

Abstract
Pathologically activated neutrophils (PMN) with immune suppressive activity, which are termed myeloid-derived suppressor cells (PMN-MDSC), play a critical role in regulating tumor progression. These cells have been implicated in promoting tumor metastases by contributing to pre-metastatic niche formation. This effect was facilitated by enhanced spontaneous migration of PMN from bone marrow to the pre-metastatic niches during the early-stage of cancer development. The molecular mechanisms underpinning this phenomenon remained unclear. In this study, we found that syntaphilin (SNPH), a cytoskeletal protein previously known for anchoring mitochondria to the microtubule in neurons and tumor cells, could regulate migration of PMN. Expression of SNPH was decreased in PMN from tumor-bearing mice and cancer patients as compared to PMN from tumor-free mice and healthy donors, respectively. In Snph-knockout (SNPH-KO) mice, spontaneous migration of PMN was increased and the mice showed increased metastasis. Mechanistically, in SNPH-KO mice, the speed and distance travelled by mitochondria in PMN was increased, rates of oxidative phosphorylation and glycolysis were elevated, and generation of adenosine was increased. Thus, our study reveals a molecular mechanism regulating increased migratory activity of PMN during cancer progression and suggests a novel therapeutic targeting opportunity.
Introduction
Pathologically activated neutrophils (PMN) with immune suppressive functions, termed myeloid-suppressor cells (PMN-MDSC), have emerged as critical contributors to tumor progression including formation of pre-metastatic niches and regulation of immune responses in cancer (1–4). Migration of PMN-MDSC to distant sites is a critical component of their ability to promote tumor metastasis. Previously, we found that during early stages of tumor progression bone marrow (BM) PMN display potent ability to spontaneously migrate to distant sites and facilitate tumor metastasis (5). These cells lack the full biochemical and functional characteristics of MDSC but have up-regulated genes associated with ER stress response and elevated rates of oxidative phosphorylation and glycolysis (5). However, the molecular mechanisms that regulate the increased migratory ability of the BM PMN have yet to be defined.
Leukocyte migration requires the generation of a polarized morphology characterized by the formation of leading-edge pseudopods where F-actin is concentrated, and a highly contractile cell rear known as the uropod where most cellular organelles and the microtubule organizing center (MTOC) are located (6,7). This process requires energy in the form of ATP. Mitochondria are the most efficient source of ATP. Mitochondria cluster sites of high ATP demand in many cell types, and previous studies have suggested a possible direct functional interaction between these ATP-producing organelles and ATP-consuming cellular structures (8). Although PMN have relatively low abundance of mitochondria and historically the mitochondria in PMN were considered as largely to be involved in apoptosis (9), more recent studies have demonstrated a role for mitochondria in many PMN functions, including chemotaxis and antibacterial activity (10,11). Furthermore, chemokine-induced redistribution of mitochondria toward the cellular uropod can activate actomyosin contraction during lymphocyte migration (12).
Rapid rearrangement of the mitochondrial network to facilitate the supply of energy for specialized functions has been described in several cell types (13). Syntaphilin (SNPH) is a cytoskeletal protein that tethers and immobilizes axonal mitochondria to microtubules (14). Overexpression of SNPH reduces the number of motile mitochondria in axons (15). We previously found that tumor progression is associated with downregulation or loss of SNPH in tumor cells, and this correlates with shortened patient survival, increased mitochondrial trafficking to the cortical cytoskeleton, greater membrane dynamics, and heightened cell invasion (16). Loss of SNPH in tumor cells or expression of an SNPH mutant lacking the mitochondrial localization sequence results in increased metastatic dissemination in xenograft or syngeneic tumor models in vivo. Tumor cells that acquire the ability to metastasize in vivo constitutively downregulate SNPH and exhibit higher oxidative stress, reduced cell proliferation, and increased cell motility (17). In this study, we tested the hypothesis that SNPH is involved in regulation of myeloid-cell migration. We identified a regulatory role for SNPH in PMN migration that directly impacted tumor metastasis. SNPH deficiency increased PMN migration by increasing the speed and distance travelled by mitochondria through a mechanism involving phosphorylation of actomyosin, elevated metabolic activity of PMN, and increased production of adenosine. This mechanism is different to the mechanism by which SNPH deficiency enhanced mitochondria mobility in tumor cells and helps to understand increased motility of PMN in cancer with possible implications for therapeutic targeting.
Materials and Methods
Human subjects and samples.
Samples of peripheral blood were collected from 12 patients with previously untreated stage II–IV non-small cell lung cancer being cared for at the Helen F. Graham Cancer Center. Samples were collected before the start of treatment and processed on the day of collection. This cohort included 8 females and 4 males 48–74 years of age. 12 healthy donors were enrolled. The study was conducted in accordance to ethical standards of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of the Christiana Care Health System at the Helen F. Graham Cancer Center and The Wistar Institute. All patients and healthy donors signed IRB-approved consent forms.
Mouse models.
Mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of The Wistar Institute or AstraZeneca. BALB/c and C57BL/6 mice (female, 6–8 weeks old) were obtained from Charles River, OT-I TCR transgenic mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J, RRID:IMSR_JAX:003831) (female, 6–8 weeks old), Pmel mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J, PRID: IMSR_JAX:005023), and CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ, RRID:IMSR_JAX:002014) were purchased from Jackson Laboratory. Mice with a targeted deletion in the Snph gene Snphtm1Vlcg on a C57BL/6N background were described previously and are referred to here as Snph-knockout (SNPH-KO) mice (18). Mice were obtained from Mutant Mouse Resources & Research Centers (MMRRC). Transgenic Ret melanoma (19) and KPC pancreatic carcinoma tumor models (20) were described previously. BM chimera were generated by i.v. transplantation of 5×106 BM cells from wild-type (WT) or SNPH KO into lethally irradiated (1,000 rad) CD45.1 host mice. After 8 wks, mice were bled to check reconstitution by flow cytometry for CD45.1 and CD45.2 and only mice with > 87% chimerism were used for the experiments.
Cell lines.
The LL2 (Lewis lung carcinoma) tumor cell line expressing luciferase was a gift from R. Ramakrishnan (H. Lee Moffitt Cancer Center) received in 2014. EL-4 (ATCC Cat# TIB-39, RRID:CVCL_0255), B16F10 (ATCC Cat# CRL-6475, RRID:CVCL_0159), and LLC (ATCC Cat# CRL-1642, RRID:CVCL_4358) cell lines were purchased from ATCC in 2018. No additional authentication of the cell lines was performed. Cell lines were used for up to 8 passages and tested for mycoplasma contamination by using the Universal Mycoplasma detection kit (ATCC, Cat#30–1012K). These cells were maintained in DMEM (Sigma-Aldrich, Cat#56436C) supplemented with 10% FBS (Sigma-Aldrich, Cat#12103C) and 1% penicillin-streptomycin (ThermoFisher Scientific Cat#15140122) at 37 °C with 5% CO2.
Detection of metastasis by luciferase activity.
0.5×106 LL2-luciferase cells were injected s.c.. Three weeks later Luciferin (PerkinElmer, Cat#122799) was injected i.p. in mice before the imaging according to the manufacturer’s instruction. Mice were sacrificed, and bioluminescent images of the harvested lung were taken with IVIS Spectrum In Vivo Imaging System (IVIS, PerkinElmer) to detect luminescent signal coming from cancer cells. Analysis and quantification were performed with IVIS imaging system Living Image Software 4.7.4 (PerkinElmer). Photon counts in the area of interest were reported after subtraction of background.
Reagents.
A list of key reagents is provided in Supplemental Table S1.
Isolation of mouse neutrophils.
For the isolation of BM Ly6G+ neutrophils, cells were labeled with anti-Ly-6G MicroBeads UltraPure (Miltenyi Biotec Cat#130-120-337) and separated on MACS columns (Miltenyi Biotec Cat#130-042-401). For isolation of splenic neutrophils, spleens were put in a 70 μm strainer placed on a conical 50 mL Falcon tube and cut into small pieces. These pieces are then grinded against the cell strainer using the plunger of a 5 mL syringe and washed several times with cold CSB. Tubes were then centrifugated at 1,500 rpm at 4°C, the supernatant was removed and red blood cells were lysed by resuspending the cell pellet in ammonium chloride lysis buffer for 5 minutes at room temperature. Single-cell suspensions from lungs were prepared using mouse lung dissociation kit (Miltenyi Biotec, Cat#130-095-927) according to the manufacturer’s recommendations with an additional red blood cell lysis step as described above. Ly6G+ cells were then isolated using anti-Ly-6G MicroBeads UltraPure as described above.
Isolation of mouse monocytes.
CD11b+Ly6ChiLy6G− monocytes were sorted from BM of mice using MoFlo Astrios high-speed cell sorter.
Transwell assay for migration and chemotaxis.
The migration of unstimulated neutrophils or monocyte and chemotaxis of cells were measured as previously reported (5). Briefly, advanced RPMI (Thermo Fisher, Cat#12633–012) with the chemoattractants CXCL1 (BioLegend Cat#573702), CCL2 (R&D Systems, Cat#479-JE), or N-Formylmethionyl-leucyl-phenylalanine (fMLP; Sigma-Aldrich Cat#F3506) was placed in the bottom of the 3-μm-pore Transwell system (Neuro Probe Cat#101–3). Neutrophils were incubated with PSB12379 (Tocris) or 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (Sigma-Aldrich, Cat#C101) in advanced RPMI for 10 min before being plated on top of the Transwell (0.1×106 cells) and were incubated at 37°C, 5% CO2 for 1 h. 10μl medium was taken from the bottom wells and cells were counted by a hemocytometer. The quantification of migrated neutrophils was done with the formula N=n×104×0.029 (reflective of 29μl medium in the bottom well).
Seahorse assay.
Metabolic rates were determined using Seahorse XFe96 Flux Analyzers (Agilent Technologies) according to the manufacturer’s protocol. In brief, the microplate was coated with 22.4 μg ml−1 Cell-Tak (Fisher Cat#354240) using 200 mM sodium bicarbonate. Approximately 400,000 neutrophils were seeded per well immediately after isolation 80 μl of unbuffered RPMI (Sigma-Aldrich, Cat#R6504)) for XF96 analyzer. The microplate was incubated for 30 min at 37°C to allow the cells to settle into a monolayer. Unbuffered RPMI was gently added to the wells without disturbing the monolayer to bring the assay volume to 180 μl. The basal oxygen-consumption rate (OCR) and extracellular acidification rate (ECAR) were measured, in addition to rate changes after treatment with 5 μM oligomycin (SigmaAldrich Cat#O4876), 1 μM FCCP (Sigma-Aldrich Cat#C2920), and 0.75 μM rotenone (Sigma-Aldrich Cat#R8875) and 1 μM antimycin A (Sigma-Aldrich Cat#A8674).
Metabolomics.
Neutrophils from naïve tumor-free mice and PMN-MDSC from tumor-bearing mice were isolated from the BM, as described in Isolation of mouse neutrophils, and resuspended in culture medium which contains nutrients at physiological concentrations as was previously described (21). Cells were incubated for 1 hr in the presence of 5.5 mM of 13C6-glucose (Sigma-Aldrich, Cat#389374) and then harvested. For harvesting, cell pellets were washed twice in ice-cold PBS and extracted in a solution of LC-MS grade methanol, acetonitrile, and ultrapure water. Samples were centrifuged and the resulting cleared supernatant was transferred to a silanized MS vial and run by LC-MS.
LC-MS analysis was performed on a Q Exactive Hybrid Quadrupole-Orbitrap HF-X MS (Thermo Fisher Scientific) equipped with a HESI II probe and coupled to a Vanquish Horizon UHPLC system (Thermo Fisher Scientific). A total of 0.002 ml of sample was injected and separated by HILIC chromatography on a ZIC-pHILIC 2.1-mm column. Samples were separated by ammonium carbonate, 0.1% ammonium hydroxide, pH 9.2, and mobile phase B was acetonitrile. The LC was run at a flow rate of 0.2 ml/min and the gradient used was as follows: 0 min, 85% B; 2 min, 85% B; 17 min, 20% B; 17.1 min, 85% B; and 26 min, 85% B. The column was maintained at 45 °C and the mobile phase was also pre-heated at 45 °C before flowing into the column. The relevant MS parameters were as listed: sheath gas, 40; auxiliary gas, 10; sweep gas, 1; auxiliary gas heater temperature, 350 °C; spray voltage, 3.5 kV for the positive mode and 3.2 kV for the negative mode. Capillary temperature was set at 325 °C, and funnel RF level at 40. Samples were analyzed in full MS scan with polarity switching at scan range 65–975 m/z; 120,000 resolution; automated gain control (AGC) target of 1E6; and maximum injection time (max IT) of 100 ms. Identification and quantitation of metabolites was performed using an annotated compound library and TraceFinder 4.1 software for LC-MS data processing. Metabolite measurements were normalized based upon protein concentration determined from cell pellets.
F-actin measurement.
Total BM cells were stained with AQUA, CD11b-BV421 (Supplemental Table 1), and Ly6G-APC (BD Bioscience Cat#560599). Cells were washed and stimulated with doses of CXCL1 and fMLP in Advanced RPMI for the time points indicated in the figure legends. Cells were immediately fixed and permeabilized using Cytofix/Cytoperm Solution (BD Biosciences Cat#554722) and washed in Perm/Wash Buffer (BD) following the manufacturer’s protocol. Cells were stained with 1 unit of Phalloidin-AF488 (Thermo Fisher Cat#A12379) for 20 min at 4°C, washed in Perm/Wash Buffer, run on a LSRII flow cytometer, and analyzed using FlowJo (Tristar version 10.8.0).
RNA-sequencing (RNA-seq).
Neutrophils were isolated as described in Isolation of mouse neutrophils using Ly6G beads with purity was >95%. Total RNA was extracted from cell pellets using the Direct-zol RNA Miniprep (Zymo Research, Cat#R2051). RNA quality was validated using the TapeStation RNA ScreenTape (Agilent, Cat#5067–5579). 100 ng of total RNA was used to prepare a library for Illumina Sequencing using the Quant-Seq 3’mRNA-Seq Library Preparation Kit (Lexogen, Cat#015.96). Library quantity was determined using qPCR kit and absolute quantification with standard curve method (KAPA Biosystem). Overall library size was determined using the Agilent TapeStation and the DNA High Sensitivity D5000 ScreenTape (Agilent, Cat# G2992AA). Equimolar amounts of each sample library were pooled, denatured and Mid-Output, Paired-End, 150 cycle Next Generation Sequencing was done on a NextSeq 500 (Illumina, San Diego, CA, USA). Three samples of wild-type and SNPH deficient BM neutrophils were evaluated. No technical replicates were used. Lexogen Quantseq Library Preparation Kit was used for library preparation. Single-end read runs were used, with read lengths up to 75 bp in high output mode, with 14M per sample total read counts. Raw data was aligned against mm10 genome and gene-level read counts and FPKM values were estimated with Ensemble transcriptome information using RSEM v1.2.12 software (22). SNPH KO vs WT sample groups were compared using DESeq2 (23) software and normalized by the DESeq2 values were used for heatmap visualization. Overall changes were considered significant if passed FDR<5% threshold. Gene set enrichment analysis for genes that passed nominal p<0.05 threshold was performed using QIAGEN’s Ingenuity® Pathway Analysis software (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) based on “Canonical Pathways” and “Networks” options. RNA-seq data were deposited to NCBI GEO data repository under accession number GSE190505.
Real-time quantitative PCR.
RNA was extracted from mouse and human neutrophils using the Quick-RNA Miniprep Kit (Zymo Research Cat#R1055) according to the manufacturer’s instructions using 100 ng of the template. Reverse transcription was generated with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems Cat#4374967). Quantitative PCR was then performed using Power SYBR Green PCR Master Mix (Applied Biosystems Cat#4368708) on an ABI 7500 Fast instrument. Each experiment was performed in triplicates. Expression of mouse genes; Snph, Mfn1, Mfn2, and Drp1 iwas normalized to the expression of 18s and measured using ΔΔCT. Expression of human SNPH gene was normalized to GAPDH and measured using ΔΔCT. Primers are described in Supplementary Table S2.
Isolation of human neutrophils and PMN-MDSCs.
PMN-MDSCs and neutrophils were isolated from peripheral blood using double-density gradient of Histopaque-1077 and Histopaque-1119 (Sigma Aldrich Cat#10771 and Cat#11191, respectively). Then CD15-beads (Miltenyi, Cat#130-046-601) were used to isolate PMN-MDSCs from the low-density peripheral blood mononuclear cells and neutrophils from the high-density gradient. The purity of neutrophils and PMN-MDSCs with CD14−CD15+ phenotype was checked by flow cytometry and was > 95%.
Flow cytometry.
Antibody incubations were performed at 4°C for 15 min. Usually up to 1×106 cells were incubated with Fc-block (BD Biosciences, Cat#553142) for 10 min and surface staining was performed at 4°C for 15 min. For intracellular staining, cells were then fixed with FOXP3/transcription factor staining buffer set (eBioscience Cat#00-5523-00) according manufacturer instructions and incubated for 45 min at 4°C. Samples were analyzed using an LSRII or Symphony flow cytometer (BD Biosciences) and data were analyzed by FlowJo software 10.8.0 (Tristar RRID:SCR_008520). A list of antibodies used is in Supplementary Table S1. Gating strategy is shown in Supplementary Fig. S1.
Assessment of mitochondrial membrane potential was performed by staining with tetramethylrhodamine ethyl ester (TMRE) (Thermo Fisher, Cat#T669) (20 nM) in complete seahorse media (2mM glutamine and 20mM glucose) for 20 min at 37 °C. Excess mitochondrial dye was removed by washing and cells were further acquired by flow cytometry.
T-cell functional assay.
To measure proliferation, CD8+ T cells were purified from spleens from WT and SNPH KO mice using easysep Mouse CD8+ T cell isolation Kit (Stemcell Cat#19853), stained with Cell trace Far Red (Thermo Fisher Cat#C34564) and cultured with anti-CD3/CD28 beads (Thermo Fisher Cat#11452D) and IL2 (100U/ml) (PeproTech Cat#212-12) for 3 days. After 3 days, T-cell proliferation was assessed by flow cytometry. To measure IFNγ, CD8+ T cells were activated with anti-CD3/CD28 beads and IL2 (100U/ml) then restimulated with PMA/IONO (eBioscience™ Cell Stimulation Cocktail (plus protein transport inhibitors) (500X) Cat#00-4975-93) for 6 hours and intracellular IFNγ was measured by flow cytometry.
Western blot.
Neutrophils from BM were lysed in RIPA buffer (Sigma-Aldrich Cat#R0278) in the presence of protease inhibitor cocktail (Sigma-Aldrich Cat#P8340) and stored at −80°C. Whole-cell lysates were prepared and subjected to 10% SDS–PAGE and transferred to PVDF membrane. The membranes were probed overnight at 4°C with primary antibody. Membranes were then washed and incubated for 1 h at room temperature with secondary antibody conjugated with peroxidase and visualized with ECL (Amersham Biosciences, Cat# RPN2232). A list of antibodies used is in Supplementary Table S1.
Mitochondrial dynamic imaging.
Changes in mitochondrial morphology were investigated as described previously (24). High-optical-quality glass-bottom 35-mm plates (MatTek Corporation) were pre-coated with poly-L-lysine (Sigma-Aldrich, Cat#P4707) for 30 min at 37°C. WT and SNPH-KO PMN (1 × 104) were seeded and stained with 100nM MitoTracker Deep Red (Thermo Fisher, Cat#M46753) for 30 min and washed with complete medium. Cells were imaged on a Leica TCS SP8 X inverted confocal laser scanning microscope using a 63×, 1.40-numerical-aperture (NA) oil objective. Live imaging time-lapse was performed using a Tokai Hit incubation chamber equilibrated to bidirectional scanning at 8,000 Hz at 37°C with 5% CO2. Images were acquired every 3 sec for 1 min. Individual 12-bit images were acquired using a white-light supercontinuum laser (0.2% at 633 nm) and hybrid detectors at a 5× digital zoom with a pixel size of 70 nm by 70 nm and a step size of 0.260 μm. At least 6 single cells under each condition were collected for analysis. Initial postprocessing of the 3D sequences was carried out with Hyugens software to deconvolve the images. Images were imported into Leica LAS X software to study fission and fusion events and mitochondrial movements.
Time-lapse migration assay.
The wells of μ-Slide 4 Well Ph+ (ibidi) were coated with 50 μg/ml fibronectin (Sigma-Aldrich, Cat#F1141). Cells were plated (0.5 × 106 cells per well) in advanced RPMI without FBS and were incubated at 37°C and 5% CO2 for 10–15 min to allow cell attachment. The wells were gently washed with 2 × PBS to remove non-adherent cells, and Advanced RPMI was added to the wells. Cells were placed onto a motorized stage and were observed with a Nikon Eclipse TE300 fluorescence microscope maintained in an environment of 37°C and 5% CO2. A 10 × or 20 × objective was used to capture images during the course of the time lapse. Images were captured every 30 s over the course of 30 min from at least 6 different fields of view. Cell movement was tracked and analyzed using the ImageJ plugin Manual Tracking and the Chemotaxis tool respectively. ImageJ and the plugin are both freely available through the NIH website (http://rsbweb.nih.gov/ij/).
Suppression assays.
BM Ly6G+ neutrophils were prepared as described in Isolation of mouse neutrophils. Then cells were plated in U-bottomed 96-well plates in triplicates in complete RPMI and co-cultured at various ratios with total splenocytes from Pmel or OT-I mice in the presence of cognate peptides (OT-I, SIINFEKL, Anaspec Cat#AS-60193-1; Pmel, EGSRNQDWL, Cat# Anaspec AS-64752) for 48h. Then [3H] thymidine (PerkinElmer, Cat#NET027X001MC) was added (1μl/well), followed by incubation overnight. Proliferation was measured with a TopCount NXT instrument (PerkinElmer).
Statistics.
Statistical analysis was done using unpaired two-tailed Student’s t-tests, two-way ANOVA with correction for multiple comparisons, or Fisher Exact Test. Statistical tests were performed using GraphPad Prism version 8.0 (GraphPad Prism (RRID:SCR_002798)). P values of 0.05 were considered significant.
Data availability.
RNA-seq data were deposited to NCBI GEO data repository under accession number GSE190505. All other data are available in the main text or the supplementary materials or are available upon request from the corresponding author.
Results
Syntaphilin regulates spontaneous neutrophil migration
Naïve, tumor free SNPH-KO mice had a substantially lower proportion of CD11b+Ly6CloLy6G+ PMN among CD45+ cells in BM and spleens than WT mice. No differences in the proportion of CD11b+Ly6ChiLy6G– monocytes (MON) were observed (Fig. 1A). Although no differences in the proportion of PMN and MON in peripheral blood between WT and SNPH-KO mice were found (Supplementary Fig. S2A), we observed markedly higher proportion of PMN among CD45+ cells in lungs of SNPH-KO mice than in WT mice (Fig. 1B). The proportions of MON and dendritic cells (DC) were not different, whereas the proportion of macrophages was decreased (Fig. 1B). We investigated whether differences in the proportions of PMN in BM and lungs of WT and SNPH-KO mice could be the result of differences in the survival of these cells. To address this question, we isolated PMN from BM, spleen, and lung of WT and SNPH-KO mice and cultured them overnight in complete medium without cytokines. The absence of SNPH did not affect the proportion of recovered cells Supplementary Fig. S2B.
Figure 1. SNPH deficiency promotes PMN migration.
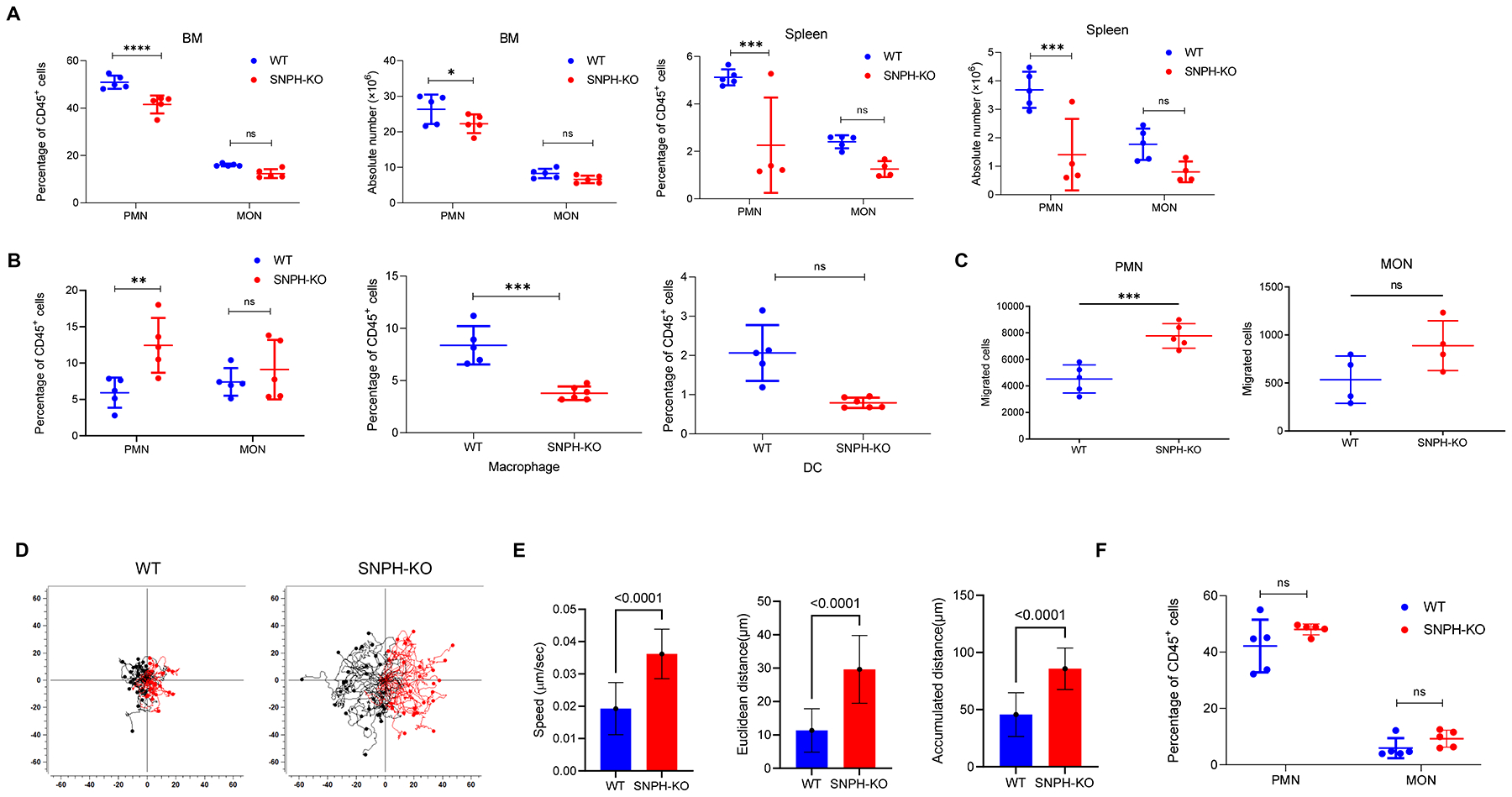
(A) Percentage (of CD45+ cells) and absolute numbers of CD11b+Ly6ClowLy6G+ PMN and CD11b+Ly6C+Ly6G– MON in BM and spleen of WT and SNPH-KO mice (n=5 per group), as determined by flow cytometry. The data shown are representative of two experiments. (B) Percentage of PMN, MON, CD11b+Ly6ClowF4/80+ macrophages and CD11C+Ly6C–MHCⅡ+ DC in the lung of WT and SNPH-KO mice (n=5 per group), as determined by flow cytometry. The data are representative of two experiments. (C) Spontaneous migration of PMN and MON from WT and SNPH-KO mice was determined in Transwell assays. (D, E). Time-lapse video analysis of the migration of BM PMN from WT and SNPH-KO mice, presented as cell traces (D), speed and distance (E). Each tracing corresponds to an individual cell (n=52–59). Two experiments with the same results were performed. (F). The presence of PMN and MON in lungs of tumor-bearing mice. C57BL/6 mice were injected s.c. with 5×105 LLC tumor cells. Lungs were collected three weeks later and the proportion of cells was evaluated (n=5). Data are shown as mean ± SD. Two-way ANOVA test with correction for multiple comparisons was used. P values are shown on graphs. *-p<0.05; **-p<0.01; ***-p<0.001; **** - p<0.0001.
Taken together, these results suggested a possible effect of SNPH on the migration of PMN to the tissues. To address this possibility directly, we evaluated migration of PMN and MON in a standard Transwell membrane assay. BM PMN but not BM MON from SNPH-KO mice had markedly higher spontaneous migration than their WT counterparts (Fig. 1C). Increased spontaneous migration of SNPH-deficient PMN was confirmed by time-lapse microscopy (Fig. 1D,E).
We investigated whether SNPH deletion would affect PMN presence in the lungs of tumor-bearing mice. To address this question, mice were injected s.c. with 0.5×106 Lewis Lung Carcinoma (LL2) cells, which are known to spontaneously metastasize to lungs. The presence of PMN and MON was evaluated three weeks after tumor injection (tumor size >1.5 cm in diameter). We observed large accumulation of PMN in lungs of WT and SNPH-KO tumor-bearing mice (Fig. 1F), with no statistical difference. These results support the concept that once tumor is established and produces a large number of chemokines (as could be the case in 3 week LLC tumors with propensity to metastasize to the lung), the effect of SNPH deletion is lost.
To further investigate SNPH involvement in PMN migration, we assessed PMN migration in response to chemotactic signals. Although PMN from SNPH-KO mice migrated better than PMN from WT mice in response to fMLP, no increase in the ratio of the responses to the chemokine was found (Fig. 2A). PMN from SNPH-KO mice migrated better than PMN from WT mice in response to the chemokine CXLC1. However, as was the case with fMLP, no changes in the ratio of response were observed (Fig. 2B). Expression of CXCR2, which is the main receptor responsible for CXCL1-mediated chemotaxis, was the same in PMN from WT and SNHP-KO mice (Fig. 2C). Although no statistically significant differences in spontaneous migration of MON between WT and SNPH-deficient cells was observed, we tested the possibility that the response of MON to chemokines could be different. However, MON from WT and SNPH-KO mice responded equally well to stimulation with the monocyte specific chemokine CCL2 (Fig. 2D). Thus, deletion of SNPH enhanced spontaneous migration of PMN, which also resulted in higher migration in the presence of chemokines.
Figure 2. SNPH effect on PMN migration.
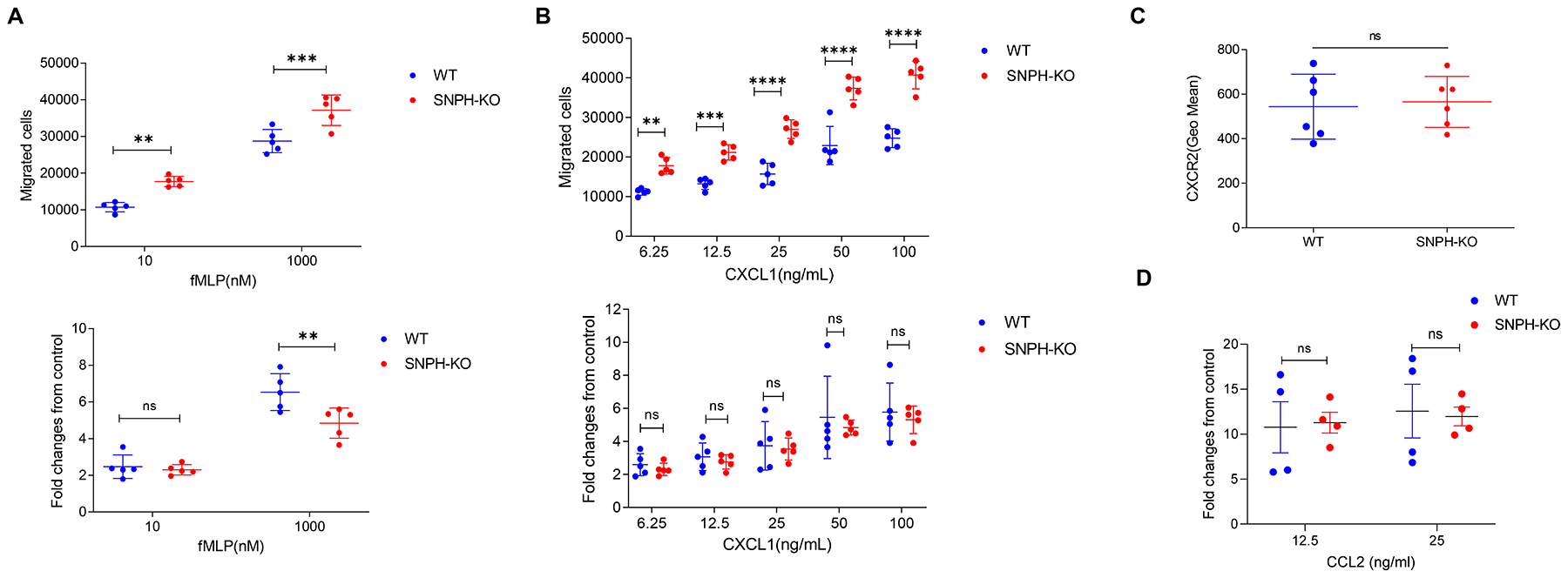
(A,B). Transwell assays evaluating the ability of PMN from the BM of WT and SNPH-KO mice to migrate in response to fMLP (A) or CXCL1 (B) stimuli (n=5). The data are representative of two experiments. Fold increase is calculated as the ratio of the number of cells that migrate in response to a stimulus to the number of cells that spontaneously migrate. (C) CXCR2 expression on the cell surface of PMN from the BM of WT and SNPH-KO mice (n=6 per group), as determined by flow cytometry. (D) Chemotaxis of monocytes in response to CCL2. Monocytes isolated from WT and SNPH-KO mice were used in a Transwell migration assay using CCL2 at indicated concentrations as stimulus. Fold changes over spontaneous migration in the absence of chemokine are shown. n=4. Data are shown as mean ± SD. ns - not significant. Unpaired, two-sided Student’s t-test was used.
Biological role of Syntaphilin in PMN in tumor-bearing mice
Next, we studied the biological relevance of SNPH expression in PMN. We evaluated Snph expression in BM CD11b+Ly6CloLy6G+ PMN from transgenic and transplantable tumor models that previously demonstrated different pattern of PMN migration (5). BM PMN collected from genetically engineered RET melanoma and pancreatic cancer KPC models (after tumors were developed) had substantially reduced expression of Snph. In contrast, BM PMN from s.c. injected EL4 and LLC tumor-bearing (TB) mice showed no difference in Snph expression from naive tumor-free mice (Fig. 3A). These data paralleled the differences in BM PMN migration previously observed in mice bearing transgenic (high spontaneous PMN migration) and subcutaneous transplantable models (no differences in migration activity from control PMN) (5). PMN from cancer patients had substantially lower expression of SNPH (Fig. 3B) than PMN from healthy donors. In paired samples from the same patients, PMN had lower expression of SNPH than PMN-MDSC (Fig. 3C). These results were also consistent with the pattern of migration of PMN and PMN-MDSC from cancer patients described previously showing substantially higher migration of cancer patients PMN than PMN-MDSC (5).
Figure 3. SNPH expression in PMN.
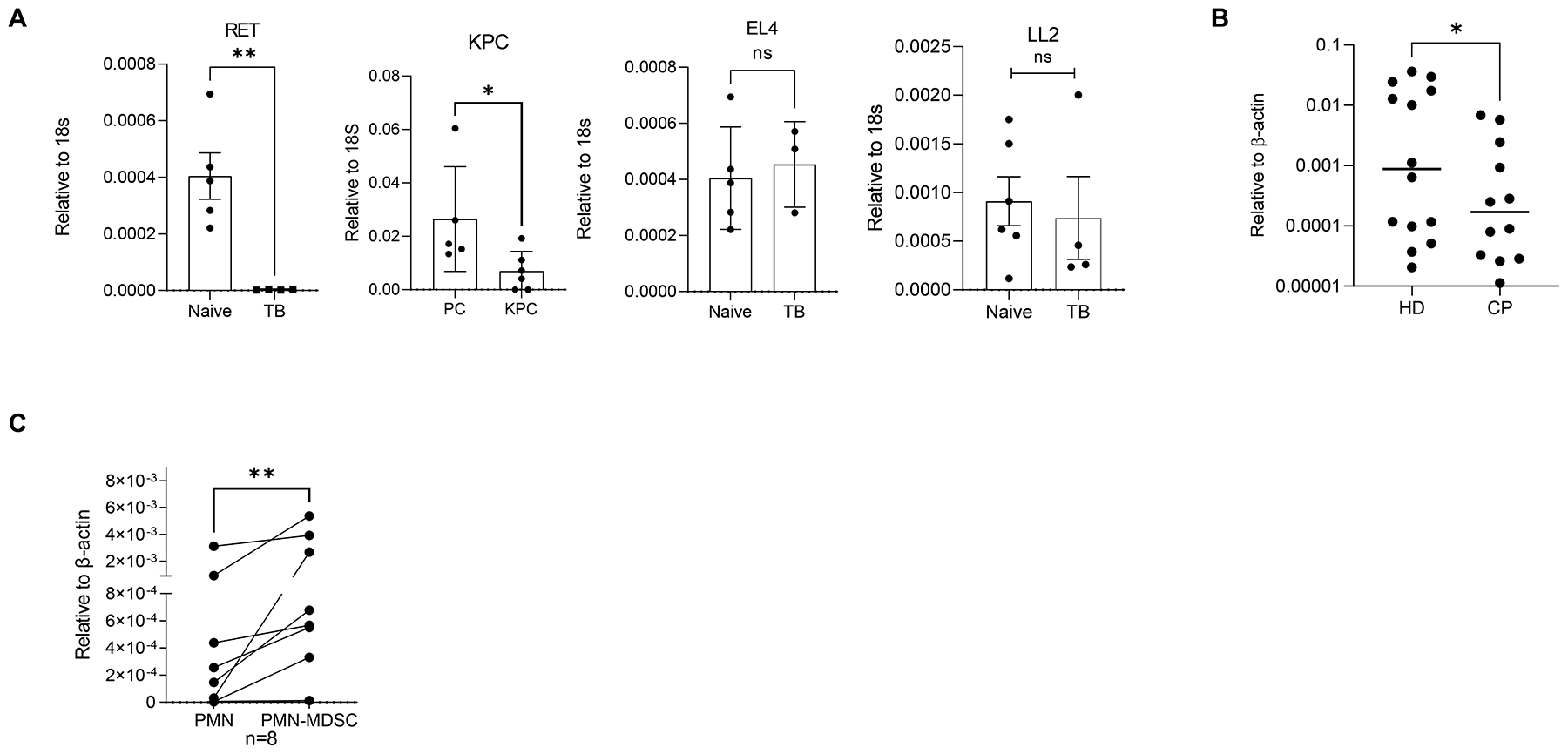
(A) Snph expression in PMN from the BM of tumor-free mice (n=5) or mice bearing the indicated tumors (RET (n=4), KPC (n=5), EL4 (n=3), or LL2 (n=4)). (B) SNPH expression in PMN from healthy donors (HD) and patients with cancer (CP). Individual results in 14 healthy donors and 12 cancer patients are shown. (C) SNPH expression in PMN and PMN-MDSC from the same patients with cancer (n=8). *P < 0.05, **P < 0.01, ns (not significant), in unpaired two-sided Student’s t-test except (C) - Wilcoxon test
Next, we investigated the effect of SNPH deletion on tumor metastases. Luciferase-expressing LL2 was established subcutaneously in WT and SNPH-KO mice. Mice were evaluated three weeks after tumor inoculation when tumor size reached 1.5 cm in diameter. No differences in size of primary tumors were observed (Fig. 4A). In contrast, SNPH-KO mice had significantly more lung metastases than WT mice (Fig. 4B). To further investigate a possible effect of SNPH deletion in hematopoietic cells on metastatic spread, we established BM chimeras by reconstituting lethally irradiated WT congenic mice with BM cells from WT or SNPH-KO mice. Six weeks after reconstitution and confirmation of >87% of donor hematopoietic cell phenotype (Fig. 4C), we evaluated the growth of LL2 tumors established subcutaneously. No differences in growth of primary tumors between mice reconstituted with WT and SNPH-KO BM was observed (Fig. 4D). However, mice reconstituted with BM from SNPH-KO donors showed marked increase in the presence of lung metastases. Whereas only 26% of mice reconstituted with WT BM had developed lung metastasis 3 weeks after s.c. tumor inoculation, tumor lesions in lungs were found in 60% of mice reconstituted with SNPH-KO BM (p=0.022) (Fig. 4E).
Figure 4. Effect of SNPH deletion on tumor metastases.
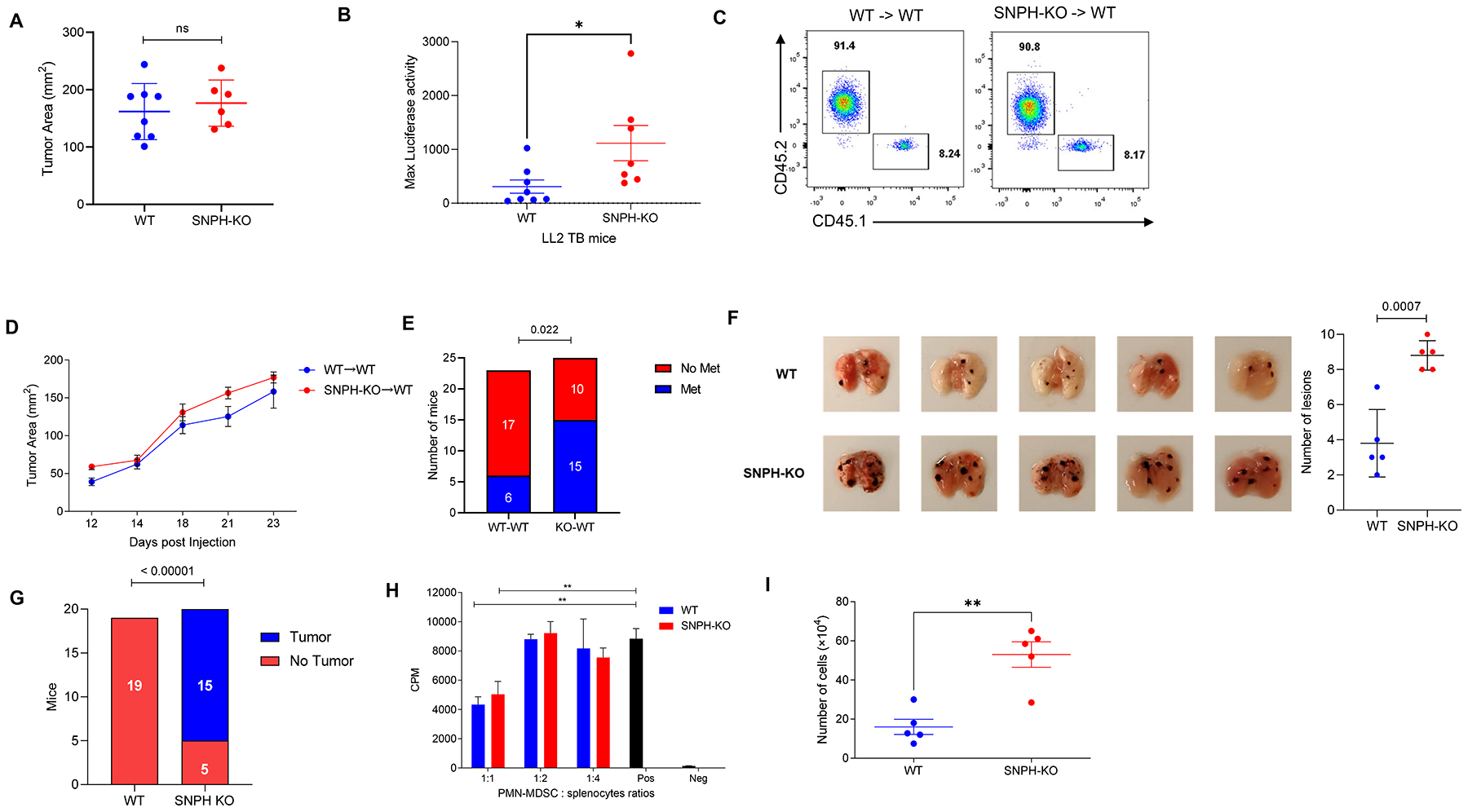
(A). 0.5×106 LL2 cells were injected s.c. and primary tumor size was measured 20 days later in WT and SNPH-KO mice (n=7–8). (B) Luciferase activity in excised lungs from the WT and SNPH-KO tumor-bearing mice described in (A) (n=7–8). (C). The presence of donor CD45.2 hematopoietic cells after reconstitution of lethally irradiated WT CD45.1 congenic mice was determined by flow cytometry. (D) s.c. LL2 tumor growth in WT CD45.2 congenic mice reconstituted with BM from WT or SNPH-KO mice (n=5 per group). The data are representative of three experiments. (E) Lung metastases quantified by luciferase activity. The number of mice with detectable metastases was scored. Detection was performed 3 weeks after the s.c injection of LL2 tumor cells in the chimeric mice. N= 23 for WT to WT group and N=25 in SNPH KO to WT group. (F) Lung metastasis in B16F10-bearing mice. C57BL/6 mice were reconstituted with BM from WT or SNPH-KO mice. After 8 weeks 5×105 B16F10 melanoma cells were injected s.c. and the number of lung metastases were evaluated three weeks later (n = 5 per group). On the left, pictures of lungs with metastases, on the right, the number of lesions. (G) Tumor development in B16F10-bearing mice. C57BL/6 mice were reconstituted with BM from WT and SNPH-KO mice. After 8 weeks 7×104 B16F10 melanoma cells were injected s.c. and tumor development was evaluated for 4 weeks. (H) Suppressive activity of PMN-MDSC from spleens from WT and SNPH-KO LL2 tumor-bearing mice (n=3). OT-1 splenocytes stimulated with specific peptide were used as responders. Proliferation was measured by 3H thymidine uptake. Two experiments with the similar results were performed. (I) Spontaneous migration of PMN-MDSC from WT and SNPH-KO LL2 tumor-bearing mice (n=5 per group) was assessed in Traswell assays. Mean and SD are shown. *P < 0.05, **P < 0.01, ns (not significant), in unpaired two-sided Student’s t-test except (E and G) where Fisher Exact Test was used.
To validate these observations, we tested a different tumor model. WT and SNPH KO mice were challenged s.c. with B16F10 melanoma. Three weeks later lung metastasis were evaluated. We observed a significantly higher number of lung metastases in SNPH KO mice than in WT mice (Fig. 4F). Since these results suggested that the deletion of SNPH promoted spontaneous migration of PMN to tissues and tumor metastasis, we investigated whether this would also affect the initial development of the tumors. To test this hypothesis, we used BM chimeras, established as described above. However, in these experiments we injected small amount of tumor cells—7×104 cells. In mice reconstituted with WT BM no tumors were visible four weeks after injection in all 19 mice tested. In contrast, 15 out of 20 mice reconstituted with SNPH-KO BM had developed tumors (Fig. 4G).
Next, we tested the possibility that SNPH could affect the suppressive activity of PMN-MDSC. Splenic PMN-MDSC from WT and SNPH-KO LL2 tumor-bearing mice demonstrated similar levels of immune suppressive activity (Fig. 4H). SNPH deletion didn’t alter the proportion of PMN in BM or spleens (Supplementary Fig. S2C). There was a trend in decreased presence of MON but it did not reach statistical significance (Supplementary Fig. S2D). In the transwell assays, spontaneous migration of PMN-MDSC from SNPH-KO mice bearing LL2 tumors was markedly higher than from WT mice (Fig. 4I).
We also assessed the effect of SNPH deletion on T cells. No difference in the presence of CD8+ T cells, CD4+FOXP3+ Treg cells, and CD4+Foxp3– T cells was found between WT and SNPH-KO mice (Supplementary Fig. S3A–C). Stimulation of CD8+ T cells with anti-CD3/anti-CD28 resulted in equivalent proliferation of WT and SNPH-deficient cells (Supplementary Fig. S3D). Similarly, no differences were observed in IFNγ production by T cells in response to PMA/Ionomycin stimulation (Supplementary Fig. S3E). Thus, the data suggest increased migration of SNPH-deficient PMN can contribute to tumor metastases.
Mechanism of SNPH effect on PMN migration
To understand the possible mechanisms by which SNPH regulates PMN, we performed whole-transcriptome RNA-seq of BM PMN isolated from wild-type and SNPH-KO mice. 113 genes were found to be significantly differentially expressed (false discovery rate FDR<5%). Among those genes, 48 were up-regulated in PMN from SNHP-KO mice and 65 were down-regulated. The twenty-five most changed genes are shown in Supplementary Fig. S4A. Cellular assembly and organization, cellular function and maintenance, energy production, and free radical scavenging were among the most changed gene networks (Supplementary Fig. S4B). Paxillin and ephrin receptor signaling, which is involved in cell motility, were among the most activated pathways in SNPH-deficient PMN (Supplementary Fig. S4C). These differences suggested that signaling involved in cell motility is the biggest effect of loss of SNPH.
Neutrophil migration requires Rac-1–dependent actin polymerization at the leading edge to push the front of the cell forward and Rho-dependent actomyosin contraction at the uropod to retract the trailing edge (6,25). We evaluated actin polarization (F-actin) in SNPH-deficient PMN. Stimulation with CXCL1 resulted in an increase in F-actin. However, we found no difference between PMN from wild-type mice and SNPH-KO mice (Fig. 5A). The retraction of the cell membrane at the uropod is accomplished through contractile forces generated by myosin motor proteins and the consequent interactions with actin filaments at lateral and posterior regions of the cell cortex (6). This process is regulated by Rho-A–mediated phosphorylation of myosin light chain2 (MLC2). We found that PMN from SNPH-KO mice had substantial upregulation of pMLC2 relative to WT PMN (Fig. 5B).
Figure 5. SNPH regulation of mitochondrial bioenergetics and mitochondrial trafficking.
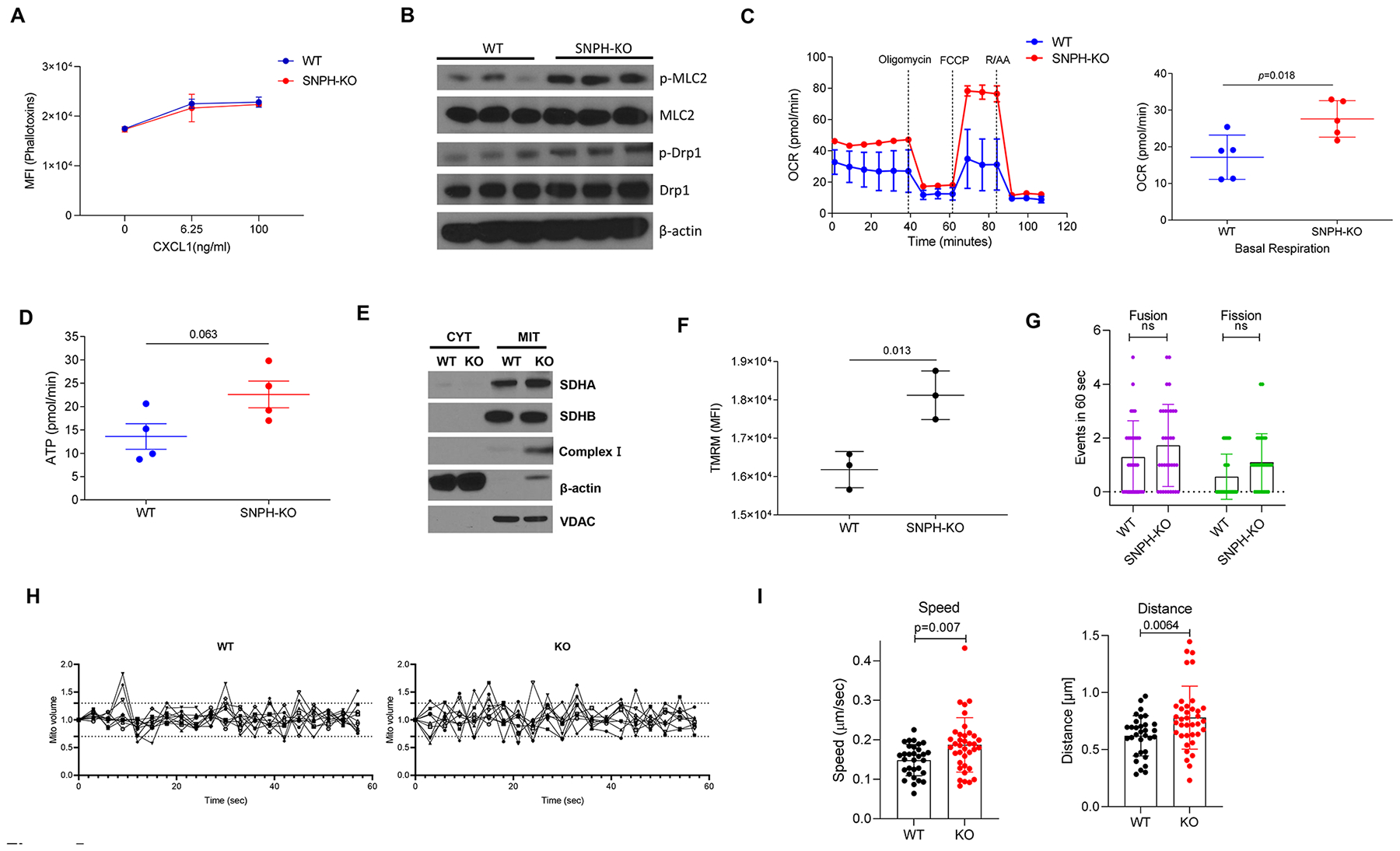
(A) F-actin was measured using flow cytometry before and after stimulation with the indicated concentrations of CXCL1 in BM PMN from WT and SNPH-KO mice (n=2 per group). (B) Immunoblot analysis of phosphorylated MLC2 (p-MCL2), MLC2, phosphorylated Drp1 (p-Drp1), Drp1 and β-actin (loading control) in BM PMN isolated from WT and SNPH-KO mice (one mouse per lane). (C-D) OCR (C, left) and basal OCR (C, right) (n=5) of BM PMN from WT and SNPH-KO mice. The data are representative of three experiments (C, left). (D). ATP production (n=4). (E) Immunoblot analysis of mitochondrial complex SDHA, SDHB, β-actin and VDAC in BM PMN from WT and SNPH-KO mice. CYT cytosolic, MIT mitochondrial. One mouse per line. Two experiments with the same results were performed. (F) Median fluorescent intensity (MFI) of tetramethylrhodamine ethyl ester (TMRE) staining demonstrating mitochondrial membrane potential (n=3 per group). (G-I) WT and SNPH-KO PMN from one mouse stained with MitoTracker™ Deep Red FM were imaged by time lapse microscopy. Computational analysis of fusion and fission events in 6 cells (G) and mitochondrial fission and fusion events of the first 60 frames (60 seconds) (H) of time lapse are shown. Individual mitochondria were tracked through the stack to calculate the speed (I, left) and distance travelled by individual mitochondria (I, right). Two experiments with the same results were performed. P values were calculated in two-sided unpaired Student’s t-test.
How can deficit of SNPH enhance phosphorylation of MLC2 and PMN migration? Allocating mitochondria to the uropod of a migrating cell can provide ATP to sustain actomyosin contraction (12). We observed that BM PMN from SNPH-KO mice had a markedly higher OCR than WT PMN, which is indicative of increased oxidative phosphorylation (Fig. 5C). Those changes were associated with a trend toward an increase in ATP production (Fig. 5D). Consistent with a greater capacity for oxidative phosphorylation, PMN from SNPH-KO mice had increased expression of mitochondrial complex I, but not complex II subunits succinate dehydrogenase A (SDHA) and B (SDHB) compared with PMN from wild-type mice (Fig. 5E). Furthermore, the mitochondrial membrane potential, evaluated by staining with tetramethylrhodamine methyl ester (TMRM), was increased in SNPH-deficient PMN (Fig. 5F). Consistent with these findings, SNPH deletion didn’t affect cytoplasmic- or mitochondria-derived superoxide production by PMN nor mitochondrial mass in these cells (Supplementary Fig. S5A–C).
A previous study demonstrated that downregulation of SNPH in tumor cells promoted cell motility by increasing the speed and distance travelled by individual mitochondria and facilitating organelle fusion and fission (16). Although slight upregulation of phosphorylated dynamin-related protein 1 (pDRP1) in SNPH deficient PMN (Fig. 5B) could suggest possible changes in mitochondria fission, direct evaluation of mitochondria fusion and fission did not demonstrate significant differences (Fig. 5 G,H). This was consistent with a lack of difference in the expression of molecules involved in mitochondrial dynamics (Supplementary Fig. S5D). In contrast, mitochondria in SNPH-deficient PMN exhibited faster speed of movement and greater distance travelled compared with WT PMN (Fig. 5I and Supplemental Video S1). These data suggest that increased mitochondrial movement can facilitate contracting of actomyosin and facilitate PMN movement.
In addition to oxidative phosphorylation, PMN from SNPH-KO mice also had a higher ECAR indicative of increased glycolysis (Fig. 6A). To better assess changes in glycolysis in SNPH-deficient PMN, we performed metabolomics analysis of 13C6-glucose through glycolysis. PMN from SNPH-KO mice had higher rates of incorporation of the 13C into the glycolytic intermediates glucose-6-phosphate, glyceraldhedyde-3-phosphate, 3-phosphoglycerate and phosphoenolpyruvate than WT PMN (Fig. 6B). Only slight increases in intracellular pyruvate and lactate were found in SNPH-deficient PMN (Fig. 6B). The 13C-labeling patterns strongly indicated that loss of SNPH caused greater flux of glucose carbons through glycolysis in PMN. More detailed analysis of metabolites is shown in Supplementary Fig. S6. Consistent with the result obtained during the analysis of oxidative phosphorylation, there was higher ATP synthesis by SNPH deficient PMN as compared to WT PMN (Supplementary Fig. S6). In addition, we found a considerably higher amount of adenosine in SNPH-deficient PMN than in WT PMN (Fig. 6C). It has been long appreciated that adenosine, by activating the A1 and A3 receptors, promotes migration of PMN (26). We hypothesized that increased production of adenosine contributed to enhanced migration of SNPH-deficient PMN. The pathway of adenosine generation involves the hydrolysis of ATP by ectonucleotidases such as CD39 (ectonucleoside triphosphate diphosphohydrolase 1) and CD73 (5-nucleotidase) (27,28). We assessed the effect of inhibition of the generation of adenosine on the ability of SNPH-deficient PMN to migrate. WT and SNPH-deficient PMN were treated with PSB 12379, an inhibitor of CD73, and we observed that the spontaneous migration of SNPH-deficient PMN was completely abolished (Fig. 6D). To further validate these observations, we performed experiments with the selective adenosine A1 receptor (A1AR) antagonist DPCPX. Inhibition of A1AR completely abrogated increased spontaneous migration of SNPH-deficient PMN (Fig. 6E). Thus, SNPH-deficient PMN had increased oxidative phosphorylation, TCA-cycle flux, and glycolysis, which resulted in modest increases in ATP and substantially greater production of adenosine than in PMN from WT mice, which resulted in increased migratory ability of these cells.
Figure 6. SNPH-KO PMN have increased metabolic flux through glycolysis and have more adenosine than that of WT PMN.
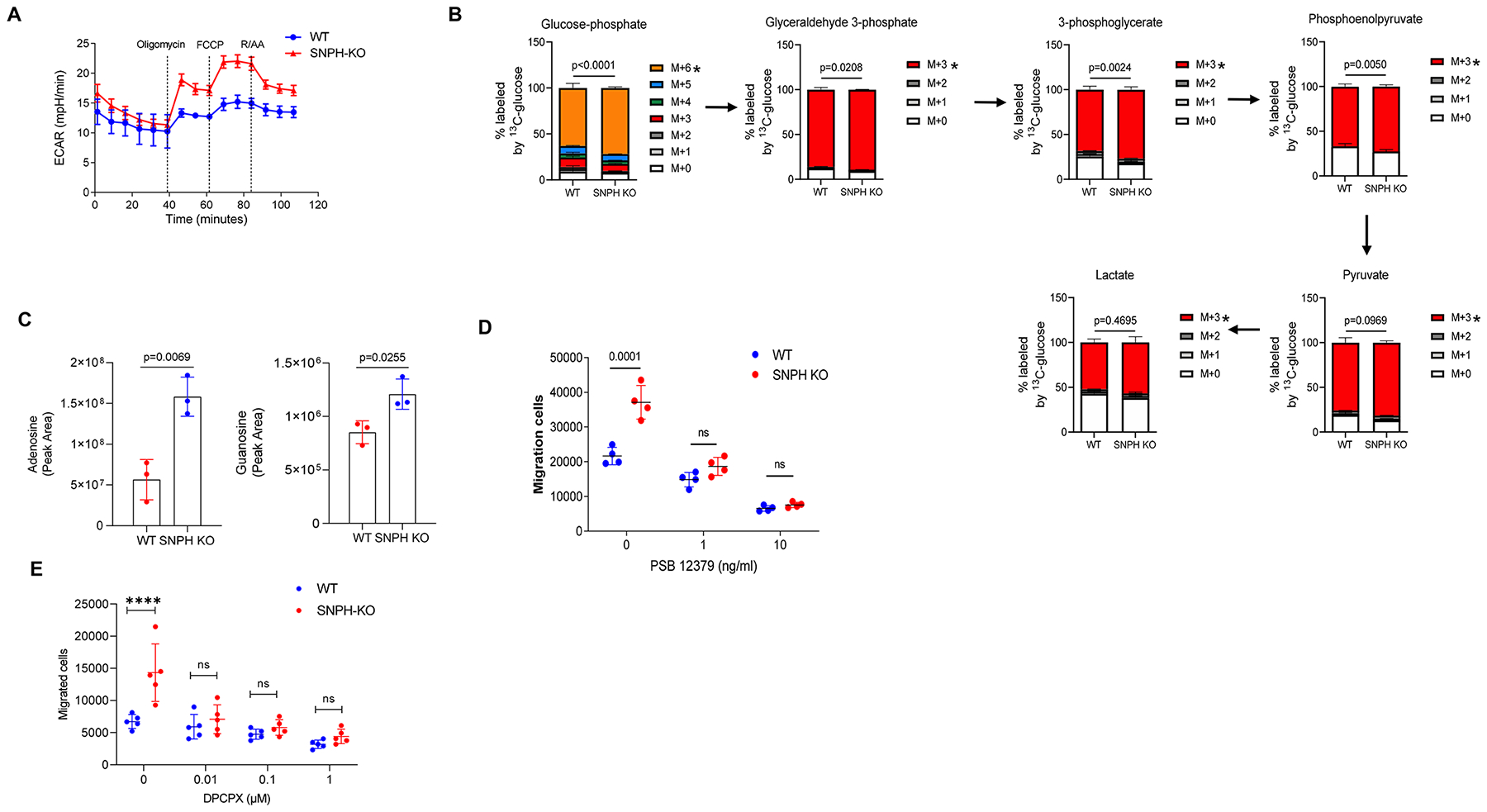
(A) ECAR of BM PMN from WT and SNPH-KO mice. Representative of three independent experiments from three mice. (B) LC-MS of 13C6-glucose through glycolysis in BM PMN from WT and SNPH-KO mice (n = 3 per group), assessed as the change in isotopologue distribution (i.e., 13C-labeling pattern) of the metabolites, normalized to protein content, and presented as ex vivo tracing (mean and s.d.): M + 0, M + 1 and so on (keys) indicate the number of 13C atoms in each metabolite; arrows between plots indicate the flow of carbon. *P < 0.05 (two-way analysis of variance (Tukey correction) with multiple comparisons). (C) LC-MS-based measurement of intracellular adenosine (left) and guanosine (right) in BM neutrophils from WT and SNPH-KO mice (n = 3 per group), normalized to protein content and presented as peak area (mean ± s.d.). unpaired, two-sided Student’s t-test. (D) Transwell assay of the ability of neutrophils from WT and SNPH-KO mice (n = 4 per group) to spontaneously migrate without treatment or after treatment with various concentration (horizontal axis) of the CD73 inhibitor PSB12379. ns - not significant. (E). Spontaneous migration of PMN from WT and SNPH-KO mice in the presence of various concentrations of Adenosine A1 receptor antagonist DPCPX (n = 5). Data are shown as mean ± SD. Two-way ANOVA with correction for multiple comparisons was performed. **** - p<0.0001
Discussion
In this study, we described the association of SNPH loss with the enhanced ability of PMN to spontaneously migrate. Loss of SNPH increased the speed and distance travelled by individual mitochondria within neutrophils, elevated metabolic activity in neutrophils, which together promoting these cells migration.
Mitochondria have recently emerged as a key regulator of leukocyte migration. Chemoattractants induce the redistribution of mitochondria toward the uropod of polarized migrating leukocytes through a mechanism involving microtubules and mitochondrial fission (29). A study in tumor cells demonstrated that SNPH can down-regulate mitochondrial fission and fusion, buffer oxidative stress and maintain complex II-dependent bioenergetics (30). Tumor cells that constitutively downregulate SNPH exhibit higher oxidative stress and increased cell motility (17). However, in PMN, no effect of SNPH deletion on mitochondrial dynamics was found. Herein, we have shown that elevated oxidative phosphorylation and glycolytic rates drove the increased migration of SNPH-deficient PMN. SNPH deficiency in PMN resulted in increased glycolysis and oxidated phosphorylation and there was no increase in mitochondria-produced superoxide. Although the precise mechanism of these differences is not clear, the explanation could be in the nature of mitochondrial involvement in PMN function. Under steady state conditions, PMN don’t rely on OXPHOS and mitochondria for their function and have a relatively small number of mitochondria. The main source of superoxide in PMN is NADPH oxidase. It is conceivable that increased mitochondrial movement as a result of loss of SNPH is sufficient to cause strong functional effect on PMN. In leukocytes, oxidative phosphorylation is required for mitochondrial dependent migration (31). These findings suggest that increased metabolic activity could provide ATP to sustain actomyosin contraction, thereby enabling retraction of the trailing edge and cell advance.
Mitochondria supply ATP essential for synaptic transmission. In neurons, SNPH immobilizes mitochondrial transport and causes local energy deficits. Enhancing mitochondrial transport via genetic manipulation of SNPH facilitated regenerative capacity of axons by replenishing healthy mitochondria in injured axons, thereby rescuing energy deficits (32). Functional micro-compartmentalization of intracellular metabolites (including adenine nucleotide) and substrates has long been recognized. Our data suggest that accumulation of mitochondria at the uropod of a migrating cell might provide ATP in this strategic position, where class II myosin proteins are localized. Our data suggest a contribution of adenosine to increased migration of SNPH-deficient PMN. Intracellular adenosine is generated by stepwise dephosphorylation of ATP or by the hydrolysis of S-adenosylhomocysteine (33). Adenosine formed extracellularly can also be transported into cells via nucleoside transporters (34). Neutrophils can both release adenosine and respond to it via expression of all four adenosine receptor subtypes. At low concentrations, adenosine can act via the A1 and A3 adenosine receptor subtypes to promote neutrophil chemotaxis and phagocytosis (26). Thus, decrease of SNPH in neutrophils may facilitate their spontaneous migration via combined effect of increased local concentration of ATP and adenosine signaling.
It is widely recognized that an increased flux of neutrophils contributes to the formation of the pre-metastatic niche. Depletion of PMN and PMN-MDSC in mice decreases the formation of metastasis (2,35,36). Our data with SNPH-KO mice are consistent with these observations. PMN-MDSC are not the only possible contributors to metastasis. Monocytes/macrophages are plying a major role in the development of tumor metastasis (37,38). However, our data suggest that deletion of SNPH is probably unlikely to exert its effect via recruitment of monocytes and accumulation of macrophages. First, the number of monocytes in BM and spleen was only marginally affected by SNPH deletion, whereas the number of PMN was substantially reduced. Second, in lungs, in contrast to PMN, the presence of monocytes and macrophages was not different between SNPH-KO and WT mice. Third, migration of SNPH-deficient monocytes was only marginally affected.
We believe the reason why neutrophils accumulate in the tissues in SNPH-KO mice is that in the absence of SNPH, neutrophils acquire a potent ability to spontaneously migrate. Normal tissues produce baseline amount of chemokines, which attract neutrophils patrolling the tissues. We hypothesize that SNPH-deficient neutrophils become much more active in response to the low chemokine gradient in tissues, which causes the accumulation of PMN in tissues not yet affected by growing tumors. This is clinically relevant in early-stage tumors prone to metastasis. Downregulation of SNPH in PMN would promote their migration to unaffected tissues and the development of distant metastases.
Our data suggest one possible molecular mechanism that regulates this flux. Increased PMN migration to lung was associated with increased metastatic spread to lung. SNPH deficiency did not affect suppressive activity of PMN-MDSC, presence or function of T cells. Thus, SNPH loss may supply distant tissues with immune suppressive PMN-MDSC able to facilitate tumor metastases.
Supplementary Material
Synopsis.
The authors identify syntaphilin as a regulator of spontaneous migration of neutrophils in cancer. Syntaphilin deletion promotes PMN spontaneous migration and metastasis via increased mitochondria motility, elevated rates of oxidative phosphorylation and glycolysis, and increased generation of adenosine.
Funding Information:
This work was supported by NCI grants P01 (CA140043) and R35 (CA220446) as well as Wistar Cancer Center support grant CA010815 to DCA.
Footnotes
Conflict of interest statement: ES, AM, and DIG are employee and shareholders of AstraZeneca. All other authors declare no relevant conflict of interest.
References
- 1.Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 2020;20:485–503 [DOI] [PubMed] [Google Scholar]
- 2.Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin Immunol 2018;35:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer 2016;16:431–46 [DOI] [PubMed] [Google Scholar]
- 4.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 2021;21:485–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol 2018;19:1236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hind LE, Vincent WJ, Huttenlocher A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Developmental cell 2016;38:161–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang HW, Collins SR, Meyer T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat Cell Biol 2016;18:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, et al. ATP synthesis and storage. Purinergic Signal 2012;8:343–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell death and differentiation 2004;11:143–53 [DOI] [PubMed] [Google Scholar]
- 10.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016;22:146–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol 2003;170:1964–72 [DOI] [PubMed] [Google Scholar]
- 12.Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med 2006;203:2879–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurz FT, Aon MA, O’Rourke B, Armoundas AA. Functional Implications of Cardiac Mitochondria Clustering. Adv Exp Med Biol 2017;982:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008;132:137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 2012;32:4145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nature communications 2016;7:13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caino MC, Seo JH, Wang Y, Rivadeneira DB, Gabrilovich DI, Kim ET, et al. Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J Clin Invest 2017;127:3755–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol 2003;21:652–9 [DOI] [PubMed] [Google Scholar]
- 19.Kato M, Takahashi M, Akhand AA, Liu W, Dai Y, Shimizu S, et al. Transgenic mouse model for skin malignant melanoma. Oncogene 1998;17:1885–8 [DOI] [PubMed] [Google Scholar]
- 20.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83 [DOI] [PubMed] [Google Scholar]
- 21.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015;27:57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertolini I, Keeney F, Altieri DC. Protocol for assessing real-time changes in mitochondrial morphology, fission and fusion events in live cells using confocal microscopy. STAR Protoc 2021;2:100767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junger WG. Purinergic regulation of neutrophil chemotaxis. Cell Mol Life Sci 2008;65:2528–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arteriosclerosis, thrombosis, and vascular biology 2012;32:856–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem 1996;271:33116–22 [DOI] [PubMed] [Google Scholar]
- 28.Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol 2020;17:611–29 [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Madrid F, Serrador JM. Mitochondrial redistribution: adding new players to the chemotaxis game. Trends Immunol 2007;28:193–6 [DOI] [PubMed] [Google Scholar]
- 30.Seo JH, Agarwal E, Bryant KG, Caino MC, Kim ET, Kossenkov AV, et al. Syntaphilin Ubiquitination Regulates Mitochondrial Dynamics and Tumor Cell Movements. Cancer Res 2018;78:4215–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biol 2014;2:206–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 2016;214:103–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boison D Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 2013;65:906–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loffler M, Morote-Garcia JC, Eltzschig SA, Coe IR, Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arteriosclerosis, thrombosis, and vascular biology 2007;27:1004–13 [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Guoqiang L, Sun M, Lu X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol Med 2020;17:32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groth C, Weber R, Lasser S, Ozbay FG, Kurzay A, Petrova V, et al. Tumor promoting capacity of polymorphonuclear myeloid-derived suppressor cells and their neutralization. Int J Cancer 2021;149:1628–38 [DOI] [PubMed] [Google Scholar]
- 37.Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol 2022;23:1148–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cotechini T, Atallah A, Grossman A. Tissue-Resident and Recruited Macrophages in Primary Tumor and Metastatic Microenvironments: Potential Targets in Cancer Therapy. Cells 2021;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data were deposited to NCBI GEO data repository under accession number GSE190505. All other data are available in the main text or the supplementary materials or are available upon request from the corresponding author.