

Abstract
Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome is a congenital condition characterized by aplasia or hypoplasia of the uterus and vagina in women with a 46,XX karyotype. This condition can occur as type I when isolated or as type II when associated with extragenital anomalies including kidney and skeletal abnormalities. The genetic basis of MRKH syndrome remains unexplained and several candidate genes have been proposed to play a role in its etiology, including HNF1B, LHX1 and WNT4. Here, we conducted a microarray analysis of 13 women affected by MRKH syndrome, resulting in the identification of chromosomal changes, including the deletion at 17q12, which contains both HNF1B and LHX1. We focused on HNF1B for further investigation due to its known association with, but unknown etiological role in, MRKH syndrome. We ablated Hnf1b specifically in the epithelium of the Müllerian ducts in mice and found that this caused hypoplastic development of the uterus, as well as kidney anomalies, closely mirroring the MRKH type II phenotype. Using single-cell RNA sequencing of uterine tissue in the Hnf1b-ablated embryos, we analyzed the molecules and pathways downstream of Hnf1b, revealing a dysregulation of processes associated with cell proliferation, migration and differentiation. Thus, we establish that loss of Hnf1b function leads to an MRKH phenotype and generate the first mouse model of MRKH syndrome type II. Our results support the investigation of HNF1B in clinical genetic settings of MRKH syndrome and shed new light on the molecular mechanisms underlying this poorly understood condition in women’s reproductive health.
Introduction
Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome (OMIM #277000) affects 1 in 4500–5000 women and features a spectrum of phenotypes, characterized by absent or incomplete development of the uterus, cervix and upper two-thirds of the vaginal canal in an otherwise typical 46,XX female. MRKH syndrome is classified into two main groups: type I, which is characterized by aplasia of uterus and/or vagina, and type II, which is associated with additional malformations. A subset of type II consists of Müllerian duct (MD) aplasia, renal aplasia and cervicothoracic somite dysplasia association (OMIM #601076) (1). The most common extragenital malformations include renal (30–57% of cases), skeletal (30–44% of cases), auditory (10–25% of cases) and, less frequently, heart anomalies (2,3). However, ovarian development and function are usually unaffected, allowing conception of biological children through gestational surrogacy or uterine transplantation (4–6). Because women with MRKH syndrome develop otherwise typical female secondary sex characteristics, most are not diagnosed until adolescence due to primary amenorrhea. The diagnosis typically causes significant psychological distress for these women and their families (7).
The genetic cause(s) of MRKH syndrome remains unknown. The situation is complicated by the variability of phenotypes, and the polygenic or multifactorial nature of the condition (8–11). As a result, it has not been possible to provide affected women with diagnostic strategies to elucidate the cause of their condition or satisfactory genetic counseling advice about the potential risk for their future children.
Both sporadic and familial cases have been reported, and analyses of familial clustering have suggested the existence of genetic predisposition (12–14). To date, more than 50 genes have been proposed as candidates for MRKH syndrome, including GREB1L, WT1, TBX6 and SHOX (15–19). However, due to the lack of functional studies, the specific role of these proposed candidate genes in MD development is still unclear. Consequently, most gene changes associated with MRKH syndrome are classified as variants of uncertain significance and their diagnostic potential remains undefined.
In addition to clinical investigations, animal studies have identified a number of genes that are involved in MD development and can also be considered potential candidates for a role in MRKH syndrome. Wnt4 was found to be necessary for urogenital development in the mouse (20), and mutations of WNT4 in humans have been associated with uterine malformations (21). However, due to hyperandrogenism and virilization of the phenotype, this has been proposed as a separate entity from MRKH syndrome (OMIM #158330) (22). In addition, other genes known to play critical roles in MD development of the mouse have yet to be associated with MRKH syndrome. These include Pax2, Digh1, Dach1/2, Hoxa11 and Wnt5a (16,17).
Several human chromosomal imbalances have been described in MRKH syndrome, including 17q12, 16p11.2 and 22q11.21 (23–26). Deletions of 1.4–1.8 Mb in 17q12 represent the most frequent chromosomal rearrangements, accounting for up to 9% of MRKH cases (27,28). This region contains the genes LHX1 and HNF1B, which are both expressed in the epithelium of the developing genitourinary tract (29,30). Ablation of Lhx1 in the mouse results in the block of MD elongation and uterine hypoplasia, demonstrating the critical role of this factor in the development of the female reproductive tract (29,31). Hnf1b has been shown to be an important factor for kidney function and development (32–34), and HNF1B mutations have been associated with complex genitourinary malformations including MRKH syndrome (19,35–37). Constitutive deletion of Hnf1b in the mouse epiblast resulted in the absence of the MD secondary to premature degeneration of the Wolffian ducts (WD) (34). However, early lethality of Hnf1b knock out embryos and the dependency of MD formation on WD integrity have precluded the specific examination of Hnf1b in MD development.
In this study, we performed single nucleotide polymorphism (SNP) microarray in a cohort of 13 women with MRKH syndrome, and identified several copy number variants (CNVs) including 17q12 deletion. We selected HNF1B for in vivo functional analysis due to the known association of 17q12 deletions with MRKH syndrome and the need to establish a functional contribution of Hnf1b to the pathophysiology of MRKH. By ablating Hnf1b specifically in the MD epithelium in mice, we generated a novel model of MRKH type II, characterized by uterine hypoplasia associated with kidney anomalies. Using single-cell RNA sequencing, we identified the molecules and pathways that were affected by Hnf1b ablation in the main cell populations of the uterus. Our translational approach shows that loss of HNF1B disrupts MD development and uterine differentiation, revealing its role in MRKH syndrome.
Results
Identification of CNVs in women with MRKH syndrome
Our cohort comprised 13 participants, and SNP microarray analysis identified CNVs in five of them, one affected by MRKH type I (MRKH06) and four by MRKH type II (MRKH01, MRKH03, MRKH08 and MRKH10) (Table 1).
Table 1.
Summary of variants identified by SNP microarray
| Patient | MRKH Type | Clinical Phenotype | CNV | Size | Location |
|---|---|---|---|---|---|
| MRKH01 | II | Uterovaginal agenesis; moderate renal impairment | Duplication: 1q21.1 Deletion: 17q12 |
1.3 Mb 1.4 Mb |
145 394 955–146 762 959 34 815 551–36 220 373 |
| MRKH03 | II | Uterovaginal agenesis; unilateral renal agenesis | Duplication: 3q13.12q13.13 | 0.4 Mb | 107 822 130–108 214 218 |
| MRKH06 | I | Uterine agenesis | Duplication: 2q24.2 | 0.5 Mb | 160 822 423–161 294 055 |
| MRKH08 | II | Uterovaginal agenesis; fused vertebrae (C2–3), osteophytic foraminal narrowing (right C4–5, right C5–6, right C6–7). | LCSH: Chr4 | >5 Mb | 140 586 701–145 900 002 |
| MRKH10 | II | Uterovaginal agenesis; hearing impairment | Deletion: Xp22.33 LCSH: Chr11 |
0.15 Mb > 5 Mb |
167 755–182 276 101 153 216–107 428 694 |
We found a 1.4 Mb deletion at 17q12 in proband MRKH01. This is one of the most common chromosomal changes found in MRKH syndrome and contains the candidate genes LHX1 and HNF1B. The same participant displayed an interstitial duplication at 1q21.1. Duplications in this area have been reported in intellectual disability with incomplete penetrance and variable phenotypes, although these features did not apply to our case. A 1q21.1 duplication was reported in a woman with Müllerian aplasia but no signs of intellectual disability (24); similar to our case, she had a second CNV, at 22q11.21, which has been associated with Müllerian aplasia (24,26,38).
The other CNVs have not previously been described in MRKH syndrome. The novel duplication at 3q13.12q13.13 in proband MRKH03 contains three main genes—IFTP57, HHLA2 and MYH15—with no known association with Müllerian anomalies. Proband MRKH10 had a microdeletion at Xp22.33 within the upstream conserved noncoding elements enhancer region of SHOX, a gene associated with MRKH syndrome (18). The novel duplication at 2q24.2 of proband MRKH06 involved PLA2R1, ITGB6 and RBMS1, which are all expressed in the reproductive tract. Strikingly, disruption of Rbms1 in the mouse impairs uterine development (39). Finally, we found long continuous stretches of homozygosity (LCSH) in two MRKH08 and MRKH10. Although LCSH can be associated with recessive Mendelian disorders, they usually represent ancestral haplotypes of low-recombination regions and are unlikely to be of clinical significance (40).
In vivo functional testing of candidate gene HNF1B
The deletion at 17q12 is well known for being associated with MRKH syndrome. In our cohort, we found it in 1/13 cases (7%), in accord with the reported prevalence of 2–9% (24,25,27,38,41,42).
To identify the specific function of HNF1B and its contribution to the pathogenesis of MRKH syndrome, we generated and studied a ‘conditional’ loss-of-function model in mice using a Cre/lox recombination system. This strategy requires a mouse line expressing a tissue specific Cre recombinase and a line harboring a gene of interest flanked (‘floxed’) by loxP sequences. When the two lines are crossed, the Cre protein recombines the loxP sites only in the tissue where it is expressed causing deletion of the gene of interest. To ablate Hnf1b specifically in the epithelial cells of the MD, we crossed a Wnt7a-Cre line with a mouse line carrying loxP sites flanking Hnf1b exon 4 (Hnf1bfl/fl) (31,32). We first used the ROSA26-LacZ mouse reporter line to verify the expression of the Cre recombinase in the Wnt7a-Cre mouse line. We found that Cre was expressed throughout the entire period of MD formation, as expected (31). At 11.5 days post coitum (dpc), a few mesonephric epithelial cells begin differentiation into MD epithelial cells (Supplementary Fig. S1A). This is followed by the formation of the tubular structure of the MDs, which elongate posteriorly until they reach the urogenital sinus at 13.5 dpc (Supplementary Material, Fig. S1A). As demonstrated by β-gal activity, Cre expression was detected in the epithelium of the MD but not the WD (Supplementary Material, Fig. S1B).
To confirm specific ablation of Hnf1b in Wnt7a-Cre+;Hnf1bfl/fl embryos, we performed immunofluorescence staining at 13.5 dpc, when both the MD and WD are normally present, and birth (P0), when only the MD remains in female animals. In non-Cre expressing littermates (Wnt7a-Cre−;Hnf1bfl/fl mice, referred to as controls), HNF1B protein was expressed in both the MD and WD at 13.5 dpc and in the MD at P0, as expected (Fig. 1A and B). However, the MD of Wnt7a-Cre+;Hnf1bfl/fl mice (referred to as mutants) were negative for HNF1B at both stages (Fig. 1A and B). We confirmed these results by quantitative real-time polymerase chain reaction (qRT-PCR) analysis, showing a reduction of Hnf1b mRNA levels by 40% compared with controls, consistent with loss of expression in the MD but not WD (Fig. 1C). At P0, however, when the WD has regressed, Hnf1b expression was abolished in mutants (Fig. 1C). These results indicate successful ablation of Hnf1b in the developing MD of mutant animals.
Figure 1.
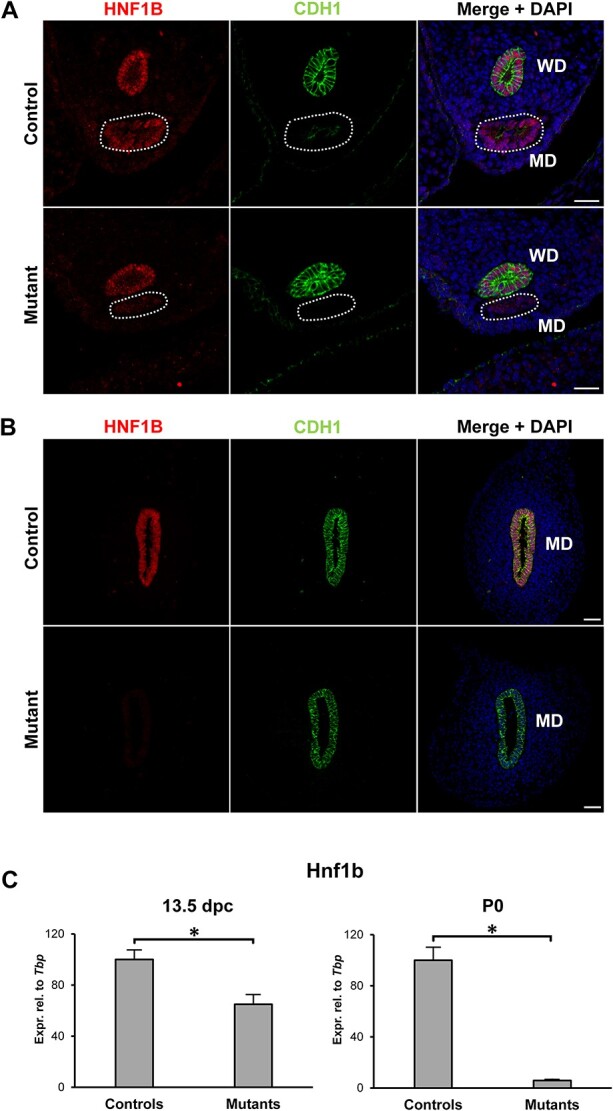
Hnf1b ablation in Mullerian duct epithelium. (A) In control animals, HNF1B was expressed in both the Wolffian ducts (WD) and the Müllerian ducts (MD, dashed line) at 13.5 dpc. In mutants, HNF1B expression was lost specifically in the MD. (B) At P0, only the MD remains in female animals, and immunohistochemistry showed no expression in the mutants. The epithelial marker CDH1/E-cadherin was used to visualize the WD at 13.5dpc and the MD at P0. Scale bar = 25 μm. (C) Real-time PCR of whole mesonephric tissues showed a reduction in Hnf1b expression at 13.5 dpc in mutant samples, consistent with the presence of the Hnf1b-expressing WD. At P0, expression of Hnf1b was lost in the mutants. * = P < 0.05.
Conditional ablation of Hnf1b results in uterine hypoplasia and renal anomalies
We examined the development of the female reproductive tract in Wnt7a-Cre+;Hnf1bfl/fl embryos to determine whether deletion of Hnf1b in mice has any consequences that mirror the phenotype of MRKH women. At embryonic stages, there were no gross differences between mutant and control MDs. However, by P0, the uteri of mutant mice appeared smaller, less developed and were ~20% shorter than controls (Fig. 2A–C). Hematoxylin and eosin staining revealed that the mutant epithelium failed to organize in the typical pseudostratified columnar fashion of controls (Fig. 2D). At P0, the stromal compartment showed reduced cell density, and overall, cross-sections of the mutant uterus were smaller compared with controls (Fig. 2D).
Figure 2.

Phenotype of the Hnf1b mutant genitourinary system. (A and B) Gross morphology of reproductive tracts showed uterine horns of Hnf1b mutant mice that appeared smaller than controls. (C) Measurement of the uterine horn length at P0. n = 5. (D) Hematoxylin and eosin staining at P0 and 4 M. Both epithelial and stromal compartments of the endometrium appeared hypoplastic in the mutant uteri; 4 M control samples showed uterine glands (arrows) that were lacking in the mutants. (E) Unilateral kidney agenesis in a mutant animal. (F) Hematoxylin and eosin staining of kidney samples showing a case of cystic kidney in the mutant. Scale bars = 2 mm (A, E, F), 1 mm (B), 20 μm (D, P0 and 4 M right), 200 μm (D, 4 M left). * = P < 0.05.
In adult mutant mice at 4 months of age, the uterus remained smaller, characterized by a thin epithelium, a stroma with rare endometrial glands (89.53 ± 4.03 versus 6.66 ± 1.56 in controls and mutant uteri, respectively), and a thicker outer longitudinal myometrium (OLM, Fig. 2D, Supplementary Material, Fig. S2). Despite the uterine hypoplasia, and similar to MRKH syndrome, the ovaries of mutant animals did not show any difference compared with controls, displaying follicles at every stage of folliculogenesis. Consistent with normal follicle dynamics, mutant female mice mated without apparent problems as evidenced by the presence of regular plugs. As expected, however, they failed to achieve pregnancy (Supplementary Material, Figure S3).
Surprisingly, we found that the Hnf1b mutant phenotype was associated with renal anomalies similar to those described for MRKH. We detected unilateral kidney agenesis in 6 of 36 mutant mice (16.7%), whereas it was never observed in controls (n = 35, P = 0.0249) or heterozygous animals (n = 64, P = 0.0016) (Figure 2E). In addition to agenesis, we found cystic kidneys as an additional renal association (Figure 2F) but did not assess their frequency due to the more subtle phenotype, which could not be identified a priori. Immunofluorescence staining of 13.5 dpc kidney sections showed diminished expression of HNF1B in several developing branches (Supplementary Material, Figure S4). These results show that Hnf1b ablation results in uterine hypoplasia associated with kidney anomalies, providing a mouse model for MRKH syndrome type II.
Hnf1b regulates female reproductive tract differentiation
To investigate the molecular mechanisms by which the reproductive tract anomalies in Wnt7a-Cre+;Hnf1bfl/fl mice arise, we next examined the expression of several known markers of MD development. At 13.5 dpc, we found a significant dysregulation of the genes Lhx1 and Dach2 (Figure 3A). However, several other factors including Wnt4, Wnt5a, Wnt7, Wnt9, Dach1, Hoxa11 and Pax2 were not differentially expressed between mutants and controls (Supplementary Material, Figure S5).
Figure 3.

Ablation of Hnf1b cause dysregulation of few key genes involved in MD development. (A) Real-time PCR of control and Hnf1b mutant samples at 13.5 dpc showing dysregulation of Lhx1 and Dach2. n = 4. (B) Volcano plot of the Wnt pathway RT2 Profiler PCR Array. Upregulated genes are in red, downregulated in blue. n = 3; * = P < 0.05.
Because of the importance of the Wnt pathway in tissue morphogenesis, we further analyzed Wnt gene expression using an RT2 Profiler PCR Array. The upregulated genes were Wnt2, and the Wnt-inhibitors Wif1 and Sfrp4, whereas the Wnt target Fosl1 was downregulated (Figure 3B). As Wnt signaling plays critical roles in cell proliferation, we performed immunofluorescence staining of the MD using PH3 as proliferation marker. At 12.5 dpc, there was no difference in MD epithelial cell proliferation between mutants and controls (Figure 4A and B). However, at 13.5 dpc, proliferation started to decrease in the mutants compared with controls (P = 0.048) and was strikingly reduced by P0 (Figure 4A and B), which marks the start of the differentiation of the epithelial, stromal and smooth muscle compartments. These results are consistent with the reduced proliferation of the MD epithelium in Hnf1b mutant mice, resulting in the observed shorter uterine length. Furthermore, epithelial cell proliferation was almost completely absent in young (P7) and adult (4-month-old, 4 M) uterine tissues (Figure 4C), similar to histologic observations of uterine rudiments in women with MRKH syndrome (43).
Figure 4.

Decrease in cell proliferation in Hnf1b mutants. (A, C) Immunofluorescence staining of the proliferation marker PH3 in samples at 12.5 and 13.5 dpc, and P0, P7 and 4 M. (B) Quantification of PH3-positive cells in 12.5-, 13.5 dpc and P0 samples. n = 3. Scale bar = 25 μm. * = P < 0.05, ** = P < 0.01.
We further characterized the uterine hypoplasia in Hnf1b mutants by analyzing the expression of specific markers, including the epithelial PAX2, the stromal FOXL2 and VIM, and smooth muscle actin (SMA) from P0 to 4 M. In controls, PAX2 remained highly expressed throughout development and complete maturation of the uterus. However, immunostaining of PAX2 in P0 mutants did not appear uniform (Figure 5A, arrowheads), and by P7, loss of PAX2 expression was evident and limited to few, sparse clusters of epithelial cells (Figure 5A, arrows). Similarly, at 4 months, PAX2 was expressed only in some cells of the uterine epithelium (Figure 5A, arrows). At P0, FOXL2 was not expressed in control or mutant mice. However, while it was expressed as early as P7 in controls, it failed to be properly turned on in the mutant uterus, where it was virtually undetectable by 4 months (Figure 5A). VIM and SMA were both downregulated in the mutants at P0, consistent with underdevelopment of the mutant uterus compared with control (Figure 5B). At P7, however, they were upregulated in mutant samples (Figure 5B). By 4 M, SMA staining showed the formation of a thicker but less organized myometrium, especially in its inner circular layer, which lacked a longitudinal pattern of expression (Figure 5B). Interestingly, VIM remained upregulated in the stromal endometrium but showed dysregulation in the myometrium (Figure 5B, arrow). In the control uterus, VIM co-localized with SMA in the inner circular and OLM (Figure 5B, asterisks; Supplementary Material, Figure S6). In the mutant, however, both myometrial layers lost expression of VIM (Figure 5B, arrowheads; Supplementary Material, Figure S6).
Figure 5.

Dysregulation of epithelial and stromal markers in Hnf1b mutant uteri. (A) Immunofluorescence staining of FOXL2 and PAX2 in samples at P0, P7 and 4 M. After birth, FOXL2 failed to be upregulated whereas PAX2 lost expression in mutant samples. (B) Immunofluorescence staining of SMA and VIM in samples at P0, P7 and 4 M. In mutants, SMA and VIM had reduced expression at P0 but were upregulated at P7. By 4 M, SMA showed the development of a thicker myometrium and VIM continued to be upregulated in the stroma (arrow) of mutant animals. VIM expression co-localized with SMA in both the inner circular (ICM) and OLM of control (asterisks) but not mutant (arrowheads) samples. Scale bar = 25 μm.
We also checked the expression of laminin, an important component of the basal lamina of epithelial and endothelial layers (Figure 6). Laminin showed high and uniform expression in control mice. In the mutants at P0, laminin showed a lower expression at the base of the MD epithelium while remaining normally expressed around blood vessels (Figure 6, arrowheads). By P7, and showing also in 4 M samples, the mouse uterus had completely lost expression of laminin underneath the MD epithelium (Figure 6, dashed lines).
Figure 6.
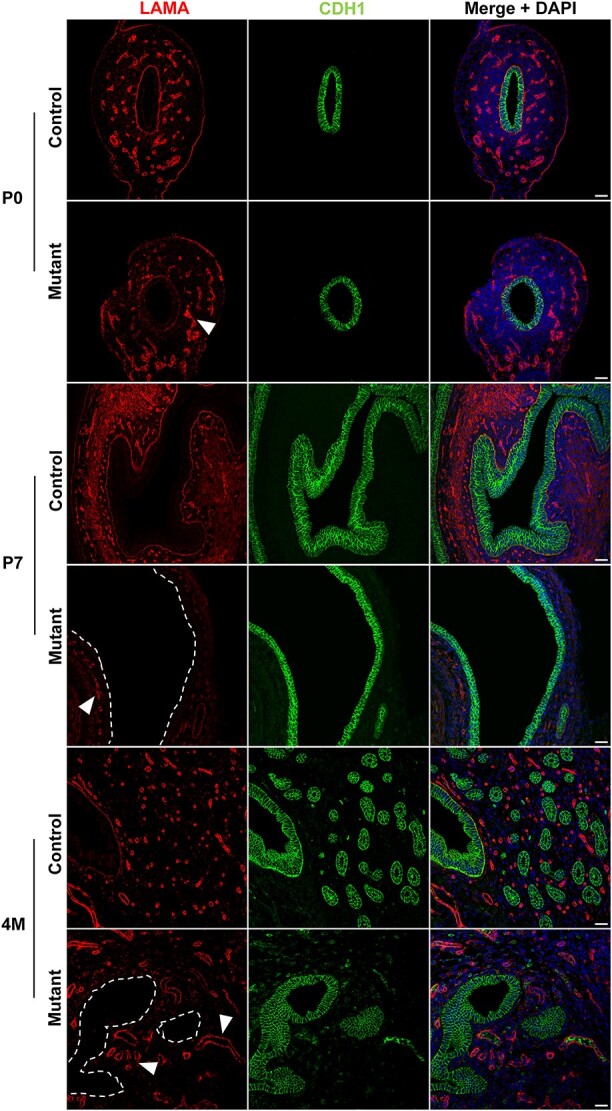
Laminin is lost in the uterine epithelium basal lamina. Immunofluorescence staining of LAMA and CDH1 in control and mutant samples at P0, P7 and 4 M. Laminin is gradually lost underneath the uterine epithelium (dashed lines represent the location of the missing LAMA staining). In mutant samples, LAMA was expressed around the vasculature (arrowheads) similar to controls. Scale bar = 25 μm.
Altogether, these results show that key epithelial, stromal and smooth muscle markers are dysregulated as early as P0, consistent with the uterine hypoplasia observed in mutant mice.
Single cell transcriptomics of the differentiating uterus
Although Hnf1b was ablated specifically in the MD epithelium, uterine hypoplasia extended to the stromal and muscular compartments. To better understand the specific molecular and cellular changes in each cell type, we performed single-cell RNA-Seq analysis at P0, the time when immature MD cells start differentiating into specialized uterine cells (44). Single cell transcriptomic analysis identified nine distinct cell populations in both control and Hnf1b mutants (Figure 7A). We assigned identities to these clusters based on reported expression enrichment of specific markers and the Mouse Cell Atlas single cell RNA-Seq database (45).
Figure 7.

Cell population of the newborn uterus. (A) UMAP plot from sc-RNA Seq analysis of uterine samples at P0 reveals 9 cell clusters (C1–C9 from largest to smallest). (B) UMAP plot of the C1 stromal population after sub-clustering. (C) Violin plots of the top upregulated and (D) downregulated genes in the epithelial cell cluster.
Cluster C1 represented the most abundant cell population expressing stromal markers such as Des, Htra3, Cnrip1 and Csrp1. Cluster C2 expressed genes that are characteristic of proliferating cells, including Stmn1, Top2a, Mki67, Prc1 and Kif11. Cluster C3 showed expression of genes shared by stromal and epithelial cells. This cluster may represent cells undergoing epithelial–mesenchymal transition, a critical process during MD development (46). Alternatively, C3 could be an unidentified cell type with similar gene expression profile to both stromal and epithelial cells. Cluster C4 represented pericytes, which expressed the markers Cspg4, Abcc9, Rgs5 and Cald1. Cluster C5 consisted of epithelial cells, which expressed markers including Cdh1 and Cldn4. Cluster C6 showed enriched expression of genes associated with endothelial cells, including Cd93, Tie1 and Esam. Cluster C7 expressed genes characteristic of the mesothelium such as Msln, Dpp4, Muc16 and Fbln2. Cluster C8 represented the erythroid lineage, expressing markers Hba.a1, Hba.a2, Hbb and Alas2. Finally, cluster C9 comprised myeloid cells, which expressed Lyz2, Cybb and Aif1.
To better characterize the large population of stromal cells, we performed further sub-clustering of C1, revealing four sub-types (Figure 7B). Cluster C1.1 expressed markers of the inner stroma Vcan, Plac8, Cpxm2 and Axin2 (47). Cluster C1.2 represented smooth muscle cells, which expressed Ptn, Lum, Col1a1 and Gpc3. Cluster C1.3 showed enrichment of outer stroma genes such as Smoc2, Apoe and Fbn2. Cluster C1.4 expressed genes associated with cell cycle, including Hmgb2, Tubb5, Cenpf and Tmpo, and represented proliferating stromal cells.
Single cell transcriptomic profile reveals dysregulation of key pathways during reproductive tract differentiation
To gain further insight into the molecular processes that were disrupted by Hnf1b ablation, we analyzed the gene expression profiles of the three main clusters of epithelial, stromal and proliferating cells (Supplementary Material, Tables S1–S3). Consistent with our histological findings, DAVID Gene Ontology analysis revealed that the most affected biological processes involved gene expression, cell differentiation and development (Table 2). In addition, ingenuity pathway analysis (IPA) showed that the most enriched functional annotations for the differentially expressed genes in our datasets were organismal development, survival, embryonic development and organ morphology (Table 3). Pathway analysis showed that the most dysregulated signaling pathways were the estrogen receptor, TGFβ,□WNT and PI3K/AKT pathways (Table 4).
Table 2.
Gene ontology analysis of differentially expressed genesa
| Epithelial cells | Stromal cells | Proliferating cells | ||||
|---|---|---|---|---|---|---|
| Rank | GO class | # molecules | GO class | # molecules | GO class | # molecules |
| 1 | Positive regulation of transcription from RNA polymerase II promoter | 97 | Regulation of transcription from RNA polymerase II promoter | 319 | Regulation of transcription from RNA polymerase II promoter | 172 |
| 2 | Cell differentiation | 74 | Positive regulation of transcription from RNA polymerase II promoter | 265 | Positive regulation of transcription from RNA polymerase II promoter | 158 |
| 3 | Multicellular organism development | 70 | Negative regulation of transcription from RNA polymerase II promoter | 223 | Regulation of transcription, DNA templated | 137 |
| 4 | Negative regulation of transcription | 69 | Cell differentiation | 198 | Negative regulation of transcription from RNA polymerase II promoter | 133 |
| 5 | Lipid metabolic process | 60 | Multicellular organism development | 185 | Multicellular organism development | 126 |
aTop 5 Biological Process classes in epithelial, stromal and proliferating cell clusters.
Table 3.
Pathway analysis of differentially expressed genes in epithelial, stromal and proliferating cell clustersa
| Epithelial cells | Stromal cells | Proliferating cells | ||||||
|---|---|---|---|---|---|---|---|---|
| Category | No. of molecules | P-value range | Category | No. of molecules | P-value range | Category | No. of molecules | P-value range |
| Organismal development | 226 | 1.72E−05–1.57E−14 | Organismal development | 80 | 4.29E−03–2.16E−05 | Organismal development | 70 | 6.68E−03–2.66E−05 |
| Organismal survival | 162 | 9.42E−06–1.75E−16 | Organismal survival | 66 | 5.51E−07–5.51E−07 | Cardiovascular system development and function | 36 | 6.68E−03–2.66E−05 |
| Embryonic development | 127 | 1.02E−05–4.67E−12 | Organ morphology | 51 | 4.50E−03–2.16E−05 | Connective system development and function | 35 | 6.68E−03–2.64E−05 |
| Cardiovascular system development and function | 117 | 1.86E−0–3.58E−13 | Cardiovascular system development and function | 47 | 3.96E−03– 1.08E−05 | Tissue morphology | 28 | 6.68E−03–2.64E−05 |
| Digestive system development and function | 70 | 1.33E−05– 9.99E−12 | Reproductive system development and function | 28 | 4.06E−03– 9.78E−06 | Skeletal and muscular system development and function | 23 | 6.68E−03–8.29E−07 |
aMost enriched ‘System Development and Function’ categories.
Table 4.
Dysregulated signaling pathways in Hnf1b mutant uteri
| Cell type | Signaling pathways | P-value |
|---|---|---|
| Epithelial | TGFb Estrogen WNT |
5.60E−03 1.90E−02 3.50E−02 |
| Stromal | WNT PI3K-AKT TGFb |
1.30E−03 2.10E−02 4.90E−02 |
| Proliferating | WNT PI3K-AKT |
3.50E−02 4.90E−02 |
The specific molecular role of Hnf1b during MD development remains unknown. To address this issue, we focused on the epithelial cluster, where Hnf1b functions cell-autonomously. Analysis of the most differentially expressed genes, shown in Figure 7C–D, revealed that Hnf1b controls critical processes of MD development. Dysregulated factors included Erdr1, Prap1, Hdc, Ifitm1, Tgf2b, Lgals7 and Ripply3, which are involved in cell proliferation, migration and patterning; Krt5, Krt14, Krt15 and Krt79, which regulate epithelial cell development; Ndufa4l2, involved in the estrogen receptor pathway; and Calb1, which plays a role in kidney development. In addition, 66 dysregulated genes were involved in epithelial apico-basal and baso-lateral polarity, suggesting that Hnf1b has a role in establishing MD epithelial polarization similar to its function in the kidney (48) (Supplementary Material, Table S4).
We then looked for potential targets by interrogating the Ingenuity Knowledge Database and the ToppGene Suite for factors that are annotated as either having HNF1B transcription factor binding sites or a direct downstream relationship with HNF1B. We identified a total of 37 genes that were differentially expressed in the Hnf1b mutant uterine epithelium (Table 5). These factors included known targets such as Lgals3, Socs3, Crb3 and Glis3 (48–51). Other particularly relevant genes included Ppargc1a, which is regulated by estrogen in the uterus; Cldn10, which is required to maintain epithelial integrity; Axin2, which is a target of WNT and is necessary for epithelial homeostasis; Mafb, which is directly activated by HNF1B in response to retinoic acid; and the estrogen pathway gene Pcbd1, whose product is a binding partner of HNF1B (52–56). Strikingly, several of these genes have been identified as targets of HNF1B in ChIP-Seq analyses of renal cells, including 18 factors that are involved in cell polarization (Table 5, Supplementary Material, Table S4) (57).
Table 5.
Known and putative targets of HNF1B
| Gene | Log Fold Change | FDR |
|---|---|---|
| Anxa4 | 0.73 | 6.55E−03 |
| Aspa | -2.16 | 1.52E−02 |
| Atp6v0a4 | 1.59 | 2.79E−02 |
| Axin2 | -0.92 | 2.97E−02 |
| Birc5 | -0.76 | 1.82E−02 |
| Cldn10 | -2.11 | 5.09E−04 |
| Cluh | -0.82 | 3.27E−02 |
| Col4a5 | 1.06 | 2.79E−03 |
| Crb3 | 0.64 | 2.54E−03 |
| Ddr1 | 1.15 | 5.63E−05 |
| Dpp4 | 2.61 | 8.87E−04 |
| Enpp3 | 1.25 | 2.64E−02 |
| Eps8 | 2.37 | 1.54E−03 |
| Ganc | 1.63 | 9.85E−03 |
| Glis3 | 0.94 | 7.85E−03 |
| Gnas | 0.43 | 3.67E−03 |
| Hax1 | 0.86 | 2.42E−03 |
| Igfbp3 | 1.36 | 7.36E−04 |
| Igsf5 | 1.73 | 3.05E−02 |
| Itpk1 | 1.14 | 4.69E−03 |
| Kcnk1 | 0.39 | 1.85E−02 |
| Lgals3 | -0.37 | 1.34E−02 |
| Mafb | -1.85 | 4.93E−04 |
| Mmp10 | 1.21 | 4.23E−02 |
| Napsa | 2.57 | 2.03E−03 |
| Ppargc1a | -1.29 | 3.02E−02 |
| Pcbd1 | 1.23 | 1.96E−04 |
| Plau | -2.32 | 5.26E−04 |
| Prom1 | 0.97 | 3.48E−03 |
| Slc6a6 | -0.62 | 3.16E−02 |
| Slc7a5 | -0.99 | 4.13E−02 |
| Slc12a2 | 0.79 | 1.24E−02 |
| Slc39a4 | 3.46 | 5.85E−04 |
| Slc40a1 | -0.96 | 3.84E−02 |
| Socs3 | 0.52 | 2.52E−02 |
| Srsf7 | -0.77 | 5.85E−04 |
| Tgfbr1 | 1.10 | 1.65E−04 |
Underlined genes have been reported in HNF1B ChIP-Seq experiments (57).
These results show that Hnf1b regulates key processes of MD development including epithelial cell proliferation, patterning and differentiation. Consistent with our histological findings, they also show that Hnf1b ablation disrupts epithelial polarity leading to loss of basal membrane.
Discussion
MRKH syndrome is a significant women’s reproductive health issue. Like other Müllerian anomalies, this congenital condition is characterized by variable presentations and a complex, undefined genetics (58). MRKH syndrome denies affected women the possibility of conceiving and bearing children, and its almost invariably unexpected diagnosis can result in distress, confusion, depression and shame (7). The situation is exacerbated by the current inability of healthcare professionals to provide a molecular diagnosis and hence an explanation for how the condition arose in any given affected woman. In this light, defining the genetic causes of MRKH syndrome is an important goal but remains an ongoing challenge in clinical genomics. Several approaches have been applied including microarray analysis and genome-wide DNA sequencing (59–62). These investigations have revealed a long list of candidate genes, but confirmation of their role in MD development, and most importantly MRKH syndrome, is lacking.
In this study, we combined SNP microarray in a cohort of women with MRKH syndrome and functional genomics in the mouse model to investigate the role of candidate gene HNF1B. We identified a heterozygous deletion at 17q12, which is the most frequent chromosomal change associated with MRKH syndrome (19,27,28). Genitourinary anomalies, including MRKH syndrome, are associated with systemic conditions including 17q12 deletion syndrome (16). Affected patients may show congenital abnormalities of the kidney and urinary tract, or maturity-onset diabetes of the young type 5, sometimes in association with kidney cysts (renal cysts and diabetes syndrome) (16,37). Clinical diagnosis of these conditions is often accompanied by genetic testing. However, if MRKH syndrome presents in isolation, genetic testing may not be performed, and an accurate, molecular diagnosis may be missed.
The 17q12 region contains two genes—LHX1 and HNF1B—that are expressed in the genitourinary tract and are considered strong candidates for MRKH syndrome. Conditional deletion studies in the mouse have demonstrated the critical role of Lhx1 in MD development (31). However, the specific function of Hnf1b has not been investigated. Disruptions of HNF1B are associated with complex presentations involving the female reproductive tract and the renal system (36,63,64). Whole gene deletions account for roughly half of cases, whereas the remainder are due to point mutations, and no genotype–phenotype differences have been found between patients with 17q12 deletions and HNF1B point mutations (65–70).
By specific ablation in the MD epithelium, we demonstrated that Hnf1b is necessary for the differentiation of the female reproductive tract. We found several similarities, but also striking differences, with the phenotype of Lhx1 conditional knockouts (31). Hnf1b loss-of-function caused a decrease in MD epithelial cell proliferation, which started as early as 13.5 dpc, leading to the development of a shorter uterus. This was similar to the reduced cell proliferation in the Lhx1 mutant MD epithelium. Hnf1b mutant mice showed upregulation of Wif1, a gene controlled by AMH and involved in MD regression in males (71). Although a few additional genes were dysregulated during embryonic development, including Lhx1 and factors of the Wnt pathway, morphological differences between Hnf1b mutant and control animals became evident around the time of birth when uterine differentiation takes place (44). Hnf1b mutant mice displayed a hypoplastic uterus, characterized by a simple cuboidal epithelium and reduced stromal thickness that failed to properly develop endometrial glands. This phenotype was less severe than that of the Lhx1 conditional knockout, which displayed a lack of the entire endometrial layer (31). This was attributed to the loss of the epithelial compartment and therefore its inability to induce postnatal mesenchymal differentiation. In the Hnf1b mutant uterus, the epithelial layer did not regress but was unable to differentiate beyond a vestigial state, consequently affecting the postnatal development of the stromal lineages. This is similar to the role Hnf1b shows in the kidney, where it is not required for initial nephrogenesis, but is necessary for subsequent differentiation of the proximal and distal tubules (33). Overall, the Hnf1b mutant uterus resembled the histology of uterine rudiments found in MRKH syndrome, including a lower cell proliferation capacity and a simple, less differentiated epithelium (43).
Our transcriptomic analysis provided new insight into the molecular mechanisms responsible for uterine hypoplasia in mutant mice. Processes affected by Hnf1b loss included cell proliferation and differentiation in the epithelial, stromal and proliferative cell clusters. Analysis of the epithelial lineage revealed key cell-autonomous functions of Hnf1b and factors previously identified as HNF1B targets, including Lgals3, Crb3 and Glis3. Loss of Hnf1b resulted in the downregulation of important genes regulating epithelial integrity including Cldn10, Axin2 and several keratin members. In addition, Socs3 was upregulated in the mutant epithelium, confirming previous results (49). Repression of Socs3 by HNF1B is necessary for proper tubulogenesis of the kidney (49).
Several genes with apico-basal and baso-lateral localization were dysregulated in the MD of Hnf1b mutants (Supplementary Materials, Table S4). This was similar to the role HNF1B displays in the kidney, where it directly regulates genes involved in epithelial apico-basal polarity and was consistent with the loss of laminin we observed in the mutant uteri (48,72). Laminin is a component of the basement membrane, required for maintaining polarization of the epithelial layer and regulating morphogenesis and patterning (73). The Hnf1b mutant uterine epithelium failed to maintain expression of PAX2, which is necessary to maintain epithelial integrity and polarity (74). As the basal lamina is also important for the functional coordination between epithelium and the underlying stroma, loss of laminin could explain the dysregulated expression of stromal markers including VIM and SMA, which were downregulated at birth but upregulated at later stages. In addition, stromal FOXL2, which is turned on after birth, was not expressed, leading to a phenotype similar to that of PgrCre/+;Foxl2fl/fl mutant mice and characterized by a smaller stromal compartment and a thicker myometrial layer (75). This could be due to the proposed role of FOXL2 in regulating Wnt7a, which in turn maintains the expression of other Wnt and Hoxa factors inducing differentiation of the stroma into the endometrial and myometrial compartments (75).
Pathway analyses showed that several main signaling pathways of MD development were disrupted. In uterine epithelial cells, where Hnf1b is normally expressed, the estrogen receptor pathway regulates proliferation as well as postnatal development of uterine glands (76). In addition, we found the downregulation of critical factors including Lef1, Wnt6 and Wnt10a, suggesting inhibition of the Wnt pathway, which plays critical roles in cell differentiation and epithelial–mesenchymal transition, both necessary for the development of the reproductive tract. The PI3K/AKT pathway is involved in the breaking down of the extracellular matrix to allow for MD elongation (77). Finally, TGFb signaling was dysregulated in epithelial and stromal cells. This pathway is involved in maintaining uterine integrity and function and may also play a role in early MD development (78,79).
Although Hnf1b plays important roles in kidney development, the finding of kidney anomalies following Hnf1b ablation in the MD epithelium was surprising (32,34,48). It is unclear why this phenotype occurred only in some mutant animals. This may be due to the Cre-directed ablation of Hnf1b in a subset of cells of the developing kidney (Supplementary Material, Figure S4). Maybe the activity of the Wnt7a promoter is variable in some mesonephric or metanephric cells leading to inconsistent Cre expression. However, the Wnt7a-Cre mouse line in our study was the same used to ablate Lhx1, which did not result in kidney anomalies (31). This suggests a specific contribution of Hnf1b in urogenital conditions consistently with the observation of frequent kidney anomalies in HNF1B ablations or modifications (36,63,64). The prevalence of kidney agenesis in Hnf1b mutant mice was similar to that of MRKH type II, and this was reminiscent of the incomplete penetrance and variable expressivity characteristic of this condition (2).
Our study demonstrates the benefit of modeling MRKH syndrome in the mouse following identification of candidate genes in humans. In addition to the known 17q12 region, our genomic analyses detected several novel CNVs, providing additional clues to the already complex genetics of MRKH syndrome. The identified chromosomal locations 1q21.1, 2q24.2, 3q13.12q13.13 and Xp22.33 contain several genes involved in developmental processes, including Ift57, which regulates embryo patterning (80); and Rbms1, whose ablation in the mouse is associated with underdevelopment of the uterus (39). Future functional studies may elucidate the potential role of these genes in MD development and genitourinary anomalies.
Conclusions
A diagnosis of MRKH syndrome has a major impact on the lives of affected individuals. There are likely to be several pathogenetic pathways, involving a number of genes, that can result in MRKH syndrome but, for the present, these remain unknown. Our study shows the potential of in vivo functional genomic analyses, following genetic testing and candidate gene identification, to provide information about syndrome causation. Our research has demonstrated that Hnf1b inactivation causes MRKH features in the mouse and provides further evidence supporting the genetic investigation of HNF1B in girls and women with amenorrhea, especially if renal disease or anomalies are present (81). These outcomes will aid efforts to provide informative molecular diagnosis for MRKH syndrome, improve general care and genetic counseling options, and lead to better psychological support and increased awareness of a condition that remains enigmatic for clinicians, scientists, affected women and the community.
Materials and Methods
Participants
Inclusion criteria for recruitment of participants to this study were MRKH type I or II diagnosed clinically or radiographically. The cohort comprised 13 participants affected by MRKH syndrome and recruited by their primary clinician. For two participants, we were unable to retrieve information related to their specific type of MRKH syndrome. Each participant was de-identified and assigned a case number (e.g. MRKH01, MRKH02). Written informed consent was received prior to participation.
Genetic analysis
DNA was extracted from peripheral blood by the Victorian Clinical Genetics Services (VCGS) Molecular Genetics laboratory or local laboratories using standard protocols. DNA quality was assessed by a TapeStation 2200 (Agilent, California, USA) and concentration was measured using Qubit dsDNA Broad Range (Thermofisher, Massachusetts, USA). Cytogenetics confirmed that all women had a 46,XX karyotype.
SNP microarray
DNA samples were analyzed for CNVs by SNP microarray undertaken by the VCGS cytogenetics laboratory using the Omni 2.5–8 (Illumina, California, USA) technology. This platform provides high-level SNP coverage of exonic regions (1–2 SNP probes per 10 kb) through the entire genome. The assay detects copy number changes to an average resolution of 10–20 Kb, LCSH regions >2 Mb, and mosaicism above 10–20%. Library preparation and sequencing was carried out according to the manufacturer’s instructions. CNV coordinates were based on the National Center for Biotechnology Information Human Genome Build 37.
Mouse strains and husbandry
Hnf1bfl/fl mice were a generous gift from Dr S. Cereghini (32), and Wnt7a-Cre mice were a generous gift from Dr R. Behringer (31). ROSA26-LacZ (B6;129S-Gt(ROSA)26Sor/J) mice were obtained from the Jackson Laboratory (82). All mouse strains were maintained on a C57BL/6 genetic background. Samples were collected at 12.5- and 13.5-dpc, 0- and 7-days post-partum and 4 months of age. All animal procedures were approved by the University of Queensland Animal Ethics Committee.
RNA expression analysis
Samples were stored in RNAlater (Invitrogen, Massachusetts, USA) and total RNA was extracted using either an RNAeasy mini or RNAeasy micro kit (Qiagen, Germany). Following RNA amplification into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Massachusetts, USA), real-time PCR assays were performed on three biological replicates per genotype using a SYBR Green PCR master mix (Applied Biosystems). Quantitative real-time PCR was performed using a QuantStudio 3 system (Applied Biosystems). Expression was normalized to Tbp. Primers used are listed in Supplementary Material, Table S5.
Histological preparation and immunohistochemistry
Samples were fixed overnight in 4% paraformaldehyde-phosphate buffered saline (PBS). After fixation, samples were embedded in paraffin and sectioned at 5 μm. For immunohistochemistry, ovary sections were deparaffinized in xylene and rehydrated using sequential washes and decreasing alcohol concentrations. Heat-mediated antigen retrieval was performed in 10 mM citrate buffer or Tris-based buffer (Vector, California, USA). Immunofluorescence was done as previously described (83) using the following primary antibodies: CDH1 (1:100, BD Biosciences, New Jersey, USA, #610182), HNF1B (1:100, Thermo Fisher Scientific #PA-50531), FOXL2 (1:100, Novus Biologicals, Colorado, USA, #NB100–1277), PAX2 (1:100, BioLegend, California, USA, #901001), PH3 (1:100, Abcam, United Kingdom, #5176), LAMA (1:100, Sigma, Missouri, USA, #L9393), VIM (1:200, Abcam #92547), SMA (1:100, Sigma #A2547) and FOXA2 (1:200, Cell Signaling, Massachusetts, USA, # 8186S). Images were taken on a LSM710 confocal microscope (Zeiss, Germany) and processed using Adobe Photoshop.
Single-cell RNA sequencing
Dissected reproductive tracts were cut into small pieces and incubated in a solution of DMEM/F12/Collagenase/Hyaluronidase/FBS (Stemcell) for 60 min at 37°C. Following centrifugation at 300 × g for 5 min, cells were resuspended in 0.25% Trypsin–EDTA and incubated for 5 min in Hanks’ Balanced Salt Solution Modified (HBSS, Stemcell) supplemented with 10% FBS. After centrifugation, cells were resuspended in 1 mg/mL DNase I (Stemcell) with continuous gentle pipetting for 1 min. Cells were washed three times in HBSS, resuspended in PBS, and cell count was performed to determine viability and cell concentration. The single-cell suspension was partitioned and barcoded using the 10X Genomics Chromium Controller (10X Genomics, California, USA) and the Single Cell 3’ Library and Gel Bead Kit v3.1 (V3.1; 10X Genomics; PN-1000121). The cells were loaded onto the Chromium Single Cell Chip G (10X Genomics; PN-1000120) to target 10 000 cells. GEM generation and barcoding, cDNA amplification and library construction were performed according to the 10X Genomics Chromium User Guide. The resulting single-cell transcriptome libraries contained unique sample indices for each sample. The libraries were quantified on the Agilent BioAnalyzer 2100 using the High Sensitivity DNA Kit (Agilent, 5067–4626). Libraries were pooled in equimolar ratios, and the pool was quantified by qRT-PCR using the KAPA Library Quantification Kit—Illumina/Universal (KAPA Biosystems, Massachusetts, USA, KK4824) in combination with the Life Technologies ViiA 7 real-time PCR instrument. Denatured libraries were loaded onto an Illumina NextSeq-500 and sequenced over 2 runs using a 150-cycle High-Output Kit as follows: 28 bp (Read1), 8 bp (i7 index) and 111 bp (Read2). For additional sequencing, denatured libraries were loaded onto an Illumina NovaSeq6000 and sequenced using one SP 100 cycle flow cell and one S1 100 cycle flow cell as follows: 28 bp (Read1), 8 bp (i7 index) and 91 bp (Read2). Read1 supplies the cell barcode and UMI, i7 the sample index and Read2 the 3′ sequence of the transcript. Data pre-processing was performed using cellranger 3.0.2 (10x Genomics) and a mouse GRCm38 reference. Final read depth was ~18 000–26 000 reads per cell across all samples. Library preparation and NextSeq sequencing were performed at the Institute for Molecular Bioscience Sequencing Facility (University of Queensland). NovaSeq sequencing was performed at Microba, based at the Translational Research Institute in Brisbane.
Analysis of single-cell RNA sequencing
Raw sequencing data in BCL format were processed into the single-cell feature counts using Cell Ranger software from 10X Genomics. For the downstream analysis, we used Seurat package version 4.0.3 to load the six Cell Ranger count matrices into Seurat objects. Cells with fewer than 200 genes and more than 5% of the reads mapped to mitochondria were filtered during the pre-processing step. To remove batch effects, each dataset was normalized with SCTranform (84). We used the function SelectIntegrationFeatures() to choose the top 5000 variable genes across six datasets prior to running the FindIntegrationAnchors() function to identify integration anchors. These anchors were subsequently used to merge all six samples into one Seurat object using the IntegrateData() function.
The integrated data were then used for dimensionality reduction and cell clustering, which used the top 5000 most variable genes of the integrated data. Dimensionality reduction was performed using Principal Component Analysis to project the high dimensional space with 5000 genes to the reduced space of 50 PCs. Uniform manifold approximation and projection (UMAP) plot was generated using the first 30 PCs as input, and 30 PCs were determined as a good threshold by using the ElbowPlot function (Supplementary Material, Figure S7). Visualization of the integrated data with cells was produced (Supplementary Material, Figure S8). Next, we clustered the cells using the Louvain algorithm (85) by running the FindNeighbors() and FindClusters() functions. Using the resolution 0.02 for the FindClusters() function, we identified nine clusters of cell types from the integrated data. Differential expression analysis was performed using the Wilcoxon Rank Sum test to find the top differentially expressed genes of each cluster. Cluster annotation was based on differentially expressed genes as described in the results section. Stromal cell subclusters were determined by subsetting the respective clusters, rescaling the data and repeating unsupervised clustering on the subset of the larger Stromal Cells cluster. We obtained four subclusters using a higher resolution parameter when running the FindClusters() function. Markers for the clusters were found by performing differential expression analysis using Wilcoxon Rank Sum; cluster annotation was then done based on the gene markers. Differential gene expression between conditions was performed with an ‘pseudo-bulk’ expression approach (86). For each cell type, the pseudo-bulk expression profile was generated by summing the gene counts of all cells with the same condition (controls and Hnf1b mutants). Genes with an expression lower than the threshold of log count per million were filtered. To normalize library size differences for each pseudo-bulk samples, we used the calcNormFactors() function from EdgeR (87). The normalized pseudo-bulk expression of each sample was weighted to the condition from which it originated and fed to a linear model for differential expression analysis. Finally, empirical Bayes statistic through eBayes() function was applied to rank the genes in order of significance (88). Further analyses including functional annotations, enrichment lists and pathway analysis were done using DAVID Bioinformatics Resources, ToppGene Suite and IPA (89,90).
Statistical analyses
For gene expression using qRT-PCR, statistical analysis was performed by the unpaired t-test (GraphPad). Data were represented as mean expression levels with standard error of the mean. For comparison of unilateral kidney agenesis occurrence, significance was calculated using Fisher’s exact test.
Supplementary Material
Acknowledgements
We thank MRKH Australia for their support in recruiting women, Dr Richard Behringer for providing the Wnt7-Cre mouse line and Enya Longmuss for assistance with managing mouse colonies.
Conflict of Interest statement.The authors have no conflict of interest to declare.
Contributor Information
Ella Thomson, Centre for Clinical Research, The University of Queensland, Brisbane, QLD, Australia; Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Minh Tran, Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Gorjana Robevska, Reproductive Development, Murdoch Children's Research Institute, Melbourne, VIC, Australia.
Katie Ayers, Reproductive Development, Murdoch Children's Research Institute, Melbourne, VIC, Australia; Department of Paediatrics, The University of Melbourne, Melbourne, VIC, Australia.
Jocelyn van der Bergen, Reproductive Development, Murdoch Children's Research Institute, Melbourne, VIC, Australia.
Prarthna Gopalakrishnan Bhaskaran, Centre for Clinical Research, The University of Queensland, Brisbane, QLD, Australia.
Eric Haan, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA, Australia.
Silvia Cereghini, Institut de Biologie Paris-Seine, Sorbonne Université, Paris, France.
Alla Vash-Margita, Division of Pediatric and Adolescent Gynecology, Department of Obstetrics, Gynecology and Reproductive Sciences, Yale University School of Medicine, New Haven, CT, USA.
Miranda Margetts, Center for American Indian and Rural Health Equity, Montana State University, Bozeman, MT, USA.
Alison Hensley, MRKH Australia, Melbourne, VIC, Australia.
Quan Nguyen, Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Andrew Sinclair, Reproductive Development, Murdoch Children's Research Institute, Melbourne, VIC, Australia; Department of Paediatrics, The University of Melbourne, Melbourne, VIC, Australia.
Peter Koopman, Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Emanuele Pelosi, Centre for Clinical Research, The University of Queensland, Brisbane, QLD, Australia; Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Funding
National Health and Medical Research Council of Australia.
References
- 1. Duncan, P.A., Shapiro, L.R., Stangel, J.J., Klein, R.M. and Addonizio, J.C. (1979) The MURCS association: Mullerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia. J. Pediatr., 95, 399–402. [DOI] [PubMed] [Google Scholar]
- 2. Morcel, K., Camborieux, L., Programme de Recherches sur les Aplasies Müllériennes and Guerrier, D. (2007) Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Orphanet J. Rare. Dis., 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rall, K., Eisenbeis, S., Henninger, V., Henes, M., Wallwiener, D., Bonin, M. and Brucker, S. (2015) Typical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome. J. Pediatr. Adolesc. Gynecol., 28, 362–368. [DOI] [PubMed] [Google Scholar]
- 4. Beski, S., Gorgy, A., Venkat, G., Craft, I.L. and Edmonds, K. (2000) Gestational surrogacy: a feasible option for patients with Rokitansky syndrome. Hum. Reprod., 15, 2326–2328. [DOI] [PubMed] [Google Scholar]
- 5. Brännström, M., Johannesson, L., Bokström, H., Kvarnström, N., Mölne, J., Dahm-Kähler, P., Enskog, A., Milenkovic, M., Ekberg, J., Diaz-Garcia, C. et al. (2015) Livebirth after uterus transplantation. Lancet, 385, 607–616. [DOI] [PubMed] [Google Scholar]
- 6. Johannesson, L., Richards, E., Reddy, V., Walter, J., Olthoff, K., Quintini, C., Tzakis, A., Latif, M., Porrett, P., O'Neill, K. et al. (2022) The first 5 years of uterus transplant in the US: a report from the United States uterus transplant consortium. JAMA Surg., 157, e222612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsarna, E., Eleftheriades, A., Eleftheriades, M., Kalampokas, E., Liakopoulou, M.K. and Christopoulos, P. (2022) The impact of Mayer-Rokitansky-Küster-Hauser syndrome on psychology, quality of life, and sexual life of patients: a systematic review. Child. Aust., 9, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wottgen, M., Brucker, S., Renner, S.P., Strissel, P.L., Strick, R., Kellermann, A., Wallwiener, D., Beckmann, M.W. and Oppelt, P. (2008) Higher incidence of linked malformations in siblings of Mayer-Rokitansky-Küster-Hauser-syndrome patients. Hum. Reprod., 23, 1226–1231. [DOI] [PubMed] [Google Scholar]
- 9. Duru, U.A. and Laufer, M.R. (2009) Discordance in Mayer-von Rokitansky-Küster-Hauser Syndrome noted in monozygotic twins. J. Pediatr. Adolesc. Gynecol., 22, e73–e75. [DOI] [PubMed] [Google Scholar]
- 10. Rall, K., Eisenbeis, S., Barresi, G., Rückner, D., Walter, M., Poths, S., Wallwiener, D., Riess, O., Bonin, M. and Brucker, S. (2015) Mayer-Rokitansky-Kuster-Hauser syndrome discordance in monozygotic twins: Matrix metalloproteinase 14, low-density lipoprotein receptor-related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific mosaicism. Fertil. Steril., 103, 494–502. [DOI] [PubMed] [Google Scholar]
- 11. Milsom, S.R., Ogilvie, C.M., Jefferies, C. and Cree, L. (2015) Discordant Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome in identical twins – a case report and implications for reproduction in MRKH women. Gynecol. Endocrinol., 31, 684–687. [DOI] [PubMed] [Google Scholar]
- 12. Herlin, M., Højland, A.T. and Petersen, M.B. (2014) Familial occurrence of Mayer-Rokitansky-Kuster-Hauser syndrome: a case report and review of the literature. Am. J. Med. Genet. A, 164A, 2276–2286. [DOI] [PubMed] [Google Scholar]
- 13. Herlin, M., Bjørn, A.M., Rasmussen, M., Trolle, B. and Petersen, M.B. (2016) Prevalence and patient characteristics of Mayer-Rokitansky-Kuster-Hauser syndrome: a nationwide registry-based study. Hum. Reprod., 31, 384–2390. [DOI] [PubMed] [Google Scholar]
- 14. Ma, X., Yao, B., Pan, Q., Xu, W., Xu, K. and Ma, F. (2016) Familial occurrence of Mayer-Rokitansky-Kuster-Hauser syndrome. J. Obstet. Gynaecol., 36, 817–818. [DOI] [PubMed] [Google Scholar]
- 15. Kyei-Barffour, I., Margetts, M., Vash-Margita, A. and Pelosi, E. (2021) The embryological landscape of Mayer-Rokitansky-Kuster-Hauser syndrome: genetics and environmental factors. Yale J. Biol. Med., 94, 657–672. [PMC free article] [PubMed] [Google Scholar]
- 16. Jacquinet, A., Millar, D. and Lehman, A. (2016) Etiologies of uterine malformations. Am. J. Med. Genet. A, 170, 2141–2172. [DOI] [PubMed] [Google Scholar]
- 17. Mullen, R.D. and Behringer, R.R. (2014) Molecular genetics of Müllerian duct formation, regression and differentiation. Sex. Dev., 8, 281–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gervasini, C., Grati, F.R., Lalatta, F., Tabano, S., Gentilin, B., Colapietro, P., De Toffol, S., Frontino, G., Motta, F., Maitz, S. et al. (2010) SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet. Med., 12, 634–640. [DOI] [PubMed] [Google Scholar]
- 19. Fontana, L., Gentilin, B., Fedele, L., Gervasini, C. and Miozzo, M. (2017) Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin. Genet., 91, 233–246. [DOI] [PubMed] [Google Scholar]
- 20. Vainio, S., Heikkilä, M., Kispert, A., Chin, N. and McMahon, A.P. (1999) Female development in mammals is regulated by Wnt-4 signalling. Nature, 397, 405–409. [DOI] [PubMed] [Google Scholar]
- 21. Biason-Lauber, A., Konrad, D., Navratil, F. and Schoenle, E.J. (2004) A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46, XX woman. N. Engl. J. Med., 351, 792–798. [DOI] [PubMed] [Google Scholar]
- 22. Biason-Lauber, A., De Filippo, G., Konrad, D., Scarano, G., Nazzaro, A. and Schoenle, E.J. (2007) WNT4 deficiency--a clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: a case report. Hum. Reprod., 22, 224–229. [DOI] [PubMed] [Google Scholar]
- 23. Sundaram, U.T., McDonald-McGinn, D.M., Huff, D., Emanuel, B.S., Zackai, E.H., Driscoll, D.A. and Bodurtha, J. (2007) Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am. J. Med. Genet. A, 143A, 2016–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheroki, C., Krepischi-Santos, A.C.V., Szuhai, K., Brenner, V., Kim, C.A.E., Otto, P.A. and Rosenberg, C. (2008) Genomic imbalances associated with mullerian aplasia. J. Med. Genet., 45, 228–232. [DOI] [PubMed] [Google Scholar]
- 25. Nik-Zainal, S., Strick, R., Storer, M., Huang, N., Rad, R., Willatt, L., Fitzgerald, T., Martin, V., Sandford, R., Carter, N.P. et al. (2011) High incidence of recurrent copy number variants in patients with isolated and syndromic Müllerian aplasia. J. Med. Genet., 48, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ledig, S., Tewes, A.C., Hucke, J., Römer, T., Kapczuk, K., Schippert, C., Hillemanns, P. and Wieacker, P. (2018) Array-comparative genomic hybridization analysis in patients with Mullerian fusion anomalies. Clin. Genet., 93, 640–646. [DOI] [PubMed] [Google Scholar]
- 27. Edghill, E.L., Bingham, C., Ellard, S. and Hattersley, A.T. (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J. Med. Genet., 43, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernardini, L., Gimelli, S., Gervasini, C., Carella, M., Baban, A., Frontino, G., Barbano, G., Divizia, M.T., Fedele, L., Novelli, A. et al. (2009) Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: Two case reports. Orphanet J. Rare Dis., 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kobayashi, A., Shawlot, W., Kania, A. and Behringer, R.R. (2004) Requirement of Lim1 for female reproductive tract development. Development, 131, 539–549. [DOI] [PubMed] [Google Scholar]
- 30. Coffinier, C., Barra, J., Babinet, C. and Yaniv, M. (1999) Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech. Dev., 89, 211–213. [DOI] [PubMed] [Google Scholar]
- 31. Huang, C.C., Orvis, G.D., Kwan, K.M. and Behringer, R.R. (2014) Lhx1 is required in Mullerian duct epithelium for uterine development. Dev. Biol., 389, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heliot, C., Desgrange, A., Buisson, I., Prunskaite-Hyyryläinen, R., Shan, J., Vainio, S., Umbhauer, M. and Cereghini, S. (2013) HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Development, 140, 873–885. [DOI] [PubMed] [Google Scholar]
- 33. Massa, F., Garbay, S., Bouvier, R., Sugitani, Y., Noda, T., Gubler, M.C., Heidet, L., Pontoglio, M. and Fischer, E. (2013) Hepatocyte nuclear factor 1beta controls nephron tubular development. Development, 140, 886–896. [DOI] [PubMed] [Google Scholar]
- 34. Lokmane, L., Heliot, C., Garcia-Villalba, P., Fabre, M. and Cereghini, S. (2010) vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development, 137, 347–357. [DOI] [PubMed] [Google Scholar]
- 35. Lindner, T.H., Njolstad, P.R., Horikawa, Y., Bostad, L., Bell, G.I. and Sovik, O. (1999) A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet., 8, 2001–2008. [DOI] [PubMed] [Google Scholar]
- 36. Oram, R.A., Edghill, E.L., Blackman, J., Taylor, M.J.O., Kay, T., Flanagan, S.E., Ismail-Pratt, I., Creighton, S.M., Ellard, S., Hattersley, A.T. et al. (2010) Mutations in the hepatocyte nuclear factor-1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am. J. Obstet. Gynecol., 203, e1–e5. [DOI] [PubMed] [Google Scholar]
- 37. Haumaitre, C., Fabre, M., Cormier, S., Baumann, C., Delezoide, A.L. and Cereghini, S. (2006) Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1β/MODY5 mutations. Hum. Mol. Genet., 15, 2363–23375. [DOI] [PubMed] [Google Scholar]
- 38. Ledig, S., Schippert, C., Strick, R., Beckmann, M.W., Oppelt, P.G. and Wieacker, P. (2011) Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil. Steril., 95, 1589–1594. [DOI] [PubMed] [Google Scholar]
- 39. Fujimoto, M., Matsumoto, K., Iguchi-Ariga, S.M. and Ariga, H. (2001) Disruption of MSSP, c-myc single-strand binding protein, leads to embryonic lethality in some homozygous mice. Genes Cells, 6, 1067–1075. [DOI] [PubMed] [Google Scholar]
- 40. Sund, K.L. and Rehder, C.W. (2014) Detection and reporting of homozygosity associated with consanguinity in the clinical laboratory. Hum. Hered., 77, 217–224. [DOI] [PubMed] [Google Scholar]
- 41. Sandbacka, M., Laivuori, H., Freitas, E., Halttunen, M., Jokimaa, V., Morin-Papunen, L., Rosenberg, C. and Aittomäki, K. (2013) TBX6, LHX1 and copy number variations in the complex genetics of Müllerian aplasia. Orphanet J. Rare Dis., 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morcel, K., Dallapiccola, B., Pasquier, L., Watrin, T., Bernardini, L. and Guerrier, D. (2012) Clinical utility gene card for: Mayer-Rokitansky-Küster-Hauser syndrome. Eur. J. Hum. Genet., 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rall, K., Barresi, G., Wallwiener, D., Brucker, S.Y. and Staebler, A. (2013) Uterine rudiments in patients with Mayer-Rokitansky-Küster-Hauser syndrome consist of typical uterine tissue types with predominantly basalis-like endometrium. Fertil. Steril., 99, 1392–1399. [DOI] [PubMed] [Google Scholar]
- 44. Stewart, C.A., Wang, Y., Bonilla-Claudio, M., Martin, J.F., Gonzalez, G., Taketo, M.M. and Behringer, R.R. (2013) Ctnnb1 in mesenchyme regulates epithelial cell differentiation during Mullerian duct and postnatal uterine development. Mol. Endocrinol., 27, 1442–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Han, X., Wang, R., Zhou, Y., Fei, L., Sun, H., Lai, S., Saadatpour, A., Zhou, Z., Chen, H., Ye, F. et al. (2018) Mapping the Mouse Cell Atlas by Microwell-Seq. Cell, 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- 46. Klattig, J. and Englert, C. (2007) The Müllerian duct: recent insights into its development and regression. Sex. Dev., 1, 271–278. [DOI] [PubMed] [Google Scholar]
- 47. Saatcioglu, H.D., Kano, M., Horn, H., Zhang, L., Samore, W., Nagykery, N., Meinsohn, M.C., Hyun, M., Suliman, R., Poulo, J. et al. (2019) Single-cell sequencing of neonatal uterus reveals an Misr2+ endometrial progenitor indispensable for fertility. elife, 8, e46349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Desgrange, A., Heliot, C., Skovorodkin, I., Akram, S.U., Heikkilä, J., Ronkainen, V.P., Miinalainen, I., Vainio, S.J. and Cereghini, S. (2017) HNF1B controls epithelial organization and cell polarity during ureteric bud branching and collecting duct morphogenesis. Development, 144, 4704–4719. [DOI] [PubMed] [Google Scholar]
- 49. Ma, Z., Gong, Y., Patel, V., Karner, C.M., Fischer, E., Hiesberger, T., Carroll, T.J., Pontoglio, M. and Igarashi, P. (2007) Mutations of HNF-1beta inhibit epithelial morphogenesis through dysregulation of SOCS-3. Proc. Natl. Acad. Sci. U. S. A., 104, 20386–20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verdeguer, F., Le Corre, S., Fischer, E., Callens, C., Garbay, S., Doyen, A., Igarashi, P., Terzi, F. and Pontoglio, M. (2010) A mitotic transcriptional switch in polycystic kidney disease. Nat. Med., 16, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Vas, M., Kopp, J.L., Heliot, C., Sandar, M., Cereghini, S. and Haumaitre, C. (2015) Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development, 142, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Macari, C., Teyssier, C., Tribollet, V., Mouzat, K., Forcet, C., Horard, B., Lobaccaro, J.M. and Vanacker, J.M. (2010) Estrogens repress PGC1-α expression in the uterus. Mol. Cell. Endocrinol., 330, 33–40. [DOI] [PubMed] [Google Scholar]
- 53. Hadj-Rabia, S., Brideau, G., Al-Sarraj, Y., Maroun, R.C., Figueres, M.L., Leclerc-Mercier, S., Olinger, E., Baron, S., Chaussain, C., Nochy, D. et al. (2018) Multiplex epithelium dysfunction due to CLDN10 mutation: The HELIX syndrome. Genet. Med., 20, 190–201. [DOI] [PubMed] [Google Scholar]
- 54. Syed, S.M., Kumar, M., Ghosh, A., Tomasetig, F., Ali, A., Whan, R.M., Alterman, D. and Tanwar, P.S. (2020) Endometrial axin2+ cells drive epithelial homeostasis, regeneration, and cancer following oncogenic transformation. Cell Stem Cell, 26, 64–80.e13. [DOI] [PubMed] [Google Scholar]
- 55. Sturgeon, K., Kaneko, T., Biemann, M., Gauthier, A., Chawengsaksophak, K. and Cordes, S.P. (2011) Cdx1 refines positional identity of the vertebrate hindbrain by directly repressing Mafb expression. Development, 138, 65–74. [DOI] [PubMed] [Google Scholar]
- 56. Ferrè, S., de Baaij, J.H.F., Ferreira, P., Germann, R., de Klerk, J.B.C., Lavrijsen, M., van Zeeland, F., Venselaar, H., Kluijtmans, L.A.J., Hoenderop, J.G.J. et al. (2014) Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J. Am. Soc. Nephrol., 25, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aboudehen, K., Kim, M.S., Mitsche, M., Garland, K., Anderson, N., Noureddine, L., Pontoglio, M., Patel, V., Xie, Y., DeBose-Boyd, R. et al. (2016) Transcription factor hepatocyte nuclear factor–1β regulates renal cholesterol metabolism. J. Am. Soc. Nephrol., 27, 2408–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pfeifer, S.M., Attaran, M., Goldstein, J., Lindheim, S.R., Petrozza, J.C., Rackow, B.W., Siegelman, E., Troiano, R., Winter, T., Zuckerman, A. et al. (2021) ASRM Müllerian anomalies classification 2021. Fertil. Steril., 116, 1238–1252. [DOI] [PubMed] [Google Scholar]
- 59. Backhouse, B., Hanna, C., Robevska, G., van den Bergen, J., Pelosi, E., Simons, C., Koopman, P., Juniarto, A.Z., Grover, S., Faradz, S. et al. (2019) Identification of candidate genes for Mayer-Rokitansky-Küster-Hauser syndrome using enomic Approaches. Sex. Dev., 13, 26–34. [DOI] [PubMed] [Google Scholar]
- 60. Nodale, C., Ceccarelli, S., Giuliano, M., Cammarota, M., D'Amici, S., Vescarelli, E., Maffucci, D., Bellati, F., Benedetti Panici, P., Romano, F. et al. (2014) Gene expression profile of patients with Mayer-Rokitansky-Küster-Hauser syndrome: New insights into the potential role of developmental pathways. PLoS One, 9, e91010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mikhael, S., Dugar, S., Morton, M., Chorich, L.P., Tam, K.B., Lossie, A.C., Kim, H.G., Knight, J., Taylor, H.S., Mukherjee, S. et al. (2021) Genetics of agenesis/hypoplasia of the uterus and vagina: narrowing down the number of candidate genes for Mayer-Rokitansky-Küster-Hauser syndrome. Hum. Genet., 140, 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pontecorvi, P., Bernardini, L., Capalbo, A., Ceccarelli, S., Megiorni, F., Vescarelli, E., Bottillo, I., Preziosi, N., Fabbretti, M., Perniola, G. et al. (2021) Protein-protein interaction network analysis applied to DNA copy number profiling suggests new perspectives on the aetiology of Mayer-Rokitansky-Küster-Hauser syndrome. Sci. Rep., 11, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Faguer, S., Decramer, S., Chassaing, N., Bellanné-Chantelot, C., Calvas, P., Beaufils, S., Bessenay, L., Lengelé, J.P., Dahan, K., Ronco, P. et al. (2011) Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int., 80, 768–776. [DOI] [PubMed] [Google Scholar]
- 64. Heidet, L., Decramer, S., Pawtowski, A., Morinière, V., Bandin, F., Knebelmann, B., Lebre, A.S., Faguer, S., Guigonis, V., Antignac, C. and Salomon, R. (2010) Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol., 5, 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Decramer, S., Parant, O., Beaufils, S., Clauin, S., Guillou, C., Kessler, S., Aziza, J., Bandin, F., Schanstra, J.P. and Bellanné-Chantelot, C. (2007) Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J. Am. Soc. Nephrol., 18, 923–933. [DOI] [PubMed] [Google Scholar]
- 66. Bellanné-Chantelot, C., Clauin, S., Chauveau, D., Collin, P., Daumont, M., Douillard, C., Dubois-Laforgue, D., Dusselier, L., Gautier, J.F., Jadoul, M. et al. (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes, 54, 3126–3132. [DOI] [PubMed] [Google Scholar]
- 67. Edghill, E.L., Oram, R.A., Owens, M., Stals, K.L., Harries, L.W., Hattersley, A.T., Ellard, S. and Bingham, C. (2008) Hepatocyte nuclear factor-1beta gene deletions--a common cause of renal disease. Nephrol. Dial. Transplant., 23, 627–635. [DOI] [PubMed] [Google Scholar]
- 68. Clissold, R.L., Hamilton, A.J., Hattersley, A.T., Ellard, S. and Bingham, C. (2015) HNF1B-associated renal and extra-renal disease—an expanding clinical spectrum. Nat. Rev. Nephrol., 11, 102–112. [DOI] [PubMed] [Google Scholar]
- 69. Dubois-Laforgue, D., Cornu, E., Saint-Martin, C., Coste, J., Bellanné-Chantelot, C. and Timsit, J. (2017) Monogenic diabetes study group of the Société Francophone du Diabète. Diabetes, associated clinical spectrum, long-term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1b (HNF1B) molecular defects. Diabetes Care, 40, 1436–1443. [DOI] [PubMed] [Google Scholar]
- 70. Duval, H., Michel-Calemard, L., Gonzales, M., Loget, P., Beneteau, C., Buenerd, A., Joubert, M., Denis-Musquer, M., Clemenson, A., Chesnais, A.L. et al. (2016) Fetal anomalies associated with HNF1B mutations: report of 20 autopsy cases. Prenat. Diagn., 36, 744–751. [DOI] [PubMed] [Google Scholar]
- 71. Park, J.H., Tanaka, Y., Arango, N.A., Zhang, L., Benedict, L.A., Roh, M.I., Donahoe, P.K. and Teixeira, J.M. (2014) Induction of WNT inhibitory factor 1 expression by Müllerian inhibiting substance/antiMullerian hormone in the Müllerian duct mesenchyme is linked to Müllerian duct regression. Dev. Biol., 386, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang, T., Kwon, S.H., Peng, X., Urdy, S., Lu, Z., Schmitz, R.J., Dalton, S., Mostov, K.E. and Zhao, S. (2020) A qualitative change in the transcriptome occurs after the first cell cycle and coincides with lumen establishment during MDCKII cystogenesis. iScience, 23, 101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matlin, K.S., Myllymäki, S.M. and Manninen, A. (2017) Laminins in epithelial cell polarization: Old questions in search of new answers. Cold Spring Harb. Perspect. Biol., 9, a027920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Soofi, A., Levitan, I. and Dressler, G.R. (2012) Two novel EGFP insertion alleles reveal unique aspects of Pax2 function in embryonic and adult kidneys. Dev. Biol., 365, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bellessort, B., Bachelot, A., Heude, E., Alfama, G., Fontaine, A., Le Cardinal, M., Treier, M. and Levi, G. (2015) Role of Foxl2 in uterine maturation and function. Hum. Mol. Genet., 24, 3092–3103. [DOI] [PubMed] [Google Scholar]
- 76. Nanjappa, M.K., Medrano, T.I., March, A.G. and Cooke, P.S. (2015) Neonatal uterine and vaginal cell proliferation and adenogenesis are independent of estrogen receptor 1 (ESR1) in the mouse. Biol. Reprod., 92, 78. [DOI] [PubMed] [Google Scholar]
- 77. Fujino, A., Arango, N.A., Zhan, Y., Manganaro, T.F., Li, X., MacLaughlin, D.T. and Donahoe, P.K. (2009) Cell migration and activated PI3K/AKT-directed elongation in the developing rat Müllerian duct. Dev. Biol., 325, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li, Q., Agno, J.E., Edson, M.A., Nagaraja, A.K., Nagashima, T. and Matzuk, M.M. (2011) Transforming growth factor β receptor type 1 is essential for female reproductive tract integrity and function. PLoS Genet., 7, e1002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Roly, Z.Y., Major, A.T., Fulcher, A., Estermann, M.A., Hirst, C.E. and Smith, C.A. (2020) Adhesion G-protein-coupled receptor, GPR56, is required for Müllerian duct development in the chick. J. Endocrinol., 244, 395–413. [DOI] [PubMed] [Google Scholar]
- 80. Houde, C., Dickinson, R.J., Houtzager, V.M., Cullum, R., Montpetit, R., Metzler, M., Simpson, E.M., Roy, S., Hayden, M.R., Hoodless, P.A. et al. (2006) Hippi is essential for node cilia assembly and Sonic hedgehog signaling. Dev. Biol., 300, 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Faguer, S., Chassaing, N., Bandin, F., Prouheze, C., Garnier, A., Casemayou, A., Huart, A., Schanstra, J.P., Calvas, P., Decramer, S. et al. (2014) The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int., 86, 1007–1015. [DOI] [PubMed] [Google Scholar]
- 82. Friedrich, G. and Soriano, P. (1991) Promoter traps in embryonic stem cells: A genetic screen to identify and mutate developmental genes in mice. Genes Dev., 5, 1513–1523. [DOI] [PubMed] [Google Scholar]
- 83. Pelosi, E., Omari, S., Michel, M., Ding, J., Amano, T., Forabosco, A., Schlessinger, D. and Ottolenghi, C. (1843) Constitutively active Foxo3 in oocytes preserves ovarian reserve in mice. Nat. Commun., 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hafemeister, C. and Satija, R. (2019) Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol., 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Traag, V.A., Waltman, L. and van Eck, N.J. (2019) From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep., 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tung, P.Y., Blischak, J.D., Hsiao, C.D., Knowles, D.A., Burnett, J.E., Pritchard, J.K. and Gilad, Y. (2017) Batch effects and the effective design of single-cell gene expression studies. Sci. Rep., 7, 39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Robinson, M.D., McCarthy, D.J. and Smyth, G.K. (2010) edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Law, C.W., Chen, Y., Shi, W. and Smyth, G.K. (2014) Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol., 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Huang, D.W., Sherman, B.T. and Lempicki, R.A. (2009) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res., 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen, J., Bardes, E.E., Aronow, B.J. and Jegga, A.J. (2009) ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acid Res., 37, W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.