

Abstract
The zinc finger-containing transcription factor GATA4 has been implicated as a critical regulator of multiple cardiac-expressed genes as well as a regulator of inducible gene expression in response to hypertrophic stimulation. Here we demonstrate that GATA4 is itself regulated by the mitogen-activated protein kinase signaling cascade through direct phosphorylation. Site-directed mutagenesis and phospho-specific GATA4 antiserum revealed serine 105 as the primary site involved in agonist-induced phosphorylation of GATA4. Infection of cultured cardiomyocytes with an activated MEK1-expressing adenovirus induced robust phosphorylation of serine 105 in GATA4, while a dominant-negative MEK1-expressing adenovirus blocked agonist-induced phosphorylation of serine 105, implicating extracellular signal-regulated kinase (ERK) as a GATA4 kinase. Indeed, bacterially purified ERK2 protein directly phosphorylated purified GATA4 at serine 105 in vitro. Phosphorylation of serine 105 enhanced the transcriptional potency of GATA4, which was sensitive to U0126 (MEK1 inhibitor) but not SB202190 (p38 inhibitor). Phosphorylation of serine 105 also modestly enhanced the DNA binding activity of bacterially purified GATA4. Finally, induction of cardiomyocyte hypertrophy with an activated MEK1-expressing adenovirus was blocked with a dominant-negative GATA4-engrailed-expressing adenovirus. These results suggest a molecular pathway whereby MEK1-ERK1/2 signaling regulates cardiomyocyte hypertrophic growth through the transcription factor GATA4 by direct phosphorylation of serine 105, which enhances DNA binding and transcriptional activation.
The zinc finger-containing transcription factor GATA4 is an important regulator of tissue-specific gene expression in multiple mesodermally and endodermally derived tissues. In cardiac myocytes, GATA4 regulates expression of diverse genes including α-myosin heavy chain (α-MHC), β-myosin heavy chain (β-MHC), cardiac troponin-C, atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), cardiac troponin-I, sodium/calcium exchanger, cardiac-restricted ankyrin repeat protein, A1 adenosine receptor, m2 muscarinic receptor, and the myosin light chain 1/3 (reviewed in reference 22). In response to agonist or stress stimulation, many of the above listed genes are upregulated in cardiomyocytes as part of the hypertrophic response, suggesting that a common regulatory factor such as GATA4 would be an ideal factor for coordinating uniform alterations in stress-induced gene expression.
Analysis of the β-MHC promoter in aortic-banded rats (pressure overload) revealed a proximal GATA binding site that mediated hypertrophy-induced expression in vivo (15). GATA4 was also implicated in regulating pressure overload-induced expression of the angiotensin type-1A receptor promoter in the adult rat heart (17). In neonatal cardiomyocyte cultures, electrical pacing-induced hypertrophy was associated with increased GATA4 mRNA levels (41), although GATA4 protein levels are not likely affected by hypertrophic stimulation (16, 20, 24). Alternatively, GATA4 transcriptional activity and DNA binding activity have been shown to be upregulated in response to hypertrophic stimuli (16, 17, 20, 24). In one report, as little as 15 min of arginine-vasopressin infusion in the adult rat was associated with enhanced GATA4 DNA binding activity in the heart (16). In cultured cardiomyocytes, phenylephrine (PE)-induced upregulation of the endothelin-1 promoter was associated with increased phosphorylation of GATA4, which was sensitive to PD98059, suggesting a role for MEK1/2-ERK1/2 in regulating GATA4 (24). Collectively, these various studies have suggested the hypothesis that GATA4 is regulated in the heart by stress-responsive signaling pathways such as the mitogen-activated protein kinase (MAPK) cascade.
The MAPK cascade consists of a series of successively acting protein kinases that include three well-characterized branches, the extracellular signal-regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38 MAPKs (reviewed in reference 13). Signaling through each of these MAPK branches is initiated by diverse stress and mitogenic stimuli localized to the cell membrane or within the cytoplasm. Activation of ERKs, JNKs, and p38 MAPKs facilitates the phosphorylation of multiple transcriptional regulators such as myocyte enhancer factor-2, activating transcription factor-2, Elk-1, p53, nuclear factor of activated T cells, Max, c-Jun, and c-Myc (13). MAPK-mediated phosphorylation of these and other transcriptional regulators profoundly influences adaptive and inducible gene expression in many cell types. Members of the MAPK signaling cascade are also important regulators of cardiomyocyte hypertrophy (reviewed in reference 34), yet the downstream transcriptional mechanisms that alter cardiac gene expression have not been well characterized.
Here we demonstrate that GATA4 contains a conserved MAPK phosphorylation site at serine 105 within the transcriptional activation domain. Serine 105 of GATA4 is phosphorylated in response to agonist stimulation through MEK1-ERK1/2, but only weakly through JNK1/2 or p38 MAPKs. Phosphorylation-specific antisera directed against serine 105 of GATA4 implicated ERK1/2 as the relevant kinase. Mutagenesis of serine 105 in GATA4 attenuated agonist-induced transcriptional upregulation, as did the MEK1 inhibitor U0126. Finally, purified ERK2 protein directly phosphorylated serine 105 of bacterially purified GATA4. That GATA4 is a critical downstream effector of MEK1-ERK1/2 signaling was demonstrated through the inhibition of MEK1-induced cardiomyocyte hypertrophy with a dominant-negative engrailed-GATA4-expressing adenovirus.
MATERIALS AND METHODS
Western blotting.
Protein extracts were generated from cultured cardiomyocytes or whole hearts and subjected to polyacrylamide gel electrophoresis and Western blotting as described previously (10). Primary antibodies and secondary antibodies were incubated overnight at room temperature in 5% milk and for 1 h at room temperature in 5% milk, respectively. Quantitative chemiluminescent detection was performed with Vistra enhanced chemifluorescence (Amersham) and scanned utilizing a Storm 860 (Molecular Dynamics, Sunnyvale, Calif.). Commercial antibodies used included anti-phospho-p38, anti-p38, anti-phospho-ERK1/2, anti-ERK1/2 (Cell Signaling, Beverly, Mass.), anti-GAPDH (Research Diagnostics Inc., Flanders, N.J.), and anti-GATA4 (Santa Cruz, Santa Cruz, Calif.).
Anti-phospho-GATA4(105) antibody generation.
Phospho-specific rabbit antiserum was prepared against a GATA4 peptide consisting of YTPPPV-phospho-serine-PRFSFP (amino acids 99 to 111) at Research Genetics (Huntsville, Ala.). The resulting antiserum was cross-absorbed with the nonphosphorylated peptide YTPPPVSPRFSFP followed by immunoaffinity purification.
GST-GATA4 fusion proteins.
To generate fusion proteins between glutathione S-transferase (GST) and GATA4, DNA sequences encoding amino acids 80 to 250 or 241 to 378 of GATA4 were amplified by PCR and subcloned into the EcoRI-XhoI sites of pGEX-4T-1 (Amersham Pharmacia Biotech). The full-length GST-GATA4 fusion cDNA (or the S105A mutant) encodes amino acids 1 to 441 of GATA4, which was subcloned into pGEX-4T-1 as an EcoRI fragment. All fusion proteins were expressed in Escherichia coli BL21 cells, precipitated with glutathione-Sepharose beads, and eluted with reduced glutathione (10 mM in 50 mM Tris, pH 8.0). The purity and concentration of each fusion protein were determined by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis using bovine serum albumin standards.
In vitro phosphorylation assays.
In vitro phosphorylation of GST-GATA4 fusions was carried out at 30°C for 20 min using 25 ng of activated ERK2, p38α, or JNK1 (Upstate Biotechnology) in a buffer containing 20 mM MOPS (pH 7.2), 25 mM β-glycerol phosphate, 5 mM EGTA, 1 mM sodium orthovanadate, and 1 mM dithiothreitol supplemented with 75 mM MgCl2, 500 μM ATP, and 10 μCi of [γ-32P]ATP. Three hundred nanograms of each GATA4 fusion protein or 10 μg of myelin basic protein (MBP) (positive control) or recombinant c-Jun was used to examine ERK2-, p38α- or JNK1-induced phosphorylation. Reactions were separated by SDS-polyacrylamide gel electrophoresis. Parallel phosphorylation reactions were performed in the absence of [γ-32P]ATP and subjected to Western blot analysis with phospho-specific GATA4 antiserum.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed using a double-stranded oligonucleotide containing two GATA motifs from the α-MHC promoter as previously described (20). Briefly, 300 ng of bacterially generated GST-GATA4 (full-length) or GST-GATA4 S105A (full-length) were incubated with 50,000 cpm of 32P-labeled double-stranded oligonucleotide, 1 μg of poly(dI-dC)-(dI-dC), and EMSA buffer (12 mM HEPES [pH 7.9], 4 mM Tris [pH 7.9], 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 12% glycerol, and 2 μg of aprotinin, leupeptin, and pepstatin/ml) at room temperature for 20 min in a 20-μl volume. A nondenaturing 5% polyacrylamide gel with 0.5× Tris-borate-EDTA was used to resolve the bound protein complexes from the free probe.
Cell culturing and transfections.
Primary neonatal rat cardiomyocytes derived from the ventricles of 1- to 2-day-old Sprague-Dawley rat neonates were prepared as described previously (10). Cardiomyocytes were plated on gelatinized dishes and cultured in serum-free M199 medium containing 100 U of penicillin-streptomycin/ml and 2 mmol of l-glutamine/liter. Both cardiomyocytes and Cos-7 cells were transfected with Fugene 6 (Roche Diagnostics Corporation, Indianapolis, Ind.). Cultures were harvested 48 h after transfection and luciferase assays were performed as described previously (23). PE and MAPK inhibitors were added 24 h prior to harvest.
Plasmids.
Plasmids used to generate AdGATA4, AdG4-Engr, and AdEngr were described previously (20). The activated MKK6b (Ser 207,211 Glu) and dominant-negative MKK3b (K-A) were a gift from Roger Davis, University of Massachusetts, Worcester, while MKK7 was a gift from E. Nishida, Kyoto University. Dominant-negative MEK1 (Ser 221 Ala) was a gift from C. J. Marshall, Institute of Cancer Research, London, United Kingdom. Each cDNA was subcloned as a HindIII fragment into pACCMCpLpA for subsequent viral production. To generate GAL4-GATA4, PCR-amplified GATA4 (amino acids 33 to 227) was ligated into pM1-GAL4 (amino acids 1 to 147) as an EcoRI fragment. Generation of the site-specific mutation at amino acid 105 was performed as previously described (23). The BNP luciferase reporter (−116 to +83) was previously described and was a gift from Chris Glembotski, San Diego State, San Diego, Calif. (37).
Replication-deficient adenovirus infection.
The procedures for generating and plaque purifying replication-deficient adenovirus were performed as previously described (10). Cardiomyocytes were infected at a multiplicity of infection of 100 PFU per cell for a single infection or at 50 PFU per cell for coinfections. Infection occurred for 2 h at 37°C in a humidified, 6% CO2 incubator, and then the cardiomyocytes were placed in serum-free M199 medium for an additional 24 h prior to treatments or harvesting. Under these conditions approximately 98% of cells showed expression of each of the different viral gene inserts. The constitutively active MEK1- and dominant-negative MKK4-expressing adenoviruses were previously described (5, 6).
Immunocytochemistry.
Cultured cardiomyocytes were prepared for immunocytochemistry against sarcomeric α-actinin and ANF as described previously (10). Quantitation of cardiomyocyte cell surface area was performed on α-actinin-stained cardiomyocytes using NIH Image software on a Sun system workstation. At least 100 cardiomyocytes in 15 to 30 fields at ×400 magnification were examined in three independent experiments. For quantitation of ANF expression, cardiomyocytes showing intense perinuclear staining were counted in at least 25 fields (n = 3).
Leucine incorporation assay.
Determination of protein synthesis rates in cultured cardiomyocytes by [3H]leucine incorporation was described previously (31). Briefly, cardiomyocytes were infected with adenovirus, incubated 24 h to allow adequate expression, preincubated with leucine-free RPMI medium for 1 h followed again by incubation with 2.5 μCi of [3H]leucine/ml for 24 h. Plates were washed with phosphate-buffered saline and then 10% trichloroacetic acid was added at 4°C and incubated for 60 min to precipitate protein. The precipitate was washed twice in 95% ethanol and resuspended in 0.5 N NaOH and was measured by scintillation counting.
Statistical analysis.
Data are expressed as means ± standard errors of the means. Differences between groups were evaluated for statistical significance using Student's t test for unpaired data or one-way analysis of variance followed by Bonferroni's posttest. P values of <0.05 were considered statistically significant. All statistical analyses were performed by using Instat 3.0 software (GraphPad, San Diego, Calif.).
RESULTS
GATA4 is phosphorylated at serine 105.
We previously demonstrated that GATA4 DNA binding activity was up-regulated in pressure-overloaded rat hearts, suggesting that GATA4 is activated by stress responses in the heart (17). One potential mechanism involved in mediating this increase in GATA4 DNA binding activity is through direct phosphorylation by stress-activated kinases, which are important regulators of cardiomyocyte hypertrophy. Indeed, Morimoto et al. demonstrated that GATA4 was phosphorylated in response to α-adrenergic stimulation of cardiomyocytes (24). Consistent with this finding, we also observed in vivo phosphorylation of GATA4 in response to hypertrophic agonist stimulation in cardiomyocytes; however, the relevant site(s) and potential modifying kinases are not known.
Examination of the GATA4 primary amino acid sequence identified a putative MAPK phosphorylation site at position 105 that lies within a PXSP high-affinity motif conserved between mouse, human, and chicken GATA4 and found within many other stress-activated transcription factors (9) (Fig. 1A). To characterize the potential physiological relevance of this site and its ability to undergo phosphorylation, phospho-specific antiserum was generated. Cardiomyocyte protein extracts were generated from PE (10 μM)-stimulated cultures after 1, 3, and 24 h and subjected to Western blotting with phospho-specific GATA4 antiserum. PE is also a potent inducer of cardiomyocyte hypertrophy and MAPK activation in culture (34). The data demonstrate increased phosphorylation of endogenous GATA4 in response to PE stimulation, without a change in GATA4 protein levels (Fig. 1B). Cultured cardiomyocytes were also infected with a GATA4-expressing adenovirus (AdGATA4) to increase total protein levels for detection purposes. AdGATA4-infected cardiomyocytes stimulated with PE also demonstrated a significant increase in GATA4 phosphorylation at serine 105 (Fig. 1B). GATA4 serine 105 phosphorylation was also observed in response to endothelin-1, angiotensin II, isoproterenol, and fetal bovine serum stimulation (data not shown). Collectively, these data indicate that hypertrophic agonists induce GATA4 phosphorylation at serine 105 in cultured cardiomyocytes.
FIG. 1.

GATA4 contains a conserved MAPK phosphorylation site at serine 105. (A) Diagram of GATA4 showing the position of the zinc finger domains (Zn), the nuclear localization sequence and basic domain (nls), and the transcriptional activation domain (TAD). A MAPK recognition site containing serine 105 is conserved between mouse, human, and chicken GATA4. (B) Phospho-105-GATA4-specific antiserum was generated and used to examine GATA4 phosphorylation in response to α-adrenergic stimulation with PE in cultured cardiomyocytes. Phosphorylation of endogenous GATA4 was upregulated by PE in control Adβgal-infected cultures, while a similar pattern of up-regulation was observed when GATA4 was overexpressed by AdGATA4 infection. (C) Endogenous GATA4 was also phosphorylated at serine 105 in the hearts of PE-injected mice (3 h), compared to saline-treated control mice (n = 3 in each group). GATA4 protein levels did not vary.
It was also of interest to determine whether hypertrophic agonist stimulation could promote GATA4 serine 105 phosphorylation within a mouse heart. Accordingly, neonatal mice were injected subcutaneously with PE (10 mg/kg of body weight) and the hearts were harvested 3 h afterwards and processed for Western blotting. The data demonstrate an approximately threefold increase in GATA4 serine 105 phosphorylation in the hearts of PE-injected mice compared to saline controls (three mice in each group) (Fig. 1C). These results indicate that GATA4 is phosphorylated in the mouse heart in response to agonist stimulation.
ERK1/2 mediates phosphorylation of serine 105.
The broad range of agonists identified above that promote GATA4 phosphorylation suggested the involvement of MAPK signaling effectors since they mediate diverse stress stimuli in many cell types (13). Cultured cardiomyocytes were coinfected with AdGATA4 and specific MAPK kinase-encoding adenoviruses in an attempt to implicate one or more kinase effectors in the phosphorylation of serine 105. The data demonstrate that coinfection with a constitutively active MEK1 adenovirus, which directly activates ERK1/2, promoted robust phosphorylation of GATA4 compared to Adβgal-coinfected cardiomyocyte cultures (Fig. 2A). However, infection with a constitutively active MKK6 (activates p38) or MKK7 (activates JNK1/2) adenovirus did not result in the same degree of activation (Fig. 2A). GATA4 protein levels did not significantly vary in any of the coinfected cultures (Fig. 2A). Similar results were observed for three independent experiments. These data suggest that the MEK1-ERK1/2 pathway promotes phosphorylation of serine 105 in GATA4.
FIG. 2.

MEK1-ERK1/2 signaling promotes phosphorylation of serine 105 in GATA4. (A) Cardiomyocyte cultures were coinfected with AdGATA4 and either Adβgal, AdMEK1, AdMKK7, or AdMKK6. Twenty-four hours later, cultures were harvested and Western blotted with phospho-specific GATA4 antibody and then reprobed with GATA4 antibody. (B) Cardiomyocyte cultures were coinfected with AdGATA4 and either Adβgal, Ad-dnMEK1, Ad-dnMKK4, or Ad-dnMKK3 and stimulated with PE for 3 h. Western blotting demonstrated reduction in PE-induced serine 105 phosphorylation with dominant-negative MEK1 (asterisk) but not with other dominant-negative MAPK kinase factors. (C) Quantitation of three independent experiments demonstrates a significant inhibition of PE-induced serine 105 phosphorylation only with Ad-dnMEK1 infection. *, P < 0.05 compared to Adβgal infection.
Conversely, dominant negative-encoding adenoviruses for each of the three MAPK signaling branches were coinfected with AdGATA4 and subjected to PE stimulation. The data demonstrate that dominant-negative MEK1 significantly antagonized PE-induced phosphorylation of GATA4 in cardiomyocytes, while dominant-negative MKK4 (blocks JNK1/2) and dominant-negative MKK3 (blocks p38) had no inhibitory effect (Fig. 2B and C). Quantitation of data from three independent experiments demonstrated that only dominant-negative MEK1 blocked PE-induced GATA4 phosphorylation (Fig. 2C). We also verified that the dominant-negative MEK1 and MKK3-encoding adenovirus inhibited ERK1/2 and p38 phosphorylation, respectively (Fig. 2B), while the dominant-negative MKK4 adenovirus was previously shown to inhibit JNK1/2 (6). Collectively, these data implicate MEK1 and ERK1/2 as regulators of GATA4 phosphorylation at serine 105.
While the data described above demonstrate an association between MEK1-ERK1/2 signaling and GATA4 serine 105 phosphorylation, it is uncertain if ERK1/2 directly or indirectly regulates GATA4 phosphorylation. In this regard, in vitro phosphorylation assays were performed with purified GATA4 and ERK2 to examine direct phosphorylation. GATA4 amino acids 80 to 250 or 241 to 378 were each expressed in bacteria as GST fusion proteins and subsequently purified (Fig. 3A). Purified ERK2 protein (activated) was incubated with either GST alone, GST-GATA4 80-250, or GST-GATA4 241-378 in the presence of [32P]ATP. The data demonstrate that ERK2 efficiently phosphorylates GST-GATA4 80-250, but not GST alone or GST-GATA4 241-378 (Fig. 3B). As a postitive control, ERK2 was also incubated with purified MBP, which demonstrated efficient phosphorylation (Fig. 3B). More importantly, Western blotting with phospho-specific GATA4 antisera in parallel “cold” ATP labeling assays demonstrated efficient ERK2-mediated phosphorylation of serine 105 in vitro (Fig. 3C). As a final control, the membrane was stripped and reprobed with GST antiserum, which demonstrated equal sample loading (data not shown). Collectively, these results indicate that ERK2 directly phosphorylates GATA4 at serine 105.
FIG. 3.

ERK2 directly phosphorylates GATA4 in vitro. (A) Schematic representation of full-length GATA4 and the two different fusion proteins that were generated and purified from bacteria. (B) Polyacrylamide gel of [32P]ATP-labeled proteins. Three hundred nanograms of GST-GATA4 fusion protein, GST alone, or MBP was incubated with 25 ng of activated ERK2 protein for the labeling reaction. Only MBP and the GST-G4 fusion protein containing GATA4 amino acids 80 to 250 were efficiently labeled. (C) Western blot with GATA4 phospho-specific antiserum and ERK2-mediated in vitro-labeled GST and GST-GATA4 fusion proteins (300 ng). The asterisk shows the specific increase in ERK2-mediated phosphorylation of GST-GATA4 80-250.
Phosphorylation of serine 105 enhances transcriptional activation and DNA binding.
MAPK-mediated phosphorylation of transcription factors can influence either DNA binding activity or transcriptional activation. To isolate effects specific for transcriptional activation apart from DNA binding, the GATA4 activation domain (amino acids 33 to 227) containing either the wild-type sequence or an S105A mutation was fused to the GAL4 DNA binding domain (Fig. 4A, bottom). These constructs were cotransfected into cultured cardiomyocytes along with the GAL4-luciferase reporter (G5E1b-luciferase). The data demonstrate that the S105A mutant had twofold less activity in unstimulated cardiomyocytes than in the wild-type fusion construct (Fig. 4A). Furthermore, PE induced a greater than fourfold activation of wild-type GAL4-GATA4, but not of the GAL4-GATA4 S105A mutant (P < 0.05) (Fig. 4A). PE-induced transcriptional activation of wild-type GAL4-GATA4 was blocked with the MEK1-ERK1/2 inhibitor U0126, but not with the p38 inhibitor SB202190 (n = 3) (Fig. 4A). Both the wild-type and S105A mutant constructs were expressed at comparable levels as assessed by Western blotting of protein extracts from transfected Cos-7 cells (data not shown). Collectively, these data suggest that serine 105 phosphorylation directly enhances the intrinsic transcriptional activation properties of GATA4 through a MEK1-ERK1/2-sensitive mechanism.
FIG. 4.

Serine 105 in GATA4 mediates PE transcriptional responsiveness. (A) A plasmid encoding the GAL4 DNA binding domain fused to the wild-type (Wt) or S105A mutant GATA4 transcriptional activation domain (amino acids 33 to 227) was cotransfected into cardiomyocytes with the GAL4-luciferase reporter, G5E1b-luciferase. Cultures were either PE stimulated, left unstimulated, or treated with U0126 or SB202190 (SB) for 24 h. *, P < 0.05 versus GAL4; #, P < 0.05 versus wild type). (B) The BNP minimal promoter luciferase reporter was cotransfected into cultured cardiomyocytes with either wild-type GATA4 (Wt), S105A mutant GATA4, or empty vector (pCDNAI) and PE stimulated for 24 h and/or treated with U0126. *, P < 0.05 versus pCDNAI; #, P < 0.05 versus wild-type GATA4; †, P < 0.05 versus PE plus wild-type GATA4.
It was also of interest to determine if MEK1-ERK1/2 signaling could regulate GATA4 transcriptional activity in the context of a physiologically relevant promoter. Accordingly, the minimal BNP promoter (−116 to +83), which contains three GATA binding elements, was cotransfected into cardiomyocytes with or without a GATA4 expression vector. Both PE treatment and GATA4 transfection induced a fivefold activation of the hypertrophy-responsive BNP promoter (P < 0.05) (Fig. 4B). In addition, GATA4 transfection together with PE stimulation synergistically activated BNP promoter activity (P < 0.05) (Fig. 4B). More significantly, MEK1-ERK1/2 inhibition with U0126 blocked GATA4 and GATA4-PE synergistic activation of the BNP promoter, and the S105A mutant GATA4 construct showed a significant reduction in PE-induced activity (P < 0.05). Both the wild-type and S105A mutant GATA4 expression vectors produced comparable expression levels as assessed by Western blotting of protein extracts from transfected Cos-7 cells (data not shown). Taken together, these data indicate that MEK1-ERK1/2 signaling up-regulates GATA4 transcriptional activity through phosphorylation of serine 105 on a hypertrophy-responsive promoter.
Hypertrophic or agonist stimulation has been previously associated with an increase in GATA4 DNA binding activity, without a change in protein levels (16, 17, 20, 24). Such results suggest that ERK-mediated phosphorylation might also regulate, in part, GATA4 DNA binding activity. To investigate this possibility, full-length GST-GATA4(Wt) and full-length GST-S105A mutant GATA4 were purified from bacteria, subjected to in vitro phosphorylation with ERK2, and subsequently assayed for effects on DNA binding. The data demonstrate that ERK2 enhances wild-type GATA4-DNA binding by 1.5-fold ± 0.02 (P < 0.05) (Fig. 5A). In addition, S105A mutant GATA4 demonstrated reduced basal DNA binding activity compared to wild-type GATA4 and also failed to demonstrate ERK-enhanced DNA binding (Fig. 5A). Such a result suggests that serine 105 might potentially regulate the conformation of GATA4 and accessibility of the DNA binding domain (see Discussion).
FIG. 5.

ERK2-mediated phosphorylation of GATA4 enhances its DNA binding activity. (A) EMSA demonstrates that ERK2 enhances the DNA binding activity of a bacterially purified GST-GATA4 (full-length) wild-type (Wt) fusion protein but not GST-S105A mutant GATA4 (full-length). (B) Western blot analysis with phospho-specific GATA4 (serine 105) antiserum using the same protein reactions used for panel A demonstrates efficient ERK2-mediated phosphorylation of full-length GATA4, but not the S105A mutant GATA4 or GST. (C) Western blot analysis with GATA4 antibody using the same extracts from panel A demonstrates equal protein levels.
Western blotting was also performed with the same extracts used in the EMSA experiments to control for protein content and to show ERK2-mediated phosphorylation. Full-length wild-type GATA4 purified as a GST fusion from bacteria was efficiently phosphorylated with purified ERK2 protein, but the S105A mutant GATA4 showed no ERK2-mediated phosphorylation of serine 105 (Fig. 5B). These data demonstrate the specificity of the phospho-specific GATA4 antibody. As a final control, the phospho-GATA4 Western blot was stripped and reprobed for total GATA4 protein levels, which demonstrated equal amounts of protein (Fig. 5C). Collectively, these data indicate that ERK2-mediated phosphorylation of serine 105 in GATA4 directly influences DNA binding activity.
JNK and p38 only weakly phosphorylate serine 105 in GATA4.
To determine if other MAPK terminal effectors can phosphorylate serine 105 in GATA4, purified wild-type and S105A mutant proteins were incubated with purified ERK2, p38α, and JNK1 proteins in the presence of [32P]ATP. Similar to the results shown in Fig. 3 with truncated GATA4, ERK2 efficiently phosphorylated full-length wild-type GATA4 (Fig. 6). However, full-length S105A mutant GATA4 was not significantly phosphorylated by ERK2, further demonstrating that serine 105 is the only relevant site for ERK-mediated phosphorylation (Fig. 6). As a control, ERK2 and p38α were each capable of phosphorylating MBP, while JNK1 showed efficient phosphorylation of recombinant c-Jun protein (Fig. 6). Incubation of both wild-type and S105A mutant GATA4 with either p38α or JNK1 revealed detectable phosphorylation by each kinase, although the stoichiometry of phosphorylation was not assessed. Coomassie brilliant blue staining of each gel revealed similar levels of loaded protein (data not shown). These results suggest that both p38α and JNK1 are capable of phosphorylating GATA4 in vitro. However, phosphorylation mediated by p38α and JNK1 showed only a minor preference for serine 105, suggesting that other serine-proline motifs within GATA4 are likely targeted by these two kinases (see Discussion). Taken together, these results indicate that ERK2 specifically targets serine 105 for phosphorylation, while p38α and JNK1 show only a minor effect.
FIG. 6.

ERK2, but not p38 and JNK, mediates phosphorylation of serine 105. (A) In vitro kinase reaction with bacterially purified wild-type (Wt) full-length GATA4 or the S105A mutant GATA4 incubated with purified ERK2 in the presence of [32P]ATP and subjected to SDS-polyacrylamide gel electrophoresis. Both proteins were also incubated with purified p38α (B) and JNK1 (C) protein to assess phosphorylation. MBP and recombinant c-Jun were used as positive controls in the kinase assays. The arrow shows the migration of the full-length GST-GATA4 fusion protein, although degradation products of faster migration were also visible.
GATA4 is required for mediating MEK1-ERK1/2-induced hypertrophy.
We previously demonstrated that expression of activated MEK1 in the hearts of transgenic mice or by adenoviral gene transfer in cultured cardiomyocytes induces cardiac hypertrophy (5). However, the mechanism whereby ERK1/2 activation might promote cardiomyocyte hypertrophy has not been characterized. To determine if GATA4 is a necessary component of MAPK-induced cardiac hypertrophy, a dominant-negative GATA4 adenovirus was generated. A fusion protein was constructed consisting of N-terminal Flag epitope, the engrailed repressor domain (amino acids 2 to 298), and the GATA4 zinc finger DNA binding domain (amino acids 211 to 329) (20). A similar strategy was previously used to generate a dominant-negative Nkx2.5 transcription factor which blocked heart development in vivo (12). Western blotting of protein extracts from AdG4-Engr-infected cardiomyocytes confirmed the integrity and stability of the fusion protein (Fig. 7A). The engrailed-GATA4 fusion functions by binding DNA and blocking GATA-dependent transcriptional responses in cardiomyocytes, while the engrailed domain alone has no effect (20).
FIG. 7.

GATA4-engrailed blocks MEK1-ERK1/2-induced cardiomyocyte hypertrophy. (A) Western blotting with Flag monoclonal antibody demonstrates a protein of the predicted size from AdG4-Engr-infected cardiomyocytes. (B) Cell surface areas were quantified from each of the indicated adenoviral infected cardiomyocyte cultures. *, P < 0.05 versus Adβgal; #, P < 0.05 versus AdMEK1 plus AdEngr. (C) The percentage of cells expressing ANF protein was quantified from each of the indicated adenoviral infected cardiomyocyte cultures. *, P < 0.05 versus Adβgal; #, P < 0.05 versus AdMEK1 plus AdEngr. (D) [3H]leucine incorporation was measured in Adβgal-, AdMEK1-, and AdMEK1-plus-AdG4-Engr-infected cardiomyocytes. *, P < 0.05 versus Adβgal. All data represent the averages of three independent experiments.
To examine the importance of GATA4 as a downstream mediator of MEK1-induced hypertrophy, AdG4-Engr and AdMEK1 were coinfected into cultured cardiomyocytes. Consistent with a previous report (5), AdMEK1 infection increased cardiomyocyte cell surface area, sarcomeric organization, ANF protein expression, and [3H]leucine incorporation (Fig. 7B, C, and D and Fig. 8). As an important control, an engrailed-only-expressing adenovirus was also generated to control for any deleterious effect associated with overexpression of this transcriptional repressor domain (20). Coinfection with AdMEK1 and AdEngr (each at a mulitplicity of infection of 50 PFU/cell) did not diminish the ability of MEK1 to induce a hypertrophic response (Fig. 7B and C). However, coinfection with AdG4-Engr blocked MEK1-induced increases in cell surface area, [3H]leucine incorporation, ANF expression, and sarcomeric organization (Fig. 7B, C, and D and Fig. 8). To determine if the inhibitory effect of AdG4-Engr was specific to MEK1, an MKK7-expressing adenovirus which was previously characterized to induce a robust hypertrophic response in cultured cardiomyocytes through JNK1/2 (40) was also coinfected. Surprisingly, AdG4-Engr did not inhibit MKK7-induced cardiomyocyte hypertrophy (Fig. 7B and C and Fig. 8). Such a result may suggest that MKK7 (JNK1/2) activation utilizes a GATA4-independent transcriptional pathway or simply that MKK7 is a more potent stimulator of cardiomyocyte hypertrophy than MEK1. Taken together, these data suggest that MEK1-ERK1/2 signaling utilizes GATA4 as a necessary transcriptional effector of the cardiac hypertrophic growth response.
FIG. 8.
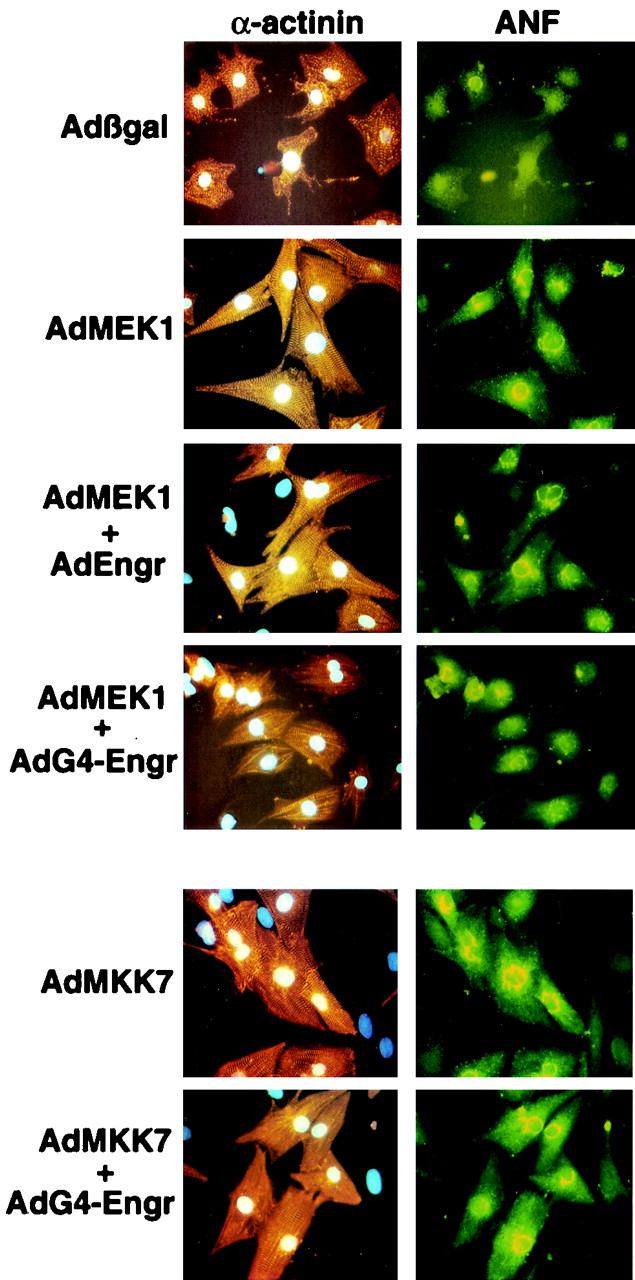
GATA4-engrailed blocks MEK1-ERK1/2-induced sarcomeric organization and hypertrophy. Representative images of immunostained cardiomyocyte cultures infected with each of the indicated adenoviral constructs are shown. The left panels show anti-α-actinin antibody (orange) reactivity to demonstrate cardiomyocyte sarcomeres and gross morphology. The right panels show the same cells double immunostained for ANF (green), which appears in a perinuclear pattern.
DISCUSSION
Cardiac myocytes respond to a wide array of neural-humoral stimuli through G-protein-coupled receptors and/or receptor tyrosine kinases. Membrane receptors in turn activate discrete intermediate signal transduction pathways, which ultimately modify intracellular physiology and mediate inducible gene expression within the nucleus. However, the mechanisms whereby intracellular signaling cascades mediate alterations in cardiac gene expression are poorly understood. Here it is demonstrated that the cardiac-expressed transcription factor GATA4 is phosphorylated by the MEK1-ERK1/2 signaling pathway as a mechanism involved in altering hypertrophic gene expression within the heart.
Role of serine 105 phosphorylation in GATA4.
GATA4 is a member of a subfamily that includes the closely related GATA5 and GATA6 genes. All three genes are expressed within cardiomyocytes of the embryonic heart, yet as development progresses, GATA5 expression is lost from the heart (28). While it is not known if other cardiac-expressed GATA factors are also posttranslationally modified in response to hypertrophic stimulation, the consensus phosphorylation site identified within GATA4 (serine 105) is not present in GATA5 or GATA6, suggesting alternative mechanisms of regulation. Noncardiac-expressed GATA family members, namely GATA1 and GATA2, have also been shown to undergo site-specific phosphorylation (8, 38). In fact, ERK MAPK was shown to directly phosphorylate GATA2 in hematopoietic progenitor cells and COS cells, although effects on DNA binding or transcriptional activation were not noted (38).
Protein domain deletion analysis of GATA4 demonstrated that serine 105 lies within a region encompassing the transcriptional activation domain (amino acids 1 to 204) (27). This site, PXSP, has been implicated as a preferred MAPK phosphorylation site in other regulatory proteins, such as c-Myc, c-Jun, cyclin B, Elk-1, MAPK activating protein (MAPKAP) kinase 2, and MBP (9). While GATA4 contained other potential MAPK phosphorylation sites (XXS/TP, where X = a basic or neutral residue), mutagenesis of serine 105 prevented in vivo labeling of GATA4 in response to agonist stimulation, implicating it as a primary site of physiologic regulation. Consistent with this interpretation, in vitro labeling of purified full-length GATA4 with ERK2 revealed direct phosphorylation at serine 105 but not at other potential sites. Indeed, the full-length S105A mutant of GATA4 was refractory to ERK2-mediated phosphorylation in vitro. Analysis of other MAPK signaling factors demonstrated detectable phosphorylation induced by recombinant p38α and JNK1 in vitro. However, S105A mutant GATA4 showed only a partial reduction in p38α- or JNK1-mediated phosphorylation in vitro. Adenoviral coinfection experiments suggested a prominent role for MEK1-ERK1/2 signaling in mediating serine 105 phosphorylation, although p38 appeared to weakly induce phosphorylation of serine 105 in MKK6 adenoviral infected cardiomyocytes (Fig. 2A). However, inhibition of p38 with dominant-negative MKK3 or the pharmacologic agent SB202190 did not significantly reduce GATA4 activity or phosphorylation at serine 105. Taken together, these results suggest that serine 105 phosphorylation is regulated primarily by ERK1/2 in cardiomyocytes.
MEK1-ERK1/2-mediated phosphorylation of GATA4 might regulate transcriptional potency by enhancing interactions between the GATA4 activation domain and other transcription accessory proteins, or alternatively, phosphorylation might enhance the DNA binding activity of GATA4. The results of this study suggest that ERK2-mediated phosphorylation of GATA4 enhances both transcriptional and DNA binding properties. Indeed, PE-induced transcriptional activation of the GAL4-GATA4 fusion construct (amino acids 33 to 227), which lacks the GATA4 DNA binding domain, was sensitive to serine 105 mutagenesis (Fig. 4A). In addition, U0126 blocked PE-induced transcriptional activation of the wild-type GAL4-GATA4 fusion construct in cardiomyocytes, further suggesting that ERK regulates the transcriptional activating properties of GATA4.
Hypertrophic agonists are also known to regulate GATA4 DNA binding activity without affecting GATA4 protein levels (16, 17, 20, 24). To this end, ERK2-mediated phosphorylation of bacterially purified full-length GATA4 slightly, albeit significantly, enhanced DNA binding activity (Fig. 5). Consistent with this observation, U0126 treatment prevented the normal increase in GATA4 DNA binding activity that is associated with PE stimulation (data not shown). Since GATA4 was purified from a prokaryotic source, the observed ERK2-dependent increase in DNA binding activity is likely a direct effect and not attributable to cofactor interaction or the actions of another downstream kinase such as MAPKAP kinase. Also in support of this notion, MAPKAP kinase recognizes a completely different consensus phosphorylation motif from that recognized by ERK1/2 (33). In any event, the mechanism whereby phosphorylation of serine 105 within the transcriptional activation domain might regulate DNA binding characteristics of GATA4 is uncertain. However, at least one potential mechanism is suggested by the observation that phosphorylation of serum response factor, which occurs with the N-terminal transcriptional activation domain, enhances DNA binding activity through a conformational shift within the entire protein (21).
While ERK2-mediated phosphorylation of GATA4 plays an important role in up-regulating GATA4 transcriptional potency in response to agonist stimulation, it is possible that other posttranslational modifications also regulate GATA4 activity. For example, p300 was shown to directly acetylate GATA1 resulting in enhanced DNA binding activity, suggesting that other members of the GATA family might be subjected to such regulation (4). In addition, GATA4 is known to interact with other cardiac-expressed transcription factors such as Nkx2.5, serum response factor, and myocyte enhancer factor-2, suggesting additional mechanisms for affecting GATA4 activity in the heart (2, 11, 25, 26). A final consideration is that our data do not exclude a potential role for other kinases in the posttranslational modification of GATA4 at serine 105 or other sites (i.e., p38, JNK, protein kinase C, calmodulin-dependent kinases). Indeed, both p38α and JNK1 were capable of phosphorylating purified GATA4 in vitro, although the relative stoichiometry of phosphorylation was not assessed. The fact that multiple mechanisms might influence GATA4 potency underscores its potential importance as a regulator of hypertrophic transcriptional responses.
Evidence implicating GATA4 as a hypertrophic regulator.
The identification of an ERK MAPK phosphorylation site in GATA4 suggests a transcriptional mechanism whereby MEK1-ERK1/2 signaling might mediate hypertrophy. If such a hypothesis is correct, GATA4 alone should also induce cardiomyocyte hypertrophy. Indeed, it has recently been demonstrated that adenoviral-mediated overexpression of GATA4 in cultured cardiomyocytes induces hypertrophy characterized by enhanced sarcomeric organization, increased cell surface area, and increased protein accumulation (20). In addition, transgenic mice with 2.5-fold GATA4 overexpression in the heart demonstrated cardiac hypertrophy, collectively suggesting that GATA factors are sufficient regulators of cardiomyocyte hypertrophy in vitro and in vivo (20).
While the hypothesis that GATA4 participates in the hypertrophic response has been previously proposed, the data in this report extend our understanding in several significant ways. First, GATA4 was shown to be phosphorylated at serine 105 in both cultured cardiomyocytes and in the mouse heart in response to agonist stimulation. Second, serine 105 in GATA4 was shown to be directly phosphorylated by ERK2, suggesting a novel transcriptional target of ERK1/2 signaling. Third, phosphorylation of serine 105 was shown to enhance the intrinsic transcriptional activation and DNA binding properties of GATA4 in cardiomyocytes. Finally, engrailed-GATA4 was shown to block MEK1-ERK1/2-induced cardiomyocyte hypertrophy, suggesting at least one mechanism whereby MEK1-ERK1/2 signaling might induce hypertrophy.
Evidence implicating MEK1-ERK1/2 signaling in cardiac hypertrophy.
The role that MEK1-ERK1/2 signaling plays in regulating the cardiac hypertrophic response is an area of ongoing investigation. ERK1/2 has been shown to be activated in cultured neonatal rat cardiomyocytes by agonist stimulation and cell stretching, suggesting a functional role in hypertrophy (3, 35). In support of this hypothesis, Glennon et al. demonstrated that ERK signaling was necessary for PE-induced cardiomyocyte hypertrophy using antisense oligonucleotides (14). Similarly, using the MEK1 inhibitor PD98059, a number of studies have concluded a role for ERK signaling in mediating aspects of cardiomyocyte hypertrophy (1, 7, 18, 19, 42). More significantly, adenoviral gene transfer of dominant-negative MEK1 in cultured cardiomyocytes was shown to block PE-, endothelin-1-, isoproterenol-, and stretch-induced hypertrophy, providing convincing evidence without the use of pharmacologic inhibitors (39). In vivo, transgenic mice expressing activated MEK1 in the heart showed constitutive ERK1/2 activation and prominent cardiac hypertrophy, demonstrating that the MEK1-ERK1/2 signaling pathway can initiate the hypertrophic response in vivo (5). Similar results were also reported in cultured cardiomyocytes infected with an adenovirus expressing activated MEK1 (5, 39).
While the studies discussed above have implicated the MEK1-ERK1/2 pathway as a hypertrophic regulator, other studies utilizing the inhibitor PD98059 have concluded no significant role for this pathway in mediating the hypertrophic response (6, 29, 30, 32, 36, 43). These discordant reports likely reflect differences in experimental conditions such as cardiomyocyte culture densities and medium composition, concentration and time course of PD98059 treatment, and the type and timing of agonist stimulation. Additional genetic strategies utilizing dominant-negative or gene knockout approaches in the mouse heart will be instrumental for unequivocally resolving the importance of MEK1-ERK1/2 signaling in the heart.
The downstream mechanisms whereby MEK-ERK1/2 signaling might regulate cardiac hypertrophic growth are only partially understood. In this report, GATA4 was shown to be an important downstream target of MEK1-ERK1/2 signaling, suggesting a novel molecular response pathway that results in altered gene expression. Such an observation is also consistent with previous reports implicating GATA4 as a regulator of β-MHC, angiotensin type 1A, and ANF promoter activity in response to hypertrophic stimuli (15, 17, 25). Thus, GATA4 functions as an important terminal effector of MEK1-ERK1/2 signaling initiated by a wide array of neural-humoral stimuli in cardiac myocytes. Such an effector function suggests a transcriptional mechanism whereby global alterations in hypertrophic gene expression are coordinated in the heart.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (to J.D.M. [HL60562] and B.E.M. [HL43662]) and by a Pew Charitable Trust Scholar Award (J.D.M.).
REFERENCES
- 1.Aoki H, Richmond M, Izumo S, Sadoshima J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem J. 2000;347:275–284. [PMC free article] [PubMed] [Google Scholar]
- 2.Belaguli N S, Sepulveda J L, Nigam V, Charron F, Nemer M, Schwartz R J. Cardiac tissue-enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol. 2000;20:7550–7558. doi: 10.1128/mcb.20.20.7550-7558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogoyevitch M A, Glennon P E, Andersson M B, Clerk A, Lazou A, Marshall C J, Parker P J, Sugden P H. Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the integration of two signaling pathways leading to myocyte hypertrophy. J Biol Chem. 1994;269:1110–1119. [PubMed] [Google Scholar]
- 4.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594–598. doi: 10.1038/25166. [DOI] [PubMed] [Google Scholar]
- 5.Bueno O F, De Windt L J, Tymitz K M, Witt S A, Kimball T R, Klevitsky R, Hewett T E, Jones S P, Lefer D J, Peng C-F, Kitsis R N, Molkentin J D. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choukroun G, Hajjar R, Kyriakis J M, Bonventre J V, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Investig. 1998;102:1311–1320. doi: 10.1172/JCI3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clerk A, Michael A, Sugden P H. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol. 1998;142:523–535. doi: 10.1083/jcb.142.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crossley M, Orkin S H. Phosphorylation of the erythroid transcription factor GATA-1. J Biol Chem. 1994;269:16589–16596. [PubMed] [Google Scholar]
- 9.Davis R J. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 10.De Windt L J, Lim H W, Haq S, Force T, Molkentin J D. Calcineurin promotes protein kinase C and c-Jun NH2-terminal kinase activation in the heart. Cross-talk between cardiac hypertrophic signaling pathways. J Biol Chem. 2000;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- 11.Durocher D, Charron F, Warren R, Schwartz R J, Nemer M. The cardiac transcription factors Nkx2–5 and GATA-4 are mutual cofactors. EMBO J. 1997;16:5687–5696. doi: 10.1093/emboj/16.18.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Y, Yan W, Mohun T J, Evans S M. Vertebrate tinman homologues XNkx2–3 and XNkx2–5 are required for heart formation in a functionally redundant manner. Development. 1998;125:4439–4449. doi: 10.1242/dev.125.22.4439. [DOI] [PubMed] [Google Scholar]
- 13.Garrington T P, Johnson G L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 14.Glennon P E, Kaddoura S, Sale E M, Sale G J, Fuller S J, Sugden P H. Depletion of mitogen-activated protein kinase using an antisense oligodeoxynucleotide approach downregulates the phenylephrine-induced hypertrophic response in rat cardiac myocytes. Circ Res. 1996;78:954–961. doi: 10.1161/01.res.78.6.954. [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa K, Lee S J, Jobe S M, Markham B E, Kitsis R N. cis-Acting sequences that mediate induction of beta-myosin heavy chain gene expression during left ventricular hypertrophy due to aortic constriction. Circulation. 1997;96:3943–3953. doi: 10.1161/01.cir.96.11.3943. [DOI] [PubMed] [Google Scholar]
- 16.Hautala N, Takola H, Luodonpaa M, Puhakka J, Romppanen H, Vuolteenaho O, Ruskoaho H. Pressure overload increases GATA4 binding activity via endothelin-1. Circulation. 2001;103:730–735. doi: 10.1161/01.cir.103.5.730. [DOI] [PubMed] [Google Scholar]
- 17.Herzig T C, Jobe S M, Aoki H, Molkentin J D, Cowley A W, Jr, Izumo S, Markham B E. Angiotensin II type 1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc Natl Acad Sci USA. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kodama H, Fukuda K, Pan J, Sano M, Takahashi T, Kato T, Makino S, Manabe T, Murata M, Ogawa S. Significance of ERK cascade compared with JAK/STAT and PI3-K pathway in gp130-mediated cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279:H1635–H1644. doi: 10.1152/ajpheart.2000.279.4.H1635. [DOI] [PubMed] [Google Scholar]
- 19.Liang F, Lu S, Gardner D G. Endothelin-dependent and -independent components of strain-activated brain natriuretic peptide gene transcription require extracellular signal-regulated kinase and p38 mitogen-activated protein kinase. Hypertension. 2000;35:188–192. doi: 10.1161/01.hyp.35.1.188. [DOI] [PubMed] [Google Scholar]
- 20.Liang Q, De Windt L J, Witt S A, Kimball T R, Markham B E, Molkentin J D. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 21.Manak I J, Prywes R. Mutation of serum response factor phosphorylation sites and the mechanism by which its DNA-binding activity is increased by casein kinase II. Mol Cell Biol. 1991;11:3652–3659. doi: 10.1128/mcb.11.7.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molkentin J D. The zinc finger-containing transcription factors GATA-4, -5, and -6: ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 23.Molkentin J D, Kalvakolanu D V, Markham B E. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Mol Cell Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morimoto T, Hasegawa K, Kaburagi S, Kakita T, Wada H, Yanazume T, Sasayama S. Phosphorylation of GATA-4 is involved in alpha 1-adrenergic agonist-responsive transcription of the endothelin-1 gene in cardiac myocytes. J Biol Chem. 2000;275:13721–13726. doi: 10.1074/jbc.275.18.13721. [DOI] [PubMed] [Google Scholar]
- 25.Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morin S, Paradis P, Aries A, Nemer M. Serum response factor-GATA ternary complex required for nuclear signaling by a G-protein-coupled receptor. Mol Cell Biol. 2001;21:1036–1044. doi: 10.1128/MCB.21.4.1036-1044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrisey E E, Ip H S, Tang Z, Parmacek M S. GATA-4 activates transcription via two novel domains that are conserved within the GATA-4/5/6 subfamily. J Biol Chem. 1997;272:8515–8524. doi: 10.1074/jbc.272.13.8515. [DOI] [PubMed] [Google Scholar]
- 28.Morrisey E E, Ip H S, Tang Z, Lu M M, Parmacek M S. GATA5: a transcriptional activator expressed in a novel temporally and spatially restricted pattern during embryonic development. Dev Biol. 1997;183:21–36. doi: 10.1006/dbio.1996.8485. [DOI] [PubMed] [Google Scholar]
- 29.Post G R, Goldstein D, Thuerauf D J, Glembotski C C, Brown J H. Dissociation of p44 and p42 mitogen-activated protein kinase activation from receptor-induced hypertrophy in neonatal rat ventricular myocytes. J Biol Chem. 1996;271:8452–8457. doi: 10.1074/jbc.271.14.8452. [DOI] [PubMed] [Google Scholar]
- 30.Ramirez M T, Sah V P, Zhao X L, Hunter J J, Chien K R, Brown J H. The MEKK-JNK pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J Biol Chem. 1997;272:14057–14061. doi: 10.1074/jbc.272.22.14057. [DOI] [PubMed] [Google Scholar]
- 31.Sadoshima J-I, Jahn L, Takahashi T, Kulik T J, Izumo S. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. J Biol Chem. 1992;267:10551–10560. [PubMed] [Google Scholar]
- 32.Silberbach M, Gorenc T, Hershberger R E, Stork P J, Steyger P S, Roberts C T., Jr Extracellular signal-regulated protein kinase activation is required for the anti-hypertrophic effect of atrial natriuretic factor in neonatal rat ventricular myocytes. J Biol Chem. 1999;274:24858–24864. doi: 10.1074/jbc.274.35.24858. [DOI] [PubMed] [Google Scholar]
- 33.Stokoe D, Caudwell B, Cohen P T, Cohen P. The substrate specificity and structure of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem J. 1993;296:843–849. doi: 10.1042/bj2960843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugden P H, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;24:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 35.Sugden P H, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal. 1997;9:337–351. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- 36.Thorburn J, Frost J A, Thorburn A. Mitogen-activated protein kinases mediate changes in gene expression, but not cytoskeletal organization associated with cardiac muscle cell hypertrophy. J Cell Biol. 1994;126:1565–1572. doi: 10.1083/jcb.126.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thuerauf D J, Hanford D S, Glembotski C C. Regulation of rat brain natriuretic peptide transcription. A potential role for GATA-related transcription factors in myocardial cell gene expression. J Biol Chem. 1994;269:17772–17775. [PubMed] [Google Scholar]
- 38.Towatari M, May G E, Marais R, Perkins G R, Marshall C J, Cowley S, Enver T. Regulation of GATA-2 phosphorylation by mitogen-activated protein kinase and interleukin-3. J Biol Chem. 1995;270:4101–4107. doi: 10.1074/jbc.270.8.4101. [DOI] [PubMed] [Google Scholar]
- 39.Ueyama T, Kawashima S, Sakoda T, Rikitake Y, Ishida T, Kawai M, Yanashita T, Ishido S, Hotta H, Yaokyama M. Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J Mol Cell Cardiol. 2000;32:947–960. doi: 10.1006/jmcc.2000.1135. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Su B, Sah V P, Heller-Brown J, Han J, Chien K R. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem. 1998;272:5423–5426. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 41.Xia Y, McMillin J B, Lewis A, Moore M, Zhu W G, Williams R S, Kellems R E. Electrical stimulation of neonatal cardiac myocytes activates the NFAT3 and GATA4 pathways and up-regulates the adenylosuccinate synthetase 1 gene. J Biol Chem. 2000;275:1855–1863. doi: 10.1074/jbc.275.3.1855. [DOI] [PubMed] [Google Scholar]
- 42.Yue T L, Gu J L, Wang C, Reith A D, Lee J C, Mirabile R C, Kreutz R, Wang Y, Maleeff B, Parsons A A, Ohlstein E H. Extracellular signal-regulated kinase plays an essential role in hypertrohic agonists, endothelin-1, and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 43.Zechner D, Thuerauf D J, Hanford D S, McDonough P M, Glembotski C C. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J Cell Biol. 1997;139:115–127. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]