

Abstract
Diabetic kidney disease (DKD) is recognized as an important public health challenge. However, its genomic mechanisms are poorly understood. To identify rare variants for DKD, we conducted a whole-exome sequencing (WES) study leveraging large cohorts well-phenotyped for chronic kidney disease and diabetes. Our two-stage WES study included 4372 European and African ancestry participants from the Chronic Renal Insufficiency Cohort and Atherosclerosis Risk in Communities studies (stage 1) and 11 487 multi-ancestry Trans-Omics for Precision Medicine participants (stage 2). Generalized linear mixed models, which accounted for genetic relatedness and adjusted for age, sex and ancestry, were used to test associations between single variants and DKD. Gene-based aggregate rare variant analyses were conducted using an optimized sequence kernel association test implemented within our mixed model framework. We identified four novel exome-wide significant DKD-related loci through initiating diabetes. In single-variant analyses, participants carrying a rare, in-frame insertion in the DIS3L2 gene (rs141560952) exhibited a 193-fold increased odds [95% confidence interval (CI): 33.6, 1105] of DKD compared with noncarriers (P = 3.59 × 10−9). Likewise, each copy of a low-frequency KRT6B splice-site variant (rs425827) conferred a 5.31-fold higher odds (95% CI: 3.06, 9.21) of DKD (P = 2.72 × 10−9). Aggregate gene-based analyses further identified ERAP2 (P = 4.03 × 10−8) and NPEPPS (P = 1.51 × 10−7), which are both expressed in the kidney and implicated in renin–angiotensin–aldosterone system modulated immune response. In the largest WES study of DKD, we identified novel rare variant loci attaining exome-wide significance. These findings provide new insights into the molecular mechanisms underlying DKD.
Introduction
Over one-third of patients with diabetes will develop chronic kidney disease (CKD) (1,2), contributing substantially to the overall burden of CKD and its sequelae (3,4). As one of the most common complications of diabetes, diabetic kidney disease (DKD) is responsible for approximately 50% of end-stage kidney disease (ESKD) in developed countries (5) and deemed as a major risk factor for cardiovascular disease and premature mortality (4,6,7). Despite this public health burden, strategies for the treatment and prevention of DKD remain suboptimal (8). As a complex phenotype influenced by environmental as well as genetic factors (9–12), the molecular characterization of DKD may provide novel insights into biological pathways that could be targeted for therapeutic development. Furthermore, such work could identify individuals at high risk for disease who might benefit from early, targeted prevention efforts.
Genome-wide association studies and candidate gene studies, which focused primarily on common genetic variants, have discovered multiple loci associated with DKD (13–15). However, identified variants tend to have modest effect sizes and cumulatively explain only a limited portion of the estimated heritability of this condition (13,16,17). Few studies have assessed rare variants which may have large effects and play an important role in susceptibility to DKD (18–20). The previous sequencing studies include just one whole-genome sequencing (WGS) study, which examined 76 Finnish twins with type 1 diabetes who were discordant for kidney disease (18), and one whole-exome sequencing (WES) study, which investigated diabetes-attributed ESKD among 456 cases and 936 controls (20). Although both of these works identified promising signals for DKD, sample sizes were relatively small and no findings achieved statistical significance after stringent correction for multiple testing. Additional WGS and WES studies leveraging larger samples sizes may further elucidate the genetic architecture of this condition.
We conducted a WES study of DKD leveraging data from 4372 participants of the Chronic Renal Insufficiency Cohort (CRIC) and Atherosclerosis Risk in Communities (ARIC) studies. Using an extreme case control study design, we compared participants with diabetes and the most rapidly progressing kidney disease from the CRIC study to controls without DKD from the ARIC study. In our primary analysis, DKD cases were compared with healthy controls. Our rationale for this comparison was to identify variants associated with the full spectrum of the DKD disease course, from the development of diabetes to the progression to kidney disease. To discern whether identified signals exerted their influence through the development of diabetes and/or through the initiation of kidney disease, which may not be mutually exclusive, secondary analyses compared DKD cases with diabetes patients without CKD (diabetes controls). Furthermore, to assess whether identified variants may act as a more general risk factor for CKD, regardless of diabetes status, secondary analysis additionally compared DKD cases with CKD patients without diabetes (CKD controls). Suggestive signals identified in the stage-1 WES study were confirmed among 11 487 Trans-Omics for Precision Medicine (TOPMed) participants with existing WGS data (stage 2). The overall study design is illustrated in Figure 1.
Figure 1.
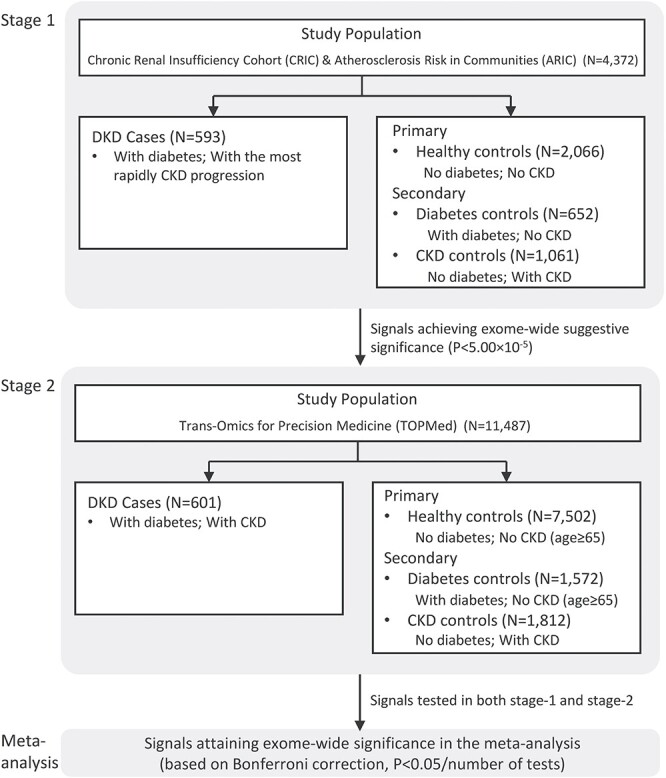
Study design. In this two-stage whole-exome sequencing study of DKD, an extreme case–control study design was adopted by leveraging 4372 participants of the Chronic Renal Insufficiency Cohort (CRIC) and Atherosclerosis Risk in Communities (ARIC) studies in the stage-1 analysis. Participants with DKD from the CRIC study were compared with one primary and two secondary control groups without DKD from the ARIC study. Suggestive signals identified in the stage-1 WES study were confirmed among 11 487 Trans-Omics for Precision Medicine (TOPMed) participants with existing WGS data (stage-2). Signals achieving an exome-wide significance based on Bonferroni correction in the meta-analysis were reported.
Results
Characteristics of study participants
Characteristics of the stage-1 and stage-2 study participants are provided in Table 1 and Supplementary Material, Tables S1 and S2.
Table 1.
Characteristics of study participants
| Stage 1 | Stage 2 | |||||||
|---|---|---|---|---|---|---|---|---|
|
DKD Cases (N = 593) |
Healthy Controls (N = 2066) |
Diabetes Controls (N = 652) |
CKD Controls (N = 1061) |
DKD Cases (N = 601) |
Healthy Controls (N = 7502) |
Diabetes Controls (N = 1572) |
CKD Controls (N = 1812) |
|
| Age, mean (SD), year | 58.02 (10.08) | 74.75 (4.8) | 74.54 (4.67) | 77.35 (5.34) | 68.15 (8.83) | 70.87 (4.57) | 70.62 (4.33) | 70.71 (9.83) |
| Male, no. (%) | 363 (61.21) | 775 (37.51) | 295 (45.25) | 440 (41.47) | 236 (39.27) | 2162 (28.82) | 528 (33.59) | 753 (41.56) |
| Non-European ancestry, no. (%) | 297 (50.08) | 406 (19.65) | 210 (32.21) | 210 (19.79) | 419 (69.72) | 2626 (35.0) | 977 (62.15) | 704 (38.85) |
| BMI, mean (SD) | 34.41 (8.17) | 27.78 (5.29) | 30.92 (5.99) | 27.91 (5.34) | 31.5 (6.16) | 27.78 (5.2) | 30.69 (5.66) | 27.8 (5.3) |
| SBP, mean (SD), mm Hg | 137.04 (21.43) | 129.06 (17.0) | 127.9 (15.85) | 132.08 (19.21) | 141.72 (24.33) | 133.66 (19.81) | 138.18 (19.86) | 135.97 (22.46) |
| DBP, mean (SD), mm Hg | 71.18 (12.66) | 67.27 (10.27) | 65.75 (9.83) | 66.55 (11.41) | 73.12 (11.41) | 72.99 (10.36) | 72.19 (10.61) | 72.27 (11.71) |
| Hypertension, no. (%) | 587 (99.32) | 1635 (79.22) | 610 (93.7) | 967 (91.14) | 539 (89.83) | 4517 (60.32) | 1280 (81.68) | 1355 (74.86) |
| FPG, mean (SD), mg/dL | 149.9 (72.24) | 102.44 (9.88) | 139.35 (32.85) | 102.96 (11.02) | 158.14 (70.58) | 98.78 (11.52) | 157.3 (53.37) | 97.57 (11.22) |
| eGFR, mean (SD), ml/min/1.73 m2 | 39.47 (13.44) | 77.66 (10.6) | 79.52 (11.81) | 55.53 (16.23) | 45.88 (12.84) | 81.51 (11.86) | 83.81 (13.38) | 49.53 (9.73) |
Abbreviations: BMI, body mass index; CKD, chronic kidney disease; DBP, diastolic blood pressure (BP); DKD, diabetic kidney disease; eGFR, estimated glomerular filtration rate; FPG, fasting plasma glucose; SBP, systolic BP; SD, standard deviation.
Results of stage-1 single-marker analyses
Stage-1 single-variant analyses of multi-ancestry and ancestry-specific samples identified 196 novel exonic loci achieving suggestive significance (P < 5.00 × 10−5) when comparing DKD cases with healthy controls. An additional 84 suggestive signals were identified in secondary analyses comparing DKD cases with diabetes controls or CKD controls. Genomic control lambda values for the stage-1 single-variant analyses ranged from 0.92 to 1.01 (Supplementary Material, Figs S1–S3). Among the identified signals from stage-1 analysis, 101 achieved exome-wide significance (maximum P ranging from 3.66 × 10−7 to 9.28 × 10−7; Fig. 2A–C and Supplementary Material, Tables S4–S12). Reassuringly, there were an additional 126 suggestive loci that were located in close proximity (<500 kb) to previously reported kidney disease variants (21). Among them, 47 reached exome-wide significance. Identified signals were generally consistent across comparisons of cases with the three sets of controls (Fig. 2A–C).
Figure 2.
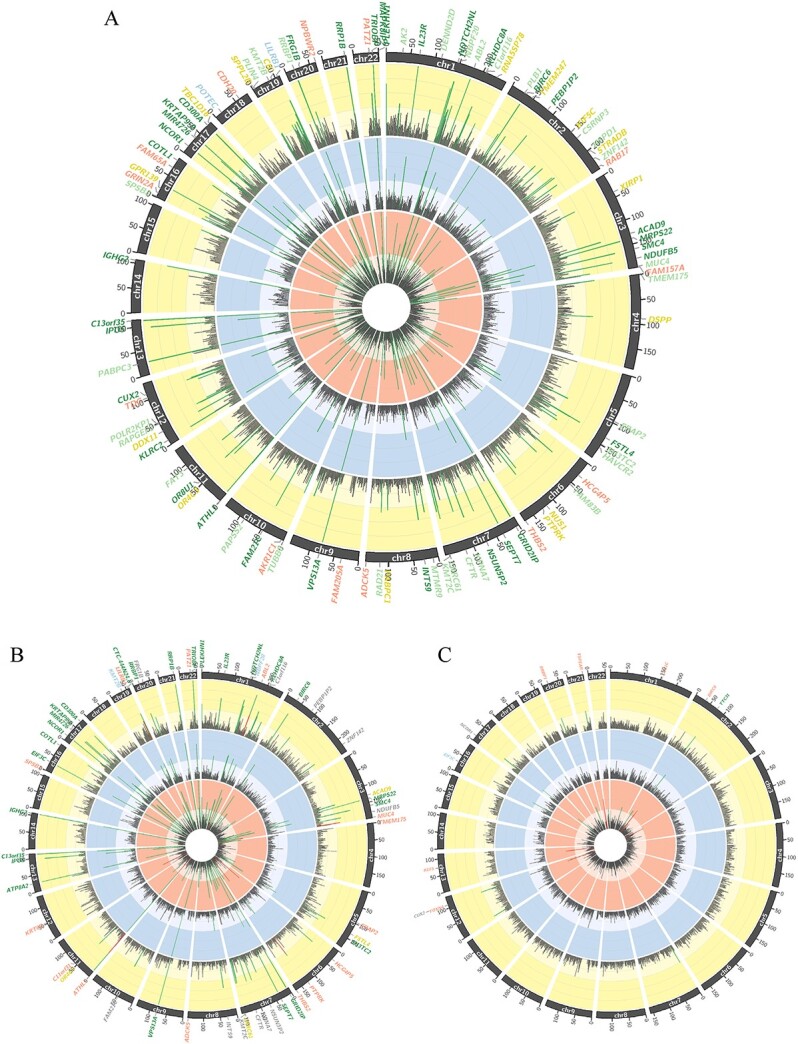
Circos plots display stage-1 signals from analyses comparing DKD cases with healthy controls (inner orange ring), diabetes controls (middle blue ring) and kidney disease controls (outer yellow ring) for the (A) multi-ancestry participants, (B) European ancestry participants and (C) African ancestry participants. Inside each ring are peaks of representing the –log P-value of the smallest P-value within a 500 kb-region on genome. The vertical axis of each ring shows –log P-value (ranging from 0 to 15), with suggestive multi-ancestry signals (P < 1.00 × 10−6) indicated by green coloring and suggestive ancestry-specific signals indicated by red coloring. Suggestive gene loci are annotated on the outside of the circos plots, with loci shared by two and all three case–control comparisons colored in light green and dark green, respectively. Loci identified by comparing DKD cases with healthy, diabetes and kidney disease controls are indicated in orange, blue and yellow, respectively.
Results of stage-1 gene-based aggregate rare variant analyses
Stage-1 gene-based aggregate rare variant analyses of predicted loss-of-function (pLOF) variants, missense variants and protein-altering insertion-deletions (indels) identified 12 suggestive gene signals when comparing DKD cases with healthy controls and an additional 3 signals when comparing DKD cases with diabetes and CKD controls. Among the identified signals, five achieved exome-wide significance in stage-1 analyses (Supplementary Material, Tables S13–S15). No gene signals were identified in aggregate analyses limited to high-confidence pLOF variants, likely owing to limited statistical power when restricted to these variants. In Leave-one-out (LOO) analyses of suggestive genes, removal of 20 rare variants attenuated the optimized sequence kernel association test (SKAT-O) P-values from identified aggregate units by at least one order of magnitude (Supplementary Material, Tables S16–S18).
Stage-2 and meta-analysis of suggestive DKD signals from stage-1 single-variant analyses
Two novel variants achieved exome-wide significance and exhibited consistent effect directions in meta-analyses of stage-1 and stage-2 results comparing DKD cases with healthy controls (Table 2). In meta-analysis of the multi-ancestry sample, 37 carriers of a rare DIS3L2 in-frame insertion (rs141560952 AGGG allele) had significantly increased odds of DKD compared with noncarriers [odds ratio (95% confidence interval [CI]) =193 (34, 1105), P = 3.59 × 10−9; Fig. 3A and Table 2]. This association was also identified in the analysis of European ancestry participants [odds ratio (95% CI) =100 (20, 510), P = 3.01 × 10=8; Fig. 3B and Table 2]. Note that this rare variant was not found in African ancestry participants in the stage-1 analysis, indicating a European ancestry-specific association. Furthermore, a low-frequency splice site variant, rs425827 (minor allele frequency [MAF] = 0.038), at the novel KRT6B locus strongly associated with DKD in European ancestry participants [odds ratio (95% CI) = 5.31 (3.06, 9.21), P = 2.72 × 10−9; Fig. 3C and Table 2]. A suggestive stage-1 signal, in IGHE, failed to achieve exome-wide significance but demonstrated consistent effect directions in the stage 2 and achieved a maximum P-value < 5.00 × 10−5 in meta-analyses of the stage-1 and stage-2 findings (Supplementary Material, Table S19). This promising variant was identified in multi-ancestry analyses comparing DKD cases with healthy controls.
Table 2.
Exome-wide significant signals for diabetic kidney disease identified in meta-analyses of stage-1 and -2 analyses
| Chr:Pos (hg19) | HGNC Symbol | rsID | Alt/Ref | Variant Type | Phase | MAC | DKD Cases vs. Healthy Controls | DKD Cases vs. Diabetes Controls | DKD Cases vs. CKD Controls | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | (95% CI) | P | OR | (95% CI) | P | OR | (95% CI) | P | |||||||
| Multi-ancestry analysis | |||||||||||||||
| 2:233208123 | DIS3L2 | rs141560952 | AGGG/A | Inframe insertion | Stage 1 | 11 | 6569 | (185.5232588) | 1.36E-06 | 34.99 | (4.33282.9) | 8.57E-04 | 88.83 | (8.34945.7) | 2.01E-04 |
| Stage 2 | 26 | 63.23 | (8.53469.0) | 1.13E-03 | 6.02 | (1.24,29.35) | 2.63E-02 | 25.25 | (3.92162.8) | 6.82E-04 | |||||
| Meta-analysis | - | 192.6 | (33.581105) | 3.59E-09 | 11.44 | (3.24,40.43) | 1.54E-04 | 40.88 | (9.46176.7) | 6.73E-07 | |||||
| European-ancestry analysis | |||||||||||||||
| 2:233208123 | DIS3L2 | rs141560952 | AGGG/A | Inframe insertion | Stage 1 | 11 | 641.4 | (33.1912396) | 1.89E-05 | 21.28 | (3.1145.9) | 1.85E-03 | 23.87 | (3.31172.2) | 1.65E-03 |
| Stage 2 | 25 | 44.69 | (6.35314.4) | 1.38E-03 | 4.51 | (0.92,22.22) | 6.40E-02 | 34.2 | (4.99234.5) | 1.27E-03 | |||||
| Meta-analysis | - | 100.1 | (19.62510.4) | 3.01E-08 | 8.48 | (2.48,28.95) | 6.44E-04 | 28.71 | (7.23114.0) | 1.82E-06 | |||||
| 12:52843534 | KRT6B | rs425827 | A/T | Splice region | Stage 1 | 108 | 5.55 | (3.11,9.88) | 6.10E-09 | 2.98 | (1.58,5.61) | 7.24E-04 | 3.89 | (2.2,6.88) | 2.80E-06 |
| Stage 2 | 32 | 3.47 | (0.57,21.35) | 0.18 | 6.09 | (0.52,71.37) | 0.15 | 4.27 | (0.63,29.11) | 0.14 | |||||
| Meta-analysis | - | 5.31 | (3.06,9.21) | 2.72E-09 | 3.11 | (1.69,5.75) | 2.81E-04 | 3.92 | (2.27,6.77) | 8.97E-07 | |||||
Abbreviations: AAF, alternative allele frequency; Alt, alternative; Chr, chromosome; CI, confidence interval; CKD, chronic kidney disease; DKD, diabetic kidney disease; OR, odds ratio; P, P-value; Ref, reference.
Figure 3.
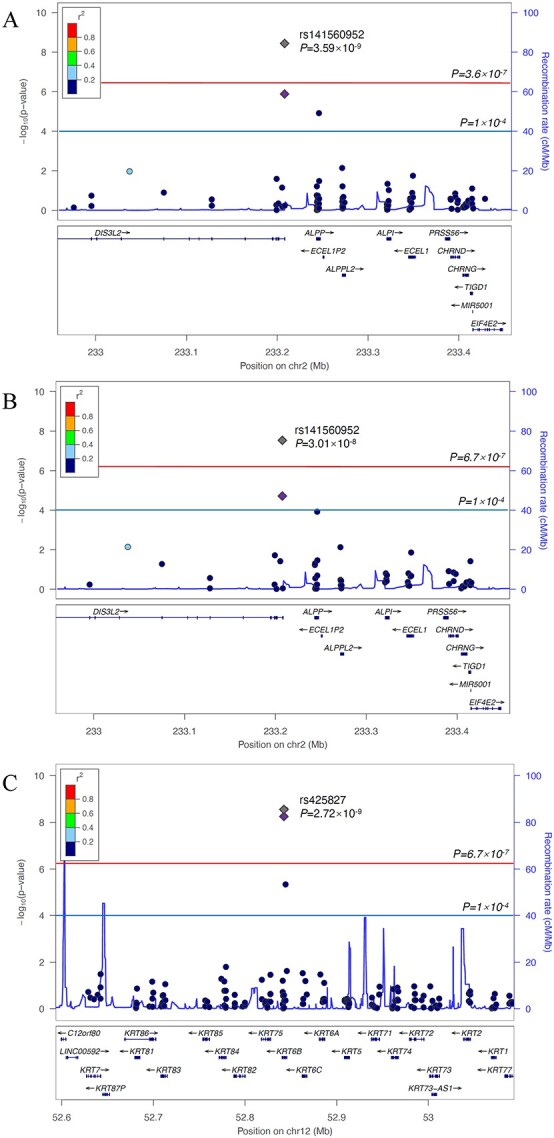
Regional association plots displaying significant signals at (A) DIS3L2 in multi-ancestry analyses comparing DKD cases with healthy controls, (B) DIS3L2 in European ancestry analyses comparing DKD cases with healthy controls and (C) KRT6B in European ancestry analyses comparing DKD cases with healthy controls.
Stage-2 and meta-analysis of suggestive genes from stage-1 aggregate rare variant analyses
Two suggestive gene signals identified in stage-1 aggregate rare variant analyses, ERAP2 and NPEPPS, were confirmed in stage-2 analyses and attained exome-wide significance (P = 4.03 × 10−8 and 1.51 × 10−7, respectively) in meta-analyses of multi-ancestry results comparing those with DKD to healthy controls and kidney disease controls, respectively (Table 3). Apparent signal-driving variants for ERAP2 and NPEPPS were identified by LOO analyses and included rs75364820 and rs1809279, respectively (Fig. 4A and B). However, neither of these variants were available for assessment in the stage-2 sample. Furthermore, none of the variants identified by LOO analyses of suggestive stage-1 gene-based signals were confirmed in stage-2 nor did they achieve exome-wide significance in meta-analyses (Supplementary Material, Tables S16–S18).
Table 3.
Exome-wide significant genes for diabetic kidney disease identified in meta-analyses of stage-1 and -2 analyses
| HGNC Symbol | Location (hg19) | Phase | DKD Cases vs. Healthy Controls | DKD Cases vs. Diabetes Controls | DKD Cases vs. CKD Controls | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rare Variant Sites (N) | Rare Alleles (N) | SKAT-O P | Rare Variant Sites (N) | Rare Alleles (N) | SKAT-O P | Rare Variant Sites (N) | Rare Alleles (N) | SKAT-O P | |||
| ERAP2 | 5:96211643-96255420 | Stage 1 | 5 | 76 | 9.60E-08 | 3 | 22 | 0.70 | 3 | 21 | 1.00 |
| Stage 2 | 8 | 138 | 1.99E-02 | 4 | 46 | 4.44E-02 | 3 | 48 | 5.51E-03 | ||
| Meta-analysis | - | - | 4.03E-08 | - | - | 0.14 | - | - | 3.42E-02 | ||
| NPEPPS | 17:45600308-45700642 | Stage 1 | 7 | 65 | 8.89E-09 | 4 | 20 | 0.19 | 5 | 43 | 1.55E-07 |
| Stage 2 | 2 | 18 | 0.65 | 1 | 6 | 0.48 | 1 | 2 | 4.95E-02 | ||
| Meta-analysis | - | - | 1.16E-07 | - | - | 0.31 | - | - | 1.51E-07 | ||
Abbreviations: CKD, chronic kidney disease; DKD, diabetic kidney disease; P, P-value; SKAT-O, optimal sequence kernel association test. Based on multi-ancestry analyses.
Figure 4.
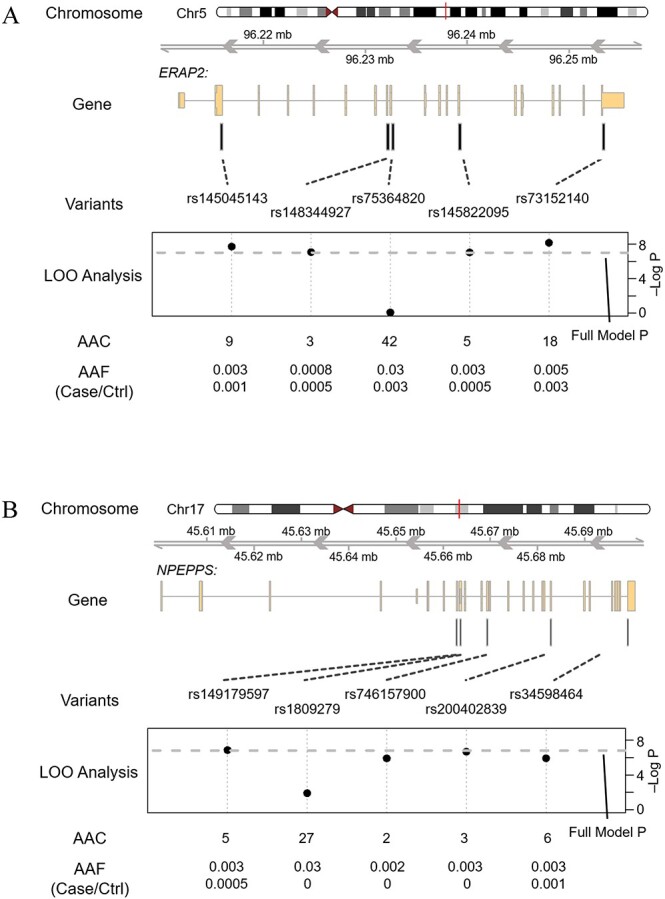
Gene structure plot illustrating the LOO analysis of aggregate signals at gene (A) ERAP2 and (B) NPEPPS.
Discussion
In the largest WES study of DKD conducted to date, we identified four novel gene loci, DIS3L2, KRT6B, ERAP2 and NPEPPS, that achieved exome-wide significance and may play an important role in the DKD disease course. The rare and low-frequency variants robustly identified by our single-variant analyses, DIS3L2 rs141560952 and KRT6B rs425827, respectively, demonstrated large effect sizes and, as an in-frame insertion and splice site variant, respectively, may have causal functional effects on DKD. Furthermore, ERAP2 and NPEPPS were identified in gene-based aggregate rare variant analyses. Interestingly, both genes encode aminopeptidases that are thought to be involved in the regulation of the renin–angiotensin–aldosterone–system (RAAS) (22,23) and have been associated with various autoimmune conditions (24,25). These results shed additional insights into the molecular mechanisms and biological pathways involved in the pathogenesis of DKD. Reassuringly, the stage-1 analysis also identified 126 suggestive associations within known kidney disease loci from previous GWAS studies. In total, our findings provide some of the first empirical evidence for a role of rare and low-frequency variants in the genetic architecture of DKD and highlight the added value of aggregate rare variant analyses in gene discovery.
Thirty-seven carriers of a 3 bp in-frame insertion in exon 14 (rs141560952) of the DIS3L2 gene exhibited a remarkable 193-fold increased odds of DKD compared with noncarriers in meta-analyses. While exome-wide significance was achieved only when comparing DKD cases with healthy controls, effect sizes were consistent in analyses of CKD controls. These findings suggest that DIS3L2 may be a risk factor for DKD, but not other CKD subphenotypes. Furthermore, the moderate but incomplete attenuation of the association signal when comparing DKD cases with diabetes controls indicates that diabetes status may not completely mediate the observed association between DIS3L2 and DKD. DIS3L2 might instead promote diabetes while also influencing the development of kidney disease among patients with diabetes. We note caution in this interpretation as the possible mediation effect was based on separated case control analyses instead of a formal mediation analysis, which is not suitable for this study population and for rare variants. DIS3L2 encodes a 3′-5′ endonuclease that helps to mediate the degradation of both messenger RNA (mRNA) and microRNA (miRNA) and has been linked to abnormal mitosis (26–28). Functional studies of DIS3L2 have demonstrated that germline mutations to the DIS3L2 gene were responsible for Perlman syndrome, (27) an autosomal recessive disorder resulting in kidney enlargement and high risk of Wilms tumor. A recent study further described the underlying molecular mechanism of renal abnormalities of the Perlman syndrome. A defective DIS3L2 function could cause dysregulations in endoplasmic reticulum-associated mRNA translation and intracellular calcium homeostasis, both are critical for proper function of kidney and other organs (26). In addition, the DIS3L2 locus has been associated with body mass index (BMI) by previous GWAS studies (21). However, in post-hoc analyses, adjustment for BMI did not attenuate our findings (data not shown). Although experimental studies are warranted to confirm the functional effects of the rs171560952 variant, these results provide compelling evidence for a role of DIS3L2 in DKD.
A low-frequency variant in a splicing region of the KRT6B gene, rs425827, also achieved exome-wide significance in analyses comparing DKD cases with healthy controls. In meta-analysis, each copy of the minor A allele of this variant conferred 5.3-fold higher odds of DKD. Although findings did not achieve exome-wide significance in analyses comparing DKD cases with diabetes and CKD controls, the effect sizes were consistent to those of the main analysis of the larger sample of healthy controls. As an isoform of keratin, KRT6B is expressed in the skin, esophagus and minor salivary gland, as well as in female tissues, according to gene expression data from Genotype-Tissue Expression project (29). The rs425827 variant has also been identified as an expression quantitative trait locus for KRT76 (29), a nearby gene encoding a keratin protein that is also expressed in the digestive tract. Given the predicted functional relevance of this variant in both KRT6B and KRT76, along with the high density of genes in this region of chromosome 12, future studies will be needed to better localize this promising signal.
Stage-1 gene-based analyses identified a suggestive signal at ERAP2 which was successfully confirmed and achieved exome-wide significance in meta-analysis of stage-1 and stage-2 results comparing DKD cases with healthy controls. Of note, a high-confidence pLOF variant in ERAP2 (rs75364820) appeared to drive the gene-based signal in the stage-1 analysis. This same variant was identified in stage-1 single-variant analysis, where it achieved genome-wide significance. Unfortunately, its lack of availability in the stage-2 sample prohibited confirmation of the relevance of rs75364820 to DKD. Nonetheless, ERAP2, which encodes a zinc-metallo aminopeptidase, is itself a promising DKD gene candidate. ERAP2 is expressed in the kidney and known to convert angiotensin II to angiotensin III, helping to regulate the RAAS (22,30). Furthermore, ERAP2 has been previously associated with a number of autoimmune conditions (24,25,31,32). These findings lend further credence to emerging evidence suggesting a role for RAAS-related immune activation in promoting renal injury and fibrosis (33). The ERAP2 locus was additionally reported to associate with 2-h oral glucose tolerance in GWAS study (34). This latter finding suggests that diabetes might mediate the relationship between ERAP2 and DKD. Indeed, the stage-1 signal was completely attenuated when comparing DKD cases with diabetes controls; however, the association remained nominally significant in corresponding analyses in the stage-2 study, limiting our interpretation of this finding. Hence, further research to elucidate whether ERAP2 influences DKD through the initiation of diabetes and/or the promotion of CKD among diabetes patients is needed.
In gene-based aggregate rare variant analyses, NPEPPS achieved exome-wide significance when comparing DKD cases to controls with CKD but free from diabetes. Because the signal was completely attenuated in analyses comparing DKD cases with diabetes controls, our data suggest that the association may be mediated by diabetes status. However, we note caution in this interpretation given the small number of participants with rare variants in the NPEPPS gene. NPEPPS encodes a puromycin-sensitive aminopeptidase which is ubiquitously expressed in 27 tissues, including the kidney (25,35). Like ERAP2, NPEPPS is thought to be involved in the RAAS (23) and has been linked to autoimmune conditions (24,25,36) as well lipid and hematologic phenotypes (37). However, we are the first to report an association with DKD. Functional studies to validate NPEPPS in diabetes-related kidney disease and identify relevant functional variants are warranted to follow-up on these promising findings.
Our study has several important strengths. By using a powerful extreme case–control design and limiting the study to a more homogeneous DKD phenotype we were able to identify robust signals for DKD in a relatively small sample (38,39). Furthermore, our unique analysis strategy, which leveraged three sets of controls, allowed us to better interpret whether identified genes and variants were related to DKD specifically or CKD more generally, promoted DKD among diabetes patients, or associated with DKD by increasing the risk of diabetes as an intermediate variable. Other strengths include sophisticated statistical modeling to handle the case–control imbalances in single and aggregate rare variant analysis and the use of multi-ancestry samples to identify genomic signals relevant to diverse populations. Several limitations of this study also warrant mention. Although our work represents one of the largest sequencing studies focused on DKD to date, the sample size likely restricted our ability to detect genetic associations, particularly in ancestry-stratified analysis as well as the secondary analysis of DKD vs. diabetes controls. In addition, kidney biopsy is the gold standard method for the identification of DKD (40–42), which was not feasible for defining cases in this large-scale epidemiologic study. However, to minimize misclassification, which could reduce statistical power, DKD cases were stringently defined in the stage-1 sample as patients with diabetes and CKD who not only had the most rapidly declining estimated glomerular filtration rate (eGFR) but also a baseline 24-hour urinary albumin excretion ≥30 mg/day, another important indicator of DKD (40). In stage-2, this criterion was relaxed due to a lack of availability of urinary albumin measurements in TOPMed participants. Additional limitations of this study include an inability to distinguish type 1 diabetes from type 2 diabetes and adjust for diabetes duration in the analysis, which again was due to a lack of available data for these variables. Furthermore, despite using sophisticated analytic methods to improve estimation of P-values for rare variants in the case–control analysis (43), the penalized quasi-likelihood followed by the saddlepoint approximation (SPA) in SAIGE could result in poorly estimated parameters for rare variants. In addition, the estimated parameters for rare variants might be influenced by adjustments of included covariates. While we attempted to mitigate this phenomenon by performing stage-2 analyses using GENESIS software, a careful and conservative interpretation of the odds ratios derived from the meta-analysis is recommended. In post-hoc evaluations, the odds ratios of the two identified loci were largely consistent across software (SAIGE and GENESIS) and covariable adjustments in the stage-1 analyses (data not shown).
In summary, our study identified two potentially functional variants, a rare DIS3L2 indel and low-frequency KRT6B single-nucleotide variant (SNV), which were robustly associated with the DKD disease course in a diverse sample of study participants. DIS3L2 rs141560952 and KRT6B rs425827 demonstrated strikingly large effects on DKD, providing compelling support for a role of these loci in DKD, which may, in part, be explained by their promotion of diabetes. In addition, aggregate gene-based analyses of rare variants implicated ERAP2 and NPEPPS, which are aminopeptidases believed to play a role in RAAS activation and immune response. These data provide further support for the role of RAAS-related immune activation not just in kidney damage but in the development of diabetes that precedes DKD. In total, our findings provide new insights into the molecular mechanisms underlying DKD, which could potentially be leveraged to aid in the prevention of DKD, from the development of diabetes to related kidney damage.
Materials and Methods
Study population
The overall study design is illustrated in Figure 1. Our stage-1 study employed an extreme case–control design, which included the selection of the 593 most rapidly progressing DKD cases from the CRIC study. DKD was defined as: (1) a history of diabetes at baseline; (2) 24-h urinary albumin excretion ≥30 mg/day; and (3) eGFR < 60 mL/min/1.73m2 (40). Among CRIC participants with DKD, we selected 297 African ancestry and 296 European ancestry participants with the most rapid decline in kidney function based on eGFR measured annually in up to 12 years of follow-up. Post-hoc analyses demonstrated that the selected DKD cases have a hazard ratio of 4.50 for DKD progression incident compared with other participants with diabetes in CRIC (Supplementary Material, Table S20). Cases were compared with three sets of controls selected from the ARIC study, including 2066 healthy controls in our primary analysis, as well as 652 diabetes controls and 1061 diabetes-free CKD controls in secondary analyses. ARIC study controls included ancestrally African and European participants, who at the fifth study visit [means (SD) age = 75.44 (5.09)] had (1) neither diabetes nor CKD (healthy controls); (2) diabetes but no CKD (diabetes controls); and (3) CKD but no diabetes (CKD controls). Diabetes was defined as a fasting plasma glucose (FPG) ≥126 mg/dL, non-fasting glucose (fasting time < 8 h) ≥200 mg/dL or the use of glucose lowering medications. CKD was defined as an eGFR < 60 mL/min/1.73 m2 or spot urine albumin-to-creatinine ratio ≥ 30 μg/mg.
Stage-2 analyses leveraged WGS data from participants of 11 TOPMed program studies (44) (TOPMed sequencing methods, freeze 8 [https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8]), which included the Amish Complex Disease Research program, Cardiovascular Health Study, Framingham Heart Study, Genetic Studies of Atherosclerosis Risk, Genetic Epidemiology Network of Arteriopathy, Genetic Epidemiology Network of Salt Sensitivity, Hypertension Genetic Epidemiology Network, Jackson Heart Study, Multi-ethnic Study of Atherosclerosis, Hispanic Community Health Study-Study of Latinos and Women’s Health Initiative. Among 32 733 multi-ancestry participants with kidney function and diabetes phenotypes, we selected 11 487 who met the criteria for categorization into the DKD case or one of the three control groups. In the stage-2 study, DKD cases were defined as participants with both diabetes and CKD (N = 601). Controls included participants with: (1) no evidence of diabetes and no evidence of CKD by 65 years of age (healthy controls; N = 7502); (2) diabetes but no evidence of CKD by 65 years of age (diabetes controls; N = 1572); and (3) participants with no evidence of diabetes but with CKD (CKD controls; N = 1812). Diabetes was defined as FPG ≥126 mg/dL, non-fasting glucose (fasting time < 8 h) ≥200 mg/dL or the use of glucose lowering medications, and CKD was defined as an eGFR < 60 mL/min/1.73 m2. TOPMed study cases and controls included participants with African ancestry, European ancestry and other ancestry groups. Descriptions of stage-1 and -2 studies are detailed in Supplementary Methods.
This study was approved by the Tulane University Biomedical Institutional Review Board.
Exome sequencing and variant calling
DNA samples were prepared and sequenced by Illumina HiSeq platforms in pair-end mode, followed by sequencing data processing using the Mercury pipeline with human reference genome GRCh37 (Supplementary Methods). Variant discovery (SNVs and indels) and genotype calling were conducted jointly across CRIC and ARIC study samples.
Quality control
Each variant call (SNV and indel) was filtered to produce a high-quality variant list using standard criteria (Supplementary Methods). This was followed by variant-level and sample-level quality controls, which filtered variants outside the exon capture regions, multi-allelic sites, missing rate > 5%, differential missingness case–control status, mapping score < 0.8, mean depth of coverage > 500-fold, Hardy–Weinberg equilibrium tests (P < 5 × 10−6) in ancestry-specific groups, sample-level missingness > 5%, excessive heterozygosity, etc. (Supplementary Methods).
Association analysis
Stage-1 multi-ancestry and ancestry-stratified single-variant association tests comparing cases with each of the three control groups were performed using SAIGE (V0.41) (43). SAIGE employs a generalized mixed model association test that accounts for sample relatedness and case–control imbalance. Variants with minor allele count (MAC) ≥ 10 were individually tested for association with DKD in all analyses. An empirical kinship matrix was used in SAIGE to control for population structure and relatedness within the sample. Covariates including baseline age, sex and the top 11 ancestry principal components (PCs) were included in the model (Supplementary Material, Fig. S4). BMI was additionally adjusted in sensitivity analyses. Identified variants achieving exome-wide suggestive significance (P < 5.00 × 10−5) (20,45) were moved forward for stage-2 analysis. Novel loci were defined as those with lead variants that were neither in close proximity (>500 KB) nor correlated (r2 < 0.1) with previously reported DKD or kidney function-related variant identified through curation of the NHGRI-EBI GWAS Catalog (21).
For rare variants, gene-based aggregate association tests were conducted. First, functional annotation was performed on all variants using the WGS Annotator (WGSA) (46). Then, rare variants with MAC ≥2 and MAF < 1% were aggregated into gene units using two design motifs: (1) variants were annotated as pLOF variants, missense variants and protein-altering indels in Variant Effect Predictor (VEP) ensemble consequences and have a FATHMM-XF score > 0.5 (47) and (2) high-confidence pLOF variants. The pLOF variants were defined by the Ensembl VEP as likely to have a loss of function consequence (48), which included belonging to any of the following categories: ‘transcript_ablation’, ‘splice_acceptor_variant’, ‘splice_donor_variant’, ‘stop_gained’, ‘frameshift_variant’, ‘stop_lost’, ‘start_lost’ and ‘transcript_amplification’. High-confidence pLOF variants are defined by the VEP annotation of ‘HC’ LOF. Multi-ancestry and ancestry-specific aggregate association analyses of rare variants were then performed using SAIGE-GENE (49), a generalized mixed model similar to SAIGE but designed for aggregate association tests. We used the SKAT-O (50) implemented in SAIGE-GENE to evaluate associations between aggregated rare variant gene units and DKD employing the same analytical frameworks described for single-variant analyses. LOO analyses were conducted to identify signal-driving variants among aggregate units achieving suggestive significance (P < 5.00 × 10−5). Any aggregate gene unit or variant whose removal attenuated the SKAT-O P-value by one order of magnitude or more was moved forward for stage-2 analysis.
Stage 2 and meta-analysis
Stage-2 analyses of multi-ancestry and ancestry-specific single and aggregate rare variant signals were performed on the Analysis Commons cloud-based platform following an analytical framework similar to that of the stage-1 (51). The GENESIS package (52) was used to perform association tests with the ‘SPA.score’ testing option enabled, which corresponds to the same statistical method in SAIGE/SAIGE-GENE packages employed in the stage-1 analysis pipeline. In rare instances of convergence failure, the default ‘Score’ test was used. All analyses again adjusted for sex, age, study and the top 11 ancestry PCs. For gene-based aggregate association analysis, WGSA annotation was again used to form gene-based units for stage-2 testing.
Results of single-variant analyses were meta-analyzed across stage-1 and stage-2 using standard inverse-variance-weighted methods (53). Exome-wide significance, which has a larger P-value threshold than the GWAS analysis due to less multiple-testing for exome-region variants, was determined based on a Bonferroni correction for the number of single-variant tests in each analysis, with maximum P-values for discerning exome-wide significance ranging from 3.66 × 10−7 (0.05/136454 tests) to 9.28 × 10−7 (0.05/53904 tests). Robustly identified signals included those that achieved exome-wide significance in meta-analysis, demonstrated consistent effect sizes across the stage-1 and -2 analyses (based on effect directions and a conservative heterogeneity P > 1.00 × 10−3) and achieved a minimum P-value in the pooled analysis of stage-1 and -2 results.
For meta-analyses of gene-based signals, Fisher’s method was employed (using the stats function in SciPy), which does not require the same variants to be tested across the stage-1 and stage-2. A standard P < 2.00 × 10−6 was used for discerning exome-wide significance, which assumes up to 25 000 gene-based tests. Robust gene-based signals were identified as those achieving nominal significance in the stage-2 analysis and exome-wide significance in meta-analysis.
Data and resource availability
The data supporting the findings of this study are available in the following repositories: the phenotype and genotype data of the ARIC study are available through dbGaP (accession no. phs000280). The phenotype data of the CRIC study are available through dbGaP (accession no. phs000524). The variant calling data and summary statistics of the CRIC WES data generated in this study will be deposit to the CRIC study at phs000524. The phenotype and genotype data of the TOPMed data used in this manuscript are available through dbGaP. The dbGaP accession numbers for each TOPMed studies are referenced in the TOPMed main paper. The TOPMed genotype data can be also accessed through Analysis Commons platform upon approval.
Conflict of interest statement
B.K. reports receiving personal fees from Reatta Pharmaceuticals. B.D.M.: the Amish Research Program receives partial support from Regeneron Pharmaceuticals. L.A.C. spends part of her time consulting for Dyslipidemia Foundation, a non-profit company, as a statistical consultant. B.M.P. serves on the DSMB of a clinical trial funded by the manufacturer (Zoll LifeCor) and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. R.A.G. is an employee of Baylor College of Medicine that receives revenue from Genetic Testing. H.F. reported personal fees from the National Kidney Foundation as the Editor-in-Chief of the American Journal of Kidney Diseases, personal fees from InMed Inc., personal fees from Kyowa Hakko Kirin Inc., travel expenses from Rogosin Institute, personal fees from DLA Piper, personal fees from AAKP, travel expenses from KDIGO (Kidney Disease Improving Global Outcomes), personal fees from the University of California Los Angeles and travel expenses from ISN outside the submitted work.
Funding
This research was supported by funds from the National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under award numbers R01 DK105050.
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program were supported by the National Heart, Lung and Blood Institute (NHLBI). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health or the U.S. Department of Health and Human Services.
A full list of CRIC, ARIC and TOPMed Project members and funding information is found in Supplemental Appendix 1. Grants supporting individual investigators are listed below.
R.V. was supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine.
L.M.R. was also supported by T32HL129982. The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR002490 (L.M.R).
M.F.. was supported by the University of Georgia Research Foundation.
Authors’ Contributions
T.N.K., J.E.H. and J.H. contributed to the conception or design of the project. Y.H. and T.N.K. contributed to study management. E.B., H.F., R.P.T., B.M.P., A.P.R., C.K., J.I.R., K.E.N., R.C.K., D.L., L.A.C., R.S.V., L.M.R., A.C., D.K.A., S.L.R.K., P.A.P., B.D.M., J.H., R.A.M, L.H., D.M.L.-J., M.F. and M.R.I. provided phenotype data. G.M., D.M, R.A.G., T.N.K. and E.B. contributed to study sequencing or provided whole-exome sequencing data. Y.P., X.S., X.M., Z.H. and T.N.K. processed the sequencing data, harmonized phenotype data and performed the analysis for this paper. R.A.G., A.C.M., H.F., E.B., Y.H., J.E.H., D.M., G.M., A.T., J.H. and T.N.K. contributed to the WES study of CRIC and ARIC data. R.P.T., B.M.P., A.P.R., C.K., J.I.R., K.E.N., R.C.K., D.L., L.A.C., R.S.V., L.M.R., A.C., D.K.A., S.L.R.K., P.A.P., B.D.M., J.H., R.A.M., L.H., D.M.L.-J., M.F., Y.I.C., D.V., L.R.Y., J.A.S., M.R.I., S.J.H., X.G., L.A.L., H.K., J.C.M., C.M.S., B.K., J.A.B., M.F., N.F., A.K. and the TOPMed Kidney Function Working group contributed to the analysis of TOPMed WGS data. Y.P., X.S., X.M., Z.H., J.H. and T.N.K. contributed to the interpretation of the results. T.N.K. and Y.P. drafted the manuscript and revised the paper according to co-author suggestions. T.N.K. and Y.P are guarantors of this work. All authors reviewed the manuscript, suggested revisions as needed and approved the final version.
Supplementary Material
Contributor Information
Yang Pan, Division of Nephrology, Department of Medicine, College of Medicine, University of Illinois at Chicago, Chicago, IL 60612, USA.
Xiao Sun, Department of Epidemiology, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA.
Xuenan Mi, Department of Epidemiology, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA.
Zhijie Huang, Department of Epidemiology, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA.
Yenchih Hsu, Center for Clinical Epidemiology and Biostatistics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA.
James E Hixson, Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Donna Munzy, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Ginger Metcalf, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Nora Franceschini, Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27516, USA.
Adrienne Tin, University of Mississippi Medical Center, Jackson, MS 39216, USA.
Anna Köttgen, Institute of Genetic Epidemiology, Faculty of Medicine and Medical Center – University of Freiburg, Freiburg 79106, Germany; Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA.
Michael Francis, Institute of Bioinformatics, University of Georgia, Athens, GA 30602, USA.
Jennifer A Brody, Cardiovascular Health Research Unit, Departments of Medicine, University of Washington, Seattle, WA 98195, USA.
Bryan Kestenbaum, University of Washington, Department of Medicine, Division of Nephrology, Kidney Research Institute, Seattle, WA 98195, USA.
Colleen M Sitlani, Cardiovascular Health Research Unit, Departments of Medicine, University of Washington, Seattle, WA 98195, USA.
Josyf C Mychaleckyj, Center for Public Health Genomics, University of Virginia, Charlottesville, Charlottesville, VA 22903, USA.
Holly Kramer, Department of Public Health Sciences, Loyola University Chicago, Maywood, IL 60153, USA.
Leslie A Lange, Division of Biomedical Informatics and Personalized Medicine, School of Medicine, University of Colorado Denver, Aurora, CO 80045, USA.
Xiuqing Guo, The Institute for Translational Genomics and Population Sciences, Department of Pediatrics, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Centre, Torrance, CA 90502, USA.
Shih-Jen Hwang, Framingham Heart Study, Framingham, MA 01702, USA; Department of Biostatistics, Boston University School of Public Health, Boston, MA 02118, USA.
Marguerite R Irvin, Department of Epidemiology, University of Alabama at Birmingham School of Public Health, Birmingham, AL 35233, USA.
Jennifer A Smith, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Lisa R Yanek, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Dhananjay Vaidya, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Yii-Der Ida Chen, The Institute for Translational Genomics and Population Sciences, Department of Pediatrics, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Centre, Torrance, CA 90502, USA.
Myriam Fornage, Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; Brown Foundation Institute of Molecular Medicine, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Donald M Lloyd-Jones, Department of Preventive Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Lifang Hou, Department of Preventive Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Rasika A Mathias, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Braxton D Mitchell, Department of Medicine, University of Maryland School of Medicine, Baltimore, MD 21201, USA; Geriatrics Research and Education Clinical Center, Baltimore Veterans Administration Medical Center, Baltimore, MD 21201, USA.
Patricia A Peyser, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Sharon L R Kardia, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Donna K Arnett, Department of Epidemiology, University of Kentucky, Lexington, KY 40506, USA.
Adolfo Correa, University of Mississippi Medical Center, Jackson, MS 39216, USA.
Laura M Raffield, Department of Genetics, University of North Carolina, Chapel Hill, NC 27516, USA.
Ramachandran S Vasan, Framingham Heart Study, Framingham, MA 01702, USA; Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA.
L Adrienne Cupple, Framingham Heart Study, Framingham, MA 01702, USA; Department of Biostatistics, Boston University School of Public Health, Boston, MA 02118, USA.
Daniel Levy, Framingham Heart Study, Framingham, MA 01702, USA; Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA; Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20814, USA.
Robert C Kaplan, Department of Epidemiology & Population Health, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Kari E North, Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27516, USA.
Jerome I Rotter, The Institute for Translational Genomics and Population Sciences, Department of Pediatrics, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Centre, Torrance, CA 90502, USA.
Charles Kooperberg, Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Alexander P Reiner, Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA; Department of Epidemiology, University of Washington, Seattle, WA 98195, USA.
Bruce M Psaty, Cardiovascular Health Research Unit, Departments of Medicine, University of Washington, Seattle, WA 98195, USA; Department of Epidemiology, University of Washington, Seattle, WA 98195, USA; Department of Health Services, University of Washington, Seattle, WA 98195, USA.
Russell P Tracy, Departments of Pathology & Laboratory Medicine and Biochemistry, Larner College of Medicine, University of Vermont, Burlington, VT 05405, USA.
Richard A Gibbs, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Alanna C Morrison, Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Harold Feldman, Center for Clinical Epidemiology and Biostatistics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA.
Eric Boerwinkle, Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Jiang He, Department of Epidemiology, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA.
Tanika N Kelly, Division of Nephrology, Department of Medicine, College of Medicine, University of Illinois at Chicago, Chicago, IL 60612, USA.
References
- 1. De Boer, I.H., Rue, T.C., Hall, Y.N., Heagerty, P.J., Weiss, N.S. and Himmelfarb, J. (2011) Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA, 305, 2532–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas, M.C., Brownlee, M., Susztak, K., Sharma, K., Jandeleit-Dahm, K.A.M., Zoungas, S., Rossing, P., Groop, P.H. and Cooper, M.E. (2015) Diabetic kidney disease. Nat. Rev. Dis. Primers., 1, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zelnick, L.R., Weiss, N.S., Kestenbaum, B.R., Robinson-Cohen, C., Heagerty, P.J., Tuttle, K., Hall, Y.N., Hirsch, I.B. and De Boer, I.H. (2017) Diabetes and CKD in the United States population, 2009–2014. Clin. J. Am. Soc. Nephrol., 12, 1984–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Afkarian, M., Sachs, M.C., Kestenbaum, B., Hirsch, I.B., Tuttle, K.R., Himmelfarb, J. and Boer, I.H.D. (2013) Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol., 24, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tuttle, K.R., Bakris, G.L., Bilous, R.W., Chiang, J.L., De Boer, I.H., Goldstein-Fuchs, J., Hirsch, I.B., Kalantar-Zadeh, K., Narva, A.S., Navaneethan, S.D. et al. (2014) Diabetic kidney disease: a report from an ADA Consensus Conference. Am. J. Kidney Dis., 64, 510–533. [DOI] [PubMed] [Google Scholar]
- 6. Bikbov, B., Purcell, C.A., Levey, A.S., Smith, M., Abdoli, A., Abebe, M., Adebayo, O.M., Afarideh, M., Agarwal, S.K., Agudelo-Botero, M. et al. (2020) Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet, 395, 709–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins, A.J., Foley, R.N., Herzog, C., Chavers, B., Gilbertson, D., Ishani, A., Kasiske, B., Liu, J., Mau, L.W., McBean, M. et al. (2011) US renal data system 2010 annual data report. Am. J. Kidney Dis., 57, A8. [DOI] [PubMed] [Google Scholar]
- 8. Krolewski, A.S., Skupien, J., Rossing, P. and Warram, J.H. Fast renal decline to end-stage renal disease: an unrecognized feature of nephropathy in diabetes. Kidney Int., 91, 1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seaquist, E.R., Goetz, F.C., Rich, S. and Barbosa, J. (1989) Familial clustering of diabetic kidney disease. N. Engl. J. Med., 320, 1161–1165. [DOI] [PubMed] [Google Scholar]
- 10. The Diabetes Control and Complications Trial Research Group (1997) Clustering of long-term complications in families with diabetes in the Diabetes Control and Complications Trial. Diabetes, 46, 1829–1839. [PubMed] [Google Scholar]
- 11. Quinn, M., Angelico, M.C., Warram, J.H. and Krolewski, A.S. (1996) Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia, 39, 940–945. [DOI] [PubMed] [Google Scholar]
- 12. Harjutsalo, V., Katoh, S., Sarti, C., Tajima, N. and Tuomilehto, J. (2004) Population-based assessment of familial clustering of diabetic nephropathy in type 1 diabetes. Diabetes, 53, 2449–2454. [DOI] [PubMed] [Google Scholar]
- 13. Thomas, M.C., Groop, P.H. and Tryggvason, K. (2012) Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr. Opin. Nephrol. Hypertens., 21, 195–202. [DOI] [PubMed] [Google Scholar]
- 14. Van Zuydam, N.R., Ahlqvist, E., Sandholm, N., Deshmukh, H., Rayner, N.W., Abdalla, M., Ladenvall, C., Ziemek, D., Fauman, E., Robertson, N.R. et al. (2018) A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes, 67, 1414–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iyengar, S.K., Sedor, J.R., Freedman, B.I., Kao, W.H.L., Kretzler, M., Keller, B.J., Abboud, H.E., Adler, S.G., Best, L.G., Bowden, D.W. et al. (2015) Genome-wide association and trans-ethnic meta-analysis for advanced diabetic kidney disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet., 11, e1005352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Seaghdha, C.M. and Fox, C.S. (2012) Genome-wide association studies of chronic kidney disease: What have we learned? Nat. Rev. Nephrol., 8, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palmer, N.D. and Freedman, B.I. (2012) Insights into the genetic architecture of diabetic nephropathy. Curr. Diab. Rep., 12, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo, J., Rackham, O.J.L., Sandholm, N., He, B., Österholm, A.M., Valo, E., Harjutsalo, V., Forsblom, C., Toppila, I., Parkkonen, M. et al. (2020) Whole-genome sequencing of finnish type 1 diabetic siblings discordant for kidney disease reveals DNA variants associated with diabetic nephropathy. J. Am. Soc. Nephrol., 31, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lazaro-Guevara, J., Fierro-Morales, J., Wright, A.H., Gunville, R., Simeone, C., Frodsham, S.G., Pezzolesi, M.H., Zaffino, C.A., Al-Rabadi, L., Ramkumar, N. et al. (2021) Targeted next-generation sequencing identifies pathogenic variants in diabetic kidney disease. Am. J. Nephrol., 52, 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guan, M., Keaton, J.M., Dimitrov, L., Hicks, P.J., Xu, J., Palmer, N.D., Wilson, J.G., Freedman, B.I., Bowden, D.W. and Ng, M.C.Y. (2018) An exome-wide association study for type 2 diabetes-attributed end-stage kidney disease in African Americans. Kidney Int. Reports, 3, 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buniello, A., Macarthur, J.A.L., Cerezo, M., Harris, L.W., Hayhurst, J., Malangone, C., McMahon, A., Morales, J., Mountjoy, E., Sollis, E. et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res., 47, D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paladini, F., Fiorillo, M.T., Tedeschi, V., Mattorre, B. and Sorrentino, R. (2020) The multifaceted nature of aminopeptidases ERAP1, ERAP2, and LNPEP: from evolution to disease. Front. Immunol., 11, 1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kopf, P.G., Park, S.-K., Herrnreiter, A., Krause, C., Roques, B.P. and Campbell, W.B. (2018) Obligatory metabolism of angiotensin II to angiotensin III for zona glomerulosa cell-mediated relaxations of bovine adrenal cortical arteries. Endocrinology, 159, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellinghaus, D., Jostins, L., Spain, S.L., Cortes, A., Bethune, J., Han, B., Park, Y.R., Raychaudhuri, S., Pouget, J.G., Hübenthal, M. et al. (2016) Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet., 48, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortes, A., Hadler, J., Pointon, J.P., Robinson, P.C., Karaderi, T., Leo, P., Cremin, K., Pryce, K., Harris, J., Lee, S. et al. (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet., 45, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pirouz, M., Wang, C.H., Liu, Q., Ebrahimi, A.G., Shamsi, F., Tseng, Y.H. and Gregory, R.I. (2020) The Perlman syndrome DIS3L2 exoribonuclease safeguards endoplasmic reticulum-targeted mRNA translation and calcium ion homeostasis. Nat. Commun., 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Astuti, D., Morris, M.R., Cooper, W.N., Staals, R.H.J., Wake, N.C., Fews, G.A., Gill, H., Gentle, D., Shuib, S., Ricketts, C.J. et al. (2012) Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet., 44, 277–284. [DOI] [PubMed] [Google Scholar]
- 28. Chang, H.M., Triboulet, R., Thornton, J.E. and Gregory, R.I. (2013) A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature, 497, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., Hasz, R., Walters, G., Garcia, F., Young, N. et al. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu, X., Eales, J.M., Akbarov, A., Guo, H., Becker, L., Talavera, D., Ashraf, F., Nawaz, J., Pramanik, S., Bowes, J. et al. (2018) Molecular insights into genome-wide association studies of chronic kidney disease-defining traits. Nat. Commun., 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li, Y.R., Li, J., Zhao, S.D., Bradfield, J.P., Mentch, F.D., Maggadottir, S.M., Hou, C., Abrams, D.J., Chang, D., Gao, F. et al. (2015) Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med., 21, 1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsoi, L.C., Spain, S.L., Knight, J., Ellinghaus, E., Stuart, P.E., Capon, F., Ding, J., Li, Y., Tejasvi, T., Gudjonsson, J.E. et al. (2012) Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet., 44, 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roscioni, S.S., Heerspink, H.J.L. and Zeeuw, D.d. (2013) The effect of RAAS blockade on the progression of diabetic nephropathy. Nat. Rev. Nephrol., 10, 77–87. [DOI] [PubMed] [Google Scholar]
- 34. Scott, R.A., Lagou, V., Welch, R.P., Wheeler, E., Montasser, M.E., Luan, J., MäGi, R., Strawbridge, R.J., Rehnberg, E., Gustafsson, S. et al. (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet., 44, 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thul, P.J., Akesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., Alm, T., Asplund, A., Björk, L., Breckels, L.M. et al. (2017) A subcellular map of the human proteome. Science, 356, eaal3321. [DOI] [PubMed] [Google Scholar]
- 36. Goris, A., van Setten, J., Diekstra, F., Ripke, S., Patsopoulos, N.A., Sawcer, S.J., van Es, M., Andersen, P.M., Melki, J., Meininger, V. et al. (2014) No evidence for shared genetic basis of common variants in multiple sclerosis and amyotrophic lateral sclerosis. Hum. Mol. Genet., 23, 1916–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang, B., Xiong, X., Zhang, L., Liu, X., Wang, Y., Gong, X., Sang, Q., Lu, Y., Qu, H., Zheng, H. et al. (2021) PSA controls hepatic lipid metabolism by regulating the NRF2 signaling pathway. J. Mol. Cell Biol., 13, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emond, M.J., Louie, T., Emerson, J., Zhao, W., Mathias, R.A., Knowles, M.R., Wright, F.A., Rieder, M.J., Tabor, H.K., Nickerson, D.A. et al. (2012) Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet., 44, 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shtir, C., Aldahmesh, M.A., Al-Dahmash, S., Abboud, E., Alkuraya, H., Abouammoh, M.A., Nowailaty, S.R., Al-Thubaiti, G., Naim, E.A., ALYounes, B. et al. (2016) Exome-based case–control association study using extreme phenotype design reveals novel candidates with protective effect in diabetic retinopathy. Hum. Genet., 135, 193–200. [DOI] [PubMed] [Google Scholar]
- 40. Ekinci, E.I., Jerums, G., Skene, A., Crammer, P., Power, D., Cheong, K.Y., Panagiotopoulos, S., McNeil, K., Baker, S.T., Fioretto, P. et al. (2013) Renal structure in normoalbuminuric and albuminuric patients with type 2 diabetes and impaired renal function. Diabetes Care, 36, 3620–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fineberg, D., Jandeleit-Dahm, K.A.M. and Cooper, M.E. (2013) Diabetic nephropathy: diagnosis and treatment. Nat. Rev. Endocrinol., 9, 713–723. [DOI] [PubMed] [Google Scholar]
- 42. Suarez, M.L.G., Thomas, D.B., Barisoni, L. and Fornoni, A. (2013) Diabetic nephropathy: is it time yet for routine kidney biopsy? World J. Diabetes, 4, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou, W., Nielsen, J.B., Fritsche, L.G., Dey, R., Gabrielsen, M.E., Wolford, B.N., LeFaive, J., VandeHaar, P., Gagliano, S.A., Gifford, A. et al. (2018) Efficiently controlling for case–control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet., 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M. et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Torres, G.G., Nygaard, M., Caliebe, A., Blanché, H., Chantalat, S., Galan, P., Lieb, W., Christiansen, L., Deleuze, J.F., Christensen, K. et al. (2021) Exome-wide association study identifies FN3KRP and PGP as new candidate longevity genes. J Gerontol.A Biol. Sci. Med. Sci., 76, 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu, X., White, S., Peng, B., Johnson, A.D., Brody, J.A., Li, A.H., Huang, Z., Carroll, A., Wei, P., Gibbs, R. et al. (2015) WGSA: an annotation pipeline for human genome sequencing studies. J. Med. Genet., 53, 111–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rogers, M.F., Shihab, H.A., Mort, M., Cooper, D.N., Gaunt, T.R. and Campbell, C. (2018) FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics, 34, 511–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McLaren, W., Gil, L., Hunt, S.E., Riat, H.S., Ritchie, G.R.S., Thormann, A., Flicek, P. and Cunningham, F. (2016) The Ensembl Variant Effect Predictor. Genome Biol., 17, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou, W., Zhao, Z., Nielsen, J.B., Fritsche, L.G., LeFaive, J., Gagliano Taliun, S.A., Bi, W., Gabrielsen, M.E., Daly, M.J., Neale, B.M. et al. (2020) Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nat. Genet., 52, 634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee, S., Wu, M.C. and Lin, X. (2012) Optimal tests for rare variant effects in sequencing association studies. Biostatistics, 13, 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brody, J.A., Morrison, A.C., Bis, J.C., O’Connell, J.R., Brown, M.R., Huffman, J.E., Ames, D.C., Carroll, A., Conomos, M.P., Gabriel, S. et al. (2017) Analysis commons, a team approach to discovery in a big-data environment for genetic epidemiology. Nat. Genet., 49, 1560–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gogarten, S.M., Sofer, T., Chen, H., Yu, C., Brody, J.A., Thornton, T.A., Rice, K.M. and Conomos, M.P. (2019) Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics, 35, 5346–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Willer, C.J., Li, Y. and Abecasis, G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.