

Abstract
To address antigen escape and loss of T-cell functionality, we report a phase-1 clinical trial (NCT04007029) evaluating autologous naïve and memory T (TN/MEM) cells engineered to express a bispecific anti-CD19/CD20 CAR (CART19/20) for patients with relapsed/refractory NHL, with safety as the primary end point. Ten patients were treated with 36–165 × 106 CART19/20 cells. No patient experienced neurotoxicity of any grade, or over grade-1 cytokine release syndrome. One case of dose-limiting toxicity (persistent cytopenia) was observed. Nine of ten patients achieved objective response (90% ORR), with seven achieving complete remission (70% CR rate). One patient relapsed after 18 months in CR, but returned to CR after receiving a second dose of CART19/20 cells. Median progression-free survival and overall survival were not reached with a 17-month median follow-up. In conclusion, CART19/20 TN/MEM cells are safe and effective in patients with relapsed/refractory NHL, with durable responses achieved at low dosage levels.
Keywords: CAR-T cell therapy, cancer immunotherapy, non-Hodgkin lymphoma
INTRODUCTION
Effective chimeric antigen receptor (CAR)-T cell therapy exerts a strong selective pressure against cancer cells that express the CAR-targeted antigen, and downregulation or loss of expression is the natural escape route for target antigens that are not critical to cell survival. Accurate quantification of relapse rate attributable to CD19 antigen escape is complicated by lack of tissue acquisition following relapse, and reported CD19-negative relapse frequencies range from 27% to 100% of relapsed cases among patients with leukemia and lymphoma (1–5). The frequency of cases with CD19-negative relapse demonstrates the susceptibility of CD19 to antigen loss, and points to the identification of alternative target antigens that are more resistant to gene-expression downregulation as a potential remedy.
To address the problem of CD19 antigen escape, we developed a CD19/CD20 bispecific CAR-T cell therapy, and previously demonstrated its ability to eradicate B-cell lymphoma with heterogenous CD19 expression and prevent relapse in mouse models of human lymphoma (6,7). CD19/CD20 bispecific CAR-T cells outperformed single-input CD19 CAR-T cells in achieving long-term, progression-free survival in a lymphoma xenograft model (6,7). CD20, like CD19, is pan–B-cell marker, and the first-line therapy for B-cell malignancies typically includes an anti-CD20 antibody such as rituximab (8). In fact, rituximab is commonly included in each subsequent line of chemotherapy administered to patients with NHL, yet CD20 antigen loss is a low-frequency event despite repeated cycles of CD20-targeted therapies (9), suggesting CD20 may be a suitable CAR target with low propensity for antigen escape. However, the clinical outcomes of CD20 CAR-T cell therapy have been uneven to date (10–13), resulting in more limited clinical advancement compared to CD19 CAR-T cell therapy. We hypothesized that simultaneously targeting CD19 and CD20 would enable both high initial response rate and increased resistance to antigen escape. Importantly, dual targeting of CD19 and CD20 would not increase on-target, off-tumor toxicity compared to either CD19 or CD20 single-input CAR-T cell therapy because both CD19 and CD20 are B-cell–specific markers, thus limiting the off-tumor toxicity to healthy B cells whose aplasia is a clinically manageable condition (14).
Multiple trials have reported association of CD19-positive relapses with T-cell exhaustion and lack of CAR-T cell persistence, and patients with durable response to therapy have relatively elevated levels of naïve and memory T cells (4,5,15–18). The concept of infusing T cells enriched in naïve and memory phenotypes is consistent with prior reports indicating naïve and/or memory T cells (TN/MEM cells) enhance in vivo CAR-T cell function (19–21). Therefore, we hypothesized that engineering TN/MEM cells to express our molecularly optimized CD19/CD20 bispecific CAR would yield a therapy, termed CART19/20, with strong efficacy coupled to improved CAR-T cell persistence and reduced CAR-T cell exhaustion. Here, we report the dose-escalation phase-1 trial evaluating CART19/20 in adults with relapsed/refractory NHL, and demonstrate durable responses and strong safety profile in patients treated with CART19/20 cells.
RESULTS
CAR construct and clinical trial design.
We generated a CD19/CD20 bispecific CAR consisting of a single-chain variable fragment (scFv) derived from the anti-CD20 monoclonal antibody Leu16 fused to a second scFv derived from the anti-CD19 monoclonal antibody FMC63, followed by fusion of the scFv domains to the hinge domain of human IgG4, the transmembrane domain of human CD28, and the cytoplasmic signaling domains of human 4–1BB and CD3ζ (Fig. 1A) (6,7). The bispecific CAR was encoded by a third-generation self-inactivating lentiviral vector under the control of an elongation factor 1 alpha (EF1-α) promoter (22). We planned a phase-1 cell dose escalation trial with a fixed lymphodepletion chemotherapy of fludarabine 30 mg/m2 daily for 3 days on Day –5 to Day –3 and cyclophosphamide 500 mg/m2 daily for 3 days on Day –5 to Day –3, followed by CART19/20 infusion on Day 0 with dose levels of 50 × 106 CAR+ T cells (DL1), 200 × 106 CAR+ T cells (DL2), and 600 × 106 CAR+ T cells (DL3), with each DL allowing ±30% range (Fig. 1B).
Fig. 1. Design of phase-1 clinical trial evaluating CD19/CD20 bispecific CAR-T cell therapy (CART19/20) in patients with non-Hodgkin lymphoma, chronic lymphocytic leukemia, and small lymphocytic lymphoma.
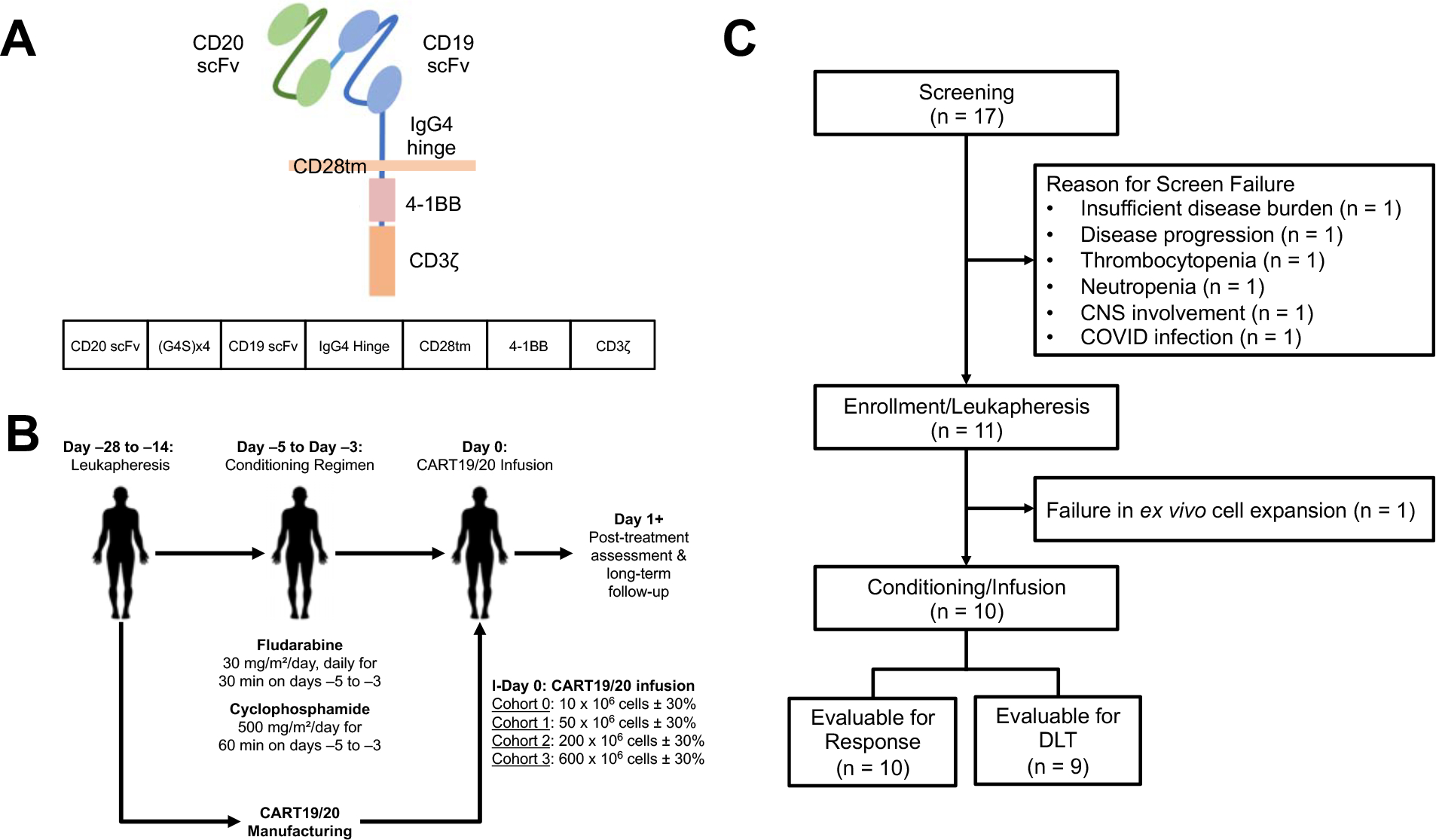
(A) Schematic of CD19/CD20 bispecific CAR construct. (B) Schematic of phase-1 dose escalation trial design. (C) Consolidated Standards of Reporting Trials (CONSORT) diagram of CART19/20 trial.
Patient characteristics.
Seventeen patients were screened, 11 patients went onto leukapheresis (Fig. 1C), and ten patients received CART19/20 infusion in two cohorts (DL1, n=7; DL2, n=3) (Fig. 1C). The median age at the time of CART19/20 infusion was 59 (range, 29–70) (Table 1). The diagnoses were mantle-cell lymphoma (MCL; n=1), follicular lymphoma (FL; n=3), de novo diffuse large B-cell lymphoma (DLBCL; n=1), transformed FL to DLBCL (n=3), primary mediastinal B-cell lymphoma (PMBCL; n=1), and high-grade B-cell lymphoma (HGBCL; n=1) with BCL6 and cMYC double-hit rearrangement. All patients with FL had progression of disease within 24 months after front-line treatment (POD24). The median lines of prior therapy were 3.5 (range, 2–4). One patient (Patient 004) was refractory to prior anti-CD19 bispecific T-cell engager (BiTE) therapy. All patients were CAR naïve and had stage-4 disease at the time of CART19/20 treatment. Nine patients were given bridging therapy prior to infusion due to progressive disease (Table 1).
Table 1.
Patient demographics and treatment history
| Patient ID | Age | Sex | Diagnosis | Prior Therapies | Disease Stage | Tumor Burden (SPD in cm2) | ALC at Screening (×10^3/μL) | Bridging Therapy | Dosing Level | CAR+ T Cells Received | CRS (Grade) | ICANS (Grade) | Best Response (PFS) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # of Lines | Therapies | Notes | |||||||||||||
| 001 | 60 | M | MCL | 4 | Bendamustine/rituximab | IV | 467.8 | 0.40 | Cyclophosphamide | DL1 | 53 × 10^6 | Yes (1) | No | CR (30+) | |
| Rituximab | |||||||||||||||
| Acalabrutinib | |||||||||||||||
| Umbralisib/ublituximab | |||||||||||||||
| 002 | 58 | M | FL | 4 | Bendamustine/rituximab | Primary Refractory | IV | 350 | 0.96 | Cyclophosphamide R-EPOCH | DL1 | 58 × 10^6 | Yes (1) | No | CR (24+) |
| R-CHOP | |||||||||||||||
| R-ICE | |||||||||||||||
| Lenalidomide/rituximab | |||||||||||||||
| 003 | 29 | F | DLBCL (PMBCL) | 2 | R-EPOCH | IV | 187 | 0.17 | R-GEMOX | DL1 | 52 × 10^6 | Yes (1) | No | PD | |
| R-ICE | |||||||||||||||
| 004 | 62 | M | FL | 4 | Bendamustine/rituximab | POD24; Refractory to CD19 BiTE | IV | 88.4 | 1.02 | R-GEMOX (did not tolerate/complete rituximab) | DL1 | 56 × 10^6 | Yes (1) | No | CR (18) |
| ROR1-Targeting Antibody-Drug Conjugate | |||||||||||||||
| Anti-CD19 BiTE | |||||||||||||||
| Rituximab/gemcitabine/oxaliplatin | |||||||||||||||
| 005 | 35 | F | FL | 3 | Bendamustine/rituximab | POD24 | IV | 150 | 1.12 | None | DL2 | 165 × 10^6 | Yes (1) | No | CR (18+) |
| Rituximab | |||||||||||||||
| Rituximab/dexamethasone | |||||||||||||||
| 009 | 67 | M | DLBCL (NOS) | 4 | Rituximab/cyclophosphamide/ etoposide | Primary Refractory | IV | 29.2 | 2.09 | GEMOX | DL2 | 158 × 10^6 | Yes (1) | No | CR (9+) * |
| R-CHOP | |||||||||||||||
| R-ICE | |||||||||||||||
| ASCT | |||||||||||||||
| 010 | 56 | M | DLBCL (tFL) | 3 | Bendamustine/rituximab | IV | 812 | 0.63 | ICE | DL1 | 42 × 10^6 | No | No | CR (12+) | |
| R-CHOP | |||||||||||||||
| R-ICE | |||||||||||||||
| 014 | 64 | M | DLBCL (tFL) | 2 | Rituximab/gemcitabine/dexamethasone/carboplatin | Primary Refractory | IV | 404.8 | 2.60 | ICE | DL2 | 146 × 10^6 | No | No | CR (9+) |
| R-CHOP | |||||||||||||||
| 016 | 70 | M | DLBCL (tFL) | 4 | R-CHOP | IV | 44 | 1.20 | GEMOX + R-GEMOX | DL1 | 37 × 10^6 | Yes (1) | No | PR (2) | |
| BP + polatuzumab | |||||||||||||||
| R-ICE | |||||||||||||||
| R-GEMOX | |||||||||||||||
| 017 | 39 | M | DLBCL (HGBCL double-hit) | 2 | R-CHOP | Primary Refractory | IV | 701.5 | 1.88 | ICE | DL1 | 36 × 10^6 | No | No | PR (2+) |
| R-EPOCH | |||||||||||||||
Patient 009 elected to transition to comfort care shortly prior to the scheduled 12-month follow-up. Biopsy performed at that time showed no evidence of lymphoma. However, CR at 12 month was not verified with PET scan.
Abbreviations: SPD: sum of the products of diameters; ALC: absolute lymphocyte count; CRS: cytokine release syndrome; ICANS: immune-effector cell associated neurotoxicity syndrome; PFS: progression-free survival; MCL, mantle-cell lymphoma; FL, follicular lymphoma; PMBCL, primary mediastinal B-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; tFL, transformed follicular lymphoma; R-CHOP: rituximab/cyclophosphamide/doxorubicin/hydrochloride/vincristine sulfate/prednisone; R-ICE: rituximab/ifosfamide/carboplatin/etoposide; R-EPOCH: rituximab/etoposide/prednisone/vincristine/cyclophosphamide/doxorubicin; BR: bendamustine/rituximab; BiTE: bispecific T-cell engager; ASCT: autologous stem-cell transplant; R-GEMOX: Rituximab/gemcitabine/oxaliplatin; ICE: ifosfamide/carboplatin/etoposide; CR: complete response; PR: partial response; PD: progressive disease
As of the data cutoff on July 11, 2022, a total of ten patients were evaluable for response. Nine patients were evaluable for dose-limiting toxicity (DLT), including six treated at DL1 and three treated at DL2. A decision was made to not escalate to DL3 based on the strong efficacy outcomes observed at the two lower dosing levels. The maximum tolerated dose was not reached.
CART19/20 cell manufacturing and product characterization.
Patient leukocytes harvested from leukapheresis were enriched for cells expressing CD62L, a marker for TN/MEM cells, by magnetic bead-based cell separation. Leukapheresis products with greater than 5% CD14+ and/or CD25+ cells among viable singlets based on flow cytometry analysis were subjected to an additional CD14/CD25 depletion step to remove myeloid and regulatory T cells (Tregs), respectively, prior to CD62L enrichment. TN/MEM cells were activated with an anti-CD3/CD28 colloidal nanomatrix-based activation reagent, lentivirally transduced to express the CD19/CD20 bispecific CAR, and expanded ex vivo for a total of 12 days (n=9),14 days (n=1, Patient 016), or 16 days (n=1, Patient 007) in the presence of IL-2 and IL-15 prior to cryopreservation (Fig. 2A). One manufacturing failure occurred due to low CAR+ T-cell counts that did not meet dose requirements (Patient 007).
Fig. 2. CART19/20 cells manufactured from naïve/memory T cells are enriched in memory phenotype.
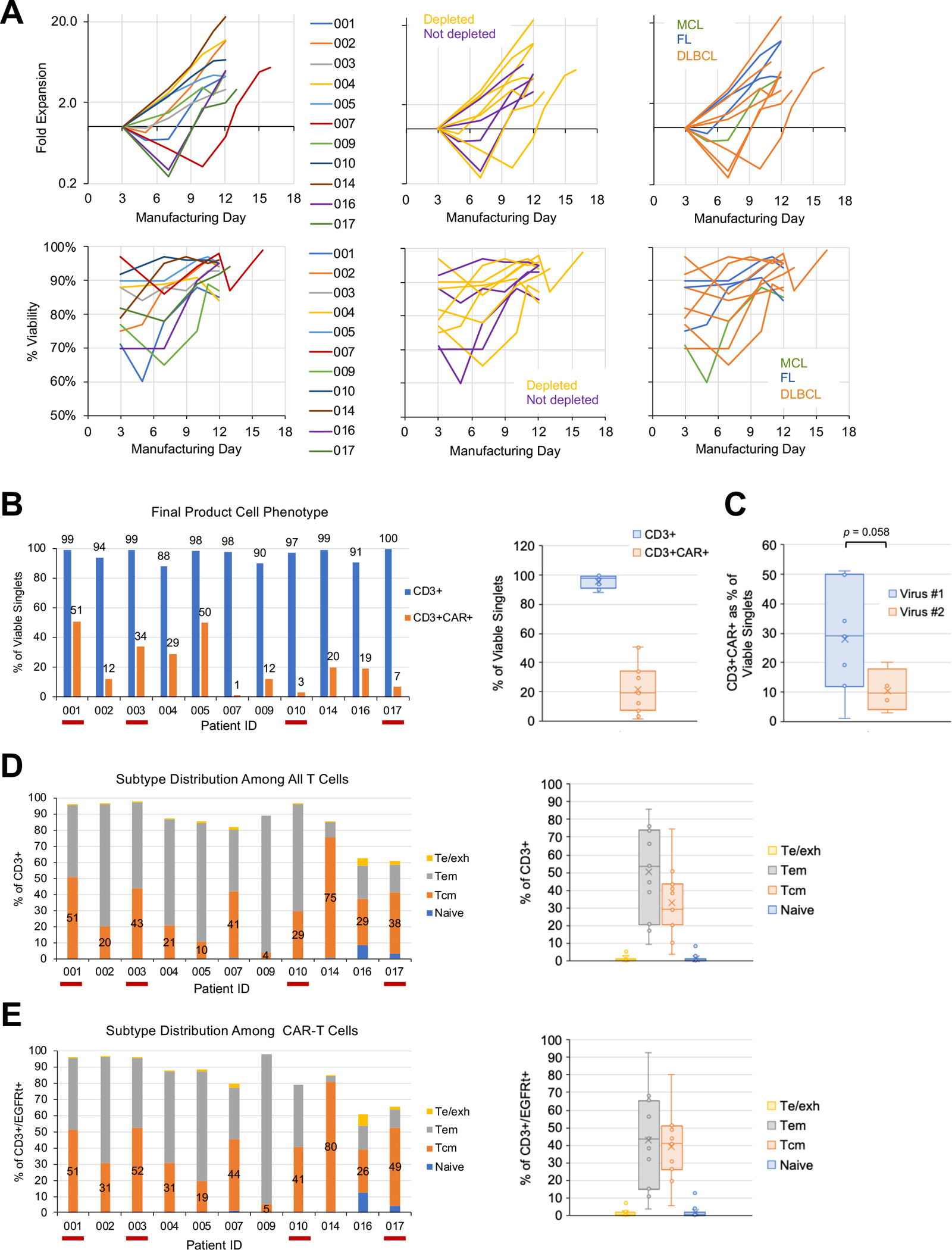
(A) Fold expansion (top) and viability (bottom) of cell product during ex vivo manufacturing. Cell counts were normalized to counts on the day of transduction (day 3). Data are shown with color coding by patient (left), by whether starting cell population underwent CD14/CD25 depletion (middle), and by disease indication (right). (B–E) Flow cytometry performed on cryopreserved cell aliquots post thaw to characterize (B) CD3+ purity and transduction efficiency of final cell product, (C) transduction efficiency grouped by batch of lentivirus used in manufacturing, (D) T-cell subtype distribution among all CD3+ T cells, and (E) T-cell subtype distribution among CAR-expressing T cells. Te/exh: effector/exhausted T cells, CD45RA+/CD45RO−/CD62L−; Tem: effector-memory T cells, CD45RA−/CD45RO+/CD62L−; Tcm: central-memory T cells: CD45RA−/CD45RO+/CD62L+; naïve: CD45RA+/CD45RO−/CD62L+. In panels B, D, and E, red underscoring of patient ID indicates products that did not undergo CD14/CD25 depletion.
The CART19/20 cell products have a high level of CD3+ purity, but wide range of transduction efficiency was observed (Fig. 2B). In addition to substantial donor-to-donor variability, the specific lot of GMP-grade lentivirus used also affected transduction efficiency, although the difference was below statistical significance (Fig. 2C; p = 0.058 by two-tailed Student’s t test). Despite the variability in transduction efficiency, only one manufacturing campaign failed to generate a sufficient dose for infusion. Patient 007 had a low absolute lymphocyte count (ALC) of 0.16 × 103 cells/μL at the time of screening, and a low white blood cell (WBC) count (32.79 × 103/μL) in the leukapheresis product. Furthermore, the patient’s cells transduced and expanded poorly during ex vivo manufacturing (Fig. 2A and B), resulting in failure to meet the required CART19/20 cell dose for infusion.
Overall, CART19/20 cell products contained a substantial fraction of central-memory T (Tcm) cells (median: 29.3%, range: 3.6%–74.9%; Fig. 2D), indicating the retention of memory phenotype in cell products manufactured from TN/MEM cells. Of note, CAR-expressing T cells tend to have slightly higher Tcm content (median: 40.9%, range: 5.3%–80.1%) compared to the overall T-cell population (Fig. 2E). A breakdown of CD4+ vs. CD8+ subtype distribution reveals that CAR+ T cells tend to have higher % CD4+ than the total T-cell population (Supplementary Fig. S1A), and CD4+ T cells tend to be more enriched in the Tcm phenotype compared to CD8+ T cells, although the difference is just under statistical significance (Supplementary Fig. S1B; p = 0.055 by two-tailed Student’s t test).
We chose to incorporate CD14 depletion in order to minimize the presence of myeloid cells, which had been reported to reduce T-cell activation through phagocytosis of activation agents (23) and could potentially reduce transduction efficiency by competing with T cells for lentivirus uptake. The removal of immunosuppressive Tregs through CD25 depletion (24) aimed to further enhance the anti-tumor efficacy of CART19/20 products. The minimum threshold of ≥5% CD14+ and/or CD25+ cells for depletion was based on the empirical observation that up to 5% of antigen-positive cells can remain even after depletion during preclinical process development. Following this criterion, the leukapheresis products of Patients 001 and 003 were not subjected to depletion and proceeded directly to CD62L enrichment (Supplementary Fig. S1C). However, the post-isolation cell population for these two patients showed a notable increase in percent CD14+ (from 3% and 2% to 48% and 74%, respectively), together with an uptick in percent CD25+ (from 0.9% and 0.3% to 8% and 4%, respectively; Supplementary Fig. S1D). This unintended enrichment is consistent with the fact that a substantial fraction of CD62L+ cells in patient leukapheresis products are CD14+ and/or CD25+ (Supplementary Fig. S1E), thus the CD62L enrichment step would simultaneously result in selective retention of CD14+ and/or CD25+ cells. Furthermore, the adherent nature of myeloid cells may also facilitate the retention of CD14+ cells during bead-based cell sorting in the absence of a depletion step. Based on these observations, the protocol was amended to trigger CD14/CD25 depletion when ≥5% of CD62L+ cells (as opposed to ≥5% of viable singlets) were CD14+ and/or CD25+, starting with the product for Patient 004.
Despite the fact that the post-isolation cell population for patients 001 and 003 contained a substantial number of myeloid cells, the CART19/20 cultures for these two patients showed typical fold expansion and viability levels during the manufacturing process (Fig. 2A). Both final products exhibited high levels of CD3+ purity and CAR transduction efficiency (Fig. 2B), and had similar T-cell subtype distribution at the time of cryopreservation as products made from CD14/CD25-depeleted cells (Fig. 2D and E). The only clear difference was that products generated from non-depleted starting material were CD4 dominant, whereas products generated from CD14/CD25-depleted cells were CD8 dominant (Supplementary Fig. S1A), potentially due to myeloid cells’ ability to stimulate CD4+ cells through MHC-II antigen presentation. Taken together, these results indicate the culture conditions succeeded in selectively expanding T cells while avoiding the negative impacts of myeloid cells observed in the manufacturing processes described in previous reports (23,25,26). Given that the starting percent CD25+ was <1% in both Patients 001 and 003, there was minimal concern regarding Treg enrichment. Indeed, FOXP3 intracellular staining showed no clear correlation between CD25 depletion or lack thereof with the level of FOXP3 expression among CD4+ T cells in CART19/20 final products (Supplementary Fig. 2).
Among the subsequent products, two more were manufactured in the absence of CD14/CD25 depletion (Patients 010 and 017). Both products were enriched in CD4+ T cells, with no distinctive pattern in ex vivo expansion rate, transduction efficiency, or T-cell subtype distribution compared to CD14/CD25-depleted products, consistent with initial observations made with Patients 001 and 003 (Fig. 2).
Safety and adverse events.
Ten patients received CART19/20 cell products. One patient treated at DL1 (Patient 003) progressed prior to the end of DLT-monitoring period and was replaced per protocol and excluded from DLT evaluation. Infusions were generally well tolerated; 2 patients experienced a grade-1 infusion-related reaction (IRR) (Table 2). Grade-1 cytokine release syndrome (CRS) occurred in 6 patients; no grade 2 or higher CRS was observed in any patient. The median time from infusion to CRS was 8 days (range, 1–11) and the median duration was 2.5 days (range, 1–3). One dose of tocilizumab was given to Patient 009 for grade-1 CRS lasting greater than 48 hours. There were no cases of immune-effector cell associated neurotoxicity syndrome (ICANS), and no steroids were administered in the study for CRS or ICANS management. All patients experienced grade-3 or above adverse events consisting of generalized pain (n=2, 22%), hypotension (n=1, 11%), anemia (n=3, 33%), neutropenia (n=5, 56%), and thrombocytopenia (n=5, 56%). The one episode of grade-3 hypotension occurred during bridging therapy prior to CART19/20 infusion for Patient 002. The only grade ≥3 adverse events attributable to CART19/20 cells were anemia, thrombocytopenia, and neutropenia experienced by Patient 009.
Table 2.
treatment emergent adverse events
| TEAE | All Grades (%) | Grade 1 (%) | Grade 2 (%) | Grade 3 (%) | Grade 4 (%) | Grade 5 (%) |
|---|---|---|---|---|---|---|
| Any | 9 (100%) | 9 (100%) | 9 (100%) | 8 (89%) | 7 (78%) | 1 (11%) |
| IRR | 2 (22%) | 2 (22%) | 0 | 0 | 0 | 0 |
| CRS | 6 (67%) | 6 (67%) | 0 | 0 | 0 | 0 |
| ICANS | 0 | 0 | 0 | 0 | 0 | 0 |
| Infection | 0 | 0 | 0 | 0 | 0 | 0 |
| Pain | 4 (44%) | 2 (22%) | 0 | 2 (22%) | 0 | 0 |
| Fatigue | 8 (89%) | 8 (89%) | 0 | 0 | 0 | 0 |
| Hypotension | 2 (22%) | 0 | 1 (11%) | 1 (11%) | 0 | 0 |
| Headache | 2 (22%) | 2 (22%) | 0 | 0 | 0 | 0 |
| ALT increased | 1 (11%) | 1 (11%) | 0 | 0 | 0 | 0 |
| AST increased | 1 (11%) | 0 | 1 (11%) | 0 | 0 | 0 |
| Anemia | 9 (100%) | 1 (11%) | 5 (56%) | 3 (33%) | 0 | 0 |
| Neutropenia | 8 (89%) | 1 (11%) | 2 (22%) | 0 | 5 (56%) | 0 |
| Thrombocytopenia | 8 (89%) | 3 (33%) | 0 | 1 (11%) | 4 (44%) | 0 |
| Nausea | 6 (67%) | 6 (67%) | 0 | 0 | 0 | 0 |
| Vomiting | 2 (22%) | 2 (22%) | 0 | 0 | 0 | 0 |
| Diarrhea | 2 (22%) | 2 (22%) | 0 | 0 | 0 | 0 |
| Hypocellular marrow | 1 (11%) | 0 | 0 | 0 | 0 | 1 (11%) |
TEAE: Treatment emergent adverse event; IRR: infusion-related reaction; CRS: cytokine release syndrome; ICANS: immune effector cell-associated neurotoxicity syndrome; ALT: alanine transaminase; AST: aspartate transaminase
In contrast to the other grade ≥3 hematologic adverse events, the cytopenia in Patient 009 persisted beyond the expected recovery time from lymphodepletion chemotherapy and resulted in the only DLT observed in this study. Patient 009 had received autologous stem-cell transplant (ASCT) 11 months prior to receiving CART19/20 cell infusion, and exhibited elevated levels of multiple cytokines pre-infusion (Supplementary Fig. S3A) as well as a sustained increase in C-reactive protein (CRP) and ferritin levels post-infusion (Supplementary Fig. S3B and S3C). A bone marrow biopsy performed five months post CART19/20 cell infusion revealed increased in frequency of a pre-existing TET2 mutation and a new ASXL1 mutation, suggestive of evolution of a myeloid neoplasm in the bone marrow related to prior therapy. Patient 009 elected to cease medical treatment shortly before the 12-month follow-up assessment, and the death from grade-5 hypocellular marrow is considered possibly related treatment and possibly related to secondary malignancy.
Response.
Primary response assessment was performed 60 days after CART19/20 infusion by PET/CT scan. Nine out of ten patients responded to therapy (ORR = 90%). Seven patients achieved a CR by the first disease assessment at day 60 (CR rate = 70%), and two additional patients had a PR at day 60 (Fig. 3 and 4A). Bridging therapy did not result in significant reduction of tumor burden in the majority of cases (Fig. 4A and Supplementary Fig. S4), and there was no correlation between patient response and application of bridging therapy (Table 1). With a median follow-up of 17 months from time of CART19/20 cell infusion (range 2–30 months), the median overall survival (OS) and progression-free survival (PFS) were not reached (Fig. 4B).
Fig. 3. Patients refractory to multiple prior lines of treatment respond to CART19/20 cell therapy.
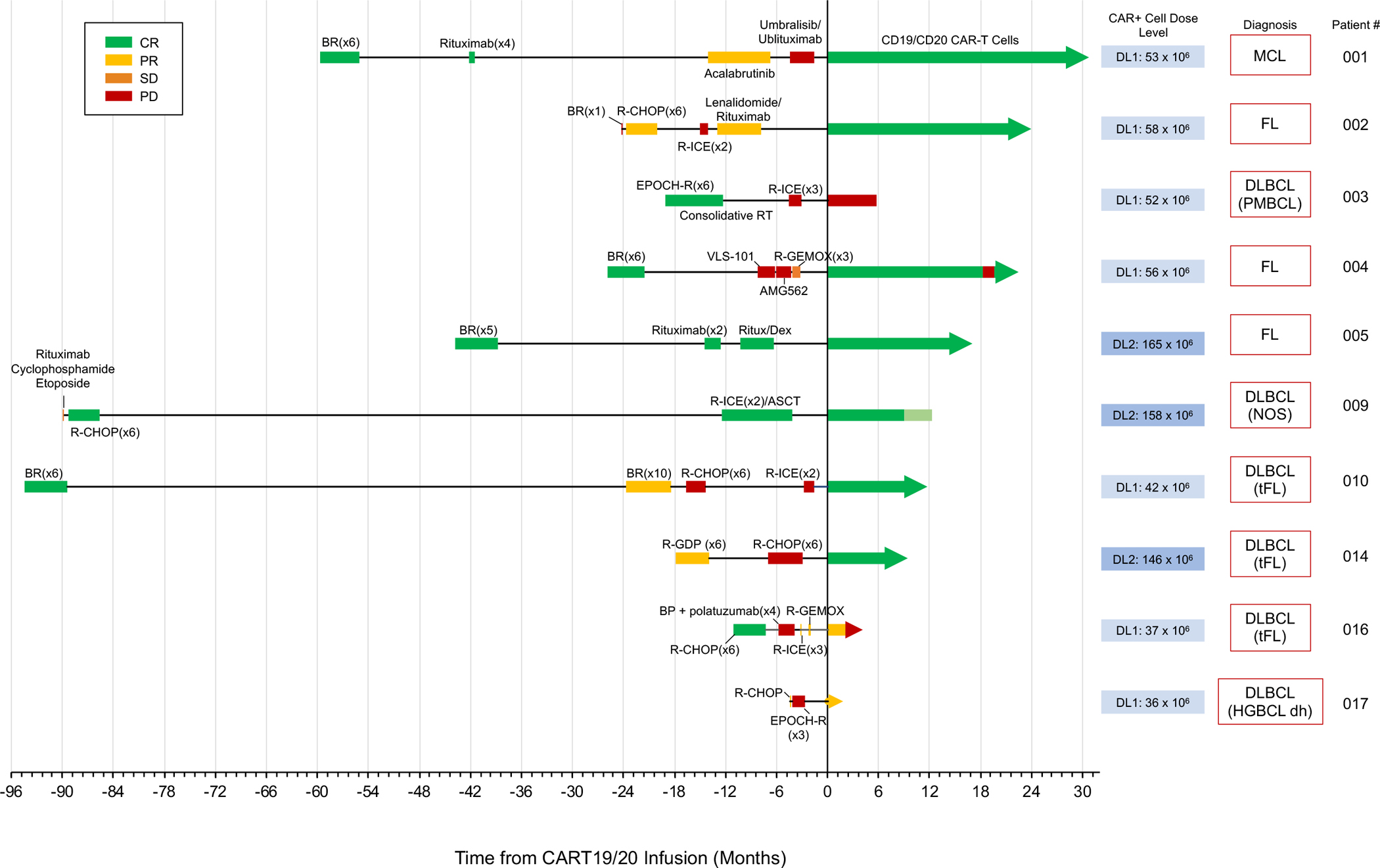
Timeline of individual patients’ response to prior treatment and to CART19/20 cell therapy. The disease indication and dose of CART19/20 cells received are also indicated for each patient. Light green box for patient 009 post CAR-T cell infusion denotes time between the last CR confirmed by PET scan and the time of patient death. The patient had no detectible lymphoma based on IHC analysis performed on bone-marrow biopsy shortly before death due to grade-5 hypocellular marrow. MCL, mantle-cell lymphoma; FL, follicular lymphoma; PMBCL, primary mediastinal B-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; tFL, transformed follicular lymphoma; HGBCL dh, high-grade B-cell lymphoma double-hit.
Fig. 4. CART19/20 cell therapy is highly effective in treating relapsed/refractory non-Hodgkin lymphomas.

(A) Representative PET scans of patients treated with CART19/20. (B) Overall survival and progression-free survival curves from the time of CART19/20 cell infusion. (C) PET scan obtained at screening for Patient 003, indicating pulmonary involvement of PMBCL. (D) Immunohistochemistry (IHC) analysis of Patient 003 tissue biopsies; original magnification x160. Supraclavicular lymph node biopsy obtained at screening and bone-marrow biopsy obtained 14 days post CART19/20 infusion were analyzed by IHC. Results reveal rapid emergence of a CD19−CD20−BCL6−cMYC− tumor population within 14 days of CART19/20 treatment.
The one patient who did not respond to therapy (Patient 003) was diagnosed with PMBCL (Fig. 4C), and had low ALC at the time of screening (0.17 × 103 cells/μL). The patient exhibited marked clinical improvements, including reduced dyspnea and pain, in the week after CART19/20 cell infusion and was discharged from the hospital. However, disease progression was detected 14 days post CART19/20 infusion in a bone-marrow biopsy performed as part of routine evaluation for CAR-T cell infiltration and expansion. The screening biopsy of a supraclavicular lymph node from Patient 003 was positive for CD19, CD20, CD30 (patchy), BCL2, BCL6, and cMYC (Fig. 4D), with kappa light chain restriction; the screening biopsy was negative for CD10 and BCL1. In contrast, the bone-marrow sample obtained 14 days after CART19/20 infusion expressed CD30 and weak BCL2, and was negative for CD10, CD19, CD20, BCL1, BCL6 and cMYC (Fig. 4D), indicating a clonal shift in the tumor population. A biopsy of the post-relapse lung mass analyzed by bulk RNA sequencing (RNA-seq) showed loss of gene expression for CD19, CD20 and BCL6 (reads per kilobase transcript, per million mapped reads (RPKM) = 0.12, 0.11, and 0.72, respectively), low cMYC expression (RPKM = 2.04), and a lack of CD22 expression (RPKM = 0.07). The concomitant loss of BCL6 and cMYC—two antigens not under selective pressure from the CD19/20 bispecific CAR—within 14 days of CART19/20 cell infusion suggests the possibility of heterogenous tumor that contained a pre-existing antigen-negative subclone.
The seven patients who achieved a CR include one patient diagnosed with MCL, three patients with DLBCL (one de novo and two tFL) and three patients with FL (Fig. 3). All patients with FL were POD24, and the majority of patients were characterized by high tumor burdens (Supplementary Fig. S4). Among the seven patients who achieved a CR, three (Patients 002, 009, and 014) had primary refractory disease, and a fourth (Patient 004) was refractory to anti-CD19 BiTE therapy (Table 1). In addition to anti-CD19 BiTE, Patient 004 was also refractory to ROR1-targeted antibody-drug conjugate therapy and progressed through the 2nd–4th lines of therapy within 5 months (Fig. 3). Flow cytometry analysis of Patient 004’s peripheral blood at the time of screening indicated the presence of CD20+CD19dim/− cells (Fig. 5A). This population was substantially reduced within 7 days of CART19/20 infusion, suggesting CART19/20’s ability to target tumor cells that have downregulated or lost CD19 expression. Patient 004 achieved a CR within 60 days and remained in CR until progressive disease with biopsy-proven CD20+CD19+ FL was detected at the month-18 evaluation (Fig. 5B). Given the patient’s history of therapy resistance and 18-month response to CART19/20, permission was obtained from the FDA to re-dose Patient 004 with 126 × 106 CART19/20 cells, using extra aliquots of the same cell product as the first infusion. At day 60 post re-dosing, the patient again achieved a CR and remains in CR as of data cutoff (Fig. 5B).
Fig. 5. CART19/20 cells exhibit sustained persistence and efficacy with strong safety.
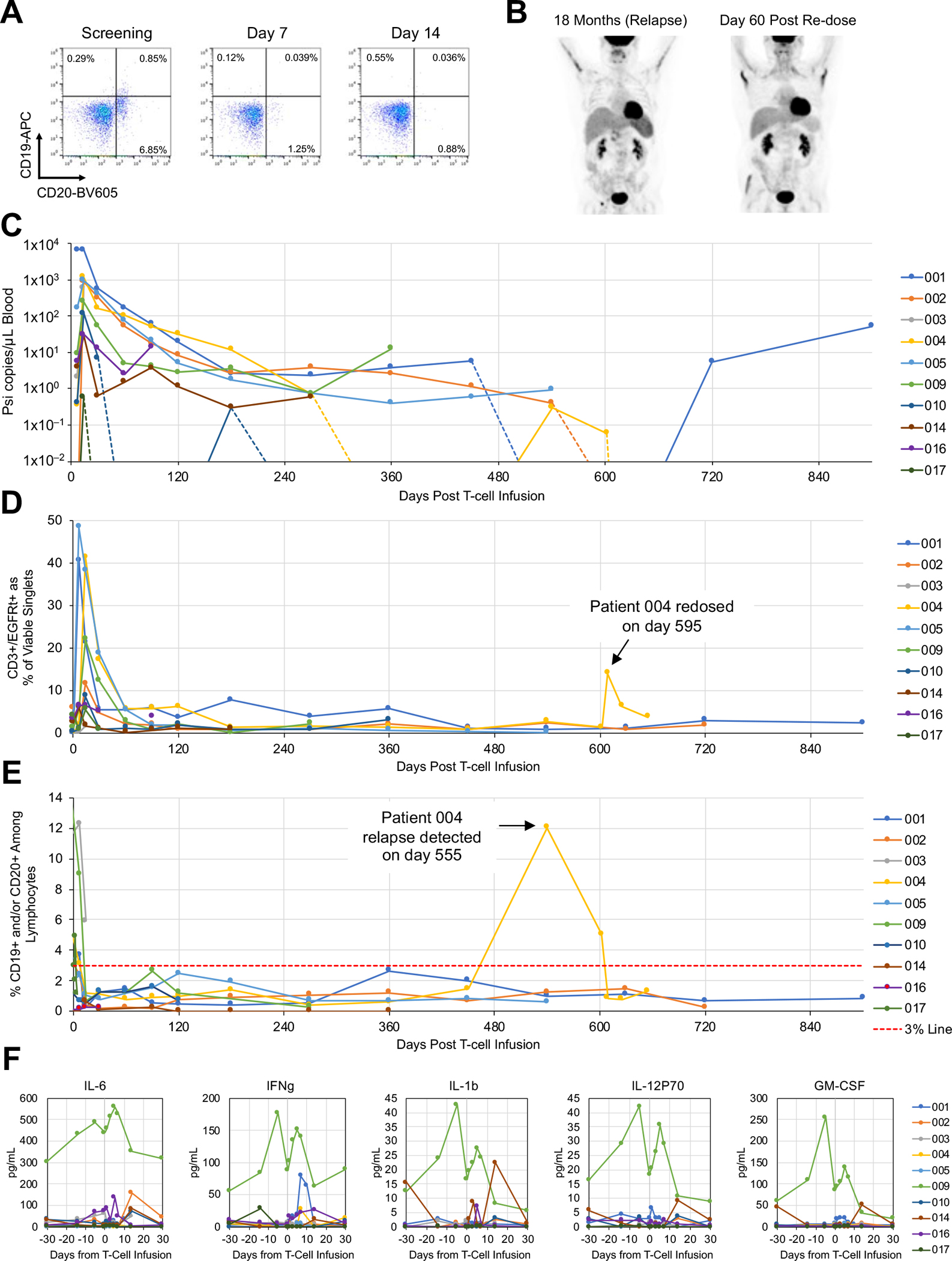
(A) Flow cytometry analysis on peripheral blood samples collected from Patient 004 at screening as well as 7- and 14-days post CART19/20 infusion. CD19 and CD20 surface staining results indicate the presence of CD20+CD19dim/– cells in Patient 004 prior to CART19/20 cell treatment. (B) PET scans of Patient 004 at the time of relapse and at 60 days post second dose of CART19/20 cells. (C) Presence of CAR transgene as quantified by droplet digital PCR. The psi signal integrated through lentiviral transduction was quantified. Inset shows zoomed-in data from the first 30 days post CART19/20 infusion. (D) Presence of CAR-expressing T cells among peripheral blood mononuclear cells (PBMCs) as quantified by flow cytometry. (E) Presence of CD19+ and/or CD20+ cells among lymphocytes as quantified by flow cytometry. A 3% line is shown to denote threshold for B-cell aplasia. (F) Serum cytokine levels as qualified by Luminex multiplex assay.
CART19/20 cell persistence and B-cell aplasia.
The presence of CART19/20 cells in peripheral blood post infusion were detected by both flow cytometry and droplet digital PCR (ddPCR). CAR copy number quantified by ddPCR consistently peaked at 14 days post infusion (Fig. 5C). Six of ten patients had detectible CAR-T cells as of the last available data point, and all six patients with follow-up periods of 12 months of more had detectible CAR-T cells at or past 12 months (Fig. 5C). At peak expansion, up to 48% of all viable singlets in PBMCs were CAR+ T cells (median 11.6%; range 1.5%–48.3%; Fig. 5D, Supplementary Fig. S5A, and S5B). At the time of data cutoff, all seven patients who were in PR (n=1) or CR (n=6) remained in B-cell aplasia based on flow cytometry analysis of peripheral blood (Fig. 5E), indicating functional persistence of CART19/20 cells.
For Patient 004, who remained in CR for 18 months before relapsing, CAR+ T cells dropped below ddPCR detection at 12 months but reemerged at the time of relapse (Fig. 5C). A second peak of CAR-T cell signal was detected by flow cytometry in the patient’s peripheral blood after the second dose of CART19/20 cells (Fig. 5D). B cells were undetectable in the peripheral blood of Patient 004 after the first CART19/20 cell infusion, was detected at the time of relapse, and dropped below detection again after receiving the second dose of CART19/20 cells, consistent with response to therapy (Fig. 5E).
As previously noted, CART19/20 cell products were either CD4- or CD8-dominant at the time of cryopreservation, depending on whether CD14/CD25 depletion was performed (Supplementary Fig. S1A). Regardless of the CD4:CD8 ratio in the cryopreserved cell product, both CAR-expressing T cells and total T cells rapidly became CD8-dominant after CART19/20 cell infusion in all but one patient (Supplementary Fig. S6A and S6B). The CD8 dominance declines over time, mirroring recently reported findings in a decade-long follow up of two patients with leukemia treated with CD19 CAR-T cell therapy (27).
Cytokine assessments.
Consistent with clinical presentation of grade 1 CRS for over 48 hours, Patient 009 showed elevated levels of most cytokines in peripheral blood compared to other patients in the trial (Fig. 5F; Supplementary Fig. S3A). As a whole, cytokine levels observed in patients treated with CART19/20 cells are similar to or substantially lower than values reported in earlier trials for single-input CD19 CAR-T cell therapies (3,5,28–30) and CD19/CD20 bispecific CAR-T cell therapies (18,31). The relatively low peak cytokine levels in patients treated with CART19/20 may be a contributing factor to the strong safety profile observed in this trial to date. Taken together, results of this phase-1 clinical trial indicate CART19/20 cell therapy is safe and effective for the treatment of NHL (Supplementary Table S1).
DISCUSSION
CD19 CAR-T cell therapy became the first FDA-approved gene-modified cell therapy in 2017, and is making rapid progress toward incorporation in earlier lines of treatment for B-cell malignancies. However, antigen escape and lack of T-cell expansion and persistence remain key factors that limit the frequency as well as durability of response in patients treated with CD19 CAR-T cell therapy (1–5,15–17,32,33). Early results of our phase-1 trial on CART19/20 cell therapy demonstrate that dual-targeting of CD19 and CD20 using a molecularly optimized single-chain bispecific CAR (6,7) is safe and effective in patients with relapsed/refractory NHL.
In this trial, ten patients were treated with a median dose of 55 × 106 CART19/20 cells, with no patient receiving more than 165 × 106 cells. These dosing levels are substantially lower than the median dosage level used in clinical trials evaluating other CD19/CD20 bispecific CAR clinical candidates (18,34) as well as in pivotal trials for single-input CD19 CAR-T cell products including axicabtagene ciloleucel (35,36), tisagenlecleucel (37,38), and lisocabtagene maraleucel (39). Despite the low dosage used, strong efficacy was observed in this trial (90% ORR; 70% CR rate), with a heavily pretreated patient population carrying high tumor burden and highly aggressive disease. To date, all but one patient who achieved a CR has remained in CR. The only patient to experience a relapse did so after 18 months, and the patient subsequently returned to CR after receiving a second dose of CART19/20 cells. With a median follow-up of 17 months, median PFS has not been reached in this trial.
Importantly, the strong efficacy observed in this trial was achieved with no ICANS of any grade and no CRS above grade 1 after CART19/20 infusion. This is in contrast to the FDA approved single-input CD19 CAR-T cells products where the rate of grade 3 or higher ICANS reached as high 32% in the ZUMA-1 trial (35) and rate of grade 3 or higher CRS as high as 23% in the JULIET trial (38). To date, the vast majority of grade ≥3 adverse events experienced by patients treated with CART19/20 were toxicities attributable to bridging and lymphodepletion chemotherapies in patients with substantial previous chemotherapy exposure. One patient death (Patient 009) from grade-5 hypocellular marrow is considered possibly related to treatment, and possibly related to myeloid neoplasm consequent to therapies received prior to CART19/20. Although no evidence of lymphoma was detected in the patient at the time of elected transition to comfort care, this case underscores the potential benefit of providing CAR-T cell therapy as an earlier line of treatment, which could reduce the total amount of chemotherapy exposure and related toxicities to the patient. Overall, the safety profile observed in this trial compares favorably with prior reports of CD19-targeted as well as CD19/CD20-targeted CAR-T cell therapies (18,34–40), and supports the possibility of combining strong efficiency with a high level of safety.
The trial reported here included one patient with MCL, three patients with FL, and five patients with DLBCL of various subtypes. It is plausible that different DLBCL subtypes have different response rates to CAR-T cell therapy, although no trial reported to date has been statistically powered to evaluate the responses of different DLBCL subtypes to CD19 CAR-T cell therapy (35,38,39). FL is often described as an indolent disease, and has shown favorable response rates to therapy (37). It should be noted that in this trial, all three patients with FL were POD24, including one patient who was primary refractory (Patient 002), and another patient who was refractory to the three lines of therapy immediately before CART19/20, including CD19-targeted BiTE (Patient 004). All patients with FL achieved a CR post CART19/20 cell infusion. In addition to four other patients with MCL or DLBCL who also achieved a CR, one patient with primary refractory, double-hit HGBCL (Patient 017) achieved a PR at day 60. This patient will continue to be monitored for the possibility of a delayed CR. Overall, CART19/20 cell therapy has shown robust efficacy in a highly pretreated patient population with challenging disease profiles.
The only patient who did not respond to CART19/20 therapy to date (Patient 003) had PMBCL refractory to R-ICE salvage chemotherapy. Despite notable clinical improvement in the week post CART19/20 infusion, this patient experienced a rapid emergence of CD19−CD20− tumor cells that had also lost BCL6 and cMYC expression within 14 days of CART19/20 treatment. The number of protein-expression changes combined with the rapidity of clonal shift suggests a pre-existing tumor subpopulation that was able to swiftly expand after CART19/20 cells eliminated the originally dominant CD19+CD20+ tumor cells. The emergence of CD19− tumor was previously reported in the relapse of a patient with PMBCL treated with CD19 CAR-T cell therapy, and sequencing analysis indicated the likely cause was a pre-existing clone that expanded under CD19-targeted selective pressure (41).
Promoting T-cell persistence and function through the generation of cell products enriched in naïve and memory cell types was a key element of the CART19/20 therapy design. CART19/20 products were generated from a TN/MEM cell population obtained through bead-based enrichment of CD62L expression, and the final products retained substantial central-memory T-cell content. Contrary to our original expectations, the presence of the CD14+ cells did not adversely impact our ability to successfully manufacture CART19/20 cell products with high T-cell purity and clinical efficacy. The only clearly measurable impact of CD14+ cell presence was the CD4:CD8 ratio of the final CART19/20 product, with the presence of CD14+ cells leading to CD4-dominant products while the depletion of CD14+ cells from the starting material led to CD8-dominant products. However, based on the results to date, there is no correlation between the CD4:CD8 ratio and clinical outcome. Similarly, the lack of Treg depletion through CD25 did not show measurable impact on treatment outcome, which is consistent with prior reports (42). We note that TN/MEM cell population isolated by CD62L enrichment could also include CD45RA+CD62L+ stem-cell memory T cells (TSCM) cells that have been shown to exhibit increased proliferation and anti-tumor activities (43,44). The markers CD45RA and CD62L alone cannot distinguish between purely naïve and TSCM cells, but phenotype profiling of infusion products indicates the cryopreserved cells are predominantly CD45RA−CD45RO+, consistent with exposure to activating agents during the manufacturing process (Fig. 2D and E). The 91% manufacturing success rate in this trial is comparable to those reported for other CAR-T cell therapy trials targeting CD19 or CD19/CD20 (Supplementary Table S1), demonstrating feasibility of using TN/MEM cells as starting material for CAR-T cell manufacturing. However, consistency in transduction efficiency is an area of improvement, with a need to reduce donor-to-donor variation as well as variability in the potency of GMP-grade lentivirus used in manufacturing.
A key question of interest is how the CART19/20 cells evaluated in this trial are able to achieve the high level of efficacy at low dosage level and without incurring the type of toxicities observed in comparable trials. As previously reported, the CD19/CD20 bispecific CAR used here had been optimized at an amino-acid sequence level to maximize efficacy (6). Safety was not a consideration in the CAR engineering process. However, the robust efficacy enabled the use of a very low cell dose to achieve therapeutic benefit, and the low cell dose may in turn have contributed to the favorable safety profile observed in this trial. The use of TN/MEM-derived cells may further contribute to the potency and safety profile of CART19/20 cells by reducing peak cytokine levels while retaining long-term anti-tumor efficacy.
In summary, early results from this phase-1 trial indicate that autologous, CD19/CD20 dual-targeting CAR-T cells enriched in naïve and memory phenotypes are safe and highly efficacious in the treatment of relapsed/refractory NHL. Our results suggest potent clinical efficacy can be achieved while avoiding severe toxicities typically associated with CAR-T cell therapy, and highlight the utility of dual-antigen targeting cell-based immunotherapy.
METHODS
Trial Design.
A prospective, first-in-human phase I clinical trial assessing CART19/20 in adult patients with relapsed/refractory NHL and CLL/SLL was initiated at the University of California, Los Angeles (UCLA) Medical Center. The study was approved by the UCLA Institutional Review Board (IRB) and registered with ClinicalTrials.gov (NCT04007029). Informed written consent was obtained in accordance with the Declaration of Helsinki, International Conference on Harmonization (ICH) Good Clinical Practice (GCP), US Code of Federal Regulations for Protection of Human Subjects, the Health Insurance Portability and Accountability Act, and local regulations. Data monitoring was conducted by the UCLA Jonsson Comprehensive Cancer Center (JCCC) Data Safety and Monitoring Board (DSMB). The primary endpoint was safety, defined by the incidence and severity of dose-limiting toxicities (DLTs), as well as determination of the maximum tolerated dose (MTD). Secondary endpoints included clinical efficacy measures, and analysis of CAR T-cell persistence and B-cell aplasia. Exploratory endpoint was cytokine release syndrome analysis.
Patient enrollment and eligibility.
Patients eligible for the clinical trial were ≥18 years old with diffuse large B-cell lymphoma (DLBCL) or primary mediastinal B-cell lymphoma (PMBCL) after ≥2 prior lines of therapy, or with mantle-cell lymphoma (MCL), follicular lymphoma (FL), chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) after ≥3 prior lines of therapy. Transformed indolent lymphomas, including Richter transformation, were eligible and previous lines of therapy were considered from the time of transformation. Autologous stem cell transplant (ASCT) recipients were allowed in the study. Prior CAR-T cell therapy was an exclusion criterion, but other forms of CD19- or CD20-targeted therapies were allowed. Patients were required to have greater than 30% positivity in malignant cells of CD19 and/or CD20, as well as measurable tumor burden on PET/CT. Any NHL- or CLL/SLL-directed therapy, including corticosteroids, within 14 days of initiation of lymphodepletion chemotherapy was exclusionary. After leukapheresis, bridging therapy was permitted at the investigator’s discretion. Lymphodepletion chemotherapy, consisting of fludarabine 30 mg/m2/day and cyclophosphamide 500 mg/m2/day, was administered for 3 days from Day −5 to Day −3 prior to infusion. Study representation of underserved communities is summarized in Supplementary Table S2.
Toxicity assessment.
Adverse events were recorded for all treated patients until disease relapse or death, with incidence and severity graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. CRS was graded according to the ASTCT and Lee criteria, with the former guiding treatment (45), For neurotoxic events, the ASTCT criteria was for scoring and treatment, with specific guidance to key disorders outlined by Neelapu et al. (46).
Response assessment.
The clinical response in lymphoma was evaluated with the criteria defined by The Revised Cheson Response Criteria and Lugano Classification (47,48). The overall response rate (ORR) was defined as the total of complete responses (CR) and partial responses (PR).
CART19/20 cell manufacturing.
Fresh patient leukapheresis products were analyzed by flow cytometry to determine the CD3+, CD62L+, CD14+, and CD25+ cell frequency. When needed, cells were labeled with anti-CD14 and anti-CD25 CliniMACS microbeads and depleted using the CliniMACS Plus system (Miltenyi Biotec). Remaining cells were subsequently enriched for CD62L using the same system to yield TN/MEM cells. TN/MEM cells were activated with TransAct (Miltenyi Biotec) and transduced with GMP-grade lentivirus. Patient cells were expanded ex vivo for a total of 12–16 days in the presence of IL-2 and IL-15 prior to cryopreservation. Cells were never frozen prior to the manufacturing process, and were only frozen and thawed once before infusion into patients.
Flow cytometry analysis of lymphocytes.
For pre- and post-isolation leukapheresis product analysis, samples were stained with antibodies for CD3, CD14, CD25, and CD62L. For CART19/20 final product analysis, cryopreserved products were thawed, washed with PBS, and stained with antibodies for CD3 and epidermal growth factor receptor (EGFR). The CD19/CD20 bispecific CAR is co-expressed with a truncated, non-signaling EGFR (EGFRt), thus EGFRt serves as a proxy for CAR expression. For patient peripheral blood analysis, blood samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes, and peripheral blood mononuclear cells (PBMCs) were collected using the SepMate system (STEMCELL Technologies) following manufacturer’s protocol. Isolated PBMCs were frozen until use. Thawed cells were surface stained with antibody panels for T-cell phenotype (CD3, CD4, CD8, CD62L, CD45RA, CD45RO, and EGFR), or B-cell quantification (CD19, CD20, CD56, CD3, CD14, and SYTOX Blue). Flow cytometry was performed on an Attune NxT flow cytometer (ThermoFisher), and data were analyzed using FlowJo v.10.7.1 (FlowJo, LLC). Gating strategies are shown in Supplementary Fig. S4.
Cytokine analysis.
Patient peripheral blood was collected into red-top tubes containing no anti-coagulant or preservative, allowed to clot for 30 minutes in the upright position at room temperature, transferred to a conical tube, and centrifuged at 900 × g for 10 minutes. The supernatant was frozen in aliquots until use. Cytokine analysis was performed by the UCLA Immune Assessment Core Facility using the Luminex 38-plex human cytokine chemokine panel, following manufacturer’s protocols.
Statistical analysis.
Descriptive statistics were measured by median and range for continuous variables and counts and percentages for categorical variables. Patients were censored at the time of the last follow up. Duration of remission (DOR), progression-free survival (PFS), and overall survival (OS) were estimated by Kaplan-Meier method. The statistical software packages used was IBM SPSS statistics. DOR was defined as the time of the first documented CR/PR until the first date that recurrent or progressive disease is objectively documented, or until death. PFS was defined as the time of CART19/20 infusion until documentation of objective disease progression or death due to any cause. OS was measured from the date of CART19/20 infusion in the clinical trial until death.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Autologous CD19/CD20 bispecific CAR-T cell therapy generated from naïve/memory T cells for patients with non-Hodgkin lymphoma is safe (no neurotoxicity, maximum grade-1 cytokine release syndrome) and demonstrates strong efficacy (90% overall response rate, 70% complete response rate) in a first-in-human phase-1 dose-escalation trial.
ACKNOWLEDGEMENTS
We thank the study participants and all members of the Cellular Therapy Program at UCLA. This work was supported by the Parker Institute for Cancer Immunotherapy (grant no. 20163828 to Y.Y.C) and Jean and Stephen Kaplan (gift to Y.Y.C.). This trial was additionally supported by the Aramont Clinical/Translational Research Program in Hematologic Malignancies and the Hornsey Foundation. A.R. is supported by NIH grants R35CA197633 and P30CA016042. J.M.T. is supported by the Jaime Erin Follicular Lymphoma Research Consortium. This study utilized the UCLA Jonsson Comprehensive Cancer Center (JCCC) Flow Cytometry Shared Resource and Technology Center for Genomics & Bioinformatics, which are supported by the NIH Cancer Center Support Grant (grant no. P30CA016042 to Michael A. Teitell). We thank the UCLA Human Gene and Cell Therapy Facility (HGCTF) for performing Quality Assurance review of our Good Manufacturing Practice (GMP) cell-manufacturing process. We thank Dr. Bea Fernandez her assistance in performing vector copy number (VCN) and lentivirus titer analyses. We thank Dr. Xiangzhi Meng and Dr. Ximin Chen for assistance in performing bulk RNA-seq data analysis. We thank the UCLA Immune Assessment Core for performing cytokine analysis.
Footnotes
COMPETING INTERESTS
Y.Y.C. is an inventor on a patent application for CART19/20 and holds several patent applications in the area of CAR-T cell therapy. Y.Y.C. is a founder of, holds equity in, and receives consulting fees from ImmPACT Bio. She is a member of the scientific advisory board of and holds equity in Catamaran Bio, Notch Therapeutics, Pluto Immunotherapeutics, Prime Medicine, Sonoma Biotherapeutics, and Waypoint Bio. She has consulted for Novartis and Gritstone Bio. S.M.L. holds equity in 1200 Pharma and TORL BioTherapeutics, and has received research funding from Abbvie, Bioline, Bristol Myers Squibb (BMS), Janssen, Novartis, Pfizer, and Sanofi. C.M.W. is a current employee of and holds equity in Orca Bio. J.T. is a current employee of and holds equity in ImmPACT Bio. M.R. is a current employee of and holds equity in Fate Therapeutics. G.J.S. holds equity in Amgen, BMS, and Johnson & Johnson; has consulted for or received honoraria from Kite, Astellas, AbbVie, Incyte, BMS, Stemline, Karyopharm, Agios, Amgen, AstraZenecca, Novartis, Ono Pharma, Celgene, and Jazz; and has received research funding from Actinium, Actuate, AbbVie, AltruBio, Arog, Astellas, AVM Biopharma, Cellectis, Celgene, Cellerant, Constellation, CTI, Forma, Cyclacel, Daiichi-Sankyo, Deciphera, Ifly, FujiFilm, Gamida, Gilead, Genetech/Roche, Geron, Glycomimetics, Incyte, Janssen, Karyopharm, Kite, Mateon, Medimmune, Millenium, Novartis, Onconova, Pfizer, PreCOG, Regimmune, Samus, Sellas, Sangamo, Semline, Takeda, Tolero, and Trovagene. A.R. has received honoraria from consulting with Cstone, Merck, and Vedanta, is or has been a member of the scientific advisory board and holds stock in Advaxis, Appia, Apricity, Arcus, Compugen, CytomX, Highlight, ImaginAb, ImmPact, ImmuneSensor, Inspirna, Isoplexis, Kite-Gilead, Lutris, MapKure, Merus, PACT, Pluto, RAPT, Synthekine and Tango, has received research funding from Agilent and from Bristol-Myers Squibb through Stand Up to Cancer (SU2C), and patent royalties from Arsenal Bio. The other authors declare no conflicts of interest.
Data Availability Statement:
Data involving patient confidentiality generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.
REFERENCES
- 1.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17 doi 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531–44 doi 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28 doi 10.1016/S0140-6736(14)61403-3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129(25):3322–31 doi 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 doi 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 2016;4(6):498–508 doi 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. ADDENDUM: T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 2016;4(7):639–41 doi 10.1158/2326-6066.CIR-16-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salles G, Barrett M, Foa R, Maurer J, O’Brien S, Valente N, et al. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv Ther 2017;34(10):2232–73 doi 10.1007/s12325-017-0612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson NA, Leach S, Woolcock B, deLeeuw RJ, Bashashati A, Sehn LH, et al. CD20 mutations involving the rituximab epitope are rare in diffuse large B-cell lymphomas and are not a significant cause of R-CHOP failure. Haematologica 2009;94(3):423–7 doi 10.3324/haematol.2008.001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008;112(6):2261–71 doi 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results. Blood 2012;119(17):3940–50 doi 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 2014;155(2):160–75 doi 10.1016/j.clim.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Zhang WY, Wang Y, Guo YL, Dai HR, Yang QM, Zhang YJ, et al. Treatment of CD20-directed Chimeric Antigen Receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther 2016;1:16002 doi 10.1038/sigtrans.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J 2020;10(8):79 doi 10.1038/s41408-020-00346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh EM, Scherer LD, Rouce RH. Replacing CAR-T cell resistance with persistence by changing a single residue. J Clin Invest 2020;130(6):2806–8 doi 10.1172/JCI136872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126(6):2123–38 doi 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med 2020;26(10):1569–75 doi 10.1038/s41591-020-1081-3. [DOI] [PubMed] [Google Scholar]
- 19.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 2008;118(1):294–305 doi 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014;123(24):3750–9 doi 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat Commun 2020;11(1):2283 doi 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yam PY, Li S, Wu J, Hu J, Zaia JA, Yee JK. Design of HIV vectors for efficient gene delivery into human hematopoietic cells. Mol Ther 2002;5(4):479–84 doi 10.1006/mthe.2002.0558. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Borquez-Ojeda O, Stefanski J, Du F, Qu J, Chaudhari J, et al. Depletion of high-content CD14(+) cells from apheresis products is critical for successful transduction and expansion of CAR T cells during large-scale cGMP manufacturing. Mol Ther Methods Clin Dev 2021;22:377–87 doi 10.1016/j.omtm.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer 2007;7(11):880–7 doi 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 25.Kunkele A, Brown C, Beebe A, Mgebroff S, Johnson AJ, Taraseviciute A, et al. Manufacture of Chimeric Antigen Receptor T Cells from Mobilized Cyropreserved Peripheral Blood Stem Cell Units Depends on Monocyte Depletion. Biol Blood Marrow Transplant 2019;25(2):223–32 doi 10.1016/j.bbmt.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy 2016;18(7):893–901 doi 10.1016/j.jcyt.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 2022;602(7897):503–9 doi 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 2010;18(4):666–8 doi 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther 2017;25(1):285–95 doi 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012;119(12):2709–20 doi 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood 2020;136(14):1632–44 doi 10.1182/blood.2020005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plaks V, Rossi JM, Chou J, Wang L, Poddar S, Han G, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood 2021;138(12):1081–5 doi 10.1182/blood.2021010930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov 2018;8(10) doi 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Wang Y, Liu Y, Tong C, Wang C, Guo Y, et al. Long-term activity of tandem CD19/CD20 CAR therapy in refractory/relapsed B-cell lymphoma: a single-arm, phase 1–2 trial. Leukemia 2022;36(1):189–96 doi 10.1038/s41375-021-01345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 2019;20(1):31–42 doi 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 2022;23(1):91–103 doi 10.1016/S1470-2045(21)00591-X. [DOI] [PubMed] [Google Scholar]
- 37.Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 2022;28(2):325–32 doi 10.1038/s41591-021-01622-0. [DOI] [PubMed] [Google Scholar]
- 38.Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2021;22(10):1403–15 doi 10.1016/S1470-2045(21)00375-2. [DOI] [PubMed] [Google Scholar]
- 39.Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020;396(10254):839–52 doi 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 40.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 2019;25(9):1341–55 doi 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 41.Yu H, Sotillo E, Harrington C, Wertheim G, Paessler M, Maude SL, et al. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am J Hematol 2017;92(1):E11–E3 doi 10.1002/ajh.24594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beider K, Besser MJ, Schachter J, Grushchenko-Polaq AH, Voevoda V, Wolf I, et al. Upregulation of Senescent/Exhausted Phenotype of CAR T Cells and Induction of Both Treg and Myeloid Suppressive Cells Correlate with Reduced Response to CAR T Cell Therapy in Relapsed/Refractory B Cell Malignancies. Blood 2019;134:3234. [Google Scholar]
- 43.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med 2011;17(10):1290–7 doi 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016;128(4):519–28 doi 10.1182/blood-2015-11-683847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 2019;25(4):625–38 doi 10.1016/j.bbmt.2018.12.758.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 2018;15(1):47–62 doi 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32(27):3059–68 doi 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol 2007;25(5):579–86 doi 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data involving patient confidentiality generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.