milano; francesca.ingegnoli@unimi.it); Iuliano Anna Maria (AO San Camillo Forlanini, Roma; annamariaiuliano@hotmail.it); Lazzaroni Maria Grazia (Spedali Civili and University of Brescia; mariagrazialazzaroni@gmail.com); Lepri Gemma (University of Florence; lepri.gemma@gmail.com); Lubrano Ennio (Università del Molise, Campobasso; ennio.lubrano@unimol.it); Lumetti Federica (University of Modena & Reggio Emilia; fedelumetti@gmail.com); Magnani Luca (Arcispedale S. Maria Nuova, Reggio Emilia; luca.magnani@ausl.re.it); Mennillo Gianna (AOR San Carlo di Potenza; giannaangelamennillo@virgilio.it); Murdaca Giuseppe Ospedale (Policlinico S Martino-Universita’ di Genova; giuseppe.murdaca@unige.it); Pagano Mariano Giuseppa (Ospedale Bianchi-Melacrino-Morelli, Reggio Calabria; giusypaganomariano@libero.it); Parisi Simone (AOU Città della Salute e della Scienza, Torino; simone.parisi@hotmail.it); Pellegrino Greta (Sapienza, Università di Roma; greta.pellegrino01@gmail.com); Peroni Clara Lisa (AOU Città della Salute e della Scienza, Torino; claralisaperoni@gmail.com); Pigatto Erika (Università degli Studi di Padova; erika.pigatto@gmail.com); Riccieri Valeria (Sapienza Università di Roma; valeria.riccieri@uniroma1.it); Romeo Nicoletta (ASO S. Croce e Carle, Cuneo; romeo.n@ospedale.cuneo.it); Rosato Edoardo (Università degli Studi di Roma “La Sapienza” Policlinico Umberto I; edoardo.rosato@uniroma1.it); Sambataro Gianluca (Azienda Ospedaliera Cannizzaro, Catania); Saracco Marta (AO Ordine Mauriziano, Torino; marta.saracco@gmail.com); Sebastiani Giandomenico (AO San Camillo Forlanini, Roma; gsebastiani@scamilloforlanini.rm.it); Spinella Amelia (University of Modena & Reggio Emilia; amelia.spinella@gmail.com); Talotta Rossella (L. Sacco Hospital, Milan; talotta1@virgilio.it); Visalli Elisa (AOU Policlinico San Marco, Catania; elivisa21@gmail.com); Vultaggio Licia (AOU S. Anna, Ferrara, licia.vultaggio@unife.it); Zanatta Elisabetta (Università degli Studi di Padova; elisabetta.zanatta@yahoo.it); Zanframundo Giovanni (Policlinico San Matteo, Pavia; gio.zanframundo@gmail.com); Study Center of the Italian Society of Rheumatology (SIR): Carlo Scirè (Università degli Studi, Milano-Bicocca, Milan; c.scire@reumatologia.it); Greta Carrara (Epidemiology Unit, Italian Society for Rheumatology, Milan, Italy; g.carrara@reumatologia.it); Gianpiero Landolfi (Epidemiology Unit, Italian Society for Rheumatology, Milan, Italy; g.landolfi@reumatologia.it); Davide Rozza (Epidemiology Unit, Italian Society for Rheumatology, Milan, Italy; d.rozza@reumatologia.it); Anna Zanetti (Epidemiology Unit, Italian Society for Rheumatology, Milan, Italy; a.zanetti@reumatologia.it).
Contributors: RDA conceived the idea for the study, contributed to the study design, supervised data analysis, interpreted the results, reviewed the literature, co-wrote the first draft of the manuscript and critically reviewed the manuscript. CF and MMC contributed to the study design, supervised data analysis, interpreted the results, and critically reviewed the manuscript. EC supervised data analysis, interpreted the results, and critically reviewed the manuscript. DR performed data analysis. DG, GB, LD, SBR, GZ, RF, FC, GC, AA, ER, GL, FG, VR, EZ, SB, IC, FI, MDS, GM, GA, NR, ADR, MC, AI, GC, LB, GB, EL, IDA, AG, MS, CA, FL, AS, LM, CC, GDL, VC, EV, CDV, AG, GP, EP, MGL, FF, EG, GM, SB, GPM, FF, LV, SP, CLP, AZ, GC, GL, CAS, GB, EF, GDS, MG, SDA, FC, SG, AD, CS, FI, collected clinical data and critically reviewed the manuscript. All authors approved the submitted manuscript. RDA is responsible for the overall content as guarantor and attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Contributor Information
SPRING-SIR (Systemic Sclerosis PRogression INvestiGation group of the Italian Society of Rheumatology):
Clodoveo Ferri, Marco Matucci-Cerinic, Abignano Giuseppina, Agnes Cecilia, Amato Giorgio, Ariani Alarico, Bagnato Gianluca, Bajoicchi Gianluigi, Barsotti Simone, Bellando-Randone Silvia, Benenati Alessia, Beretta Lorenzo, Bianchi Gerolamo, Bosello Silvia, Cacciapaglia Fabio, Calabrese Francesca, Caminiti Maurizio, Campochiaro Corrado, Carignola Renato, Ciano Giovanni, Cipolletta Edoardo, Codullo Veronica, Cozzi Franco, Cuomo Giovanna, D’Angelo Salvatore, Dagna Lorenzo, Dall’Ara Francesca, De Andres Ilenia, De Angelis Rossella, De Cata Angelo, De Luca Giacomo, De Santis Maria, Della Rossa Alessandra, Di Vico Claudio, Doria Andrea, Doveri Marica, Foti Rosario, Furini Federica, Fusaro Enrico, Generali Elena, Gigante Antonietta, Giollo Alessandro, Girelli Francesco, Giuggioli Dilia, Govoni Marcello, Guiducci Serena, Iannone Florenzo, Ingegnoli Francesca, Iuliano Anna Maria, Lazzaroni Maria Grazia, Lepri Gemma, Lubrano Ennio, Lumetti Federica, Magnani Luca, Mennillo Gianna, Murdaca Giuseppe Ospedale, Pagano Mariano Giuseppa, Parisi Simone, Pellegrino Greta, Peroni Clara Lisa, Pigatto Erika, Riccieri Valeria, Romeo Nicoletta, Rosato Edoardo, Sambataro Gianluca, Saracco Marta, Sebastiani Giandomenico, Spinella Amelia, Talotta Rossella, Visalli Elisa, Vultaggio Licia, Zanatta Elisabetta, Zanframundo Giovanni, Carlo Scirè, Greta Carrara, Gianpiero Landolfi, Davide Rozza, and Anna Zanetti
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by reference number OSS 15.10, Azienda Ospedaliera Universitaria Careggi-Firenze. Participants gave informed consent to participate in the study before taking part.
References
- 1. Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002;81:139–53. 10.1097/00005792-200203000-00004 [DOI] [PubMed] [Google Scholar]
- 2. Nevskaya T, Pope JE, Turk MA, et al. Systematic analysis of the literature in search of defining systemic sclerosis subsets. J Rheumatol 2021;48:1698–717. 10.3899/jrheum.201594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lafyatis R, Valenzi E. Assessment of disease outcome measures in systemic sclerosis. Nat Rev Rheumatol 2022;18:527–41. 10.1038/s41584-022-00803-6 [DOI] [PubMed] [Google Scholar]
- 4. Johnson SR, Soowamber ML, Fransen J, et al. There is a need for new systemic sclerosis subset criteria. A content analytic approach. Scand J Rheumatol 2018;47:62–70. 10.1080/03009742.2017.1299793 [DOI] [PubMed] [Google Scholar]
- 5. RODNAN GP, FENNELL RH. Progressive systemic sclerosis sine scleroderma. JAMA 1962;180:665–70. 10.1001/jama.1962.03050210027006 [DOI] [PubMed] [Google Scholar]
- 6. Toya SP, Tzelepis GE. The many faces of scleroderma sine scleroderma: a literature review focusing on cardiopulmonary complications. Rheumatol Int 2009;29:861–8. 10.1007/s00296-009-0878-7 [DOI] [PubMed] [Google Scholar]
- 7. Kucharz EJ, Kopeć-Mędrek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med 2017;26:875–80. 10.17219/acem/64334 [DOI] [PubMed] [Google Scholar]
- 8. Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum 2000;43:444–51. 10.1002/1529-0131(200002)43:2<444::AID-ANR27>3.0.CO;2-G [DOI] [PubMed] [Google Scholar]
- 9. Hunzelmann N, Genth E, Krieg T, et al. The registry of the German network for systemic scleroderma: frequency of disease subsets and patterns of organ involvement. Rheumatology (Oxford) 2008;47:1185–92. 10.1093/rheumatology/ken179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simeón-Aznar CP, Fonollosa-Plá V, Tolosa-Vilella C, et al. Registry of the Spanish network for systemic sclerosis: clinical pattern according to cutaneous subsets and immunological status. Semin Arthritis Rheum 2012;41:789–800. 10.1016/j.semarthrit.2011.10.004 [DOI] [PubMed] [Google Scholar]
- 11. Marangoni RG, Rocha LF, Del Rio APT, et al. Systemic sclerosis sine scleroderma: distinct features in a large Brazilian cohort. Rheumatology (Oxford) 2013;52:1520–4. 10.1093/rheumatology/ket163 [DOI] [PubMed] [Google Scholar]
- 12. Diab S, Dostrovsky N, Hudson M, et al. Systemic sclerosis sine scleroderma: a multicenter study of 1417 subjects. J Rheumatol 2014;41:2179–85. 10.3899/jrheum.140236 [DOI] [PubMed] [Google Scholar]
- 13. Tolosa-Vilella C, Morera-Morales ML, Simeón-Aznar CP, et al. Digital ulcers and cutaneous subsets of systemic sclerosis: clinical, immunological, nailfold capillaroscopy, and survival differences in the spanish RESCLE registry. Semin Arthritis Rheum 2016;46:200–8. 10.1016/j.semarthrit.2016.04.007 [DOI] [PubMed] [Google Scholar]
- 14. De Almeida Chaves S, Porel T, Mounié M, et al. Sine scleroderma, limited cutaneous, and diffused cutaneous systemic sclerosis survival and predictors of mortality. Arthritis Res Ther 2021;23:295. 10.1186/s13075-021-02672-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferri C, Giuggioli D, Guiducci S, et al. Systemic sclerosis progression investigation (spring) Italian registry: demographic and clinico-serological features of the scleroderma spectrum. Clin Exp Rheumatol 2020;38 Suppl 125:40–7. [PubMed] [Google Scholar]
- 16. De Angelis R, Giuggioli D, Bajocchi G, et al. Sex-Related differences in systemic sclerosis: a multicenter cross-sectional study from the National Registry of the Italian Society for rheumatology. J Rheumatol 2022;49:176–85. 10.3899/jrheum.210794 [DOI] [PubMed] [Google Scholar]
- 17. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of rheumatology/european League against rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. 10.1002/art.38098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith V, Herrick AL, Ingegnoli F, et al. EULAR study group on microcirculation in rheumatic diseases and the scleroderma clinical trials consortium group on capillaroscopy standardisation of nailfold capillaroscopy for the assessment of patients with raynaud’s phenomenon and systemic sclerosis. Autoimmun Rev 2020;19:102458. 10.1016/j.autrev.2020.102458 [DOI] [PubMed] [Google Scholar]
- 19. Weatherald J, Montani D, Jevnikar M, et al. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev 2019;28:190023. 10.1183/16000617.0023-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American rheumatism association diagnostic and therapeutic criteria Committee. Arthritis Rheum 1980;23:581–90. 10.1002/art.1780230510 [DOI] [PubMed] [Google Scholar]
- 21. Ferri C, Arcangeletti M-C, Caselli E, et al. Insights into the knowledge of complex diseases: environmental infectious/toxic agents as potential etiopathogenetic factors of systemic sclerosis. J Autoimmun 2021;124:102727. 10.1016/j.jaut.2021.102727 [DOI] [PubMed] [Google Scholar]
- 22. Ferri C, De Angelis R, Giuggioli D, et al. Geographical heterogeneity of clinical and serological phenotypes of systemic sclerosis observed at tertiary referral centres. the experience of the italian SIR-SPRING registry and review of the world literature. Autoimmun Rev 2022;21:103159. 10.1016/j.autrev.2022.103159 Available: 10.1016/j.autrev.2022.103159 [DOI] [PubMed] [Google Scholar]
- 23. Allanore Y, Distler O, Matucci-Cerinic M, et al. Review: defining a unified vascular phenotype in systemic sclerosis. Arthritis Rheumatol 2018;70:162–70. 10.1002/art.40377 [DOI] [PubMed] [Google Scholar]
- 24. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum 2013;65:1953–62. 10.1002/art.37988 [DOI] [PubMed] [Google Scholar]
- 25. Hughes M, Heal C, Henes J, et al. Digital pitting scars are associated with a severe disease course and death in systemic sclerosis: a study from the EUSTAR cohort. Rheumatology (Oxford) 2022;61:1141–7. 10.1093/rheumatology/keab510 [DOI] [PubMed] [Google Scholar]
- 26. Cappelli S, Bellando Randone S, Camiciottoli G, et al. Interstitial lung disease in systemic sclerosis: where do we stand? Eur Respir Rev 2015;24:411–9. 10.1183/16000617.00002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frantz C, Huscher D, Avouac J, et al. Outcomes of limited cutaneous systemic sclerosis patients: results on more than 12,000 patients from the EUSTAR database. Autoimmun Rev 2020;19:102452. 10.1016/j.autrev.2019.102452 [DOI] [PubMed] [Google Scholar]
- 28. Simeón-Aznar CP, Tolosa-Vilella C, Gabarró-Juliá L, et al. Systemic sclerosis sine scleroderma and limited cutaneous systemic sclerosis: similarities and differences. Clin Exp Rheumatol 2014;32:S33–40. [PubMed] [Google Scholar]
- 29. Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther 2013;15:215. 10.1186/ar4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nihtyanova SI, Denton CP. Pathogenesis of systemic sclerosis associated interstitial lung disease. J Scleroderma Relat Disord 2020;5:6–16. 10.1177/2397198320903867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tabib T, Huang M, Morse N, et al. Myofibroblast transcriptome indicates sfrp2hi fibroblast progenitors in systemic sclerosis skin. Nat Commun 2021;12:4384. 10.1038/s41467-021-24607-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Senécal J-L, Hoa S, Yang R, et al. Pathogenic roles of autoantibodies in systemic sclerosis: current understandings in pathogenesis. J Scleroderma Relat Disord 2020;5:103–29. 10.1177/2397198319870667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinchcliff M, Mahoney JM. Towards a new classification of systemic sclerosis. Nat Rev Rheumatol 2019;15:456–7. 10.1038/s41584-019-0257-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.