

Abstract
Cellular differentiation entails the coordination of cell cycle arrest and tissue-specific gene expression. We investigated the involvement of basic helix-loop-helix (bHLH) factors in differentiation of osteoblasts using the human osteoblastic cell line MG63. Serum starvation induced growth arrest at G1 phase, accompanied by expression of cyclin-dependent kinase inhibitor p21WAF1/Cip1. Reporter assays with the p21 gene promoter demonstrated that the combination of E2A (E12 or E47) and coactivator CBP was responsible for p21 induction independent of p53. Twist inhibited E2A-CBP-dependent activation of the exogenous and endogenous p21 promoters. Ids similarly inhibited the exogenously transfected p21 promoter; however less antagonistic effect on the endogenous p21 promoter was observed. Twist was predominantly present in nuclei in MG63 cells growing in complete medium, while it localized mainly in the cytoplasm after serum starvation. The fibroblast growth factor receptor 3 gene (FGFR3), which generates signals leading to differentiation of osteoblasts, was found to be controlled by the same transcriptional regulation as the p21 gene. E2A and Twist influenced alkaline phosphatase expression, a consensus marker of osteoblast differentiation. Expression of E2A and FGFR3 was seen at the location of osteoblast differentiation in the calvaria of mouse embryos, implicating bHLH molecules in physiological osteoblast differentiation. These results demonstrate that a common regulatory system is involved in at least two distinct steps in osteoblastic differentiation. Our results also provide the molecular basis of Saethre-Chotzen syndrome, caused by mutations of the TWIST and FGFR3 genes.
The development and remodeling of bone require the differentiation of osteoblasts from undifferentiated proliferating mesenchymal osteoprogenitor cells. Unregulated differentiation of osteoblasts causes craniofacial and limb anomalies, such as Saethre-Chotzen syndrome (acrocephalosyndactyly type III; OMIM 101400), one of the most common genetic conditions with craniosynostosis. The molecular basis of mechanisms that induce the differentiated osteoblastic phenotype is poorly understood.
Cell differentiation is a consequence of a coordinated sequence of biochemical events associated with morphological changes, including arrest in G1 phase followed by irreversible exit from the cell cycle and a timely ordered expression of tissue-specific genes. Cell proliferation and terminal differentiation are usually mutually exclusive. Among multiple mechanisms involved in the control of terminal differentiation, cell cycle arrest through inactivation of cyclin-dependent kinases (CDKs) is likely to be a central feature (46). p21WAF1/Cip1 is an inhibitor of CDKs (26) and is transcriptionally regulated in p53-dependent (17) and -independent manners (46).
The E2A proteins belong to the basic helix-loop-helix (bHLH) family of transcriptional regulatory proteins, functioning as dimers via a helix-loop-helix (HLH) domain (40). The E2A gene encodes two alternatively spliced products, E12 and E47, which differ in their bHLH domains and hence their DNA-binding properties (40, 64). E47 has been demonstrated to activate p21 gene expression in HeLa cells (52). In addition, E2A proteins have been shown to be involved in regulation of growth arrest (50) and to recruit coactivator p300/CBP (cyclic AMP-responsive element binding factor CREB-binding protein) (11, 16). The dominant-negative-type HLH Id proteins, which contain functional HLH dimerization motifs but which lack the DNA-binding basic region, have been shown to interact with other bHLH proteins and block their DNA-binding activity (4).
The bHLH protein Twist is expressed in mesodermal and cranial neural crest cells during embryogenesis in both invertebrate and vertebrate development (3, 19, 65, 69). Expression of Twist has been implicated in the inhibition of differentiation of multiple cell lineages including muscle (27, 62), cartilage (27), and bone cells (35, 39, 55). Twist directly interacts with E12 and inhibits the expression of the muscle creatine kinase gene (MCK) (24, 62), suggesting its involvement in myogenesis.
The fibroblast growth factor receptor 3 (FGFR3) signaling pathway results in upregulation of genes that are related to osteoblast differentiation, including the genes for osteopontin, osteonectin, and osteocalcin (10). FGFR3 is detected in sutural osteogenic fronts, and Twist expression and FGFR3 expression are mutually exclusive (55). After birth cranial sutures are the primary site of osteoblast differentiation and bone formation in the calvaria. Saethre-Chotzen syndrome is characteristic of skull deformity due to craniosynostosis, the premature fusion of the cranial sutures. This syndrome is caused by mutations in the gene encoding Twist or FGFR3 (18, 28, 56). The question, then, is whether Twist and FGFR3 are in the same or parallel developmental pathways in calvarial bone development.
To this end, we examined how these bHLH proteins are involved in transcriptional regulation of cell cycle arrest (p21) and differentiation (FGFR3) in a human osteoblastic cell line MG63. Our results clearly show that E12 and E47 induce transcription of the p21 gene, resulting in cell cycle arrest in a p53-independent manner. This induction is inhibited by Twist and Ids. The same is true for the regulation of FGFR3 expression, demonstrating that common transcriptional regulation controls cell cycle arrest and differentiation of osteoblasts. Our results also provide a molecular basis for the pathogenesis of Saethre-Chotzen syndrome.
MATERIALS AND METHODS
Cell culture.
The human osteosarcoma osteoblast-like cell line MG63, which is negative for p53 (9), was cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C under a humidified atmosphere of 5% CO2. For induction of cell cycle arrest and differentiation, cells were cultured in DMEM containing 0.1 or 2% FBS with or without 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3; 10−8 M] (Biomol Research Laboratories).
Cell cycle analysis.
MG63 cells (105 cells/dish 60 mm in diameter) were cultured in DMEM containing either 0.1, 2, or 10% FBS and treated with or without 1,25-(OH)2D3 (10−8 M). Cells were harvested by trypsinization, fixed with a fluorescence-activated cell sorter (FACS) lysis solution (Becton Dickinson), and permeabilized with a FACS permeabilization solution (Becton Dickinson). After being washed with phosphate-buffered saline (PBS), the cells were resuspended in staining solution containing propidium iodide (50 μg/ml) and RNase A (50 μg/ml) and analyzed for their DNA content with a FACScan (Becton Dickinson).
Plasmid construction.
An expression plasmid for Twist was constructed by cloning the XhoI-XbaI fragment of human Twist cDNA into pcDNA3 (Invitrogen). A point mutation (Y103X) was introduced into the Twist coding region by site-directed mutagenesis, generating pCMV-Twist (Y103X). The mutation was confirmed by DNA sequencing. Full-length cDNAs encompassing the respective coding regions of human E12 and E47 in pSP64 vector and human vitamin D3 receptor (hVDR) in p91023B were cloned into pcDNA3, generating pCMV-E12, pCMV-E47, and pCMV-hVDR, respectively. Mammalian expression vectors for Id1, Id2, and the E12 N-terminal deletion mutant Δ1-507 (amino acids [aa] 508 to 654) were described elsewhere (25). p21 cDNA was ligated in the sense or antisense orientation, generating pCMV-p21 and pCMV-p21as. The expression plasmid for CBP (pRSV-CBP) was a generous gift from T. Nakajima of the University of Tsukuba (42). The p21-luc reporter construct (a gift from X.-F. Wang, Duke University) carries the 2.4-kb HindIII fragment of the p21 promoter in the pGL3-basic vector (Promega) (13). p21 promoter mutants p21-1.8kb-luc, p21-220bp-luc, and p21-del PvuII-luc were produced by deletion of the XhoI and TthIII1, XhoI and PstI, and PvuII and PvuII fragments from the p21-luc construct. The 1.5-kb NcoI-SphI fragment of the FGFR3 promoter from cosmid clone pc385.12 containing the human FGFR3 genomic fragment (47) was inserted into the pGL2-basic vector (Promega), thus generating FGFR3-luc. FGFR3 promoter mutants FGFR3-1.0kb-luc, FGFR3-0.7kb-luc, and FGFR3-0.5kb-luc were produced by deletion of the NcoI and HincII, NcoI and XhoI, and NcoI and PvuII fragments from the FGFR3-luc construct. The simian virus 40 nuclear localization signal sequence was fused to pEGFP-C2 (Clontech), generating vector pnGFP, expressing green fluorescent protein localized in the nucleus.
Transient transfection and luciferase assay.
Subconfluent cultures of MG63 cells (2 × 105 cells/dish 60 mm in diameter) were transfected with a total of 10 μg of expression and reporter plasmids by the calcium phosphate method together with β-galactosidase (β-Gal) expression vector pCMV-β-gal, which was used as an internal control to monitor the transfection efficiency. Total amounts of transfected DNA were equalized by the addition of empty vectors pcDNA3 and pRc/RSV. After incubation overnight with the DNA precipitate, cells were then cultured in DMEM containing 0.1, 2, or 10% FBS for a further 48 h for luciferase assay and for a further 72 h for immunostaining. Luciferase activities were assayed using the luciferase assay system (Promega) and normalized to β-Gal activities, which were determined by the method of Rose and Botstein (57). All assays were performed at least three times in duplicate, and representative data are presented. The results are the means of different experiments ± standard errors.
Immunofluorescence staining.
Cells were fixed in PBS with 3.7% formaldehyde, permeabilized with Triton X-100 (0.1%) in PBS, and then treated with serum to block nonspecific binding sites. Polyclonal rabbit antibodies against E2A (sc-349; Santa Cruz Biotechnology), Id1 (sc-488; Santa Cruz Biotechnology), HEB (sc-357; Santa Cruz Biotechnology), β-Gal (Zymed), and FGFR3 (sc-123; Santa Cruz Biotechnology), polyclonal goat antibodies raised against a peptide corresponding to an amino acid sequence at either the amino terminus (sc-6070; Santa Cruz Biotechnology) or the carboxyl terminus (sc-6269; Santa Cruz Biotechnology) of human Twist, and a mouse monoclonal antibody to human p21 (sc-817; Santa Cruz Biotechnology) were used. After permeabilization, cells were incubated for 1 h at room temperature with the above-mentioned primary antibodies at the respective dilutions of 1:50 to 1:1,200. To discriminate the exogenous expression of E2A from the endogenous one, different concentrations of the anti-E2A antibody were used; endogenous expression of E2A could be detected by 1:100-diluted antibody, while overexpressed E2A derived from the exogenous gene was detected by 1:300-diluted antibody. Immune complexes containing E2A, Twist, Id1, HEB, FGFR3, and p21 were detected with rhodamine-conjugated anti-rabbit immunoglobulin G (IgG) (Chemicon), rhodamine-conjugated anti-goat IgG (Santa Cruz Biotechnology), and fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Chemicon). DNA in nuclei was detected with 4′, 6′-diamidino-2-phenylindole (DAPI). DNA synthesis was determined by measuring the 1-h uptake of thymidine analog bromodeoxyuridine (BrdU). After additional fixation in PBS and DNA denaturation in 2 N HCl, BrdU was detected using a commercial kit (Boehringer) according to the manufacturer's instructions, except that a FITC-conjugated antimouse antibody was substituted in the secondary-antibody step. In the quantitative analysis, a minimum of 150 positive cells were evaluated in each transfection. Cells were examined with an Olympus fluorescence microscope (BX-FLA) or a Zeiss confocal laser-scanning fluorescence microscope (LSM510UV). The intensity of fluorescence was quantified by image analyzing computer software NIH Image.
Western blotting.
Cell extracts were prepared by trichloroacetic acid precipitation as described previously (51). Proteins were separated by electrophoresis with a 10% acrylamide gel and then analyzed by immunoblotting. Antibodies were detected using the ECL method (Amersham Life Science).
Immunohistochemistry.
Whole heads of mice at embryonic day 15 were fixed overnight in 4% paraformaldehyde. Sections were prepared as described previously (20). Tissue sections were stained with a rabbit polyclonal antibody raised against E2A, HEB, or FGFR3 and then incubated in biotinylated goat anti-rabbit IgG (Vector Laboratories). Immunostains were visualized by using a peroxidase substrate system (Vectastain-elite ABC kit; Vector). Irrelevant rabbit IgG was used for control experiments. After immunostaining, sections were counterstained by immersion in 0.5% methyl green in 0.1 M sodium acetate solution (pH 4.0) and then observed under a light microscope.
ALP activity assay.
Alkaline phosphatase (ALP) activity in cell supernatants was assayed as outlined in detail previously (6). Briefly, at the end of the culture periods, the cells were washed with PBS and scraped into 10 mM Tris-HCl (pH 7.5)–0.5 mM MgCl2–0.1% Triton X-100. The ALP activity in thawed and sonicated samples was measured using an ALP diagnostic kit (Sigma; 104-LS). Absorbance at 410 nm was read, and the enzyme activity was calculated. Protein concentrations were determined with the Bio-Rad protein assay kit. ALP specific activity was calculated as nanomoles of p-nitrophenylphosphate per microgram of protein.
RESULTS
Cell cycle arrest and p21 induction in differentiating MG63 cells.
MG63 cells used in the present study are p53 deficient (9) and retain common features of normal human osteoblasts, such as the ability to differentiate in culture by serum starvation with 1,25-(OH)2D3, an active form of vitamin D3 (8). First, we investigated how serum starvation and 1,25-(OH)2D3 influence cell cycle arrest of MG63 cells. MG63 cells, when cultured in medium with 0.1% FBS for 72 h, showed a great reduction in the proportion of cells in S and G2/M phases (from 33 to 7%) (Table 1; Fig. 1A). This result indicates that low-serum culture induces G1 arrest of the cell cycle preceding differentiation of MG63 cells. The arrest was accelerated by the addition of 1,25-(OH)2D3 (Table 1).
TABLE 1.
Cell cycle distribution of serum-starved and 1,25-(OH)2D3-treated MG63 cellsa
| Time (h) | Percentage of cells in S and G2/M at indicated FBS concn (%) and 1,25-(OH)2D3 statusb
|
||||
|---|---|---|---|---|---|
| 10
|
2
|
0.1c | |||
| − | + | − | + | ||
| 24 | 32.9 | 32.4 | 26.1 | 22.7 | nd |
| 48 | 25.5 | 20.5 | 13.7 | 10.2 | 8.3 |
| 72 | 15.7 | 15.1 | 13.6 | 12.1 | 7.6 |
MG63 cells were cultured in DMEM with FBS at the concentrations indicated. The cells were treated with or without 1,25-(OH)2D3 for the indicated times and analyzed for DNA content by propidium iodide staining with a flow cytometer. Data were processed using Cell Quest. nd, not determined.
+, present; −, absent.
1,25-(OH)2D3 was absent at this FBS concentration.
FIG. 1.

Effects of serum starvation on cell cycle arrest and p21 expression in MG63 cells. (A) G1 arrest by serum starvation. MG63 cells were cultured in DMEM containing 10% FBS (a) or 0.1% FBS (b) for 48 h and analyzed for their DNA content by flow cytometry. (B) Activation of p21 promoter by serum starvation. MG63 cells were transfected with 2.5 μg of p21-luc, cultured in DMEM containing either 10, 2, or 0.1% FBS for 48 h, and collected for luciferase assay. Luciferase activity is expressed as fold increase over that for cells cultured in DMEM containing 10% FBS. (C) Inhibition of cell growth by p21 as measured by BrdU incorporation. MG63 cells were transfected with pCMV-p21 and pCMV-β-gal plasmids and maintained in DMEM containing 10% FBS for 48 h. BrdU was added 1 h before fixation. Arrowheads, positions corresponding to cells. The cells were stained for β-Gal (a), BrdU (b), and DNA (c). DAPI was used for counterstaining to visualize the nucleus. Scale bar, 20 μm.
To gain insight into the molecular mechanism of G1 arrest, we examined whether low-serum culture induced expression of p21. Culture in low-serum medium induced expression of endogenous p21 in nuclei of MG63 cells (Fig. 2A, a and b), suggesting that induction of the p21 gene is involved in cell cycle arrest induced by low-serum culture. To monitor the promoter activity of the p21 gene, the reporter plasmid (p21-luc) carrying the 2.4-kb fragment encompassing the p21 promoter region was transfected into MG63 cells. Then the cells were cultured for 48 h in medium with 10, 2, or 0.1% FBS, and luciferase activity of cell lysates was determined. A slight increase (approximately twofold) in luciferase activity was seen by culture in medium with 2% FBS compared with that in medium with 10% FBS; culture in medium with 0.1% FBS greatly enhanced luciferase activity by up to six-to eightfold (Fig. 1B). These results indicate that serum starvation induces activation of the p21 gene promoter.
FIG. 2.
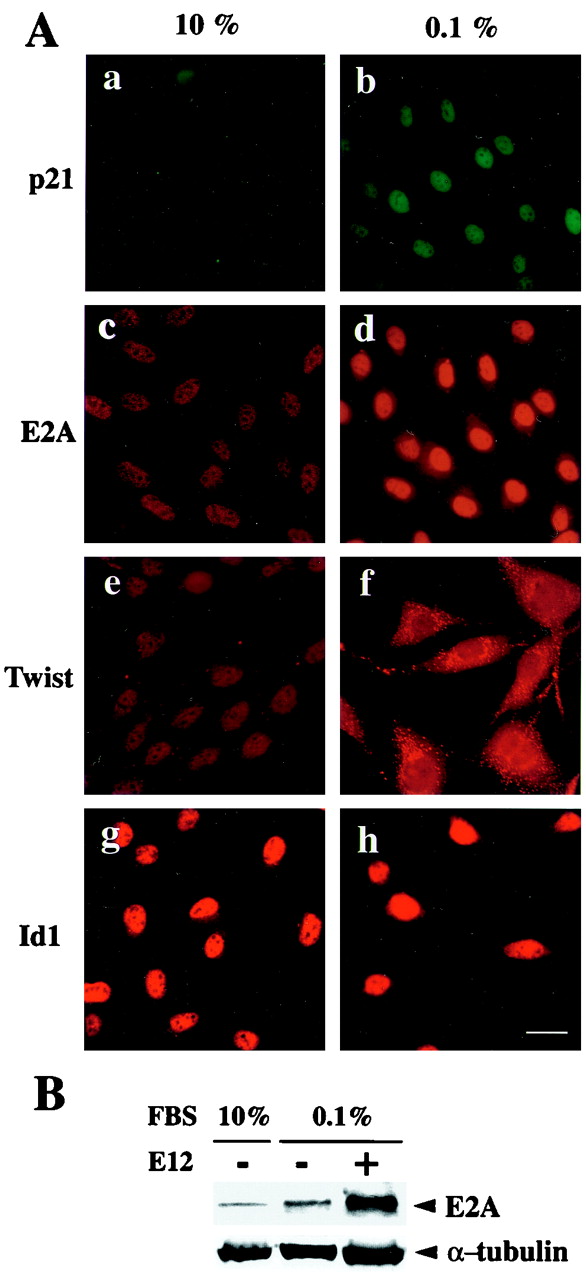
Effects of serum starvation on expression of p21, E2A, Twist, and Id1 in MG63 cells. (A) Proliferating MG63 cells in DMEM containing 10% FBS (a, c, e, and g) and G1-arrested MG63 cells in DMEM containing 0.1% FBS (b, d, f, and h) were stained with antibodies for p21 (fluorescein; a and b), E2A (rhodamine; c and d), Twist (rhodamine; e and f), and Id1 (rhodamine; g and h) and observed under a fluorescence microscope. Scale bar, 20 μm. (B) MG63 cells with (+) or without (−) the E12-expressing plasmid were cultured in indicated concentrations of FBS. Equal amounts of whole-cell extracts were analyzed by Western blotting with antibodies against E2A and α-tubulin, Lane +, expression of E12 in cells exogenously transfected.
We directly tested if p21 could induce cell cycle arrest in MG63 cells. MG63 cells were transfected with an expression plasmid encoding p21 as well as the β-Gal expression vector and examined for BrdU incorporation by immunostaining. The incorporation of BrdU was impaired in 95.2% of p21-positive MG63 cells cultured in high serum (Fig. 1C), supporting a close link between p21 induction and cell cycle arrest in MG63 cells. Importantly, both cell cycle arrest and p21 induction by serum starvation are independent of p53 in MG63 cells.
Activation of the p21 promoter by ectopic E2A.
To determine whether E2A proteins play a part in growth arrest of MG63 cells, we examined the expression of endogenous E2A proteins. Our investigation revealed that E2A was present in nuclei of MG63 cells cultured in high serum (10% FBS). Low-serum culture (0.1% FBS) enhanced the expression of E2A (Fig. 2A, c and d). Western blot analysis demonstrated that the average level of E2A expression in MG63 cells cultured in low serum was significantly higher than that in the cells cultured in high serum (Fig. 2B). It should be noted that induction of endogenous p21 expression paralleled the enhanced expression of endogenous E2A (Fig. 2A, a to d).
To determine whether E2A proteins regulate the expression of p21, we examined the effects of the E2A gene products, E12 and E47, on p21 promoter activity in MG63 cells. MG63 cells were cotransfected with p21-luc and an E2A expression plasmid (pCMV-E12 or pCMV-E47), and luciferase activities were determined after 48 h of culture. Overexpression of E12 induced activation of the p21 promoter up to 400-fold in an E12 dose-dependent manner (Fig. 3A). The E12 N-terminal deletion mutant (aa 508 to 654; del E12) lacking the transcription activation domain (1) failed to induce p21 promoter activation (Fig. 3A). Similarly, the p21 promoter was activated by the ectopic expression of E47, although the magnitude of activation was lower than that of E12. Additive effects of E12 with E47 were not evident (Fig. 3A). These results demonstrate that the E2A products, E12 and E47, induce p21 promoter activity in MG63 cells.
FIG. 3.

Activation of p21 promoter by E12 and E47 independent of p53. (A) Transcriptional activation of p21 promoter by E12 and E47. MG63 cells were cotransfected with 2.0 μg of the p21-luc reporter plasmid in combination with pCMV-E12, pCMV-E47, or pCMV-del E12 (Δ1-507). The cells were cultured in DMEM containing 2% FBS for 48 h and collected for luciferase assay. Luciferase activity mediated by pcDNA3 is arbitrarily set at 1. (B) Schematic diagram of luc reporter constructs. The 2.4-kb promoter sequence of the p21 gene (solid line) was cloned at the site upstream of the luciferase gene in p21-luc. Possible binding sites for E2A (inverted triangles) and p53 (boxes) in the promoter are shown. Dashed lines, internal deletions. (C) Two micrograms of p21 promoter deletion mutant reporter plasmids with or without the E12 or E47 expression plasmid (2 μg) along with the pCMV-β-gal plasmid was transfected into MG63 cells. The cells were cultured in DMEM containing 2% FBS for 48 h and collected for luciferase assay. Luciferase activities were normalized against β-Gal activities. Activities relative to that mediated by the empty pcDNA3 are shown.
The p21 gene is shown to have seven putative E2A-binding sequences (designated E1 to E7) spreading over the 2.4-kb sequence upstream of the transcriptional initiation site (Fig. 3B). These binding sequences are expected to mediate the E2A-dependent activation of the p21 promoter. To examine this assumption, truncated fragments containing E2A-binding sites were linked to the luc reporter plasmids and tested for their ability to respond to E2A in MG63 cells. A profound reduction in E2A responsiveness was seen in a mutant carrying only three proximal E2A-binding sequences (E1 to E3) (Fig. 3C). Our results suggest the involvement of multiple elements in the p21 promoter region in E2A-induced activation of p21 gene transcription.
Endogenous p21 induction and repression of BrdU incorporation by ectopic E12 expression.
We further tested if E12 could stimulate expression of the endogenous p21 gene. MG63 cells were transfected with pCMV-E12 and examined for p21 expression by immunostaining. Upon introduction of E12, endogenous p21 expression was induced in cells expressing high levels of E12 even when cultured in high serum (Fig. 4A; Table 2). These observations strongly support a close link between p21 induction and E12 expression in MG63 cells (Fig. 2). The incorporation of BrdU, conversely, was decreased in E12-positive MG63 cells cultured in high serum (Fig. 4B; Table 2). Similar experiments were carried out by introduction of β-Gal expression vector pCMV-β-gal in addition to E12 expression vector pCMV-E12 to identify cells transfected. Cells expressing β-Gal produced p21 with E12 expression at high levels and were impaired in incorporation of BrdU (data not shown). Upon introduction of pCMV-β-gal with a control empty vector, neither endogenous p21 induction nor inhibition of incorporation of BrdU was observed (data not shown). No appreciable induction of p21 was seen, even in MG63 cells expressing E12, when the antisense p21 sequence was overexpressed (Fig. 4C). In addition, introduction of the p21 antisense sequence inhibited E12-mediated cell cycle attenuation; all cells expressing E12 in Fig. 4D were BrdU positive, unlike cells expressing E12 in Fig. 4B. The proportion of E12-positive MG63 cells expressing p21 was approximately equivalent to that of cells incapable of synthesizing new DNA (Table 2). The forced expression of E12 thus presumably causes MG63 cells to arrest at G1 phase through the induction of p21.
FIG. 4.
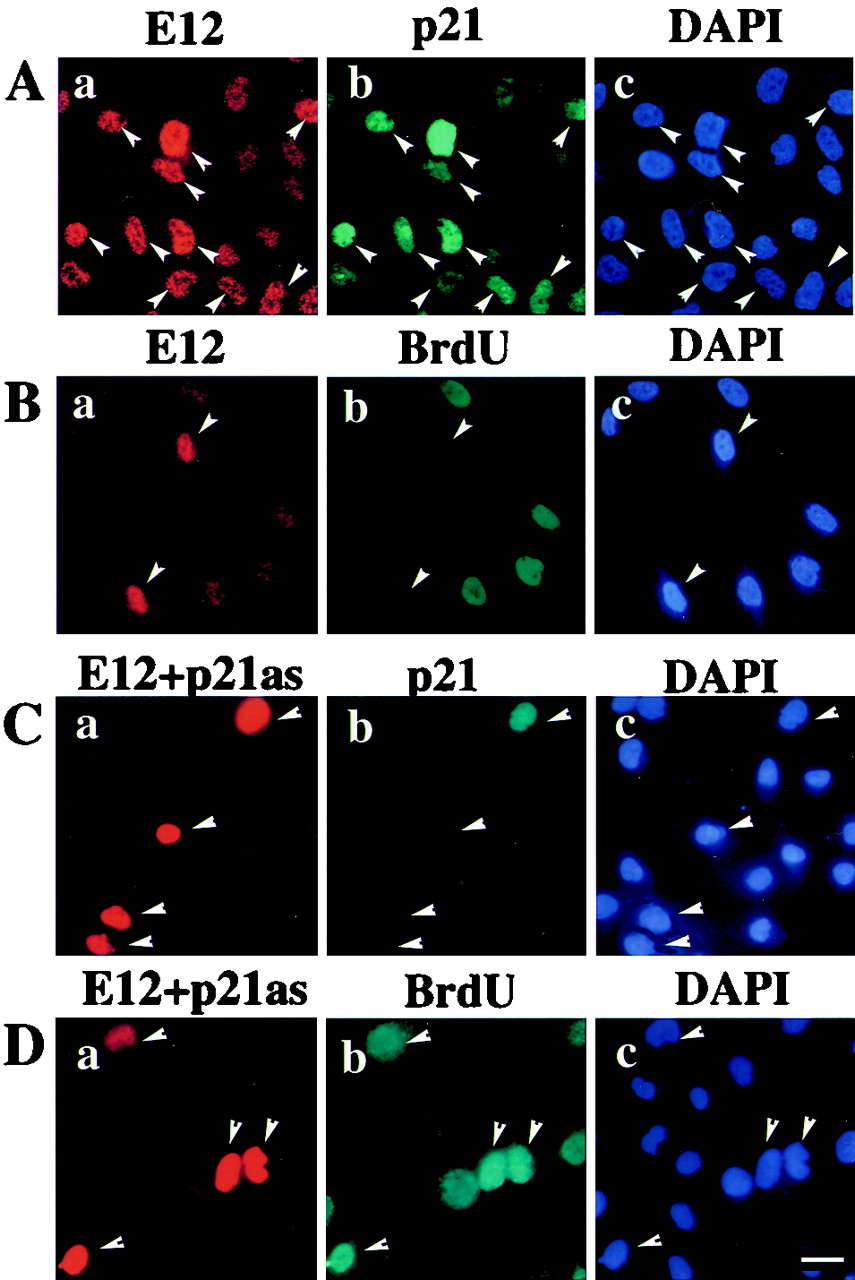
Effects of E12 on endogenous p21 expression and BrdU incorporation in MG63 cells. (A) Activation of endogenous p21 by E12 overexpression. MG63 cells were transfected with pCMV-E12 and maintained in DMEM containing 10% FBS for 72 h. The cells were fixed and stained for E12 (rhodamine; a), p21 (fluorescein; b), and DNA (DAPI; c). DAPI was used for counterstaining to visualize the nucleus. Arrowheads, cells transfected with E12. (B) Effects of E12 on cell proliferation. MG63 cells were treated by the same procedure as for panel A. BrdU was added 1 h before fixation. Arrowheads, positions corresponding to cells. The cells were stained for E12 (rhodamine; a), BrdU (fluorescein; b), and DNA (DAPI; c). (C) Impairment of E12-dependent p21 induction by the p21 antisense plasmid. MG63 cells were transfected with pCMV-E12 and an antisense p21 construct (pCMV-p21as) and maintained in DMEM containing 10% FBS for 72 h. The cells were fixed and stained for E12 (rhodamine; a), p21 (fluorescein; b), and DNA (DAPI; c). Arrowheads, cells expressing E12. (D) Impairment of E12-mediated cell cycle attenuation by the p21 antisense plasmid. MG63 cells were treated by the same procedure as for panel C. BrdU was added 1 h before fixation. Arrowheads, positions corresponding to cells. The cells were stained for E12 (rhodamine; a), BrdU (fluorescein; b), and DNA (DAPI; c). Scale bar, 20 μm.
TABLE 2.
Effects of E12 expression on DNA synthesis and p21 expressiona
| Transfection | % of cells that were:
|
|||
|---|---|---|---|---|
| E12+ and:
|
E12− and:
|
|||
| p21+ | BrdU− | p21+ | BrdU− | |
| E12 | 88.3 | 81.4 | 0.1 | 43.7 |
| E12 + p21as | 15.0 | 27.6 | 0.1 | 51.0 |
MG63 cells were transiently transfected with 5.0 μg of pCMV-E12 and either an empty vector or pCMV-p21 antisense (p21as). The cells were maintained in DMEM containing 10% FBS. The expression of p21 was monitored by immunostaining of E12 and p21 (Fig. 4A and C). DNA synthesis was evaluated by measuring the uptake of BrdU for 1 h in a parallel dish of cells, and cells were stained for E12 (Fig. 4B and D). At least 150 cells were counted in each category, and results are representative of multiple experiments performed.
Transcriptional inhibition of the p21 gene by Twist and Ids.
Twist has been shown to interact with E12 (24, 62) and to be expressed in early preosteoblasts (55). We therefore examined by immunostaining with a polyclonal antibody raised against the amino terminus of Twist whether Twist was expressed in MG63 cells. Twist was predominantly present in nuclei of MG63 cells cultured in medium containing either 2 (data not shown) or 10% FBS (Fig. 2A, e). Interestingly, we found a peculiar localization of Twist in serum-starved MG63 cells; it was mainly localized in the cytoplasm with a granular appearance and was weakly positive in nuclei (Fig. 2A, f). Similar results were obtained using a polyclonal antibody raised against the carboxyl terminus of Twist (data not shown).
To further analyze the biological significance of the interaction of Twist with E2A for p21 induction in MG63 cells, we tested the role of Twist in E2A-dependent activation of p21 transcription. When p21-luc was transfected along with the Twist expression vector, a great reduction in luciferase activity in both E12- and E47-containing cells was observed (Fig. 5A and B), indicating antagonistic effects of Twist on E2A-induced transcriptional activation. Some patients with Saethre-Chotzen syndrome have mutations in the TWIST gene (17, 28). A nonsense mutation (Y103X) found in a patient was examined for its effect on E2A-induced p21 activation. Unlike the wild type, the Twist mutant failed to inhibit p21 promoter activation in response to E2A (Fig. 5A and B). The inhibitory effect of Twist on E12-dependent p21 promoter activation was dose dependent (Fig. 5C). We similarly attempted to determine whether exogenous Twist suppressed the p21 transcriptional activation induced by serum starvation. MG63 cells were transfected with or without the Twist expression vector along with p21-luc and then cultured in medium containing either 0.1, 2, or 10% FBS. As expected, the exogenous expression of Twist suppressed the stimulatory effect of low-serum culture on p21-luc expression, while the Twist (Y103X) mutant had little, if any, effect on low-serum-mediated p21 promoter activation (Fig. 5D).
FIG. 5.

Effects of Twist, Id1, Id2, and CBP on E2A-dependent transactivation of p21. (A) Inhibition of E12-dependent transactivation of p21 by Twist, Id1, and Id2. MG63 cells were cotransfected with 2.5 μg of p21-luc in combination with 2.0 μg of either pCMV-E12, pCMV-Twist, pCMV-Twist (Y103X), pCMV-Id1, or pCMV-Id2. The cells were cultured in DMEM containing 2% FBS for 48 h and collected for luciferase assay. Activation mediated by pcDNA3 is arbitrarily set at 1. (B) Inhibition of E47-dependent transactivation of p21 by Twist, Id1, and Id2. MG63 cells were cotransfected as for panel A except for pCMV-E12, which was replaced with 2.0 μg of pCMV-E47. (C) CBP-mediated suppression of inhibition of E12-dependent p21 transcription by Twist. MG63 cells were cotransfected with 1.0 μg of p21-luc and 0.25 μg of either pcDNA3 or pCMV-E12. The cells were cultured in DMEM containing 2% FBS for 48 h and collected for luciferase assay. Activation mediated by pcDNA3 is arbitrarily set at 1. (D) Twist-mediated inhibition of p21 gene expression induced by serum starvation. MG63 cells were transfected with 2.5 μg of p21-luc and 2.5 μg of pCMV-Twist or pCMV-Twist (Y103X). The cells were cultured in DMEM containing either 10, 2, or 0.1% FBS for 48 h and collected for luciferase assay. Luciferase activity is expressed as fold increase over that for vehicle-transfected cells cultured in DMEM containing 10% FBS. Empty vector pcDNA3 was included to adjust DNA amounts.
Id proteins are dominant-negative-type HLH molecules which contain functional HLH dimerization motifs but which lack the DNA-binding basic region (4). Id1 is expressed in early preosteoblasts, similar to Twist (39, 55). Our investigation revealed that Id1 was present in the nuclei of MG63 cells cultured in high serum (10% FBS) and low serum (0.1% FBS) (Fig. 2A, g and h). Therefore, the effects of expression of Id proteins, in addition to Twist, with respect to p21 promoter activation were examined. Both Id1 and Id2 reduced luciferase activities driven by the p21 promoter in response to E12 and E47 (Fig. 5A and B). The inhibitory levels achieved by Id1 and Id2 expression were equivalent to those achieved by Twist expression. These results are compatible with the report that overexpression of Id1 in NIH 3T3 cells accelerates cell growth and inhibits p21 expression (52). Ids have been shown to act as dominant-negative antagonists of other bHLH transcription factors (48, 52), but the effects of Ids on Twist function are not known. We thus investigated how Id expression influenced the function of Twist. The inhibition of E2A-dependent p21 promoter activation by Twist was further enhanced by addition of either Id1 or Id2 (Fig. 5A and B), suggesting that Twist, Id1, and Id2 inhibit p21 expression in a cooperative manner.
Inhibition of endogenous p21 induction by ectopic Twist and Ids.
We further tested if Twist and Id proteins could inhibit expression of the endogenous p21 gene. MG63 cells were transfected with expression plasmids for E12 and Twist and then examined for p21 expression by immunostaining. Upon introduction of E12, endogenous p21 expression was induced in cells expressing high levels of E12 (Fig. 4A). Overexpression of Twist abolished the endogenous p21 expression induced by E12 (Fig. 6A and Table 3). Interestingly, overexpression of Id1 or Id2 showed little effect on E12-dependent endogenous p21 expression (Fig. 6B and C and Table 3), in contrast to the profound inhibition of the p21 promoter in reporter plasmids (Fig. 5A and B).
FIG. 6.
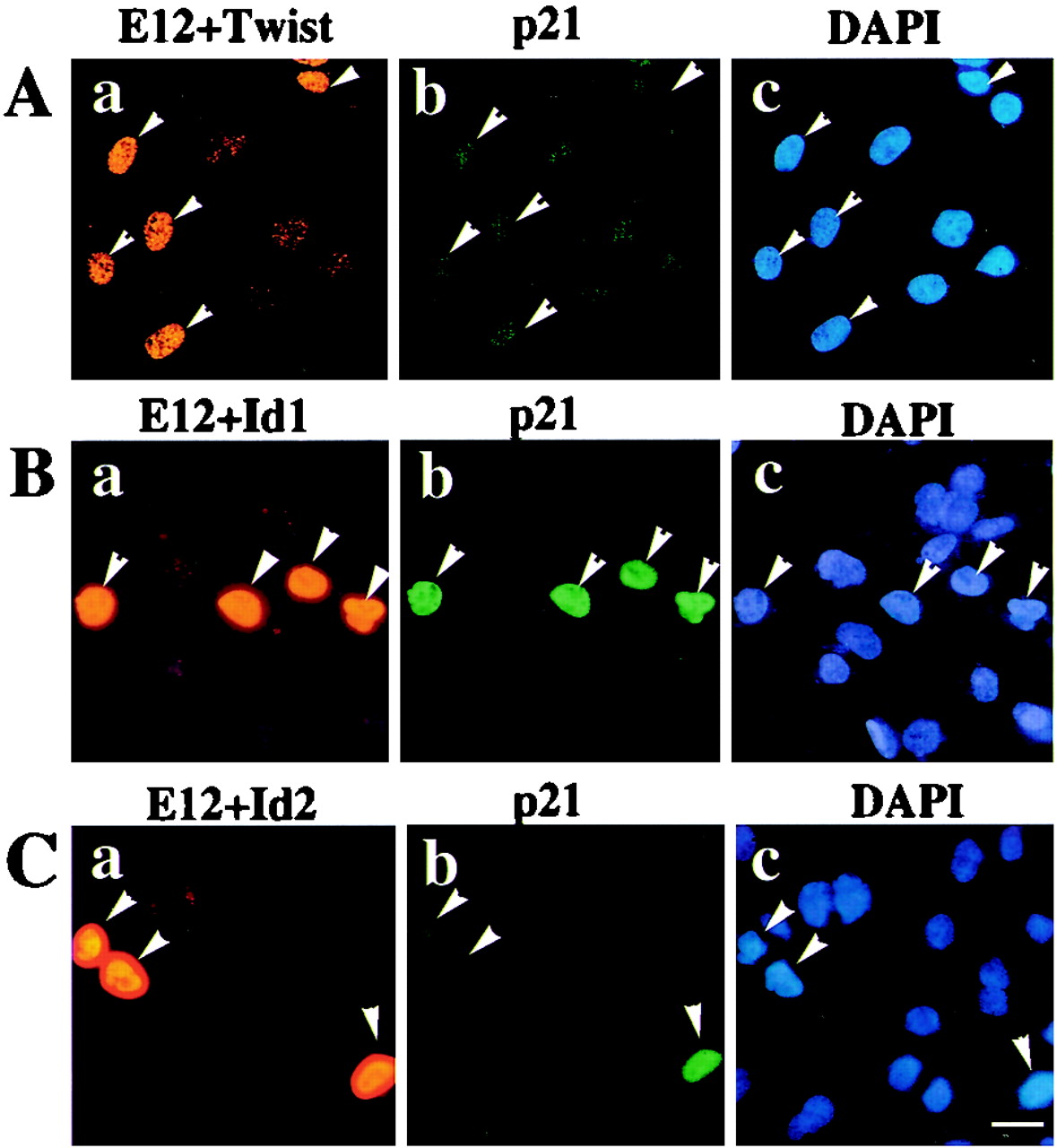
Inhibition of E12-dependent p21 induction by Twist and Ids overexpression. MG63 cells were transfected with pCMV-E12 and either pCMV-Twist (A), pCMV-Id1 (B), or pCMV-Id2 (C). DNA amounts used for transfection were adjusted by empty vectors. The cells were maintained in DMEM containing 10% FBS for 72 h. The cells were fixed and stained for E12 (rhodamine; a), p21 (fluorescein; b), and DNA (DAPI; c). Arrowheads, cells expressing E12. Scale bar, 20 μm.
TABLE 3.
Effects of Twist and Ids on E12-dependent p21 expressiona
| Transfection | % of cells that were:
|
|||
|---|---|---|---|---|
| E12+ and:
|
E12− and:
|
|||
| p21+ | p21− | p21+ | p21− | |
| E12 | 88.3 | 11.7 | 0.1 | 99.9 |
| E12 + Twist | 19.9 | 80.1 | 1.9 | 98.1 |
| E12 + Id1 | 45.5 | 54.5 | 1.6 | 98.4 |
| E12 + Id2 | 42.6 | 57.4 | 2.6 | 97.4 |
MG63 cells were transiently cotransfected with 5.0 μg of pCMV-E12 and either pCMV-Twist, pCMV-Id1, or pCMV-Id2. DNA amounts used for transfection were adjusted by an empty vector. The cells were maintained in DMEM containing 10% FBS. The expression of p21 was monitored by immunostaining of E12 and p21 (Fig. 4A and 6). At least 150 cells were counted in each category, and results are representative of multiple experiments performed.
Competition of E12-dependent transcriptional activity between CBP and Twist.
The coactivator p300/CBP harboring histone acetyltransferase (HAT) activity has been shown to interact with E12 (16). HAT activity is thought to be important for decondensing the chromatin and facilitating the binding of the RNA polymerase II transcription complex to core promoters (2, 44). To analyze the functional implications of the interactions between E12 and the HAT coactivator, we studied the effects of CBP on E12-dependent transcriptional activation of the p21 gene. Overexpression of CBP significantly enhanced the E12-mediated transactivation of the p21 promoter in the absence of Twist (Fig. 5C). Twist-induced inhibition of p21 promoter activation in response to E12 was overcome by CBP in a dose-dependent manner (Fig. 5C). Taken together, these findings support the view that CBP acts as a coactivator of the E12-mediated transcription of the p21 promoter and suggest that Twist suppresses transcriptional activity conferred by a combination of E12 and CBP.
Transcriptional regulation of the FGFR3 gene by E2A, Twist, Ids, and CBP.
We were interested in investigating the transcriptional regulatory mechanism that involves E2A, Twist, and Ids in osteoblast differentiation beyond cell cycle arrest. Thus these molecules were tested for their effects on expression of the FGFR3 gene. FGFR3 is expressed in differentiating osteoblasts at the osteogenic front (55), presumably leading to the upregulation of genes related to osteoblast differentiation (10). MG63 cells were cotransfected with luciferase reporter construct FGFR3-luc containing the 1.5-kb promoter sequence of the FGFR3 gene, along with one of E2A expression vectors pCMV-E12 and pCMV-E47. Overexpression of either E12 or E47 activated the FGFR3 promoter in a dose-dependent manner (Fig. 7A). We searched potential sequences for E2A binding in the FGFR3 gene promoter and found six candidates spreading over the 1.5-kb sequence upstream of the transcriptional initiation site (Fig. 7B). To examine whether these binding sequences mediate the E2A-dependent activation of the FGFR3 promoter, 5′-end-truncated fragments of the FGFR3 promoter were linked to the luc reporter plasmids and tested for their ability to respond to E2A in MG63 cells. All mutant fragments, even those lacking any putative E2A-binding sites, were found to be activated as highly as the wild type (Fig. 7C).
FIG. 7.

Effects of E2A, Twist, Id1, Id2, and CBP on FGFR3 transcription in MG63 cells. (A) Transcriptional activation of the FGFR3 promoter by E12 and E47. MG63 cells were cotransfected with 2.5 μg of the FGFR3-luc reporter plasmid along with pCMV-E12 or pCMV-E47. Cells were harvested 48 h after transfection and then assayed for their luciferase activities. (B) Schematic diagram of luc reporter constructs. The 1.5-kb promoter sequence of the FGFR3 gene (solid line) was cloned at the site upstream of the luciferase gene in FGFR3-luc. Possible binding sites for E2A (inverted triangles) in the promoter are shown. (C) Two micrograms of FGFR3 promoter deletion mutant reporter plasmids with or without the E47 expression plasmid (2 μg) along with the pCMV-β-gal plasmid was transfected into MG63 cells, and luciferase assays were performed. Luciferase activities were normalized against β-Gal activities. Activities relative to that mediated by the empty pcDNA3 are shown. (D) Inhibition of E2A-dependent transactivation of FGFR3 by Twist, Id1, and Id2. MG63 cells were cotransfected with 2.5 μg of p21-luc in combination with 2.0 μg of pCMV-E12, pCMV-E47, pCMV-Twist, pCMV-Twist (Y103X), pCMV-Id1, and pCMV-Id2. The cells were harvested 48 h after transfection and then assayed for reporter gene expression. (E) CBP-mediated suppression of inhibition of E12-dependent FGFR3 transcription by Twist. MG63 cells were cotransfected with 1.0 μg of FGFR3-luc and 0.25 μg of either pcDNA3 or the pCMV-E12 expression vector. Activation mediated by pcDNA3 is arbitrarily set at 1. (F) Additive effect of E2A and 1,25-(OH)2D3 on FGFR3 expression. MG63 cells were transfected with 2.5 μg of FGFR3-luc and 2.5 μg of pCMV-E12. The cells were cultured in DMEM containing either 10, 2, or 0.1% FBS with or without 1,25-(OH)2D3 for 48 h and collected for luciferase assay. Luciferase activity is expressed as fold increase over that for vehicle-transfected cells cultured in DMEM containing 10% FBS. DNA amounts were adjusted by empty vectors.
We next tested the role of Twist, Id1, and Id2 in E2A-dependent activation of FGFR3 transcription. A reduction in luciferase activity from FGFR3-luc was observed by expression of either Twist, Id1, or Id2, while no significant change was induced by expression of the Twist (Y103X) mutant (Fig. 7D). Twist-induced inhibition of FGFR3 promoter activation in response to E12 was overcome by CBP in a dose-dependent manner (Fig. 7E). These results demonstrate that FGFR3 is a transcriptional target of E2A, and this expression could be regulated by a coordination of Twist, Ids, and CBP, similar to what was found for the p21 gene.
Expression of E2A and FGFR3 during calvarial bone development in vivo.
To address the implications of E2A and FGFR3 for in vivo calvarial bone development, we examined the localization of the two molecules in calvarial bone tissue of mouse fetus by immunostaining. An examination of fetuses at embryonic day 15 showed that E2A was expressed at bordering areas of condensing calvarial mesenchyme just lateral to the temporal cartilage (Fig. 8A, a and b). Expression of FGFR3 was found at the same sites (Fig. 8A, c). Ubiquitous bHLH factor HEB was also expressed in the developing calvarial bones (data not shown). The border positive for E2A, HEB, and FGFR3 is known to be an osteogenic area where osteoprogenitors differentiate into the parietal bones through osteoblasts (55). Interestingly, E2A and FGFR3 were also expressed in differentiated neurons of the cortical plate, whereas very low levels of FGFR3 and E2A were detected in undifferentiated cells of the ventricular zone (Fig. 8A). These findings strongly support the possibility that bHLH molecules E2A and presumably HEB play roles in regulating the expression of FGFR3.
FIG. 8.

Osteoblast differentiation in vivo and in vitro. (A) E2A and FGFR3 expression in the developing calvarial bones. Frontal sections were stained with antibodies for E2A (a and b) and FGFR3 (c). Solid arrowheads (a to c) and open arrowhead (a), cortical plate and the ventricular zone, respectively. The region between arrows (b and c) indicate parietal bones with osteoblast differentiation. Box (a), magnified area shown in panels b and c. cc, cerebral cortex; pb, parietal bone; tc, temporal cartilages; ep, epithelium. Scale bar, 200 μm. (B) Activation of endogenous FGFR3 by E47 overexpression. MG63 cells were transfected with pCMV-E47 and pnGFP and maintained in DMEM containing 2% FBS for 72 h. The cells were fixed and stained for FGFR3 (rhodamine; b) and DNA (DAPI; c). pnGFP (fluorescein; a) was used as a marker to visualize the transfected cells. DAPI was used for counterstaining to visualize the nucleus. Arrowheads, cells transfected with E47. Scale bar, 20 μm. (C) Effects of 1,25-(OH)2D3 with serum starvation on expression of endogenous FGFR3 in MG63 cells. Proliferating MG63 cells in DMEM containing 10% FBS (a) and G1-arrested MG63 cells in DMEM containing 0.1% FBS with 1,25-(OH)2D3 (b) were stained with antibodies for FGFR3 and observed under a confocal laser microscope. Scale bar, 20 μm. (D) ALP activity in MG63 cells. Cells were transfected with either pCMV-E12, pCMV-Twist, or empty vector and cultured with or without 1,25-(OH)2D3 for 96 h. The cells were harvested, and cell extracts were prepared for determining ALP activities. Values are means with standard errors of three independent experiments.
Endogenous FGFR3 induction by ectopic E47 expression.
We further tested if E47 could stimulate expression of the endogenous FGFR3 gene. MG63 cells were cotransfected with pCMV-E47 and pnGFP and examined for FGFR3 expression by immunostaining. Upon introduction of E47, endogenous FGFR3 expression in cells cultured in 2% FBS was greatly enhanced (Fig. 8B). Quantitative analysis of the integrated density indicated that MG63 cells transfected with E47 expressed endogenous FGFR3 at a level approximately twofold that at which it was expressed in untransfected cells. These observations are consistent with the reporter assay (Fig. 7A) and strongly support the implication of E47 in FGFR3 promoter activation in MG63 cells.
Effects of 1,25-(OH)2D3 on expression of p21 and FGFR3.
1,25-(OH)2D3 stimulates MG63 cells to differentiate into cells with osteoblastic phenotypes under low-serum conditions (6). We therefore examined the effects of 1,25-(OH)2D3 on the bHLH-regulated transcriptional regulatory network. Low-serum-induced G1 cell cycle arrest of MG63 cells was enhanced by the addition of 1,25-(OH)2D3 (Table 1). The p21 promoter was slightly activated (approximately 1.5-fold) in response to 1,25-(OH)2D3 with 2% FBS when the vitamin D3 receptor was introduced into the MG63 cells (data not shown).
The influence of 1,25-(OH)2D3 on the expression of FGFR3 in MG63 cells further examined. 1,25-(OH)2D3 with 2% FBS showed little effect (approximately 1.8-fold enhancement) on E12-induced activation of the FGFR3 promoter (Fig. 7F). Western blot analysis indicated that amounts of E2A molecules did not appear to be changed greatly in response to 1,25-(OH)2D3 (data not shown). Thus the effects of 1,25-(OH)2D3 and E2A on the FGFR3 promoter seemed to be independent and additive (Fig. 7F). The 1,25-(OH)2D3 effect was presumably exerted through two possible vitamin D response elements in the FGFR3 promoter region. We then examined by immunostaining with a polyclonal antibody raised against FGFR3 whether FGFR3 was induced by 1,25-(OH)2D3 in low-serum medium. Immunofluorescence staining revealed that 1,25-(OH)2D3 in low-serum culture profoundly enhanced the expression of endogenous FGFR3 in the cytoplasm (Fig. 8C). However it was hard to see any difference in endogenous FGFR3 expression between 0.1%-serum cultures with and without 1,25-(OH)2D3 (data not shown).
Osteoblast differentiation by bHLH proteins.
We finally wished to examine the possibility that bHLH-mediated coordination of p21 and FGFR3 expression induced osteoblast differentiation. This issue was assessed by examining induction of ALP, which is considered a marker of the final differentiation of osteoblasts. MG63 cells cultured in low serum (0.1%) produced a significant amount of ALP (Fig. 8D), similar to the results of low-serum-induced activation of E2A, as shown in Fig. 2. No appreciable change between 10 and 2% serum culture was observed (Fig. 8D). Either addition of 1,25-(OH)2D3 or overexpression of E12 slightly enhanced expression of ALP in 0.1%-FBS culture; additive effects were seen when both treatments were done together. Introduction of Twist into MG63 cells cultured in 0.1% FBS with 1,25-(OH)2D3 remarkably reduced the level of ALP. These results directly demonstrate the involvement of bHLH molecules in physiological osteoblast differentiation.
DISCUSSION
The present study demonstrates that E2A transcription factors (E12 and E47), HLH transcriptional inhibitors (Twist, Id1, and Id2), and coactivator CBP regulate the expression of the p21 and FGFR3 genes, which are closely associated with cell cycle arrest and differentiation of osteoblasts, respectively. Cellular differentiation involves at least two major and distinct steps: (i) commitment, which irreversibly withdraws proliferating cells from the cell cycle, and (ii) induction of tissue-specific genes, which promote differentiation resulting in expression of characteristic phenotypes. This is the first report showing the involvement of common transcriptional regulation in two genes expressed at distinct steps in the course of osteoblast differentiation. The finding that these transcription factors are involved in regulation of differentiation through gene expression related to cell cycle arrest and tissue-specific gene expression, particularly regulation of the p21 gene, may be applicable to other cell lineages besides osteoblasts. This assumption may be supported by observations that MyoD induces terminal cell cycle arrest during skeletal muscle differentiation by increasing the expression level of p21 (22) and that Twist inhibits the expression of the MCK gene by interacting with MyoD (24, 62).
p53 has been shown to be responsible for cell cycle arrest at the G1 phase through direct induction of the p21 gene (15, 18). In contrast, p53-independent induction of p21 has been demonstrated in several cell lineages (60, 63). It should be noted that mice that are homozygous for a targeted disruption of the p53 gene develop normally (14). There is a paucity of information on p53 expression during differentiation from preosteoblasts to mature osteocytes via osteoblasts. Our results show that the p53-deficient osteoblasts remain arrested in G1 phase when cultured in low-serum medium, suggesting the possibility that cell cycle withdrawal and induction of p21 occur independently of p53 in osteoblasts. p53 may act synergistically with E2A, to activate p21 expression in wild-type cells. Alternatively, regulation of p21 gene expression by p53 may be independent of regulation by E2A. HEB, a ubiquitous bHLH factor with biochemical and functional properties similar to those of E2A (29), was expressed in the developing calvarial bones and the nuclei of MG63 cells (data not shown). These results may support the observation that E2A-deficient mice do not show bone formation defects (71); these transcription factors presumably compensate each other.
This study presents several aspects of the mechanism by which low-serum culture induces p21. An increase in the amount of E2A in the nucleus due to low-serum culture (Fig. 2A, c and d, and B) is, at least in part, responsible for induction of the p21 gene. E47 has also been reported to be increased during erythroid differentiation by the addition of stem cell factor (70). The p21 promoter region contained multiple binding sites for p53 and E2A (Fig. 3B), and two proximal E2A-binding sites (E1 and E2) were demonstrated to be able to mediate E47-dependent activation in HeLa cells (52). Our deletion analysis of the p21 promoter indicates that the two proximal E2A-binding sites are not sufficient for E47-dependent activation of the promoter in osteoblast cell line MG63 (Fig. 3C). The discrepancy in results between the two groups may be due to differences in the experimental systems, including the different cell lines used. It is possible that E2A activates the p21 promoter indirectly. Deletion analysis of the FGFR3 promoter also indicates that the putative E2A binding sites are not sufficient for E47-dependent activation of the promoter in MG63 cells (Fig. 7C). Further studies on the implications of bHLH proteins in these promoters would be useful for our understanding of osteoblastic differentiation and Saethre-Chotzen syndrome.
We demonstrate in this study that overexpression of coactivator CBP transcriptionally enhances E12-dependent activation of the p21 promoter in osteoblasts (Fig. 5C). This result is similar to previous reports that p300/CBP is required for the expression of cell cycle inhibitors p21 and p27 in F9 cells (31). The induction of p21 expression in keratinocyte differentiation is dependent on p300, which alone has been shown to be unable to stimulate the p21 promoter to any significant extent (37). E2A proteins recruit p300/CBP to activate target genes via nuclear HAT activity (16). HAT activity seems to play a role in the induction of p21 gene expression during osteoblast differentiation.
Our results further show that Twist, Id1, and Id2 inhibit transcription of the p21 gene induced by the combination of E2A and CBP. Twist and Ids are involved in the suppression of differentiation through inhibition of p21 expression, preventing unnecessary G1 arrest. We assume that growing cells are inhibited in expressing p21 presumably by formation of a multicomponent complex consisting of an activator complex of E2A with p300/CBP and the inhibitor Twist. Physical interactions between E2A and E2A (40), E2A and Twist (24, 62), E2A and Ids (25), E2A and p300/CBP (16, 54), and Twist and p300/CBP (23) have been reported. In particular, overexpression of Twist completely suppressed E12-dependent induction of endogenous p21 expression (Fig. 6A and Table 3), while overexpression of Ids was less effective in suppression of endogenous p21 expression in response to E12 (Fig. 6B and C and Table 3). Ids may be present in insufficient amounts in our experiment. Alternatively, the difference may be due to the functional properties of Twist and Ids. Very recently Twist has been reported to bind directly to p300/CBP and p300/CBP-associated factor through two independent HAT domains, resulting in inhibition of HAT activities (23). HAT activity is important for decondensing the chromatin (2, 44), changing the accessibility of the transcription machinery (67). In contrast, it has not been reported that Ids have the ability to inhibit HAT activities. Transcription from ectopic reporter plasmids lacking a condensed chromatin structure is inhibited by Ids; however Ids ineffectively inhibit the endogenous p21 gene promoter with the chromatin structure. These ideas might be consistent with the observation that Saethre-Chotzen syndrome results from mutations of the TWIST gene; nevertheless no mutations in the Id genes of patients with Saethre-Chotzen syndrome have been found so far, even though both molecules are expressed in early preosteoblasts (55). Our observations firmly imply that the inhibition of HAT activities may be a mechanism underlying the Twist-mediated inhibition of the E2A-CBP-dependent transcription.
Dimerization is essential for bHLH proteins to bind DNA and exert transcriptional activity (34), and, in general, the ubiquitously expressed E2A proteins heterodimerize with tissue-specific bHLH proteins such as MyoD and myogenin in myoblasts (34, 68), LYL1 in T cells (38), and eHAND in trophoblasts (12). Cell-type-specific bHLH proteins can directly interact with the p300/CBP HAT (16, 41, 53, 54, 59) when bound as a heterodimer with an E2A protein. Although there has been no report concerning bone-specific bHLH factors, it is interesting to speculate that E2A heterodimerizes with an unidentified bone-specific bHLH factor. This speculation may be supported by our and other groups' observations on the transactivation activity of the bHLH proteins. For example, E12 activates transcriptional activity of the p21 gene more highly than E47 (Fig. 3A); nevertheless E12 is shown to efficiently heterodimerize with tissue-specific bHLH factors and not to homodimerize, unlike E47 (5, 64).
Low-serum culture of MG63 cells induces Twist expression. Interestingly, we found that, in cells cultured in low-serum medium, the Twist protein is predominantly and characteristically present in the cytoplasm in a granular pattern, in contrast to nuclear localization of Twist protein in cells cultured in high serum (Fig. 2A, e and f). bHLH proteins have been shown to be posttranslationally modified in terms of phosphorylation and dephosphorylation (30, 61). Granular localization in the cytoplasm may result from phosphorylation that may be mediated by a kinase activated upon low-serum culture. It has been shown in yeast that phosphorylation alters the localization of bHLH protein Pho4 (45). There is a possibility that Twist in the cytoplasm blocks nuclear import of other bHLH factors. The bHLH inhibitor I-mfa binds to MyoD family members and blocks their nuclear import (32). Heterodimerization of bHLH proteins has been proposed to take place in the cytoplasm prior to nuclear import (21). An increase in the amount of Twist due to low-serum culture may make the effect complete.
Without affecting the expression of genes participating in proliferation, such as the E2F-1, E2F-2, cyclin E, and myc genes (data not shown), Twist may make cells remain in an undifferentiated state by regulating p21 expression, thereby increasing the proliferation rate, and may have a role in the genesis of some tumors. This is consistent with the fact that enhanced expression of Twist is observed in rhabdomyosarcomas, which originate from undifferentiated mesenchymal cells (36).
The present study paves the way for analyzing the molecular etiology of craniosynostosis. Craniosynostosis syndromes are characterized by early fusion of cranial sutures, presumably based on an increase in osteoblast activity as a result of precocious osteoblast differentiation. Saethre-Chotzen syndrome has been attributed to mutations of the gene for Twist or FGFR3, and phenotypes in patients with mutations of TWIST and FGFR3 are indistinguishable (56). The craniosynostosis induced by a loss-of-function mutation of TWIST is a haploinsufficiency phenotype (7). Craniosynostosis associated with FGFR3 with a Pro250Arg mutation is thought to be due to a gain-of-function mechanism. Inhibition of transcription by Twist would thus be greatly significant in determining the timing of initiation of osteoblast differentiation. Twist inhibits the terminal differentiation of osteoprogenitors into osteoblasts by inhibiting p21 and FGFR3 expression, while the Twist mutant fails to inhibit p21 and FGFR3 promoter activation (Fig. 5 and 7D). These observations provide evidence that Twist and FGFR3 are included in the same developmental pathway in osteoblast differentiation (Fig. 9). Mutations in TWIST could lead to a growth defect of osteoprogenitors in the early fusion of the cranial suture by decreasing proliferation activity and accelerating differentiation. Other types of FGFR molecules may also be under the transcriptional regulation by the HLH inhibitors. In addition, Twist probably functions as an inhibitor of chondrogenesis by inhibiting expression of FGFR3, which inhibits cell growth in the cartilaginous growth plates; expression of Twist and terminal differentiation markers for chondrocyte precursors are mutually exclusive (27, 58).
FIG. 9.

Regulation of p21 and FGFR3 expression by E2A, Twist, Ids, and CBP during osteoblast differentiation. Based on our results, we propose a regulatory mechanism by which expression of p21 and FGFR3 is controlled by E2A proteins, inhibitory HLH proteins, and coactivator CBP. E2A proteins are activators for both the p21 and FGFR3 genes. Twist inhibits the function of the E2A-CBP complex. The model provides the integrated regulation of the expression of p21 necessary for G1 arrest of the cell cycle and the expression of FGFR3 as a signal molecule for maturation of osteoblasts. Dysfunction of Twist and overactivation of FGFR3 signaling result in craniosynostosis because of the acceleration of bone differentiation. Twist alters the expression of FGFR3, thereby providing a direct link between the Twist and FGFR3 pathways.
Expression of FGFR3 is reported to be seen in neural crest, limb, and head mesenchyme as well as bone (19, 33, 43, 49, 69). We have shown that FGFR3 and E2A are expressed in differentiated neurons, whereas very low levels of FGFR3 and E2A were detected in undifferentiated cells of the ventricular zone (Fig. 8A). In addition, FGFR3 functions in the neuronal differentiation of PC12 cells (66). Some Saethre-Chotzen syndrome patients show not only synostosis of the coronal suture but also limb abnormalities (brachydactyly and cutaneous syndactyly), mental retardation, conductive deafness, and cleft plates. These suggest that Twist and FGFR3 are presumably implicated in the development of the epithelial cells and neurons other than osteoblasts.
However it seems unlikely that all cells expressing p21 express FGFR3. Indeed we observed that the FGFR3 promoter was not activated in rat embryonic fibroblasts, in contrast to promoters of the MCK genes (data not shown). It is intriguing to note that E2A overexpression induces p21 gene expression to a greater degree than that of the FGFR3 gene (Fig. 3A versus 7A). We thus speculate that there is a difference in gene expression between p21 and FGFR3 in terms of transcriptional regulation.
In a broader context, our study suggests that HLH factors and coactivator CBP play multiple roles in development via the cell cycle and differentiation of osteoblasts. The determination of components of these developmental pathways could lead to the identification of additional candidate genes for different genetic diseases.
ACKNOWLEDGMENTS
We thank C. Murre for the E12 and E47 clones, H. Hara for the Id1, Id2, and del E12 (aa 508 to 654) clones, E. Lees for the cosmid clone pc385.12 containing the human FGFR3 gene, B. Vogelstein for the p21 promoter, T. Nakajima for the expression plasmids for CBP, and E. Bingman for the hVDR clones. We are also indebted to T. Gridley, H. Hara, and H. Funato for critical reading and useful suggestions.
This study was supported in part by Grants-in-Aid for Scientific Research and Cancer Research from the Ministry of Education, Science, Sports and Culture of Japan, by a grant from the Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Corp., and by the Exceptional Research Subsidy from Tokyo Medical and Dental University.
REFERENCES
- 1.Aronheim A, Shiran R, Rosen A, Walker M D. The E2A gene product contains two separable and functionally distinct transcription activation domains. Proc Natl Acad Sci USA. 1993;90:8063–8067. doi: 10.1073/pnas.90.17.8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bannister A J, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 3.Bate M, Rushton E, Currie D A. Cells with persistent twist expression are the embryonic precursors of adult muscles in Drosophila. Development. 1991;113:79–89. doi: 10.1242/dev.113.1.79. [DOI] [PubMed] [Google Scholar]
- 4.Benezra R, Davis R L, Lockshon D, Turner D L, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 5.Blackwell T K, Weintraub H. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science. 1990;250:1104–1110. doi: 10.1126/science.2174572. [DOI] [PubMed] [Google Scholar]
- 6.Bonewald L F, Kester M B, Schwartz Z, Swain L D, Khare A, Johnson T L, Leach R J, Boyan B D. Effects of combining transforming growth factor b and 1,25-dihydroxyvitamin D3 on differentiation of a human osteosarcoma (MG-63) J Biol Chem. 1992;267:8943–8949. [PubMed] [Google Scholar]
- 7.Bourgeois P, Bolcato B A, Danse J M, Bloch Z A, Yoshiba K, Stoetzel C, Perrin S F. The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Hum Mol Genet. 1998;7:945–957. doi: 10.1093/hmg/7.6.945. [DOI] [PubMed] [Google Scholar]
- 8.Boyan B D, Schwartz Z, Bonewald L F, Swain L D. Localization of 1,25-(OH)2D3-responsive alkaline phosphatase in osteoblast-like cells (ROS 17/2.8, MG 63, and MC 3T3) and growth cartilage cells in culture. J Biol Chem. 1989;264:11879–11886. [PubMed] [Google Scholar]
- 9.Chandar N, Billig B, McMaster J, Novak J. Inactivation of p53 gene in human and murine osteosarcoma cells. Br J Cancer. 1992;65:208–214. doi: 10.1038/bjc.1992.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Adar R, Yang X, Monsonego E O, Li C, Hauschka P V, Yayon A, Deng C X. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Investig. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrivia J C, Kwok R P, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 12.Cserjesi P, Brown D, Lyons G E, Olson E N. Expression of the novel basic helix-loop-helix gene eHAND in neural crest derivatives and extraembryonic membranes during mouse development. Dev Biol. 1995;170:664–678. doi: 10.1006/dbio.1995.1245. [DOI] [PubMed] [Google Scholar]
- 13.Datto M B, Li Y, Panus J F, Howe D J, Xiong Y, Wang X F. Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donehower L A, Harvey M, Slagle B L, McArthur M J, Montgomery C A, Jr, Butel J S, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 15.Dulic V, Kaufmann W K, Wilson S J, Tlsty T D, Lees E, Harper J W, Elledge S J, Reed S I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 16.Eckner R, Yao T P, Oldread E, Livingston D M. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 1996;10:2478–2490. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- 17.el-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 18.el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin A L, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- 19.Fuchtbauer E M. Expression of M-twist during postimplantation development of the mouse. Dev Dyn. 1995;204:316–322. doi: 10.1002/aja.1002040309. [DOI] [PubMed] [Google Scholar]
- 20.Funato N, Moriyama K, Saitoh M, Baba Y, Ichijo H, Kuroda T. Evidence for apoptosis signal-regulating kinase 1 in the regenerating palatal epithelium upon acute injury. Lab Investig. 1998;78:477–483. [PubMed] [Google Scholar]
- 21.Goldfarb A N, Lewandowska K. Nuclear redirection of a cytoplasmic helix-loop-helix protein via heterodimerization with a nuclear localizing partner. Exp Cell Res. 1994;214:481–485. doi: 10.1006/excr.1994.1285. [DOI] [PubMed] [Google Scholar]
- 22.Halevy O, Novitch B G, Spicer D B, Skapek S X, Rhee J, Hannon G J, Beach D, Lassar A B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 23.Hamamori Y, Sartorelli V, Ogryzko V, Puri P L, Wu H Y, Wang J Y, Nakatani Y, Kedes L. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell. 1999;96:405–413. doi: 10.1016/s0092-8674(00)80553-x. [DOI] [PubMed] [Google Scholar]
- 24.Hamamori Y, Wu H Y, Sartorelli V, Kedes L. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol Cell Biol. 1997;17:6563–6573. doi: 10.1128/mcb.17.11.6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara E, Hall M, Peters G. Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J. 1997;16:332–342. doi: 10.1093/emboj/16.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 27.Hebrok M, Wertz K, Fuchtbauer E M. M-twist is an inhibitor of muscle differentiation. Dev Biol. 1994;165:537–544. doi: 10.1006/dbio.1994.1273. [DOI] [PubMed] [Google Scholar]
- 28.Howard T D, Paznekas W A, Green E D, Chiang L C, Ma N, Ortiz de Luna R I, Garcia D C, Gonzalez R M, Kline A D, Jabs E W. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- 29.Hu J S, Olson E N, Kingston R E. HEB, a helix-loop-helix protein related to E2A and ITF2 that can modulate the DNA-binding ability of myogenic regulatory factors. Mol Cell Biol. 1992;12:1031–1042. doi: 10.1128/mcb.12.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson S E, Wang X, Hardy S, Taparowsky E J, Konieczny S F. Casein kinase II increases the transcriptional activities of MRF4 and MyoD independently of their direct phosphorylation. Mol Cell Biol. 1996;16:1604–1613. doi: 10.1128/mcb.16.4.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawasaki H, Eckner R, Yao T P, Taira K, Chiu R, Livingston D M, Yokoyama K K. Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature. 1998;393:284–289. doi: 10.1038/30538. [DOI] [PubMed] [Google Scholar]
- 32.Kraut N, Snider L, Chen C M, Tapscott S J, Groudine M. Requirement of the mouse I-mfa gene for placental development and skeletal patterning. EMBO J. 1998;17:6276–6288. doi: 10.1093/emboj/17.21.6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubota Y, Ito K. Chemotactic migration of mesencephalic neural crest cells in the mouse. Dev Dyn. 2000;217:170–179. doi: 10.1002/(SICI)1097-0177(200002)217:2<170::AID-DVDY4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Lassar A B, Davis R L, Wright W E, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 35.Lee M S, Lowe G N, Strong D D, Wergedal J E, Glackin C A. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem. 1999;75:566–577. doi: 10.1002/(sici)1097-4644(19991215)75:4<566::aid-jcb3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 36.Maestro R, Dei Tos A P, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach D H, Hannon G J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999;13:2207–2217. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Missero C, Calautti E, Eckner R, Chin J, Tsai L H, Livingston D M, Dotto G P. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc Natl Acad Sci USA. 1995;92:5451–5455. doi: 10.1073/pnas.92.12.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyamoto A, Cui X, Naumovski L, Cleary M L. Helix-loop-helix proteins LYL1 and E2a form heterodimeric complexes with distinctive DNA-binding properties in hematolymphoid cells. Mol Cell Biol. 1996;16:2394–2401. doi: 10.1128/mcb.16.5.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray S S, Glackin C A, Winters K A, Gazit D, Kahn A J, Murray E J. Expression of helix-loop-helix regulatory genes during differentiation of mouse osteoblastic cells. J Bone Miner Res. 1992;7:1131–1138. doi: 10.1002/jbmr.5650071004. [DOI] [PubMed] [Google Scholar]
- 40.Murre C, McCaw P S, Vaessin H, Caudy M, Jan L Y, Jan Y N, Cabrera C V, Buskin J N, Hauschka S D, Lassar A B. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 41.Mutoh H, Naya F J, Tsai M J, Leiter A B. The basic helix-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes Dev. 1998;12:820–830. doi: 10.1101/gad.12.6.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakajima T, Fukamizu A, Takahashi J, Gage F H, Fisher T, Blenis J, Montminy M R. The signal-dependent coactivator CBP is a nuclear target for pp90RSK. Cell. 1996;86:465–474. doi: 10.1016/s0092-8674(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 43.Noji S, Koyama E, Myokai F, Nohno T, Ohuchi H, Nishikawa K, Taniguchi S. Differential expression of three chick FGF receptor genes, FGFR1, FGFR2 and FGFR3, in limb and feather development. Prog Clin Biol Res. 1993;383B:645–654. [PubMed] [Google Scholar]
- 44.Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 45.O'Neill E M, Kaffman A, Jolly E R, O'Shea E K. Regulation of PHO4 nuclear localization by the PHO80-PHO85 cyclin-CDK complex. Science. 1996;271:209–212. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- 46.Parker S B, Eichele G, Zhang P, Rawls A, Sands A T, Bradley A, Olson E N, Harper J W, Elledge S J. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- 47.Perez-Castro A V, Wilson J, Altherr M R. Genomic organization of the human fibroblast growth factor receptor 3 (FGFR3) gene and comparative sequence analysis with the mouse Fgfr3 gene. Genomics. 1997;41:10–16. doi: 10.1006/geno.1997.4616. [DOI] [PubMed] [Google Scholar]
- 48.Pesce S, Benezra R. The loop region of the helix-loop-helix protein Id1 is critical for its dominant negative activity. Mol Cell Biol. 1993;13:7874–7880. doi: 10.1128/mcb.13.12.7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- 50.Peverali F A, Ramqvist T, Saffrich R, Pepperkok R, Barone M V, Philipson L. Regulation of G1 progression by E2A and Id helix-loop-helixproteins. EMBO J. 1994;13:4291–4301. doi: 10.1002/j.1460-2075.1994.tb06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piatti S, Bohm T, Cocker J H, Diffley J F, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- 52.Prabhu S, Ignatova A, Park S T, Sun X H. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888–5896. doi: 10.1128/mcb.17.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puri P L, Avantaggiati M L, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu Y, Sharma A, Stein R. p300 mediates transcriptional stimulation by the basic helix-loop-helix activators of the insulin gene. Mol Cell Biol. 1998;18:2957–2964. doi: 10.1128/mcb.18.5.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rice D P, Aberg T, Chan Y, Tang Z, Kettunen P J, Pakarinen L, Maxson R E, Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- 56.Rose C S, Patel P, Reardon W, Malcolm S, Winter R M. The TWIST gene, although not disrupted in Saethre-Chotzen patients with apparently balanced translocations of 7p21, is mutated in familial and sporadic cases. Hum Mol Genet. 1997;6:1369–1373. doi: 10.1093/hmg/6.8.1369. [DOI] [PubMed] [Google Scholar]
- 57.Rose M, Botstein D. Construction and use of gene fusions to lacZ (β-galactosidase) that are expressed in yeast. Methods Enzymol. 1983;101:167–180. doi: 10.1016/0076-6879(83)01012-5. [DOI] [PubMed] [Google Scholar]
- 58.Sandell L J, Morris N, Robbins J R, Goldring M B. Alternatively spliced type II procollagen mRNAs define distinct populations of cells during vertebral development: differential expression of the amino-propeptide. J Cell Biol. 1991;114:1307–1319. doi: 10.1083/jcb.114.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheikh M S, Li X S, Chen J C, Shao Z M, Ordonez J V, Fontana J A. Mechanisms of regulation of WAF1/Cip1 gene expression in human breast carcinoma: role of p53-dependent and independent signal transduction pathways. Oncogene. 1994;9:3407–3415. [PubMed] [Google Scholar]
- 61.Sloan S R, Shen C P, McCarrick-Walmsley R, Kadesch T. Phosphorylation of E47 as a potential determinant of B-cell-specific activity. Mol Cell Biol. 1996;16:6900–6908. doi: 10.1128/mcb.16.12.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spicer D B, Rhee J, Cheung W L, Lassar A B. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- 63.Steinman R A, Hoffman B, Iro A, Guillouf C, Liebermann D A, el-Houseini M E. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene. 1994;9:3389–3396. [PubMed] [Google Scholar]
- 64.Sun X H, Baltimore D. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell. 1991;64:459–470. doi: 10.1016/0092-8674(91)90653-g. [DOI] [PubMed] [Google Scholar]
- 65.Thisse B, Stoetzel C, Gorostiza T C, Perrin S F. Sequence of the twist gene and nuclear localization of its protein in endomesodermal cells of early Drosophila embryos. EMBO J. 1988;7:2175–2183. doi: 10.1002/j.1460-2075.1988.tb03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thompson L M, Raffioni S, Wasmuth J J, Bradshaw R A. Chimeras of the native form or achondroplasia mutant (G375C) of human fibroblast growth factor receptor 3 induce ligand-dependent differentiation of PC12 cells. Mol Cell Biol. 1997;17:4169–4177. doi: 10.1128/mcb.17.7.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ura K, Kurumizaka H, Dimitrov S, Almouzni G, Wolffe A P. Histone acetylation: influence on transcription, nucleosome mobility and positioning, and linker histone-dependent transcriptional repression. EMBO J. 1997;16:2096–2107. doi: 10.1093/emboj/16.8.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell T K, Turner D, Rupp R, Hollenberg S. The myoD gene family: nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- 69.Wolf C, Thisse C, Stoetzel C, Thisse B, Gerlinger P, Perrin S F. The M-twist gene of Mus is expressed in subsets of mesodermal cells and is closely related to the Xenopus X-twi and the Drosophila twist genes. Dev Biol. 1991;143:363–373. doi: 10.1016/0012-1606(91)90086-i. [DOI] [PubMed] [Google Scholar]
- 70.Zhang M Y, Clawson G A, Olivieri N F, Bell L L, Begley C G, Miller B A. Expression of SCL is normal in transfusion-dependent Diamond-Blackfan anemia but other bHLH proteins are deficient. Blood. 1997;90:2068–2074. [PubMed] [Google Scholar]
- 71.Zhuang Y, Kim C G, Bartelmez S, Cheng P, Groudine M, Weintraub H. Helix-loop-helix transcription factors E12 and E47 are not essential for skeletal or cardiac myogenesis, erythropoiesis, chondrogenesis, or neurogenesis. Proc Natl Acad Sci USA. 1992;89:12132–12136. doi: 10.1073/pnas.89.24.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]