

Abstract
The intermediate filament cytoskeleton is composed of keratins in all epithelial cells and imparts mechanical integrity to these cells. However, beyond this shared function, the functional significance of the carefully regulated tissue- and differentiation-specific expression of the large keratin family of cytoskeletal proteins remains unclear. We recently demonstrated that expression of keratin K10 or K16 may regulate the phosphorylation of the retinoblastoma protein (pRb), inhibiting (K10) or stimulating (K16) cell proliferation (J. M. Paramio, M. L. Casanova, C. Segrelles, S. Mittnacht, E. B. Lane, and J. L. Jorcano, Mol. Cell. Biol. 19:3086–3094, 1999). Here we show that keratin K10 function as a negative modulator of cell cycle progression involves changes in the phosphoinositide 3-kinase (PI-3K) signal transduction pathway. Physical interaction of K10 with Akt (protein kinase B [PKB]) and atypical PKCζ causes sequestration of these kinases within the cytoskeleton and inhibits their intracellular translocation. As a consequence, the expression of K10 impairs the activation of PKB and PKCζ. We also demonstrate that this inhibition impedes pRb phosphorylation and reduces the expression of cyclins D1 and E. Functional and biochemical data also demonstrate that the interaction between K10 and these kinases involves the non-α-helical amino domain of K10 (NTerm). Together, these results suggest new and essential roles for the keratins as modulators of specific signal transduction pathways.
Keratins form the intermediate filament cytoskeleton of all epithelial cells. Although it has been demonstrated that this structure is essential for providing cell resilience in epithelia (11, 13), there is little information on individual keratin-specific functions that can account for the complex cell type- and differentiation-specific expression patterns observed in this protein family. Keratin expression is highly regulated in the epidermis. Proliferative basal cells express the keratin pair comprising K5 and K14 (K5-K14 pair). However, when keratinocytes begin terminal differentiation, they move upward, become postmitotic, and switch to the expression of keratin pair K1-K10. Under hyperproliferative conditions (e.g., tumors and wound healing), keratinocytes downregulate K1-K10 and express K6-K16. We recently showed that the ectopic expression of keratin K10 inhibits the proliferation of human keratinocytes, while K16 expression stimulates the process (23). In agreement with this, K10 impairs skin tumor development when ectopically expressed in transgenic mice (29) and K16 overexpression, or ectopic expression, in transgenic mice leads to aberrant epidermal keratinization and hyperproliferation (19, 30).
Keratin K10-induced inhibition of cell proliferation occurs through a process linked to the retinoblastoma protein (pRb) and the molecular machinery controlling cell cycle progression during G1 (23). The different cellular distributions of keratins (cytoplasmic) and pRb (nuclear) nonetheless suggest that this effect must take place through the K10-mediated impairment of a pathway leading to the functional inactivation of pRb rather than by the direct physical interaction of these two molecules. We selected the ras oncogene-dependent pathway on the basis of a number of findings that imply that K10 and ras could have antagonistic functions in keratinocyte proliferation and differentiation. First, Ha-ras signaling has been implicated in the control of cell cycle progression in a pRb-dependent manner (18, 28), similar to that described for K10 (23). Second, whereas mutations in Ha-ras are early and essential events in mouse skin carcinogenesis protocols (3), K10 is lost during early stages of tumor development. Third, although K10 is expressed in postmitotic, terminally differentiating epidermal cells, transgenic mice expressing a mutant Ha-ras gene from a K10 promoter develop generalized hyperkeratosis of the skin as well as skin tumors (2). Since ras-dependent mitogenic signals diverge through different pathways (see for reviews references 7 and 17), such as those for small GTPases, raf, and phosphoinositide 3-kinase (PI-3K), we studied which of these pathways is specifically affected by K10 expression. The collected data clearly demonstrate that keratin K10 impairs cell cycle progression through the sequestration and inhibition of protein kinase B (PKB; Akt), and atypical PKCζ.
MATERIALS AND METHODS
Plasmids and transfections.
The plasmids coding for keratins K1, K10, and K16 have previously been described (23). Plasmids coding for wild-type (wt) and dominant-negative activated Ha-ras, v-raf, and PKCζ were provided by J. Moscat (Centro de Biologia Molecular, Madrid, Spain). Those plasmids coding for activated forms of RhoA, Rac1, and Cdc42Hs were kindly provided by J. C. Lacal (Instituto de Investigaciones Biomédicas, Madrid, Spain), while those coding for wt and dominant-negative Akt and p110CAAX were from J. Downward (Imperial Cancer Research Fund, London, United Kingdom). The plasmid coding for PDK1 was provided by K. Anderson (Babraham Institute, Cambridge, United Kingdom). Hemagglutinin (HA)-tagged forms of Akt and PKCζ, as well as myrAkt, were provided by J. S. Gutkind (National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, Md.). Plasmid pCDNApRb coding for human wt pRb cDNA (a generous gift from S. Mittnacht) under the control of the cytomegalovirus (CMV) promoter has been previously described (22, 23). pVM6NTermK10 (NTerm) was generated by inserting the HindIII-SacI fragment coding for NTerm (23) into the pVM6 vector (Boehringer Mannheim) in frame with the vesicular stomatitis virus G (VSV-G) tag under the control of the CMV promoter. pRasNTerm was generated by inserting the NTerm-encoding fragment in frame with the sequence encoding Ha-ras at the carboxyl terminus-encoding sequence of pRas[61]ΔF (6). The integrity of the constructs was confirmed by sequencing. Transfections using calcium phosphate or Fugene 6 (Boehringer Mannheim) were performed as previously described (22–27). After selection for 15 to 25 days in the presence of appropriate antibiotics (G418 [0.5 mg/ml] or hygromycin [0.1 mg/ml] or both for cotransfections) colony-forming efficiency experiments were performed at least in triplicate (22–24). Unless otherwise indicated, cells were cotransfected with the same quantity of plasmids coding for K10 and for the different signaling molecules (5 μg of each per p90 dish). The total DNA amount was kept constant in all the transfection experiments, including appropriate amounts of pcDNA3 empty vector. The relative number of colonies was calculated, considering as 100% the number of colonies obtained with empty vectors in each cell line. Data are shown as means ± standard deviations (SD).
Cell culture and immunofluorescence.
HaCaT, PB, and MCA3D keratinocytes and C33A and BMGE+H cells were cultured as previously described (21–25). For immunofluorescence analysis after transfection, cells were cultured, fixed, and stained as described previously (22–24) using antibodies against keratin K10 (undiluted supernatants of mouse monoclonal antibody [MAb:] LH1, LH2, or LH3 or 1/40-diluted K8.60 mouse MAb) (21–25). PKCζ (Santa Cruz Biotechnology or Boehringer Mannheim; both rabbit polyclonal antibodies used at 1/200 and 1/400 dilutions, respectively), Akt (Upstate Biotechnology or Santa Cruz Biotechnology; both goat polyclonal antibodies used at 1/250 and 1/300 dilutions, respectively), phosphorylated Akt (Upstate Biotechnology or BioLabs; both rabbit polyclonal antibodies used at 1/200 and 1/100 dilutions, respectively), and a mouse MAb against VSV-G tag diluted 1/100 (Boehringer Mannheim). Controls, including incubation with preimmune serum, omitting the primary antibody, and preincubation with the immunizing peptide, were routinely performed. Secondary antibodies specific for multiple-labeling purposes were purchased from Jackson Immunoresearch Labs and used as described previously (21–25). Bromodeoxyuridine (BrdU) incorporation and terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays were performed essentially as previously described (23, 24) using a rat MAb against BrdU (23) and a fluorescein isothiocyanate (FITC)-labeled cell death detection kit (Boehringer Mannheim) in parallel with K10 immunofluorescence (1/40 dilution of antibody K8.60). Specimens were analyzed using a Bio-Rad C600 confocal microscope or an Axiophot conventional immunofluorescence microscope (22–27). Experiments were performed in triplicate; at least 200 transfected cells were scored in each. For triple immunofluorescence, AMCA-labeled antimouse, Texas-red-conjugated antigoat, and FITC-conjugated antirabbit antibodies were simultaneously used.
Immunoprecipitation, immunoblotting, and kinase assays.
For coimmunoprecipitation, cell extracts were obtained in phosphoprotein buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4, 1 μg of leupeptin/ml, 1 μg of aprotinin/ml, 1 mM phenylmethylsulfonyl fluoride). Protein extracts (50 μg in 500 μl of phosphoprotein buffer) were incubated with agitation (overnight at 4°C) in the presence of 2 μl of antibodies against K10 (mouse MAb K8.60), Akt and PKCζ (goat and rabbit polyclonal antibodies, respectively, from Santa Cruz Biotechnology), or anti-VSV-G tag (mouse MAb from Boehringer Mannheim). Soluble and keratin-enriched fractions were obtained essentially as described previously (25). Total protein extracts, cytoskeletal enriched fractions, and immunoprecipitates were analyzed by Western blotting as described previously (22–27) using the same antibodies against Akt, PKCζ, phosphorylated Akt, and VSV-G tag described in “Cell culture and immunofluorescence.” Mouse MAb LH3 was used to detect simultaneously K10 and ΔNΔC. Antibodies against pRb, cycD1, and cycE have been previously described (22, 23). LI0025 was used to detect human K16. The antibodies used in immunoblotting were diluted 1/1,000 to 1/10,000 in Tris-buffered saline containing 0.5% bovine serum albumin and 0.05% Tween 20. To normalize loading, immunoblots were stained with Ponceau S prior to antibody incubations. The kinase activities of Akt and PKCζ from HaCaT cells cotransfected with 5 μg of K10 or K16 and 2 μg of HA epitope-tagged Akt or PKCζ were determined upon immunoprecipitation with HA-specific MAb 12CA5 (Babco), using histone 2B (H2B) or myelin basic protein (MBP) as the substrate. Briefly, cells were washed once in cold phosphate-buffered saline and lysed on ice with 1 ml of lysis buffer containing protease and phosphatase inhibitors (1% Triton X-100, 10% glycerol, 137 mM NaCl, 20 mM Tris-HCl [pH 7.5], 1 μg of aprotinin and leupeptin/ml, 1 mM phenylmethylsulfonyl fluoride, 20 mM NaF, 1 mM disodium pyrophosphate, and 1 mM Na3VO4). After samples were precleared by centrifugation, lysates were immunoprecipitated with 1 μl of an anti-HA MAb using γ-binding beads (Amersham Pharmacia Biotech) to sediment immunocomplexes. After three 1-ml washes with lysis buffer, one 1-ml wash with water, and one 1-ml wash with kinase buffer (20 mM HEPES [pH 7.4], 10 mM MgCl2, 10 mM MnCl2), reactions were performed (30 min at 25°C) in 30 μl of kinase buffer containing 0.05 mg of the appropriate substrate/ml, 5 μM ATP, 1 mM dithiothreitol, and 10 μCi of [γ-32P]ATP. The products of the kinase reactions were fractionated in sodium dodecyl sulfate–15% polyacrylamide gels and radiographically exposed. In some cases, cells were serum starved overnight prior to lysis. When necessary, the same membranes were subsequently probed by Western blotting using the mouse anti-HA (Babco, 1:500) or AE1 (23) MAb to assess the expression level of cotransfected proteins.
Two-cell hybrid experiments.
The assay of the interaction between myrAkt and NTerm by the two-cell hybrid approach was performed using the ras rescue system, a modification of the CytoTrap two-hybrid system (Stratagene), essentially as described previously (6). Briefly, yeast temperature-sensitive cdc25-2 cells were cotransformed with the plasmids shown in Fig. 8B. Transformants were cultured on selectable plates and incubated at 25 or 37°C for 5 days. Positive interaction was determined by growth at 37°C in galactose-containing plates. Negative controls included plates with minimal galactose and plates with glucose (not shown).
FIG. 8.

NTerm interacts with Akt in vitro and in vivo. (A) Immunoprecipitation using an anti-VSV-G MAb of extracts from pooled clones of VSV-G-tagged NTerm- and vector-transfected HaCaT cells, followed by Western blotting against VSV-G, Akt, and PKCζ showing that both Akt and PKCζ coprecipitate with NTerm. (B) Two-cell hybrid experiments using the ras rescue system (6). cdc25-2 yeast cells were cotransformed with the indicated plasmids and plated at the permissive (25°C) or the restrictive (37°C) temperature onto galactose-containing plates. Note that, besides the positive controls including RasCAAX expression (1 and 3) and pMyrMAFB plus pSOSMAFB expression (6), growth at the restrictive temperature was obtained with the cotransformation of pMyrAkt plus RasNTerm (4), indicating that protein-protein interaction between myrAkt and NTerm has occurred. The absence of growth in 2 demonstrates that NTerm by itself cannot override the mutation of cdc25-2 yeast cells. As a negative control, a myristoylated form of phosduccin D (PhosD) was cotransformed with pRasNTerm (5). No colonies were observed at 37°C in glucose-containing, galactose-depleted plates, indicating that myrAkt alone does not allow yeast cell growth (not shown).
RESULTS
The K10-induced inihibition of cell proliferation is reversed by Akt and PKCζ.
We have previously demonstrated that the expression of keratin K10, but not a mutant K10 lacking both amino and carboxyl ends (ΔNΔC), induces growth arrest in human HaCaT keratinocytes (23). Although we have also demonstrated that this effect is independent of p53 (23), given that this protein is mutated in HaCaT cells, we analyzed whether keratin K10 was able to inhibit cell growth in other cell types. Figure 1A shows that keratin K10 also compromises cell growth in mouse skin keratinocytes (MCA3D and PB cells) and bovine mammary gland (BMGE+H) cells. However, in agreement with our previous results (23), this effect was not observed in human cervical carcinoma C33A cells lacking functional pRb (Fig. 1A).
FIG. 1.

Keratin-induced modulation of cell proliferation is related to signaling effectors belonging to the PI-3K pathway. (A) Keratin K10 (open bars) but not the ΔNΔC form, which lacks amino and carboxyl termini (solid bars), inhibits cell growth in a variety of epithelial cell lines including human (HaCaT) and mouse (MCA3D and PB) keratinocytes and bovine mammary gland cells (BMGE+H), but not in Rb-deficient human cervical carcinoma (C33A) cells. (B) Keratin K10 also mediates inhibition of HaCaT cell growth when coexpressed with human keratin K1, its natural partner in suprabasal, differentiating keratinocytes of the epidermis (see the introduction). (C) Keratin K10-induced HaCaT keratinocyte cell growth arrest is rescued by coexpression of Ha-ras, PDK1, Akt, and PKCζ but not by v-raf, RhoA, Rac1, Cdc42, or the activated catalytic subunit of PI-3K (p110CAAX). (D) Western blot using mouse MAb K8.60 (1/10,000 diluted) in lysates of cells cotransfected with K10 and the cited plasmids, demonstrating that the K10 expression levels in these transfections are similar. (E) Colony-forming experiments showing that Akt rescues cell growth inhibition induced by K10 in HaCaT cells in a dose-dependent manner. As a negative control, transfection of increasing amounts of the v-raf-encoding plasmid does not significantly improve the ability of this protein to reverse the K10 inhibitory effect. (F) Akt and PKCζ functions are necessary to allow normal growth of HaCaT keratinocytes as demonstrated by the reduction in the number of colonies obtained after transfection with 10 μg of dominant-negative forms of Akt or PKCζ (DNAkt and DNPKCζ, respectively) relative to the numbers from similar transfection experiments with an empty vector (pcDNA3). A dominant-negative form of Ha-ras (rasN17) was used as a positive control for cell growth inhibition. (G) Transfection of keratin K10 inhibits BrdU incorporation (solid bars) but not apoptosis (TUNEL assay; open bars) in HaCaT. (H) Western blots from protein extracts of pRb-deficient C33A cells transiently cotransfected with K10 or its inactive mutant ΔNΔC, pRb, and p110CAAX, Akt, PKCζ, or PDK1, demonstrating that K10-induced repression of pRb hyperphosphorylation and repression of cyclin D1 and E expression are restored by coexpression of PDK1, Akt, or PKCζ but not by p110CAAX. Numbers, apparent molecular weights (in thousands). Data in panels A to C and E to G are from at least triplicate independent experiments and are shown as means ± SD.
Keratin K10 is coexpressed in the epidermis with keratin K1, with which it associates to form the intermediate filaments characteristic of differentiating keratinocytes. Figure 1B demonstrates that K10-induced cell growth arrest cannot be attributed to the expression of this keratin in the absence of its natural partner, K1, since K1-plus-K10 cotransfection showed a similar or even stronger inhibitory effect (Fig. 1B).
To study the mechanism by which K10 alters the cell cycle, we performed rescue experiments in which effectors belonging to the ras signaling pathway were cotransfected with K10 by employing a colony-forming assay previously used to establish the K10 antiproliferative effect (23). Cotransfection of Ha-ras (V12), PDK-1, Akt, or PKCζ rescued the inhibitory effect of K10, whereas v-raf, RhoA, Rac1, Cdc42, and p110CAAX (a permanently active PI-3K catalytic subunit) had only a partial effect (Fig. 1C). Parallel experiments showed that K10 was expressed at similar levels in these cotransfections, indicating that the effect observed was not due to differences in the level of transfected-K10 expression (Fig. 1D). Equal amounts of the plasmids coding for K10 and for the different effector proteins (5 μg) were employed in these experiments. To confirm the specificity of these rescues, colony-forming experiments in which increasing amounts of Akt or v-raf were cotransfected in HaCaT cells with a fixed amount of K10 were performed. The results showed that Akt rescues the inhibitory effect of K10 in a dose-dependent manner (Fig. 1E). In contrast, and as expected, increasing the amount of v-raf-encoding plasmid did not significantly change the capacity of this kinase to rescue K10-induced cell growth inhibition. The fact that Akt and PKCζ, but not the active form of PI-3K, rescued the growth inhibition promoted by K10 expression suggests that this inhibition acts at the level of these downstream kinases rather than affecting the overall PI-3K-dependent signaling. In addition, these results indicate that the activity of Akt and PKCζ is required for normal proliferation in HaCaT cells. To test this, colony-forming experiments were performed with HaCaT cells transfected with the empty vector (as a control) or with dominant-negative forms of Akt (DNAkt) or PKCζ (DNPKCζ). We also included a dominant-negative form of ras (rasN17) as a positive control of growth inhibition. It was found that both Akt and PKCζ activities are required for HaCaT cell proliferation since the dominant-negative forms of these kinases inhibited colony formation to an extent similar to the extent to which it was inhibited by rasN17 (Fig. 1F).
We have previously shown that keratin K10 expression inhibits cell cycle progression by decreasing cyclin D1 expression, thus impairing pRb phosphorylation (23). However, given the well-established role of Akt in the protection of cells against apoptosis, we studied the possible induction of apoptosis in keratin K10-transfected HaCaT cells by TUNEL analysis. In parallel, we analyzed the capacity of these transfected cells to incorporate BrdU. The results demonstrate that K10-expressing cells display a strong inhibition of BrdU incorporation (Fig. 1G), whereas their degree of apoptosis was similar to that of nontransfected cells (Fig. 1G). These data clearly confirm the previous conclusion that K10 expression causes cell cycle arrest and indicate that, in the absence of additional external stimuli, K10 expression cannot induce apoptosis by itself.
Finally, we studied whether cell growth rescue promoted by PI-3K signaling intermediates reverses these effects on the cell cycle machinery. For this, Rb-deficient C33A cells were cotransfected with plasmids encoding K10 and pRb and either p110CAAX, Akt, PKCζ, or PDK1. We previously showed that, following pRb transfection of C33A cells, this protein is efficiently phosphorylated and the endogenous cyclin D1 protein is induced (23, 24) and that these effects were suppressed by cotransfecting K10 (23). In the present work, efficient hyperphosphorylation of transfected pRb and increased levels of endogenous cyclin D1 and cyclin E were observed when K10 was cotransfected with PDK1, Akt, or PKCζ but not with p110CAAX (Fig. 1H) (see references in references 23 and 24). In addition, ΔNΔC, an inactive K10 mutant lacking both amino and carboxyl termini (23), did not inhibit pRb phosphorylation or the expression of cyclin D1 or cyclin E (Fig. 1H). Together these results demonstrate that effectors of the PI-3K signaling pathway, but not PI-3K itself, reverse the effects of K10 on proliferation and cell cycle regulatory molecules.
Unlike K10, K16 stimulates keratinocyte proliferation and reverses K10-induced growth arrest (23). To determine whether this stimulation also involves the PI-3K pathway, we analyzed the ability of transfected K16 to reverse the effect of specific inhibitors of MEK (PD098059) or PI-3K (wortmannin) (Fig. 2A). When used at defined concentrations (5 μM and 100 nM, respectively) these drugs inhibit MEK1 and PI-3K, respectively. Both caused a severe growth inhibition of vector (pcDNA3)-transfected HaCaT cells as well as nontransfected parental HaCaT keratinocytes (not shown). K16-transfected cells were also sensitive to the MEK inhibitor, but K16 appears to reverse the inhibition promoted by wortmannin. Western blot analysis showed similar K16 levels in all cases (Fig. 2B), indicating that the effects observed could not be attributed to differences in K16 expression.
FIG. 2.

K16 expression rescues the growth inhibition promoted by PI-3K inhibition. (A) Colony-forming experiments demonstrating that the growth inhibition promoted by wortmannin (Calbiochem; 100 nM), but not by MEK inhibitor PD098059 (Calbiochem; 5 μM), is efficiently rescued by K16 expression. (B) Western blot showing that the expression level of transfected K16 in cells treated with wortmannin is similar to that in cells treated with PD098059. DMSO, K16-transfected cells treated with dimethyl sulfoxide alone.
Transfected K10 interacts with endogenous Akt and PKCζ and prevents their activation in human keratinocytes.
The results showing that effectors of the PI-3K pathway, but not active PI-3K itself, reverse the K10-induced cell cycle arrest suggest that K10 may inhibit the function of such effectors. PI-3K-dependent signals induce changes in the subcellular localization of specific kinases, including Akt and PKCζ (1, 9). A possible explanation for the inhibitory effect of K10 might be that the presence of this keratin prevents such translocations. This might also explain why the overexpression of the wt forms of these kinases reverses K10-induced cell cycle arrest. To test this hypothesis, we transfected K10 into HaCaT cells and studied the localization of endogenous Akt and PKCζ by double immunofluorescence and confocal microscopy. In nontransfected cells, these kinases have a diffuse nuclear and cytoplasmic localization (Fig. 3A′, B′, E, and F). In contrast, in cells expressing transfected K10, this keratin is assembled into the endogenous keratin intermediate filaments (Fig. 3A and B) and most Akt (Fig. 3A′) and PKCζ (Fig. 3B′) clearly colocalize along these filaments. Such colocalization was not detected when HaCaT cells were transfected with cell cycle-inactive K10 mutant ΔNΔC (Fig. 3C, C′, D, and D′) although this mutant keratin is also integrated into the intermediate filaments formed by the endogenous keratins. Western blot experiments also demonstrated that this effect cannot be attributed to different expression of K10 and ΔNΔC, since these constructs are expressed to similar levels upon transfection in HaCaT cells (Fig. 3G).
FIG. 3.

Akt and PKCζ colocalize with the keratin cytoskeleton in cells transfected with K10 but not with a mutant form lacking both the amino and carboxyl termini (ΔNΔC). HaCaT cells were transiently transfected with K10 (A and B) or ΔNΔC (C and D) and analyzed by double immunofluorescence and confocal microscopy to visualize the transfected keratin (using the LH3 mouse MAb; A to D) and the endogenous Akt (using a goat polyclonal antibody; A′ and C′) or PKCζ (using a rabbit polyclonal antibody; B′ and D′). (E and F) Distribution of endogenous PKCζ (E) and Akt (F) in nontransfected HaCaT cells (see also arrows in panels A′ and B′). Note that K10, but not ΔNΔC, changes the distribution of the endogenous kinases from a dispersed to a filamentous shape, such that the distribution colocalizes with that of K10. Similar results were obtained with two different antibodies raised against different epitopes of Akt or PKCζ (not shown). Arrows (A′ and B′), nontransfected cells. Representative examples from three independent experiments are shown. (G) Western blot using MAb LH3 demonstrating that K10 and ΔNΔC are expressed to similar extents in transfected HaCaT cells (see also Fig. 7B). Bars, 10 μm.
Altered Akt and PKCζ distribution as a consequence of K10 expression suggests that K10 interferes with the translocation of these molecules to the cell membrane, thus preventing their activation by phosphorylation (1, 9, 10, 15). Double-immunofluorescence analysis of phosphorylated Akt in cells transfected with K10 or ΔNΔC showed a strong decrease in phospho-Akt in cells expressing K10 (Fig. 4A). However, in transfections with inactive mutant ΔNΔC, significant amounts of phospho-Akt were clearly observed to persist in transfected cells (Fig. 4B). Finally, to determine whether keratin expression does, in fact, inhibit the kinase activity of Akt and PKCζ, HaCaT cells were cotransfected with 2 μg of plasmids encoding HA-tagged Akt or PKCζ and 5 μg of plasmids encoding K10 or K16. The activity of the corresponding cotransfected kinase was assayed upon immunoprecipitation, exploiting the HA epitope and using H2B and MBP as substrates for Akt and PKCζ, respectively. The results (Fig. 4C and D) showed that K10 reduces the kinase activity of both Akt (Fig. 4C) and PKCζ (Fig. 4D) to levels similar to those obtained upon transfection of the kinase in serum-starved cells. In contrast, cotransfection of K16 stimulated both kinase activities (Fig. 4C and D). The transfected kinases and keratins were consistently expressed at similar levels in these experiments, as determined by Western blotting (Fig. 4C and D and data not shown).
FIG. 4.
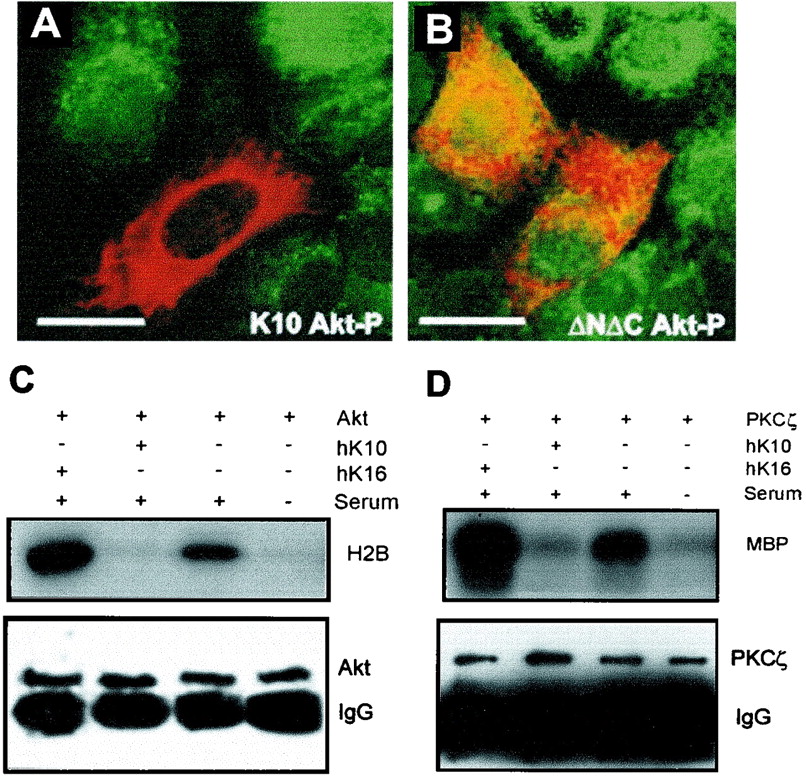
Impaired activation of PI-3K signaling effectors as a consequence of K10, but not ΔNΔC, expression. HaCaT cells synchronized in G0 by serum starvation were transfected with K10 (A) or ΔNΔC (B). After 48 h cells were allowed to reenter the cell cycle by serum addition. After 3 h, expression of the transfected construct (in red) and endogenous phosphorylated Akt (Akt-P; in green) was analyzed by indirect double immunofluorescence. The images shown are derived by merging the two immunofluorescence channels. Note that the expression of K10 leads to nondetectable Akt-P levels, whereas in cells transfected with ΔNΔC at least some Akt-P (green and yellow) is present. (C and D) Keratin expression alters Akt (C) and PKCζ (D) activities. HaCaT cells were transfected with K10 or K16 and HA-tagged Akt (C) or PKCζ (D), and the kinase activities were determined following immunoprecipitation against the HA epitope using H2B and MBP, respectively, as substrates (see Materials and Methods). The lower portions of panels C and D show anti-HA tag immunoblots, demonstrating that similar amounts of immunoprecipitated Akt and PKCζ are found in the corresponding samples. Representative examples from three independent experiments are shown. Bars, 15 μm.
Akt and PKCζ interact with K10 during keratinocyte differentiation.
The above-described results were obtained upon transfection of keratin K10 in cultured keratinocytes. To determine whether K10 also interacts with Akt and PKCζ in vivo, we analyzed the cellular distribution of these kinases during keratinocyte differentiation. Since a small proportion HaCaT cells spontaneously differentiate and induce K10 expression (4, 21, 27), we first analyzed the distribution of endogenous Akt and PKCζ in these spontaneously differentiating cells by double immunofluorescence. It was found that both Akt (Fig. 5A) and PKCζ (not shown) changed their cellular distributions and colocalized with the endogenous keratin K10-containing filaments (Fig. 5A′). To biochemically confirm these data, confluent HaCaT cultures were induced to differentiate by serum starvation as previously reported (22, 26, 27). The soluble and keratin-enriched fractions were obtained (25) and probed by Western blotting. It was observed that, in undifferentiated cultures, Akt and PKCζ were mostly present in the soluble fraction (Fig. 5B). In addition, small amounts of these enzymes were observed in the keratin-enriched, insoluble fraction, probably due to the presence of some spontaneously differentiating cells in the culture, as demonstrated by the presence of keratin K10 in this fraction (Fig. 5B). However, in differentiated HaCaT cells, both Akt and PKCζ were found in the keratin fraction. Finally, the phosphorylated, i.e., active, Akt was detected exclusively in the soluble pool of undifferentiated cells. These results demonstrate that, as observed in transfection (Fig. 3), both Akt and PKCζ interact with K10 during the differentiation of HaCaT keratinocytes.
FIG. 5.
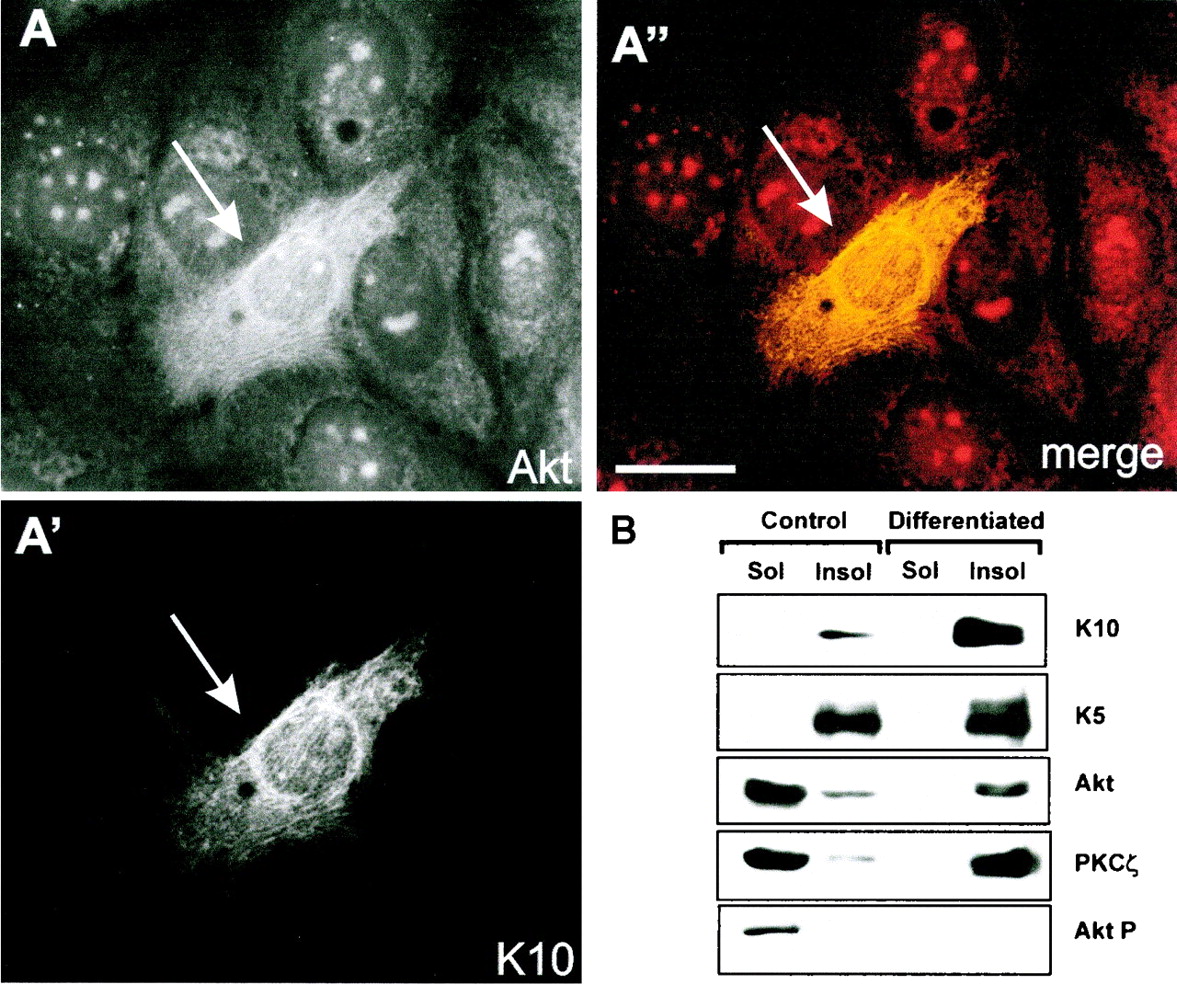
Akt and PKCζ interact with K10 during HaCaT keratinocyte differentiation. Spontaneously differentiated HaCaT keratinocytes (4, 27) were fixed and analyzed by indirect immunofluorescence to detect endogenous Akt (A) and keratin K10 (A′). Note that in differentiating cells (arrow), characterized by the presence of K10, the Akt staining pattern changes, becoming filamentous and coincident with that of keratin K10 (A′′). (B) Akt and PKCζ are associated with keratins in differentiated HaCaT keratinocyte cultures. HaCaT cells were induced to differentiate by serum starvation for 12 days (22, 26, 27), and the soluble (Sol) and keratin-enriched fractions were obtained as described previously (25) and analyzed by Western blotting using antibodies against keratin K5, keratin K10, Akt, PKCζ, and Akt-P. Soluble and keratin-enriched fractions from control nondifferentiated cultures were analyzed in parallel. Note the presence of most Akt, PKCζ, and phosphorylated Akt in the soluble fraction in nondifferentiated cultures (K10 negative), whereas in differentiated cultures both Akt and PKCζ are in the insoluble (Insol)-keratin fraction, which contains keratin K10. The K5 signal demonstrates similar loadings in the samples from control and differentiating cells.
We next studied the distribution of Akt, phospho Akt, and keratin K10 in newborn mouse skin. Triple-immunofluorescence analyses demonstrated that Akt (Fig. 6A) is present in both the basal and suprabasal compartments of mouse epidermis. Phosphorylated, active Akt was restricted to the basal proliferative layer (Fig. 6B), whereas K10, as expected, was exclusively expressed in the suprabasal nonproliferative layers (Fig. 6C). To further confirm the possible interaction between Akt and PKCζ with K10 in vivo, total skin extracts were immunoprecipitated with antibodies against Akt, PKCζ, keratin K5 (characteristic of basal cells), and keratin K10 (present in suprabasal cells) and subsequently probed by Western blotting. K10 was found in both Akt and PKCζ immunoprecipitates, and similarly both Akt and PKCζ were present in K10 immunoprecipitates but not in K5 immunoprecipitates (Fig. 6E). Phosphorylated Akt was detected in Akt and PKCζ immunoprecipitates but not in K10 immunoprecipitates (Fig. 6E), indicating that keratin K10 interacts only with inactive Akt. Finally, it is worth noting that Akt and PKCζ coprecipitate, indicating that these two kinases interact in the epidermis in vivo. Collectively, these results demonstrate that, as in the K10 transfection experiments, Akt and PKCζ interact in vivo with K10 (present in differentiating, postmitotic keratinocytes) but not with K5 (present in proliferation-competent keratinocytes) and that, at least for Akt, this interaction may prevent its activation.
FIG. 6.
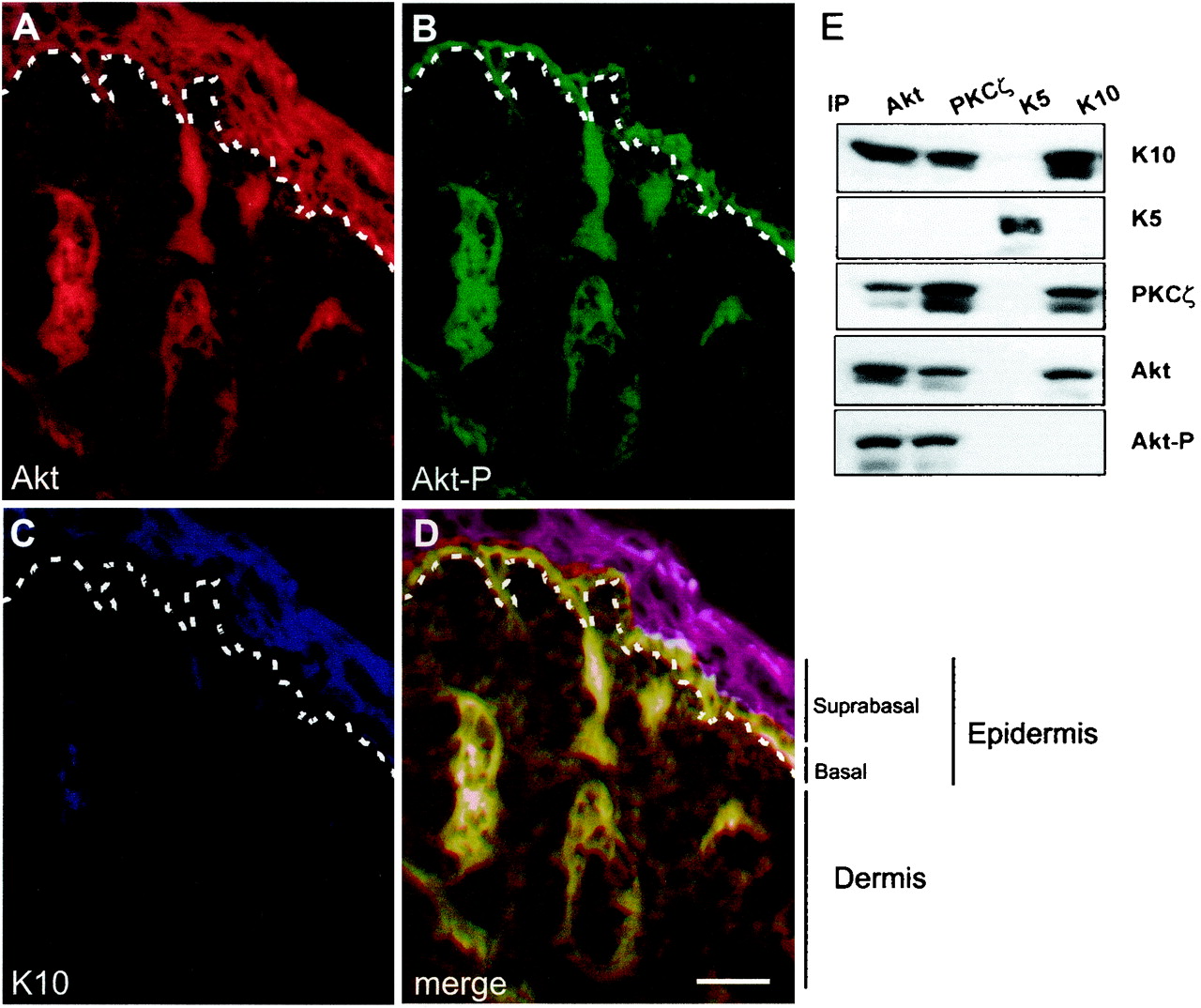
Distribution of Akt, Akt-P, and K10 in newborn mouse skin. (A to D) Frozen sections of newborn mouse skin were processed for immunofluorescence using antibodies against Akt (A), Akt-P (B), and K10 (C). The three immunofluorescence channels (Texas red, fluorescein isothiocyanate, and AMCA, respectively) are shown merged in panel D. Note that Akt is present throughout all the epidermal layers, whereas phosphorylated Akt is present exclusively in the basal layer and K10 is restricted to differentiated suprabasal layers. This shows that Akt and K10 are coexpressed in suprabasal cells but that only basal, K10-negative cells have active, phosphorylated Akt. Dashed lines, epidermal-dermal boundary. (E) Immunoprecipitation-Western blot experiments to biochemically confirm the K10-Akt-PKCζ interaction in skin. Whole-skin extracts were immunoprecipitated (IP) with the indicated antibodies and probed by Western blotting with the antibodies to proteins indicated at the right. Note the presence of K10, but not K5, in Akt and PKCζ immunoprecipitates and vice versa, whereas phosphorylated Akt is absent from K10 immunoprecipitates. Also note the absence of the kinases in K5 immunoprecipitates. Bar, 100 μm.
The interaction between Akt and PKCζ and K10 involves the non-α-helical amino terminus domain of K10.
We have previously shown that the non-α-helical amino and carboxyl terminus domains of K10 are involved in the capacity of K10 to arrest the cell cycle and that the mutant ΔNΔC form, which lacks these domains, is inactive (Fig. 1A) (23). These ends protrude from the filament core and may mediate interaction with other proteins. We analyzed whether the amino terminus domain of K10 (NTerm) was involved in the interaction with Akt or PKCζ. We first analyzed whether it has an effect on cell proliferation by itself. For this, the coding sequence for this fragment was subcloned under the control of the CMV promoter in the pVM6 plasmid to provide a VSV-G tag and Neo resistance. Colony-forming experiments showed that NTerm does not inhibit cell proliferation as K10 does (not shown). On the other hand, cotransfection of increasing amounts of NTerm reverses the inhibitory effect of K10 in HaCaT keratinocytes (Fig. 7A) and restores pRb hyperphosphorylation and cyclin D1 expression in C33A cells (Fig. 7B) without affecting the expression of K10 (Fig. 7B). This suggests that this fragment interacts with the same proteins as does intact K10 and that NTerm, present in increasing amounts, competes with filamentous K10.
FIG. 7.
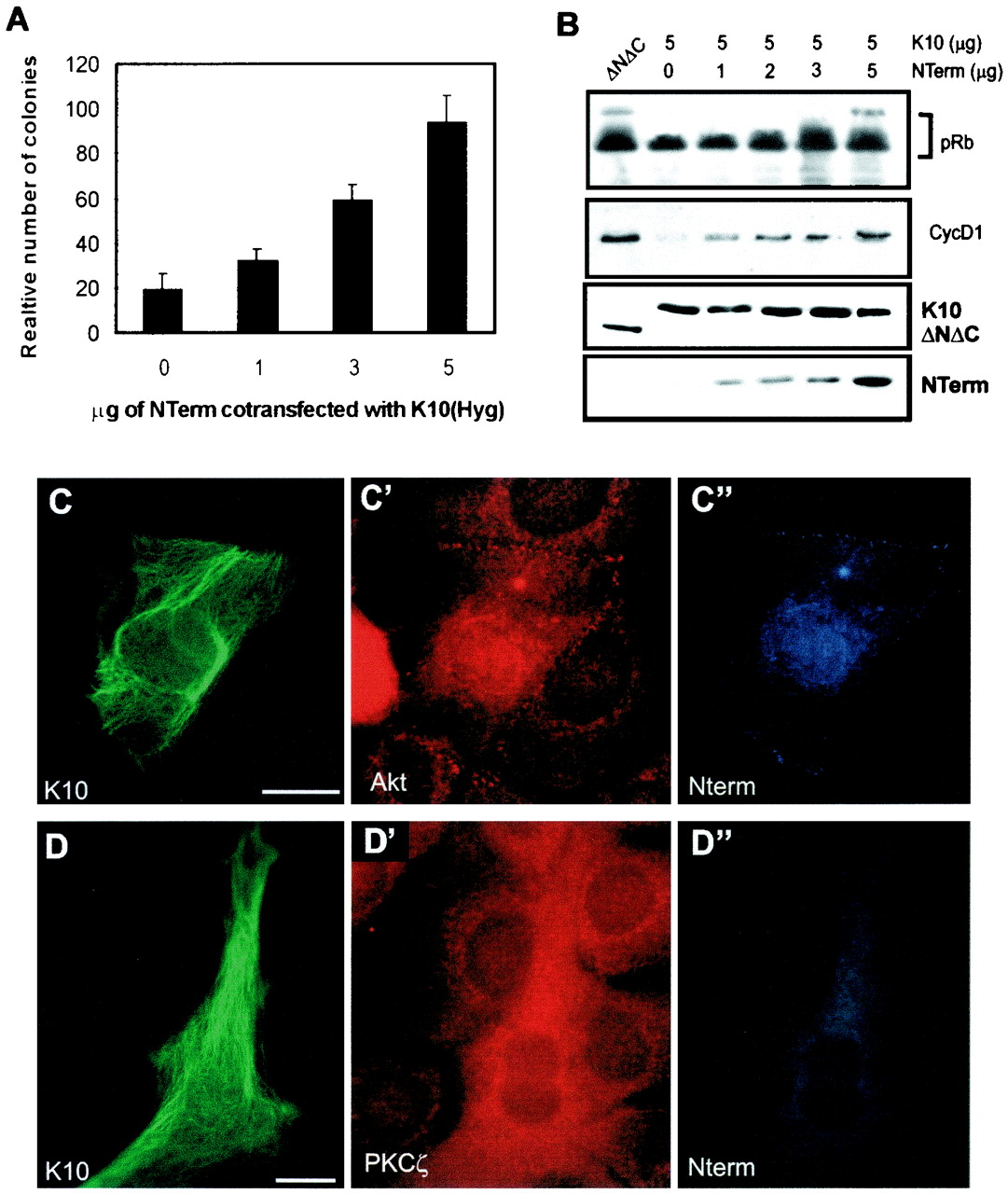
Expression of NTerm alleviates K10-induced cell growth arrest. (A) Colony-forming experiments with HaCaT cells demonstrate that the cotransfection of increasing amounts of an NTerm-encoding plasmid with 5 μg of the K10-encoding plasmid rescues K10-promoted inhibition of cell proliferation. (B) Similar cotransfection experiments with C33A cells in the presence of co transfected pRb demonstrate that NTerm also restores transfected pRb phosphorylation and endogenous cyclin D1 levels. Lower two sections demonstrate that the levels of K10 and ΔNΔC expression are similar and that the expression of increasing amounts of NTerm does not affect the expression of K10. Note also that, unlike K10, ΔNΔC has no effect on pRb phophorylation and cyclin D1 levels. (C to D′′) Examples of triple immunofluorescence after transient cotransfection of 5 μg of K10 and 5 μg of NTerm in HaCaT cells, demonstrating that the colocalization of K10 and endogenous Akt and PKCζ is abolished in cells expressing NTerm. (C and D) Immunofluorescence visualizing K10. (C′ and D′) Immunofluorescences visualizing Akt and PKCζ, respectively. (C" and D") Immunofluorescences visualizing NTerm. For comparison, see Fig. 3A, A′, B, and B′. Bars, (C and D), 15 μm. Data in panel A are from triplicate independent experiments and are means ± SD.
Since NTerm is soluble and is not anchored to filaments, proteins interacting with it can undergo translocation and activation. To test this, we cotransfected equal amounts (5 μg) of NTerm- and K10-encoding plasmids in HaCaT cells. The distribution of K10 (Fig. 7C and D), endogenous Akt (Fig. 7C′) or PKCζ (Fig. 7 D′), and NTerm (Fig 7C" and D") was analyzed by immunofluorescence. As predicted, it was found that the presence of NTerm disrupted the interaction between K10 and the endogenous Akt (compare Fig. 7C and C′ with Fig. 3A and A′) or PKCζ (compare Fig. 7D and D′ with Fig. 3B and B′) and that the kinases do not colocalize with the keratin.
We next studied the interaction of transfected NTerm with endogenous Akt and PKCζ by immunoprecipitation using the anti-VSV-G tag followed by Western blotting. NTerm was found to interact with Akt and PKCζ (Fig. 8A). Finally, to further confirm the physical interaction between Akt and NTerm in vivo, we performed two-cell hybrid experiments using the ras rescue system (6). For this, the NTerm coding sequence was subcloned in frame into plasmid pRas[61]ΔF (6) replacing the CAAX signal and the six combinations of plasmids shown in Fig. 8B were cotransformed in cdc25-2 yeast cells. In this system (6) the cells can grow at 25°C but not at 37°C unless rescued by protein-protein interaction or expression of RasCAAX. In fact we found growth at the restrictive temperature only in the positive controls (Fig. 8B) and in cells cotransformed with a myristoylated Akt (myrAkt) and RasNTerm. No growth was detected with either the negative controls (Fig. 8B) or cells transformed only with pMyrAkt (not shown). This confirms that the non-α-helical amino terminus domain of K10 interacts in vivo with Akt.
DISCUSSION
Keratin function was long been thought to be mainly structural, until the recent realization that keratin expression may influence cell proliferation and differentiation (19, 23). We have previously demonstrated that K10 inhibits proliferation by reducing cyclin D1 expression and thus pRb phosphorylation (23). Using several approaches, we here show here that these effects are due to the interaction of K10 with Akt and PKCζ, critical effectors of the PI-3K pathway. We also show that this interaction occurs during the differentiation of human and mouse keratinocytes and involves the non-α-helical amino terminus of this protein, although we did not determine whether similar interactions also take place through the K10 non-α-helical carboxyl terminus, which is also involved in cell cycle arrest (23). The binding of Akt and PKCζ to K10-containing filaments prevents the translocation of these kinases to the membrane and thus their activation. We also demonstrate that this inhibition is responsible for K10-induced cell cycle arrest.
PI-3K activity, for which Akt and PKCζ are the main specific effectors, has important roles in the control of cell cycle progression. For instance, PI-3K activation is sufficient for cell cycle entry in fibroblasts (14), and the activation of PI-3K and Akt in T lymphocytes promotes pRb phosphorylation and E2F activation (5). Similarly, expression of tumor suppressor PTEN, which dephosphorylates phosphoinositides and thus acts in opposition to PI-3K, inhibits cell cycle progression in a pRb-dependent manner in fibroblasts and keratinocytes (24, 31). In addition, Akt has been identified as a key regulator of cell survival and therefore of oncogenesis. The phosphorylation of cytoskeletal elements, which implies interaction with protein kinases, has been widely reported and appears to control cytoskeleton dynamics (12, 16, 25). Here we show, however, that the interaction of keratin K10 with Akt and PKCζ also controls the activity of these enzymes by restraining their translocation and subsequent activation. Through this mechanism, keratins affect cell proliferation, differentiation, and apoptosis. Previously reported data also support the possible involvement of the keratins in signal transduction processes. For instance, ectopic expression of K16 in the mouse epidermis, which leads to increased proliferation, also increases the tyrosine phosphorylation of the EGF receptor (19). Further, the overexpression of K8 in the pancreases of transgenic mice leads to phenotypic alterations that mimic the lack of transforming growth factor β signaling (8). In addition, proteosomes, which are involved in the control of cell cycle and signal transduction processes, interact with keratins in a cell cycle-dependent manner (20). The present data demonstrate unexpected and new functional roles for the intermediate filament cytoskeleton and provide evidence for its involvement in the regulation of critical signaling molecules such as Akt and PKCζ, which may result in the control of cell cycle progression. Further studies will be required to fully understand the increasingly relevant functions that keratins appear to perform in epithelial cells.
ACKNOWLEDGMENTS
We acknowledge K. Anderson, J. Downward, J. S. Gutkind, J. C. Lacal, D. P. Lane, and J. Moscat for their generous gifts of materials used in these studies and C. Mark for editorial assistance. We are also grateful for the expertise and help of J. Vázquez-Prado during the two-cell hybrid analysis and C. Murga for help with kinase assays.
This work was supported by grants SAF98-0047 and PB94-1230 from the DGICYT.
REFERENCES
- 1.Anderson K E, Caldwell J, Stephens L E, Hawkins P T. Translocation of PDK1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr Biol. 1998;8:684–691. doi: 10.1016/s0960-9822(98)70274-x. [DOI] [PubMed] [Google Scholar]
- 2.Bailleul B, Surani M A, White S, Barton S C, Brown K, Blessing M, Jorcano J L, Balmain A. Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell. 1990;62:697–708. doi: 10.1016/0092-8674(90)90115-u. [DOI] [PubMed] [Google Scholar]
- 3.Balmain A, Ramsden M, Bowden G T, Smith J. Activation of the mouse cellular Harvey ras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–660. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- 4.Boukamp P, Petrussevska R T, Breltkreutz D, Hornung J, Markham A, Fusenig N E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan P, Babbage J W, Burgering B M T, Groner B, Reifand K, Cantrell D A. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- 6.Broder Y C, Katz S, Aronheim A. The Ras recruitment system, a novel approach to the study of protein-protein interactions. Curr Biol. 1998;8:1121–1124. doi: 10.1016/s0960-9822(98)70467-1. [DOI] [PubMed] [Google Scholar]
- 7.Campbell S L, Khosravi-Far R, Rossman K L, Clark G J, Der C J. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 8.Casanova M L, Bravo A, Ramírez A, Morreale de Escobar G, Were F, Merlino G, Vidal M, Jorcano J L. Exocrine pancreatic disorders in transgenic mice expressing human keratin 8. J Clin Investig. 1999;103:1587–1595. doi: 10.1172/JCI5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou M M, Hou W, Johnson J, Graham L K, Lee M H, Chen C-S, Newton A C, Schaffhausen B S, Toker A. Regulation of protein kinase Cζ by PI 3-kinase and PDK1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 10.Dutil E M, Toker A, Newton A C. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK1) Curr Biol. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs E V. Of mice and men: genetic disorders of the cytoskeleton. Mol Biol Cell. 1997;69:899–902. doi: 10.1091/mbc.8.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gómez J, Martínez de Aragón A, Bonay P, Pitton C, García A, Silva A, Fresno M, Alvarez F, Rebollo A. Physical association and functional relationship between protein kinase Cζ and the actin cytoskeleton. Eur J Immunol. 1995;25:2673–2678. doi: 10.1002/eji.1830250941. [DOI] [PubMed] [Google Scholar]
- 13.Irvine A D, McLean W H I. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br J Dermatol. 1999;140:815–828. doi: 10.1046/j.1365-2133.1999.02810.x. [DOI] [PubMed] [Google Scholar]
- 14.Klippel A, Escobedo M A, Wachowicz M S, Apell G, Brown T W, Giedlin M, Kavanaugh W M, Williams L T. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol. 1998;18:5699–5711. doi: 10.1128/mcb.18.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kulik G, Klippel A, Weber M J. Antiapoptotic signalling by the insulin growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehrich R W, Forrest J N., Jr Protein kinase Cζ is associated with the mitotic apparatus in primary cell cultures of the shark rectal gland. J Biol Chem. 1994;269:32446–32450. [PubMed] [Google Scholar]
- 17.Marshall C J. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 18.Mittnacht S, Paterson H, Olson M F, Marshall C J. Ras signaling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr Biol. 1997;7:219–221. doi: 10.1016/s0960-9822(97)70094-0. [DOI] [PubMed] [Google Scholar]
- 19.Paladini R D, Coulombe P A. Directed expression of keratin K16 to the progenitor basal cells of transgenic mouse skin delays skin maturation. J Cell Biol. 1998;142:1035–1051. doi: 10.1083/jcb.142.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer A, Mason G F G, Paramio J M, Knecht E W, Rivett A J. Changes in proteosome during the cell cycle. Eur J Cell Biol. 1994;64:163–175. [PubMed] [Google Scholar]
- 21.Paramio J M, Jorcano J L. Role of protein kinases in the in vitro differentiation of human epidermal HaCaT cells. Br J Dermatol. 1997;137:44–50. [PubMed] [Google Scholar]
- 22.Paramio J M, Lain S, Segrelles C, Lane E B, Jorcano J L. Differential expression and functionally co-operative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene. 1998;17:949–958. doi: 10.1038/sj.onc.1202031. [DOI] [PubMed] [Google Scholar]
- 23.Paramio J M, Casanova M L, Segrelles C, Mittnacht S, Lane E B, Jorcano J L. Modulation of cell proliferation by cytokeratins k10 and k16. Mol Cell Biol. 1999;19:3086–3094. doi: 10.1128/mcb.19.4.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paramio J M, Navarro M, Segrelles C, Gómez-Casero E, Jorcano J L. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene. 1999;18:7462–7468. doi: 10.1038/sj.onc.1203151. [DOI] [PubMed] [Google Scholar]
- 25.Paramio J M. A role for phosphorylation in the dynamics of keratin intermediate filaments. Eur J Cell Biol. 1999;78:33–43. doi: 10.1016/S0171-9335(99)80005-3. [DOI] [PubMed] [Google Scholar]
- 26.Paramio J M, Segrelles C, Casanova M L, Jorcano J L. Opposite functions for E2F1 and E2F4 in human epidermal keratinocyte differentiation. J Biol Chem. 2000;275:41219–41226. doi: 10.1074/jbc.M004973200. [DOI] [PubMed] [Google Scholar]
- 27.Paramio J M, Segrelles C, Laín S, Gómez-Casero E, Lane D P, Lane E B, Jorcano J L. p53 is phosphorylated at the carboxyl terminus and promotes the differentiation of human HaCaT keratinocytes. Mol Carcinogenesis. 2000;29:251–262. doi: 10.1002/1098-2744(200012)29:4<251::aid-mc1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 28.Peeper D S, Upton T M, Ladha M H, Neumann E, Zalvide J, Bernards R, DeCaprio J A, Ewen M E. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature. 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- 29.Santos M, Ballestín C, García-Martín R, Jorcano J L. Delays in malignant tumor development in transgenic mice by forced epidermal keratin K10 in mouse skin carcinomas. Mol Carcinogenesis. 1997;20:3–9. doi: 10.1002/(sici)1098-2744(199709)20:1<3::aid-mc2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi K, Folmer J, Coulombe P A. Increased expression of keratin K16 causes anomalies in cytoarchitecture and keratinization in transgenic mice. J Cell Biol. 1994;127:505–520. doi: 10.1083/jcb.127.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng L-P, Brown J L, Eng C. PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]