

Abstract
Termination of transcription by RNA polymerase II usually requires the presence of a functional poly(A) site. How the poly(A) site signals its presence to the polymerase is unknown. All models assume that the signal is generated after the poly(A) site has been extruded from the polymerase, but this has never been tested experimentally. It is also widely accepted that a “pause” element in the DNA stops the polymerase and that cleavage at the poly(A) site then signals termination. These ideas also have never been tested. The lack of any direct tests of the poly(A) signaling mechanism reflects a lack of success in reproducing the poly(A) signaling phenomenon in vitro. Here we describe a cell-free transcription elongation assay that faithfully recapitulates poly(A) signaling in a crude nuclear extract. The assay requires the use of citrate, an inhibitor of RNA polymerase II carboxyl-terminal domain phosphorylation. Using this assay we show the following. (i) Wild-type but not mutant poly(A) signals instruct the polymerase to stop transcription on downstream DNA in a manner that parallels true transcription termination in vivo. (ii) Transcription stops without the need of downstream elements in the DNA. (iii) cis-antisense inhibition blocks signal transduction, indicating that the signal to stop transcription is generated following extrusion of the poly(A) site from the polymerase. (iv) Signaling can be uncoupled from processing, demonstrating that signaling does not require cleavage at the poly(A) site.
It has become clear in recent years that RNA polymerase II (RNAPII) not only transcribes the mRNA but shepherds it through the stages of processing as well (42, 67). An early indicator of this coupling between transcription and processing was the finding that the poly(A) signal for cleavage and polyadenylation of pre-mRNA directs not only 3′-end processing of the transcript but also termination of transcription by the polymerase (20, 32, 50, 61, 77, 79). A major challenge has been to explain how the poly(A) signal communicates with the polymerase.
The core poly(A) signal in vertebrates consists of two recognition elements flanking a cleavage-polyadenylation site (76, 82). Typically, an almost invariant AAUAAA hexamer lies 20 to 50 nucleotides (nt) upstream of a more variable element rich in U or GU residues. Cleavage of the nascent transcript occurs between these two elements and is coupled to the addition of up to 250 adenosines to the 5′ cleavage product. The cleavage is mediated in vitro by a large, multicomponent protein complex that can be separated into five distinguishable factors. Two of these factors are the cleavage and polyadenylation specificity factor (CPSF), which binds the AAUAAA motif, and the cleavage stimulation factor (CstF), which binds the downstream U-rich element. In vitro studies suggest that CPSF (26) and probably CstF as well (55, 71) are recruited to the polymerase at the promoter. Presumably they then ride with the polymerase during transcription, scanning the extruding transcript so as to snare the poly(A) site when it emerges. The situation in yeast may be similar (8, 29). Strictly speaking, the term “poly(A) site” refers only to the point at which cleavage occurs and the poly(A) tail is appended, but we use the term here to refer to the poly(A) signal as a whole when this helps to distinguish between the poly(A) signal as an entity and poly(A) signal transduction [or “poly(A) signaling”] as a process.
Models that attempt to explain transduction of the signal from the poly(A) site to the polymerase can be divided into two categories (50): (i) cleavage-dependent models, in which the poly(A) site is recognized by virtue of the processing that it carries out, and (ii) cleavage-independent models, in which the poly(A) site is recognized directly by a component of the transcription elongation complex. Cleavage-independent models were initially favored (50) and recently gained renewed support from the finding that CPSF and CstF may be components of the transcription elongation complex (55). As such, these factors would presumably be positioned to recognize the poly(A) site as soon as it emerges so as to transduce a termination signal to the polymerase. This is the mechanism used by the vaccinia virus RNA polymerase (27, 40), which closely resembles RNAPII in many aspects of structure and mechanism (see reference 28). Moreover, elongating polymerases not only carry factors that can recognize signals in the extruding RNA (see also reference 23), but they also have considerable intrinsic potential to recognize sequence elements at both the DNA and RNA levels prior to extrusion (4, 58). Thus, cleavage-independent models readily offer plausible scenarios for poly(A) signaling based on recognition of the signal either prior to, during, or following extrusion.
However, the preferred model for poly(A) signaling has for many years been a cleavage-dependent model in which the signal is generated by the new, uncapped RNA 5′ end resulting from poly(A) site cleavage (20, 68, 81). According to this model the new 5′ end recruits a signal-transducing 5′ exonuclease which chases the polymerase by processively degrading the RNA, thereby to deliver the signal. Consistent with this hypothesis, Edwalds-Gilbert et al. (32) and Birse et al. (14) reported a close correlation between processing efficiencies and termination efficiencies for a variety of mutated poly(A) sites and processing factors, suggesting that some aspect of the process leading to poly(A) site cleavage is involved in generating the signal to terminate.
A different point of view has been expressed by Batt et al. (10) and Osheim et al. (61), who favored a cleavage-independent mechanism for transcription termination. However, neither of these groups actually assayed termination itself. The data in the work of Batt et al. (10) reflected variations in processing efficiency rather than termination, while the polymerase stacks in the electron micrographs of Osheim et al. (61) may well have resulted from polymerase pausing or arrest. Osheim et al. (61) pointed out that the stacked-up polymerases in their micrographs carried mostly uncleaved, full-length transcripts although the stacks extended across the poly(A) site. This is actually consistent with current cleavage-dependent models in which cleavage triggers termination as part of a single, concerted reaction (11, 30, 67, 80). This underscores the difficulty until now of distinguishing experimentally between cleavage-dependent and cleavage-independent models and has even led to the suggestion that there is no mechanistic common denominator to poly(A) signaling, termination being cleavage dependent, cleavage independent, or both cleavage and 5′-exonuclease dependent according to circumstance (81).
Transcriptional pause elements in the DNA downstream of the poly(A) signal have long played a prominent role in models for poly(A)-dependent termination (2, 13, 41, 68, 81). Pausing of the polymerase is thought to be necessary to allow the poly(A) site time to act and, thereby, to signal the polymerase. Consistent with this hypothesis pausing is indeed frequently observed to occur downstream of poly(A) sites (7, 13, 16, 33, 41, 53, 74, 78). Also consistent with this hypothesis, some poly(A) sites are known to be accompanied by downstream elements that enhance cleavage and polyadenylation and/or termination or arrest (2, 6, 13, 21, 31, 33, 72). Although these elements are usually referred to as pause elements because of a presumption about their mechanism of action, it has not been confirmed that they actually cause the pausing seen downstream of poly(A) sites in vivo. Moreover, pausing per se does not enhance cleavage and polyadenylation in vitro (80). Indeed, unlike confirmed pause elements (24, 45, 62, 65, 70), these downstream elements are not associated with pausing in a consistent way—pausing being found variously upstream, downstream, or coincident with the elements (2, 13, 33, 41). This has been attributed to DNA sequence artifacts related to the nascent transcript analysis used to detect pausing and termination (2). However, the widespread occurrence of broad zones of apparent pausing downstream of poly(A) sites suggests that the pausing itself is genuine and that it is the mechanistic relationship of this pausing to the downstream elements that is obscure. Thus, it is not known whether elements in downstream DNA enhance processing and termination through pausing or by some other mechanism such as by a direct effect on the poly(A) signal itself (19, 52). Nor is it known whether the pausing commonly seen downstream of poly(A) sites is due to additional elements in the downstream DNA or to an effect of the poly(A) signal itself on events that occur downstream.
Given the uncertainties concerning the number and nature of elements in a typical poly(A)-dependent terminator together with the uncertainties concerning the basis of poly(A) dependence itself, we have adopted a parsimonious approach to the study of poly(A)-dependent termination. First, we have concentrated exclusively on the core of the poly(A) signal in order to determine its unique contribution to termination. To do this we have capitalized on our previous finding that the core of the simian virus 40 (SV40) early poly(A) signal is capable of inducing efficient termination in vivo without the assistance of any downstream elements in the DNA (79). Thus, we eliminate ambiguities related to the role of such elements in termination.
Second, we have limited our attention to the signaling step of poly(A)-dependent termination—i.e., how the poly(A) site makes its presence known to the polymerase. The immediate consequence of signaling need not be transcript release as is generally assumed. It could be pausing, consistent with the apparent collection of paused transcription complexes seen in the striking electron micrographs of Osheim et al. (61) and as suggested by the broad pausing profiles commonly seen downstream of poly(A) sites following nascent transcript run-on analysis (7, 13, 16, 33, 41, 53, 74, 78). We have therefore designed an assay that measures the collective effects of the poly(A) signal on the efficiency of transcription elongation. Thus, we detect the first effect of the poly(A) site on transcription [i.e., poly(A) signaling], whether that effect is complete termination, halting of the polymerase, or merely slowing down transcription.
Finally, wishing to address questions of mechanism in a direct way, we have devised an in vitro system for studying poly(A) signaling. There have been prior reports suggesting successful coupling in vitro of polyadenylation to termination. However, one such report (57) actually described measurements not of termination but of poly(A) site cleavage, while another (81) failed to show that the termination under study was dependent on the presence of a poly(A) signal. In contrast, we now describe an in vitro system that faithfully reproduces signaling from the poly(A) site to the polymerase, and we validate this in vitro assay by reference to an in vivo assay that is free of the potential artifacts associated with conventional hybridization-based nascent transcript analysis (79). We show below that signaling in vitro does not require pause elements in the DNA and does not require cleavage of the transcript but does require extrusion of the poly(A) site from the polymerase. This is the first successful demonstration of poly(A) signaling in vitro.
MATERIALS AND METHODS
Plasmids.
In our nomenclature, subscripts (as in APw and APm) refer to plasmids with wild-type and mutant poly(A) signal hexamers, respectively. Often the subscripts are omitted when the context is clear or when mutant and wild-type versions of the plasmid are being referred to collectively or generically. Brackets (as in 〈cat〉) refer to G-less cassettes of 120 and 261 bp flanking the indicated sequence unless otherwise specified, as in 〈117cat〉. Plasmids not described elsewhere were constructed as follows. For pAPw〈117cat〉, a BamHI site was introduced into the 377-bp precassette of pAP〈cat〉 (79) by site-directed mutagenesis, and the resulting 323-bp BamHI fragment was removed, leaving a 117-bp precassette. pAPm〈117cat〉 was constructed as described above except that the poly(A) signal was first inactivated by mutating the hexamer (AATAAA→AgTAct [lowercase letters indicate mutations]) using site-directed mutagenesis. For pAP〈cat〉, SmaI and HindIII sites were added at the 3′ end of the upstream cassette (simultaneously lengthening it to 120 bp) in the pAP〈117cat〉 plasmids by use of site-directed mutagenesis (gcttggcgagatt→cccgggcaagctt). For pAP〈377〉, the pAP〈cat〉 plasmids were cut with HindIII and EcoRV and the BamHI-RsaI fragment containing the 377-bp G-less cassette from pAP〈cat〉 was inserted. The construction of pAP〈C9〉 was like that for pAP〈377〉, but the insert, C9, was a 1,012-bp piece of DNA consisting primarily of nine copies of an arbitrarily selected 102-bp sequence from the middle of the C2 region of β-globin 3′-flanking DNA (79), containing no known functional elements. For p377A174P〈C9〉, the 377-bp G-less cassette (above) and a 174-bp cassette were inserted into pAP〈C9〉 at the StuI and HpaI sites, respectively. The 174-bp cassette was obtained by cutting pAP〈cat〉 with BglII and HinfI after site-directed mutagenesis (atattt→aGatCt) of the 261-bp cassette. For pAP〈C9377〉, the 1,012-bp C9 fragment was inserted into SmaI-cut pAP〈377〉.
Poly(A) signaling assay.
HeLa nuclear extract was prepared exactly as described by Flaherty et al. (34) except that the final centrifugation was at 13,000 × gav for 30 min. The extract was dialyzed against buffer D until the conductances of the extract and the buffer were equal (5 to 7 h). The extract was then aliquoted and stored in liquid nitrogen. Different extracts vary somewhat in their properties.
A typical signaling assay began with 6 to 12.5 μl of nuclear extract, which was brought to a total volume of 12.5 μl with buffer D. This was then mixed with sodium citrate, dithiothreitol, creatine phosphate, MgCl2, anti-RNase (Ambion), DNA, and water up to a volume of 22 μl. The mixture was preincubated at 30°C for 30 min, and then 3 μl of nucleoside triphosphate mix containing 20 μCi of [α-32P]CTP (800 Ci/mmol) was added and transcription was allowed to continue for 15 min. Final concentrations in a typical transcription mixture were as follows: 10 mM HEPES (pH 7.9); 10% glycerol; 50 mM KCl; 0.1 mM EDTA; 2 mM dithiothreitol; 8 mM sodium citrate (pH 6.7); 20 mM creatine phosphate; 5 mM MgCl2; 15 to 30 U of anti-RNase; 0.6 μg of DNA; a 200 μM concentration (each) of ATP, GTP, and UTP; and 5 μM unlabeled CTP. Any variations from this standard procedure are noted in the figure legends. Because of some variations between experiments, including the use of several different extract preparations, comparisons of absolute signaling efficiency should be made only within, not between, figures.
For most of the experiments reported in this work, transcription was stopped by addition of 1 μl each of α-amanitin (1 mg/ml; Sigma) and DNase I (2 U/μl; Ambion). After 10 min at room temperature, 1 μl each of 50 mM EDTA and RNase T1 (200 to 250 U/μl; Ambion) was added for 15 min at 30°C. Then 1 μl of proteinase K (20 mg/ml) was added and, after 10 min at room temperature, the reaction was extracted with 350 μl of TRIzol (Gibco BRL) and 70 μl of chloroform. However, a simpler procedure is often adequate, in which transcription is stopped using 2 μl of 250 mM EDTA containing 200 U of RNase T1, followed, after 15 min, by TRIzol extraction. The RNA was precipitated with 1.5 to 2 μl of Saccharomyces cerevisiae tRNA (10 mg/ml; Sigma) and 350 μl of isopropanol (10 min, room temperature), spun in a microcentrifuge (10 min, 4°C), washed with 150 μl of 75% ethanol, resuspended in 15 μl of 7 M urea-loading dye, heated at 90°C for 5 min, chilled immediately on ice for 1 min, and loaded onto an 8% polyacrylamide gel to separate the G-less transcription products. For analyzing transcripts containing antisense sequences, one or two additional digestions with T1 under denaturing conditions were carried out, as follows. Instead of resuspending in urea after the first precipitation, the RNA was resuspended in 32 μl of 50% formamide–0.5 mM Tris–0.5 mM EDTA (pH 8) and then heated to 90°C for 5 min. After cooling to 37°C, 7 μl of RNase T1 (1,000 U/μl) was added and the samples were placed in an oven at 63°C for 1 h. Extraction with TRIzol and subsequent steps were then carried out as described earlier.
Following electrophoresis, results were recorded and analyzed using a PhosphorImager with ImageQuant software (Molecular Dynamics). Unless otherwise indicated, results are reported as the average of two separate experiments carried out on different days. Error bars indicate the range of values obtained in the two individual experiments. For most individual experiments, reactions were carried out in duplicate and the averages of the duplicates were taken as the outcome of the experiment.
Nascent transcript G-less cassette analysis of poly(A)-dependent termination in vivo (79).
COS cells were grown in 35-mm-diameter wells and transfected with plasmid DNA using Fugene 6 (Roche). After 48 h cells were rinsed twice with cold phosphate-buffered saline and lysed with a solution containing 0.5% IGEPAL (Sigma), 10 mM Tris (pH 7.4), 3 mM MgCl2, and 10 mM NaCl. Nuclei were pelleted and then resuspended in 15 μl of 50 mM Tris (pH 8.3)–0.1 mM EDTA–40% glycerol–5 mM MgCl2. Run-on transcription was carried out for 45 min at 30°C following addition of 16 μl of a solution containing 10 mM Tris (pH 8), 5 mM MgCl2, 600 mM (NH4)2SO4, 1 mM ATP, 1 mM UTP, 0.2 mM 3′-OMeGTP, 6 mM dithiothreitol, 20 U of anti-RNase, and 1 μl of 180 μM CTP containing 30 μCi of [α-32P]CTP. After a 10-min cold chase with 1 μl of 40 mM CTP, 1 μl (15 U) of T1 RNase and 1 μl of 50 mM EDTA were added for an additional 30 min at 30°C. Following digestion with 2 μl of DNase I (20 U) for 15 min at 30°C, 1.8 μl of 10% sodium dodecyl sulfate was added. The sample was then extracted with 500 μl of TRIzol and 140 μl of chloroform, and 20 μg of carrier tRNA and 500 μl of isopropanol were added to the aqueous phase to precipitate the RNA. The RNA was resuspended in 32 μl of 10 mM Tris–1 mM EDTA (pH 7), heated for 2 min at 90°C, and cooled on ice for 1 min, and then 1 μl (250 U) of T1 was added for 30 min at 30°C. TRIzol (350 μl) extraction was then carried out in a manner similar to that described above.
RNase protection assay.
RNA from the equivalent of four standard signaling assays (with cold CTP replacing [α-32P]-CTP) or transfected RNA was analyzed as described (18) with hybridization carried out at 63.5°C. The probe used was a T7 RNA polymerase transcript of BglI-digested pAP〈cat〉 into which the T7 promoter (HincII-PvuII fragment from pBluescript II SK [Stratagene]) had been inserted at the HindIII site.
RESULTS
Poly(A) signaling in vitro mimics poly(A)-dependent termination in vivo.
Poly(A) signaling in our system is detected as a decrease in the elongation efficiency of RNAPII after crossing a poly(A) site. To measure signaling we modified a G-less cassette assay previously used to monitor the efficiency of transcriptional elongation in vitro (48, 51). Figure 1 illustrates the use of this assay to study signaling during transcription in nuclear extracts and compares the results obtained for signaling in vitro with parallel results obtained for termination in vivo.
FIG. 1.

Poly(A) signaling in vitro. (A) The pAP〈C9377〉 construct drawn to scale. The arrows indicate the start point of transcription for the SV40 early-early promoter (9) and the position of poly(A) site cleavage (20). (B) G-less cassette transcripts from a signaling assay carried out with mutant and wild-type versions of pAP〈C9377〉. The line graphs showing signal intensities for the two gel lanes were superimposed, but offset slightly for clarity, and then aligned with the gel images. (C) Quantitative comparison of poly(A) signaling in vitro with poly(A)-dependent termination in vivo on the pAP〈C9377〉 template. The efficiency of elongation (% wild-type readthrough) downstream of the wild-type poly(A) site is expressed (after normalizing all signals on each template to that for the 120-nt cassette) as the ratio of the intensity for each cassette on the wild-type template to that for the same cassette on the mutant template. (The images in panel A were also normalized with respect to the 120-nt cassette for purposes of presentation.)
Figure 1A shows a diagram of pAP〈C9377〉, the DNA template used for these experiments. It contains the core SV40 early poly(A) signal followed immediately by a short, 120-nt G-less cassette. Two additional cassettes, distinguishable in size, lie farther downstream, separated from the upstream cassette by a spacer sequence (called C9). The efficiency of elongation downstream of the poly(A) site is indicated by the extent to which the two downstream cassettes (postcassettes) are transcribed relative to the upstream cassette (precassette). Poly(A) signaling is evident when this elongation efficiency is less for templates with wild-type poly(A) sites than for templates with mutant poly(A) sites (hereafter called mutant or wild-type templates).
Figure 1B, lane 1, shows the G-less RNAs that survive RNase T1 digestion of the transcripts produced in vitro from the wild-type version of the pAP〈C9377〉 template. Lane 2 shows the results, obtained in parallel, for the mutant version of the template which differs only in that the AATAAA hexamer of the poly(A) signal has been mutated to AgTAct. Clearly the downstream cassettes are transcribed less efficiently from the wild-type template than from the mutant template. The results are presented quantitatively to the right of the gel images. The molar ratio of 261-nt postcassette transcripts to 120-nt precassette transcripts is 0.51 for the mutant but only 0.19 for the wild-type template. Thus, transcriptional elongation on the wild-type template between the 120- and the 261-nt cassettes is only 37% as efficient as on the mutant template. In Fig. 1B and C, and throughout this report, we refer to this as the percent wild-type readthrough.
We note that the postcassette/precassette molar ratio is less than one even for the mutant template. This may reflect, in part, frequent pause and/or arrest of RNAPII induced by the low concentrations of CTP in our transcription mixtures (66) combined with protein binding to the DNA in our extracts and an insufficient concentration of TFIIS to efficiently override these nonspecific impediments to transcription (47). The low postcassette/precassette ratio also reflects our use of relatively short transcription times for these experiments. We have chosen to concentrate our attention on the forward wave of polymerases that elongate most efficiently down the template, which means that many stragglers and late-initiating polymerases will have transcribed the precassette but not yet arrived at the postcassette by the time that we stop the reaction. However, these effects of transcription time and CTP concentration do not compromise our conclusions, because they apply to both wild-type and mutant templates equally and are thus normalized away when we take the wild-type/mutant ratios. Control experiments show that none of our conclusions depends on the particular transcription time or concentration of CTP chosen.
The ability of the wild-type poly(A) signal to decrease recovery of the downstream cassettes relative to the upstream cassette reflects a decrease in downstream transcription on the wild-type template. The reduced recovery is not a consequence of preferential degradation of the downstream cassettes following 3′ end processing of the wild-type transcript. This conclusion follows from the design of the template: all three cassettes in pAP〈C9377〉 lie downstream of the poly(A) signal (Fig. 1A). Thus, although processing would indeed destabilize downstream RNA, this would include all of the cassettes. Moreover, the upstream cassette would, if anything, be more, not less vulnerable to degradation since degradation is probably mediated by a 5′-specific exonuclease (60, 81). Therefore, the observed effect must be transcriptional, not posttranscriptional, in nature (see also below).
Next we asked whether the poly(A) signaling observed in vitro (Fig. 1B) is related to poly(A)-dependent termination as measured in vivo (79). To measure termination in vivo we transfected mutant and wild-type pAP〈C9377〉 into COS cells. We then harvested nuclei after 2 days and carried out nascent-transcript analysis to determine the steady-state distribution of polymerases over the three G-less cassettes in vivo (79). It should be emphasized that this nascent-transcript G-less-cassette analysis reflects actual termination in vivo (not pausing) and is free of many of the potential artifacts associated with conventional run-on transcription-hybridization analyses (see reference 79 for a discussion). Figure 1C is a plot of the efficiency with which polymerases reach the downstream cassettes on the wild-type compared to the mutant templates both in vivo and in vitro. Clearly the signaling that occurs in vitro closely parallels the termination that occurs in vivo. We conclude that signaling either yields termination directly, or it triggers an event such as pausing that leads to termination with high efficiency.
Poly(A) signaling commences at the poly(A) site.
The results of Fig. 1 show that transcription is impaired downstream of a wild-type poly(A) site. The simplest interpretation is that the polymerases become transcriptionally impaired as a consequence of crossing the poly(A) site. However, it is formally possible that the wild-type template differs from the mutant template in some global property (e.g., supercoiling) that impairs transcription throughout the plasmid. To distinguish between these possibilities, we assayed transcription both upstream and downstream of the poly(A) site on mutant and wild-type versions of the p377A174P〈C9〉 template. This plasmid contains four cassettes, two upstream and two downstream of the poly(A) site (Fig. 2).
FIG. 2.

Transcription is impaired only downstream of the poly(A) site. (A) Cassettes are transcribed sequentially from the SV40 early promoter. Transcription of the mutant version of p377A174P〈C9〉 was carried out as described in Materials and Methods except that 7 mM Mg2+ and 7 mM citrate were used, and transcription times were varied from 1 to 32 min. The top panel shows molar amounts of each cassette produced plotted on an arbitrary scale. The bottom panel shows molar ratios of each cassette produced relative to the 377-nt cassette. (B) Elongation efficiency drops after crossing the poly(A) site. The percent readthrough for each cassette on the wild-type template relative to that of the mutant is plotted, as for Fig. 1C, above a scale map of the template.
First, we confirmed that the four cassettes in this plasmid are transcribed successively, as predicted for polymerases that initiate at the SV40 early promoter, and then traverse each cassette in turn. To avoid any effect on transcription from an active poly(A) site we used the mutant version of p377A174P〈C9〉 for these preliminary experiments. Figure 2A (top panel) shows that the cassette RNAs do indeed appear sequentially during transcription and then build in concentration. In the bottom panel of Fig. 2A the cassette RNA concentrations are expressed relative to that of the first cassette in the series to emphasize their sequential appearance. Figure 2B shows that the cassettes upstream of the poly(A) site are transcribed similarly from the mutant and wild-type templates but that, downstream of the poly(A) site, transcription on the wild-type template rapidly falls off. Thus, transcription on the wild-type template diminishes relative to the mutant only after the polymerases cross the poly(A) site.
Different poly(A) sites signal similarly.
Figure 3 summarizes results showing that in addition to P (the core SV40 early site), both S (SPA of reference 49) and L (the complete SV40 late site) carry out signaling in our assay. Thus, pAPw〈C9377〉 exhibits impaired elongation compared to its mutant (Fig. 3, constructs 1 and 2) as we have already shown (Fig. 1). However, when the mutant poly(A) site of pAPm〈C9377〉 was replaced, following HpaI-BamHI digestion, by either wild-type S (construct 3) or wild-type L (construct 4), signaling was restored. Thus, the ability of the system described here to support signaling in vitro is quite general.
FIG. 3.

Poly(A) signaling by different poly(A) sites. Construct pASw〈C9377〉 was obtained by replacing the mutant poly(A) site of pAPm〈C9377〉 with an HpaI-BamHI fragment differing only at the ends from the SPA sequence used previously (18): 5′-aacaataa… tgtgtctagaactagtg-3′. Similarly, for pALw〈C9377〉 the mutant poly(A) site was replaced with a SmaI-BamHI-trimmed PCR copy of the SV40 late site differing only at the ends from that used previously (18): 5′-ggggatctggac… tgggag-3′. The histogram shows the percent readthrough for the 261-nt cassette of each template relative to that of pAPm〈C9377〉. For the extract preparation used in these assays the citrate and Mg2+ concentrations were both 7 mM.
Characteristics of the signaling reaction.
The most striking characteristic of the in vitro poly(A) signaling reaction is the citrate requirement. This is illustrated in Fig. 4A where the efficiency of signaling (i.e., 100 − % readthrough) is plotted as a function of citrate concentration. Signaling is not observed in the absence of citrate, but as the citrate concentration is increased (in the presence of 7 mM Mg2+ for this extract) signaling rises and goes through a maximum at a citrate concentration of about 8 mM (Fig. 4A). Citrate curtails RNAPII carboxyl-terminal domain (CTD) phosphorylation and reduces the elongation efficiency of transcription in vitro (64). We were drawn to the use of citrate in the signaling reaction because of the role of citrate in allowing the transactivation response (TAR) element in nascent human immunodeficiency virus (HIV) transcripts to mediate an increase in HIV transcription elongation efficiency (64). In the absence of citrate, apparently, the intrinsic elongation efficiency of HIV transcription is too high to be augmented by TAR. Similarly, after a long period of fruitless experimentation, we reasoned that perhaps the intransigence of our system reflected an intrinsic transcription elongation efficiency that was too high in vitro to be abrogated by the poly(A) signal.
FIG. 4.

Citrate and Mg2+ titrations of the signaling reaction. Total transcription refers to the yield, in arbitrary PhosphorImager units, of the 120-nt cassette from the mutant template. It is called total transcription because both initiation and elongation efficiencies contribute to the yield. Elongation efficiency is defined here as the efficiency of 120- to 261-nt cassette readthrough, in this case when signaling is absent (i.e., the molar ratio of 261- to 120-nt cassette transcription on the mutant template). Signaling is defined as 100% − % wild-type readthrough (i.e., 100 minus the wild type-versus-mutant elongation efficiency ratio, expressed as a percentage). Points without error bars correspond to values taken from a single experiment only. Several different extract preparations were used for the experiments reported in this paper. Therefore, the citrate and Mg2+ optima exhibited in this figure differ slightly from the concentrations used for other experiments reflecting the use of a different extract preparation here. Also note in panels B and C that equivalent signaling efficiencies can be obtained using different combinations of citrate and Mg2+ concentrations, further accounting for the use of different concentrations of these constituents in different experiments.
The results in Fig. 4A show how the relationship between signaling and elongation efficiency varies as a function of citrate concentration. The hypothesized inverse correlation between these two parameters is evident between 4 and 8 mM citrate as the signaling curve rises to its maximum. At higher citrate concentrations (10 and 12 mM) signaling declines. This decline may reflect, in part, the ability of citrate to chelate Mg2+, since the detection of signaling in our system is impaired at low [Mg2+] (Fig. 4B and C).
Figure 4A also confirms that the effect of citrate on transcription is principally at the level of elongation rather than initiation, since the elongation efficiency curve closely matches the curve for total transcription in the range of 4 to 12 mM citrate. It is not surprising that elongation efficiency is an important determinant of cassette yield since, for templates such as pAP〈C9377〉, even the first cassette lies nearly 800 bp downstream of the start point of transcription. The curve for total transcription drops below that for elongation efficiency at 0 mM citrate, probably because initiation is impaired at the higher free Mg2+ concentration that exists in the absence of chelation by citrate. This [Mg2+] is considerably higher than the optimum for the SV40 early promoter (<2 mM) in our extracts (data not shown). A similar effect is apparent in Fig. 4B and C, where the curve for total transcription decreases at high Mg+ concentrations despite continuing increases in elongation efficiency.
In addition to citrate, the signaling reaction at its present state of development requires that the nuclear extract be very crude. As yet we have not subjected this parameter to careful scrutiny, but consistently we have obtained the best signaling with extracts subjected only to a relatively low-speed spin following extraction of the nuclei (see Materials and Methods). The state of the DNA (linear or supercoiled), its method of purification (spin column, gel, or CsCl), and the presence or absence of DNA or RNA carrier are unimportant to signaling. Cassette signals are completely α-amanitin sensitive at 0.2 μg/ml (data not shown) and therefore arise entirely from RNAPII transcription. In summary, poly(A) signaling in our in vitro system is robust and reproducible and therefore suitable for use in mechanistic studies.
Poly(A) signaling resembles a stochastic process and requires no special element in the downstream DNA.
The data in Fig. 1C suggest that the ability to elongate is lost increasingly with distance after crossing the poly(A) site. This trend is illustrated further in Fig. 5, which shows examples of experiments that are representative of our experience with the system. In Fig. 5 the histogram peaks have been aligned with the distal ends of their corresponding cassettes to reflect the distances that must be traveled by the individual polymerases to produce the various cassette transcripts. The data show that transcription gradually decreases across more than 1.8 kb of DNA downstream of the core SV40 early poly(A) site. We have shown previously that commitment of the SV40 early poly(A) site to cleavage and polyadenylation in vivo is a stochastic process (18). In this respect, signaling by this poly(A) site to stop transcription appears to be similar.
FIG. 5.

Transcription diminishes gradually with distance following the poly(A) site. Poly(A) signaling on the pAP〈C9377〉 template (Fig. 1B) is compared with that of two other templates having cassettes separated by shorter distances. Only single examples of reactions with pAP〈377〉 and pAP〈C9〉 are presented here. These reactions were carried out under conditions similar to those described for Fig. 1B and could therefore be directly compared. However, the conclusions for these constructs are consistent with numerous other experiments carried out under a variety of slightly different conditions (that prevent their being plotted on the same graph).
Significantly, the poly(A)-dependent decrease of transcription for the templates in Fig. 5 appears to be relatively insensitive to the nature of the downstream DNA. Thus, for the pAP〈377〉 template, substantial poly(A)-dependent loss of transcription occurs across a region that contains no eukaryotic DNA. In this template the DNA contains, for more than 1 kb downstream of the poly(A) signal, only a mixture of artificial sequences (G-less cassettes and multiple cloning sites) and prokaryotic DNA (chloramphenicol acetyltransferase and pBR322). In contrast, the DNA between the cassettes in pAP〈C9〉 is mostly eukaryotic in origin, having been obtained from the transcription termination region of the chicken βH globin gene. Yet both templates fit the same trend for decrease of transcription with distance following the poly(A) site. This lack of sensitivity to the nature of the underlying DNA is further underscored by the results for pAP〈cat〉 of Fig. 6. Here the poly(A)-dependent loss of transcription takes place across yet a different downstream DNA sequence, but the effect still fits the trend shown in Fig. 5. Since DNA from eukaryotic termination regions, prokaryotic coding regions, and artificial constructs all support signaling similarly, we conclude that signaling in vitro does not require any special element in the downstream DNA.
FIG. 6.

Signaling is not an early event following poly(A) site transcription. (A) Signaling can be inhibited by an antisense transcript placed 126 nt downstream of the poly(A) site. The antisense segment (open arrow) is the HinfI-BamHI fragment of the sequence in panel B inserted backwards into either HindIII- or EcoRV-cut pAP〈cat〉. The region targeted by the antisense sequence is denoted by the grey arrow. The sense-antisense separations for constructs 2 and 3 are 126 and 686 nt, respectively. (B) Sequence for the region denoted by the square bracket over construct 2 in panel A. The sequence for the wild-type version, pAPw〈αcat〉, is shown. The complete sense region (underlined) and part of the antisense region (also underlined) are presented. The poly(A) signal and its downstream complement are shown in boldface type. The G-less cassette is shown in lowercase type. (C) Diagram, drawn linearly to scale, of the stem-loop structure predicted to form in the sense-antisense region of transcripts from the mutant version, pAPm〈αcat〉, of construct 2. The tick marks above and below the duplex indicate the positions of G residues. Note that since it is the mutant version of the stem-loop that is shown, there is a G in the hexamer region of the sense strand reflecting the presence of the hexamer mutation (see Materials and Methods). Accompanying this mutation is a small interruption of base pairing in the stem at the position of the hexamer since the wild-type sequence was used for the antisense module in all of our constructs. The loop is comprised of 117 nt from the 120-nt G-less cassette plus 9 nt of non-G-less sequence at its 3′ end. Three nucleotides at the 5′ end of the G-less cassette invade the sense-antisense duplex. Multifold secondary structure analysis (44) predicts that the three G residues flanking the 3′ end of the cassette interact with a patch of C's (shown in white) near its 5′ end. This secondary structure model for the sense-antisense stem-loop predicts more efficient cutting by RNase T1 in sequences flanking the base of the stem (dark arrow) than within its loop (open arrow) and slower cutting still (dotted arrow) at the mispaired G residue in the mutated hexamer (38). (D) RNase T1 digestion confirms the predicted secondary structure of the pAPm〈αcat〉 stem-loop. Transcription of pAPm〈cat〉 and pAPm〈αcat〉 was carried out for either 15 or 2 min as indicated. Citrate (6 mM) was used. Following transcription one-fourth volume of RNase T1 (1,000 U/μl) was added for 0.25 min or 1.25 min as indicated, and then the mixture was extracted with TRIzol and the RNA was analyzed by gel electrophoresis. The interpretive drawings in the middle of the panel refer to the stem-loop structure shown in part C cut by T1 at one or more of the indicated positions. The mobilities of G-less markers and their nominal sizes are shown on the left. The true sizes of the G-less markers are greater by one than the nominal sizes owing to the single G that remains at the 3′ end following cutting by T1.
Poly(A) signaling does not occur until after poly(A) site extrusion.
It is generally assumed that poly(A) signaling occurs after the poly(A) site has been extruded from the polymerase. This supposition has never been tested experimentally and is based on the presumption that the poly(A) signal is too complex to be recognized directly by the transcription apparatus (32). However, Escherichia coli RNAP is able to monitor DNA sequence throughout its 35-bp footprint to receive regulatory input for both pausing and termination (4, 58). The DNA contacts for RNAPII are presumably even more extensive (25). Additional opportunities for intrinsic regulatory input come from interactions of the RNAP with the DNA-RNA hybrid and with the RNA itself in the exit tunnel (58). Moreover, the efficiency of poly(A)-dependent termination is not, as previously thought (32), directly related to processing efficiency (1, 79), indicating the existence of additional sources and/or sources of regulatory input different from simply the efficiency of processing. Although processing factors are known to affect signaling (8), their role may be limited to maintaining the transcription complex in a responsive conformation. Thus, current data are not inconsistent with a model in which poly(A) signaling reflects an intrinsic ability of the transcription apparatus to recognize the poly(A) site. We therefore decided to test this possibility directly.
If the transcriptional apparatus is intrinsically capable of recognizing the poly(A) site, then signaling may occur before the poly(A) site is extruded from the polymerase. To determine whether poly(A) signaling occurs before or after extrusion of the poly(A) site, we used the cis-antisense approach. This method involves cloning an inverted copy of a poly(A) signal downstream of the properly oriented parent and has been used to study the timing of cleavage and polyadenylation complex assembly in vivo (18). Transcription of the inverted poly(A) signal generates an antisense transcript to the authentic poly(A) signal upstream. When this antisense transcript emerges from the polymerase, it forms a duplex with the previously extruded poly(A) signal and prevents cleavage and polyadenylation (18). If signaling to the polymerase, like assembly of the cleavage and polyadenylation apparatus, occurs following extrusion of the poly(A) site, then signaling-like processing may be blocked by the antisense transcript. However, if signaling occurs earlier—during transcription and extrusion—then the antisense should be unable to block an event that has already occurred.
To determine whether an antisense transcript to the poly(A) site can block signaling to the polymerase, we prepared the constructs diagrammed in Fig. 6A. As before, each construct shown refers to a mutant and wild-type pair. Construct 1, pAP〈cat〉, was the parent construct for the plasmids of this experiment. The separation between the cassettes in pAP〈cat〉 is slightly greater than in pAP〈377〉 of Fig. 5, and pAP〈cat〉 exhibits a slightly greater poly(A)-dependent decrease in transcription; readthrough for the wild type is ∼60% of that for the mutant (Fig. 6A, construct 1). Construct 2 contains, just downstream of the 120-nt cassette, a 218-nt antisense module (α) which is an inverted repeat of the poly(A) signal and its upstream flanking sequences. The antisense transcript is targeted not only to the poly(A) signal (whose cleavage site is 19 nt upstream of the 120-nt G-less cassette [Fig. 6B]) but also to some upstream flanking sequence to ensure adequate duplex stability when the stem-loop (Fig. 6C) forms in the RNA (18).
Figure 6A shows that construct 2 exhibits no signaling: wild-type transcription is indistinguishable (99% readthrough ratio) from that of the mutant. Thus, the presence of the antisense transcript completely blocks signaling by the poly(A) site. This is despite the fact that the separation between the cassettes in construct 2 is significantly greater than in construct 1 so that, other things being equal, signaling for construct 2 would have been expected to be greater than for construct 1 (Fig. 5).
Notice, for construct 2, that the nascent RNA must be extended more than 126 nt beyond full extrusion of the poly(A) signal from the polymerase before the antisense transcript even begins to appear (Fig. 6A, construct 2, and B and C). Moreover, since the antisense must be extruded a further 10 to 20 nt before sense-antisense duplex formation can even be initiated, we conclude that the signaling normally detected by our assay does not occur until at least 140 nt after extrusion of the poly(A) site. In fact, the estimate of 10 to 20 nt as the minimum length for initiation of duplex formation is almost certainly a considerable underestimate. Not only must the initiating duplex be stable enough to form across a large and fairly unstructured G-less RNA loop, but it must also be stable enough to compete with factors seeking to bind the poly(A) site. It is, therefore, likely that the bulk of the signaling detected in these assays actually does not occur until beyond 140 nt following poly(A) site extrusion.
To determine whether the antisense module might be exerting some unspecified negative effect on signaling for this template, we also assayed signaling using construct 3 of Fig. 6A. This construct differs from construct 2 only in that the antisense module is located several hundred base pairs farther downstream, near the end of the interval over which signaling is measured. Since construct 3 is identical to construct 1 up to the beginning of the antisense module, signaling for the two constructs should be similar if the presence of the antisense module in the template is benign. As shown in the histogram of Fig. 6A, signaling for construct 3 did indeed resemble that for construct 1, confirming that the mere presence of the antisense sequence in the template is not detrimental to the signaling assay.
Taken together the above cis-antisense inhibition results demonstrate for the first time that the signal to stop transcription occurs at the level of the RNA and that signaling follows extrusion of the poly(A) site from the polymerase. Indeed, signaling occurs more than 140 nt after the poly(A) signal has been completely extruded (Fig. 6A), indicating that signal transduction occurs sometime in the interval between 140 nt postextrusion and the time at which the transcript is cleaved.
Confirmation of sense-antisense duplex formation during transcription in vitro.
The interpretation of the above cis-antisense results assumes sense-antisense duplex formation. Numerous control experiments in vivo support this mechanism (18). Below we confirm, by means of RNase T1 secondary structure mapping, that sense-antisense duplex formation also occurs rapidly during transcription in vitro. Indeed, the G-less cassette analysis for Fig. 6A required RNase T1 digestion under denaturing conditions (T1 is single-strand specific) in order to digest the duplexed RNA produced by constructs 2 and 3 (see Materials and Methods).
To assess sense-antisense duplex formation during transcription we carried out the secondary structure mapping directly in the transcription mixtures (Fig. 6D). For this mapping we used pAPm〈cat〉 and pAPm〈αcat〉, which are the mutant versions of constructs 1 and 2. The mutants were chosen in order to eliminate any differences in transcription efficiency between the constructs due to signaling. Immediately following transcription we subjected the transcription mixtures to a 1.25-min pulse of RNase T1 digestion. Figure 6C shows the structure of the sense-antisense stem-loop that is predicted to form in transcripts of pAPm〈αcat〉. RNase T1 is expected to cleave rapidly in the unstructured RNA to the right of the stem-loop, more slowly in the loop itself, and slowest of all at the mismatched G in the mutated hexamer (see Fig. 6C legend). Thus, T1 digestion is predicted initially to excise an intact stem-loop and then to process it further by cutting between the cassette and the antisense sequence and at the mismatched G (Fig. 6C). The cassette can be cleaved from the sense sequence only when fraying at the end of the duplex, to which it is anchored, permits. On a denaturing gel these products would be expected to run as 564-, 336-, 222-, 175-, and 161-nt species, accompanied by a small amount of 120-nt G-less cassette (see interpretive drawings in Fig. 6D). The results in Fig. 6D, lane 2, confirm these predictions. The presence of each predicted species is apparent, together with a low yield of the 120-nt cassette. In contrast, lane 1 shows that for the homologous plasmid without the antisense no species other than the 261-nt and 120-nt G-less cassettes appear. When we reduced the T1 pulse to 0.25 min, the 564-nt stem-loop band increased and the 120-nt G-less cassette band decreased in intensity (Fig. 6D, lane 4) as expected for a precursor-product relationship. The vanishing 120-nt band in lane 4 suggests that stem-loop formation is quantitative. The shorter T1 pulse was also accompanied by incomplete trimming of the cassette bands themselves (lane 3). Similar results have been obtained using 1/10 the concentration of T1 and longer digestion times.
The band assignments in Fig. 6D have been confirmed based on several independent criteria. First, the bands appear only for constructs that produce transcripts containing antisense. Second, the bands resulting from secondary structure are selectively eliminated by T1 digestion under denaturing conditions. Third, the mobilities of the bands are consistent with the sizes predicted from Fig. 6C. Finally, the mobilities of the individual bands can be selectively varied by targeted sequence alterations in the predicted stem-loop (data not shown). Thus, (i) the positions of the bands at 336 and 161 nt can be selectively varied by altering the size of the G-less cassette; (ii) the bands at 175 and 161 nt are selectively absent when the experiment is done with pAPw〈αcat〉, which has no mismatched G at the hexamer position; (iii) the position of the band at 222 nt varies with the length of the antisense module used; and (iv) the G-less cassette band at 120 nt is no longer underrepresented if a G-containing insert (which can be cut by T1) is placed between the sense strand of the predicted duplex and the cassette.
The data above confirmed the efficient formation of the predicted sense-antisense stem-loops by the end of a 15-min transcription. To determine whether these structures form continuously during transcription we repeated the 0.25-min T1 analysis, but after only 2 min of transcription. Figure 6D, lane 6 shows that the same spectrum of stem-loop digestion products is produced within this short period of transcription as is produced during the standard reaction (lane 4). Taken together these control experiments show that sense-antisense duplex formation occurs efficiently and continuously during transcription in vitro, as required by a cis-antisense inhibition mechanism.
The signal to stop transcription is generated before transcript cleavage.
As pointed out earlier, the differential cassette recovery that results from attenuated transcription (i.e., reduced production of postcassette) is the opposite of that expected from the degradation that accompanies 3′-end processing (i.e., preferential loss of precassette). Thus, by using the differential recovery of cassettes as a criterion in the development of the in vitro signaling assay, we simultaneously selected for conditions that would uncouple processing from signaling. This is apparent in Fig. 4B and C, where signaling peaks at relatively high Mg2+ concentrations and is severely depressed at the low Mg2+ concentrations that favor cleavage in vitro (56, 59). If signaling and processing have indeed been uncoupled in our system, that would indicate that signaling does not depend on transcript cleavage.
To determine directly whether signaling and processing are uncoupled in our system, we made a new construct for studying signaling, pA〈G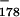 catC9〉, as shown in Fig. 7A. This construct was explicitly designed so that the assay could work only if processing is not a prerequisite for signaling. The essence of the design is a new 178-bp upstream cassette containing an imbedded G-less poly(A) signal (Fig. 7A). This cassette provides not only the upstream cassette for signaling analysis but also the poly(A) site itself. Since the poly(A) site is imbedded in the G-less cassette, any instance of poly(A) site cleavage will also result in cassette cleavage. Thus, if poly(A) signaling depends on prior poly(A) site cleavage, then every failure of the 261-nt cassette to be transcribed (because of signaling) will be matched by a failure of the 178-nt cassette to be recovered (because of cleavage). That is, if cleavage is required for poly(A) signaling, no decrease in 261-nt cassette recovery relative to recovery of the 178-nt cassette will be apparent as a result of signaling, and the basis for detection of signaling in our assay will be eliminated. Therefore, if any signaling at all can be detected as a decreased 261-nt cassette/178-nt cassette ratio for wild-type (AATAAA) compared to mutant (TTTAAA) versions of pA〈G
catC9〉, as shown in Fig. 7A. This construct was explicitly designed so that the assay could work only if processing is not a prerequisite for signaling. The essence of the design is a new 178-bp upstream cassette containing an imbedded G-less poly(A) signal (Fig. 7A). This cassette provides not only the upstream cassette for signaling analysis but also the poly(A) site itself. Since the poly(A) site is imbedded in the G-less cassette, any instance of poly(A) site cleavage will also result in cassette cleavage. Thus, if poly(A) signaling depends on prior poly(A) site cleavage, then every failure of the 261-nt cassette to be transcribed (because of signaling) will be matched by a failure of the 178-nt cassette to be recovered (because of cleavage). That is, if cleavage is required for poly(A) signaling, no decrease in 261-nt cassette recovery relative to recovery of the 178-nt cassette will be apparent as a result of signaling, and the basis for detection of signaling in our assay will be eliminated. Therefore, if any signaling at all can be detected as a decreased 261-nt cassette/178-nt cassette ratio for wild-type (AATAAA) compared to mutant (TTTAAA) versions of pA〈G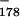 catC9〉, then that signaling cannot be cleavage dependent.
catC9〉, then that signaling cannot be cleavage dependent.
FIG. 7.

Signaling is not cleavage dependent. (A) Signaling by a G-less poly(A) site is not accompanied by processing. The first step of construction for pA〈G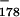 catC9〉 was similar to that for pASw〈C9377〉 of Fig. 3 except that a G-less version of S, the sequence shown in Fig. 7A, was inserted into pAPm〈117cat〉. The G of the blunted BamHI site remaining after ligation was converted to a T by means of site-directed mutagenesis (AATCGATCC→AATattTCC), thereby fusing the G-less poly(A) signal with the 117-nt G-less cassette. To enhance the detection of signaling, the distance between the cassettes was increased by inserting the previously described C9 fragment into the unique EcoRV site (Fig. 4A) of the cat sequence. The cleavage site indicated by the arrow in the wild-type G-less poly(A) sequence is the cleavage position for the original SPA (5). A representative experiment is shown. Citrate was added with the nucleotides for these assays. (B) RNase protection analysis reveals negligible processing of the in vitro transcription products. The same RNA probe was used for all samples in the figure. This probe spans the identical poly(A) sites of pAPw〈C9〉 (lanes 2 and 3) and pIAPw〈cat〉 (lane 4). The latter plasmid, used as a positive control, was obtained from the former by insertion of a PstI-RsaI fragment from pRL-SV40 (Promega) at the StuI site. This adds an intron to allow stable expression of RNA in vivo. The negative control (lane 1) was pAL〈117cat〉, which has identical upstream sequences to pAPw〈C9〉 but a different poly(A) site. Note that the bands marked with asterisks are present irrespective of the presence or absence of the homologous poly(A) site in the in vitro transcribed construct. The smudge in lane 1 at the position of uncleaved RNA is the edge of a halo from an intense band that was in the lane to the left on the gel.
catC9〉 was similar to that for pASw〈C9377〉 of Fig. 3 except that a G-less version of S, the sequence shown in Fig. 7A, was inserted into pAPm〈117cat〉. The G of the blunted BamHI site remaining after ligation was converted to a T by means of site-directed mutagenesis (AATCGATCC→AATattTCC), thereby fusing the G-less poly(A) signal with the 117-nt G-less cassette. To enhance the detection of signaling, the distance between the cassettes was increased by inserting the previously described C9 fragment into the unique EcoRV site (Fig. 4A) of the cat sequence. The cleavage site indicated by the arrow in the wild-type G-less poly(A) sequence is the cleavage position for the original SPA (5). A representative experiment is shown. Citrate was added with the nucleotides for these assays. (B) RNase protection analysis reveals negligible processing of the in vitro transcription products. The same RNA probe was used for all samples in the figure. This probe spans the identical poly(A) sites of pAPw〈C9〉 (lanes 2 and 3) and pIAPw〈cat〉 (lane 4). The latter plasmid, used as a positive control, was obtained from the former by insertion of a PstI-RsaI fragment from pRL-SV40 (Promega) at the StuI site. This adds an intron to allow stable expression of RNA in vivo. The negative control (lane 1) was pAL〈117cat〉, which has identical upstream sequences to pAPw〈C9〉 but a different poly(A) site. Note that the bands marked with asterisks are present irrespective of the presence or absence of the homologous poly(A) site in the in vitro transcribed construct. The smudge in lane 1 at the position of uncleaved RNA is the edge of a halo from an intense band that was in the lane to the left on the gel.
Figure 7A shows that signaling, to the extent of 28% impaired transcription in the wild type, was indeed detected upon comparison of wild-type and mutant versions of the pA〈G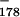 catC9〉 template. Although this is less signaling than obtained with our other plasmids, we were nevertheless surprised at the vigor of the effect, since the poly(A) signal in this template had been extensively mutated (all of its G residues). Despite the unusual sequence of the G-less poly(A) signal, there can be no doubt that the impaired transcription of the wild-type plasmid is a poly(A)-dependent effect because the transcriptional impairment is abolished by mutating only two of the A's in the poly(A) hexamer (Fig. 7A). These results unambiguously establish that poly(A) signaling is cleavage independent in vitro.
catC9〉 template. Although this is less signaling than obtained with our other plasmids, we were nevertheless surprised at the vigor of the effect, since the poly(A) signal in this template had been extensively mutated (all of its G residues). Despite the unusual sequence of the G-less poly(A) signal, there can be no doubt that the impaired transcription of the wild-type plasmid is a poly(A)-dependent effect because the transcriptional impairment is abolished by mutating only two of the A's in the poly(A) hexamer (Fig. 7A). These results unambiguously establish that poly(A) signaling is cleavage independent in vitro.
Although it was clear from the data discussed above that the observed poly(A) signaling is cleavage independent, it remained possible that some processing at the G-less poly(A) site, unrelated to signaling, may also have occurred. If so the 5′ polyadenylation product, and perhaps also the 3′ cleavage and partial degradation products, should be detectable on the gel (81). However, the lane containing the wild type in Fig. 7A reveals no evidence of any G-less poly(A) site processing. Poly(A) site cleavage in the 178-nt precassette would yield 5′ 28-nt and 3′ 150-nt subcassettes. Polyadenylation of the 5′ product would yield a heterogeneous collection of G-less RNAs on the gel ranging from perhaps 180 to 280 nt. Partial degradation of the 3′ product would yield a heterogeneous collection of G-less RNAs on the gel of less than 150 nt. There is no evidence in the wild type-containing lane of Fig. 7A for any of these products of processing. Thus, not only is signaling cleavage independent in this assay but, at least for the G-less poly(A) site, there does not seem to be any processing at all under these experimental conditions.
The G-less poly(A) site offers the advantage that any signaling detected by our assay must unambiguously have occurred by a cleavage-independent mechanism. However, the poly(A) site used for most of the experiments in the present study was the SV40 early poly(A) site. In order to determine whether signaling is also cleavage independent for this site, we carried out RNase protection assays on the products remaining after signaling reactions. Figure 7B shows that essentially no cleavage at the SV40 early poly(A) site occurred under the conditions of our assays. Lane 2 shows the result of an RNase protection analysis of the in vitro transcription products of a typical 15-min signaling reaction. Lane 3 shows the result for a signaling reaction that was allowed to incubate three times as long. Lane 4 shows the RNase protection result for authentic cellular mRNA containing the same poly(A) site. For the cellular RNA (lane 4) a band at the expected position for cleaved RNA (107 nt) is obtained, plus a subband. In contrast, for the in vitro RNA these bands are either scarcely visible (15-min reaction, lane 2) or only faintly apparent (45-min reaction, lane 3). Instead, the in vitro RNA yields almost exclusively a protected species migrating near the 129-nt position expected for uncleaved RNA. Additional bands flanking the position for cleaved RNA in lanes 2 and 3 are not related to 3′ end formation as they appear also in assays of RNA from a plasmid bearing an unrelated poly(A) site (lane 1).
Quantitative analysis indicates an upper limit of less than 2% poly(A) site cleavage of RNA transcribed in vitro for 15 min (Fig. 7B, lane 2). In contrast, poly(A) signaling can lead to an elongation deficit of more than 60% under the same conditions (Fig. 1B). Thus, signaling has occurred in the virtual absence of cleavage. Even after 45 min of incubation in vitro the proportion of poly(A) site cleavage reaches only 6% (maximum estimate from analysis of Fig. 7B, lane 3). We conclude that poly(A) signaling for the SV40 early poly(A) site has been uncoupled from poly(A) site cleavage under our reaction conditions. Therefore, signaling is independent of cleavage for both a natural and an artificial poly(A) site.
DISCUSSION
Poly(A) signaling in vitro.
Poly(A)-dependent termination is presumably a complex process in which poly(A) signaling is but the first of several mechanistic steps. For example, signaling may lead first to pausing and then to transcript release. To focus on signaling per se, therefore, we have used in this work an assay that does not require transcript release in order to produce a measurable outcome. Rather, the assay detects any downstream effect of signaling that impedes transcription.
Our results show that poly(A) signaling in vitro faithfully reflects three important aspects of poly(A)-dependent termination in vivo (79). First, of course, signaling is dependent on a functional poly(A) site. Second, signaling exerts its effect regardless of the nature of the downstream DNA: no special pause site or other element is required. Third, the effect increases with distance, suggesting a stochastic process. Thus, the in vitro events characterized here recapitulate important aspects of the in vivo process that leads to poly(A)-dependent termination.
Detection of signaling under our assay conditions requires citrate. The molecular target of citrate is not known, but citrate has been shown to inhibit phosphorylation of the RNAPII CTD (64) and is said to be an HIV-specific depressant of elongation (63). Here we confirm, using an assay for elongation efficiency, that citrate preferentially impairs elongation (Fig. 4A). Our results also show that the effect is not restricted to HIV since our constructs are driven by the SV40 early promoter. Moreover, our elongation efficiency assay (Fig. 4) measures the efficiency of elongation between 895 and 2,757 bp downstream of the promoter, showing that the effect of citrate is not limited to promoter-proximal events. Thus, the reduction of elongation efficiency imposed by citrate includes the region of the template containing the poly(A) site. It is possible that, by reducing the phosphorylation of the CTD, citrate renders the polymerases more responsive to the poly(A) signal. Alternatively, citrate may reduce a background of efficient elongation contributed by nonresponding polymerases that otherwise would mask signaling. It is also possible that citrate affects signaling directly, by acting on some component of the transcription complex independently of its effect on elongation efficiency.
Using this poly(A) signaling assay we have shown here that signaling occurs well after extrusion of the poly(A) site, but before cleavage, and that it does not depend on the presence of pause elements in the DNA. Assembly of the cleavage and polyadenylation apparatus occurs in distinguishable steps in vivo (18). The results reported here support our earlier suggestion (18) that signaling may occur during one of these intermediate steps, being generated by new interactions among known factors (3, 8,14, 17) or by the recruitment of new factors responsible for termination.
Poly(A) signaling is not coupled to extrusion of the poly(A) site.
Before 3′-end processing can occur, the poly(A) site must first undergo extrusion and then stepwise assembly of the cleavage and polyadenylation apparatus (18). Cleavage-independent models for poly(A) signaling may accordingly be divided into intrinsic and extrinsic models according to whether recognition is by the transcriptional apparatus itself or by additional factors that recognize the signal following extrusion. Here we have addressed the intrinsic class of models which propose that the poly(A) site is recognized as it is being transcribed and extruded, thereby leading to termination. A precedent for such a mechanism is E. coli RNA polymerase, which can recognize signals for pausing and termination either directly in the DNA or in the nascent RNA during extrusion and whose responses to these signals can be modulated by factors (4, 58). Accordingly, in eukaryotes, RNAPII may recognize the poly(A) site during transcription, and its response to the poly(A) site may be modulated by the polymerase-associated poly(A) site cleavage factors that are known to be required for termination (3, 8,14, 17). This model has never been tested but is now ruled out by the experiment of Fig. 6, which establishes that signaling cannot have taken place during transcription of the poly(A) site, since all signaling can still be blocked by antisense after extrusion of the poly(A) site is complete.
Our data also address the simplest of the extrinsic models for poly(A) signaling. This model, an extension of the above scenario, is the CPSF-CstF recognition model, which proposes that the signal to terminate is generated as the poly(A) site is propelled past the CTD-bound CPSF and CstF after emerging from the polymerase (55). The initial interaction between these factors and the poly(A) site is envisioned as the event that generates the signal. Both functional and structural considerations (see below) suggest that CPSF and CstF interact promptly with the poly(A) site when it emerges from the polymerase. Yet the results of Fig. 6 show that signaling has still not occurred 140 or more nt after extrusion is complete, despite the presumed efficiency with which the poly(A) signal is captured by CPSF and CstF. Accordingly, our results disfavor the CPSF-CstF-recognition model and suggest that continued association of CPSF and CstF with the poly(A) site is required in order to participate in generating the signal to terminate at a later step of cleavage and polyadenylation complex assembly.
Prompt targeting of CPSF and CstF to the poly(A) site following extrusion is suggested by analogy to the mechanism whereby capping enzyme is targeted to the RNA 5′ end. Like CPSF and CstF, capping enzyme binds to the polymerase CTD, which then delivers it to the emerging RNA (43, 54). This delivery is so efficient that the RNA is quantitatively capped in vitro before it is 50 nt long (46). Additional studies in vivo suggest that capping occurs when the RNA is between 25 and 30 nt long (69). Since approximately 18 nt of RNA is contained within the structure of the RNAPII ternary complex (39), capping must occur on 5′ ends that lie only about 10 nt beyond the point of extrusion. Thus, functionally, the CTD appears to be designed to deliver capping enzyme and possibly other proteins to a region very close to the point at which the RNA emerges from the polymerase. This functional conclusion is consistent with the recently published crystal structure of RNAPII, which indicates that the newly extruded RNA emerges at the base of the CTD (25, 36).
Given the juxtaposition of the CTD to the emerging RNA, the presumed flexibility of the CTD (25), the role of the CTD in directing virtually immediate capping of emergent RNA, and the role of the CTD in facilitating 3′ end processing, it is likely that the CPSF and the CstF on the CTD gain rapid access to the nascent poly(A) site. In this they would resemble the vaccinia virus transcription termination factor, which also travels with the polymerase and then accesses its cognate element in the nascent RNA as soon as it emerges (40). Unlike the vaccinia situation, however, initial recognition of the poly(A) signal by CPSF and CstF does not appear to coincide with the signal transduction event. Thus, some later event on the way to processing is apparently responsible for signaling. Nevertheless, 140 nt of RNA would be insufficient to reach the end of a hypothetical, fully extended, inflexible CTD. Therefore, the CPSF-CstF-recognition model remains a formal possibility.
Poly(A) signaling is not cleavage dependent.
Very soon after it was discovered that termination is poly(A) dependent, it was suggested that processing itself might be the signal that leads to termination (50). Subsequently, elegant models were developed (20, 68) and refined (30, 81) to explain how cleavage could be instrumental in generating the signal. Attempts to test cleavage-dependent models have centered on the relative timing of cleavage and termination in vivo (11, 30, 61). The results for several genes indicate that these events both occur at various distances downstream of the poly(A) site but that, within the resolution of the experiments, they tend to occur together. This has led to the view that cleavage and termination in vivo may both be part of a single concerted event (11, 12, 30). It has variously been suggested that this event is triggered by cleavage (30) or that it is not triggered by cleavage (61). Both possibilities are consistent with the available in vivo data, as are the additional possibilities that cleavage is triggered by termination or that both occur independently in response to a common signal. Thus, the central postulate of the cleavage-dependent models, namely, that cleavage generates the signal to the polymerase, has remained an open question.
As is often the case, the relationships among coupled events are clarified by uncoupling them in vitro. By uncoupling cleavage and signaling in our assay we have shown that signaling occurs in the essential absence of any cleavage. This was demonstrated directly for the SV40 early (Fig. 7B) and the G-less poly(A) sites (Fig. 7A). Moreover, because optimization of the signaling assay involved selecting for conditions that suppress processing, this is evidently true for the SV40 late and the synthetic poly(A) sites as well (Fig. 3). We hasten to point out, however, that our extracts do carry out efficient 3′-end processing under conventional cleavage and polyadenylation conditions (75) using exogenously prepared pre-mRNA substrates (data not shown).
Poly(A) signaling does not require downstream elements in the DNA.
We have shown here that four different poly(A) sites are capable of signaling a stop to transcription in vitro in the absence of any identifiable downstream elements. For example, the SV40 early poly(A) site transduces the signal to stop transcription across both eukaryotic and artificial DNA sequences (Fig. 5), as well as across prokaryotic DNA (Fig. 6A). The same is true for the SV40 late, the SPA, and the G-less poly(A) signals (Fig. 3, Fig. 7A, and additional data not shown). This confirms and extends our previous report that poly(A) signals can induce efficient termination in vivo without the assistance of downstream elements in the DNA (79).
Nevertheless, a number of downstream elements have been described that do enhance termination when located downstream of poly(A) sites (2, 6, 13, 21, 31, 33, 72). However, only occasionally have the effects of the presence and absence of these elements been determined in a background lacking poly(A) signals (e.g., references 22, 31, and 41). Thus, often it is not known whether a poly(A)-independent effect has simply been superimposed on a poly(A)-containing background, or whether such an element interacts synergistically with the poly(A) site to constitute a true downstream member of a bipartite terminator.
Pause elements in downstream DNA are a prominent feature of most discussions of poly(A)-dependent termination (e.g., see references 2, 6, 13, 33, 41, 73, 81, and 82). These elements are thought to be required to slow down the polymerase so that the poly(A) signal can act. Yet, with one possible exception, no termination-enhancing element has been shown to pause polymerases in the absence of a functioning poly(A) signal. For example, the nmt2 pause element of Schizosaccharomyces pombe (2) does not induce pausing if the poly(A) signal is deleted (41). Yet there are pausing and efficient termination downstream of the poly(A) signal if the element itself is deleted (41). Thus, although the element is unquestionably an enhancer of polyadenylation (2), it is not a pause element and it is not required for pausing or termination. The possible exception referred to above is an element that was accompanied by both pausing and termination downstream of a mutant poly(A) site in one experiment (33) but which gave no discernible pausing downstream of a wild-type poly(A) site in other experiments (77).
If the various downstream elements that have been described are not pause elements and are generally not required for termination, what might be their function? Since we have shown both in vivo (79) and in vitro (this report) that a poly(A) signal alone is sufficient to stop transcription, these downstream elements are unlikely to be an integral part of the poly(A)- dependent termination mechanism. The so-called pause elements discussed above, actually resemble enhancers or auxiliary downstream elements for polyadenylation (19, 35, 52). They may function at the RNA level and affect termination by modifying the pathway leading to cleavage and polyadenylation itself. A second type of element functions at the DNA level to bind a protein that enhances processing and/or termination when encountered by a transcription complex (6, 21, 81). One such element, the MAZ protein binding site, enhances cleavage and polyadenylation in vivo (6). MAZ bound to DNA causes pausing in vitro, but the pausing per se is not responsible for the enhanced cleavage and polyadenylation, since pausing by other proteins has no effect on processing unless the polymerases are paused next to MAZ (80, 81). MAZ may, in fact, be a poly(A)-independent termination factor in vivo (15), a property shared by the only other DNA-binding protein thought to be involved in termination by RNAPII (21). Significantly, both of these proteins are known primarily for their roles in transcription initiation. Thus, their roles in processing and termination may be primarily to act as fail-safe devices, not necessarily dependent on a poly(A) signal, designed to protect promoters from transcription interference (37).
ACKNOWLEDGMENTS
We thank Carol Eng in the laboratory of Arnie Berk for a constant supply of HeLa cell starter cultures; Guillaume Chanfreau for insightful comments on the manuscript; and Ian Orozco, Amir Kazerouninia, and David Tsao for contributing clones.
This work was supported by NIH grant GM50863 and by an award from the Jonsson Cancer Center Foundation.
REFERENCES
- 1.Aranda A, Perez-Ortin J E, Moore C, del Olmo M L. Transcription termination downstream of the Saccharomyces cerevisiae FBP1 poly(A) site does not depend on efficient 3′end processing. RNA. 1998;4:303–318. [PMC free article] [PubMed] [Google Scholar]
- 2.Aranda A, Proudfoot N J. Definition of transcriptional pause elements in fission yeast. Mol Cell Biol. 1999;19:1251–1261. doi: 10.1128/mcb.19.2.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aranda A, Proudfoot N. Transcriptional termination factors for RNA polymerase II in yeast. Mol Cell. 2001;7:1003–1011. doi: 10.1016/s1097-2765(01)00235-0. [DOI] [PubMed] [Google Scholar]
- 4.Artsimovitch I, Landick R. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc Natl Acad Sci USA. 2000;97:7090–7095. doi: 10.1073/pnas.97.13.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashe M P, Pearson L H, Proudfoot N J. The HIV-1 5′ LTR poly(A) site is inactivated by U1 snRNP interaction with the downstream major splice donor site. EMBO J. 1997;16:5752–5763. doi: 10.1093/emboj/16.18.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashfield R, Patel A J, Bossone S A, Brown H, Campbell R D, Marcu K B, Proudfoot N J. MAZ-dependent termination between closely spaced human complement genes. EMBO J. 1994;13:5656–5667. doi: 10.1002/j.1460-2075.1994.tb06904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker C C, Noe J S. Transcriptional termination between bovine papillomavirus type 1 (BPV-1) early and late polyadenylation sites blocks late transcription in BPV-1-transformed cells. J Virol. 1989;63:3529–3534. doi: 10.1128/jvi.63.8.3529-3534.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barilla D, Lee B A, Proudfoot N J. Cleavage/polyadenylation factor IA associates with the carboxyl-terminal domain of RNA polymerase II in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2001;98:445–450. doi: 10.1073/pnas.98.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrera-Saldana H, Takahashi K, Vigneron M, Wildeman A, Davidson I, Chambon P. All six GC-motifs of the SV40 early upstream element contribute to promoter activity in vivo and in vitro. EMBO J. 1985;4:3839–3849. doi: 10.1002/j.1460-2075.1985.tb04156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batt D B, Luo Y, Carmichael G G. Polyadenylation and transcription termination in gene constructs containing multiple tandem polyadenylation signals. Nucleic Acids Res. 1994;22:2811–2816. doi: 10.1093/nar/22.14.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baurén G, Belikov S, Wieslander L. Transcriptional termination in the Balbiani ring 1 gene is closely coupled to 3′-end formation and excision of the 3′-terminal intron. Genes Dev. 1998;12:2759–2769. doi: 10.1101/gad.12.17.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentley D. Coupling RNA polymerase II transcription with pre-mRNA processing. Curr Opin Cell Biol. 1999;11:347–351. doi: 10.1016/S0955-0674(99)80048-9. [DOI] [PubMed] [Google Scholar]
- 13.Birse C E, Lee B A, Hansen K, Proudfoot N J. Transcriptional termination signals for RNA polymerase II in fission yeast. EMBO J. 1997;16:3633–3643. doi: 10.1093/emboj/16.12.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birse C E, Minvielle-Sebastia L, Lee B A, Keller W, Proudfoot N J. Coupling termination of transcription to messenger RNA maturation in yeast. Science. 1998;280:298–301. doi: 10.1126/science.280.5361.298. [DOI] [PubMed] [Google Scholar]
- 15.Bossone S A, Asselin C, Patel A J, Marcu K B. MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc Natl Acad Sci USA. 1992;89:7452–7456. doi: 10.1073/pnas.89.16.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brackenridge S, Ashe H L, Giacca M, Proudfoot N J. Transcription and polyadenylation in a short human intergenic region. Nucleic Acids Res. 1997;25:2326–2336. doi: 10.1093/nar/25.12.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvo O, Manley J L. Evolutionarily conserved interaction between CstF-64 and PC4 links transcription, polyadenylation, and termination. Mol Cell. 2001;7:1013–1023. doi: 10.1016/s1097-2765(01)00236-2. [DOI] [PubMed] [Google Scholar]
- 18.Chao L C, Jamil A, Kim S J, Huang L, Martinson H G. Assembly of the cleavage and polyadenylation apparatus requires about 10 seconds in vivo and is faster for strong than for weak poly(A) sites. Mol Cell Biol. 1999;19:5588–5600. doi: 10.1128/mcb.19.8.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen F, Wilusz J. Auxiliary downstream elements are required for efficient polyadenylation of mammalian pre-mRNAs. Nucleic Acids Res. 1998;26:2891–2898. doi: 10.1093/nar/26.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connelly S, Manley J L. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev. 1988;2:440–452. doi: 10.1101/gad.2.4.440. [DOI] [PubMed] [Google Scholar]
- 21.Connelly S, Manley J L. RNA polymerase II transcription termination is mediated specifically by protein binding to a CCAAT box sequence. Mol Cell Biol. 1989;9:5254–5259. doi: 10.1128/mcb.9.11.5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connelly S, Manley J L. A CCAAT box sequence in the adenovirus major late promoter functions as part of an RNA polymerase II termination signal. Cell. 1989;57:561–571. doi: 10.1016/0092-8674(89)90126-8. [DOI] [PubMed] [Google Scholar]
- 23.Conrad N K, Wilson S M, Steinmetz E J, Patturajan M, Brow D A, Swanson M S, Corden J L. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 2000;154:557–571. doi: 10.1093/genetics/154.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coulon V, Veyrune J L, Tourkine N, Vie A, Hipskind R A, Blanchard J M. A novel calcium signaling pathway targets the c-fos intragenic transcriptional pausing site. J Biol Chem. 1999;274:30439–30446. doi: 10.1074/jbc.274.43.30439. [DOI] [PubMed] [Google Scholar]
- 25.Cramer P, Bushnell D A, Kornberg R D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. [Google Scholar]
- 26.Dantonel J C, Murthy K G, Manley J L, Tora L. Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature. 1997;389:399–402. doi: 10.1038/38763. [DOI] [PubMed] [Google Scholar]
- 27.Deng L, Hagler J, Shuman S. Factor-dependent release of nascent RNA by ternary complexes of vaccinia RNA polymerase. J Biol Chem. 1996;271:19556–19562. doi: 10.1074/jbc.271.32.19556. [DOI] [PubMed] [Google Scholar]
- 28.Deng L, Shuman S. Vaccinia NPH-I, a DExH-box ATPase, is the energy coupling factor for mRNA transcription termination. Genes Dev. 1998;12:538–546. doi: 10.1101/gad.12.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dichtl B, Keller W. Recognition of polyadenylation sites in yeast pre-mRNAs by cleavage and polyadenylation factor. EMBO J. 2001;20:3197–3209. doi: 10.1093/emboj/20.12.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dye M J, Proudfoot N J. Terminal exon definition occurs cotranscriptionally and promotes termination of RNA polymerase II. Mol Cell. 1999;3:371–378. doi: 10.1016/s1097-2765(00)80464-5. [DOI] [PubMed] [Google Scholar]
- 31.Dye M J, Proudfoot N J. Multiple transcript cleavage precedes polymerase release in termination by RNA polymerase II. Cell. 2001;105:669–681. doi: 10.1016/s0092-8674(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 32.Edwalds-Gilbert G, Prescott J, Falck-Pedersen E. 3′ RNA processing efficiency plays a primary role in generating termination-competent RNA polymerase II elongation complexes. Mol Cell Biol. 1993;13:3472–3480. doi: 10.1128/mcb.13.6.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enriquez-Harris P, Levitt N, Briggs D, Proudfoot N J. A pause site for RNA polymerase II is associated with termination of transcription. EMBO J. 1991;10:1833–1842. doi: 10.1002/j.1460-2075.1991.tb07709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flaherty S M, Fortes P, Izaurralde E, Mattaj I W, Gilmartin G M. Participation of the nuclear cap binding complex in pre-mRNA 3′ processing. Proc Natl Acad Sci USA. 1997;94:11893–11898. doi: 10.1073/pnas.94.22.11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilmartin G M, Hung S L, DeZazzo J D, Fleming E S, Imperiale M J. Sequences regulating poly(A) site selection within the adenovirus major late transcription unit influence the interaction of constitutive processing factors with the pre-mRNA. J Virol. 1996;70:1775–1783. doi: 10.1128/jvi.70.3.1775-1783.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gnatt A L, Cramer P, Fu J, Bushnell D A, Kornberg R D. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science. 2001;292:1876–1882. [Google Scholar]
- 37.Greger I H, Aranda A, Proudfoot N. Balancing transcriptional interference and initiation on the GAL7 promoter of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:8415–8420. doi: 10.1073/pnas.140217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greiner-Stoffele T, Foerster H H, Hahn U. Ribonuclease T1 cleaves RNA after guanosines within single-stranded gaps of any length. Nucleosides Nucleotides Nucleic Acids. 2000;19:1101–1109. doi: 10.1080/15257770008035033. [DOI] [PubMed] [Google Scholar]
- 39.Gu W, Wind M, Reines D. Increased accommodation of nascent RNA in a product site on RNA polymerase II during arrest. Proc Natl Acad Sci USA. 1996;93:6935–6940. doi: 10.1073/pnas.93.14.6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagler J, Luo Y, Shuman S. Factor-dependent transcription termination by vaccinia RNA polymerase. Kinetic coupling and requirement for ATP hydrolysis. J Biol Chem. 1994;269:10050–10060. [PubMed] [Google Scholar]
- 41.Hansen K, Birse C E, Proudfoot N J. Nascent transcription from the nmt1 and nmt2 genes of Schizosaccharomyces pombe overlaps neighbouring genes. EMBO J. 1998;17:3066–3077. doi: 10.1093/emboj/17.11.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirose Y, Manley J L. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000;14:1415–1429. [PubMed] [Google Scholar]
- 43.Ho C K, Sriskanda V, McCracken S, Bentley D, Schwer B, Shuman S. The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 1998;273:9577–9585. doi: 10.1074/jbc.273.16.9577. [DOI] [PubMed] [Google Scholar]
- 44.Jaeger J A, Turner D H, Zuker M. Improved predictions of secondary structures for RNA. Proc Natl Acad Sci USA. 1989;86:7706–7710. doi: 10.1073/pnas.86.20.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jansa P, Grummt I. Mechanism of transcription termination: PTRF interacts with the largest subunit of RNA polymerase I and dissociates paused transcription complexes from yeast and mouse. Mol Gen Genet. 1999;262:508–514. doi: 10.1007/s004380051112. [DOI] [PubMed] [Google Scholar]
- 46.Jove R, Manley J L. In vitro transcription from the adenovirus 2 major late promoter utilizing templates truncated at promoter-proximal sites. J Biol Chem. 1984;259:8513–8521. [PubMed] [Google Scholar]
- 47.Kulish D, Struhl K. TFIIS enhances transcriptional elongation through an artificial arrest site in vivo. Mol Cell Biol. 2001;21:4162–4168. doi: 10.1128/MCB.21.13.4162-4168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J M, Greenleaf A L. Modulation of RNA polymerase II elongation efficiency by C-terminal heptapeptide repeat domain kinase I. J Biol Chem. 1997;272:10990–10993. doi: 10.1074/jbc.272.17.10990. [DOI] [PubMed] [Google Scholar]
- 49.Levitt N, Briggs D, Gil A, Proudfoot N J. Definition of an efficient synthetic poly(A) site. Genes Dev. 1989;3:1019–1025. doi: 10.1101/gad.3.7.1019. [DOI] [PubMed] [Google Scholar]
- 50.Logan J, Falck-Pedersen E, Darnell J E, Jr, Shenk T. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouse βmaj-globin gene. Proc Natl Acad Sci USA. 1987;84:8306–8310. doi: 10.1073/pnas.84.23.8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.London L, Keene R G, Landick R. Analysis of premature termination in c-myc during transcription by RNA polymerase II in a HeLa nuclear extract. Mol Cell Biol. 1991;11:4599–4615. doi: 10.1128/mcb.11.9.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lou H, Helfman D M, Gagel R F, Berget S M. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol Cell Biol. 1999;19:78–85. doi: 10.1128/mcb.19.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maa M C, Chinsky J M, Ramamurthy V, Martin B D, Kellems R E. Identification of transcription stop sites at the 5′ and 3′ ends of the murine adenosine deaminase gene. J Biol Chem. 1990;265:12513–12519. [PubMed] [Google Scholar]
- 54.McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley D L. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11:3306–3318. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson S D, Wickens M, Bentley D L. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- 56.Mifflin R C, Kellems R E. Coupled transcription-polyadenylation in a cell-free system. J Biol Chem. 1991;266:19593–19598. [PubMed] [Google Scholar]
- 57.Miralles V J. Termination of transcription in an ‘in vitro’ system is dependent on a polyadenylation sequence. Nucleic Acids Res. 1991;19:3593–3599. doi: 10.1093/nar/19.13.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mooney R A, Artsimovitch I, Landick R. Information processing by RNA polymerase: recognition of regulatory signals during RNA chain elongation. J Bacteriol. 1998;180:3265–3275. doi: 10.1128/jb.180.13.3265-3275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore C L, Sharp P A. Accurate cleavage and polyadenylation of exogenous RNA substrate. Cell. 1985;41:845–855. doi: 10.1016/s0092-8674(85)80065-9. [DOI] [PubMed] [Google Scholar]
- 60.Murthy K G, Park P, Manley J L. A nuclear micrococcal-sensitive, ATP-dependent exoribonuclease degrades uncapped but not capped RNA substrates. Nucleic Acids Res. 1991;19:2685–2692. doi: 10.1093/nar/19.10.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osheim Y N, Proudfoot N J, Beyer A L. EM visualization of transcription by RNA polymerase II: downstream termination requires a poly(A) signal but not transcript cleavage. Mol Cell. 1999;3:379–387. doi: 10.1016/s1097-2765(00)80465-7. [DOI] [PubMed] [Google Scholar]
- 62.Palangat M, Meier T I, Keene R G, Landick R. Transcriptional pausing at +62 of the HIV-1 nascent RNA modulates formation of the TAR RNA structure. Mol Cell. 1998;1:1033–1042. doi: 10.1016/s1097-2765(00)80103-3. [DOI] [PubMed] [Google Scholar]
- 63.Parada C A, Yoon J B, Roeder R G. A novel LBP-1-mediated restriction of HIV-1 transcription at the level of elongation in vitro. J Biol Chem. 1995;270:2274–2283. doi: 10.1074/jbc.270.5.2274. [DOI] [PubMed] [Google Scholar]
- 64.Parada C A, Roeder R G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature. 1996;384:375–378. doi: 10.1038/384375a0. [DOI] [PubMed] [Google Scholar]
- 65.Plet A, Eick D, Blanchard J M. Elongation and premature termination of transcripts initiated from c-fos and c-myc promoters show dissimilar patterns. Oncogene. 1995;10:319–328. [PubMed] [Google Scholar]
- 66.Powell W, Reines D. Mutations in the second largest subunit of RNA polymerase II cause 6-azauracil sensitivity in yeast and increased transcriptional arrest in vitro. J Biol Chem. 1996;271:6866–6873. doi: 10.1074/jbc.271.12.6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Proudfoot N. Connecting transcription to messenger RNA processing. Trends Biochem Sci. 2000;25:290–293. doi: 10.1016/s0968-0004(00)01591-7. [DOI] [PubMed] [Google Scholar]
- 68.Proudfoot N J. How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem Sci. 1989;14:105–110. doi: 10.1016/0968-0004(89)90132-1. [DOI] [PubMed] [Google Scholar]
- 69.Rasmussen E B, Lis J T. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci USA. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reeder R H, Guevara P, Roan J G. Saccharomyces cerevisiae RNA polymerase I terminates transcription at the Reb1 terminator in vivo. Mol Cell Biol. 1999;19:7369–7376. doi: 10.1128/mcb.19.11.7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takagaki Y, Manley J L. Complex protein interactions within the human polyadenylation machinery identify a novel component. Mol Cell Biol. 2000;20:1515–1525. doi: 10.1128/mcb.20.5.1515-1525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tantravahi J, Alvira M, Falck-Pedersen E. Characterization of the mouse βmaj globin transcription termination region: a spacing sequence is required between the poly(A) signal sequence and multiple downstream termination elements. Mol Cell Biol. 1993;13:578–587. doi: 10.1128/mcb.13.1.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tollervey D, Caceres J F. RNA processing marches on. Cell. 2000;103:703–709. doi: 10.1016/s0092-8674(00)00174-4. [DOI] [PubMed] [Google Scholar]
- 74.Vandenbergh D J, James-Pederson M, Hardison R C. An apparent pause site in the transcription unit of the rabbit α-globin gene. J Mol Biol. 1991;220:255–270. doi: 10.1016/0022-2836(91)90011-t. [DOI] [PubMed] [Google Scholar]
- 75.Wahle E, Keller W. 3′ end-processing of mRNA. In: Higgins S J, Hames B D, editors. RNA processing: a practical approach. II. New York, N.Y: Oxford University Press; 1994. pp. 1–34. [Google Scholar]
- 76.Wahle E, Ruegsegger U. 3′-End processing of pre-mRNA in eukaryotes. FEMS Microbiol Rev. 1999;23:277–295. doi: 10.1111/j.1574-6976.1999.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 77.Whitelaw E, Proudfoot N. α-Thalassaemia caused by a poly(A) site mutation reveals that transcriptional termination is linked to 3′ end processing in the human α2 globin gene. EMBO J. 1986;5:2915–2922. doi: 10.1002/j.1460-2075.1986.tb04587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu M, Barnard M B, Rose S M, Cockerill P N, Huang S Y, Garrard W T. Transcription termination and chromatin structure of the active immunoglobulin kappa gene locus. J Biol Chem. 1986;261:3838–3845. [PubMed] [Google Scholar]
- 79.Yeung G, Choi L M, Chao L C, Park N J, Liu D, Jamil A, Martinson H G. Poly(A)-driven and poly(A)-assisted termination: two different modes of poly(A)-dependent transcription termination. Mol Cell Biol. 1998;18:276–289. doi: 10.1128/mcb.18.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yonaha M, Proudfoot N J. Specific transcriptional pausing activates polyadenylation in a coupled in vitro system. Mol Cell. 1999;3:593–600. doi: 10.1016/s1097-2765(00)80352-4. [DOI] [PubMed] [Google Scholar]
- 81.Yonaha M, Proudfoot N J. Transcriptional termination and coupled polyadenylation in vitro. EMBO J. 2000;19:3770–3777. doi: 10.1093/emboj/19.14.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]