

Abstract
Glycoengineering of recombinant glycans and glycoconjugates is a rapidly evolving field. However, the production and exploitation of glycans has lagged behind that of proteins and nucleic acids. Biosynthetic glycoconjugate production requires the coordinated cooperation of three key components within a bacterial cell: a substrate protein, a coupling oligosaccharyltransferase, and a glycan biosynthesis locus. While the acceptor protein and oligosaccharyltransferase are the products of single genes, the glycan is a product of a multigene metabolic pathway. Typically, the glycan biosynthesis locus is cloned and transferred en bloc from the native organism to a suitable Escherichia coli strain. However, gene expression within these pathways has been optimized by natural selection in the native host and is unlikely to be optimal for heterologous production in an unrelated organism. In recent years, synthetic biology has addressed the challenges in heterologous expression of multigene systems by deconstructing these pathways and rebuilding them from the bottom up. The use of DNA assembly methods allows the convenient assembly of such pathways by combining defined parts with the requisite coding sequences in a single step. In this study, we apply combinatorial assembly to the heterologous biosynthesis of the Campylobacter jejuni N-glycosylation (pgl) pathway in E. coli. We engineered reconstructed biosynthesis clusters that faithfully reproduced the C. jejuni heptasaccharide glycan. Furthermore, following a single round of combinatorial assembly and screening, we identified pathway clones that outperform glycan and glycoconjugate production of the native unmodified pgl cluster. This platform offers a flexible method for optimal engineering of glycan structures in E. coli.
Keywords: bioconjugation, DNA assembly, glycoconjugate vaccines, N-glycosylation, synthetic biology
Introduction
Heterologous biosynthesis of complex glycan structures involves the cloning and expression of multiple genes that encode the enzymes necessary for their assembly. Bacteria are ideal hosts for this process, known as glycoengineering, as they tolerate manipulation of glycosylation and other glycostructural pathways such as N-linked and O-linked protein glycosylation systems, capsular polysaccharide, and lipooligosaccharide (LPS) biosynthesis. These recombinant glycans can be covalently linked in vivo to other macromolecules, such as proteins and lipids (Chen et al. 2016; Gerritzen et al. 2017; Valguarnera and Feldman 2017), to generate glycoconjugate vaccines, therapeutics, and diagnostics. This burgeoning technology, known as Protein Glycan Coupling Technology (PGCT), offers the potential to custom design and combine protein and glycan structures in safe, genetically tractable bacterial strains (Kay et al. 2019). Due to their typical surface location and central role in disease, glycans are the prime candidates for diagnostic and glycoconjugate vaccine development (Ladhani 2012; Perrett et al. 2013; Micoli et al. 2018).
Traditionally, PGCT involves transforming Escherichia coli cells with three plasmids that encode the requisite components for glycoconjugate production: a substrate protein, an oligosaccharyltransferase, and a glycan biosynthesis locus. The typical approach in the PGCT vaccine field is to attempt to transfer a complete, unmodified glycan biosynthetic gene cluster from a native host into E. coli (Cuccui et al. 2013; Kay et al. 2016; Harding et al. 2019; Duke et al. 2021). In some cases, this results in functional, although usually not optimal, glycan biosynthesis. Transcription and translation of genes within these clusters are finely tuned for synthesis in the native host, and simply introducing the unmodified cluster into E. coli often results in suboptimal (or no) glycan production. Introduction of a heterologous biosynthetic pathway may be poorly tolerated, or tolerated at the expense of glycoconjugate yield (Glasscock et al. 2018). Furthermore, faithful cloning of large (frequently >15 Kbp) biosynthetic loci as a single contiguous fragment is technically challenging and the incoming pathway may conflict with the endogenous sugar metabolic pathways of the host strain (Ceroni et al. 2015).
A number of advancements have been made to improve the capabilities of PGCT and the yield of the desired glycoconjugate, including (but not limited to) the identification of custom acceptor proteins with improved immunogenicity (Reglinski et al. 2018), identification of novel oligosaccharyltransferases with alternative sugar substrate specificity (Ihssen et al. 2012; Feldman et al. 2019; Harding et al. 2019), and removal of conflicting sugar biosynthetic pathways from the E. coli host strain (Yates et al. 2019). However, the glycan biosynthesis pathway is frequently overlooked. To date, the vast majority of advancements to specifically improve glycan yield have focused on the modification of the E. coli host strain or the introduction of one or two specific genes rather than targeting the incoming glycan biosynthetic locus itself (Yates et al. 2019; Kay et al. 2022).
In recent years, synthetic biology has tackled the challenge of heterologous expression of complex multigene systems and has fundamentally changed the state of the art. Seminal work introduced “refactoring”: deconstructing complex gene clusters and rebuilding them from the bottom up in a modular format (Temme et al. 2012; Smanski et al. 2014). Combinatorial construction using well-defined parts (synthetic promoters, ribosome-binding sites [RBSs], coding sequences [CDSs], and terminators) has been enabled by new DNA assembly methods that combine multiple DNA parts in a single step (Zelcbuch et al. 2013; Smanski et al. 2014; Awan et al. 2017). The modular and hierarchical nature of these assemblies obviates the need to clone large genetic loci as a single contiguous fragment, facilitates the identification of any components causing issues, and provides a platform for the efficient systematic optimisation of expression and function through iterative cycles of engineering. To date, this approach has not been applied to the biosynthesis of complex glycan pathways.
In this study, our aim was to apply these modern synthetic biology principles to the refactoring and assembly of the Campylobacter jejuni N-linked protein glycosylation (pgl) biosynthesis pathway in E. coli. We refactored the C. jejuni pgl cluster by removing each gene from its native regulation and rebuilt it from the bottom up using combinatorial Start-Stop Assembly, a scarless modular DNA assembly system that is well-suited to the construction of combinatorial libraries of multigene expression constructs (Taylor et al. 2019). Each of the 10 CDSs within the pgl cluster responsible for biosynthesis of the glycan were assembled with mixtures of synthetic promoters and RBSs of various strengths to generate a large library of expression constructs. We established a rapid screen which, despite only being able to cover a tiny fraction of the library, identified pgl clones that outperformed glycan production compared with the native, unmodified pgl cluster cloned directly from C. jejuni. Furthermore, the improved glycan production translated to a greater glycoconjugate yield when these refactored loci were transformed into glycoengineering strains expressing the C. jejuni N-oligosaccharyltransferase, pglB, and a target protein harboring an appropriate sequon. Thus, we demonstrate the potential of this approach for improved, refactored sugar biosynthesis loci construction towards recombinant glycan biosynthesis and subsequent glycoconjugate vaccine production.
Results
Design and construction of a refactored C. jejuni pgl locus expression library
The first bacterial N-linked glycosylation system was identified in the human enteric pathogen C. jejuni (Szymanski et al. 1999; Wacker et al. 2002). Genetic and biochemical studies revealed that C. jejuni modifies at least 50 proteins post-translation with the heptasaccharide, GalNAc-α1,4-GalNAc-[Glcβ1,3-]GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,3-diBacNAc-β1 (where GalNAc is N-acetylgalactosamine, Glc is glucose, and diNacBAc is 2,4-diacetamido-2,4,6-trideoxyglucopyranose) (Linton et al. 2002). The discovery that the C. jejuni pgl cluster is fully functional when transferred to E. coli unlocked a new area of bacterial glycoengineering and the potential to construct recombinant glycans and glycoconjugates (Wacker et al. 2002). Subsequent discoveries demonstrated that pglB, which encodes the oligosaccharyltransferase within this locus, has a relaxed glycan substrate specificity and can transfer a range of glycostructures to almost any protein, provided it possesses the D/E-XNX-S/T sequon (Feldman et al. 2005; Kowarik et al. 2006).
In this pathway, the enzymes PglFED are responsible for the synthesis of the reducing end sugar UDP-diNAcBac; Gne (alternatively referred to as GalE) is an UDP-glucose 4-epimerase, which catalyzes the conversion of UDP-GlcNAc to UDP-GalNAc; the glycosyltransferase enzymes PglC, PglA, PglJH, and PglI progressively build the heptasaccharide on the lipid carrier undecaprenylpyrophosphate (UndP) on the cytoplasmic side of the inner membrane. This glycan structure is then flipped to the periplasmic side of the membrane by the flippase, PglK. Finally, the oligosaccharyltransferase, PglB, transfers the glycan “en bloc” to asparagine residues within substrate proteins that contain the acceptor sequon D/E-XNX-S/T, where X can be any amino acid except proline (Fig. 1; Linton et al. 2005).
Fig. 1.
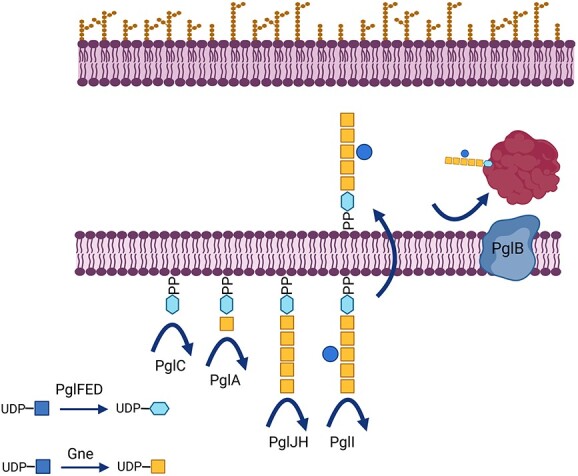
Schematic of the N-glycosylation pathway of C. jejuni. Blue squares: GlcNAc, blue hexagon, diNAcBac; yellow squares, GalNAc; and blue circle, Glc; OM, outer membrane; IM, inner membrane.
The 10 genes necessary for heptasaccharide formation in E. coli, gne, pglA, pglC, pglD, pglE, pglF, pglH, pglI, pglJ, and pglK (Fig. 2A and B) were selected for pathway refactoring. The CDS for the oligosaccharyltransferase PglB (Figs. 1 and 2A) was deliberately excluded as it would offer more flexibility as to which transferase could be used for coupling to an acceptor protein. Furthermore, the notion that the expression of pglB could be detrimental to the host cell has been previously reported (Abouelhadid et al. 2021), and we treated it as a separate engineering challenge for the purpose of this study. Each CDS was searched for BsaI, SapI, and BbsI restriction sites, and those genes which did not contain these restriction sites were amplified by PCR from C. jejuni genomic DNA. Those genes that did contain these restriction sites were chemically synthesized, with the restriction sequences removed.
Fig. 2.

A) Operon architecture of the pgl cluster from C. jejuni strain 81116. B) Design of the refactored pgl pathway with monocistronic architecture. C) Schematic showing Start-Stop assembly for the refactored pgl pathway library, which is composed of 10 genes with a standard set of set of 6 promoters, 6 RBSs, and 1 terminator. Plasmids are represented by grey boxes.
Each CDS within the pgl locus was cloned alone, without promoter, RBS, or terminator, into the level 0 Start-Stop Assembly storage vector, pStA0. These were subsequently assembled into transcription units using a standard set of 6 promoters, 6 RBSs, and a single transcriptional terminator, as shown in Fig. 2B and C. Here, equimolar mixtures of the promoter and RBS parts were combined in one-pot assembly reactions with each CDS to vary the expression of each gene in the pathway, producing 36 different expression strengths of each CDS. Blue/white screening was utilized to identify plasmids with insertions (white colonies), and all successfully assembled transformants were pooled and were used to prepare plasmid DNA. These level 1 transcription units were subsequently used to build two 5-gene pathways in level 2 vectors, pStA2, which were then combined into the level 3 vector, pStA3, containing all 10 genes required to produce the C. jejuni heptasaccharide (Fig. 2C). Plasmids containing refactored pgl libraries were transformed into NEB 10-β, which was exploited for its high transformation efficiency, particularly with high molecular weight plasmids and blue/white screening. Libraries were not further refined, and therefore, as each CDS could be expressed at up to 36 expression levels, and with 10 genes in the pathway, the library had a potential size of (6 × 6)10 = 3.615. This number is too large to screen exhaustively but instead provides a diverse pool of variants which can be sampled.
Screening assays identify successfully refactored sugar synthesis clones from a library of millions
To identify the clones in the refactored pgl expression library which were able to synthesize the C. jejuni heptasaccharide in E. coli, a screen was developed in which the glycan displayed on the surface of the cell was detected by immunoblot. Bacterial protein N-linked glycosylation (as utilized by the C. jejuni pgl locus) and O-antigen polysaccharide biosynthesis broadly share a similar assembly mechanism in which the glycans are assembled on a lipid carrier on the cytoplasmic face of the inner membrane and are then flipped to the periplasmic face. Once in the periplasm, the glycan can serve as a substrate for both a protein oligosaccharyltransferase (PglB) or the O-antigen ligase, WaaL, which traffic it to a substrate protein or the cell surface, respectively. Here, the overlapping assembly mechanisms were exploited (as they have been previously; Price et al. 2016; Strutton et al. 2019; Yates et al. 2019; Mauri et al. 2021) to harness the native WaaL and attach the preassembled C. jejuni heptasaccharide to the outer core of the LPS.
To determine whether these refactored pgl loci displayed the C. jejuni heptasaccharide on their cell surface, individual clones were cultured in 96-well plates, washed to remove culture medium, and cell suspensions were transferred to a nitrocellulose membrane. Cell surface glycan production was assessed by probing spots with soybean agglutinin (SBA) lectin, which specifically binds terminal GalNAc residues within the C. jejuni heptasaccharide. Two independent replicates of the final assembly reaction and transformation were performed (Fig. 3, replicate 1, and Supplementary Fig. S1, replicate 2). We observed a number of clones that displayed crossreactivity with SBA, which indicate the biosynthesis and surface exposure of the GlcGalNAc5diNAcBac heptasaccharide (Fig. 3A). Furthermore, two clones from the first assembly and 1 clone from the second assembly appeared to exhibit greater fluorescent signal compared with cells containing the native unmodified cluster, suggesting superior glycan production. By contrast, we observed no signal in a control strain harboring an empty plasmid.
Fig. 3.

A) Randomly picked clones probed for production of the C. jejuni heptasaccharide using an SBA, a biotinylated GalNAc-specific lectin, and green streptavidin-coupled IRDye. Clones were selected at random, grown in high throughput 200-μL cultures, and spotted on nitrocellulose membrane. Clones selected for further characterization are circled in red. B) Clones were subsequently grown in 10-mL cultures, normalised to the same OD, and probed for GalNAc production with SBA. Cells harboring plasmid-borne native unmodified pgl cluster and empty plasmid control are denoted as “+” and “-,” respectively. C) ELISA glycan quantification shows a range of glycan production from refactored clusters compared with the wildtype cluster. D and E) Glycoconjugate produced by 8 refactored pgl cluster variants from 2 independent assemblies and transformations. Glycoproteins were resolved by SDS-PAGE and were detected by immunoblot analysis probed with anti-6xHis antibody (red) and SBA lectin (green). Coordinates corresponding to position of clones in the 96 well plate are denoted on the blots.
In this dot blot screen, cell suspensions were not normalised to the same optical density (OD), therefore, variation in the signal may be due to the differential cell growth and not altered cell surface glycan display. We selected 8 clones from the first replicate which displayed either high- or low- signal for further characterisation. These clones were cultured, their ODs (600 nm) were normalised, washed, and spotted on to nitrocellulose membrane as described above (Fig. 3B). Of these 8 clones, comparable signals between the two methods were observed with the exception of clones pgl1.H2 and pgl1.A5, which exhibited high signals in the initial 96-well plate screen but considerably lower qualitative signal when OD-matched.
To determine the relative abundance of the glycan moiety present on the cell surface, whole-cell ELISA was performed on the same 8 clones. A strain containing an empty plasmid was used to establish a baseline for OD readings. Clones pgl1.C7 and pgl1.C8 exhibited 2- and 1.75-fold increased cell surface-exposed GalNAc relative to the strain containing the unmodified pgl cluster, which suggested superior heptasaccharide production in these strains (Fig. 3C). Glycan-surface abundance was further assessed by densitometry analysis performed on the OD-matched dot blots. Quantification of the spot pixel intensities revealed a linear relationship between the fluorescent signal and the OD of culture for the strain containing the unmodified pgl cluster (Supplementary Figs. S2 and S3). We observed significantly increased anti-glycan signal in clones pgl1.C7, pgl1.C8, and pgl2.F6 (1.94-, 1.88-, and 1.77-fold, respectively) relative to those containing the native locus (Supplementary Fig. S4).
Refactored pgl locus improves ExoA-heptasaccharide glycoconjugate yield in E. coli
By refactoring the pgl locus, the quantity of surface-displayed glycan could be improved in comparison to the native pgl locus. To determine whether these refactored loci would also improve the yield of glycoconjugate, the 8 clones previously selected were transformed into an E. coli strain that was specifically engineered as a glycoconjugate production platform. This chassis strain is derived from the waaL-, wecA+ strain, CLM24, and expresses a chromosomal copy of the C. jejuni OST pglB (Abouelhadid et al. 2021). The carrier protein ExoA from Pseudomonas aeruginosa was encoded on a second plasmid and harbors two PglB glycosylation sequons. WecA is the initiator glycosyltransferase for the E. coli O-antigen and Enterobacterial common antigen synthesis pathways and catalyzes the transfer of GlcNAc onto UndP. Previous studies have shown that expression of the native pgl pathway in this strain results in the synthesis of a hybrid glycan that contains a mixture of GlcNac and diNAcBac at its reducing end (Wacker et al. 2006). ExoA was purified by His-affinity chromatography and was analyzed by western blotting using a protein-specific anti-His antibody and glycan-specific SBA lectin. Glycosylation of ExoA was observed in strains transformed with the variant plasmids pgl1.C7, pgl1.C8, and pgl2.F6 (Fig. 3D and E). Here, glycosylation efficiency was broadly comparable to strains transformed with the wild-type cluster. No glycosylation was observed in strains containing plasmids pgl1.A5 and pgl1.F6, which is consistent with the previous dot blot and ELISA experiments in which they produced less cell surface glycan. However, no glycosylation was also observed in the strains containing plasmids pgl2.A5, pgl2.B8, and pgl2.F12 despite these strains producing comparable surface glycan to pgl1.F6, as determined by ELISA.
Sequencing of those plasmids which produced both LPS-linked and glycoconjugate glycans, pgl1.C7, pgl1.C8, and pgl2.F6 was undertaken and revealed a variety of combinations of promoter and RBS strengths corresponding to each CDS (Fig. 4). Plasmids pgl1.C7 and pgl2.F6 were found to have correct assembly of parts with intact CDS within the refactored loci. By contrast, sequencing of clone pgl1.C8 revealed that pglF had assembled incorrectly and contained a premature stop codon, resulting in a truncated gene product. pglF encodes a 4,6 UDP-GlcNAc dehydratase and is the first gene in the pathway for the synthesis of diNAcBac (Olivier et al. 2006). Any effect of this mutation is unlikely to be evident in the glycoproteins produced from chassis strains such as CLM24 or 10-β, where any deficiency in diNAcBac synthesis would be compensated by the endogenous wecA gene. Indeed, we observed negligible difference in the glycoconjugate yield between the pglF mutant pathway and correctly assembled loci.
Fig. 4.

Sequencing of the three characterized pgl pathway variants revealed the genes:parts combinations for each gene in the C. jejuni N-glycosylation pathway. No promoter, no RBS, and a truncated gene product were detected for the pglF gene in locus pgl1.C8.
The relaxed specificity of the PglA transferase, which recognizes both GlcNAc and diNAcBac as substrates, means that biosynthesis of the C. jejuni heptasaccharide in wecA+ strains effectively bypasses the activity of PglC, PglD, PglE, and PglF enzymes, rendering them functionally redundant. Therefore, we investigated whether our refactored loci were able to synthesize and transfer diNAcBac and thus reproduce the native C. jejuni heptasaccharide as efficiently as the unmodified cluster. We transformed our refactored loci and plasmids encoding exoA and pglB into the wecA-, waaL-, W3110-derivative, SDB1, to determine glycoconjugate production. Resulting glycoproteins were purified, normalised to the same protein concentration, and analysed by western blotting as before (Fig. 5A). An increase in the anti-glycan signal was observed in resolved glycoproteins corresponding to the strains containing plasmids pgl1.C7 and pgl2.F6, which indicates an increase in the glycan:protein ratio and improved glycoconjugate yield in these strains. Interestingly, the production of a glycoconjugate was observed in the strain containing plasmid pgl1.C8 despite this pathway being possessing a premature stop codon in the diNAcBAc biosynthesis gene, pglF. However, the SBA signal was markedly reduced compared to glycoconjugates purified from cells containing the other pgl pathways, suggesting that glycan production is severely impaired in the absence of functional PglF. PglC orthologues have been shown to demonstrate a relaxed substrate specificity and can complement the function of WecA by transferring GlcNAc to UndP when heterologously overexpressed in vivo (Harding et al. 2018), which may explain the rescue of glycan production observed here.
Fig. 5.
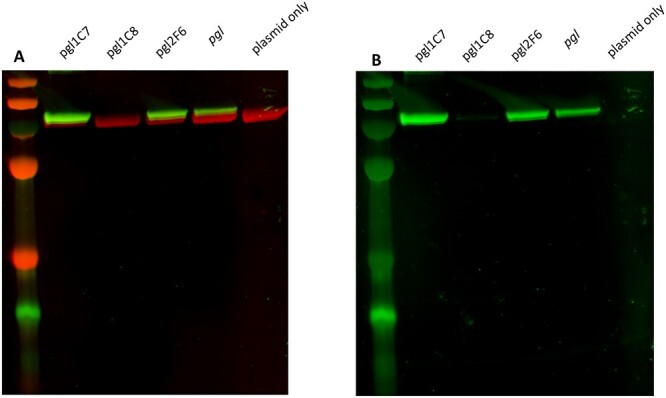
Glycoconjugate production by 3 refactored pgl cluster variants in the wecA- waaL- W3110-derivative SDB1. Hexahistidine tagged-ExoA with 2 potential glycosylation sequons was used as a substrate acceptor protein for PglB. A) Glycoproteins were resolved by SDS-PAGE and were detected by immunoblot analysis probed with anti-6xHis antibody (red) and SBA lectin (green). B) Anti-glycan SBA-only (green) channel.
Refactored pgl locus improves sequon site occupancy of AcrATag-heptasaccharide glycoconjugate in E. coli
To determine whether glycosylation sequon site occupancy was altered in glycoconjugates derived from our refactored loci, a hepta-glycotag acceptor protein was constructed based on the C. jejuni protein AcrA. This acceptor protein was modified to contain 3 internal and 4 C-terminal PglB substrate sequons and was designated as AcrATag. acrATag was coexpressed with the refactored pgl clusters and the native pgl locus as a control in CLM24 and SDB1 chassis strains, and the resulting glycoconjugate production was assessed by western blotting as described above (Fig. 6). We observed hexa- and quinta- glycosylated AcrATag when it was coexpressed with plasmids pgl1.C7 and pgl2.F6, respectively, in the chassis strain SBD1 (Fig. 6A). While we did observe a heptaglycosylated species when AcrATag was coexpressed with the native pgl pathway, the overall SBA signal was reduced compared with the glycoconjugates derived from the pgl1.C7 and pgl2.F6 pathways, indicating an inferior glycoprotein yield. Crossreactivity with anti-His (the protein alone) suggested the production of quinta-, tri-, and di- glycosylated proteins resulting from coexpression with pgl1.C7, pgl2.F6, and the native locus, respectively (Fig. 6B). Relative glycan:protein ratio was assessed by sandwich ELISA (Supplementary Fig. S5) and densitometry analysis performed with 3 independent biological replicates (Supplementary Figs. S6 and S7), which revealed that AcrATag derived from cells with the pgl1.C7 or pgl2.F6 pathways was conjugated with 2.3- (ELISA) or 3.3-fold (densitometry) and 2.3- (ELISA) or 2.2-fold (densitometry) more glycan, respectively, relative to the protein purified from cells containing the native pgl locus. Consistent with previous observations, reduced glycoprotein production was detected in cells containing the pgl1.C8 pathway.
Fig. 6.

Glycoconjugate production by 3 refactored pgl cluster variants in the A and B) wecA- waaL- W3110-derivative SDB1 and the C and D) wecA+ waaL- CLM24. A modified version of Hexahistidine tagged-AcrA with 7 glycosylation sequons was used as a PglB substrate acceptor protein. Glycoproteins were resolved by SDS-PAGE and were detected by immunoblot analysis probed with SBA lectin (green) and anti-6xHis antibody (red). Merged SBA lectin (green) and anti-6xHis (red) channels are shown in panels A and C; anti-6xHis only channel is shown in panels B and D.
By contrast, qualitatively little difference in the overall glycoconjugate yields were observed when a wecA+ strain was used as the chassis (Fig. 6C and D). Triglycosylated protein species were detected in cells containing pathways pgl1.C7, pgl2.F6, and the native locus, as indicated by crossreactivity with SBA (Fig. 6C). Diglycosylated AcrATag was observed in cells containing pathway pgl1.C8, suggesting a relatively lower glycosylation efficiency. An increased SBA signal was observed in the diglycosylated AcrATag derived from pathways pgl1.C7 and pgl2.F6 compared to the unmodified locus. However, these differences were not as pronounced as those observed in glycoproteins produced from SDB1 (Fig. 6A). Interestingly, we observed relatively reduced glycosylation of AcrATag when a wecA+ was used as a chassis strain compared with a wecA- strain. This is in contrast to the glycosylation of ExoA (Figs. 3D and 5), where we observed superior glycosylation in the wecA+ chassis, and highlights the imperative to test each glycan-acceptor protein combination empirically to determine the optimal parameters for glycoconjugate production.
Mass spectrometry confirms the structure of the glycan on ExoA
To precisely determine the N-linked glycan covalently attached to ExoA, mass spectrometry analysis was undertaken. In-gel trypsin digestion was performed, and the resulting peptides were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). ExoA was identified after the raw data were searched for a protein and a minimum of 1 peptide per protein as determined by the proteinlynx global server (PGLS). Further analysis was performed with the addition of the accurate mass of the C. jejuni heptasaccharide, GlcGalNAc5diNAcBac glycan (C56H91N7O34), to the modification list in Biopharmlynx. This analysis revealed a doubly charged peptide (HDLDIKDNNNSTPTVISHR) that was modified with the expected glycan mass (Fig. 7). The corresponding parental peak (m/z 3581) and the fragmentation peaks were analyzed using Masslynx and showed the sequential loss of 203 Da (HexNAc) and 162 Da (Hex), confirming that the expected heptasaccharide was attached to ExoA. Furthermore, we observed a peak of m/z 2404, which is consistent with the expected mass-to-charge of diNAcBac (m/z 228) attached to the peptide (m/z 2176). We observed no peaks corresponding to HexNAc attached directly to the peptide (expected m/z 2379), which indicates that diNAcBAc was the reducing end sugar. Overall, these data confirm the faithful reproduction of the native heptasaccharide from refactored cluster pgl1.C7 and its transfer to the carrier protein ExoA by PglB.
Fig. 7.
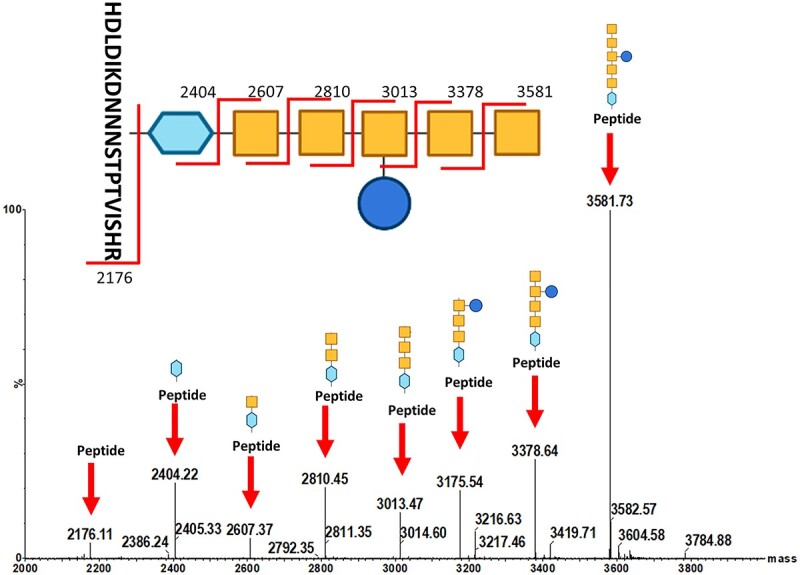
MS analysis of glycopeptides from ExoA derived from SDB1 (wecA- waaL-) with refactored pgl cluster clone, pgl1C7. Spectra were produced by fragmentation of the glycan structure-attached glycosylation sites digested by trypsin (DNNNS). Peaks indicative of fragmentation of the N-glycans are highlighted by red arrows. Blue squares, GlcNAc; blue hexagon, diNAcBac; yellow squares, GalNAc; blue circle, Glc.
Discussion
Despite the glycan moiety being the critical antigenic component of a glycoconjugate vaccine, relatively few attempts have been made to improve glycan yield by altering the biosynthetic locus itself. This is partly because “en bloc” cloning and transfer of the unmodified gene cluster can result in “adequate” glycan production, which is sufficient for the engineering of a prototype glycoconjugate vaccine. Recent developments in synthetic biology provide a tractable alternative to “en bloc” cloning by using DNA assembly for the bottom-up construction of redesigned clusters from individual parts. In this study, we employ such a synthetic biology approach to the assembly of the archetypal bacterial N-linked C. jejuni glycosylation locus in E. coli, identifying synthetic variants with improved yield of glycan and glycoconjugate production relative to the native unmodified pathway following a single combinatorial round of assembly without iterative optimisation.
In recent years, a number of strategies have been employed to rationally modify bacterial cells used for the production of glycoconjugates in a process known as “chassis engineering.” Varying flux through the phosphotransferase system (Pandhal et al. 2013), glyoxylate cycle (Pandhal et al. 2011), or blocking glycolysis and the pentose phosphate pathway (Wayman et al. 2019; Jiang et al. 2022) have been shown to improve the overall glycoconjugate yield. Similarly, modifying pathways for precursor sugar biosynthesis can result in an increased glycan and glycoconjugate yield (Pandhal et al. 2012; Kay et al. 2022). Typically, these modifications are made to the chromosome of the chassis E. coli strains and require successive targeted deletion (or introduction) of individual genes. These approaches often require significant a priori knowledge of the endogenous and incoming glycan metabolic pathways and how they may conflict. By targeting the glycan biosynthesis locus with an unbiased, empirical approach, we make minimal assumptions of the existing endogenous and incoming pathways, which makes this system ideally suited for the engineering of relatively poorly characterised glycan structures. Furthermore, the regulatory networks that drive glycan biosynthetic loci are rarely investigated, and it is frequently assumed that they have a polycistronic configuration driven by a single promoter. Such a configuration may not necessarily be optimal for the heterologous glycan production in a non-native host strain. Reconstructing the locus by systematically varying the expression level of each enzyme allows the rapid identification of which pathway variants perform well by screening. Our data show that after screening a library of <200 clones, we were able to identify an expression profile for each gene within the pgl pathway which outperformed that of the native locus. Conceivably, these assembly methods can also be combined with the rationally designed methods described above to further improve glycoconjugate yield.
Chassis engineering of the host strain by introducing PGCT components onto the chromosome is another strategy employed to improve glycoconjugate yield. For example, integration of a codon optimized, single copy of pglB into the chromosome of strain CLM24 (waaL-) resulted in the increased glycosylation of the acceptor protein with C. jejuni heptasaccharide, albeit at a cost of decreased overall glycoconjugate yield (Strutton et al. 2017). Expression of heterologous genes from the chromosome rather than multicopy plasmids is a well-recognised strategy for alleviating the toxic effects of metabolically burdensome enzymes, such as PglB, which resides in the cytoplasmic membrane (Bentley et al. 1990; Bassalo et al. 2016; Englaender et al. 2017). However, in a recent study, Terra et al. integrated the entire unmodified pgl pathway onto the chromosome of the chassis strain SDB1 (wecA- and waaL-; Terra et al. 2022) and observed greater glycoconjugate yield in 3 out of 4 acceptor proteins tested when an additional inducible copy of pglB was also provided on a plasmid. Optimal glycosylation was achieved when both the glycan biosynthesis genes and pglB were encoded on a plasmid in their native configuration. This suggests that the stoichiometry and relative expression level of each enzyme in the pathway influenced the conjugation efficiency. In our study, we observed superior glycosylation of AcrATag when pglB was encoded on a plasmid and was expressed in a wecA- strain compared with pglB on the chromosome in a wecA+ strain. Taken together, these data suggest that there is no one-size-fits-all strategy for the engineering of glycoconjugate vaccines and highlights the necessity to empirically test optimal production parameters. In this study, we focused on optimising glycan biosynthesis rather than conjugation per se. However, our data demonstrate that superior glycosylation efficiency was achieved when our refactored pathways were combined with both chromosomally and plasmid-encoded pglB. Combinatorial DNA assembly could also be applied to identify suitable expression levels of the oligosaccharyltransferase and acceptor protein to further improve glycoconjugate yield.
Improving yield of glycan biosynthesis has advantages beyond protein-based glycoconjugates. Expression of bacterial glycans in hypervesiculating strains of E. coli offers an alternative approach to protein-based glycoconjugates. Many Gram-negative bacteria produce outer membrane vesicles (OMVs), which are composed of lipopolysaccharide, outer membrane, and periplasmic proteins. Several vesicle-based based vaccines against meningococcal disease have already been developed and are licensed in many countries (Bjune et al. 1991) and there are other bacterial OMV vaccine candidates currently in development (Roberts et al. 2008; Schild et al. 2009; McConnell et al. 2011). However, current methods for producing these vaccines involve isolation of OMVs directly from the pathogenic organism of interest. By exploiting similar pathways for glycan surface display presented in this study, E. coli can be engineered to decorate lipid A with recombinant glycans from other pathogenic bacteria. Purification of OMVs from such laboratory strains of E. coli offers a safe, cheap alternative toward the production of these vaccines (Price et al. 2016).
In summary, we refactored the model C. jejuni pgl cluster using Start-Stop Assembly, constructed a combinatorial library of cluster variants, and demonstrated faithful glycan production and transfer to an acceptor protein by PglB. Despite screening a small fraction of the maximum number of possible clones within the library, we identified clones that not only produce the glycan or glycoconjugate but even outperform glycan production from the unmodified C. jejuni pgl cluster. Overall, these results demonstrate the potential of this synthetic biology approach toward an improved heterologous glycan biosynthesis and subsequent glycoconjugate vaccine production.
Materials and methods
Strains and plasmids
Tables of strains and plasmids used in this study are shown in Supplementary Tables S1 and S2, respectively. The E. coli strains were cultured on LB agar and LB broth at 37 °C. Antibiotics were added as necessary at the following concentrations: tetracycline 20 μg mL−1, ampicillin 100 μg mL−1, kanamycin 50 μg mL−1, and chloramphenicol 30 μg mL−1.
Construction of the PglB substrate protein, AcrAtag
The AcrAtag acceptor protein is a modified and truncated version of the C. jejuni lipoprotein, AcrA (amino acids 61 to 211). Amino acids 98–113 and 151–165, which flank the native AcrA PglB glycosylation sequon at Asn123, were deleted as these have previously been shown to not be required for glycosylation of Asn123. Two additional internal sequons at Asn117 and Asn147 were introduced through mutation at Phe115Asp and Thr145Asp, respectively, and 4 glycosylation sequons were added as C-terminal glycotags, which were separated by glycine-arginine-glycine linkers. Mutations at amino acids Lys97Gln and Lys191Gln were made to reduce proteolytic cleavage. An N-terminal DsbA signal sequence was included for trafficking the protein to the periplasm and a C-terminal hexahistidine tag was included for protein purification. The “acrAtag” construct was ordered for synthesis as a g-block (IDT) and was inserted into pEC415 using Gibson assembly using primers acrAtag_pEC415_f and r and the pEC415 backbone amplified using pEC415_acrAtag f and r. Amplification using Phusion polymerase and Gibson assembly using the NEB HiFi kit were both performed according to the manufacturer’s instruction.
Construction of refactored C. jejuni N-linked protein glycosylation locus (pgl) libraries
Individual parts, expression units, and biosynthetic pathways were constructed by a hierarchical Start-Stop Assembly as previously described (Taylor et al. 2019). Genetic parts were cloned in the level 0 storage plasmid pStA0 by mixing the PCR amplified or chemically synthesized fragments in a reaction with T4 DNA ligase buffer, 20 U/μL of T4 DNA ligase, and 0.5 U/μL of BsaI. The reaction mixtures were incubated in a thermocycler for 30 cycles of 37 °C for 5 min and 16 °C for 5 min before a single final step of deactivation at 65 °C for 20 min. Ligated fragments were transformed into E. coli 10β by electroporation and were selected on LB agar supplemented with ampicillin. Level 0 mixtures of 6 different promoters and 6 different RBSs (described previously, Taylor et al. 2019) were prepared as equimolar mixtures of the plasmids containing these parts. Libraries of level 1 plasmids (each containing whole expression units comprising a promoter, an RBS, a functional gene, and a transcriptional terminator) were obtained by Start-Stop Assembly reactions. For each functional gene, the reaction was performed in a total reaction volume of 20 μL in 1 × T4 DNA ligase buffer and included 20 fmol of the destination vector (pStA1AB, pStA1BC, pStA1CD, pStA1DE, or pStA1EZ), 40 fmol of the mixture of Level 0 plasmids carrying promoters, 40 fmol of the mixture of Level 0 plasmids carrying RBSs, 40 fmol of Level 0 plasmid carrying the corresponding gene, and 40 fmol of Level 0 plasmid carrying a terminator. Level 1 library reactions were performed as above, and transformants were selected on LB agar-supplemented tetracycline. Plasmid DNA was isolated from the resulting libraries by miniprep, pooled, and used in subsequent level 2 reactions. The level 2 library was generated with similar Start-Stop Assembly reactions, but the BsaI restriction enzyme was used instead of SapI and transformants were selected on LB agar supplemented with Kanamycin. Plasmids DNA was isolated from the resulting libraries as above and final level 3 libraries (which encode the full glycan biosynthetic pathway) were generated with the Start-Stop assembly reactions with BbsI restriction enzyme and selection on chloramphenicol.
Dot blot screening of refactored pgl biosynthetic pathways
Single-colony transformants were picked with a sterile toothpick and grown in 200 μL LB supplemented with chloramphenicol in 96-well polystyrene plates (Nunc) shaking at 600 rpm at 37 °C overnight. Cells were subcultured to fresh LB and chloramphenicol and were grown to stationary phase. Cultures were sedimented by centrifugation (2,800 × g, 10 min) and were washed with 300 μL of sterile phosphate buffered saline (PBS). Cell suspensions were sedimented and were washed 2 further times (3 total), and cells were resuspended in 150 μL of sterile PBS.
For low-throughput, OD-matched dot blots, cells were grown in 5 mL of LB supplemented with chloramphenicol shaking at 200 rpm at 37 °C overnight. Cultures were normalized to OD600 nm = 5 and were sedimented by centrifugation (6,000 × g, 10 min). Pellets were washed 3 times with sterile PBS.
Cell suspensions (2.5 μL) were spotted onto nitrocellulose membrane and were air dried for 30 min. Membranes were washed with PBS containing 0.1% (v/v) Tween 20 (PBST) and were probed with Biotinylated SBA lectin (Vector Laboratories) at a 1 in 10,000 dilution for 45 min. Membranes were washed 3 times with PBST and were incubated with Streptavidin IRDye 800CW (LI-COR Biosciences) at a dilution of 1 in 10,000 in PBST for 40 min. Fluorescent signal was detected with the Odyssey LI-COR detection system (LI-COR Biosciences).
Glycoconjugate expression and purification
Plasmids encoding refactored pgl loci were transformed into respective chassis strains by electroporation and were selected on LB containing appropriate antibiotics. Strains were cultured in LB broth at 200 rpm at 37 °C to early exponential phase upon which expression was induced by the addition of 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma) and 0.2% (w/v) L-arabinose (Sigma). Cultures were incubated for a further 16 h at 37 °C, sedimented by centrifugation, and resuspended in 50 mM Tris HCl pH 8, 300 mM NaCl, 10 mM imidazole. Cells were lysed using BeadBug zirconium lysing tubes (Sigma) in a FastPrep homogenizer (MP Biomedicals) and insoluble material was removed by pelleting. Hexahistidine tagged proteins were subsequently purified from cell lysates by Ni-NTA affinity chromatography and were eluted in 50 mM Tris HCl pH 8, 300 mM NaCl, and 300 mM imidazole.
Immunoblot analysis
Protein concentration was determined by Bradford assay (BioRad), samples were loaded with an equal protein content, resolved by SDS-PAGE, and transferred to nitrocellulose membranes using the iBlot 2 dry blotting system (ThermoFisher Scientific). Membranes were probed for the presence of GalNAc containing moieties as above. His-tag containing proteins were probed using mouse anti-His monoclonal primary antibody (ThermoFisher Scientific) and goat antimouse IgG IRDye 680 (LI-COR Biosciences). Fluorescent signal was detected with the Odyssey LI-COR detection system (LI-COR Biosciences).
Whole-cell ELISA
Semiquantitative analyses of surface-exposed glycan matched by OD were determined by ELISA. Cell cultures were grown overnight, washed 3 times in carbonate buffer (15 mM Na2CO3 and 35 mM NaHCO3) and were normalized to OD600nm = 1. Transparent polystyrol 96-well plates with high protein-binding capacity (F96 MaxiSorp, Nunc) were coated with OD-matched cell suspensions (50 μL per well) and were incubated overnight at 4 °C. Wells were then washed 4 times with 300 μL PBST (0.1% Tween-20) and were blocked for 1 h at room temperature with 200 μL in PBST per well, shaking at 500 rpm. After blocking, wells were washed 2 times with 300 μL PBST per well. To quantify surface-exposed terminal GalNAc, biotin-conjugated SBA lectin (Vector Laboratories) was added to each well at a 1:10,000 dilution in PBST (100 μL per well). The plate was incubated for 1 h at room temperature, 500 rpm, and unbound lectin was removed by washing the wells 4 times with 300 μL PBST per well. Streptavidin-HRP (Invitrogen) was added at 1:10,000 dilution in PBST (100 μL per well), the plate was incubated for 1 h at room temperature, and unbound antibodies were removed by 4 washes. ELISA plates were developed with 100 μL per well tetramethylbenzidine (TMB) substrate solution (Invitrogen). The oxidative reaction was stopped by adding 100 μL per well H2SO4 (2 N), and ODs at 450 nm were determined using a SpectraMax iD5 plate reader (Molecular Devices). OD450nm background values (buffer only in wells treated with biotinylated-SBA lectin and streptavidin-HRP) were subtracted from test values. Technical and biological triplicates were averaged, values represent the arithmetic mean, and error bars represent the standard deviations of biological triplicates.
Glycoconjugate sandwich ELISA
Glycoconjugates were purified as described above; concentrations were quantified using the Qubit Protein assay (Thermo Fisher Scientific) on a Qubit 2.0 instrument as per the manufacturer’s instructions. Relative glycan abundance was quantified by sandwich ELISA as previously described (Mauri et al. 2021) with minor modifications. Briefly, Polystyrene 96-well plates (F96 MaxiSorp, Nunc) were coated with mouse anti-His monocolonal antibody (Thermo Fisher Scientific), diluted 1:1,000 in PBS, and were incubated overnight at 4 °C. Wells were washed 4 times with PBST and were blocked with Carbo-free blocking buffer (Vector Laboratories) for 30 min at room temperature. Glycoconjugates were normalized to the lowest measured concentration of protein (234 ng μL−1) and were then diluted 1:50. Unbound glycoconjugate was removed by washing with PBST. Wells were incubated with biotin-conjugated SBA in a 1:1,000 dilution in PBST for 30 min at room temperature. Unbound lectin was removed by washing with PBST. Wells were then incubated with Streptavidin-HRP (Invitrogen) at 1:10,000 dilution in PBST for 30 min and unbound Streptavidin was removed as before. Plates were developed by adding 100 μL of TMB substrate solution (Invitrogen) per well, following which the reaction was stopped by addition of 100 μL H2SO4 (2 N). ODs (450 nm) were determined using a SpectraMax iD5 plate reader. Data were transformed using log10 to approximate a normal distribution and then regression analysis was used to determine the significant differences in glycan production between strains.
Densitometry of purified glycoconjugates
Glycoconjugates were purified, resolved by SDS-PAGE, and immunoblotted as described as above. Fluorescent signal was detected with the Odyssey LI-COR detection system, images were converted to greyscale, and quantification of signal intensity was performed using ImageJ software (v1.53t). A rectangle with defined area was used to gate around each band corresponding to a single glycosylated protein species within each gel lane. The same area rectangle was also used to quantify the background by taking a measurement from an empty lane. Data were transformed using log10 to approximate a normal distribution and then regression analysis was used to determine the significant differences in glycan production between strains.
Mass spectrometry analysis
In gel reduction, alkylation, and digestion with trypsin or chymotrypsin were performed on the gel sample prior to subsequent analysis by mass spectrometry. Cysteine residues were reduced with dithiothreitol and were derivatized by treatment with iodoacetamide to form stable carbamidomethyl derivatives. Trypsin digestion was carried out overnight at room temperature after initial incubation at 37 °C for 2 h.
Sample digests, resuspended in 0.1% (v/v) formic acid, were analyzed by online nano-flow reverse-phase high-performance LC with online electrospray-MS analysis with MS/MS (MSe) using a Waters SYNAPT G2-S high-definition mass spectrometer, which was coupled to a Waters ACQUITY UPLC M-Class System (Waters UK, Elstree). Separations were achieved by means of a C18 trapping column (M-Class Symmetry C18 Trap, 100 Å, 5 μm, 180 μm × 20 mm, 2G) connected in line with a 75-μm C18 reverse-phase analytical column (M-Class Peptide BEH C18, 130 Å, 1.7 μm, 75 μm × 150 mm), eluted over 90 min with a gradient of acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min. Column temperatures were maintained at 50 °C, and data were recorded in MSe “Resolution”-positive ion mode, with scan times set to 0.5 s in both the high-energy and low-energy modes of operation. The instrument was precalibrated using 10–100 fmol/μL of [Glu1]-fibrinopeptide B/5% (v/v) acetic acid (1:3, v/v) and was calibrated during analysis by means of a lockmass system using [Glu1]-Fibrinopeptide B 785.84262+ ion. The collision gas utilized was argon with collision energy ramp of 20–45 eV. Data acquisition was performed using MassLynx (Waters UK, Elstree) software and data were analyzed by means of MassLynx, BiopharmaLynx and PLGS version 3.0.2 (Waters UK, Elstree).
Supplementary Material
Acknowledgements
The authors gratefully acknowledge funding from the BBSRC (BB/W006146/1 and BB/R008124/1). Figures 1 and 2 were created with BioRender.
Conflict of interest statement: None declared.
Contributor Information
Ian J Passmore, London School of Hygiene & Tropical Medicine, Department of Infection Biology, London, WC1E 7HT, UK.
Alexandra Faulds-Pain, University of Nottingham, School of Life Sciences, Nottingham, NG7 2RD, UK.
Sherif Abouelhadid, London School of Hygiene & Tropical Medicine, Department of Infection Biology, London, WC1E 7HT, UK.
Mark A Harrison, London School of Hygiene & Tropical Medicine, Department of Infection Biology, London, WC1E 7HT, UK.
Catherine L Hall, London School of Hygiene & Tropical Medicine, Department of Infection Biology, London, WC1E 7HT, UK.
Paul Hitchen, Imperial College London, Department of Life Sciences, London, SW7 2AZ, UK.
Anne Dell, Imperial College London, Department of Life Sciences, London, SW7 2AZ, UK.
John T Heap, University of Nottingham, School of Life Sciences, Nottingham, NG7 2RD, UK.
Brendan W Wren, London School of Hygiene & Tropical Medicine, Department of Infection Biology, London, WC1E 7HT, UK.
Authors’ contributions
IJP and AF-P designed the study. IJP, AF-P, MAH, CLH, and SA performed the experiments. IJP prepared the manuscript with input from all authors.
References
- Abouelhadid S, Atkins E, Kay E, Passmore I, North SJ, Lehri B, Hitchen P, Bakke E, Rahman M, Bosse J, et al. Development of a novel glycoengineering platform for the rapid production of conjugate vaccines. 2021. 10.1101/2021.11.25.470047. [DOI] [PMC free article] [PubMed]
- Awan AR, Blount BA, Bell DJ, Shaw WM, Ho JCH, McKiernan RM, Ellis T. Biosynthesis of the antibiotic nonribosomal peptide penicillin in baker’s yeast. Nat Commun. 2017:8(1):15202. 10.1038/ncomms15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassalo MC, Garst AD, Halweg-Edwards AL, Grau WC, Domaille DW, Mutalik VK, Arkin AP, Gill RT. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth Biol. 2016:5(7):561–568. 10.1021/acssynbio.5b00187. [DOI] [PubMed] [Google Scholar]
- Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990:35(7):668–681. 10.1002/bit.260350704. [DOI] [PubMed] [Google Scholar]
- Bjune G, Høiby EA, Grønnesby JK, Arnesen Ø, Fredriksen JH, Lindbak A-K, Nøkleby H, Rosenqvist E, Solberg LK, Closs O, et al. Effect of outer membrane vesicle vaccine against group B meningococcal disease in Norway. Lancet. 1991:338(8775):1093–1096. 10.1016/0140-6736(91)91961-S. [DOI] [PubMed] [Google Scholar]
- Ceroni F, Algar R, Stan G-B, Ellis T. Quantifying cellular capacity identifies gene expression designs with reduced burden. Nat Methods. 2015:12(5):415–418. 10.1038/nmeth.3339. [DOI] [PubMed] [Google Scholar]
- Chen L, Valentine JL, Huang C-J, Endicott CE, Moeller TD, Rasmussen JA, Fletcher JR, Boll JM, Rosenthal JA, Dobruchowska J, et al. Outer membrane vesicles displaying engineered glycotopes elicit protective antibodies. Proc Natl Acad Sci U S A. 2016:113(26):E3609–E3618. 10.1073/pnas.1518311113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuccui J, Thomas RM, Moule MG, D’Elia RV, Laws TR, Mills DC, Williamson D, Atkins TP, Prior JL, Wren BW. Exploitation of bacterial N-linked glycosylation to develop a novel recombinant glycoconjugate vaccine against Francisella tularensis. Open Biol. 2013:3(5):130002. 10.1098/rsob.130002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke JA, Paschall AV, Robinson LS, Knoot CJ, Vinogradov E, Scott NE, Feldman MF, Avci FY, Harding CM. Development and immunogenicity of a prototype multivalent group B Streptococcus bioconjugate vaccine. ACS Infect Dis. 2021:7(11):3111–3123. 10.1021/acsinfecdis.1c00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englaender JA, Jones JA, Cress BF, Kuhlman TE, Linhardt RJ, Koffas MAG. Effect of genomic integration location on heterologous protein expression and metabolic engineering in E. coli. ACS Synth Biol. 2017:6(4):710–720. 10.1021/acssynbio.6b00350. [DOI] [PubMed] [Google Scholar]
- Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci U S A. 2005:102(8):3016–3021. 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MF, Mayer Bridwell AE, Scott NE, Vinogradov E, McKee SR, Chavez SM, Twentyman J, Stallings CL, Rosen DA, Harding CM. A promising bioconjugate vaccine against hypervirulent Klebsiella pneumoniae. Proc Natl Acad Sci U S A. 2019:116(37):18655–18663. 10.1073/pnas.1907833116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritzen MJH, Martens DE, Wijffels RH, van der Pol L, Stork M. Bioengineering bacterial outer membrane vesicles as vaccine platform. Biotechnol Adv. 2017:35(5):565–574. 10.1016/j.biotechadv.2017.05.003. [DOI] [PubMed] [Google Scholar]
- Glasscock CJ, Yates LE, Jaroentomeechai T, Wilson JD, Merritt JH, Lucks JB, DeLisa MP. A flow cytometric approach to engineering Escherichia coli for improved eukaryotic protein glycosylation. Metab Eng. 2018:47:488–495. 10.1016/j.ymben.2018.04.014. [DOI] [PubMed] [Google Scholar]
- Harding CM, Haurat MF, Vinogradov E, Feldman MF. Distinct amino acid residues confer one of three UDP-sugar substrate specificities in Acinetobacter baumannii Pgl C phosphoglycosyltransferases. Glycobiology. 2018:28(7):522–533. 10.1093/glycob/cwy037. [DOI] [PubMed] [Google Scholar]
- Harding CM, Nasr MA, Scott NE, Goyette-Desjardins G, Nothaft H, Mayer AE, Chavez SM, Huynh JP, Kinsella RL, Szymanski CM, et al. A platform for glycoengineering a polyvalent pneumococcal bioconjugate vaccine using E. coli as a host. Nat Commun. 2019:10(1):891. 10.1038/s41467-019-08869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihssen J, Kowarik M, Wiesli L, Reiss R, Wacker M, Thöny-Meyer L. Structural insights from random mutagenesis of Campylobacter jejunioligosaccharyltransferase PglB. BMC Biotechnol. 2012:12(1):67. 10.1186/1472-6750-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Bai J, Zhang H, Yuan J, Lu G, Wang Y, Jiang L, Liu B, Huang D, Feng L. Development of an O-polysaccharide based recombinant glycoconjugate vaccine in engineered E. coli against ExPEC O1. Carbohydr Polym. 2022:277:118796. 10.1016/j.carbpol.2021.118796. [DOI] [PubMed] [Google Scholar]
- Kay EJ, Yates LE, Terra VS, Cuccui J, Wren BW. Recombinant expression of Streptococcus pneumoniae capsular polysaccharides in Escherichia coli. Open Biol. 2016:6(4):150243. 10.1098/rsob.150243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay E, Cuccui J, Wren BW. Recent advances in the production of recombinant glycoconjugate vaccines. npj Vaccines. 2019:4(1):1–8. 10.1038/s41541-019-0110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay EJ, Mauri M, Willcocks SJ, Scott TA, Cuccui J, Wren BW. Engineering a suite of E. coli strains for enhanced expression of bacterial polysaccharides and glycoconjugate vaccines. Microb Cell Factories. 2022:21(1):66. 10.1186/s12934-022-01792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowarik M, Young NM, Numao S, Schulz BL, Hug I, Callewaert N, Mills DC, Watson DC, Hernandez M, Kelly JF, et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006:25(9):1957–1966. 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladhani SN. Two decades of experience with the Haemophilus influenzae serotype b conjugate vaccine in the United Kingdom. Clin Ther. 2012:34(2):385–399. 10.1016/j.clinthera.2011.11.027. [DOI] [PubMed] [Google Scholar]
- Linton D, Allan E, Karlyshev AV, Cronshaw AD, Wren BW. Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni. Mol Microbiol. 2002:43(2):497–508. [DOI] [PubMed] [Google Scholar]
- Linton D, Dorrell N, Hitchen PG, Amber S, Karlyshev AV, Morris HR, Dell A, Valvano MA, Aebi M, Wren BW. Functional analysis of the Campylobacter jejuni N-linked protein glycosylation pathway. Mol Microbiol. 2005:55(6):1695–1703. 10.1111/j.1365-2958.2005.04519.x. [DOI] [PubMed] [Google Scholar]
- Mauri M, Sannasiddappa TH, Vohra P, Corona-Torres R, Smith AA, Chintoan-Uta C, Bremner A, Terra VS, Abouelhadid S, Stevens MP, et al. Multivalent poultry vaccine development using Protein Glycan Coupling Technology. Microb Cell Factories. 2021:20(1):193. 10.1186/s12934-021-01682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Rumbo C, Bou G, Pachón J. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii. Vaccine. 2011:29(34):5705–5710. 10.1016/j.vaccine.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Micoli F, Costantino P, Adamo R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiol Rev. 2018:42(3):388–423. 10.1093/femsre/fuy011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier NB, Chen MM, Behr JR, Imperiali B. In vitro biosynthesis of UDP-N,N’-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system. Biochemistry. 2006:45:13659–13669. 10.1021/bi061456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandhal J, Ow SY, Noirel J, Wright PC. Improving N-glycosylation efficiency in Escherichia coli using shotgun proteomics, metabolic network analysis, and selective reaction monitoring. Biotechnol Bioeng. 2011:108(4):902–912. 10.1002/bit.23011. [DOI] [PubMed] [Google Scholar]
- Pandhal J, Desai P, Walpole C, Doroudi L, Malyshev D, Wright PC. Systematic metabolic engineering for improvement of glycosylation efficiency in Escherichia coli. Biochem Biophys Res Commun. 2012:419(3):472–476. 10.1016/j.bbrc.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandhal J, Woodruff LBA, Jaffe S, Desai P, Ow SY, Noirel J, Gill RT, Wright PC. Inverse metabolic engineering to improve Escherichia coli as an N-glycosylation host. Biotechnol Bioeng. 2013:110(9):2482–2493. 10.1002/bit.24920. [DOI] [PubMed] [Google Scholar]
- Perrett KP, Nolan TM, McVernon J. A licensed combined Haemophilus influenzae type b-Serogroups C and Y meningococcal conjugate vaccine. Infect Dis Ther. 2013:2(1):1–13. 10.1007/s40121-013-0007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Goyette-Desjardins G, Nothaft H, Valguarnera E, Szymanski CM, Segura M, Feldman MF. Glycoengineered outer membrane vesicles: a novel platform for bacterial vaccines. Sci Rep. 2016:6(1):24931. 10.1038/srep24931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reglinski M, Ercoli G, Plumptre C, Kay E, Petersen FC, Paton JC, Wren BW, Brown JS. A recombinant conjugated pneumococcal vaccine that protects against murine infections with a similar efficacy to Prevnar-13. npj Vaccines. 2018:3(1):53. 10.1038/s41541-018-0090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Moreno G, Bottero D, Gaillard ME, Fingermann M, Graieb A, Rumbo M, Hozbor D. Outer membrane vesicles as acellular vaccine against pertussis. Vaccine. 2008:26(36):4639–4646. 10.1016/j.vaccine.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Schild S, Nelson EJ, Bishop AL, Camilli A. Characterization of Vibrio cholerae outer membrane vesicles as a candidate vaccine for cholera. Infect Immun. 2009:77(1):472–484. 10.1128/IAI.01139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smanski MJ, Bhatia S, Zhao D, Park Y, B A L, Giannoukos G, Ciulla D, Busby M, Calderon J, Nicol R, et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat Biotechnol. 2014:32(12):1241–1249. 10.1038/nbt.3063. [DOI] [PubMed] [Google Scholar]
- Strutton B, Jaffé SRP, Pandhal J, Wright PC. Producing a glycosylating Escherichia coli cell factory: the placement of the bacterial oligosaccharyl transferase pglB onto the genome. Biochem Biophys Res Commun. 2017:495(1):686–692. 10.1016/j.bbrc.2017.11.023. [DOI] [PubMed] [Google Scholar]
- Strutton B, Jaffe SR, Evans CA, Fowler GJ, Dobson PD, Pandhal J, Wright PC. Engineering pathways in central carbon metabolism help to increase glycan production and improve N-type glycosylation of recombinant proteins in E. coli. Bioengineering (Basel). 2019:6(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P. Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol. 1999:32(5):1022–1030. [DOI] [PubMed] [Google Scholar]
- Taylor GM, Mordaka PM, Heap JT. Start-Stop assembly: a functionally scarless DNA assembly system optimized for metabolic engineering. Nucleic Acids Res. 2019:47(3):e17–e17. 10.1093/nar/gky1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme K, Zhao D, Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. PNAS. 2012:109(18):7085–7090. 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terra VS, Mauri M, Sannasiddappa TH, Smith AA, Stevens MP, Grant AJ, Wren BW, Cuccui J, the Glycoengineering of Veterinary Vaccines consortium (GoVV) . PglB function and glycosylation efficiency is temperature dependent when the pgl locus is integrated in the Escherichia coli chromosome. Microb Cell Factories. 2022:21(1):6. 10.1186/s12934-021-01728-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valguarnera E, Feldman MF. Glycoengineered outer membrane vesicles as a platform for vaccine development. Methods Enzymol. 2017:597:285–310. 10.1016/bs.mie.2017.06.032. [DOI] [PubMed] [Google Scholar]
- Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002:298(5599):1790–1793. 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- Wacker M, Feldman MF, Callewaert N, Kowarik M, Clarke BR, Pohl NL, Hernandez M, Vines ED, Valvano MA, Whitfield C, et al. Substrate specificity of bacterial oligosaccharyltransferase suggests a common transfer mechanism for the bacterial and eukaryotic systems. PNAS. 2006:103(18):7088–7093. 10.1073/pnas.0509207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman JA, Glasscock C, Mansell TJ, DeLisa MP, Varner JD. Improving designer glycan production in Escherichia coli through model-guided metabolic engineering. Metab Eng Commun. 2019:9:e00088. 10.1016/j.mec.2019.e00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LE, Natarajan A, Li M, Hale ME, Mills DC, DeLisa MP. Glyco-recoded Escherichia coli: recombineering-based genome editing of native polysaccharide biosynthesis gene clusters. Metab Eng. 2019:53:59–68. 10.1016/j.ymben.2019.02.002. [DOI] [PubMed] [Google Scholar]
- Zelcbuch L, Antonovsky N, Bar-Even A, Levin-Karp A, Barenholz U, Dayagi M, Liebermeister W, Flamholz A, Noor E, Amram S, et al. Spanning high-dimensional expression space using ribosome-binding site combinatorics. Nucleic Acids Res. 2013:41(9):e98. 10.1093/nar/gkt151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.