

Abstract
Targeting amyloid-β plaques and tau tangles has failed to provide effective treatments for Alzheimer’s disease and related dementias (ADRD). A more fruitful pathway to ADRD therapeutics may be the development of therapies that target common signaling pathways that disrupt synaptic connections and impede communication between neurons. In this review, we present our characterization of a signaling pathway common to several neurological diseases featuring dementia including Alzheimer’s disease, frontotemporal dementia, Lewy body dementia, and Huntington’s disease. This signaling pathway features the cleavage of tau by caspase-2 (Casp2) yielding Δtau314 (Casp2/tau/Δtau314). Through a not yet fully delineated mechanism, Δtau314 catalyzes the mislocalization and accumulation of tau to dendritic spines leading to the internalization of AMPA receptors and the concomitant weakening of synaptic transmission. Here, we review the accumulated evidence supporting Casp2 as a druggable target and its importance in ADRD. Additionally, we provide a brief overview of our initial medicinal chemistry explorations aimed at the preparation of novel, brain penetrant Casp2 inhibitors. We anticipate that this review will spark broader interest in Casp2 as a target for restoring synaptic dysfunction in ADRD.
Keywords: caspase-2, tau, Alzheimer’s disease, ADRD, Δtau314, caspase-2 inhibitors, cognition, synapse
Introduction
The promise of discovering better treatments for Alzheimer’s disease and related dementias (ADRD), among the most feared and frequent causes of debilitation in the elderly, has eluded the valiant efforts of researchers worldwide. One reason for the lack of progress is that available interventions do not treat the underlying pathophysiological mechanisms of these diseases, targeting putative pathological triggers rather than the processes that mediate their detrimental effects on the brain. Another reason is the recent recognition that most patients with dementia have multiple pathologies, potentially tempering the effects of single-target therapies. Therefore, the ability to target a unique signaling pathway that is present in multiple diseases is an increasingly important but unmet medical need. The development of therapies that repair synaptic connections and restore communication between neurons in the brain will undoubtedly improve the treatment of ADRD. There is an urgent need for better treatments since available interventions do not address the underlying mechanisms of these diseases.
Multiple experimental therapies targeting fibrillar amyloid-β (Aβ) plaques and tau tangles have failed to produce robust results. The tau protein undergoes a variety of posttranslational modifications in ADRD, resulting in the formation of tau fibrils.1,2 However, fibrillar tau has been dissociated from neuronal death and network dysfunction, implicating non-fibrillar species in mediating cognitive abnormalities.3,4 Targeting the non-fibrillar forms of Aβ and tau that are diffusely distributed throughout the brain, alone or in combination with anti-fibril therapies, may be a more successful strategy.
Caspase-2 – Role and Function
Caspases (cysteine-dependent aspartate-directed proteases) are a group of proteases that cleave target proteins after aspartic acid residues.5 They originally evolved to mediate apoptosis, a form of programmed cell death.6 Interestingly, caspases have evolved in vertebrates to also play multiple roles in brain function including synaptic plasticity, dendritic development, learning and memory.7–9 Caspases can be subdivided into three groups: initiator caspases (e.g. caspase-2, −8, −9, −10), effector caspases (e.g. caspase-3, −6, −7) and inflammatory caspases (e.g. caspase-1, −4, −5).5 All caspases are synthesized as inert zymogens or pro-caspases. Initiator caspases are activated by the assembly of zymogens into multicomponent complexes triggered by an intrinsic apoptotic pathway via cellular stress or an extrinsic apoptotic pathway by extracellular ligands via cell surface death receptors.10–12 Thus primed, the initiator caspases activate effector caspases by cleaving their corresponding zymogens.13 The activated effector caspases go on to degrade specific sets of proteins from among several hundred caspase-sensitive cell proteins to induce apoptosis.13
Caspase-2 (Casp2) is a highly conserved caspase.5 Casp2 participates in apoptosis triggered by a variety of stimuli, such as endoplasmic reticulum stress, metabolic stress, and DNA damage to name but a few.14–18 This is observed in the periphery in the process of adipocyte apoptosis which manifests as metabolic syndrome. Casp2 deficiency protected mice from diet-induced obesity, the metabolic syndrome, and nonalcoholic steatohepatitis or nonalcoholic fatty liver disease (NASH/NAFLD).19–21 In the central nervous system, Casp2 mediates retinal ganglion cell death22,23, synaptic weakening24, neurodegeneration due to ischemic damage,25,26 and the decrease in spine density found in neuronal cultures from a mouse model of AD.9 This panoply of roles has captured the attention of researchers and spurred interest in Casp2 as an exciting and promising drug target.
Two observations may temper enthusiasm for inhibiting Casp2: 1) Cascade-2 knockout mice display the complete absence of Casp2 results in aging-related loss of bone mass;27 2) Casp2 can act as a tumor suppressor in some mouse models.28–32 Arguing in favor of the safety of inhibiting Casp2 are the observations that mice carrying null mutations for Casp2 are viable and devoid of severe phenotypic abnormalities,33 have no discernable physiological abnormalities at 420 days, and exhibit a normal median life expectancy (950 days).34 Two additional observations that mitigate potential concerns related to inhibiting Casp2 are that memory deficits in mice can be reversed with a 35% reduction of Casp29,35 and intravitreal Casp2 siRNA is safe23. These observations suggest that short term inhibition of Casp2 is unlikely to cause deleterious effects.
Caspase-2 and Tau in Alzheimer’s Disease and Related Dementias
Caspases are activated by Aβ and caspase-2 has been shown to be required for Aβ1–42-induced cell death.36 These activated caspases cleave the neuronal protein tau,37 which is abnormally processed in ADRD.38 Caspase cleavage of tau is attributed mainly to Casp2, Casp3 and Casp610,35,39,40 producing tau fragments that have been shown in experimental models to induce neurodegeneration (Figure 1).41 However, it was recently demonstrated that Casp6 cleaved tau generates rare age-dependent argentophilic deposits but fails to induce tau hyperphosphorylation or aggregation, neurodegeneration, glial inflammation, or cognitive deficits.42 Casp2 was directly implicated as a key driver of synaptic dysfunction in J20 APP transgenic mice. This was proposed to be by activation of the RhoA/ROCK-II signaling pathway that leads to the collapse of dendritic spines via cleavage of ROCK-II.9
Figure 1.
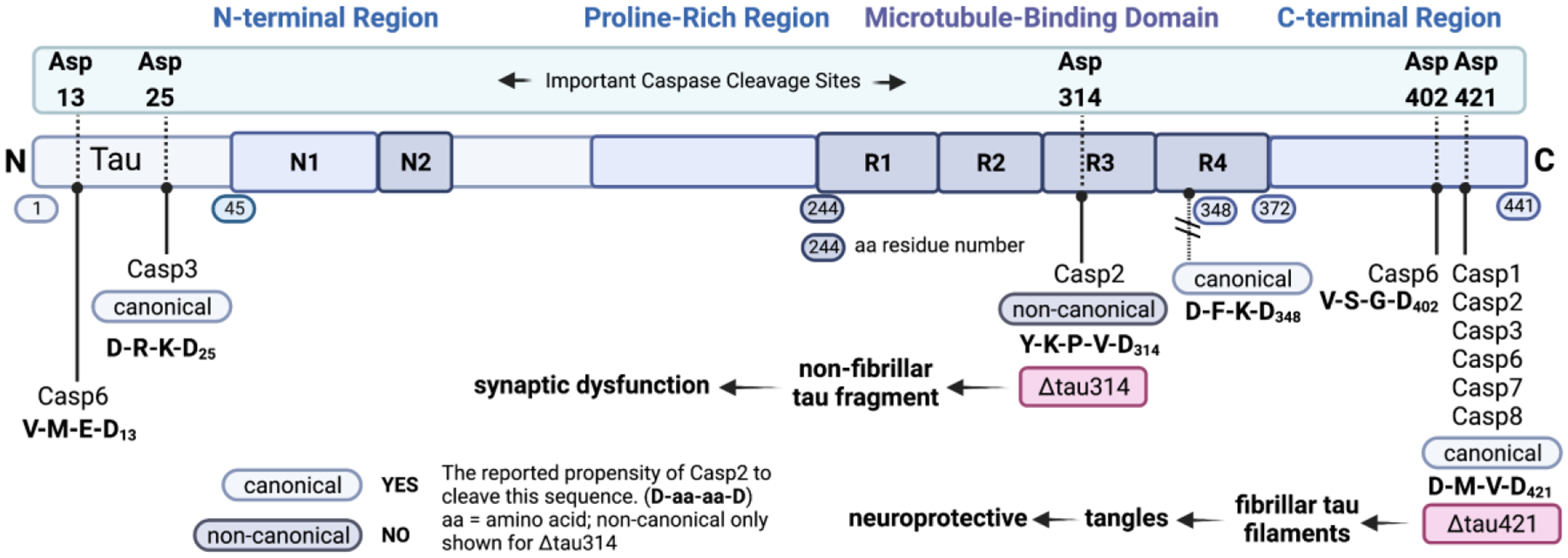
Sites in 2N/4R tau that have been observed to be cleaved by caspases. These results are typically observed in in vitro experiments with recombinant tau.46,47 (1) Asp421: cleavage site for Casp1, Casp2, Casp3, Casp6, Casp7, and Casp8. Cleavage here leads to fibrillar tau filaments and to tangles. This is a canonical cleavage site (D-aa-aa-D, where aa = amino acid) for Casp2, 3, 7, 8 and a non-canonical cleavage site for Casp1. Casp2 cleaves tau at this site only in vitro.35 (2) Asp314: Casp2 cleavage site. Cleavage here results in the formation of Δtau314, a non-fibrillar form of tau. This is a non-canonical cleavage site. Casp2 cleaves tau at this site in vitro and in vivo.35 (3) Caspase cleavage sites in the N-terminal region of tau. Asp13: cleavage site for Casp6 (canonical). Asp25: cleavage site for Casp3 (canonical). (4) Although this is a canonical cleavage site for Casp2, 3, 7, 8, no cleavage at this site has been observed. Tau image based on Xia et al.48 Created with BioRender.com.
Figure 1 shows a cartoon schematic of 2N/4R tau, indicating known sites of caspase cleavage. For clarity, post-translational modifications and cleavage by other proteases are not shown. Five sites of cleavage are known though there may be others that have not yet been detected. The tetrameric amino acid sequences for each of these sites are shown, along with a classification of the site as being a canonical sequence for Casp2. In general, Casp2 cleaves N-terminally to D-aa-aa-D (where aa = amino acid) sequences. Cleavage of tau at D13, D25, and D402 by casp2 has not been reported, but this has not been investigated fully. Four features of this schematic are noteworthy: 1. Cleavage at D421 by Casp1, Casp2, Casp3, Casp6, Casp7, and Casp8 has been well-characterized as leading to Δtau421. This tau fragment has been shown to form fibrillar filaments and tangles which have been shown to be neuroprotective;43,44 2. The cleavage of tau to give Δtau314 occurs at a non-canonical site for Casp2. We have shown that this fragment of tau is non-fibrillary, most likely because this cleavage is proximal to PHF6 which is considered essential for tau self-assembly.45 It is not known why this site is especially sensitive to Casp2 cleavage but binding of tau outside Casp2’s active site (exosites) may facilitate this cleavage; 3. Two other caspase cleavage sites are located near the N-terminus of tau and are canonical for both Casp6 and Casp3 cleavage. 4. Notably, caspase cleavage at D348 is not observed, even though this sequence is canonical for both Casp2 and Casp3 (and non-canonical for other caspases). It is possible that this proteolysis, and that at D13 and D25 occurs but is followed by other cleavage events that make it non-observable.
Our findings in Zhao et al. suggest that tau and Casp2 form a toxic partnership.49 The 35kDa tau fragment generated when Casp2 cleaves tau at Asp314 (D314) enters the dendritic spine along with full-length tau, impairing synaptic function.35 The identification of D314 as the site at which Casp2 cleaves tau to form Δtau314 was achieved by mass spectrometry.35 The ability of each of the eight known caspases expressed in the brain was also tested, and only Casp2 generated a 35-kDa tau fragment, presumably recognizing the non-canonical cleavage sequence YKPVD314 in tau.35 To verify the assumption that Casp2 cleaves tau at D314, a tau variant in which D314 was mutated to glutamate (D314E) was prepared and shown to resist Casp2 cleavage,35 firmly supporting the hypothesis that Δtau314 is formed when tau is cleaved by Casp2.
The presence of Δtau314 correlates inversely with memory function in tau transgenic mice and is elevated in humans with Alzheimer’s disease or mild cognitive impairment.35 Δtau314 reversibly impairs cognitive and synaptic function in animal and cellular models of tauopathies.35 The mislocalization and accumulation of tau in dendritic spines mediate synaptic dysfunction independent of neurodegeneration.50 In cell cultures, tau containing the P301L mutation (tauP301L) mislocalizes and accumulates in dendritic spines35, the principal loci of excitatory neurotransmission and synaptic plasticity which underlie learning and memory. Tau mislocalization resulted in decreased synaptic transmission due to the reduction of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) in the spines.50 When D314 was mutated to glutamate (D314E) to prevent caspase cleavage, tauP301L no longer mislocalized to spines.35 TauP301L also failed to mislocalize to spines in neurons cultured from Casp2 null mice.35 Conversely, recombinant Δtau314 did localize to spines even in neurons lacking Casp2.35 These data indicate that Casp2 catalyzes the mislocalization and accumulation of mutant tau and the reduction of AMPAR in spines, and also excludes the possibility that other proteases besides Casp2 induce tau mislocalization.
Under physiological conditions, Casp2 reduces functional AMPAR in dendritic spines leading to long-term depression (LTD).24 LTD is a form of synaptic plasticity that weakens excitatory neurotransmission and underlies the physiological process of forgetting, an essential component of normal cognition that is carefully regulated by neural signals (Figure 2).51 Recent studies of human brain specimens in our group have revealed elevated levels of Casp2 and Δtau314 not only in AD35,52, but also in Parkinson’s disease with dementia (PDD)53, Lewy body dementia (LBD)53, Huntington’s disease54 and frontotemporal dementia (FTD; here Δtau314 levels were elevated in the brains of rTg4510 (Zhao et al.) and rT2 mice (Steuer et al.), not in human brain specimens)35,55 (Figure 3). The elevated levels of Casp2 in these conditions suggest that the physiological Casp2-LTD signaling pathway24 is co-opted and overstimulated by pathological triggers in several neurodegenerative conditions, leading to excessive synaptic weakening and dementia. While neural signals activate the physiological Casp2-LTD pathway, the precise triggers that activate the pathological Casp2-LTD pathway are unknown. Ubiquitous features of neurodegenerative disorders such as oxidative stress, ER stress, and DNA damage have been shown to activate Casp2 and could contribute to its dysregulation.14–18 Under pathological conditions, the attenuation of excitatory postsynaptic neurotransmission is mediated by excessive Casp2 cleavage of tau leading to the accumulation of tau and reduction of AMPAR in dendritic spines. Whether the cleavage of tau by Casp2 plays a role in physiological LTD remains to be investigated. The hyper-activation of Casp2 weakens excitatory neurotransmission resulting in unregulated forgetting (Figure 3).
Figure 2.
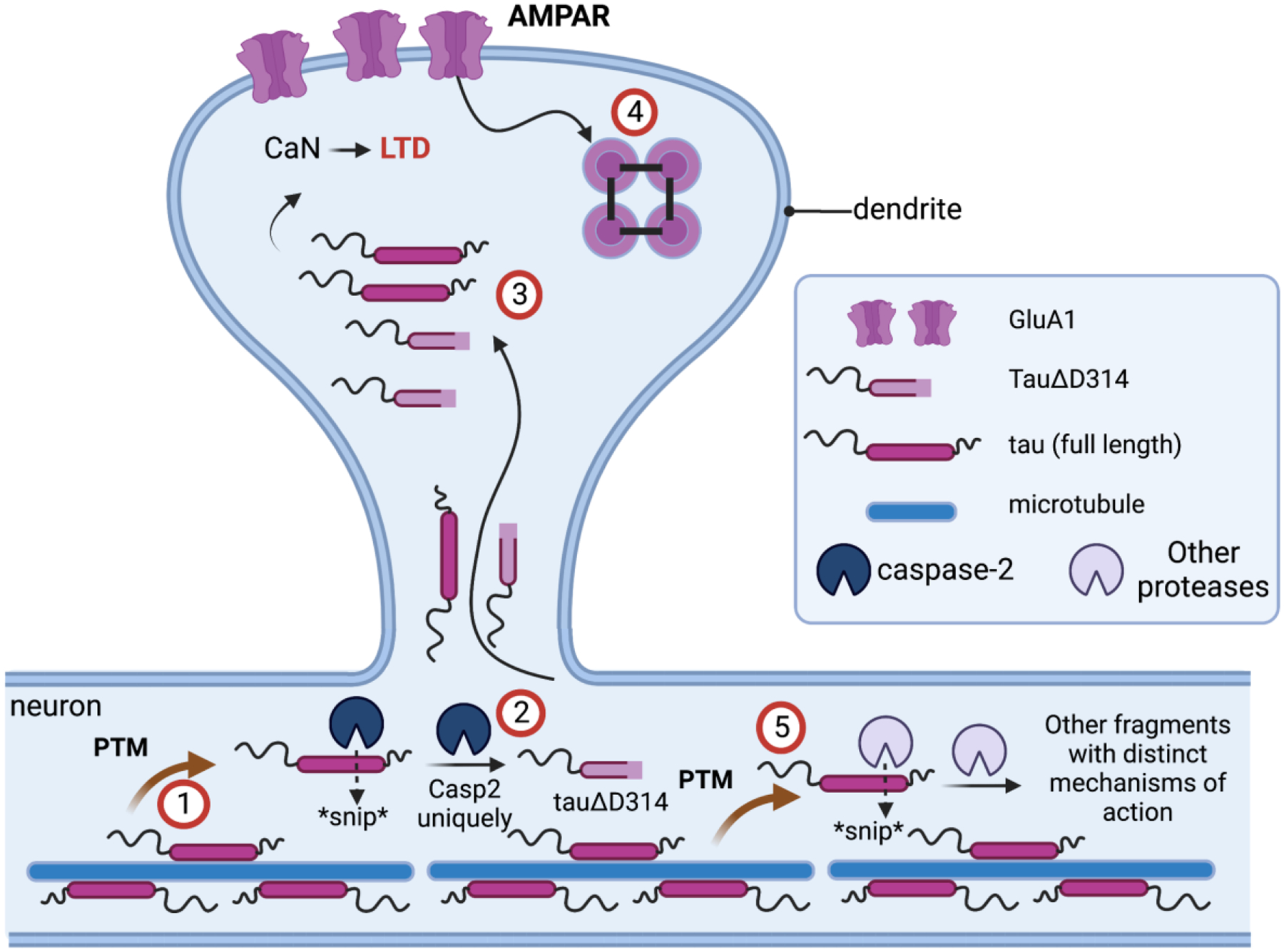
The current mechanistic understanding of the roles of Casp2 and tau in synaptic function. (1) Tau undergoes post-translational modification (PTM) and is released from microtubules. (2) PTM-tau is cleaved by active Casp2 to yield Δtau314. (3) Δtau314 catalyzes the movement of tau from the neuron into the dendrite thereby positively regulating calcineurin activity, (4) which in turn causes reversible internalization of AMPAR at the synapse. Under normal physiological conditions, neural signals regulate Casp2 activity leading to long-term depression (LTD). However, under pathological conditions various forms of cellular stress abnormally activate Casp2 independent of the neural signals leading to a dysfunctional, sustained LTD-like state that depresses excitatory neurotransmission. (5) Other proteases cleave tau leading to fragments with distinct mechanism of action. Created with BioRender.com.
Figure 3.
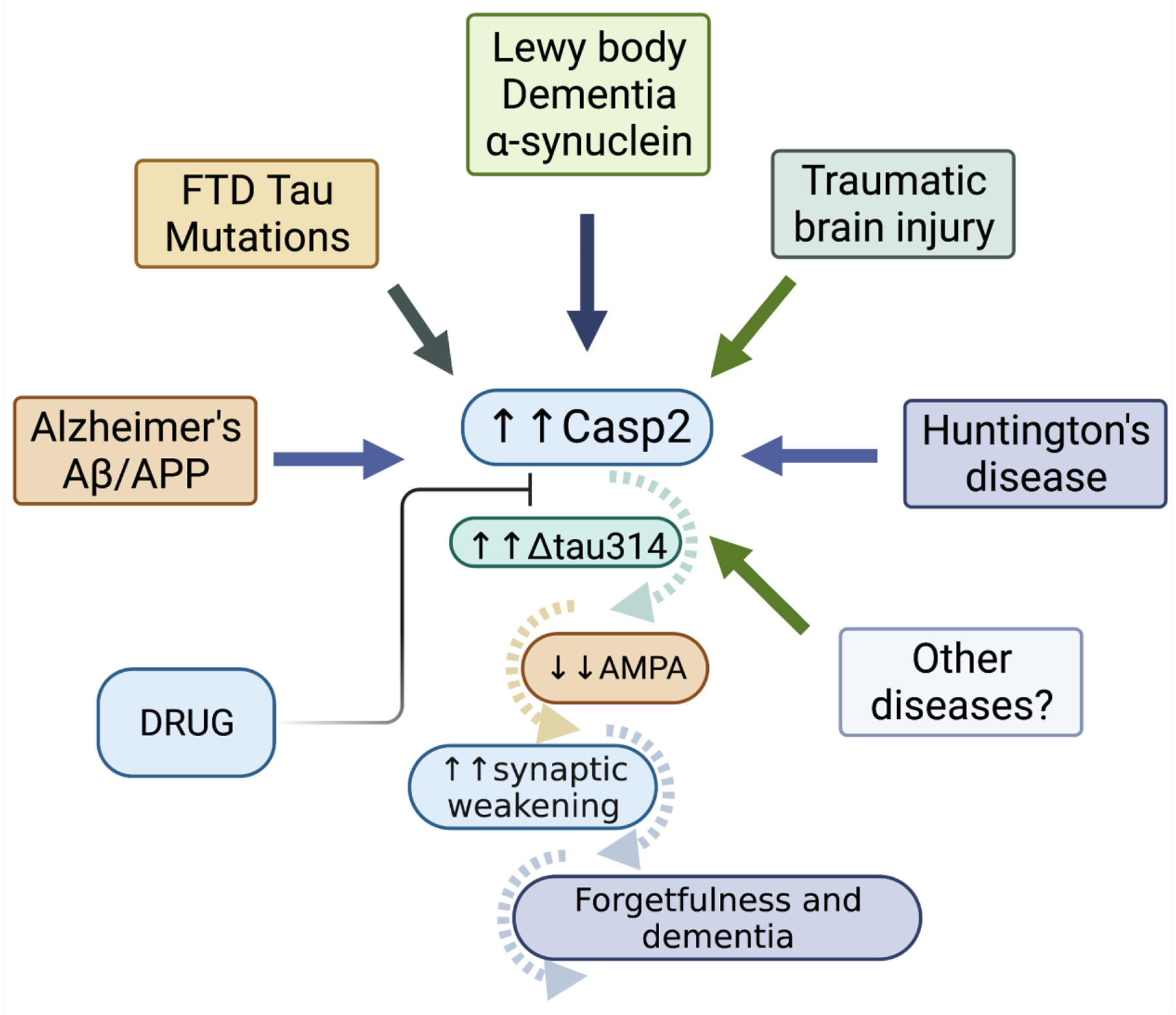
The Casp2/tau/Δtau314 cascade of events we propose to be involved in dementia. Cellular stress (typically in the form of ER stress, reactive oxygen, DNA damage, tau mutants such as tauP301L, etc.) activates Casp2, which in turn increases levels of Δtau314. This leads to the internalization of AMPAR, synaptic weakening and, ultimately, forgetfulness and dementia in multiple neurodegenerative conditions. The successful development of drugs that inhibit Casp2 could halt this insidious cascade of events and restore cognition. Created with BioRender.com.
Taken together, these results demonstrate that the pathological fragment Δtau314 which results from Casp2 cleavage of tau may be associated with synaptic dysfunction and dementia in multiple diseases.
Targeting Caspase-2 by Developing Selective Inhibitors
The identification of Casp2 cleavage of tau as a common mediator of synaptic dysfunction and dementia augments the significance and importance of developing Casp2 inhibitors. From a medicinal chemistry perspective, extensive efforts are still needed to accelerate the discovery of effective drugs. Although several studies on Casp2 inhibitors have been published in recent years,11,56,57 elementary features such as oral availability and brain penetration have not yet been achieved. Our initial studies on the development of selective Casp2 inhibitors followed an approach based on the canonical but unselective Casp2 inhibitor AcVDVAD-CHO and the tau cleavage sequence YKPVD314.58 A set of 35 pentapeptide ligands provided helpful insights into structure-affinity relationships (SAR) and achieved maximum selectivity with AcVDV(Dab)D-CHO (pKi (Casp2) = 7.26) by introducing the amino acid Dab (2,4-diaminobutyric acid) at position 2 (selectivity factor of ~30 over Casp3).58 Surprisingly, the pentapeptide AcYKPVD-CHO, derived from the tau cleavage site, showed no affinity for either Casp2 or Casp3.58 By systematically exchanging individual amino acid positions, the amino acid lysine at P4 and proline at P3 were thought to be responsible for this lack of activity.58
The development and expression of a series of recombinant tau mutants in an in vitro cleavage assay were consistent with these medicinal chemistry findings.58 In the search for new lead structures with excellent Casp2 selectivity and drug suitability, another approach was taken by our groups in Bresinsky et al.59 This approach featured the investigation of inhibitors based on cleavage sites specific to Casp2 in proteins other than tau.59 The cleavage sequences of Golgin-16060, the protein transport protein Sec16A61, transcription regulatory factor 161, the Ral GTPase-activating protein subunit alpha-161, the Holliday junction recognition protein61, and mouse double minute 2 homolog (MDM2, human and mouse)62 were selected based on the reported specificity of Casp2 cleavage and the pentapeptides AcGESPD-CHO, AcWDRAD-CHO, AcQDTRD-CHO, AcADRTD-CHO, AcLDVPD-CHO, AcFDVPD-CHO, and AcITVKD-CHO were prepared.59 This extended peptide series included another 53 new truncated molecules forming tetra- and tripeptides, but these peptides did not demonstrate enhanced Casp2 selectivity or affinity. These results support the hypothesis that a five-amino acid motif is required for efficient Casp2 inhibition.59 The inhibitors AcLDVPD-CHO (pKi (Casp2/cpCasp2) = 7.88/7.66) and AcFDVPD-CHO (pKi (Casp2/cpCasp2) = 8.15/8.03) derived from mouse and human MDM2, respectively, showed the highest affinity for Casp2 but no selectivity for Casp2 over Casp3.59 The pentapeptide AcITVKD-CHO was the only inhibitor derived from natural cleavage sequences that exhibited some selectivity for Casp2 (2-fold selectivity) with moderate affinity (pKi (Casp2) = 6.40) and, interestingly, does not contain an aspartic acid at position P4.59 Crystal structures (Casp3) of AcITV(Dab)D-CHO derived from an expanded AcITVKD-CHO-series confirmed that the P4-Thr is a good isostere for the P4-Asp and causes little change in protein conformation.59
Our subsequent study (Singh et al.) went a step further, incorporating peptidomimetic moieties and unnatural amino acids with the aim of obtaining more bioavailable structures.63 To achieve this goal, known structural elements which enhanced selectivity and efficacy from previous SAR studies57,64,65 were combined. In addition, S-33q (Figure 4) and R-33q, the pure P2 epimers of the previously published racemic 33q,65 were synthesized and pharmacologically characterized. While R-33q did not demonstrate significant affinity for Casp2 and Casp3, S-33q exhibited significant affinity for Casp2 (pKi = 7.89) with 70-fold Casp2/Casp3 selectivity.63 Alterations at amino acid positions P4 and P5 resulted in compound 3 (Figure 4) which had 794-fold Casp2 selectivity over Casp3 and Casp2 activity in the double-digit nanomolar range (pKi = 7.40).63 Further modification of the reactive warhead from aldehyde to nitrile at position P1 to improve pharmacokinetic and dynamic properties led to compound 1 (Figure 4) which exhibited high affinity (pKi (Casp2) = 8.12) and a promising selectivity profile in an in vitro caspase panel assay (123-fold selective towards Casp3 and >2000-fold selective towards Casp1, 6, 7, 9) and only weak activity towards the proteolytic enzymes thrombin and cathepsin B (IC50 = 18,620 nM, selectivity > 1600-fold).63 Especially the low activity to other cysteine proteases like cathepsin B is a promising feature for this class of inhibitors, regarding their specificity. Figure 5 shows the computationally predicted covalent docking (CovDock, Schrödinger)66,67 of compound 1 with the Casp2 apo-structure (ligand removed) from PDB:1PYO. Key interactions are shown. Although the aldehyde warhead is a prominent feature of the recently approved drug voxelotor68 for the treatment for sickle cell anemia and was an excellent warhead in our early SAR studies, this reactive group is often considered undesirable due to its high electrophilicity and susceptibility to oxidative metabolism. In contrast, nitrile-based covalent inhibitors have also been clinically validated, are known for their safety and efficacy in humans69 (e.g. nirmatrelvir a covalent inhibitor of the SARS-CoV-2 main protease), and have been used successfully in other caspase inhibitors.64 Compound 1 is a covalent reversible Casp2 inhibitor with stronger potency and higher specificity than previously reported inhibitors. More advanced biological assays confirmed that compound 1 effectively inhibits Casp2-mediated cleavage of tau and blocks the production of Δtau314.63 Using cultured rat and mouse hippocampal neurons expressing tauP301S (a commonly used model of frontotemporal dementia), it was demonstrated that compound 1 prevented the disproportionate accumulation of tauP301S in dendritic spines.63 Lastly, in an experimental electrophysiological setup, it was shown that expression of tauP301S impairs postsynaptic function of excitatory synapses, as determined by recording mEPSCs, and that compound 1 completely normalizes mEPSCs and rescues synaptic function.63 These results indicate that Casp2 mediates its pathological effects catalytically, paving the way for further efforts to develop small molecule Casp2 inhibitors to treat ADRD. Indeed, this structural evolution will be necessary because, given the high molecular weight (674.74 Da) and high TPSA (218.03 Å2) of compound 1, we do not expect it to enter the brain in significant amounts with these physicochemical properties.
Figure 4.
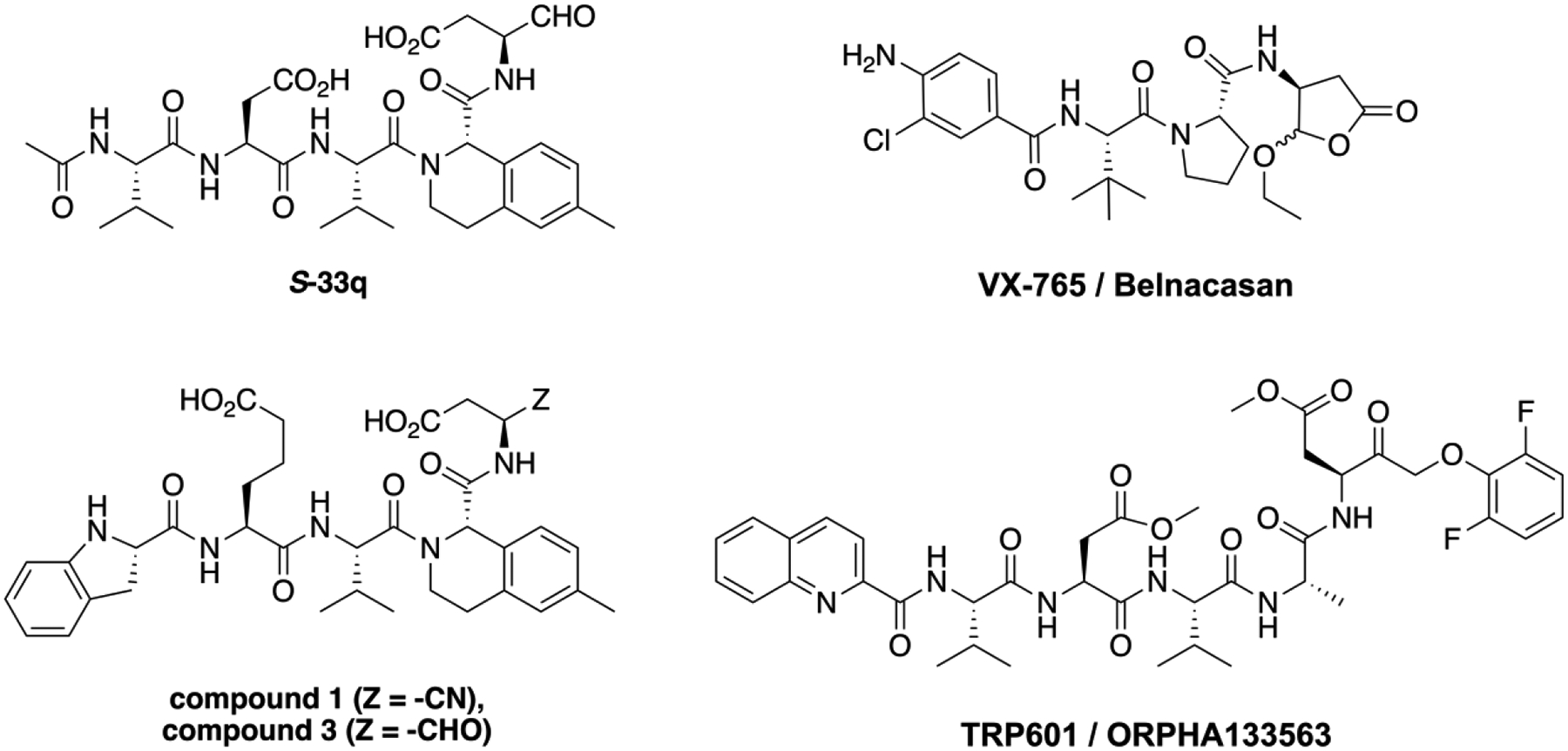
Structures of selective Casp2 inhibitors S-33q, compound 1, and compound 3, and the caspase prodrugs VX-765 (Casp1 inhibitor) and TRP601 (Casp2/3 inhibitor).
Figure 5.
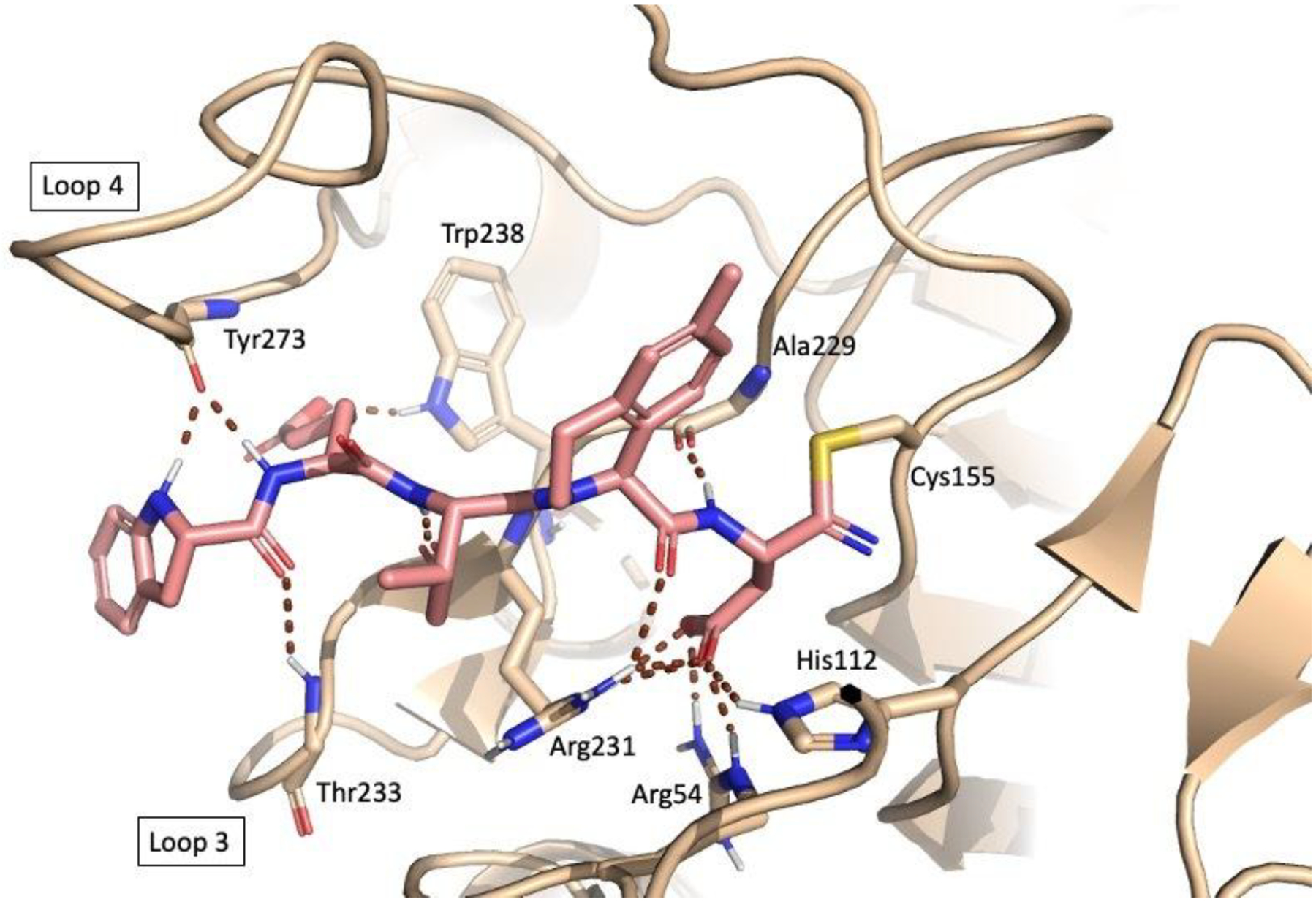
The computationally predicted (CovDock, Schrödinger)66,67 binding of compound 1 with the Casp2 apo-structure (ligand removed) from PDB:1PYO. Important interactions that confer selectivity are shown: the P5 indoline amide interaction with the amide of Y273; the P2 4-methyltetrahydroisoquinoline interaction with the S2 subpocket. (For further details on the computational methods see Bresinsky et al.58).
Summary & Outlook
The targeting of tau tangles in the treatment of ADRD has thus far been unsuccessful, warranting efforts to target other forms of tau.70–72 We expect that targeting the formation of Δtau314, a soluble fragment of tau, may be more fruitful. Several lines of evidence in humans and mice validate Casp2 as a target73: 1) Lowering Casp2 improves memory, synaptic function and neurodegeneration in mouse models of four neurodegenerative disorders – Alzheimer’s disease (AD),9 Huntington’s disease (HD)74 (newly recognized as a tauopathy)75, Parkinson’s disease (PD)76, and frontotemporal dementia (FTD)35; 2) In mice modeling FTD, reducing Casp2 levels ~35% (using a morpholino ASO) lowers brain levels of the pathological tau fragment, Δtau314, and reverses memory deficits within 6 weeks:4 Unfortunately, our attempts to produce ASO as therapeutics were clouded by unexpected challenges of toxicity. 3) Lowering Casp2 levels in these mice before neurons have died normalizes memory function;4 4) Even when 50% of hippocampal CA1 neurons have died, lowering Casp2 levels partially restores memory function;4 5) Casp2-resistant mutant tau with a D314E mutation delays memory loss and neurodegeneration in transgenic mice modeling FTD;55 6) Brain levels of Casp2 and Δtau314 are elevated in patients with AD35,52, HD54, and LBD53.
In vitro systems used to elucidate the underlying pathophysiological mechanisms of in vivo observations have shown that: 1) Tau accumulates and AMPAR are reduced in the dendritic spines of cultured primary neurons expressing mutant tau,35,49 but these processes do not occur in neurons lacking Casp2;35 2) Compound 1 prevents the accumulation of tau and reduction of AMPAR in the dendritic spines of mutant tau-expressing neurons;63 3) Rendering tau resistant to Casp2 maintains AMPAR in the dendritic spines of neurons from mouse models of FTD.52,55
The Casp2-LTD signaling pathway is a natural pathway that is dysregulated, rather than a potential artifact of an experimental disease model, as is the case in transgenic rodents with supra-abundant amyloid plaque deposition.77,78 It is therefore likely to be present in humans and clinically relevant. Because Casp2-LTD is a normal pathway, the misactivated pathway is likely not an unintended artifact of our experimental ADRD models, minimizing the possibility that a Casp2 inhibitor will target a mechanism that does not exist or plays only a minor role in the pathogenesis of actual ADRD in humans. In aggregate, these in vivo and in vitro findings suggest that uncontrolled, pathological activation of the Casp2/tau/Δtau314 signaling pathway markedly weakens excitatory neurotransmission and exaggerates forgetting, and that a Casp2 inhibitor will improve synaptic function and cognition in patients with mild and moderate ADRD. Targeting the Casp2/tau/Δtau314 pathway can prevent and reverse cognitive decline.4,35,55 One exciting clinical implication is that a Casp2 inhibitor might not only improve the ability to learn and remember new information, but also recover lost memories by fortifying the synapses in engrams that are suppressed by reductions in AMPAR.79,80 We predict that Casp2 inhibitors have the potential to improve dementia in Alzheimer’s disease and several related dementias, which is an increasingly important consideration given that most patients have more than one pathology.81–83
Several improvements of current Casp2 inhibitors are needed. Foremost is bioavailability, specifically orally administered and brain penetrant Casp2 inhibitors that restore synaptic function and reverse memory deficits in mouse models of ADRD. Our findings thus far play an important role in the design of new peptidomimetic inhibitors. It is unlikely that we can design these compounds solely based on adherence to Lipinski’s Rule of 5 (molecular weight, logP, hydrogen bond donors/acceptors, TPSA, rotatable bonds).84 Challenges to blood-brain barrier penetration have been overcome in the area of kinase inhibitors which have been a notoriously difficult class of compounds to target to the brain.85 Additionally, the development of Casp2 inhibitors as prodrug compounds may be another promising approach; brain penetrant caspase inhibitors that are prodrugs already exist. For example, TRP601/ORPHA133563 (Figure 4), an irreversible inhibitor of Casp3 and Casp2 that is a prodrug of Δ2Me-TRP601 (IC50 (Casp2) = 7.4 nM, IC50 (Casp3) = 0.42 nM), protects the newborn rodent brain against excitotoxicity, hypoxia-ischemia, and perinatal arterial stroke with a 6-hour therapeutic time window and no adverse effects on physiological parameters.86 Furthermore, TRP601 did not significantly modify the activity of a broad panel of enzyme, peptide and non-peptide receptors, nuclear receptors, ion channels or amine transporters at 10 μM.86 When administered intravenously (i.v.), TRP601 (1 mg/kg) rapidly entered the brain in adult rats (Tmax = 25 min; Cmax = 120 ng/mL), and no TRP601-related cytotoxic effects were observed when administered i.v. to adult dogs at doses up to 3 mg/kg/day for 14 days.86 Another example is VX-765 (Belnacasan) (Figure 4), an orally absorbed prodrug of desethyl-VX-765 (VRT-043198), a reversible covalent, potent Casp1 inhibitor.64,87 VX-765 has been tested in two Phase II clinical trials (treatment resistant partial epilepsy, NCT01048255; psoriasis, NCT00205465) but has not advanced further.
The uncertainties associated with developing successful peptidomimetic drugs that are bioavailable and penetrate into the brain warrant adopting additional strategies built around structures other than peptides. One potential approach involves the development of small molecule fragments that are obtained by identifying Casp2 inhibitors in libraries containing molecules < 300 daltons that possess favorable physicochemical properties. Such fragments may be used as starting points to optimize pharmacokinetic and pharmacodynamic properties. This strategy has worked well in the development of BACE (β-site cleaving enzyme; an aspartic protease) inhibitors,88 and appears to have been successfully applied to Casp6.89 Optimizing such compounds regarding to structure-activity and structure-physical properties could ultimately lead to a brain-penetrant small molecule Casp2 inhibitor.
Overall, the biological data for Casp2 as a target in ADRD is clear and compelling, and the medicinal chemistry advances for development of Casp2 inhibitors are promising for eventual use in clinical trials. Given the ever-increasing number of patients with ADRD, intensive research in this area is a vital and critical goal.
Acknowledgments
All authors have read the journal’s policy on disclosure of potential conflicts of interest and have disclosed any financial or personal relationship with organizations that could potentially be perceived as influencing the described research. All authors have read the journal’s authorship statement. The authors disclosed all potential conflicts of interests. The manuscript has been reviewed by and approved by all named authors. The authors thank Jessica Fuller for her contribution regarding Figure 5.
Funding
The authors were supported by NIH Grant R01AG0623199 (Ashe, Walters), R01AG60766 (Ashe), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Forschungsstipendium 436921318, Pockes), ADDF-Harrington Scholar Award (Repairing Neurotransmission in Alzheimer’s Disease and Related Disorders by Targeting Caspase-2, Ashe), Lucas Brothers Foundation (Caspase-2 Inhibitors to Reverse Cognitive Decline, Ashe).
Abbreviations:
- AD
Alzheimer’s disease
- ADRD
Alzheimer’s disease and related dementias
- AMPAR
α amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- APP
amyloid precursor protein
- ASO
antisense oligonucleotide
- Asp
aspartic acid
- Aβ
amyloid-β
- BACE
β-site cleaving enzyme
- CARD
caspase recruitment domain
- Casp
caspase
- Dab
2,4-diaminobutyric acid
- DED
death effector domain
- ER
endoplasmic reticulum
- FTD
frontotemporal dementia
- HD
Huntington’s disease
- LBD
Lewy body dementia
- logP
logarithmic value of the partition coefficient
- LTD
long-term depression
- MDM2
mouse double minute 2 homolog
- mEPSC
miniature excitatory synaptic current
- PD
Parkinson’s disease
- PDD
Parkinson’s disease with dementia
- PS
presenilin
- PTM
post-translational modification
- SAR
structure-affinity relationship
- Δtau314
tau cleavage product
- TPSA
topological polar surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors are founders and shareholders of Myriel, Inc.
References
- 1.Fath T, Eidenmüller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. Journal of Neuroscience. 2002;22(22):9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixit R, Ross JL, Goldman YE, Holzbaur ELF. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science. 2008;319(5866):1086–1089. doi: 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA. Pathological Tau Disrupts Ongoing Network Activity. Neuron. 2015;85(5):959–966. doi: 10.1016/j.neuron.2015.01.025 [DOI] [PubMed] [Google Scholar]
- 4.Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miles MA, Kitevska-Ilioski T, Hawkins CJ. Old and novel functions of caspase-2. International review of cell and molecular biology. 2017;332:155–212. [DOI] [PubMed] [Google Scholar]
- 6.Kurokawa M, Kornbluth S. Caspases and Kinases in a Death Grip. Cell. 2009;138(5):838–854. doi: 10.1016/j.cell.2009.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biundo F, d’Abramo C, Tambini MD, et al. Abolishing Tau cleavage by caspases at Aspartate421 causes memory/synaptic plasticity deficits and pre-pathological Tau alterations. Translational Psychiatry. 2017;7(8):e1198–e1198. doi: 10.1038/tp.2017.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salazar IL, Caldeira MV, Curcio M, Duarte CB. The Role of Proteases in Hippocampal Synaptic Plasticity: Putting Together Small Pieces of a Complex Puzzle. Neurochemical Research. 2016;41(1–2):156–182. doi: 10.1007/s11064-015-1752-5 [DOI] [PubMed] [Google Scholar]
- 9.Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nature communications. 2013;4(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochemical Journal. 2004;384(2):201–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poreba M, Groborz K, Navarro M, Snipas SJ, Drag M, Salvesen GS. Caspase selective reagents for diagnosing apoptotic mechanisms. Cell Death & Differentiation. 2019;26(2):229–244. doi: 10.1038/s41418-018-0110-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Current Opinion in Cell Biology. 2003;15(6):725–731. doi: 10.1016/j.ceb.2003.10.009 [DOI] [PubMed] [Google Scholar]
- 13.Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death & Differentiation. 2003;10(1):76–100. doi: 10.1038/sj.cdd.4401160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouchier-Hayes L, Green DR. Caspase-2: the orphan caspase. Cell death and differentiation. 2012;19(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung HH, Lynn Kelly N, Liston P, Korneluk RG. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: A role for the IAPs. Experimental Cell Research. 2006;312(12):2347–2357. doi: 10.1016/j.yexcr.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 16.Manzl C, Peintner L, Krumschnabel G, et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death And Differentiation. 2012;19:1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S. Caspase 2 in apoptosis, the DNA damage response and tumour suppression: enigma no more? Nature Reviews Cancer. 2009;9:897. [DOI] [PubMed] [Google Scholar]
- 18.Vakifahmetoglu-Norberg H, Zhivotovsky B. The unpredictable caspase-2: what can it do? Trends in Cell Biology. 2010;20(3):150–159. doi: 10.1016/j.tcb.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 19.Machado MV, Michelotti GA, Jewell ML, et al. Caspase-2 promotes obesity, the metabolic syndrome and nonalcoholic fatty liver disease. Cell Death & Disease. 2016;7(2):e2096–e2096. doi: 10.1038/cddis.2016.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehl AME, Michelotti GA, Machado MV, Kornbluth SA, Seager-Johnson E, Lindblom KR. Use of caspase-2 inhibitors to treat and prevent the metabolic syndrome. Published online 2016.
- 21.Kim JY, Garcia-Carbonell R, Yamachika S, et al. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell. 2018;175(1):133–145.e15. doi: 10.1016/j.cell.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vigneswara V, Berry M, Logan A, Ahmed Z. Pharmacological Inhibition of Caspase-2 Protects Axotomised Retinal Ganglion Cells from Apoptosis in Adult Rats. Cho KS, ed. PLoS ONE. 2012;7(12):e53473. doi: 10.1371/journal.pone.0053473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vigneswara V, Ahmed Z. Long-term neuroprotection of retinal ganglion cells by inhibiting caspase-2. Cell Death Discovery. 2016;2(1). doi: 10.1038/cddiscovery.2016.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu ZX, Tan JW, Xu H, et al. Caspase-2 promotes AMPA receptor internalization and cognitive flexibility via mTORC2-AKT-GSK3β signaling. Nature communications. 2019;10(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niizuma K, Endo H, Nito C, Myer DJ, Kim GS, Chan PH. The PIDDosome mediates delayed death of hippocampal CA1 neurons after transient global cerebral ischemia in rats. Proceedings of the National Academy of Sciences. 2008;105(42):16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlsson Y, Wang X, Schwendimann L, et al. Combined effect of hypothermia and caspase-2 gene deficiency on neonatal hypoxic–ischemic brain injury. Pediatric Research. 2012;71(5):566–572. doi: 10.1038/pr.2012.15 [DOI] [PubMed] [Google Scholar]
- 27.Callaway DA, Riquelme MA, Sharma R, Lopez-Cruzan M, Herman BA, Jiang JX. Caspase-2 modulates osteoclastogenesis through down-regulating oxidative stress. Bone. 2015;76:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoo NJ, Lee JW, Kim YJ, et al. Loss of caspase-2,-6 and-7 expression in gastric cancers. Apmis. 2004;112(6):330–335. [DOI] [PubMed] [Google Scholar]
- 29.Holleman A Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood. 2005;106(5):1817–1823. doi: 10.1182/blood-2004-11-4296 [DOI] [PubMed] [Google Scholar]
- 30.Zohrabian VM, Nandu H, Gulati N, et al. Gene expression profiling of metastatic brain cancer. Oncology reports. 2007;18(2):321–328. [PubMed] [Google Scholar]
- 31.Egorshina AYu, Zamaraev AV, Lavrik IN, Zhivotovsky BD, Kopeina GS. Caspase-2 as an Oncosupressor and Metabolism Regulator: What Life Will Bring over the Long Run? Molecular Biology. 2018;52(5):648–659. doi: 10.1134/S0026893318050060 [DOI] [PubMed] [Google Scholar]
- 32.Puccini J, Dorstyn L, Kumar S. Caspase-2 as a tumour suppressor. Cell Death & Differentiation. 2013;20(9):1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergeron L, Perez GI, Macdonald G, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes & development. 1998;12(9):1304–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Padalecki SS, Chaudhuri AR, et al. Caspase-2 deficiency enhances aging-related traits in mice. Mechanisms of ageing and development. 2007;128(2):213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Kotilinek LA, Smith B, et al. Caspase-2 cleavage of tau reversibly impairs memory. Nature medicine. 2016;22(11):1268. [DOI] [PubMed] [Google Scholar]
- 36.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by β-amyloid. Journal of Neuroscience. 2000;20(4):1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. Journal of neuropathology & experimental neurology. 2005;64(2):104–112. [DOI] [PubMed] [Google Scholar]
- 38.Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology. 2014;71(4):505–508. [DOI] [PubMed] [Google Scholar]
- 39.Jarero-Basulto JJ, Luna-Muñoz J, Mena R, et al. Proteolytic cleavage of polymeric tau protein by caspase-3: implications for Alzheimer disease. Journal of neuropathology and experimental neurology. 2013;72(12):1145–1161. [DOI] [PubMed] [Google Scholar]
- 40.Pérez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA. Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Molecular Neurobiology. 2018;55(2):1004–1018. doi: 10.1007/s12035-017-0385-x [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochemical Society Transactions. 2010;38(4):955–961. doi: 10.1042/BST0380955 [DOI] [PubMed] [Google Scholar]
- 42.Noël A, Foveau B, LeBlanc AC. Caspase-6-cleaved Tau fails to induce Tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell death & disease. 2021;12(3):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82(4):756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464(7292):1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oakley SS, Maina MB, Marshall KE, et al. Tau filament self-assembly and structure: Tau as a therapeutic target. Frontiers in Neurology. 2020;11:590754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gamblin TC, Chen F, Zambrano A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proceedings of the national academy of sciences. 2003;100(17):10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rissman RA, Poon WW, Blurton-Jones M, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. The Journal of clinical investigation. 2004;114(1):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia Y, Lloyd GM, Giasson BI. Targeted proteolytic products of τ and α-synuclein in neurodegeneration. Essays in Biochemistry. 2021;65(7):905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Troy CM, Shelanski ML. Caspase-2 and tau—a toxic partnership? Nature Medicine. 2016;22:1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoover BR, Reed MN, Su J, et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron. 2010;68(6):1067–1081. doi: 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nature reviews neuroscience. 2010;11(7):459–473. [DOI] [PubMed] [Google Scholar]
- 52.Liu P, Smith BR, Montonye ML, et al. A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Scientific reports. 2020;10(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith BR, Nelson KM, Kemper LJ, et al. A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease. Acta Neuropathologica Communications. 2019;7(1). doi: 10.1186/s40478-019-0765-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu P, Smith BR, Huang ES, et al. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta neuropathologica communications. 2019;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steuer EL, Kemper LJ, Hlynialuk CJ, et al. Blocking site-specific cleavage of human tau delays progression of disease-related phenotypes in genetically matched tau-transgenic mice modeling frontotemporal dementia. Journal of Neuroscience. 2022;42(23):4737–4754. doi: 10.1523/JNEUROSCI.0543-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poreba M, Salvesen GS, Drag M. Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nature protocols. 2017;12(10):2189–2214. [DOI] [PubMed] [Google Scholar]
- 57.Poreba M, Rut W, Groborz K, Snipas SJ, Salvesen GS, Drag M. Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells. Cell Death & Differentiation. 2019;26(12):2695–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bresinsky M, Strasser JM, Vallaster B, et al. Structure-Based Design and Biological Evaluation of Novel Caspase-2 Inhibitors Based on the Peptide AcVDVAD-CHO and the Caspase-2-Mediated Tau Cleavage Sequence YKPVD314. ACS Pharmacology & Translational Science. 2022;5(1):20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bresinsky M, Strasser JM, Hubmann A, et al. Characterization of caspase-2 inhibitors based on specific sites of caspase-2-mediated proteolysis. Archiv der Pharmazie.:e2200095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mancini M, Machamer CE, Roy S, et al. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. The Journal of cell biology. 2000;149(3):603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Julien O, Zhuang M, Wiita AP, et al. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proceedings of the National Academy of Sciences. 2016;113(14):E2001–E2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oliver TG, Meylan E, Chang GP, et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Molecular cell. 2011;43(1):57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh G, Liu P, Yao KR, et al. Caspase-2 Inhibitor Blocks Tau Truncation and Restores Excitatory Neurotransmission in Neurons Modeling FTDP-17 Tauopathy. ACS Chem Neurosci. 2022;13(10):1549–1557. doi: 10.1021/acschemneuro.2c00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boxer MB, Quinn AM, Shen M, et al. A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety. ChemMedChem. 2010;5(5):730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maillard MC, Brookfield FA, Courtney SM, et al. Exploiting differences in caspase-2 and −3 S2 subsites for selectivity: Structure-based design, solid-phase synthesis and in vitro activity of novel substrate-based caspase-2 inhibitors. Bioorganic & Medicinal Chemistry. 2011;19(19):5833–5851. doi: 10.1016/j.bmc.2011.08.020 [DOI] [PubMed] [Google Scholar]
- 66.Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O. Structure-based virtual screening approach for discovery of covalently bound ligands. Journal of chemical information and modeling. 2014;54(7):1941–1950. [DOI] [PubMed] [Google Scholar]
- 67.Zhu K, Borrelli KW, Greenwood JR, et al. Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. Journal of chemical information and modeling. 2014;54(7):1932–1940. [DOI] [PubMed] [Google Scholar]
- 68.Metcalf B, Chuang C, Dufu K, et al. Discovery of GBT440, an orally bioavailable R-state stabilizer of sickle cell hemoglobin. ACS medicinal chemistry letters. 2017;8(3):321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Owen DR, Allerton CM, Anderson AS, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–1593. [DOI] [PubMed] [Google Scholar]
- 70.Panza F, Lozupone M. The challenges of anti-tau therapeutics in Alzheimer disease. Nature Reviews Neurology. Published online 2022:1–2. [DOI] [PubMed] [Google Scholar]
- 71.Mullard A Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nature reviews Drug discovery. 2021;20(1):3–6. [DOI] [PubMed] [Google Scholar]
- 72.Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Frontiers in aging neuroscience. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu P, Ashe KH. The molecular implications of a caspase-2-mediated site-specific tau cleavage in tauopathies. Neural Regeneration Research. 2021;16(9):1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carroll JB, Southwell AL, Graham RK, et al. Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease. Molecular neurodegeneration. 2011;6(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fernández-Nogales M, Cabrera JR, Santos-Galindo M, et al. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nature medicine. 2014;20(8):881–885. [DOI] [PubMed] [Google Scholar]
- 76.Tiwari M, Herman B, Morgan WW. A knockout of the caspase 2 gene produces increased resistance of the nigrostriatal dopaminergic pathway to MPTP-induced toxicity. Experimental neurology. 2011;229(2):421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66(5):631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ashe KH. The biogenesis and biology of amyloid β oligomers in the brain. Alzheimer’s & Dementia. 2020;16(11):1561–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Ramirez S, Pang PT, et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484(7394):381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511(7509):348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boyle PA, Wilson RS, Yu L, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Annals of neurology. 2013;74(3):478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Annals of neurology. 2018;83(1):74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Annals of neurology. 2019;85(1):114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 1997;23(1–3):3–25. [DOI] [PubMed] [Google Scholar]
- 85.Keylor MH, Gulati A, Kattar SD, et al. Structure-Guided Discovery of Aminoquinazolines as Brain-Penetrant and Selective LRRK2 Inhibitors. Journal of Medicinal Chemistry. 2021;65(1):838–856. [DOI] [PubMed] [Google Scholar]
- 86.Chauvier D, Renolleau S, Holifanjaniaina S, et al. Targeting neonatal ischemic brain injury with a pentapeptide-based irreversible caspase inhibitor. Cell Death & Disease. 2011;2(9):e203–e203. doi: 10.1038/cddis.2011.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chlorophenyl)-methanoyl]-amino}-3, 3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R, 3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1β and IL-18. Journal of Pharmacology and Experimental Therapeutics. 2007;321(2):509–516. [DOI] [PubMed] [Google Scholar]
- 88.Stamford A, Strickland C. Inhibitors of BACE for treating Alzheimer’s disease: a fragment-based drug discovery story. Current opinion in chemical biology. 2013;17(3):320–328. [DOI] [PubMed] [Google Scholar]
- 89.Renslo AR, Arkin MR, Neitz RJ, et al. Caspase 6 inhibitors and uses thereof. 2021, WO/2004/002961.