

SUMMARY
Neuronal activity induces topoisomerase IIβ (Top2B) to generate DNA double strand breaks (DSBs) within the promoters of neuronal early response genes (ERGs) and facilitate their transcription, yet the mechanisms that control Top2B-mediated DSB formation are unknown. Here we report that stimulus-dependent calcium influx through NMDA receptors activates the phosphatase, calcineurin, to dephosphorylate Top2B at residues S1509 and S1511, which stimulates its DNA cleavage activity and induces it to form DSBs. Exposing mice to a fear conditioning paradigm also triggers Top2B dephosphorylation at S1509 and S1511 in the hippocampus, indicating that calcineurin also regulates Top2B-mediated DSB formation following physiological neuronal activity. Furthermore, calcineurin-Top2B interactions following neuronal activity and sites that incur activity-induced DSBs are preferentially localized at the nuclear periphery in neurons. Together these results reveal how radial gene positioning and the compartmentalization of activity-dependent signaling govern the position and timing of activityinduced DSBs and regulate gene activity patterns in neurons.
eTOC Blurb
Here we report that neuronal stimulation triggers the phosphatase, calcineurin, to dephosphorylate the topoisomerase, Top2B, and induce it to form DNA breaks within the promoters of neuronal early response genes, which in turn, promotes their rapid transcription. We further show that these events are compartmentalized at the nuclear periphery.
Graphical Abstract
INTRODUCTION
Neuronal activity triggers the rapid transcription of early response genes (ERGs) and late response genes (LRGs), and the products of activity-dependent gene transcription mediate lasting changes to neuronal morphology and synapse organization (West and Greenberg, 2011; Yap and Greenberg, 2018). Furthermore, disruptions in neuronal activity-dependent transcription programs are thought to underlie the development of various neurological disorders, including major depressive disorder, schizophrenia, addiction, intellectual disability, and autism spectrum disorders (Ebert and Greenberg, 2013; Nestler et al., 2016). A proper understanding of how neuronal activity-dependent transcription is orchestrated is therefore significant.
Mechanisms that govern the expression of ERGs have been studied in great detail, and many of the signaling events, transcription factors, epigenetic modifications, and chromatin architectural changes that facilitate activity-dependent transcription have been described (Madabhushi and Kim, 2018). Neuronal activity-dependent ERG transcription is induced by calcium influx through ligand-gated ion channels, particularly, N-methyl-D-aspartate (NMDA) receptors (NMDAR) and L-type voltage-gated calcium channels (LVGCC) (Bading, 2013; Bading et al., 1993; Morgan and Curran, 1986; Murphy et al., 1991). Calcium entry stimulates a cascade of signaling events that are transduced by mitogen-associated protein kinases (MAPKs), calcium/calmodulin-dependent protein kinases (CaMKs), and calcineurin phosphatase-mediated pathways (Deisseroth et al., 2003; Greer and Greenberg, 2008). The concerted actions of these signaling events are thought to mediate further posttranslational modifications on transcription factors, epigenetic changes, and favorable enhancer-promoter interactions that promote RNAPII pause-release and transcription elongation at ERGs (Madabhushi and Kim, 2018). However, the precise molecular mechanisms that govern these transitions are not fully understood.
A recent surprising finding in this regard has been that diverse paradigms of neuronal activity, including LTP and fear conditioning, cause the formation of DNA double strand breaks (DSBs) in neurons (Crowe et al., 2006; Madabhushi et al., 2015; Stott et al., 2021; Suberbielle et al., 2013). Activity-induced DSBs are generated by the type II topoisomerase, topoisomerase IIβ (Top2B), and are enriched within the promoters of a subset of neuronal ERGs, including Fos, Npas4, Egr1, and Nr4a1 (Madabhushi et al., 2015; Stott et al., 2021). Knockdown of Top2B attenuates neuronal activity-dependent DSB formation and ERG transcription, whereas DSB formation within ERG promoters is sufficient to induce their transcription (Madabhushi et al., 2015). While these results describe a new mechanism that facilitates rapid ERG transcription, exactly how neuronal activity causes Top2B to form DSBs at specific genomic loci is unknown.
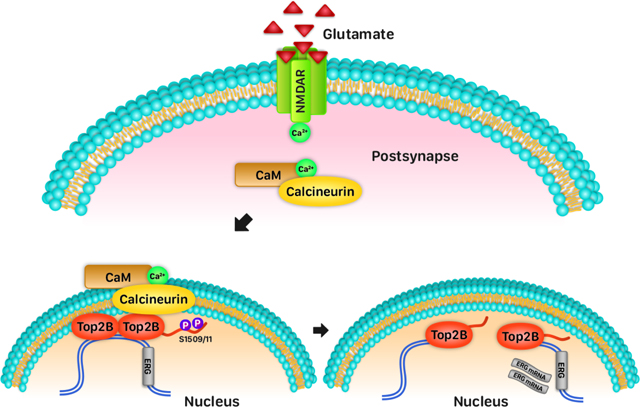
Here we report that neuronal activity-dependent dephosphorylation of Top2B by the phosphatase, calcineurin, stimulates the DNA cleavage activity of Top2B and induces it to form DNA DSBs in primary cortical neurons and in the hippocampus of mice. We show that interactions between calcineurin and Top2B are compartmentalized within the nuclear periphery and that genes that incur neuronal activity-induced DSBs also preferentially cluster at the nuclear periphery. These results provide new insights into how the spatial organization of signaling mechanisms and chromatin regulate gene activity patterns in neurons.
RESULTS
Neuronal activity-dependent DSB formation requires NMDA receptor activity
In an effort to decipher the molecular events that link neuronal stimulation to the formation of activity-induced DSBs, we began by incubating cultured primary cortical neurons with established inhibitors of various ion channels and calcium sources and assessed their effects on ERG transcription and the levels of the DSB marker, γH2AX, as a function of neuronal stimulation. We used DL-2-Amino-5-phosphonopentanoic acid to block NMDARs (NMDARi), tetrodotoxin to inhibit voltage-gated Na+ channels (VGSCi), nifedipine to block LVGCCsV (LVGCCi), 2-aminoethoxydiphenylborate to antagonize intracellular inositol 1,4,5-inositol triphosphate receptor (IP3R) (IP3Ri) and inhibit store-operated Ca2+ release from the endoplasmic reticulum, and dantrolene sodium to inhibit ryanodine receptors (RyR) and block Ca2+ release from the sarcoplasmic reticulum (RyRi).
Briefly incubating neurons with either NMDA or potassium chloride (KCl) induced rapid ERG transcription (Figure S1) and elevated levels of the DSB marker, γH2AX, as reported previously (Figures 1A and 1B) (Madabhushi et al., 2015). KCl and NMDA activate distinct routes of calcium influx and signaling to affect gene transcription (Hagenston and Bading, 2011), and these differences also manifested in stimulus-specific requirements for DSB formation. For instance, calcium entry through LVGCCs is essential for Fos transcription in response to neuronal bath stimulation with KCl, but not glutamate (Bading et al., 1997; Ginty and Greenberg, 1993; Hardingham et al., 2002). Accordingly, pre-incubating neurons with either LVGCCi or IP3Ri reduced γH2AX elevations following KCl treatment, but they affected neither ERG induction nor γH2AX increases in NMDA-treated neurons (Figures 1A, 1B, and S1). Inhibitors of other ion channels, such as VGSCi and RyRi also did not affect NMDA-dependent γH2AX formation (Figure 1A). Basal γH2AX levels in neurons were reduced by LVGCCi, IP3Ri, and RyRi, but not by either VGSCi or NMDARi, indicating that basal γH2AX formation is caused by mechanisms distinct from those that trigger activity-induced DSBs. Importantly, pre-incubating neurons with NMDARi blocked both NMDA and KCl-induced γH2AX elevations (Figures 1A and 1B), suggesting that NMDAR activity is essential for the formation of activity-induced DSBs.
Figure 1. NMDAR activity is required for DSB formation following neuronal stimulation.

(A and B) Cultured neuronswere pre-incubated with either vehicle or the indicated inhibitors. Neurons were then stimulated with either NMDA (A) or KCl (B) and the levels of γH2AX and total H2AX were then assessed by western blotting. Red and blue bars represent normalized γH2AX levels (mean ± s.e.m.) in stimulated and unstimulated neurons (controls) for each treatment expressed as fold change over vehicle control.
(C) γH2AX levels within the gene bodies of Fos and Npas4 were assessed by ChIP-qPCR in neurons that pre-incubated with NMDARi and treated with NMDA.
(D) Neurons were pre-incubated with NMDARi and then stimulated with NMDA as in (A), and the levels of Top2Bccs were assessed. Neurons incubated with etoposide (ETP) were used as a positive control.
See also Figure S1.
Neuronal activity-induced DSBs cause γH2AX to accumulate within the gene bodies of ERGs (Madabhushi et al., 2015), and chromatin immunoprecipitation (ChIP-qPCR) experiments revelated that treating neurons with NMDARi prevents the activity-dependent accrual of γH2AX within the gene bodies of Fos and Npas4 (Figure 1C), indicating that NMDAR activity is specifically important for DSB formation at ERGs. Activity-induced DSBs are generated by Top2B in which Top2B becomes covalently attached to the DNA via a phospho-tyrosyl bond and the accumulation of such covalent cleavage complexes (Top2Bccs) correlates with the propensity for DSB formation. As reported previously (Madabhushi et al., 2015), neuronal stimulation markedly elevated the levels of Top2Bccs in neurons; however, this increase was attenuated in the presence of NMDARi (Figure 1D). These results indicate that NMDAR activity predominantly regulates Top2B-mediated DSB formation in stimulated neurons.
Calcineurin (CaN) signaling mediates the formation of neuronal activity-induced DSBs
To understand how NMDAR signaling triggers activity-induced DSBs, we next incubated cultured primary cortical neurons with inhibitors of various kinases and phosphatases that transduce signals from NMDARs at the cell surface to the nucleus, and mediate excitation-transcription coupling (Deisseroth et al., 2003; Hagenston and Bading, 2011). Incubation of neurons with inhibitors of Ca2+/calmodulin-dependent protein kinase II (CaMK2i; KN-62), extracellular signal-regulated kinase (ERKi; PD98059), and protein kinase A (PKAi; KT5720) all caused a reduction in NMDA-mediated ERG transcriptional induction (Figure S2A) without affecting the formation of γH2AX following NMDA treatment (Figure 2A). Thus, activity-induced DSBs are not merely a correlate of neuronal activity-dependent transcription, rather they are induced by specific signaling events. In contrast to kinase inhibitors, pre-incubation with the PP1/PP2A phosphatase inhibitor, okadaic acid (PP1/PP2Ai), increased γH2AX levels both under basal conditions and following NMDA treatment (Figure 2A). On the other hand, pre-incubation of neurons with the selective CaN inhibitor, CN585 (CaNi) (Erdmann et al., 2010), specifically blocked the elevation in γH2AX levels following NMDA treatment, but did not alter basal γH2AX levels, suggesting that CaN promotes the formation of neuronal activity-induced DSBs (Figure 2A). Similar reductions in γH2AX levels were observed when neurons pre-incubated with a second CaN inhibitor, tacrolimus, were stimulated with NMDA (Figure S2B). CaNi pre-treatment also reduced the NMDA-dependent accumulation of γH2AX within the gene bodies of Npas4, indicating that calcineurin activity is essential for DSB formation at ERGs (Figure 2B). Previous reports indicate that CaN-mediated dephosphorylation of γCaMKII is crucial for the delivery of Ca2+/calmodulin to the nucleus for the purpose of activity-dependent gene transcription (Ma et al., 2014). However, CaMK2i failed to reduce γH2AX levels following neuronal stimulation (Figure 2A). Calcineurin-mediated dephosphorylation of the NFATc (nuclear factor of activated T cells) transcription factors causes NFATc to translocate from the cytosol to the nucleus and regulate transcription in various cell types (Crabtree and Olson, 2002; Graef et al., 1999). However, pre-incubation with the cell permeable and selective calcineurin-NFATc interaction inhibitor, INCA-6 (Roehrl et al., 2004) also failed to reduce γH2AX levels in NMDA-treated neurons (Figure S2C). Additionally, qRT-PCR assays revealed that whereas the NMDA-dependent induction of ERGs were substantially reduced in the presence of CaNi, INCA-6 treatment had no effect on ERG transcription (Figures 2C and S2D). These observations suggest that the effects of CaN signaling on activity-induced DSB formation and ERG transcription are independent of NFATc and γCaMKII in cortical neurons.
Figure 2. Calcineurin modulates Top2B and induces it to form DSBs in stimulated neurons.

(A) Cultured neurons were pre-incubated with either vehicle or the indicated inhibitors and then stimulated with NMDA, and the levels of γH2AX and total H2AX were assessed by western blotting.
(B – D) γH2AX levels within the gene body of Npas4 (B), expression of the indicated ERGs (C), and the levels of Top2Bccs (D) were assessed in neurons pre-incubated with CaNi and stimulated with NMDA using ChIP-qPCR (B), qRT-PCR (C) and slot-blotting (see STAR Methods) (D).
(E) Cultured neurons were stimulated with NMDA for 10 min and Top2B was immunoprecipitated, and Top2B and co-precipitated CaN were detected by western blotting.
(F) Supercoiled plasmids carrying sequences upstream of the Fos TSS were incubated with purified recombinant human Top2A and Top2B either in the presence or absence of purified recombinant calcineurin. The positions of supercoiled, relaxed and linear DNA are indicated. Graph shows densitometry analysis (mean ± s.e.m.) of the relaxed and linear DNA normalized to supercoiled bands and expressed as fold change relative to the corresponding levels in DNA alone.
See also Figures S2 and S3.
Calcineurin interacts with Top2B and stimulates its DNA cleavage activity
To further understand how calcineurin signaling affects the formation of neuronal activity-induced DSBs, we assessed Top2B activity in neurons stimulated in the presence of CaNi. Interestingly, whereas Top2Bccs were elevated in NMDA-treated neurons, their levels were substantially reduced in the presence of CaNi (Figure 2D). These results suggest that CaN stimulates Top2B-mediated DNA cleavage. Immunoprecipitation experiments revealed that Top2B physically interacts with CaN in a neuronal activity-dependent manner. Whereas only a weak interaction was detectable under basal conditions, either NMDA or KCl treatment significantly enriched the amount of CaN that co-precipitated with Top2B (Figure 2E and Figure S2E). These results raised the possibility that CaN could directly affect the DNA cleavage activity of Top2B. To test this idea, we incubated purified recombinant Top2B and the CaN-calmodulin (CaN/CaM) complex together with supercoiled plasmid DNA in vitro. As expected, incubation of Top2B with supercoiled plasmids increased the formation of relaxed DNA (Figure S2F). Increasing amounts of CaN further increased the amount of relaxed DNA, indicating that CaN stimulates DNA relaxation by Top2B (Figure S2F). Incubating CaN with Top2B in the absence of calmodulin did not affect Top2B activity, suggesting that the catalytic activity of CaN is essential for stimulating DNA relaxation by Top2B (Figure S2G). To determine whether CaN specifically affects the ability of Top2B to cleave DNA, we incubated recombinant Top2B-CaN reaction mixtures with supercoiled DNA in the presence of AMP-PNP. In these reactions, the presence of CaN markedly enhanced DNA cleavage by Top2B, as indicated by the increase in both linear and relaxed DNA forms (Figure 2F). Surprisingly, the effects of CaN on DNA cleavage was specific to Top2B and CaN had no effect on the DNA cleavage activity of the closely related topoisomerase II isoform, Top2A (Figure 2F). Together, these results indicate the existence of a novel physical and functional interaction between CaN and Top2B in which CaN binds and induces Top2B to form DSBs in an activity-dependent manner.
Calcineurin signaling triggers activity-induced DNA breaks at high frequency in neurons
Previous reports have demonstrated the formation of neuronal activity-induced DNA breaks by measuring levels of the surrogate DSB marker, γH2AX, by immunocytochemistry, western blotting, ChIP, and ChIP-seq (Madabhushi et al., 2015; Stott et al., 2021; Suberbielle et al., 2013), or using PCR-based methods (Madabhushi et al., 2015). Imaging γH2AX foci visualized activity-induced DSBs formation in single neurons in vivo but did not resolve where DSBs are formed (Suberbielle et al., 2013). On the other hand, while γH2AX ChIP-seq revealed that activityinduced DSBs are enriched at specific genomic loci (Madabhushi et al., 2015; Stott et al., 2021), these signals represent the average contribution from thousands of cells and do not reveal the prevalence of DSBs at ERGs within individual neurons.
To address these issues, we utilized a single-cell electrophoresis and DNA FISH-based assay (Comet-FISH) (Santos et al., 1997) to directly measure DSB formation at the Fos locus at single molecule resolution. In this assay, cells are embedded in agarose, lysed, and electrophoresed, and DSBs within a region of interest are detected using adjacent FISH probes that “split apart” during electrophoresis. For these assays, two non-overlapping BAC probes that border the murine Fos locus were selected and their adjacent disposition was verified using dual-color immuno-FISH (Figure 3A). To validate the Comet-FISH assay, cultured cortical neurons were incubated with the Top2 poison, etoposide (ETP), which generates DSBs within the promoters of ERGs, including Fos (Madabhushi et al., 2015; Wu et al., 2011). Whereas both Fos-adjacent probes were closely localized in vehicle controls, the two probes rapidly split apart in ETP-treated neurons. Splitting of the two probes was already evident after 10 min of ETP treatment when most cells showing minimal comet tails, with both the distance between the probes and the intensity of comet tails increasing sharply after 30 min of ETP treatment (Figure 3B). These results suggest that Top2B-mediated DSBs can be detected using Comet-FISH.
Figure 3. ERGs incur activity-induced DSBs in a majority of stimulated neurons.

(A) (left) UCSC genome browser view depicting the hybridizing positions of BAC probes selected to assess the formation of DSBs at the Fos locus. (Right) Cultured cortical neurons were fixed and FISH was performed using fluorescently labeled BAC probes in (A) that hybridize with regions upstream (green: RP24–368B17) and downstream (red: RP24–178I11) of Fos. A representative image shown here verifies that the probes localize adjacent to each other.
(B) Neurons were treated with ETP (10 μM) for the indicated times and assessed by Comet-FISH (see STAR Methods). Images show maximum intensity projection of z-stacks from representative cells. Splitting of the two probes indicates the formation of DSBs within intervening regions. Arrows indicate co-localized Fos-adjacent probes in control and their splitting following ETP treatment.
(C and D) Neurons were pre-incubated with either vehicle or CaNi and stimulated with NMDA for 10 min, and Comet-FISH was performed using the probes indicated in (A) and (B). (C) Maximum intensity projections of z-stacks from representative images. (D) Quantification of the percentage of loci where upstream and downstream probes were split apart.
We next incubated cultured cortical neurons with NMDA and used Comet-FISH to assess activity-induced DSB formation at the Fos locus in single cells. Consistent with previous reports that neuronal stimulation triggers DSBs at only a few genomic loci (Madabhushi et al., 2015), NMDA-treated neurons did not show significant comet tails (Figure 3C). However, Fos-adjacent FISH probes were split apart in only 17.6% of neurons under basal conditions, compared to 58.6 % of NMDA-treated neurons (Figure 3D). These results indicate that neuronal activity triggers locus-specific DSBs in a majority of neurons. Furthermore, pre-incubating neurons with CaNi largely suppressed activity-dependent DSB at the Fos locus, with only 19.8 % neurons displaying “split apart” probes (Figures 3C and 3D). These results indicate that neuronal activity-dependent CaN signaling induces Top2B to generate DSBs at specific genomic loci.
Top2B phosphorylation affects its DNA cleavage activity
Based on the specific ability of CaN to stimulate the DNA cleavage activity of Top2B, but not Top2A (Figure 2F), we wondered whether this specificity could arise from the divergent C-terminal domains (CTDs) of Top2A and Top2B, which are also sites of numerous post-translational modifications, including phosphorylation (Austin et al., 2018; Madabhushi, 2018). While global phosphoproteomic analyses have reported 49 phosphoserine sites within the CTD of Top2B (Table S1), a functional characterization of phosphorylation at these sites has not been performed. Analysis of phosphorylated sites within the CTD of Top2B using Expasy and Scansite indicated that a majority of serine motifs are likely phosphorylated by casein kinase II (CK2) (Table S1). Additionally, protein kinase C (PKC), phosphoinositide dependent protein kinase 1 (PDPK1), and ERK1 also emerged as potential kinases (Table S1).
To test how Top2B activity is affected by phosphorylation, we first performed in vitro kinase assays using recombinant CK2 and Top2B immunoprecipitated from neurons. Incorporation of radiolabeled 32P on Top2B was readily detected upon incubation with CK2 (Figure S3A), indicating that CK2 can phosphorylate Top2B. Incubation with CK2 also stimulated, whereas incubation with the non-specific calf-alkaline phosphatase (CIP) reduced, DNA relaxation by Top2B, suggesting that CK2-mediated phosphorylation enhances Top2B activity (Figure S3B). Immunoprecipitated Top2B from neurons showed marked reactivity towards antibodies that detect phospho-CK2 substrates (pS/pTDXE) under basal conditions (von Morgen et al., 2017), and this immunoreactivity was reduced in Top2B precipitated from NMDA-treated neurons (Figure S3C). These results suggest that CK2 constitutively phosphorylates Top2B in neurons and that Top2B phosphorylation is reduced following neuronal stimulation. Moreover, whereas incubating Top2B with either CK2 or CaN alone stimulated the DNA cleavage activity of Top2B, reactions containing both CaN and CK2 showed a further 32–33 % increase in the intensity of linear and relaxed bands compared to those incubated with CK2 alone (Figure S3B). The effects of CaN and CK2 on Top2B were suppressed by the inclusion of CIP (Figure S3B). These results suggest that CaN acts upon phosphorylated Top2B to induce DNA cleavage.
To determine how CaN affects Top2B, we performed targeted mass spectrometry (parallel reaction monitoring (PRM) LC-MS/MS) analysis of Top2B as a function of neuronal stimulation with NMDA, analyzing phosphopeptides containing phosphoserines reported in previous studies (Table S1 and Figure 4A). These studies revealed dynamic changes in the phosphorylation of Top2B at six distinct serine residues, S1453, S1509, S1511, S1537, S1539, and S1568, all located within the CTD of Top2B (Figure 4B). Phosphorylation of Top2B at another site, S1563, could be not distinguished from that at T1562. Comparisons between NMDA-stimulated and unstimulated neurons revealed that Top2B is dephosphorylated at a majority of these residues (Figure 4B). However, there were also some interesting deviations. For instance, whereas NMDA treatment caused a reduction in the abundance of peptides doubly phosphorylated at both S1509 and S1511, the abundance of peptides singly phosphorylated at S1511 was elevated (Figure 4B). Similarly, an increase in the abundance of peptides doubly phosphorylated at S1537 and S1539 was accompanied by a reduction in the abundance of peptides singly phosphorylated at S1539 (Figure 4B). Sequence alignments of Top2A and Top2B indicated that S1509 and S1511 are not conserved between the two enzymes (Table S2). However, S1537 in Top2B is conserved and corresponds to S1469 in Top2A (Table S2). Immunoprecipitated Top2B from NMDA-treated neurons showed greater immunoreactivity towards a phospho-Top2A (S1469) antibody compared to Top2B precipitated from controls, further validating our PRM LC-MS/MS results (Figure S3D). Whereas the CTD of Top2B is not essential for its catalytic activity, it could have a regulatory effect on Top2B (Gilroy and Austin, 2011; Meczes et al., 2008). To assess whether activity-dependent changes in Top2B phosphorylation affect the DNA cleavage activity of Top2B, we mutated each of the five differentially phosphorylated serine residues within Top2B to either alanine (Top2BS1453A/S1509A/S1511A/S15339A/S1568A, hereafter referred to as S5A for convenience) to generate phosphorylation mutants or to aspartic acid (hereafter referred to as S5D) to generate phosphomimetic mutants. Heterologous HEK293 cells were then transfected with HA-tagged wildtype Top2B, Top2BS5A, and Top2BS5D constructs, and the levels of Top2Bccs were examined. Compared to wildtype Top2B, Top2BS5A mutants displayed an increased propensity to form Top2Bccs, while Top2BS5D mutants showed reduced Top2Bccs, suggesting that dephosphorylation at these residues favors DSB formation (Figure 4C).
Figure 4. Calcineurin dephosphorylates Top2B at S1509 and S1511 in stimulated neurons.

(A) Domain arrangement of Top2B.
(B) Neurons were pre-incubated with CaNi and then stimulated with NMDA for 10 min. Neurons were collected to perform peptide targeted mass spectrometry (PRM LC-MS/MS). The normalized intensity of phosphoserines that showed changes after stimulation, expressed as log2 fold-change over control, are shown.
(C) Differentially phosphorylated serine residues identified in (B) were either mutated to alanine (S5A) or to aspartic acid (S5D) to generate phosphorylation-deficient and phosphomimetic mutants, respectively. HEK293 cells were transfected with HA-tagged WT, S5A, and S5D Top2B and the levels of Top2Bccs were assessed using an anti-HA antibody.
(D) Posttranslational modifications (PTMs) were detected by mass spectrometry on samples of Top2B immunoprecipitated from neurons pre-incubated with either vehicle or CaNi and then stimulated with either NMDA or KCl for 10 min as indicated.
(E) Top2Bccs in HEK293 cells expressing HA-tagged WT and Top2B S1509A/S1511A were assessed as in (C).
(F) Neurons were first pre-incubated with either vehicle or CaNi for 20 min and then with ETP (10 μM) for an additional 20 min, and thereafter stimulated with NMDA. The expression of the indicated ERGs was then assessed using qRT-PCR.
(G) Ten-week-old C57BL/6J mice were subjected to a contextual fear conditioning paradigm and sacrificed 30 min post-training (see STAR Methods). Top2B was immunoprecipitated from the hippocampus (4 animals per sample) and PTMs were analyzed by mass spectrometry as in (D).
See also Figure S4.
To test whether CaN dephosphorylates Top2B in activity-dependent manner, we performed targeted mass spectrometry and assessed the phosphorylation of Top2B following neuronal stimulation in the presence of CaNi. Incubation with CaNi only reduced the abundance of peptides dually phosphorylated at S1509 and S1511, but not other phosphopeptides, suggesting that Top2B could be dephosphorylated by multiple phosphatases (Figure 4B). However, CaN could also target other residues not assessed in our PRM mass spectrometry experiments. To address this possibility and a gain a deeper insight into the effects of CaN on Top2B, we immunoprecipitated Top2B from cultured primary neurons that were treated with either NMDA, KCl, or pre-incubated with CaNi prior to stimulation with NMDA. Posttranslational modifications on Top2B precipitated under these conditions were then assessed using LC-MS/MS. In agreement with previous reports (Table S1), Top2B phosphorylation was specifically enriched within its CTD (Figure 4D). Furthermore, in agreement with our PRM LC-MS/MS studies (Figure 4B), the abundance of peptides dually phosphorylated at S1509 and likely S1511, were markedly diminished following either NMDA or KCl treatment compared to untreated controls, suggesting that these residues are dephosphorylated following neuronal stimulation (Figure 4D). Importantly, activity-dependent dephosphorylation at these residues was largely inhibited in the presence of CaNi, indicating that S1509 and S1511 are dephosphorylated by CaN (Figure 4D). The majority of peptides containing S1509 and S1511 are doubly phosphorylated under basal conditions (66.7%), while peptides containing residues singly phosphorylated at S1511 comprise 15.9% and non-phosphorylated at either site comprise 14.8% of the total peptides (Figure 4D). Additionally, in agreement with PRM LC-MS/MS experiments, the abundance of peptides singly phosphorylated at S1511 increased from 15.9% to 30% and 24% after treatment with NMDA and KCl, respectively. We reason from these observations that the elevation in peptides containing singly phosphorylated S1511 in the mass spectrometry experiments (Figures 4B and 4D) is an intermediate that likely arises from activity-dependent dephosphorylation of Top2B at S1509.
To test the role of phosphorylation on S1509 and S1511, HEK293 cells were transfected with either HA-Top2B or HA-Top2BS1509/S1511A constructs and their ability to Top2Bccs was assessed. Compared to HA-Top2B, HA-Top2BS1509/S1511A mutants showed increased propensity to form Top2Bccs (Figure 4E). To assess the effects of Top2B phosphorylation mutants on activity-induced DSB formation, cultured primary neurons from conditional Top2bf/f mice were transduced with adeno-associated virus (AAV) carrying the Cre recombinase to ablate endogenous Top2b. Thereafter, the neurons were transfected with either HA-Top2B or HA-Top2BS1509A/S1511A (Figure S4A). Both HA-Top2B and HA-Top2BS1509A/S1511A showed comparable expression across all samples (Figure S4B). Neurons transfected with HA-Top2BS1509A/S1511A showed increased γH2AX immunofluorescence compared to those transfected with HA-Top2B even in the absence of neuronal stimulation, indicating that Top2BS1509A/S1511A mutants increase the propensity of Top2B to form DSBs (Figures S4A and S4C). Neuronal stimulation with NMDA caused an increase in γH2AX signals in neurons transfected with HA-Top2B but did not cause a further increase in γH2AX levels in neurons transfected with HA-Top2B1509A/1511A, indicating that Top2B phosphorylation mutants are refractory activity-dependent modulation (Figures S4A and S4C). Taken together, these results suggest that the Ca2+/CaM phosphatase, CaN, associates with Top2B in response to neuronal activity and dephosphorylates it. CaN-mediated dephosphorylation, in turn, stimulates the DNA cleavage activity of Top2B and promotes the formation of activity-induced DSBs in neurons.
To determine whether the effects of CaN on Top2B-mediated DSB formation is significant for ERG transcription, we pre-incubated neurons with both CaNi and the Top2B poison, etoposide (ETP) to induce DSBs without calcineurin activity and analyzed their effects on ERG transcription following stimulation with NMDA. As described above (Figure 2C), ERG transcriptional induction was attenuated in the presence of CaNi alone (Figure 4F). However, simultaneously treating neurons with both CaNi and ETP was able to restore ERG transcription (Figure 4F). These results suggest that CaN-mediated stimulation of DNA cleavage by Top2B is crucial for the transcriptional induction of ERGs.
Accumulating evidence suggests that DSBs are also formed in relevant brain areas in vivo following physiological neuronal stimulation (Li et al., 2019; Madabhushi et al., 2015; Navabpour et al., 2020; Stott et al., 2021; Suberbielle et al., 2013). For instance, exposing mice to a contextual fear conditioning paradigm induces Top2B-mediated DSBs at specific genomic loci in the prefrontal cortex and the hippocampus, including within ERG promoters (Li et al., 2019; Madabhushi et al., 2015; Stott et al., 2021). To test how Top2B phosphorylation relates to activity-induced DSB formation in vivo, 12-week-old C57BL/6J mice were subjected to a training paradigm for contextual fear conditioning, following which hippocampal lysates were prepared and Top2B was immunoprecipitated. Posttranslational modifications on Top2B precipitated from naïve and trained animals were then assessed using LC-MS/MS as before. Remarkably, and similar to cultured primary neurons stimulated with either NMDA or KCl, the abundance of peptides dually phosphorylated at S1509 and S1511 were rapidly reduced in mice after fear conditioning (Figure 4G). These results suggest that DSB formation following physiological neuronal stimulation in vivo also involves the dephosphorylation of Top2B at S1509 and S1511.
Calcineurin-Top2B interactions are enriched at the nuclear periphery
CaN is a heterodimer composed of a catalytic subunit (CaN A) and a regulatory subunit (CaN B). Whereas CaN is predominantly localized to the cytosol and pre- and post-synaptic terminals in neurons, previous reports in other cell types have described the stimulus-dependent translocation of calcineurin to the nucleus. Nuclear import of calcineurin is thought to be mediated through a nuclear localization signal (NLS) contained within CaN A (Hallhuber et al., 2006). Immunocytochemistry experiments using pan-CaN A antibodies detected abundant CaN A within the cytosol and neuronal processes, but CaN A was excluded from the nucleus (Figures 5A and 5B). Interestingly, NMDA treatment triggered a rapid mobilization of CaN to the nuclear periphery, where CaN clusters could be detected within 5–10 min post-stimulation (Figures 5A and 5B). CaN enrichment at the nuclear periphery persisted until 20 min, after which CaN staining again increased outside the nucleus between 40–60 min after the initial stimulus (Figure 5A). The localization of CaN at the periphery also coincided with the appearance of γH2AX foci (Figure 5A). Thus, CaN is largely localized at the nuclear periphery at times corresponding to the formation of activity-induced DSBs and its observed physical interaction with Top2B.
Figure 5. Calcineurin rapidly localizes to the nuclear periphery following neuronal stimulation.

(A) Cultured cortical neurons were stimulated with NMDA and fixed at various times post-stimulation and the indicated proteins were visualized by immunocytochemistry.
(B) Intensity profiles of calcineurin, lamin A/C, and DAPI within rectangular ROIs (15 × 0.8 μM) crossing through the center of the cell (yellow rectangles) were plotted. The peak of lamin A/C staining was used to mark the nuclear periphery (represented by “0” on the x-axis). Positive values on the x-axis indicate regions outside the nucleus, whereas negative values denote the nuclear interior. y-aixs represents the intensity of staining.
(C) Lysates from cultured primary neurons, NIH3T3, and HEK293 cells were fractionated by differential centrifugation to obtain soluble (Sol) and detergent and salt resistant (DSR) fractions. DSR fractions were further digested with Dnase I to remove genomic DNA (Dnase). The prevalence of the indicated proteins in each fraction was analyzed by western blotting.
(D) Neurons were stimulated with NMDA and fractionated at the indicated times post-stimulation. The presence of the indicated proteins in the DSR fraction was then assessed by western blotting.
(E – G) Neurons were stimulated with NMDA and Top2B-CaN interactions in single cells was analyzed using a PLA assay. (E) Representative images of Top2B-CaN interactions (green foci) as a function of time post-stimulation. (F) The average number of interactions per cell as function of time post-stimulation, (G) Lamin A/C staining was used to mark the nuclear periphery and the number of interactions at as a function of distance from lamin A/C is shown Distances were measured as shown in Figure S5C.
See also Figure S5.
CaN interacts with the cargo protein, importin β, (Hallhuber et al., 2006; Hallhuber and Ritter, 2007) to facilitate its nuclear entry, and importin β internalization involves its interaction with the nuclear pore complex (Chahine and Pierce, 2009; Lowe et al., 2015). To further determine how neuronal activity affects CaN localization, we performed subcellular fraction by differential centrifugation in the presence of detergent and high salt (DSR). Whereas both CaN and Top2B were abundant in the soluble fraction, a consistent signal for both proteins could also be detected in the DSR fraction that is enriched for lamin A/C and corresponds to the classical “nuclear matrix” fraction (Figure 5C). The enrichment of Top2B within the DSR fraction was sensitive to DNase, suggesting that Top2B in this fraction is largely DNA-bound (Figure 5C). Neither CaN nor Top2B were detected within the DSR fraction in NIH3T3 and HEK293 cells, and unlike in neurons, CaN staining was visible throughout the nucleus in NIH3T3 and HEK293 cells (Figure 5C and Figure S5A). Moreover, ectopic mobilization of calcium and stimulation of CaN activity using the calcium ionophore, ionomycin, was sufficient to induce DSBs and elevate γH2AX in neurons, but not HEK293 cells (Figure S5B). These results suggest that the CaN-Top2B pathway specifically regulates DSB formation in neurons.
Treatment of neurons with NMDA caused an elevation of both CaN and Top2B levels within the DSR fraction (Figure 5D). Both proteins were enriched by 10 min, and CaN enrichment persisted until 20 min, and diminished by 40 min after the initial stimulus, whereas Top2B levels remained elevated throughout this time (Figure 5D). The enrichment kinetics of CaN within the DSR fractions were consistent with the time-course of its localization patterns in immunocytochemistry experiments (Figure 5A). These results suggested Top2B-CaN interactions could be confined to the nuclear periphery. To test this idea, we utilized a proximity-ligation assay (PLA) that utilizes distinct secondary antibodies carrying attached oligonucleotide probes (Soderberg et al., 2006).
When these secondary antibodies recognize primary antibodies that are bound in close proximity, the attached oligonucleotide probes guide the formation of circular DNA templates for rolling circle replication and interaction sites are detected through the use of complimentary fluorescent-labeled oligonucleotide probes that bind the amplified DNA (Soderberg et al., 2006). Under basal conditions, we could not detect PLA fluorescent signals within neurons, indicating minimal Top2B-CaN interactions (Figure 5E). NMDA treatment caused a significant increase in the number of PLA fluorescent foci. The highest number of foci were detected by 10 min following stimulation, suggesting that a majority of CaN-Top2B interactions occur during this period (Figure 5E). Thereafter the number of observed foci decreased gradually and were undetectable by 60 min (Figures 5E and 5F). Visual inspection of PLA foci indicated that CaN-Top2B interactions are concentrated at the nuclear periphery (Figure 5E and Figure S5C). A majority of foci were located within 0.5μm from the nuclear membrane, as marked by staining with lamin A/C (Figures 5E, 5G, and S5C). Together these results indicate that neuronal activity-dependent interactions between Top2B and CaN are largely confined to nuclear periphery in neurons.
Sites that incur activity-induced DSBs preferentially localize to the nuclear periphery
An important characteristic of higher-order spatial chromatin organization is that chromosomes adopt non-random positions along the radial axis of the nucleus and are arranged in subnuclear domains, called chromosome territories (Cremer and Cremer, 2010; Dekker and Misteli, 2015). Even within chromosome territories, genomic loci are thought to occupy probabilistic preferential positions along the radial axis. To understand how the preferential interaction of CaN and Top2B at the nuclear periphery relates to the formation of neuronal activity-induced DSBs, bacterial artificial chromosomes (BAC) clones containing various murine loci were labeled with fluorescent nucleotides using nick translation and the labeled probes were then used in immuno-FISH experiments together with antibodies against lamin A/C. DNA-FISH signals detected at radial distances < 500 nm from lamin A/C were classified as being localized to the nuclear periphery (Figure S6A). Analysis of FISH signals from probes containing the ERGs, Fos, Npas4, and FosB, revealed surprisingly that these loci are all preferentially localized to the nuclear periphery (Figures 6A – 6D). Nearly 82% of neurons had at least one Fos locus within 0.5 μm from the nuclear envelope, marked by lamin A/C staining (Figure 6E). Additionally, Npas4 (62%) and FosB (72%) were also found to reside within the nuclear periphery in a majority of neurons (Figure 6E), and similar results were observed with probes for Malat1, a long non-coding RNA that also incurs activity-induced DSBs within its promoters (Figures 6C – 6E). Among the probed sites that incur activity-induced DSBs, only one locus, Actb, did not preferentially localize to the nuclear periphery (Figures 6B, 6D, and 6E). The preferential localization of ERGs at the nuclear periphery prompted to us to test whether these loci are clustered together within the nucleus. However, dual-color FISH experiments failed to reveal clustering of either Fos and Npas4, or Fos and FosB (Figures 6F and 6G). These results suggest that multiple ERGs are not clustered in transcription factories or similar structures within neurons.
Figure 6. Sites that incur activity-induced DSBs preferentially localize to the nuclear periphery in neurons.

DNA immuno-FISH was performed using fluorescently labeled BAC probes that hybridize with genomic regions containing the indicated genes and antibodies against lamin A/C. Probes were used to assess visualize either a single locus (A) or two distinct loci simultaneously as indicated (B, C, D, G, H, and I) Images show representative optical slices from z-stacks. (D – F) (D) Distance of the indicated loci to the nuclear rim was measured as in Figure S6A). (E) Quantification of the distance of each probe from the nuclear periphery as shown in (D). (F) Loci that were located at a distance < 0.5 μm from lamin A/C were defined as being localized to the nuclear periphery and the percentage of cells with at least one copy of the probed region located at the nuclear periphery were plotted. (J) The shortest distance to the periphery (either XY or Z) for Fos and Gm49698, which are both located on chromosome 12, were compared on a single chromosome level and plotted (linked by lines).
See also Figure S6.
As mentioned above, we performed dual-color immuno-FISH using two non-overlapping but adjacent BAC probes at the Fos locus and found that these probes to be adjacently localized (Figure 3A). Similar results were obtained using probes against Malat1 and Npas4, two closely positioned genes on chromosome 19 (Figure 6C). These observations further validate our FISH results. Finally, to gain a deeper insight into the preferential radial positioning of ERGs within the nucleus, we performed dual-color immuno-FISH experiments in which one probe labeled the Fos locus as before, whereas another probe labeled the Gm49698 locus located on the opposite side of chromosome 12 as a control. Analysis of the localization of these probes within the nucleus revealed a remarkable polarized orientation in which the probe labeling the Fos probe was largely positioned closer to the nuclear periphery compared to that labeling Gm49698 (Figures 6H – 6J). Similar results were also obtained for probes labeling Npas4 and a distally located control locus on chromosome 19, Gm50442 (Figures S6B and S6C). To determine how neuronal stimulation affects radial positioning of ERGs, cultured primary cortical neurons were incubated with NMDA (50 μM for 10 min, followed by 20 min recovery in NMDA-free media), following which immuno-FISH was utilized as before to visualize Fos, FosB, and Gm49698. Whereas the radial positions of Fos and Gm49698 were unaffected by 30 min following the initial stimulus, FosB showed increased localization towards the nuclear interior by this time (Figures S6D – S6F). Taken together, the congregation of both signaling and sites of DSBs at the nuclear periphery suggest that activity-induced DSBs are orchestrated and compartmentalized within the nuclear periphery of neurons.
DISCUSSION
Mechanism, timing, and compartmentalization of neuronal activity-induced DSB formation
Our observations provide the first molecular insights into the mechanisms that regulate DSB formation following neuronal stimulation. While Top2B catalyzes the formation of neuronal activity-induced DSBs, sites that incur such DSBs are already pre-bound by Top2B under basal conditions (Madabushi et al., 2015). These results suggest that Top2B binding alone is insufficient and that neuronal activity recruits additional mechanisms to trigger lasting DSBs. Here we report that stimulus-specific calcium signaling drives CaN to dephosphorylate Top2B, which stimulates its DNA cleavage activity, and induces it to form activity-dependent DSBs. Whereas Top2B binds to only a few hundred loci within neurons (Madabhushi et al., 2015), Top2B occupancy patterns alone do not explain how the formation of neuronal activity-dependent DSBs is restricted to select genomic loci. Our results suggest that this positional specificity is conferred through two distinct mechanisms – (1) sites that incur neuronal activity-induced DSBs preferentially localize to the nuclear periphery and Top2B is already bound at these sites under basal conditions; and (2) neuronal activity triggers rapid CaN re-localization to the nuclear periphery, which induces Top2B to form DSBs.
Recent reports suggest that the preferred radial position of genes, including neuronal activity-regulated genes can vary with cell type and activity states, and that relocating genes to distinct genome neighborhoods can affect their transcription (Dekker and Misteli, 2015). While the nuclear periphery is primarily enriched for heterochromatin that associates with the nuclear lamina, a number of poised and active genes interact with the nuclear pore complex (Sumner and Brickner, 2021; van Steensel and Belmont, 2017). Interaction with the nuclear pore complex is thought to impact the transcription of associated genes by affecting chromatin folding, including enhancer-promoter looping, and transcription burst frequency (Sumner and Brickner, 2021). Interestingly, a search of available DamID-based tagging data revealed that 17 out of the 21 loci that incur activity-induced DSBs in neurons associate with the nuclear pore complex in other cell types (Ibarra et al., 2016; Jacinto et al., 2015). While the significance of their localization to the nuclear periphery in these cell types is not known, in neurons such localization could prime them for rapid DSB formation. The activity-dependent trafficking of CaN through the nuclear pore complex could allow for the rapid dephosphorylation of pre-bound Top2B at ERGs and other sites, leading to DSB formation. CaN is also targeted to diverse intracellular compartments by association with scaffold proteins, such as the multivalent A-kinase anchoring proteins (AKAPs). In C2C12 cells and cardiac myocytes, calcineurin binding to mAKAP recruits CaN to the nuclear envelope where it, in turn, binds and stimulates the transcription factor, MEF2 (Li et al., 2013). mAKAP is also expressed in neurons, localizes to the nuclear envelope and mediates compartmentalization of signaling events at the nuclear periphery (Boczek et al., 2019).
Regulation of Top2B activity through posttranslational modifications of its CTD
A majority of Top2B phosphorylation sites are clustered within the CTD. Dephosphorylation of purified recombinant Top2B with CIP substantially reduces, whereas incubation with CK2 markedly enhances, the activity of Top2B. These results indicate that phosphorylation of the CTD of Top2B supports its basal catalytic activity, and are consistent with previously reported effects of CK2-mediated phosphorylation on the activity of yeast Top2 (Cardenas et al., 1993). Recent cryo-EM studies have elaborated the structural basis for how the CTD affects the catalytic activity of Top2A and Top2B (Vanden Broeck et al., 2021). These studies revealed that the linker between the DNA binding/cleavage domains and the CTD is positioned near the DNA segment that is captured for cleavage by Top2A (G-segment), and that its presence markedly stimulates DNA cleavage by Top2A. Importantly, the rest of the CTD is disordered and its orientation suggests that posttranslational modifications in the CTD could regulate the interactions of the linker with the G-segment and fine tune the linker’s ability to stimulate DNA cleavage by Top2A and Top2B (Vanden Broeck et al., 2021). Our observations fit well with these predictions and suggest that activity-dependent dephosphorylation of Top2B by CaN could modify interactions of the CTD linker with the G-segment to trigger DSB formation.
Analysis of chimeric proteins in which the CTDs of Top2A and Top2B were swapped revealed that the CTD of Top2B, but not Top2A, negatively regulates Top2 catalytic activity (Gilroy and Austin, 2011; Meczes et al., 2008). Whereas the CTDs of Top2A and Top2B are divergent, the CTD linker region described above is shared by the two isoforms (Vanden Broeck et al., 2021). These observations could help explain how cellular mechanisms could uniquely affect the catalytic activity of either Top2A or Top2B. Although postmitotic neurons only express Top2B, and not Top2A, serine residue S1509 within Top2B, which is targeted by CaN following neuronal stimulation is not found in Top2A and could account for these differences. Mutation of S1509 and the neighboring S1511 to alanine within Top2B markedly increases the propensity of Top2B to become trapped in covalent cleavage complexes and elevates DSB formation even in the absence of neuronal stimulation. Exposing mice to a contextual fear conditioning paradigm, which triggers DSBs in the hippocampus, causes remarkably similar changes in Top2B phosphorylation to those observed in stimulated cultured primary cortical neurons. These results suggest that mechanisms of activity-induced DSBs are broadly conserved in neurons and occur following physiological neuronal stimuli. While calcineurin inhibition alone is sufficient to prevent the formation of Top2B-mediated DSBs following neuronal activity, our targeted mass spectrometry experiments revealed additional sites within Top2B that become dephosphorylated following neuronal activity in a calcineurin-independent manner, and many of these residues, such as S1453 and S1568, are also not conserved between Top2A and Top2B. These observations indicate the existence of additional mechanisms that collaborate with calcineurin to regulate Top2B activity in neurons. The illumination of such activities in future studies should further clarify the mechanisms that control the formation of neuronal activity-induced DSBs.
Limitations of the study
One limitation of this study is that the mechanisms by which the dephosphorylation of Top2B at S1509 and S1511 stimulate its DNA cleavage activity have not been characterized. Future studies that analyze how mutations at these and other residues that undergo stimulus-dependent posttranslational modifications affect the overall structure and configuration of Top2B-DNA interactions should provide insights into this issue. Another limitation is that the mechanisms that govern the preferred radial positioning of ERGs to the nuclear periphery remain unknown. For instance, it is unclear whether regions surrounding ERGs are enriched for nuclear lamina-associated domains or whether they interact with nuclear pore complexes. Future studies that assess the dynamics of ERG localization to the nuclear periphery as a function of neural development and neuronal stimulation are required to address these issues.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ram Madabhushi (ram.madabushi@utsouthwestern.edu).
Materials Availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and Code Availability
Unprocessed blots, gels, and microscopy images are available at Mendeley Data. The mass spectrometry data have been deposited in the public proteomics repository, MassIVE. Accession numbers for the deposited data are listed in the Key Resources Table.
This manuscript does not report original code.
Any additional information required to reanalyze the reported data is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Topo IIbeta (H-8) | Santa Cruz | RRID: AB_628384 |
| Topo IIbeta | Abcam | RRID: AB_2923318 |
| Pan-Calcineurin A Antibody | Cell Signaling Technology | RRID: AB_2168458 |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | MilliporeSigma | RRID: AB_309864 |
| Calcineurin A Polyclonal Antibody | Thermo Fisher Scientific | RRID: AB_10981308 |
| Histone H2A.X antibody | Abcam | RRID: AB_297814 |
| Goat Anti-Rabbit IgG Antibody, IRDye 800CW | LI-COR | RRID: AB_10796098 |
| Lamin A/C (4C11) Mouse mAb (Alexa Fluor 488 Conjugate) | Cell Signaling Technology | RRID: AB_10997529 |
| Phospho-Topoisomerase IIα (Ser1469) (D4F5) | Cell Signaling Technology | RRID: AB_2798109 |
| Phospho-CK2 Substrate [(pS/pT) DXE] MultiMab™ | Cell Signaling Technology | RRID: AB_2797653 |
| Goat anti-Mouse IgG (H + L) Antibody, IRDye 680RD | LI-COR | RRID: AB_10956589 |
| Actin beta | Bio-Rad | RRID: AB_2923317 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific | RRID:AB_2534091 |
| Goat anti-Rabbit IgG (H+L) Highly CrossAdsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific | RRID:AB_2534095 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | RRID:AB_2535805 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | RRID:AB_2535813 |
| Lamin A/C | Cell Signaling Technology | RRID:AB_10545756 |
| Anti-gamma H2A.X (phospho S139) antibody | Abcam | RRID:AB_303388 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Calcineurin Protein | R&D systems | Cat#3160-CA |
| Calmodulin (Human) (recombinant) | Enzo | Cat#BML-SE325–0001 |
| Recombinant Human Topoisomerase II beta/TOP2B protein | Abcam | Cat#ab159736 |
| KN-62 | MilliporeSigma | Cat#422706–1MG |
| KT5720 | MilliporeSigma | Cat#420323–50UG |
| Dantrolene, sodium salt | Tocris | Cat#0507 |
| InSolution™ Okadaic Acid, Prorocentrum | MilliporeSigma | Cat#495609–25UG |
| Nifedipine | Tocris | Cat#1075 |
| 2 aminoethoxydiphenyl borate (2APB) | Tocris | Cat#1224 |
| MEK/ERK inhibitor | Tocris | Cat#PD98059 |
| Calcineurin Inhibitor VIII, CN585 - | MilliporeSigma | Cat#207003–10MG |
| Tacrolimus (FK506) | Selleckchem | Cat#S5003 |
| D-AP5 | Tocris | Cat#0106 |
| Critical commercial assays | ||
| Nick Translation DNA Labeling System | ENZO | Cat#ENZ-GEN111–0050 |
| Duolink® In Situ Detection Reagents FarRed | MilliporeSigma | Cat# DUO92013 |
| Deposited data | ||
| Targeted Mass Spectrometry (PRM LC – MS/MS) | This paper | MassIVE: MSV000088392 |
| Mass Spectrometry (LC – MS/MS of immunoprecipitated Top2B) | This paper | MassIVE: MSV000089825 |
| Mendeley Dataset - Unprocessed blots, gels, and microscopy images | This paper | DOI: 10.17632/8697s83wpv.1 |
| Experimental models: Cell lines | ||
| NIH 3T3 | ATCC | Cat#CVCL_0594 |
| HEK293 | ATCC | Cat#CVCL_0045 |
| Experimental models: Organisms/strains | ||
| Mouse C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Oligonucleotides | ||
| Fos Exon Forward: GCCAACTTTATCCCCACGGT | This paper | N/A |
| Fos Exon Reverse: TCTTCACCATTCCCGCTCTG | This paper | N/A |
| Npas4 Exon Forward: GAGCAAGAGCCTGAGCGAAAAG | This paper | N/A |
| Npas4 Exon Reverse: CCAGCAAAGAAGACACCCTTG | This paper | N/A |
| GAPDH Forward: CTCTCCTCCTCCCTGTTCC | This paper | N/A |
| GAPDH Reverse: TCCCTAGACCCGTACAGTGC | This paper | N/A |
| bglobin Forward: TGACCAATAGTCTCGGAGTCCTG | This paper | N/A |
| bglobin Reverse: AGGCTGAAGGCCTGTCCTTT | This paper | N/A |
| Fos Forward: GAACGGAATAAGATGGCTGC | This paper | N/A |
| Fos Reverse: TTGATCTGTCTCCGCTTGG | This paper | N/A |
| FosB Forward: CCCGAGAAGAGACACTTACC | This paper | N/A |
| FosB Reverse: CTCTTCAAGCTGATCAGTTTCC | This paper | N/A |
| Npas4 Forward: CTGCATCTACACTCGCAAGG | This paper | N/A |
| Npas4 Reverse: GCCACAATGTCTTCAAGCTCT | This paper | N/A |
| Egr1 Forward: AGCGAACAACCCTATGAGCA | This paper | N/A |
| Egr1 Reverse: TCGTTTGGCTGGGATAACTC | This paper | N/A |
| GAPDH Forward: CTCTCCTCCTCCCTGTTCC | This paper | N/A |
| GAPDH Reverse: TCCCTAGACCCGTACAGTGC | This paper | N/A |
| Nr4a1 Forward: ATGCCTCCCCTACCAATCTTC | This paper | N/A |
| Nr4a1 Reverse: CACCAGTTCCTGGAACTTGGA | This paper | N/A |
| ActB Forward: GGCTGTATTCCCCTCCATCG | This paper | N/A |
| ActB Reverse: CCAGTTGGTAACAATGCCATGT | This paper | N/A |
| HA-tagged mouse DNA topoisomerase IIβ | Addgene | Cat# pYLL201 |
| Recombinant DNA | ||
| Fos | CHORI (Children’s Hospital Oakland Research Institute) | RP24–208N11 |
| Fos (upstream) | CHORI (Children’s Hospital Oakland Research Institute) | RP24–368B17 |
| Fos (downstream) | CHORI (Children’s Hospital Oakland Research Institute) | RP24–178I11 |
| FosB | CHORI (Children’s Hospital Oakland Research Institute) | RP24–161J12 |
| ActB | CHORI (Children’s Hospital Oakland Research Institute) | RP24–266N |
| Malat1 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–290K10 |
| Npas4 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–238I5 |
| Gm50442 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–344E18 |
| Software and algorithms | ||
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and treatments
Dissociated cortical neurons from E16 Swiss-Webster mice were plated at a density of 0.25 million cells/cm2 in 10 cm culture dishes or 6 well plates coated beforehand by incubation with poly-D-lysine (0.5 mg/ml) or 12 well plates with coverslips coated with poly-D-lysine (0.5 mg/ml) and laminin (0.005 mg/ml) for 1 h at 37°C, followed by washing twice with dH2O. Neurons were maintained for 13 days in neurobasal media (GIBCO) and supplemented with L-alanyl-L-glutamine, penicillin/streptomycin, and B27.
Neurons (DIV13) were pre-incubated with inhibitors of ion channels, kinases, and phosphatases for either 30 or 40 min at the indicated concentrations: 50 μM DL-2-Amino-5-phosphonopentanoic acid (NMDARi), 10 μM nifedipine (LVGCCi), 50 μM 2-Aminoethoxydiphenylborate (IP3Ri), 15 μM dantrolene sodium (RyRi), 1 μM Tetrodotoxin (VGSCi), 10 μM KN-62 (CamKIIi), 50 μM PD98059 (ERKi), 4 μM KT5720 (PKAi), 25 μM okadaic acid (PP1/PP2Ai), 25 μM CN585 (CaNi), 50 μM INCA-6.
For most experiments, following the pre-incubation with the indicated inhibitors, cultured neurons were stimulated (red bars in the graphs) with 50 μM N-methyl-D-aspartate (NMDA) for 10 min followed by recovery in NMDA-free conditional media for 20 min. Neurons for specified experiments were collected either 10 min after stimulation or at the indicated times. Neuronal activity was also induced by depolarization with 20 mM of KCl for 30 min. Control neurons (blue bars in graphs) were pre-incubated with vehicle (DMSO) but were not stimulated. For the indicated experiments, neurons were stimulated with either ionomycin (5 μM final) or etoposide (ETP) (10 μM final) for 20 min.
Mouse embryonic fibroblast (NIH-3T3) (ATCC, CRL-1658) and HEK293 human embryonic kidney cells (ATCC, CRL-1573) were cultured at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, high glucose containing 10% fetal bovine serum with penicillin/streptomycin). Transient transfection of cells with expression vectors was done using polyethylenimine (PEI).
METHOD DETAILS
Immunoprecipitation
For Co-immunoprecipitation experiments, neurons were lysed in in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, phosphatase and protease inhibitors cocktail) by passing through a 25-gauge needle. All procedures were performed at 4°C. Samples were centrifuged at 20000 x g for 5 min. The supernatants were collected, and protein concentrations determined using the BCA assay. Equal amounts of protein (1000 μg) and 20 μl of Top2B antibody (Santa Cruz, sc-25330, RRID: AB_628384) were incubated overnight. Samples were incubated with 25 μl of protein G-magnetic beads (Bio-Rad) for 2 h and the beads were washed 4 times with TBS with 1% Triton, and one more time with TBS with 0.1% Triton X-100. Beads were resuspended in Laemmli buffer and analyzed by western blotting.
For analysis of Top2B phosphorylation, neurons and brain tissue were lysed in in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.2% N-Lauroylsarcosine, phosphatase and protease inhibitors cocktail) by passing through a 25-gauge needle. Lysates (300 μl) were incubated with 2 μl Benzonase® Nuclease (≥250 units/μL) for 3 h, followed by sonication. All procedures were performed at 4°C. Samples were centrifuged at 20,000 x g for 5 min. The supernatants were collected, and protein concentrations determined using the BCA assay. Equal amounts of protein and 6 μl of Top2B antibody (Abcam, ab125297, RRID: AB_2923318) per sample were incubated overnight. Samples were incubated with 25 μl of protein A/G-magnetic beads for 3 h and the beads were washed 4 times with high salt buffer (500 mM NaCl, 50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.1% SDS) and one more time with TBS with 0.1% Triton X-100. Beads were resuspended in Laemmli buffer to separate proteins by electrophoresis and analyze them by either MS or western blotting. Fluorescence was captured using either a ChemiDoc Imaging Systems (Bio-Rad), or imaged using film. The gel analyzer routine of ImageJ software was used to subtract background and quantify the optical density of the bands. The graphs show densitometry of co-immunoprecipitated CaN (mean ± s.e.m) normalized to immunoprecipitated Top2B and expressed as fold change over unstimulated neurons. Each dot in the bar graph represents the result of an independent experiment/biological replicate performed on different days using neurons cultured from independent mouse litters. The number of dots per graph represent the n for each experiment. The densitometric data were analyzed by two-tailed t-tests.
Western blotting
Samples were mixed with Laemmli buffer plus DTT, boiled and electrophoresed through 4–14% SDS–PAGE gels. Proteins were transferred to nitrocellulose membranes, blocked for 2 h at room temperature in Intercept (TBS) Blocking Buffer (LI-COR). Membranes were incubated overnight with primary antibodies: Top2B (Abcam, ab125297, RRID: AB_2923318), Pan-CaN A (Cell Signaling Technology, 2614S, RRID: AB_2168458), γH2AX (MilliporeSigma, 05–636, (RRID: AB_309864)), H2AX (Abcam, ab11175, (RRID: AB_297814)), β-actin (Bio Rad, VMA00048, (RRID: AB_2923317)), Lamin A/C (Cell Signaling Technology, 4777S, (RRID: AB_10545756)). Thereafter, the membranes were washed (4 x, 5 min/wash) in TBS-T, and incubated for 1 h with goat anti mouse or rabbit IR dye-conjugated secondary antibodies (LI-COR, 92668170, (RRID: AB_10956589) and 82708365, (RRID:AB_10796098)). Membranes were washed again and fluorescence was captured using either a ChemiDoc Imaging Systems (Bio-Rad), or imaged using film. The gel analyzer routine of ImageJ software was used to subtract background and quantify the optical density of the bands of interest (γH2AX, Top2B, CaN) and loading controls (H2AX, actin, lamin). The ratio of optical density between the band of interest and the corresponding loading control was then calculated to obtain the normalized optical density for each band of interest. To compare results across experiments, the normalized optical density for each band (treatments and vehicle controls) is divided by the average optical density for the unstimulated vehicle controls, which represents the fold change from control value depicted on the y-axis. Graphs show normalized optical density, (individual data and mean ± s.e.m.) and expressed as fold change over unstimulated vehicle controls. Red bars represent neurons stimulated with NMDA, KCl or ionomycin for each treatment. Controls (blue bars) represent neurons that were not stimulated. Each dot in the bar graph represents the result of an independent experiment/biological replicate performed on different days using neurons cultured from independent mouse litters. Number of dots per graph represent the n for every experiment. P-values < 0.05 for relevant comparisons are indicated in the graphs. The densitometric data were analyzed by either one- or two-way ANOVA with Tukey’s post-hoc test.
Immunocytochemistry
Neurons plated on coverslips were fixed with 4% paraformadehyde (PFA) for 10min, washed with PBS (4 times for 5 min) and incubated with blocking buffer 5% NGS (Normal Goat Serum) 0.3% Triton X-100 in PBS at RT for at least 4 h. Then, the neurons were incubated with the indicated primary antibodies in blocking buffer overnight at 4°C. The following day the neurons were washed and incubated with secondary antibodies goat anti-mouse and rabbit IgG-Alexa Fluor 594 and 647 (Thermo Fisher, A11032 (RRID: AB_2534091), A11037 (RRID:AB_2534095), A21236 (RRID:AB_2535805), A21245, (RRID:AB_2535813)) in blocking buffer at room temperature for 1 h and washed. Cells were subsequently incubated with Lamin A/C Alexa Fluor 488 conjugate (Cell Signaling Technology, 8617S, (RRID: AB_10997529)), DNA was stained with Hoechst 33342 for 5 min and washed again before been mounted with ProLong™ Diamond Antifade (Thermo Fisher). All the images were acquired using a Zeiss LMS800 confocal with a 64× objective (Resolution: 10.0974 pixels per micron). Intensity profiles of CaN, lamin A/C, and DAPI within rectangular ROIs (15 × 0.8 μM) crossing through the center of the cell (yellow rectangles) were plotted. The peak of lamin A/C staining was used to mark the nuclear periphery (represented by “0” on the x-axis). Positive values on the x-axis indicate regions outside the nucleus, whereas negative values denote the nuclear interior. y-aixs represents the intensity of staining. Two radial profiles corresponding to two nuclear rims were obtained from each ROI. The graph shows 52 averaged profiles per condition.
Biochemical fractionation
Biochemical fractionation experiments were performed as described (Wilson et al., 2016). Cells were rinsed with ice-cold PBS and with ice-cold CSK buffer (10 mM PIPES/KOH, 100mM NaCl, 300 mM Sucrose, 1mM EGTA, 1mM MgCl2, 1mM DTT, protease and phosphatase inhibitors). Cells were harvested in 225 μl of CSK buffer per one 10 cm dish. Triton X-100 was added to a final concentration of 0.1%. Samples was divided equally in four groups: one sample (25 μl) was saved to analyze total protein. The rest of the samples were centrifugated at 6800 × g for 2 min at 4°C. Supernatants was transferred to a different tube and the pellet was resuspend in 200 μl of CSK-DS (CSK buffer containing 0.1% Triton X-100) and high salt (0.5 M NaCl) and incubated for 5 min on ice. The samples were centrifuged again 6800 × g for 3 min and the supernatant was pooled with the previous soluble extract to analyze soluble proteins. The pellets were resuspended in 200 μl of CSK-DS again and centrifuged at 9500 × g for 3 min. Next, pellets were resuspended in 200 μl of CSK buffer with Triton X-100 and samples (100 μl) were incubated with 400 U/ml of DNase I for 50 min at 32°C. 20 μL of 5 M NaCl was added to the DNase treated sample and to the 100 μL remaining. After 5 min incubation both samples were centrifuged at 9500 x g for 5 min at 4°C. Both pellets, detergent/salt resistant fractions (DSR) and Dnase-resistant fractions, were resuspended in Laemmli buffer by sonication and analyzed by western blotting. Densitometry analysis was performed (individual values and mean ± s.e.m.) of CaN and Top2B normalized to lamin A/C and expressed as fold-change over controls (t = 0 min). Each dot in the bar graph represents the result of an independent experiment/biological replicate performed on different days using neurons cultured from independent mouse litters. Number of dots per graph represent the n for every experiment. P-values < 0.05 for relevant comparisons are indicated in the graphs. The densitometric data were analyzed by one-way ANOVA with Tukey’s post-hoc test.
DNA Fluorescence in situ hybridization (DNA-FISH)
Bacterial artificial chromosomes (BACs) were obtained from the BACPAC Resources Center (Children’s Hospital Oakland Research Institute). The following probes were used: Fos - RP24–208N11; Fos (upstream) - RP24–368B17; Fos (downstream) - RP24–178I11; Gm49698 - RP24–97I22; Npas4 - RP24–238I5; Gm50442 - RP24–344E18; Malat1 - RP24–290K10; FosB - RP24–161J12; ActB - RP24–266N. Direct-labeled BAC probes were prepared using the Nick translation DNA labeling system 2.0 (Enzo Life Sciences; ENZ-GEN111–0050) and the Gold 550 dUTP or Red 650 dUTP (Enzo Life Sciences, ENZ-42521 and ENZ-42522), according to the manufacturers recommendations. Probes were ethanol precipitated with 25 μg salmon sperm DNA (Cat. No. 15279011, Thermo Fisher) and resuspended in formamide and 1:1 volume of hybridization buffer (10% 20x SSC, 20% dextran sulfate, 1% SDS, 0.1μg/μl human Cot DNA, 1 μg/μl salmon sperm DNA).
Cells were cultured on coverslips and fixed with 4% PFA for 9 min and with 4% PFA with 0.5% Triton X-100 for 1 min. Coverslips were washed thrice with 0.1% Triton X-100 in PBS, incubated with 0.5% Triton X-100 for 10 minutes and washed thrice again with PBS. The coverslips were incubated with 100 μg/ml RNAse for 1 h at 37°C, washed thrice with PBS and incubated in 0.1 N HCl for 5 min. Coverslips were then washed with 2x SSC twice for 3 min and incubated overnight in 50% formamide/2x SSC at 4°C. The next day, samples were denatured in 70% formamide, 2x SSC at 83°C for 20 minutes. At the same time, BAC probes were denatured at 95°C for 5 minutes. Probes were then applied to the samples, sealed with incubation chamber gaskets, and incubated for 5 min at 75°C and then overnight at 37°C in a humidified chamber. Coverslips were then washed at 37°C with 2x SSC (3x, 30 min/wash) and an additional three times (30 min/wash) in 0.4x SSC with 0.2% Tween at 60°C. Samples were blocked with blocking solution (5% BSA in 2x SSC with 0.2% Tween) for 1 h at 37°C and incubated with 1:200 Lamin A/C (Alexa Fluor® 488 Conjugate) antibody (Cell Signaling 8617S, (RRID: AB_10997529)) blocking solution for at least 4 h. DNA was stained with Hoechst 33342 for 10 minutes in 2x SSC. Finally, coverslips were washed with 2x SSC (4x, 5 min/wash) and mounted with with ProLong™ Diamond Antifade. Z-stack images were captured at 0.3 μm intervals over the nucleus depth using a Zeiss LSM800 confocal microscope. Image resolution was 8.5828 pixels per micron (59.6545 microns (512), Height: 59.6545 microns (512), Depth: 7.2000 microns (24), Voxel size: 0.1165 × 0.1165 × 0.3 μm3. The size of proximity ligation and FISH spots ranged between 200 – 300 nm. ImageJ was utilized to measure distance to the nuclear periphery. Brightness and contrast were adjusted to determine the spatial position of every locus precisely. Lines were drawn from the center (approximately) of the FISH/proximity ligation spot to the edge of lamin staining. The “measure” tool in ImageJ was then used to determine the length of the line as shown in Figure 6D and S6A. To determine the distance of the FISH/proximity ligation spots to the nuclear periphery on the z-axis, we quantified the number of optical slices from the lamin staining to the FISH spots (thickness of optical slices were 300 nm) as shown in Figure 6D. The FISH spots were usually visible in 2 or 3 optical slices and the slice with the greatest signal intensity was used to approximate distance. After calculating the distance of every locus to the nuclear rim in the XY and Z planes, the shortest distance to the periphery was plotted in the graph (individual values and mean ± s.e.m.) (Figure 6E). Each dot in the graph represents the shorter distance to the periphery of one locus. Number of dots per graph represent the n. Cells from 3 or 4 independent experiments/biological replicates performed on different days using neurons cultured from independent mouse litters were analyzed. We also plotted (Figure 6F) the percentage of cells with at least one copy of the probed region located at a distance < 0.5 μm from the nuclear periphery (individual values and mean ± s.e.m.). Each dot in the bar graph represents the percentage of every independent experiment. P-values < 0.05 for relevant comparisons are indicated in the graphs. Data were analyzed by one-way ANOVA with Tukey’s post-hoc test.
Super-resolution microscopy was used to examine the distribution of adjacent Fos probes in dual-color FISH experiments. No significant differences in relative probe positions were observed between images obtained from super-resolution microscopy compared to confocal microscopy.
In situ proximity ligation assay
The in situ PLA experiments were performed using reagents and instructions found in commercially available kits from MilliporeSigma; Duolink® In Situ PLA® Probe Anti-Rabbit MINUS/PLUS (DUO92005/DUO92002), Duolink® In Situ PLA® Probe Anti-Mouse MINUS/PLUS (DUO92004/DUO92001) and Duolink® In Situ Detection Reagents Orange (DUO92007) (Soderberg et al., 2006). Neurons plated on coverslips were fixed with 4% PFA for 10 min, washed with PBS (4x, 5 min/wash) and incubated with blocking buffer (5% NGS (Normal Goat Serum), 0.3% Triton X-100 in PBS) at room temperature for at least 4 h. Following this, the neurons were incubated with an Anti-Pan-CaN A Antibody (Cell Signaling, 2614S, (RRID: AB_2168458)) and Anti-Top2B antibody (Santa Cruz Biotechnology, sc-25330, (RRID: AB_628384)) in blocking buffer overnight at 4 °C. Primary antibody solution was removed, and the slides were washed thrice in 1 × TBS, 0.05% Tween 20 (TBS-T) (5 min/wash) with gentle agitation. Secondary probes were diluted to final concentrations of 1:5 in antibody diluent (supplied in the kit) and added to each sample. The slides were incubated in a humidity chamber for 90 min at 37°C. Then slides were washed, and ligation solution was added to each sample. The slides were then incubated in a humidity chamber for 30 min at 37°C, ligation solution was removed, slides were washed, and amplification solution (Amplification/detection mix, polymerase) was added to each sample. The slides were incubated in a humidity chamber for 120 min at 37°C and then washed briefly in TBS before Hoechst dye in TBS (Life Technologies, H1399) was added to each sample to stain cell nuclei. Then the slides were washed three times with TBS and mounted. Z-stack images were captured at 0.5μm intervals over the nucleus depth. All images were acquired using a Zeiss LSM800 confocal microscope as described before. Lamin A/C staining was used to mark the nuclear periphery. ImageJ was utilized to measure distance to the nuclear periphery as described for the DNA-FISH experiments, and as shown in Figure S5C. The number of interactions (mean ± s.e.m.) as a function of distance from lamin A/C is shown in Figure 5F. The number of interactions per cell (mean ± s.e.m.) as a function of time post-stimulation is shown in Figure 5G. Three independent experiments were performed (at least 35 interactions were quantified per experiment). P-values < 0.05 for relevant comparisons are indicated in the graphs. The data were analyzed by one way ANOVA with Tukey’s post-hoc test.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed using a ChIP assay kit (MilliporeSigma) as described by the manufacturer. Briefly, cultured primary cortical neurons were crosslinked by treatment with formaldehyde (1% final) for 10 minutes at 37°C, washed twice with ice-cold PBS, scraped, and resuspended in lysis buffer containing 50 mM Tris pH 8.1, 10mM EDTA, and 1% SDS with protease and phosphatase inhibitor cocktails. Cells were sonicated to shear DNA to lengths between 200–500 bp using a bioruptor (Diagenode; high power, 30 seconds on/30 seconds off for 40 cycles). Samples were centrifuged for 10 min at 13000 rpm, the supernatant was diluted with 16.8mM Tris-HCl pH 8.1 with 1.1% Triton X-100, 1.2mM EDTA and 167mM NaCl. Sheared chromatin was incubated overnight with anti-γH2AX antibody (Abcam, ab2893, (RRID:AB_303388)) and then for 3 h with ChIP Grade Protein A/G Magnetic Beads. Beads were washed with high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) and LiCl buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid (sodium salt), 1 mM EDTA, 10 mM Tris, pH 8.1). Crosslinked immunoprecipitated proteins were then eluted from the beads in a solution containing 100 mM NaHCO3 and 1% SDS. Crosslinks were reversed by incubation at 65°C overnight with 200 mM NaCl, following which DNA was extracted using phenol/chloroform/isoamyl alcohol. Samples were then subjected to quantitative PCR using the indicated primers (Table S2). Data are shown as fold change (individual data and mean ± s.e.m) relative to unstimulated vehicle controls. Number of dots per graph represent the n for every experiment, and p-values < 0.05 for relevant comparisons are indicated in the graphs. The PCR data were analyzed by one way ANOVA with Tukey’s post-hoc test.
qRT‑PCR
Total RNA was extracted from cultured primary neurons using the Qiagen RNeasy Plus Universal Kit (Qiagen) and by following the manufacturer’s recommended protocol. cDNA was synthesized from 2 μg of total RNA using an RT-PCR EcoDry Premix (Clontech). RT-PCR reactions were performed using SsoAdvanced™ Universal SYBR® Green Supermix using the indicated primers (Table S2). Data are shown as fold change (individual data and mean ± s.e.m) relative to unstimulated vehicle controls. The number of dots per graph represent the n for every experiment, and p-values < 0.05 for relevant comparisons are indicated in the graphs. The data were analyzed by one-way ANOVA and two-way ANOVA as appropriate and using either Tukey’s or Holm-Sidak post-hoc tests.
DNA Relaxation and DNA Cleavage Assays
Topoisomerase II DNA relaxation were essentially carried out as described (Nitiss et al., 2012). Briefly, reaction mixtures (30 μl) containing 10 mM Tris (pH 8), 50 mM NaCl, 50 mM KCl, 5 mM MgCl2, 5 mM CaCl2 0.1 mM EDTA, 1 mM ATP, and 10 nM supercoiled plasmids (Fos-luc) were incubated with recombinant human Top2B and Top2A (2 units, Inspiralis). Where indicated, reactions were supplemented with 1 unit of CIP (New England Biolabs), 5 units of CK2 (NEB), 1μM calmodulin (Abcam, ab94519), and varying amounts of CaN (0.25, 0.5 and 1 unit, R&D systems). Reactions were incubated at 37°C for 30 min. For DNA cleavage assays, reactions contained 1 mM AMP-PNP instead of 1 mM ATP. Reactions were stopped by the addition of SDS (1% final) and proteinase K (10 μg final). Samples were incubated at 45°C for 1 hour. Samples were then loaded onto 1% TAE agarose gels and electrophoresed at 5 V/cm for 4 hr. Gels were stained with 1 mg/ml red gel for 30 min and visualized. Graph shows densitometry analysis (individual data and mean ± s.e.m.) of the relaxed and linear DNA normalized to supercoiled bands and expressed as fold change relative to the corresponding levels in DNA alone. Number of dots per graph represent the n for every experiment, and p-values < 0.05 for relevant comparisons are indicated in the graphs. The densitometric data were analyzed by one-way ANOVA with Tukey’s post-hoc test.
In vivo complex of enzyme assay
Assays were performed as described (Nitiss et al., 2012). Cells were lysed with 1% sarkosyl, and the DNA was sheared using a 1 ml syringe and 25G/8 gauge needle. Lysates were passed 15 times through the needle. Lysates (1.5 ml) were then layered atop a 2 ml CsCl solution in 13 × 51 mm polycarbonate centrifuge tubes (Beckman) and spun at 71,000 rpm in an SLA-100.3 rotor (Beckman) for 16–20 h at room temperature. Pellets were washed with 70% ethanol and resuspended in TE buffer (pH 7.5). Indicated amounts of DNA were applied to a nitrocellulose membrane using a slot-blot apparatus (Bio-Rad). The membranes were then assessed for the presence of Top2Bccs using standard western blotting procedures. The gel analyzer routine of ImageJ software was used to subtract background and quantify the optical density of the bands. The optical densities of the three DNA amounts were averaged. Each dot in the bar graph (individual data and mean ± s.e.m.) represents the result of an independent experiment/biological replicate performed on different days using neurons cultured from independent mouse litters. Number of dots per graph represent the n for every experiment, and p-values < 0.05 for relevant comparisons are indicated in the graphs. The densitometric data were analyzed by one-way ANOVA with Tukey’s post-hoc test.
Kinase Assays
Protein kinase assays were performed with Top2B immunoprecipitated from cultured primary cortical neurons. Phosphorylation reactions were carried out in a total volume of 30 μl kinase buffer containing 50 mM Tris-HCl (pH 7.5 at 25°C), 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij 35, 10 μM ATP, and 5 μCi of [γ−32P] ATP (Roche). Reactions were incubated for 15 min at 30°C and then stopped using SDS sample buffer and analyzed by SDS-PAGE.
Targeted Mass Spectrometry
Mass spectrometry experiments and data analysis were carried out by members of Proteomics group (Carr laboratory) at the Broad Institute of MIT and Harvard. Cultured primary cortical neurons were treated with the indicated conditions as described above, and cell pellets were collected by centrifugation and stored at −80°C. Cell pellets were lysed in 525 μl lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris, 1 mM EDTA, 10 mM NaF, PIC2, PIC3, 1 mM PMSF, Aprotinin, Leupeptin). Following this, 500 μg of the samples was reduced, alkylated, and digested using LysC and trypsin at a 1:50 ratio. Samples were then desalted in a 25 mg C18 Sep-pak plate, resuspended in 250 μl of 80% ACN/0.1% TFA (at an expected concentration of 2μg/μl), and 125 μl (~ 205 μg) was then used for IMAC enrichment using AssayMap Fe (III)-NTA cartridges. Phosphopeptide enriched samples were resuspended in 10 μl of 3% ACN/5% FA and 2μl of the samples were then injected onto a Q Exactive HF-X mass spectrometer for label free LC-MS/MS analysis. The samples were analyzed with an Orbitrap Q-Exactive HFX (Thermo Fisher Scientific) mass spectrometer coupled to a nanoflow Proxeon EASY-nLC 1200 UHPLC system (Thermo Fisher Scientific). The mass spectrometer was used in positive mode and was equipped with a nanoflow ionization source (James A. Hill Instrument Services, Arlington, MA); the spray voltage was set at 2.00 kV. The LC system, the column, and the electrospray voltage source (platinum wire) were connected via a stainless steel cross (360 μm; IDEX Health & Science; UH-906x). The column was heated to 50 °C. A volume equivalent to 500ng of protein on column was injected onto an in-house packed 20 cm × 75 μm diameter C18 silica picofrit capillary column (1.9-μm ReproSil-Pur C18-AQ beads, Dr. Maisch GmbH, r119.aq; Picofrit 10-μm tip opening, New Objective, PF360–75-10-N-5). The mobile phase had a flow rate of 200 nL/min and consisted of 3% ACN/0.1% FA (solvent A) and 90% ACN/0.1% FA (solvent B). The column was conditioned before each sample injection. Peptides were separated using the following LC gradient: 2–6% B in 1 min, 6–30% B in 74.5 min, 30–60% B in 7.5min, 60–90% B in 1 min, stay at 90% B for 5 min, and 90–50% B in 2min. For the MS1 scans, the resolution was set at 60,000 at 200 m/z and the automatic gain control (AGC) target was 3e6 with a maximum inject fill time of 20 ms. For the MS2 targeted analysis the AGC target was 1e5, the maximum inject fill time was 50ms, the loop count was 15 and the normalized collision energy was set to 28. The raw files were imported in Skyline (MacLean et al., 2010) and the statistical analysis was performed using the R package MSstats (Choi et al., 2014). The normalized intensity of phosphoserines that showed changes after stimulation, expressed as log2 fold-change over control (unstimulated), are shown in the graph.
Mass Spectrometry
Cultured primary cortical neurons (two 10 cm dishes per sample) were pre-incubated with either vehicle (DMSO) or 25 μM CN585 (CaNi) for 40 min. Then the neurons were stimulated with either 50 μM NMDA or 20mM KCl for 10 min. Control neurons were not stimulated. Neurons were lysed in in ice-cold lysis buffer (50mM Tris-HCl, pH 7.4, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, phosphatase and protease inhibitors cocktail) by sonication. Samples were centrifuged at 20000 x g for 5 min. The supernatants were collected and 2.33 volumes of lysis buffer without NaCl was added to the sample. Samples were incubated with 5 μl of an Anti-Top2B (rabbit) antibody (Abcam ab125297, (RRID: AB_2923318)) plus 25 μl of a second Anti-Top2B (mouse) antibody (Santa Cruz sc-25330, (RRID: AB_628384)) overnight. Thereafter, the samples were incubated with 25 μl of protein A/G-magnetic beads for 4 h and the beads were washed three times with 50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, and two more times with TBS with 0.1% Triton. Beads were re-suspended in Laemmli buffer and run in polyacrylamide gel. The gel was stained with Coomassie blue and the Top2B band was cut. Samples were processed on an Orbitrap Fusion Lumos mass-spectrometry platform using a short reverse-phase LC-MS/MS method. PTM were analyzed using Proteome Discoverer 2.4. The graph shows the percentage (abundance) of the phosphorylated peptides relative to the total peptides detected. The phosphorylated residues and the peptide range are shown on the x-axis.
Comet-FISH
Neurons in 6-well plates were washed three times with cold PBS and collected in 300 μl of PBS. Cell suspensions (30 μl) were quicky mixed with 270 μl of 1% molten agarose (low-melting) at 37 °C and distributed onto pre-treated glass comet-slides to allow the agarose to solidify for 1 hour at 4 °C. Slides were immersed overnight in lysis solution (Trevigen, 4250–050-01), following which the slides were incubated in alkaline electrophoresis buffer (200 mM NaOH, 1 mM EDTA) at 4 °C for 30 min and electrophoresed (1.25 V/cm for 30 min). After electrophoresis, the slides were washed (3 × 5 min) in neutralization buffer (0.4 M Tris-HCl pH 7) and dehydrated in 100% ethanol for at least 3 days. The DNA was then denatured in 0.5 M NaOH for 30 min and dehydrated in an ascending ethanol series (5 min per step): 70%, 80%, 95%, and dried at room temperature. The labeled BAC probe was prepared in hybridization buffer as described for FISH and diluted 1:1 with (50% formamide, 5% 20x SSC, 10% dextran sulfate, 1% SDS, 0.05μg/1μl Cot human DNA, 0.5 μg/1μl Salmon sperm DNA). The probe mixture (10 μL) was then applied to each well and spread with a plastic coverslip. Samples were incubated for 72 hours at 37 °C in a humidified chamber. Then the slides were washed at 37 °C with 2x SSC (3 × 30 min and then with 0.4x SSC containing 0.2% Tween at 60 °C (3 × 30 min). DNA was stained with Hoechst 33342 for 10 minutes in 2x SSC. Finally, coverslips were washed with 2x SSC (4 × 5 min) and mounted using ProLong™ Diamond Antifade. Z-stack images were captured at 0.3μm intervals over the nucleus depth. All images were acquired using a Zeiss LSM800 confocal microscope as describe for DNA-FISH. Brightness and contrast were adjusted to determinate with higher accuracy the spatial position of probes and the percentage of loci where upstream and downstream probes were split apart was quantify (individual data and mean ± s.e.m.). Three independent experiments were performed and at least 12 cells were analyzed per condition in each experiment. Each dot in the bar represents the result of every independent experiment, p-values < 0.05 for relevant comparisons are indicated in the graphs. The data was analyzed using one-way ANOVA and Tukey’s post-hoc test.
Contextual Fear Conditioning.
The experimental procedures followed the Guide for the Care and Use of Laboratory Animals (eighth edition) and were approved by the Animal Care and Use Committee at the University of Texas Southwestern Medical Center. 8-weeks-old C57/BL6 J mice were purchased from the Jackson laboratory, and were individually housed for 2–4 weeks on a 12/12 light/dark cycle with chow provided ad libitum. Fear conditioning was performed in boxes equipped with a metal grid floor connected to a scrambled shock generator (Med Associates Inc., St. Albans). Mice were individually placed in the chamber and allowed to explore the context for 2 min, following which foot shocks (3 × 0.8 mA, spaced 1 min apart) were delivered from the grid floor. The mice were allowed to remain for an additional 30 s in the chamber, and then moved to their home cages. Thirty minutes after fear conditioning, animals were sacrificed, and the hippocampus was dissected and frozen in liquid nitrogen. Hippocampi of 4 animals were pooled per sample for mass spectrometry analysis. Top2B was immunoprecipitated from the hippocampus (4 animals per sample) and PTMs were analyzed by mass spectrometry. This experiment was repeated twice with similar results. The graph shows the percentage (abundance) of the phosphorylated peptides relative to the total peptides detected. The phosphorylated residues and the peptide range are shown on the x-axis.
Quantification and Statistical Analysis
Data were generated from independent biological replicates (either a group of animals or cultured cortical neurons isolated from independent mouse litters, as described in Method Details), and the number of dots in each graph represent the n for that experiment. Descriptions of data collection, data quantification, and statistical methods used to analyze each experiment are provided within the corresponding sections in Method Details.
Supplementary Material
Highlights.
Calcineurin activates Top2B and induces it to form DSBs in response to NMDAR activity
Calcineurin dephosphorylates Top2B at residues S1509 and S1511 within its C-terminal
Calcineurin-mediated DSBs regulate the expression of neuronal early response genes
Neuronal activity-induced DSB sites preferentially localize to the nuclear periphery
Acknowledgments
We would like to thank Dr. Andrew Lemoff and the UTSW Proteomics Core for help with the analysis of mass spectrometry experiments, and Fernando Delint Ramirez for the artwork in the graphical abstract. This work was supported by CPRIT recruitment award RR170010, Welch Foundation grant I-1960-201803324, and NIMH R01-MH120132 to R.M.
Footnotes
Declaration of Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Austin CA, Lee KC, Swan RL, Khazeem MM, Manville CM, Cridland P, Treumann A, Porter A, Morris NJ, and Cowell IG (2018). TOP2B: The First Thirty Years. Int J Mol Sci 19. 10.3390/ijms19092765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H.(2013). Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 14, 593–608. 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, and Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186. 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Bading H, Hardingham GE, Johnson CM, and Chawla S.(1997). Gene regulation by nuclear and cytoplasmic calcium signals. Biochem Biophys Res Commun 236, 541–543. 10.1006/bbrc.1997.7037. [DOI] [PubMed] [Google Scholar]
- Boczek T, Cameron EG, Yu W, Xia X, Shah SH, Castillo Chabeco B, Galvao J, Nahmou M, Li J, Thakur H, et al. (2019). Regulation of Neuronal Survival and Axon Growth by a Perinuclear cAMP Compartment. J Neurosci 39, 5466–5480. 10.1523/JNEUROSCI.2752-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas ME, Walter R, Hanna D, and Gasser SM (1993). Casein kinase II copurifies with yeast DNA topoisomerase II and re-activates the dephosphorylated enzyme. J Cell Sci 104 ( Pt 2), 533–543. [DOI] [PubMed] [Google Scholar]
- Chahine MN, and Pierce GN (2009). Therapeutic targeting of nuclear protein import in pathological cell conditions. Pharmacol Rev 61, 358–372. 10.1124/pr.108.000620. [DOI] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, and Vitek O.(2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, and Olson EN (2002). NFAT signaling: choreographing the social lives of cells. Cell 109 Suppl, S67–79. 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Cremer T, and Cremer M.(2010). Chromosome territories. Cold Spring Harb Perspect Biol 2, a003889. 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SL, Movsesyan VA, Jorgensen TJ, and Kondratyev A.(2006). Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur J Neurosci 23, 2351–2361. 10.1111/j.1460-9568.2006.04768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, and Tsien RW (2003). Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13, 354–365. 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Dekker J, and Misteli T.(2015). Long-Range Chromatin Interactions. Cold Spring Harb Perspect Biol 7, a019356. 10.1101/cshperspect.a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann F, Weiwad M, Kilka S, Karanik M, Patzel M, Baumgrass R, Liebscher J, and Fischer G.(2010). The novel calcineurin inhibitor CN585 has potent immunosuppressive properties in stimulated human T cells. J Biol Chem 285, 1888–1898. 10.1074/jbc.M109.024844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilroy KL, and Austin CA (2011). The impact of the C-terminal domain on the interaction of human DNA topoisomerase II alpha and beta with DNA. PLoS One 6, e14693. 10.1371/journal.pone.0014693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty DD, and Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186. 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, and Crabtree GR (1999). L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401, 703–708. 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Greer PL, and Greenberg ME (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Hagenston AM, and Bading H.(2011). Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb Perspect Biol 3, a004564. 10.1101/cshperspect.a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallhuber M, Burkard N, Wu R, Buch MH, Engelhardt S, Hein L, Neyses L, Schuh K, and Ritter O.(2006). Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ Res 99, 626–635. 10.1161/01.RES.0000243208.59795.d8. [DOI] [PubMed] [Google Scholar]
- Hallhuber M, and Ritter O.(2007). New approach to prevent myocardial hypertrophy: the import blocking peptide. Future Cardiol 3, 91–98. 10.2217/14796678.3.1.91. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, and Bading H.(2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5, 405–414. 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Benner C, Tyagi S, Cool J, and Hetzer MW (2016). Nucleoporin-mediated regulation of cell identity genes. Genes Dev 30, 2253–2258. 10.1101/gad.287417.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto FV, Benner C, and Hetzer MW (2015). The nucleoporin Nup153 regulates embryonic stem cell pluripotency through gene silencing. Genes Dev 29, 1224–1238. 10.1101/gad.260919.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Vargas MA, Kapiloff MS, and Dodge-Kafka KL (2013). Regulation of MEF2 transcriptional activity by calcineurin/mAKAP complexes. Exp Cell Res 319, 447–454. 10.1016/j.yexcr.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Marshall PR, Leighton LJ, Zajaczkowski EL, Wang Z, Madugalle SU, Yin J, Bredy TW, and Wei W.(2019). The DNA Repair-Associated Protein Gadd45gamma Regulates the Temporal Coding of Immediate Early Gene Expression within the Prelimbic Prefrontal Cortex and Is Required for the Consolidation of Associative Fear Memory. J Neurosci 39, 970–983. 10.1523/JNEUROSCI.2024-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe AR, Tang JH, Yassif J, Graf M, Huang WY, Groves JT, Weis K, and Liphardt JT (2015). Importin-beta modulates the permeability of the nuclear pore complex in a Ran-dependent manner. Elife 4. 10.7554/eLife.04052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, and Tsien RW (2014). gammaCaMKII shuttles Ca(2)(+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294. 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R.(2018). The Roles of DNA Topoisomerase IIbeta in Transcription. Int J Mol Sci 19. 10.3390/ijms19071917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. (2015). Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605. 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, and Kim TK (2018). Emerging themes in neuronal activity-dependent gene expression. Mol Cell Neurosci 87, 27–34. 10.1016/j.mcn.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabushi R, Tewari S, Gautam SK, Agarwal A, and Agarwal A.(2015). Serratus anterior plane block: a new analgesic technique for post-thoracotomy pain. Pain Physician 18, E421–424. [PubMed] [Google Scholar]
- Meczes EL, Gilroy KL, West KL, and Austin CA (2008). The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS One 3, e1754. 10.1371/journal.pone.0001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, and Curran T.(1986). Role of ion flux in the control of c-fos expression. Nature 322, 552–555. 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, and Baraban JM (1991). L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7, 625–635. 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Navabpour S, Rogers J, McFadden T, and Jarome TJ (2020). DNA Double-Strand Breaks Are a Critical Regulator of Fear Memory Reconsolidation. Int J Mol Sci 21. 10.3390/ijms21238995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, and Akbarian S.(2016). Epigenetic Basis of Mental Illness. Neuroscientist 22, 447–463. 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, Soans E, Rogojina A, Seth A, and Mishina M.(2012). Topoisomerase assays. Curr Protoc Pharmacol Chapter 3, Unit 3 3. 10.1002/0471141755.ph0303s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, and Hogan PG (2004). Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci U S A 101, 7554–7559. 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SJ, Singh NP, and Natarajan AT (1997). Fluorescence in situ hybridization with comets. Exp Cell Res 232, 407–411. 10.1006/excr.1997.3555. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, and Landegren U.(2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Stott RT, Kritsky O, and Tsai LH (2021). Profiling DNA break sites and transcriptional changes in response to contextual fear learning. PLoS One 16, e0249691. 10.1371/journal.pone.0249691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, and Mucke L.(2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci 16, 613–621. 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner MC, and Brickner J.(2021). The Nuclear Pore Complex as a Transcription Regulator. Cold Spring Harb Perspect Biol. 10.1101/cshperspect.a039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck A, Lotz C, Drillien R, Haas L, Bedez C, and Lamour V.(2021). Structural basis for allosteric regulation of Human Topoisomerase IIalpha. Nat Commun 12, 2962. 10.1038/s41467-021-23136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Morgen P, Burdova K, Flower TG, O’Reilly NJ, Boulton SJ, Smerdon SJ, Macurek L, and Horejsi Z.(2017). MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene 36, 4943–4950. 10.1038/onc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, and Greenberg ME (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol 3. 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RH, Hesketh EL, and Coverley D.(2016). The Nuclear Matrix: Fractionation Techniques and Analysis. Cold Spring Harb Protoc 2016, pdb top074518. 10.1101/pdb.top074518. [DOI] [PubMed] [Google Scholar]
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, and Chan NL (2011). Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462. 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. 10.1016/j.neuron.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed blots, gels, and microscopy images are available at Mendeley Data. The mass spectrometry data have been deposited in the public proteomics repository, MassIVE. Accession numbers for the deposited data are listed in the Key Resources Table.
This manuscript does not report original code.
Any additional information required to reanalyze the reported data is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Topo IIbeta (H-8) | Santa Cruz | RRID: AB_628384 |
| Topo IIbeta | Abcam | RRID: AB_2923318 |
| Pan-Calcineurin A Antibody | Cell Signaling Technology | RRID: AB_2168458 |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | MilliporeSigma | RRID: AB_309864 |
| Calcineurin A Polyclonal Antibody | Thermo Fisher Scientific | RRID: AB_10981308 |
| Histone H2A.X antibody | Abcam | RRID: AB_297814 |
| Goat Anti-Rabbit IgG Antibody, IRDye 800CW | LI-COR | RRID: AB_10796098 |
| Lamin A/C (4C11) Mouse mAb (Alexa Fluor 488 Conjugate) | Cell Signaling Technology | RRID: AB_10997529 |
| Phospho-Topoisomerase IIα (Ser1469) (D4F5) | Cell Signaling Technology | RRID: AB_2798109 |
| Phospho-CK2 Substrate [(pS/pT) DXE] MultiMab™ | Cell Signaling Technology | RRID: AB_2797653 |
| Goat anti-Mouse IgG (H + L) Antibody, IRDye 680RD | LI-COR | RRID: AB_10956589 |
| Actin beta | Bio-Rad | RRID: AB_2923317 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific | RRID:AB_2534091 |
| Goat anti-Rabbit IgG (H+L) Highly CrossAdsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific | RRID:AB_2534095 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | RRID:AB_2535805 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | RRID:AB_2535813 |
| Lamin A/C | Cell Signaling Technology | RRID:AB_10545756 |
| Anti-gamma H2A.X (phospho S139) antibody | Abcam | RRID:AB_303388 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Calcineurin Protein | R&D systems | Cat#3160-CA |
| Calmodulin (Human) (recombinant) | Enzo | Cat#BML-SE325–0001 |
| Recombinant Human Topoisomerase II beta/TOP2B protein | Abcam | Cat#ab159736 |
| KN-62 | MilliporeSigma | Cat#422706–1MG |
| KT5720 | MilliporeSigma | Cat#420323–50UG |
| Dantrolene, sodium salt | Tocris | Cat#0507 |
| InSolution™ Okadaic Acid, Prorocentrum | MilliporeSigma | Cat#495609–25UG |
| Nifedipine | Tocris | Cat#1075 |
| 2 aminoethoxydiphenyl borate (2APB) | Tocris | Cat#1224 |
| MEK/ERK inhibitor | Tocris | Cat#PD98059 |
| Calcineurin Inhibitor VIII, CN585 - | MilliporeSigma | Cat#207003–10MG |
| Tacrolimus (FK506) | Selleckchem | Cat#S5003 |
| D-AP5 | Tocris | Cat#0106 |
| Critical commercial assays | ||
| Nick Translation DNA Labeling System | ENZO | Cat#ENZ-GEN111–0050 |
| Duolink® In Situ Detection Reagents FarRed | MilliporeSigma | Cat# DUO92013 |
| Deposited data | ||
| Targeted Mass Spectrometry (PRM LC – MS/MS) | This paper | MassIVE: MSV000088392 |
| Mass Spectrometry (LC – MS/MS of immunoprecipitated Top2B) | This paper | MassIVE: MSV000089825 |
| Mendeley Dataset - Unprocessed blots, gels, and microscopy images | This paper | DOI: 10.17632/8697s83wpv.1 |
| Experimental models: Cell lines | ||
| NIH 3T3 | ATCC | Cat#CVCL_0594 |
| HEK293 | ATCC | Cat#CVCL_0045 |
| Experimental models: Organisms/strains | ||
| Mouse C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Oligonucleotides | ||
| Fos Exon Forward: GCCAACTTTATCCCCACGGT | This paper | N/A |
| Fos Exon Reverse: TCTTCACCATTCCCGCTCTG | This paper | N/A |
| Npas4 Exon Forward: GAGCAAGAGCCTGAGCGAAAAG | This paper | N/A |
| Npas4 Exon Reverse: CCAGCAAAGAAGACACCCTTG | This paper | N/A |
| GAPDH Forward: CTCTCCTCCTCCCTGTTCC | This paper | N/A |
| GAPDH Reverse: TCCCTAGACCCGTACAGTGC | This paper | N/A |
| bglobin Forward: TGACCAATAGTCTCGGAGTCCTG | This paper | N/A |
| bglobin Reverse: AGGCTGAAGGCCTGTCCTTT | This paper | N/A |
| Fos Forward: GAACGGAATAAGATGGCTGC | This paper | N/A |
| Fos Reverse: TTGATCTGTCTCCGCTTGG | This paper | N/A |
| FosB Forward: CCCGAGAAGAGACACTTACC | This paper | N/A |
| FosB Reverse: CTCTTCAAGCTGATCAGTTTCC | This paper | N/A |
| Npas4 Forward: CTGCATCTACACTCGCAAGG | This paper | N/A |
| Npas4 Reverse: GCCACAATGTCTTCAAGCTCT | This paper | N/A |
| Egr1 Forward: AGCGAACAACCCTATGAGCA | This paper | N/A |
| Egr1 Reverse: TCGTTTGGCTGGGATAACTC | This paper | N/A |
| GAPDH Forward: CTCTCCTCCTCCCTGTTCC | This paper | N/A |
| GAPDH Reverse: TCCCTAGACCCGTACAGTGC | This paper | N/A |
| Nr4a1 Forward: ATGCCTCCCCTACCAATCTTC | This paper | N/A |
| Nr4a1 Reverse: CACCAGTTCCTGGAACTTGGA | This paper | N/A |
| ActB Forward: GGCTGTATTCCCCTCCATCG | This paper | N/A |
| ActB Reverse: CCAGTTGGTAACAATGCCATGT | This paper | N/A |
| HA-tagged mouse DNA topoisomerase IIβ | Addgene | Cat# pYLL201 |
| Recombinant DNA | ||
| Fos | CHORI (Children’s Hospital Oakland Research Institute) | RP24–208N11 |
| Fos (upstream) | CHORI (Children’s Hospital Oakland Research Institute) | RP24–368B17 |
| Fos (downstream) | CHORI (Children’s Hospital Oakland Research Institute) | RP24–178I11 |
| FosB | CHORI (Children’s Hospital Oakland Research Institute) | RP24–161J12 |
| ActB | CHORI (Children’s Hospital Oakland Research Institute) | RP24–266N |
| Malat1 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–290K10 |
| Npas4 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–238I5 |
| Gm50442 | CHORI (Children’s Hospital Oakland Research Institute) | RP24–344E18 |
| Software and algorithms | ||
| Other | ||