

Abstract
Endothelial nitric oxide synthase (eNOS) is critically important enzyme responsible for regulation of cardiovascular homeostasis. Under physiological conditions, constitutive eNOS activity and production of endothelial nitric oxide (NO) exert essential neurovascular protective functions. In this review, we first discuss the roles of endothelial NO in prevention of neuronal amyloid accumulation and formation of neurofibrillary tangles, hallmarks of Alzheimer’s disease pathology. Next, we review existing evidence suggesting that NO released from endothelium prevents activation of microglia, stimulates glycolysis in astrocytes, and increases biogenesis of mitochondria. We also address major risk factors for cognitive impairment including aging and apolipoprotein 4 (ApoE4) genotype with focus on their detrimental effects on eNOS/NO signaling. Relevant to this review, recent studies suggested that aged eNOS heterozygous mice are unique model of spontaneous cerebral small vessel disease. In this regard, we review contribution of dysfunctional eNOS to deposition of amyloid-β (Aβ) into blood vessel wall leading to development of cerebral amyloid angiopathy (CAA). We conclude that endothelial dysfunction manifested by the loss of neurovascular protective functions of NO may significantly contribute to development of cognitive impairment.
Keywords: BACE1, ADAM10, amyloid, tau protein, microglia, astrocyte, mitochondria
Graphical Abstract
Introduction
According to the World Health Organization, currently, more than 55 million people suffer from dementia.1 This difficult problem is aggravated by the lack of effective therapeutic interventions. Indeed, effective therapy for dementia is major unmet need in modern medicine. Prior studies established that loss of neuroprotective function of endothelium significantly contributes to vulnerability of brain to neurodegeneration and dementia.2 More precisely, impaired endothelial nitric oxide synthase (eNOS)/nitric oxide (NO) signaling appears to be an essential molecular mechanism linking cardiovascular risk factors with altered brain function and development of cognitive impairment.3,4 The focus of this review is on mechanisms underlying contribution of impaired vascular eNOS/NO signaling to initiation and progression of cognitive impairment.
Nitric oxide in the cerebrovascular endothelium
Extensive studies in experimental models and human subjects documented critically important role of eNOS in control of vascular function.5,6 Under pathological conditions, impaired production and/or biological activity of endothelial NO promotes vasoconstriction, platelets aggregation, upregulation of white blood cells adhesion molecules, and proliferation of vascular smooth muscle cells thereby significantly contributing to pathogenesis of hypertension, atherosclerosis, and vascular complications of diabetes.6 The importance of vascular protective functions of eNOS is further underscored by observations demonstrating that human subjects with a genetic predisposition to enhanced endothelial NO signaling have reduced risk of stroke.7 Whether this protective effect of eNOS polymorphisms translates into reduced risk of cognitive impairment and dementia is unknown.
Because capillaries are by far the longest segment of cerebrovascular tree, majority of eNOS in the brain is localized in capillary endothelium. Importantly, the number of endothelial cells in the brain closely matches number of neurons.8 Moreover, brain capillaries are located less than15 μm from the nearest neuronal cell body9, and therefore “cloud of NO” released from the capillary endothelium may affect function of surrounding parenchymal cells.10–13 Indeed, prior studies have demonstrated that release of NO from cerebrovascular endothelium and diffusion of NO into surrounding environment enables communication between brain vasculature and neuronal cells.14–16 Furthermore, evidence continues to accumulate that under pathological conditions, dysfunctional communication between vascular and neuronal compartments in the brain is an important component of complex pathogenesis of neurodegeneration.17
The role of endothelial NO in development of amyloid pathology
Amyloid precursor protein (APP) is highly expressed in human and murine cerebrovascular endothelium.12 Interestingly, expression of APP is significantly higher in endothelium of cerebral blood vessels compared with endothelium of systemic blood vessels.18 High expression and strong evolutionary conservation of APP implies important functional relevance. Studies in cultured human brain endothelial cells indicate that under physiological conditions, endothelial APP is for the most part cleaved by α-secretase thus resulting in the release of soluble APPα (sAPPα) from endothelium into local environment (Figure 1).19 Importantly, α-secretase cleaves APP within Aβ-sequence thereby contributing to low formation of Aβ peptides in healthy endothelium.19,20 However, in human brain endothelial cells, genetic or pharmacologic inactivation of eNOS function increases expression of APP and β-site APP-cleaving enzyme 1 (BACE1) thereby shifting APP processing towards β-cleavage and increased production of Aβ peptides (Figure 2).12,21 Reduced activity of soluble guanylate cyclase (sGC) and reduction of cyclic guanosine monophosphate (cGMP) levels are responsible for increased amyloidogenic processing of APP.12,22 Resulting high local concentration of Aβ may exert detrimental effects on endothelial function mediated by increased formation of reactive oxygen species.23 Moreover, the results of prior studies suggest that elevated production of Aβ may also adversely affect endothelium-mediated Aβ clearance.24
Figure 1.
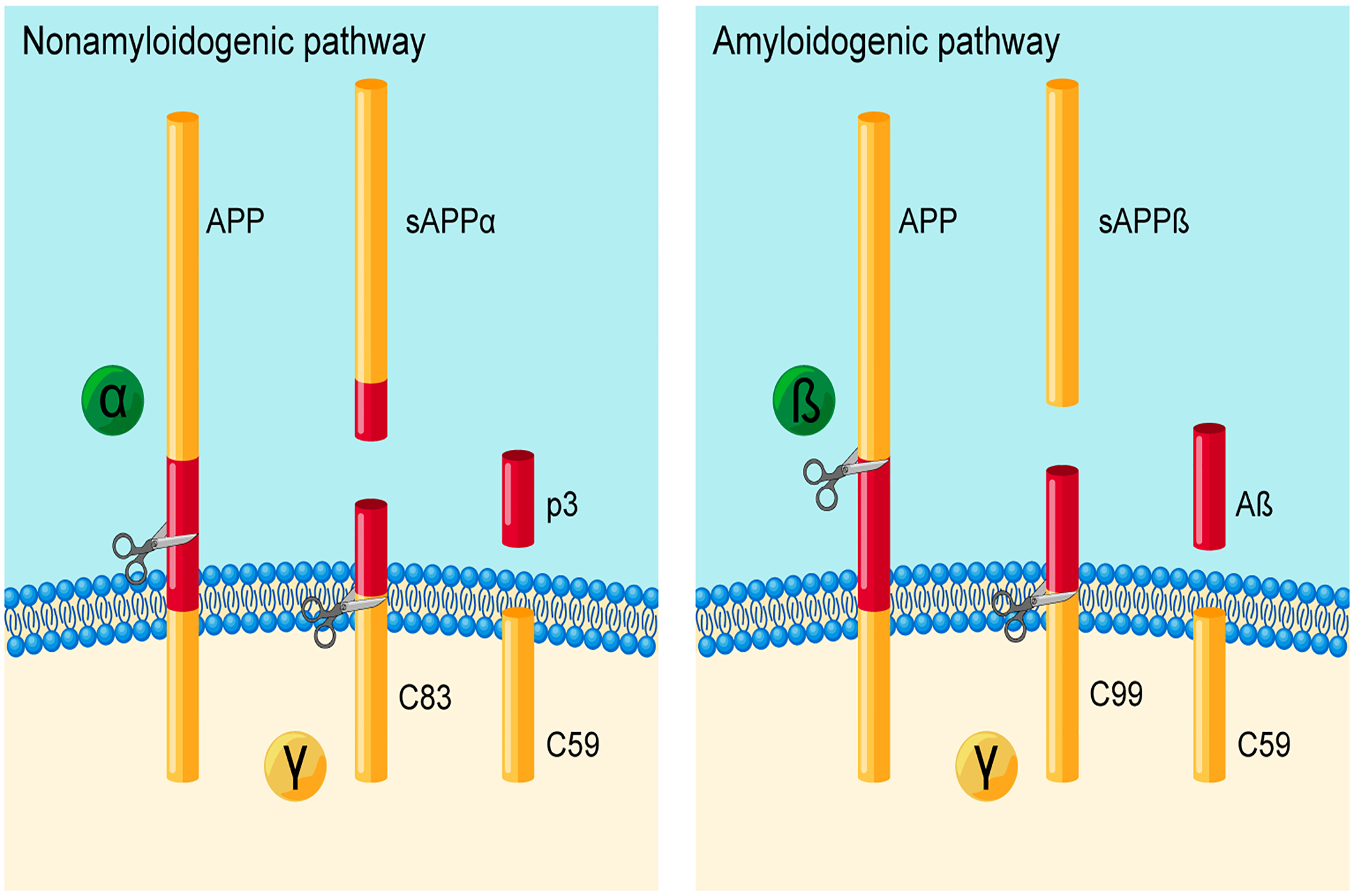
Proteolytic cleavage of amyloid precursor protein (APP) by a nonamyloidogenic pathway (left) or an amyloidogenic pathway (right). In the nonamyloidogenic pathway, APP is cleaved by α-secretase thereby releasing the sAPPα ectodomain. Following α-secretase cleavage, γ-secretase releases the p3 fragment and the APP intracellular domain C59. In the amyloidogenic pathway, β-secretase (BACE1) cleaves APP and releases the sAPPβ ectodomain. Subsequent processing by γ-secretase generates the cytotoxic peptide, Aβ, as well as the C59.
Figure 2.
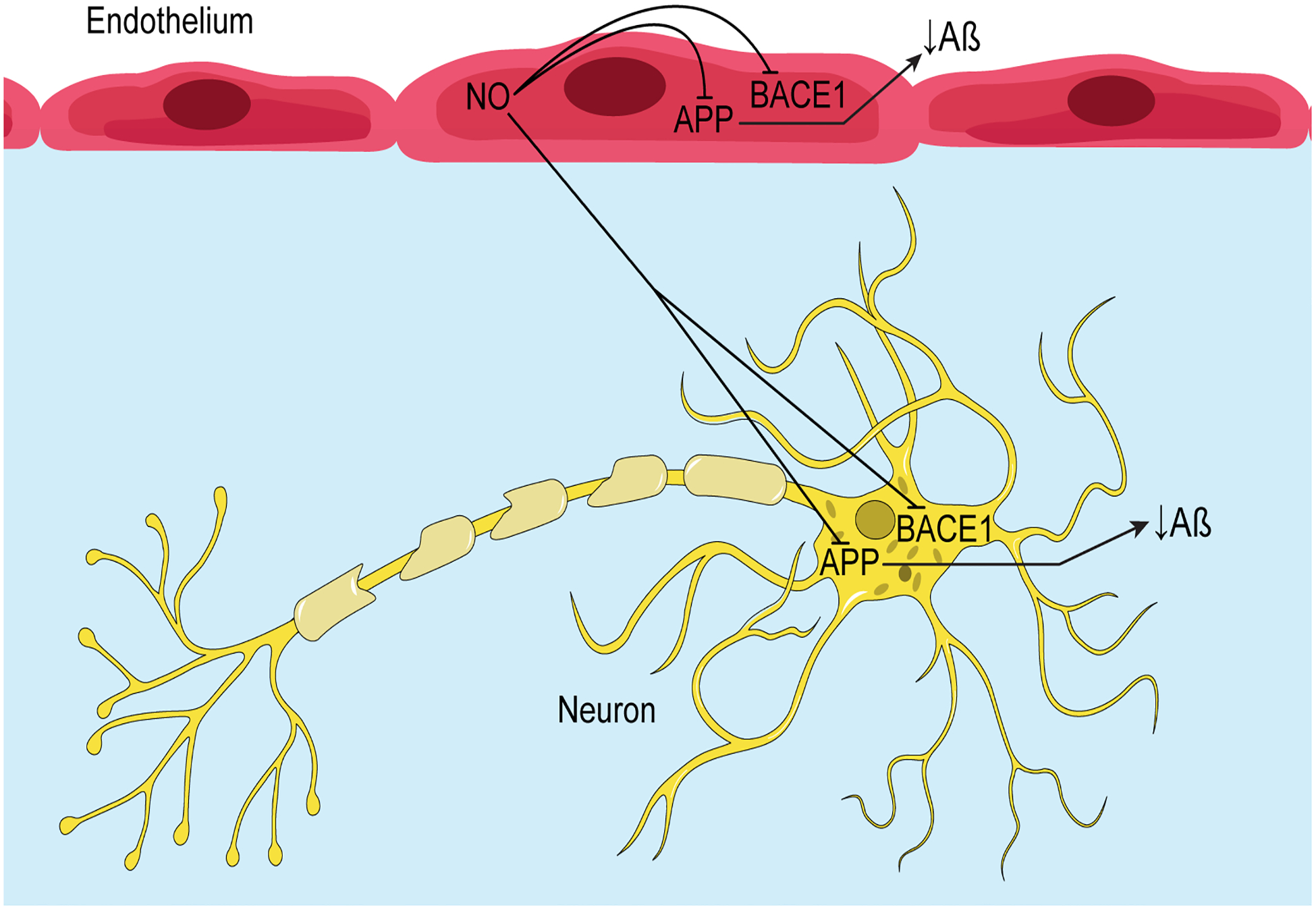
Schematic representation of the inhibitory effects of endothelial NO on expression of APP and BACE1, and production of Aβ in endothelial and neuronal cells.
Importantly, genetic inactivation of eNOS in mice (eNOS−/− mice) increases expression of APP and BACE1 as well as levels of Aβ peptides in the brain parenchyma.12 Of note, genetic inactivation of endothelial NO in mice does not affect expression of α-, and γ-secretase proteins, or Aβ degradation enzymes thereby demonstrating selectivity of endothelial NO modulatory effect for β-processing of APP.12 Agreement between data generated in brain blood vessels derived from humans and mice, demonstrates that observed endothelial NO modulation of APP and BACE1 expression is evolutionary conserved mechanism. Importantly, prior studies in eNOS−/− mice established that the loss of endothelial NO production in the cerebral circulation does not affect global cerebral blood flow.25,26 Based on these observations, we speculate that in eNOS−/− mice, increased expression of APP and BACE1 might be the primary effect of impaired endothelial NO production rather than secondary effect caused by global hypoperfusion of the brain. However, even though cerebral blood flow is not affected, eNOS−/− mice suffer from localized microvascular occlusions that apparently contribute to development of cerebral amyloid angiopathy.27 How much microvascular occlusions may contribute to development of amyloid pathology in neuronal tissue is unclear at the present time.
Because aging is a major risk factor for cognitive impairment, studies in aged eNOS−/− mice are critically important for understanding vascular and cognitive mechanisms impaired by joint loss of eNOS and aging. Indeed, new insights emerged from studies of 14–15 month [late middle aged (LMA)] eNOS−/− mice. Levels of APP, BACE1, and Aβ are significantly higher in the brains of LMA eNOS−/− mice as compared with LMA wild type mice. In addition, increased levels of APP and Aβ1–40 were detected in hippocampus of aged-matched eNOS−/− mice.28 Most notably, brain tissue derived from LMA eNOS−/− mice demonstrated significantly higher levels of microglial markers, cluster of differentiation 68 (CD68), ionized calcium-binding adaptor molecule 1 (Iba1), and major histocompatibility complex II (MHC II).28 Further analysis of brain tissue identified elevation of pro-inflammatory cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1α (IL-1α), and macrophage inflammatory protein-1β (MIP-1β). These observations were confirmed with individual ELISA essays for GM-CSF, IL-1α, and MIP-1β thereby proving that loss of endothelial NO in aged mice is associated with microglial activation. We also wish to point out that LMA eNOS−/− mice suffered from spatial memory deficit consistent with prior reports demonstrating importance of NO pathway in formation of long-term potentiation (LTP) and normal function of hippocampus.15,29 Thus, studies in LMA eNOS−/− mice revealed previously unrecognized mechanism whereby loss of endothelial NO promotes amyloid pathology and activation of microglia.
It is important to note that LMA eNOS−/− mice displayed a phenotype consistent with metabolic syndrome (elevated blood pressure, high circulating total cholesterol, density lipoprotein, triglycerides, and glucose detected in LMA eNOS−/− mice).28 The exact role of metabolic changes in development of amyloid pathology and microglial activation is unclear. However, it is unlikely that only metabolic factors and/or hypertension are responsible for development of amyloid pathology in LMA eNOS−/- mice. Indeed, pharmacological inhibition or genetic deletion of eNOS in cultured human brain microvascular endothelial cells increased expression of APP, BACE1, and Aβ levels independently of alterations of blood pressure and/or altered metabolic factors.12 In agreement with these findings, increased brain APP, BACE1, and Aβ, were detected in young eNOS−/− mice which displayed normal metabolic profile thus suggesting that loss of direct NO effects on endothelium is critical mechanism underlying alterations in cerebrovascular and neuronal expression and processing of APP.
Interestingly, several studies reported increased expression of eNOS protein in brains of AD patients and experimental animals with AD pathology.30–35 However, studies in murine model of AD indicate that despite significant upregulation of eNOS protein, eNOS enzyme activity is uncoupled from production of NO.35 Instead of NO, uncoupled eNOS generates superoxide anion and peroxynitrite thereby leading to dysfunctional NO signaling. Indeed, in cerebral microvessels of AD mice, uncoupling of eNOS results in significantly reduced levels of cGMP.35 As we have already noted, impairment of NO/cGMP signaling pathway promotes development of amyloid pathology.
Aging, a major non-modifiable risk factor for cardiovascular disease and AD, increases expression of eNOS mRNA in large cerebral arteries and brain capillaries36,37 but does not affect expression of eNOS protein.38,39 However, aging causes shift in eNOS protein phosphorylation patterns thereby promoting inhibition of eNOS activity.38 Importantly, aging also causes inactivation of endothelial NO by superoxide anion generated by upregulated NADPH oxidase and uncoupled eNOS.40,41 Similarly, increased production of superoxide anion by NADPH oxidase and uncoupled eNOS impairs NO signaling in endothelium exposed to cardiovascular risk factors including hypertension, diabetes, and hyperlipidemia.42 In aggregate, these observations suggest that loss of endothelial NO signaling in the brain may be a molecular mechanism linking cardiovascular risk factors with higher risk for development of AD pathology and cognitive impairment.43
Ongoing efforts to develop new drugs for prevention and treatment of cognitive impairment resulted in discovery of the first central nervous system (CNS)-penetrant sGC stimulators which are positive allosteric modulators of sGC.44,45 They can sensitize the sGC enzyme to NO, thereby preserving and amplifying spatiotemporal control of endogenous NO signaling.46 Preclinical studies have established that the CNS-penetrant sGC stimulators exert beneficial effects in experimental models of neuroinflammation and neurodegeneration.44,45 Currently, clinical trial with CNS-penetrant sGC stimulator, CY6463, is enrolling patients with Alzheimer’s disease (AD) and associated vascular pathology (ClinicalTrials.gov Identifier: NCT04798989). Of note, single-center, proof of concept study reported that treatment of AD patients with three medications, simvastatin, L-arginine, and tetrahydrobiopterin (known to stimulate eNOS/NO signaling) was well-tolerated and resulted in modest increase in cerebral blood flow and improved cognitive function (ClinicalTrials.gov Identifier: NCT01439555).47
Over many years, targeting cGMP/NO signaling with phosphodiesterase inhibitors have been extensively investigated in preclinical models and in patients with cognitive impairment and/or dementia including AD. While several phosphodiesterase inhibitors have shown some promising beneficial effects, none of tested phosphodiesterase inhibitors have been approved for prevention or treatment of cognitive impairment and dementia. Recently, Tropea and colleagues (2022) comprehensively reviewed therapeutic potential of phosphodiesterase inhibitors as future therapeutic approach to impaired cognition.48
The role of endothelial NO in development of tau pathology
In neuronal cells, dissociation of hyperphosphorylated tau protein from the microtubules causes formation of neurofibrillary tangles, a hallmark of AD pathology.49 Excessive phosphorylation of tau and formation of tau aggregates are critically important steps in development of cognitive impairment in AD. Existing literature supports the concept that impairment of endothelial NO production and signaling promotes tau phosphorylation thereby contributing to pathogenesis of AD.50–52 Indeed, genetic inactivation of eNOS in murine model of AD (APP/PS1/eNOS−/− mice) increases phosphorylation of tau in neuronal cells.50 This effect is driven by increased cyclin-dependent kinase 5 (Cdk5) activator, p25, and increased activity of Cdk5, enzyme responsible for phosphorylation of tau.50 These observations suggest that impaired production of NO in cerebrovascular endothelium may initiate and/or exacerbate tau pathology. Indeed, extensive analyses of vascular and cognitive function in mouse model of endothelial dysfunction induced by high salt diet, provided several new mechanistic insights into the role of eNOS in tau phosphorylation.51,52 Historically, brain hypoperfusion has been considered major mechanism by which impaired vascular function contributes to cognitive impairment.2 However, more recent findings indicate that high salt diet causes endothelial dysfunction by phosphorylation of eNOS at Threonine 495 thereby inhibiting eNOS activity and reducing endothelial production of NO.51,52 Most importantly, in this model, suppression of eNOS activity increases phosphorylation of tau thus leading to impairment of cognitive function. Comprehensive analysis of vascular and cognitive functions reinforced the conclusion that tau phosphorylation rather than cerebral hypoperfusion contributes to the cognitive dysfunction induced by high salt diet.52 More precisely, it was demonstrated that loss of endothelial NO reduces neuronal calpain nitrosylation thereby elevating levels of Cdk5 activator, p25 (Figure 3). Thus, loss of endothelial NO promotes phosphorylation of tau protein and formation of neurofibrillary tangles in murine model of endothelial dysfunction. Whether this mechanism contributes to pathogenesis of cognitive impairment in humans remains to be determined.
Figure 3.
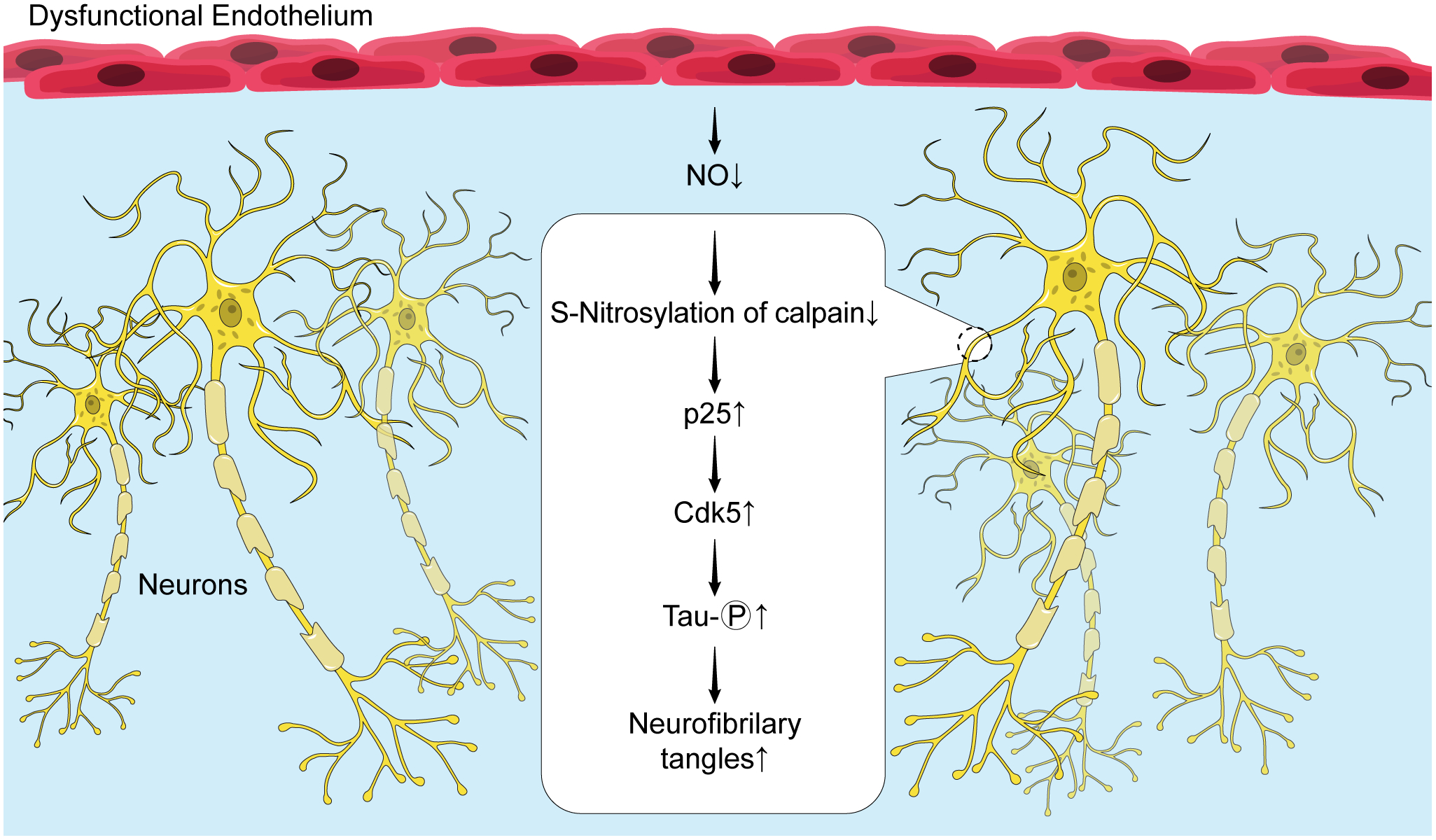
Schematic representation of the role of endothelial NO deficiency in stimulation of tau protein phosphorylation and formation of neurofibrillary tangles in neuronal cells.
Loss of endothelial NO, chronic mild hypoperfusion, and white matter damage
Recent study by Dr Liao’s group confirmed that eNOS deficiency does not reduce cerebral blood flow26, however, genetic inactivation of eNOS causes microvascular occlusions apparently leading to localized mild brain hypoperfusion.26,53 Moreover, the authors also provided evidence that loss of endothelial NO impairs mitochondrial function, increases permeability of blood-brain-barrier (BBB) and causes damage of the brain white matter.26,53 These observations significantly expanded understanding of cerebrovascular mechanisms contributing to pathogenesis of vascular cognitive impairment and dementia (VCID).54 Most notably, treatment of eNOS-deficient mice with sodium nitrate (supplementation with exogenous NO) completely prevented microvascular occlusions, leakage of BBB, damage of white matter, and normalized gait performance indicating that loss of endothelial NO is major perpetrator of vascular and central nervous system pathology.26 Impairment of BBB is entirely in agreement with reported increased expression of BACE1 in eNOS-deficient human and murine cerebrovascular endothelium.12,55 Indeed, most recent findings demonstrate that BACE1 cleaves occludin, a critically important protein responsible for preservation of intact BBB56. This observation establishes link between eNOS-deficiency and loss of BBB integrity (another major mechanism of endothelial dysfunction). It is also important to note that elevation of BACE1 exerts detrimental effect on expression and function of eNOS and therefore may further exacerbate loss of endothelial NO, leakage of BBB, and attendant cerebrovascular and neuronal pathology.56,57
Endothelial NO and Aβ cytotoxicity
Formulation of amyloid hypothesis,58,59 provided foundation for the studies designed to define exact role of expression and processing of amyloid precursor protein (APP) in pathogenesis of AD. The recognition of cytotoxic characteristics of amyloid-β (Aβ) peptides (Aβ1–40 and Aβ1–42) was followed by the studies demonstrating detrimental effects of Aβ peptides on cerebrovascular function.60–64 The mechanisms underlying these detrimental effects were recently reviewed by Cortes-Conteli and Iadecola, 2020.65 Briefly, Aβ peptides increase expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 (NOX-2) in perivascular and meningeal macrophages66. Enzyme activity of NOX-2 is major source of superoxide anion. Chemical reaction between superoxide anion and NO generates powerful oxidant, peroxynitrite,67 thereby significantly reducing local concentration of NO thus leading to impairment of endothelium-dependent vasodilatation.66 Most importantly, loss of endothelial NO increases sensitivity of endothelial cells to apoptosis.68,69 Indeed, more recent studies suggest that cerebrovascular tree is the primary target of pro-apoptotic effects of Aβ peptides in patients with AD.40,70 Moreover, and in contrast to parenchymal cells71,72, extensive loss of vascular cells including endothelium was detected in brains of patients with AD.73 Only about half as many endothelial cells were isolated from AD brain as compared to number of cells isolated from normal brain.73 Disappearance of endothelial cells in AD brain is consistent with vascular regression reported by Religa and colleagues (2013).47 We would like to emphasize that endothelial NO is a major anti-apoptotic molecule and endothelial cell survival factor.68 This vascular protective effect of endothelial NO is impaired by aging.69 Aging also impairs stimulatory effect of shear stress on expression of eNOS thereby increasing sensitivity of endothelium to apoptosis.69 The anti-apoptotic effect of shear stress is restored by overexpression of eNOS in senescent endothelial cells.40,69 Consistent with loss of eNOS signaling, patients with AD have lower levels of cyclic GMP in their cerebrospinal fluid and higher expression of phosphodiesterase 5 (PDE5) in the temporal cortex than healthy, age-matched controls.74,75 As mentioned earlier, eNOS/sGC/cGMP signaling is therapeutic target for central nervous system (CNS)-penetrant sGC stimulator, CY6463. This compound is currently being evaluated in clinical trial designed to improve cognitive function in patients with AD.
The role of endothelial NO in control of glycolysis in astrocytes
Prior studies established that NO enhances glycolysis in astrocytes but not in neurons, however, exact cellular source of NO was not identified.76,77 Relevant to this review, Brix and colleagues (2012)13 demonstrated that NO produced in endothelium enhances aerobic glycolysis in astrocytes. This observation was confirmed and extended to show that nanomolar concentration of NO is capable of modulating energy metabolism in astrocytes with the speed, reversibility, and potency of physiological signal.78 Notably, the stimulatory effect of NO on aerobic glycolysis is not mediated by activation of soluble guanylate cyclase and production of cyclic-GMP.13,76 Further analysis of signal transduction pathways demonstrated that co-culturing of astrocytes with primary brain vascular endothelial cells caused stabilization of hypoxia-inducible factor-1α (HIF-1α) and enhancement of glucose transporter-1, hexokinase-1 and monocarboxylate transporter-4 expression (MCT4).13 Most importantly, endothelium derived NO stimulated glycolysis and production of lactate in astrocytes.13 Lactate is then exported from astrocytes to neurons via MCT4 and it serves as an important substrate for production of pyruvate and adenosine triphosphate (ATP) in neurons. Moreover, lactate is signaling molecule in the brain involved in regulation of wide range of cellular and physiological homeostatic mechanisms.79 Whether impairment of endothelial NO production may reduce glycolysis in astrocytes is unknown. Of note, expression of glial fibrillary acidic protein (GFAP), marker for astrocytes, is not altered in aged eNOS−/− mice.28 The effects of eNOS deficiency on expression of glycolytic enzymes and production of lactate in aged eNOS−/− mice remain to be determined.
The role of endothelial NO in mitochondrial biogenesis
Dysfunctional mitochondria are believed to play a major role in pathogenesis of AD.80–82 Existing evidence indicate that endothelial NO activates the transcriptional mechanisms driving mitochondrial biogenesis (the process designed to increase cellular mitochondrial mass).83 Notably, the brain, kidney, liver, heart, and gastrocnemius muscle isolated from eNOS−/− mice display significantly reduced mitochondrial content associated with lower oxygen consumption and reduced levels of adenosine triphosphate (ATP).84 This effect is mediated by impairment of NO/sGC/cyclic GMP signaling pathway leading to downregulation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α).84 Importantly, the stimulatory effect of calorie restriction on mitochondrial biogenesis is abolished in eNOS−/− mice thereby demonstrating that intact eNOS function is critically important for mitochondrial adaptation to low calorie intake.85 The NO-dependent signaling responsible for mitochondrial biogenesis are not very well defined, however, stimulatory effects of NO on PGC-1α, cyclic AMP-responsive element-binding protein-1 (CREB), and nuclear respiratory factor-1 (NRF-1), and are believed to be of major importance.86 Moreover, mitochondria from hepatocytes of eNOS−/− mice have decreased markers of mitochondrial biogenesis PGC-1α, mitochondrial transcription factor A, and autophagy/mitophagy [BCL-2-interacting protein-3 (BNIP3), 1A/1Blight chain 3B (LC3)], suggesting decreased mitochondrial turnover rate.87
The stimulatory effect of endothelial NO on mitochondrial biogenesis has also been described in human cells.88 More precisely, genetic inactivation of eNOS blocks mitochondrial biogenesis and adipogenesis in human mesenchymal stem cells (hMSCs). Remarkably, the transfer of mitochondria from normal hMSCs to eNOS-deficient hMSCs restored adipogenesis thus showing that mitochondrial remodeling can be employed in treating abnormalities in energy metabolism caused by dysfunctional eNOS.88
Detrimental effects of aging and apolipoprotein 4 (ApoE4) on eNOS function
Aging –
In cerebral blood vessels, aging impairs endothelium-dependent relaxations mediated by activation of eNOS (recently reviewed by De Silva and Faraci).40 Several mechanisms have been implicated in aging-induced dysregulation of eNOS/NO signaling. The renin-angiotensin-system is responsible for production of angiotensin II, and activation of NOX-2-containing NADPH oxidase leading to increased formation of superoxide anion.89 Chemical reaction between superoxide anion NO decreases local concentration of NO and results in formation of powerful oxidant, peroxynitrite. In addition, the adaptor protein, p66Sch and activated Rho kinase exert inhibitory effects on eNOS in aged blood vessels.90,91 Interestingly, aging causes significant increase in vascular expression of APP protein.92 Increased expression of APP in aged blood vessels is associated with elevation of circulating levels of sAPPα.92 In contrast, β-processing of APP is not affected.92 Since APP and sAPPα are considered vascular protective molecules,92,93 upregulation of APP is most likely adaptive response to aging, designed to protect and preserve normal vascular function. In contrast, in cerebral microvessels derived from aged heterozygous eNOS +/− mice, impaired production of sAPPα has been detected in cerebral microvessels. Most importantly, proteolytic cleavage of APP is shifted towards β-processing and increased production of Aβ1–40.94 Thus, it appears that in mice, healthy aging of blood vessel wall is associated with increased vascular protective function of sAPPα. However, partial loss of eNOS impairs production of sAPPα and increases vascular production of cytotoxic Aβ peptides in aged arteries.94 We therefore speculate that long-lasting impairment of eNOS function associated with chronic increase in amyloidogenic processing of APP, may help explain epidemiological findings demonstrating that mid-life exposure to cardiovascular risk factors increases vulnerability to AD.95
Aging-induced impairment of eNOS signaling contributes to increased arterial stiffening,96–100 increase in pulse pressure, and mechanical stress on capillaries thereby damaging the capillary wall of strongly perfused organs such as brain, heart, and kidneys.101 Moreover, increased stiffness itself suppresses eNOS signaling thus further exacerbating endothelial dysfunction.101,102 Importantly, existing literature supports relevance of aging-induced increase in arterial stiffness to development of cognitive decline and AD.101,103–105
APOE4 –
Three different ApoE isoforms (ApoE2, ApoE3, and ApoE4) confer different risk for AD.106,107 Prior studies established that carriers of ApoE4 alleles have increased risk for AD as compared to the carriers of more common ApoE3 variant, whereas ApoE2 is protective against AD.107 Importantly, ApoE4 is considered the strongest genetic risk for late-onset AD.107 Despite intensive investigation, the exact mechanisms underlying contribution of ApoE4 to pathogenesis of AD remain incompletely defined.
In the context of this review, we wish to draw attention to the study demonstrating that circulating ApoE4 inhibits enzyme activity of eNOS.108 In addition to direct inhibitory effect on eNOS activity, ApoE4 exerts dominant-negative effect against ApoE3 thus preventing ApoE3-induced stimulation of eNOS activity in endothelium. These detrimental vascular effects of ApoE4 are mediated by binding of ApoE4 to endothelial apolipoprotein receptor 2 (ApoER2 encoded by LRP8 gene).108 Moreover, ApoE4 and genetic variant of ApoER2-R952Q attenuate eNOS activity thereby leading to impairment of reparative and anti-inflammatory capacity of endothelium.108 Since human brain endothelial cells express ApoER2,73 it is likely that circulating ApoE4 may bind to endothelial ApoER2 thereby inhibiting activity of cerebrovascular eNOS.
Prior studies in systemic arteries established that cyclophilin A (CypA), a protein secreted in response to inflammatory stimuli, promotes atherosclerosis in aorta of ApoE-deficient mice.109 This effect is mediated by CypA-induced impairment of eNOS protein expression.109 Existing evidence support the concept that ApoE4-induces activation of CypA-matrix metalloproteinase-9 signaling in cerebrovascular pericytes, thus leading to blood brain barrier (BBB) breakdown, impairment of cerebral blood flow, neurodegeneration, and cognitive dysfunction.110,111 Notably, these detrimental effects are independent of Aβ production. Given the results of prior studies demonstrating that CypA mRNA and protein are expressed in endothelium of cerebral and systemic blood vessels,73,109,112 it appears likely that circulating ApoE4 might contribute to impairment of eNOS function by increasing cerebrovascular endothelial production of CypA. Consistent with this hypothesis are the results of previous study demonstrating elevated levels of CypA in endothelial cells of ApoE4 carriers with AD.113
Endothelial NO and cerebral amyloid angiopathy (CAA)
Sporadic CAA is small vessel brain disease caused by cerebrovascular deposition of Aβ.114 CAA is common in elderly, and it is an important contributor to age-related cognitive decline.114 Importantly, CAA is detected in 90% of patients with AD. The brain injury caused by CAA is the result of cerebrovascular dysfunction leading to reduced brain blood supply and ischemia. Moreover, CAA may cause large symptomatic intracerebral hemorrhages and small (mostly asymptomatic) cerebral microbleeds.115 It is important to note that CAA makes an independent contribution to AD dementia. Indeed, AD patients suffer more severe cognitive impairment in the presence of CAA.115 Currently, there is no disease-specific treatment available to patients with CAA.
Studies on aged eNOS+/− mice provided new insights into contributions of dysfunctional eNOS to pathogenesis of cerebral small vessel disease.27,53 Traditional understanding of CAA pathogenesis is based on evidence demonstrating that Aβ deposition and accumulation in the blood vessel wall is largely driven by impaired clearance of Aβ from the brain interstitial fluid.24,116,117 More recent discoveries suggest that partial loss of endothelial NO production in aged (18-month-old) heterozygous eNOS+/− mice significantly contributes to development of CAA pathology.27,53 Indeed, aged heterozygous eNOS+/− mice exhibit CAA pathology including intracerebral hemorrhage and siderosis.27,53 Aged eNOS+/− also suffer from microinfarcts, increased permeability of blood-brain-barrier, and white matter pathology.53 In fact, aged eNOS+/− mice are currently considered a model of age-dependent, spontaneous cerebral small vessel disease including CAA.53
Increased production of Aβ in cerebral microvessels of aged eNOS+/− mice appears to be very early event in pathogenesis of CAA.94 Aβ1–40 levels are increased in microvascular tissue of aged eNOS+/− mice, and production and secretion of Aβ1–40 from cerebral microvessels is elevated before any changes in Aβ concentration could be detected in neuronal tissue.94 This suggests that cerebrovascular alterations of Aβ may occur independently of Aβ changes within the brain, and that cerebral blood vessels are more vulnerable than neurons to injury imposed by partial deficiency of eNOS. We also wish to point out that partial loss of endothelial NO is a clinically relevant as complete loss of endothelial NO does not occur in human disease.94 It is also important to emphasize that metabolic parameters body weight, glucose, total and HDL cholesterol of 18-month-old eNOS+/− mice are comparable to their littermate wild type control mice.94 In addition, arterial blood pressure is not elevated in aged eNOS+/− mice.94 These findings rule out hypertension and alterations in glucose and lipid metabolism as mechanisms responsible for increased amyloidogenic processing of APP observed in cerebral microvessels of 18-month-old eNOS+/− mice. Indeed, these findings confirm the importance of reduced local concentration of NO in blood vessel wall (rather than systemic metabolic or hemodynamic dysfunction) as an important early event in development of CAA.
Notably, adaptive responses were identified in cerebral microvessels of 18-month-old eNOS+/− mice. Expression of insulin degrading enzyme (IDE) and low-density lipoprotein receptor-related protein 1 (LRP1) are significantly upregulated in cerebral microvessels of aged eNOS−/− mice.94 This is most likely, adaptive response designed to lower elevated local concentration of Aβ in the brain blood vessel wall. In addition, significant upregulation of copper-zinc superoxide dismutase (CuZnSOD) and catalase was also present in cerebral microvessels of aged eNOS+/− mice. This upregulation of ant-oxidant enzymes system also appears to be adaptive response to oxidative stress imposed by increased Aβ.94 Of note, increased expression of IDE was detected in brain tissue in the absence of increased brain Aβ levels. These findings reinforce the concept that partial eNOS-deficiency first causes elevation of Aβ within vasculature and not in brain parenchyma.94
The importance of endothelial expression and processing of APP in pathogenesis of CAA is further supported by recent findings demonstrating that endothelium-specific expression of human APP [with the Swedish (KM670/671NL) mutation] in mice resulted in increased production of Aβ.118 Interestingly, this increased production of Aβ in endothelium did not result in Aβ deposition in the cortical blood vessels. However, crossing of these animals with AD model mice (APPNL-F/NF-F mice overexpressing APP in neuronal tissue) markedly exacerbated CAA pathology in APPNL-F/NF-F mice.118 This observation is particularly noteworthy finding because it suggests that production of Aβ in endothelium may significantly exacerbate cerebrovascular deposition of Aβ in patients with AD. Thus, inhibition of endothelial production of Aβ may offer a viable therapeutic strategy designed to reduce Aβ accumulation in the cerebral blood vessel wall.
Conclusions
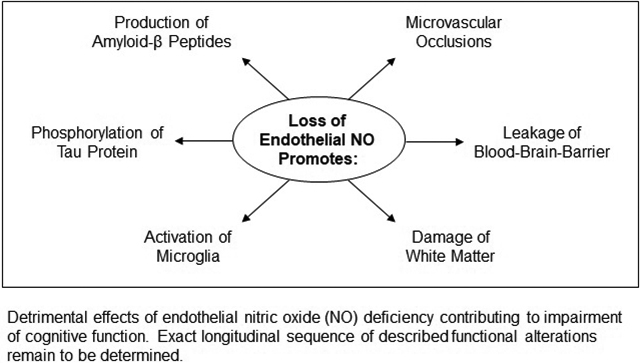
Loss of endothelial NO signaling promotes amyloidogenic processing of APP in the brain endothelial and neuronal cells. Moreover, inactivation of eNOS stimulates neuronal tau phosphorylation thereby suggesting that dysfunctional eNOS may promote formation of neurofibrillary tangles. Existing evidence also suggests that loss of endothelial NO function activates microglia and creates pro-inflammatory environment in the brain. Furthermore, loss of endothelial NO causes microvascular occlusions, leading to localized mild hypoperfusion, aberrant mitochondrial function, increased permeability of BBB, and white matter damage. In addition, it appears that endothelial NO significantly contributes to control of glycolysis in astrocytes and neuronal biogenesis of mitochondria. More recent findings support the hypothesis that partial loss of cerebrovascular NO signaling may contribute to initiation and progression of CAA. Growing recognition of dysfunctional endothelial NO signaling contribution to development of cognitive impairment provides basis for further efforts to develop, optimize, and refine therapies designed to preserve healthy cognition. Activation of NO/cGMP pathway may offer cerebrovascular protective therapy that will minimize vascular contribution to cognitive impairment. For instance, if this approach may prevent microvascular occlusions, reduce amyloid deposition in cerebral blood vessels, and protect BBB, improved vascular function may translate into significantly less severe symptoms of CAA and cognitive impairment.
Sources of Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes on Aging AG071190 and by the Mayo Foundation.
Nonstandard Abbreviations and Acronyms
- Aβ
amyloid-β
- AD
Alzheimer’s disease
- ApoE
apolipoprotein
- AAA
amyloid precursor protein
- BACE1
β-site APP-cleaving enzyme 1
- BBB
blood-brain-barrier
- CAA
cerebral amyloid angiopathy
- CD68
cluster of differentiation 68
- Cdk5
cyclin-dependent kinase 5
- CNS
central nervous system
- ELISA
enzyme-linked immunosorbent assay
- eNOS
endothelial nitric oxide synthase
- GMCSF
granulocyte-macrophage colony stimulating factor
- cGMP
cyclic guanosine monophosphate
- HDL
high density lipoprotein
- hMSCs
human mesenchymal stem cells
- Iba1
ionized calcium-binding adaptor 1
- IDE
insulin degrading enzyme
- IL-1α
interleukin-1α
- LMA
late middle aged
- LTP
long-term potentiation
- MHC II
major histocompatibility complex II
- MIP-1β
macrophage inflammatory protein-1β
- NO
nitric oxide
- NOX2
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2
- sAPPα
soluble amyloid precursor protein-α
- sGC
soluble guanylate cyclase
- tau
tubulin associated unit
Footnotes
Disclosures
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Katusic served as a consultant to Ironwood Pharmaceuticals (Massachusetts, USA).
REFERENCES:
- 1.Organization WH. Dementia https://www.who.int/news-room/fact-sheets/detail/dementia. 2021.
- 2.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35:888–894. doi: 10.1093/eurheartj/eht544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katusic ZS, Austin SA. Neurovascular Protective Function of Endothelial Nitric Oxide- Recent Advances. Circ J. 2016;80:1499–1503. doi: 10.1253/circj.CJ-16-0423 [DOI] [PubMed] [Google Scholar]
- 5.Atochin DN, Huang PL. Role of endothelial nitric oxide in cerebrovascular regulation. Curr Pharm Biotechnol. 2011;12:1334–1342. doi: 10.2174/138920111798280974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res. 2016;119:375–396. doi: 10.1161/CIRCRESAHA.116.306531 [DOI] [PubMed] [Google Scholar]
- 7.Emdin CA, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A, Haas M, Aragam K, Ardissino D, Wilson JG, et al. Phenotypic Consequences of a Genetic Predisposition to Enhanced Nitric Oxide Signaling. Circulation. 2018;137:222–232. doi: 10.1161/CIRCULATIONAHA.117.028021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Amado M, Prensa L. Stereological analysis of neuron, glial and endothelial cell numbers in the human amygdaloid complex. PLoS One. 2012;7:e38692. doi: 10.1371/journal.pone.0038692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai PS, Kaufhold JP, Blinder P, Friedman B, Drew PJ, Karten HJ, Lyden PD, Kleinfeld D. Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. J Neurosci. 2009;29:14553–14570. doi: 10.1523/JNEUROSCI.3287-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garthwaite J From synaptically localized to volume transmission by nitric oxide. J Physiol. 2016;594:9–18. doi: 10.1113/JP270297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garthwaite J NO as a multimodal transmitter in the brain: discovery and current status. Br J Pharmacol. 2019;176:197–211. doi: 10.1111/bph.14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010;107:1498–1502. doi: 10.1161/CIRCRESAHA.110.233080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brix B, Mesters JR, Pellerin L, Johren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garthwaite G, Bartus K, Malcolm D, Goodwin D, Kollb-Sielecka M, Dooldeniya C, Garthwaite J. Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci. 2006;26:7730–7740. doi: 10.1523/JNEUROSCI.1528-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006;26:11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garthwaite J Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iadecola C The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017;96:17–42. doi: 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitazume S, Tachida Y, Kato M, Yamaguchi Y, Honda T, Hashimoto Y, Wada Y, Saito T, Iwata N, Saido T, et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J Biol Chem. 2010;285:40097–40103. doi: 10.1074/jbc.M110.144626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.d’Uscio LV, He T, Santhanam AV, Katusic ZS. Endothelium-specific amyloid precursor protein deficiency causes endothelial dysfunction in cerebral arteries. J Cereb Blood Flow Metab. 2018;38:1715–1726. doi: 10.1177/0271678X17735418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583 [DOI] [PubMed] [Google Scholar]
- 21.d’Uscio LV, He T, Katusic ZS. Expression and Processing of Amyloid Precursor Protein in Vascular Endothelium. Physiology (Bethesda). 2017;32:20–32. doi: 10.1152/physiol.00021.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6:17. doi: 10.1186/1750-1326-6-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0 [DOI] [PubMed] [Google Scholar]
- 24.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab. 2003;23:1219–1226. doi: 10.1097/01.WCB.0000089601.87125.E4 [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Chen L, Lin G, Wang Z, Kodali MC, Li M, Chen H, Lebovitz SG, Ortyl TC, Li L, et al. White matter damage as a consequence of vascular dysfunction in a spontaneous mouse model of chronic mild chronic hypoperfusion with eNOS deficiency. Mol Psychiatry. 2022;27:4754–4769. doi: 10.1038/s41380-022-01701-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan XL, Xue YQ, Ma T, Wang X, Li JJ, Lan L, Malik KU, McDonald MP, Dopico AM, Liao FF. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol Neurodegener. 2015;10:24. doi: 10.1186/s13024-015-0020-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS. Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem. 2013;127:691–700. doi: 10.1111/jnc.12334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3 [DOI] [PubMed] [Google Scholar]
- 30.de la Monte SM, Lu BX, Sohn YK, Etienne D, Kraft J, Ganju N, Wands JR. Aberrant expression of nitric oxide synthase III in Alzheimer’s disease: relevance to cerebral vasculopathy and neurodegeneration. Neurobiol Aging. 2000;21:309–319. [DOI] [PubMed] [Google Scholar]
- 31.de la Monte SM, Sohn YK, Etienne D, Kraft J, Wands JR. Role of aberrant nitric oxide synthase-3 expression in cerebrovascular degeneration and vascular-mediated injury in Alzheimer’s disease. Ann N Y Acad Sci. 2000;903:61–71. [DOI] [PubMed] [Google Scholar]
- 32.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun. 1994;205:659–665. doi: 10.1006/bbrc.1994.2716 [DOI] [PubMed] [Google Scholar]
- 33.Luth HJ, Holzer M, Gartner U, Staufenbiel M, Arendt T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: evidence for an induction by amyloid pathology. Brain Res. 2001;913:57–67. doi: S0006–8993(01)02758–5 [pii] [DOI] [PubMed] [Google Scholar]
- 34.Luth HJ, Munch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–143. [DOI] [PubMed] [Google Scholar]
- 35.Santhanam AVR, d’Uscio LV, He TR, Das P, Younkin SG, Katusic ZS. Uncoupling of endothelial nitric oxide synthase in cerebral vasculature of Tg2576 mice. J Neurochem. 2015;134:1129–1138. doi: 10.1111/jnc.13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen MB, Yang AC, Yousef H, Lee D, Chen W, Schaum N, Lehallier B, Quake SR, Wyss-Coray T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020;30:4418–4432 e4414. doi: 10.1016/j.celrep.2020.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubomirov LT, Papadopoulos S, Putz S, Welter J, Klockener T, Weckmuller K, Ardestani MA, Filipova D, Metzler D, Metzner H, et al. Aging-related alterations in eNOS and nNOS responsiveness and smooth muscle reactivity of murine basilar arteries are modulated by apocynin and phosphorylation of myosin phosphatase targeting subunit-1. J Cereb Blood Flow Metab. 2017;37:1014–1029. doi: 10.1177/0271678X16649402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith AR, Visioli F, Frei B, Hagen TM. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell. 2006;5:391–400. doi: 10.1111/j.1474-9726.2006.00232.x [DOI] [PubMed] [Google Scholar]
- 39.Strosznajder JB, Jesko H, Zambrzycka A, Eckert A, Chalimoniuk M. Age-related alteration of activity and gene expression of endothelial nitric oxide synthase in different parts of the brain in rats. Neurosci Lett. 2004;370:175–179. doi: 10.1016/j.neulet.2004.08.013 [DOI] [PubMed] [Google Scholar]
- 40.De Silva TM, Faraci FM. Contributions of Aging to Cerebral Small Vessel Disease. Annu Rev Physiol. 2020;82:275–295. doi: 10.1146/annurev-physiol-021119-034338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829–1836. doi: 10.1152/ajpheart.00230.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Wallerath T, Munzel T, Forstermann U. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide. 2002;7:149–164. doi: 10.1016/s1089-8603(02)00111-8 [DOI] [PubMed] [Google Scholar]
- 43.Purnell C, Gao S, Callahan CM, Hendrie HC. Cardiovascular risk factors and incident Alzheimer disease: a systematic review of the literature. Alzheimer Dis Assoc Disord. 2009;23:1–10. doi: 10.1097/WAD.0b013e318187541c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Correia SS, Liu G, Jacobson S, Bernier SG, Tobin JV, Schwartzkopf CD, Atwater E, Lonie E, Rivers S, Carvalho A, et al. The CNS-penetrant soluble guanylate cyclase stimulator CYR119 attenuates markers of inflammation in the central nervous system. J Neuroinflammation. 2021;18:213. doi: 10.1186/s12974-021-02275-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correia SS, Iyengar RR, Germano P, Tang K, Bernier SG, Schwartzkopf CD, Tobin J, Lee TW, Liu G, Jacobson S, et al. The CNS-Penetrant Soluble Guanylate Cyclase Stimulator CY6463 Reveals its Therapeutic Potential in Neurodegenerative Diseases. Front Pharmacol. 2021;12:656561. doi: 10.3389/fphar.2021.656561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tobin JV, Zimmer DP, Shea C, Germano P, Bernier SG, Liu G, Long K, Miyashiro J, Ranganath S, Jacobson S, et al. Pharmacological Characterization of IW-1973, a Novel Soluble Guanylate Cyclase Stimulator with Extensive Tissue Distribution, Antihypertensive, Anti-Inflammatory, and Antifibrotic Effects in Preclinical Models of Disease. J Pharmacol Exp Ther. 2018;365:664–675. doi: 10.1124/jpet.117.247429 [DOI] [PubMed] [Google Scholar]
- 47.Degrush E, Shazeeb MS, Drachman D, Vardar Z, Lindsay C, Gounis MJ, Henninger N. Cumulative effect of simvastatin, L-arginine, and tetrahydrobiopterin on cerebral blood flow and cognitive function in Alzheimer’s disease. Alzheimers Res Ther. 2022;14:134. doi: 10.1186/s13195-022-01076-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tropea MR, Gulisano W, Vacanti V, Arancio O, Puzzo D, Palmeri A. Nitric oxide/cGMP/CREB pathway and amyloid-beta crosstalk: From physiology to Alzheimer’s disease. Free Radic Biol Med. 2022;193:657–668. doi: 10.1016/j.freeradbiomed.2022.11.022 [DOI] [PubMed] [Google Scholar]
- 49.Karikari TK, Ashton NJ, Brinkmalm G, Brum WS, Benedet AL, Montoliu-Gaya L, Lantero-Rodriguez J, Pascoal TA, Suarez-Calvet M, Rosa-Neto P, et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat Rev Neurol. 2022;18:400–418. doi: 10.1038/s41582-022-00665-2 [DOI] [PubMed] [Google Scholar]
- 50.Austin SA, Katusic ZS. Loss of Endothelial Nitric Oxide Synthase Promotes p25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer’s Disease. Circ Res. 2016;119:1128–1134. doi: 10.1161/CIRCRESAHA.116.309686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, Buendia I, Santisteban MM, Segarra SG, Koizumi K, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018;21:240–249. doi: 10.1038/s41593-017-0059-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faraco G, Hochrainer K, Segarra SG, Schaeffer S, Santisteban MM, Menon A, Jiang H, Holtzman DM, Anrather J, Iadecola C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature. 2019;574:686–690. doi: 10.1038/s41586-019-1688-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao FF, Lin G, Chen X, Chen L, Zheng W, Raghow R, Zhou FM, Shih AY, Tan XL. Endothelial Nitric Oxide Synthase-Deficient Mice: A Model of Spontaneous Cerebral Small-Vessel Disease. Am J Pathol. 2021;191:1932–1945. doi: 10.1016/j.ajpath.2021.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zlokovic BV, Gottesman RF, Bernstein KE, Seshadri S, McKee A, Snyder H, Greenberg SM, Yaffe K, Schaffer CB, Yuan C, et al. Vascular contributions to cognitive impairment and dementia (VCID): A report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 2020;16:1714–1733. doi: 10.1002/alz.12157 [DOI] [PubMed] [Google Scholar]
- 55.Devraj K, Poznanovic S, Spahn C, Schwall G, Harter PN, Mittelbronn M, Antoniello K, Paganetti P, Muhs A, Heilemann M, et al. BACE-1 is expressed in the blood-brain barrier endothelium and is upregulated in a murine model of Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36:1281–1294. doi: 10.1177/0271678X15606463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou H, Gao F, Yang X, Lin T, Li Z, Wang Q, Yao Y, Li L, Ding X, Shi K, et al. Endothelial BACE1 Impairs Cerebral Small Vessels via Tight Junctions and eNOS. Circ Res. 2022;130:1321–1341. doi: 10.1161/CIRCRESAHA.121.320183 [DOI] [PubMed] [Google Scholar]
- 57.He T, d’Uscio LV, Sun R, Santhanam AVR, Katusic ZS. Inactivation of BACE1 increases expression of endothelial nitric oxide synthase in cerebrovascular endothelium. J Cereb Blood Flow Metab. 2022;42:1920–1932. doi: 10.1177/0271678X221105683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2 [DOI] [PubMed] [Google Scholar]
- 59.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- 60.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715 [DOI] [PubMed] [Google Scholar]
- 61.Iadecola C Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387 [DOI] [PubMed] [Google Scholar]
- 62.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC, Alzheimer’s Disease Neuroimaging I. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de la Torre JC. Are Major Dementias Triggered by Poor Blood Flow to the Brain? Theoretical Considerations. J Alzheimers Dis. 2017;57:353–371. doi: 10.3233/JAD-161266 [DOI] [PubMed] [Google Scholar]
- 64.Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cortes-Canteli M, Iadecola C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J Am Coll Cardiol. 2020;75:942–951. doi: 10.1016/j.jacc.2019.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schaeffer S, Iadecola C. Revisiting the neurovascular unit. Nat Neurosci. 2021;24:1198–1209. doi: 10.1038/s41593-021-00904-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dimmeler S, Zeiher AM. Nitric oxide-an endothelial cell survival factor. Cell Death Differ. 1999;6:964–968. doi: 10.1038/sj.cdd.4400581 [DOI] [PubMed] [Google Scholar]
- 69.Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, Dimmeler S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89:709–715. doi: 10.1161/hh2001.097796 [DOI] [PubMed] [Google Scholar]
- 70.Religa P, Cao R, Religa D, Xue Y, Bogdanovic N, Westaway D, Marti HH, Winblad B, Cao Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci Rep. 2013;3:2053. doi: 10.1038/srep02053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4 [DOI] [PubMed] [Google Scholar]
- 73.Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603:885–892. doi: 10.1038/s41586-021-04369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hesse R, Lausser L, Gummert P, Schmid F, Wahler A, Schnack C, Kroker KS, Otto M, Tumani H, Kestler HA, et al. Reduced cGMP levels in CSF of AD patients correlate with severity of dementia and current depression. Alzheimers Res Ther. 2017;9:17. doi: 10.1186/s13195-017-0245-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ugarte A, Gil-Bea F, Garcia-Barroso C, Cedazo-Minguez A, Ramirez MJ, Franco R, Garcia-Osta A, Oyarzabal J, Cuadrado-Tejedor M. Decreased levels of guanosine 3’, 5’-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2015;41:471–482. doi: 10.1111/nan.12203 [DOI] [PubMed] [Google Scholar]
- 76.Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- 78.San Martin A, Arce-Molina R, Galaz A, Perez-Guerra G, Barros LF. Nanomolar nitric oxide concentrations quickly and reversibly modulate astrocytic energy metabolism. J Biol Chem. 2017;292:9432–9438. doi: 10.1074/jbc.M117.777243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19:235–249. doi: 10.1038/nrn.2018.19 [DOI] [PubMed] [Google Scholar]
- 80.Parodi-Rullan R, Sone JY, Fossati S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J Alzheimers Dis. 2019;72:1019–1039. doi: 10.3233/JAD-190357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mary A, Eysert F, Checler F, Chami M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol Psychiatry. 2022. doi: 10.1038/s41380-022-01631-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–633. doi: 10.1038/s41573-020-0072-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368 [DOI] [PubMed] [Google Scholar]
- 84.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728 [DOI] [PubMed] [Google Scholar]
- 86.Suliman HB, Piantadosi CA. Mitochondrial biogenesis: regulation by endogenous gases during inflammation and organ stress. Curr Pharm Des. 2014;20:5653–5662. doi: 10.2174/1381612820666140306095717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sheldon RD, Meers GM, Morris EM, Linden MA, Cunningham RP, Ibdah JA, Thyfault JP, Laughlin MH, Rector RS. eNOS deletion impairs mitochondrial quality control and exacerbates Western diet-induced NASH. Am J Physiol Endocrinol Metab. 2019;317:E605–E616. doi: 10.1152/ajpendo.00096.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee-Huang S, Huang PL, Huang PL. Endothelial Nitric Oxide Synthase Knockdown in Human Stem Cells Impacts Mitochondrial Biogenesis and Adipogenesis: Live-Cell Real-Time Fluorescence Imaging. J Clin Med. 2021;10. doi: 10.3390/jcm10040631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Modrick ML, Didion SP, Sigmund CD, Faraci FM. Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol. 2009;296:H1914–1919. doi: 10.1152/ajpheart.00300.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi Y, Savarese G, Perrone-Filardi P, Luscher TF, Camici GG. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int J Cardiol. 2014;175:446–450. doi: 10.1016/j.ijcard.2014.06.025 [DOI] [PubMed] [Google Scholar]
- 91.De Silva TM, Modrick ML, Dabertrand F, Faraci FM. Changes in Cerebral Arteries and Parenchymal Arterioles With Aging: Role of Rho Kinase 2 and Impact of Genetic Background. Hypertension. 2018;71:921–927. doi: 10.1161/HYPERTENSIONAHA.118.10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.d’Uscio LV, Katusic ZS. Endothelium-specific deletion of amyloid-beta precursor protein exacerbates endothelial dysfunction induced by aging. Aging (Albany NY). 2021;13:19165–19185. doi: 10.18632/aging.203401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.He T, Santhanam AV, Lu T, d’Uscio LV, Katusic ZS. Role of prostacyclin signaling in endothelial production of soluble amyloid precursor protein-alpha in cerebral microvessels. J Cereb Blood Flow Metab. 2017;37:106–122. doi: 10.1177/0271678X15618977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Austin SA, Katusic ZS. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J Cereb Blood Flow Metab. 2020;40:392–403. doi: 10.1177/0271678X18822474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Litke R, Garcharna LC, Jiwani S, Neugroschl J. Modifiable Risk Factors in Alzheimer Disease and Related Dementias: A Review. Clin Ther. 2021;43:953–965. doi: 10.1016/j.clinthera.2021.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kinlay S, Creager MA, Fukumoto M, Hikita H, Fang JC, Selwyn AP, Ganz P. Endothelium-derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053. doi: 10.1161/hy1101.095329 [DOI] [PubMed] [Google Scholar]
- 97.Soucy KG, Ryoo S, Benjo A, Lim HK, Gupta G, Sohi JS, Elser J, Aon MA, Nyhan D, Shoukas AA, et al. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol (1985). 2006;101:1751–1759. doi: 10.1152/japplphysiol.00138.2006 [DOI] [PubMed] [Google Scholar]
- 98.Donato AJ, Machin DR, Lesniewski LA. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res. 2018;123:825–848. doi: 10.1161/CIRCRESAHA.118.312563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ Res. 2018;123:849–867. doi: 10.1161/CIRCRESAHA.118.311378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leloup AJA, Van Hove CE, De Moudt S, De Keulenaer GW, Fransen P. Ex vivo aortic stiffness in mice with different eNOS activity. Am J Physiol Heart Circ Physiol. 2020;318:H1233–H1244. doi: 10.1152/ajpheart.00737.2019 [DOI] [PubMed] [Google Scholar]
- 101.Winder NR, Reeve EH, Walker AE. Large artery stiffness and brain health: insights from animal models. Am J Physiol Heart Circ Physiol. 2021;320:H424–H431. doi: 10.1152/ajpheart.00696.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peng X, Haldar S, Deshpande S, Irani K, Kass DA. Wall stiffness suppresses Akt/eNOS and cytoprotection in pulse-perfused endothelium. Hypertension. 2003;41:378–381. doi: 10.1161/01.hyp.0000049624.99844.3d [DOI] [PubMed] [Google Scholar]
- 103.Lim JS, Lee JY, Kwon HM, Lee YS. The correlation between cerebral arterial pulsatility and cognitive dysfunction in Alzheimer’s disease patients. J Neurol Sci. 2017;373:285–288. doi: 10.1016/j.jns.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 104.Pase MP, Beiser A, Himali JJ, Tsao C, Satizabal CL, Vasan RS, Seshadri S, Mitchell GF. Aortic Stiffness and the Risk of Incident Mild Cognitive Impairment and Dementia. Stroke. 2016;47:2256–2261. doi: 10.1161/STROKEAHA.116.013508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hanon O, Haulon S, Lenoir H, Seux ML, Rigaud AS, Safar M, Girerd X, Forette F. Relationship between arterial stiffness and cognitive function in elderly subjects with complaints of memory loss. Stroke. 2005;36:2193–2197. doi: 10.1161/01.STR.0000181771.82518.1c [DOI] [PubMed] [Google Scholar]
- 106.Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, Morris AW, Bu G. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016;131:709–723. doi: 10.1007/s00401-016-1547-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ulrich V, Konaniah ES, Herz J, Gerard RD, Jung E, Yuhanna IS, Ahmed M, Hui DY, Mineo C, Shaul PW. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function. Proc Natl Acad Sci U S A. 2014;111:13493–13498. doi: 10.1073/pnas.1402106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nigro P, Satoh K, O’Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2011;208:53–66. doi: 10.1084/jem.20101174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D, Rege SV, Grond A, Zuniga E, Barnes SR, et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-beta. Nat Aging. 2021;1:506–520. doi: 10.1038/s43587-021-00073-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. Author Correction: A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;560:E3. doi: 10.1038/s41586-018-0232-x [DOI] [PubMed] [Google Scholar]
- 113.Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36:216–227. doi: 10.1038/jcbfm.2015.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–1850. doi: 10.1093/brain/awx047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16:30–42. doi: 10.1038/s41582-019-0281-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tachida Y, Miura S, Muto Y, Takuwa H, Sahara N, Shindo A, Matsuba Y, Saito T, Taniguchi N, Kawaguchi Y, et al. Endothelial expression of human amyloid precursor protein leads to amyloid beta in the blood and induces cerebral amyloid angiopathy in knock-in mice. J Biol Chem. 2022;298:101880. doi: 10.1016/j.jbc.2022.101880 [DOI] [PMC free article] [PubMed] [Google Scholar]