

Summary
Targeted therapy and immunotherapy have revolutionized cancer treatment. However, the ability of cancer to evade the immune system remains a major barrier to effective treatment. Related to this, several targeted DNA damage response inhibitors (DDRi) are being tested in the clinic and have been shown to potentiate anti-tumor immune responses. Seminal studies have shown that these agents are highly effective in a pan-cancer class of tumors with genetic defects in key DNA repair genes such as BRCA1/2, BRCA-related genes, ATM, and others. Here, we review the molecular consequences of targeted DDR inhibition, from tumor cell death to increased engagement of the anti-tumor immune response. Additionally, we discuss mechanistic and clinical rationale for pairing targeted DDRi with immunotherapy for enhanced tumor control. We also review biomarkers for patient selection and promising new immunotherapy approaches poised to form the foundation of next generation DDRi and immunotherapy combinations.
Keywords: DNA damage repair, Mismatch Repair Deficits, PARP, ATR, Immuno-Oncology, Immune Checkpoint Blockade, Biomarkers, STING, cGAS-STING, DDRi-IO, Cellular Therapy
ETOC Blurb:
This review discusses the mechanisms of DNA damage response inhibition (DDRi) and how these therapies are leveraged to potentiate immunotherapy in cancer. Additionally, we review clinically relevant biomarkers for DDRi efficacy and promising new immunotherapy approaches which can be combined with DDRi to potentially heighten antitumoral effect.
Introduction:
Next generation sequencing studies have demonstrated that many cancers carry inactivating mutations or deletions in DNA repair genes resulting in severely compromised DNA repair1,2. This loss of DNA repair functionality induces hyper dependence on remaining repair mechanisms, creating unique therapeutic vulnerabilities that can be targeted clinically 3–5. To this end, several classes of targeted therapies have been developed to inhibit specific DNA repair effectors demonstrating promise across multiple tumor types, particularly in tumors with genetic alterations that inactivate DNA repair genes. While targeted DNA damage response inhibitor (DDRi) strategies were designed to block DNA repair and overwhelm cellular damage tolerance, additional data has shown that DDRi agents can also stimulate anti-tumor immune responses. This has led to the development of DDRi-immunotherapy combination strategies that enhance tumor control. In this review, we will describe clinically relevant approaches for targeting tumors with defective DNA damage responses, discuss strategies for pairing DDRi and immunotherapy, highlight biomarkers for patient selection, and review promising next generation immunotherapy approaches that pair with DDRi moving forward.
The DNA Repair Machinery: A Brief Overview
The cellular DNA damage response is a complex network composed of dozens of proteins functioning across several pathways. This network is responsible for repairing a variety of DNA lesions stemming from both cell-intrinsic and extrinsic sources. Generally speaking, the DNA repair machinery regulates three processes: 1) damage correction, 2) replication-based lesion bypass, and 3) damage recognition and checkpoint signaling.
Cells have evolved to incorporate repair mechanisms to facilitate efficient repair of specific types of DNA damage. Single-strand base damage is corrected by base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and direct reversal (DR) repair pathways. BER corrects DNA bases damaged by alkylation, deamination, and oxidation6. NER coordinates repair of bulky base lesions formed by carcinogen exposure (i.e. tobacco smoke), UV exposure, or certain chemotherapeutics 7. The MMR pathway senses and repairs mismatched nucleotides, as well as insertion and deletion loops 8. DR is responsible for correcting O- and N-alkylated bases9.
Cells also employ specialized pathways for repairing interstrand crosslinks and double-stranded DNA breaks. The Fanconi Anemia (FA) pathway repairs interstrand DNA crosslinks, or lesions that covalently link DNA strands together during replication10. Double-stranded DNA breaks are repaired by homologous recombination (HR), non-homologous end-joining (NHEJ), or microhomology-mediated end-joining (MMEJ) or alternative end-joining (alt-EJ) pathways11–13. Cell cycle state and repair context dictate the use of these pathways14. In G0 or G1 cell cycle states, double-stranded breaks are preferentially repaired by NHEJ. In S or G2 phases, double-stranded breaks are repaired by HR, which requires the presence of the sister chromatid for faithful repair. Mechanistically, expression of DNA resection enzymes, which are expressed during S and G2 phases, also helps govern NHEJ and HR repair choices. Resected DNA ends are poor substrates for NHEJ repair, while they are required for HR. MMEJ represents a highly error-prone mechanism for DSB repair. Microhomology sequences present near resected DNA ends are used to anneal broken strands prior to fill-in synthesis and ligation, promoting insertions, deletions, and chromosomal rearrangements 11,15. MMEJ is often used when cells have an inability to undergo faithful HR repair15.
The DNA repair machinery also coordinates replication-based lesion bypass, or translesion synthesis (TLS). TLS helps safeguard DNA replication by preventing replication fork stalling, fork collapse, and double-strand DNA break accumulation. When the replicative machinery encounters a stall-inducing lesion, specialized DNA polymerases can be employed to insert a nucleotide opposite of said lesion, extend the nascent DNA strand, and continue replication16. While this pathway carries an increased risk of DNA sequence mutation, its activity is preferred over the formation of deleterious double-strand DNA break formation.
Lastly, the DNA repair machinery uses distinct pathways for damage recognition and checkpoint signaling. Extensive research has revealed that different forms of DNA damage activate distinct damage sensing pathways17. Single-stranded DNA stretches, which are generated by stalled replication forks or through DNA resection, activate the ATR-CHK1 pathway. Double-stranded DNA breaks can activate the ATM-CHK2 or DNA-PK pathways. Following damage recognition, these pathways initiate signaling cascades that regulate DNA repair effector activity, coordinate cell cycle arrest, protect stalled replication forks, and control transcriptional responses18. Collectively, these functions are essential for facilitating accurate DNA repair.
Genetic Defects in the DNA Damage Response Create Unique Therapeutic Vulnerabilities:
Accurate DNA repair is required for faithful and efficient DNA replication. Large sequencing studies have revealed that tumors frequently carry inactivating genetic alterations in key DNA repair genes1. Seminal studies have demonstrated that these inactivating alterations can be inherited or acquired sporadically during tumor development19,20. These genetic alterations severely compromise the function of impacted DNA repair pathways resulting in increased damage accumulation, genome evolution, and cellular stress. Importantly, these genetic defects also sensitize cells to selective inhibition of remaining repair pathways. Below we review common DNA repair and DNA repair-related genes that are inactivated across cancers. Additionally, we discuss how these genetic events confer sensitivity to targeted DDRi agents. While several targeted DDRi agents have been developed, our discussion will focus on those that have demonstrated greatest clinically utility to date, such as PARP, ATR, and CHK1 inhibitors.
Genetic Defects Conferring Sensitivity to PARP Inhibition:
BRCA1 and BRCA2:
The observation that tumors with DNA repair defects are sensitive to targeted DDRi first gained traction following studies of cancer patients with germline BRCA1 or BRCA2 inactivating mutations. Patients with BRCA1/2 mutations have a significantly increased risk for developing breast, ovarian, pancreatic, gallbladder, stomach, skin, or prostate cancers during their lifetime21–25. This increased risk is due to the essential roles that BRCA1 and BRCA2 play in regulating HR repair and replication fork stability. BRCA1 plays a key role in promoting resection of double-stranded DNA breaks, which promotes their faithful repair through HR26. BRCA2, meanwhile, helps load RAD51 filaments onto resected DNA ends, promoting D-loop formation and completion of HR repair. In addition to its role in HR repair, BRCA2 also helps stabilize stalled replication forks by preventing MRE11 or DNA2-mediated resection27. When inactivated, loss of BRCA1 and/or BRCA2 severely compromises cellular capacity for HR. As a result, BRCA deficient cells become reliant on error-prone double-strand break repair mechanisms like NHEJ and MMEJ. Additionally, BRCA2 deficient cells experience decreased replication fork stability and increased double-stranded break formation. This increased double-stranded break formation and error-prone repair activity causes BRCA deficient cells to accumulate distinct genomic scars marked by telomeric allelic imbalances (NtAI), large scale transitions (LST, >10 Mb), and loss-of-heterozygosity events (HRD-LOH, >15 Mb)28. These alterations are referred to as markers of homologous recombination deficiency (HRD). Additional studies have shown that BRCA genes are also frequently dysregulated by promoter hypermethylation or homozygous deletions across cancers1.
Landmark studies discovered that BRCA deficient tumors can be successfully targeted with Poly(ADP-ribose) polymerase (PARP) inhibitors; a distinct class of targeted DDRi agents that block activity of PARP family members, primarily PARP1–429,30. PARP1 is an important mediator of single-stranded DNA break repair that is rapidly and efficiently recruited to sites of single-stranded DNA breaks. Once bound to DNA, PARP1 transfers a poly(ADP-ribose) group to itself using nicotinamide adenine dinucleotide (NAD+), in a process called “PARylation”31. This PARylation helps activate PARP1, stimulating further PARylation of histones and target DNA repair proteins. Collectively, this PARylation activity helps recruit many DDR effectors, allowing for effective repair of single-stranded DNA breaks. While several different PARP inhibitors have been developed, they all function by blocking catalytic activity of PARP, blocking PARylation and subsequent recruitment of DNA repair effectors to sites of single-stranded DNA breaks32. This is significant as unresolved single-stranded DNA breaks are processed to toxic double-stranded DNA breaks during DNA replication. Additionally, blocking PARylation prevents the auto-PARylation of PARP1 that is required to release PARP1 from DNA. As a result, inactivated PARP1 becomes persistently bound to DNA in a phenomenon referred to as “PARP-trapping”33. This is important as protein-DNA crosslinks represent strong replication impediments that promote double-strand break accumulation. This PARP inhibitor directed dual mechanism of double-strand break formation is highly lethal to BRCA deficient cells that are genetically predisposed to double-strand break formation. In these cells, BRCA loss and PARP inhibition synergize to create substantial double-strand break accumulation. This double-strand break accumulation, combined with dependence on toxic, error-prone repair mechanisms, combine to overwhelm cellular damage tolerance and cause tumor cell death (Figure 1).
Figure 1: DDRi overwhelms cellular DNA damage responses and induces cancer cell death.
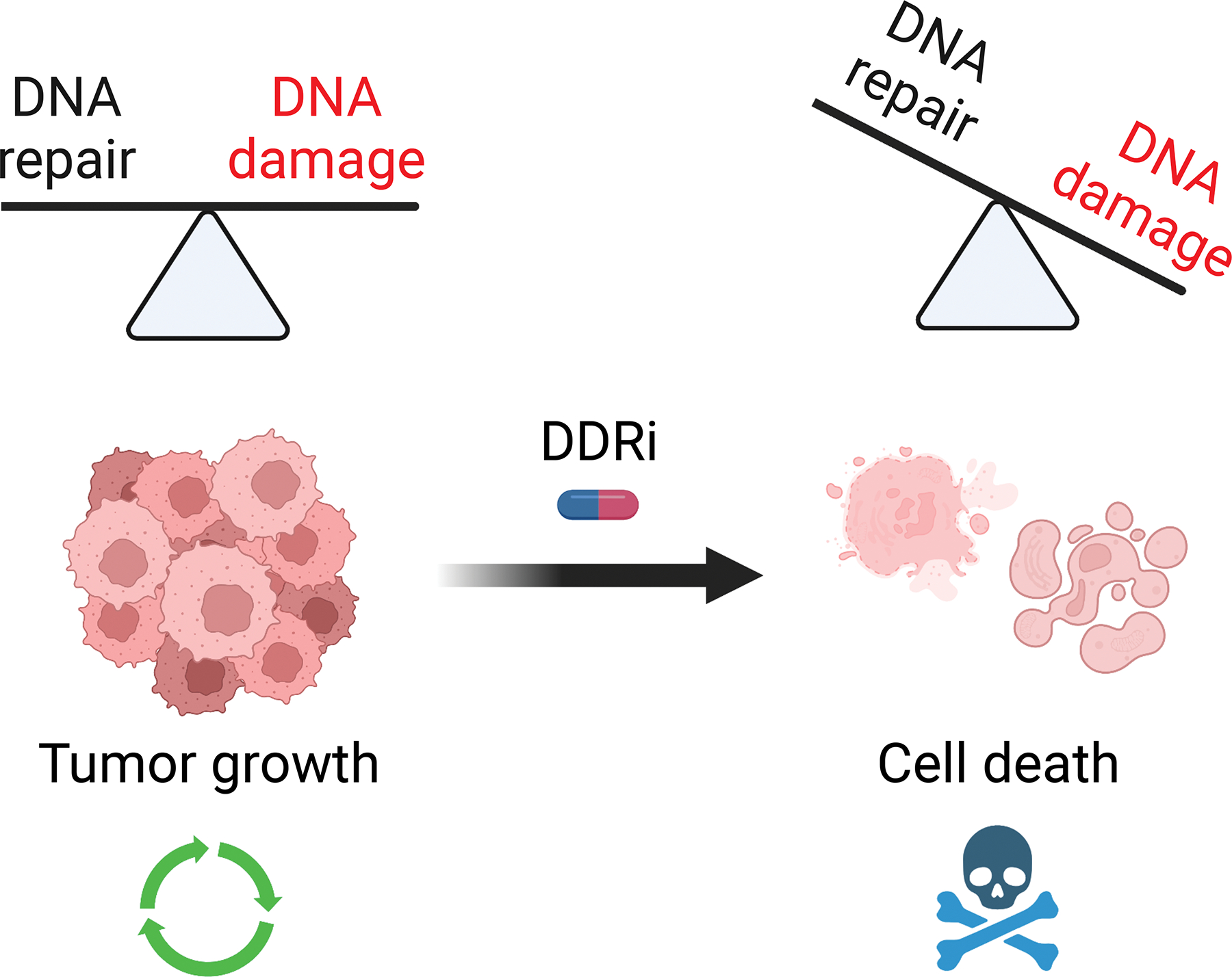
Targeted DDRi agents inactivate key DNA repair pathways, causing accumulation of double-stranded DNA breaks. This double-stranded DNA break accumulation, combined with increased dependence on error-prone repair mechanisms, overwhelms cellular damage tolerance and promotes cancer cell death.
The clinical benefit of PARP inhibition (PARPi) was first realized by treating relapsed ovarian cancer patients harboring BRCA1/2 mutations. A 2014 tumor agnostic, multicenter, single-arm, basket trial treated individuals harboring germline BRCA1/2 mutations with the olaparib (PARPi). The trial enrolled patients with platinum-resistant ovarian cancer and demonstrated a progression free survival of 7 months in an otherwise recalcitrant disease34. This led to olaparib’s approval in December of 2014, one month after the study was published.
Since 2014, there have been four PARP inhibitors—olaparib, niraparib, rucaparib, talazoparib—approved for the treatment of several cancers, typically in the context of underlying BRCA mutations35–42. Landmark trials have led to these PARPi approvals (Table 1) and more trials are currently underway testing the efficacy of PARPi in tumors with dysregulated BRCA genes and/or increased HRD.
Table 1:
Landmark Trials in DDRi Development
| Study/Author | Phase | Disease Setting | Treatments | Most Common Grade ≥3 Aes | Efficacy | Reason for Notability |
|---|---|---|---|---|---|---|
| Ledermann et al. 1,2 | II (randomized, placebo-controlled) | Platinum-sensitive, relapsed, high-grade serous ovarian cancer after ≥2 platinum-based regimens in PR or CR. | Olaparib 400mg BID vs Placebo as Maintenance. | Fatigue 7% vs 3%, anemia 5% vs 1, nausea 2% vs 0%, any 35.3% vs 20.3%. | Median PFS 8.4 mo vs 4.8mo (HR 0.35, 95% CI 0.25–0.49; P<0.001). BRCA1/2 mutant; median PFS 11.2 mo vs 4.3 mo (HR 0.18, 95% CI 0.10–0.31; P<0.0001). BRCA1/2-wild-type, median PFS 7.4 mo vs 5.5 mo (HR 0.54, 95% CI 0.34–0.85; P=0.0075). No OS difference between BRCA1/2-wild-type and mutant (HR 0.88, 95% CI 0.64–1.21; P=0.44). | Led to the first approval of a PARP inhibitor in 2014. Approved as maintenance therapy in BRCA1/2-mutated ovarian cancers in CR or PR after platinum-based chemotherapy. |
| ARIEL3; 3 | III (randomized, placebo-controlled) | Platinum-sensitive, high-grade serous/endometrioid ovarian, primary peritoneal, or fallopian tube carcinoma, after ≥2 platinum-based chemotherapy regimens, in CR or PR. | Rucaparib 600mg BID vs Placebo as Maintenance. | Anemia 19% vs 1%, increased ALT 10% vs 0%, fatigue 7% vs 3%, neutropenia 7% vs 2%, thrombocytopenia 5% vs 0%, any 54% vs 14%. | BRCA1/2-mutant; median PFS 16.6 mo vs 5.4 mo (HR 0.23, 95% CI 0.16–0.34; P<0.0001). All HR deficient; median PFS 13.6 mo vs 5.4 mo (HR 0.32, 95% CI 0.24–0.42; P<0.0001). BRCA1/2-wild-type high LOH; PFS 9.7 mo vs 5.4 mo (HR 0.44, 95% CI 0.29–0.66; P<0.0001). BRCA1/2-wild-type low LOH; PFS 6.7 mo vs 5.4 mo (HR 0.58, 95% CI 0.40–0.85; P<0.0049). | Led to FDA approval of rucaparib maintenance therapy for patients with advanced-stage ovarian cancer in CR or PR after platinum-based chemotherapy, irrespective of BRCA1/2 status. |
| OlympiAD; 4 | III (randomized, treatment vs SOC) | Germline BRCA-mutant, HER2-negative, metastatic breast cancer after ≥2 prior lines of chemotherapy | Olaparib 300mg BID vs physician's choice of single agent capecitabine, eribulin, or vinorelbine. | Anemia 16% vs 4%, neutropenia 9% vs 26%, leukopenia 3% vs 10%, any 37% vs 51% | Median PFS 7.0 mo vs 4.2 mo (HR 0.58, 95% CI 0.43–0.80; P<0.001). ORR 59.9% vs 28.8%. No OS benefit (HR 0.90, 95% CI 0.63–1.29; P=0.57). | Led to FDA approval of olaparib for metastatic HER2-negative breast cancers with BRCA1/2 mutations. |
| POLO; 5 | III (randomized, placebo-controlled) | Metastatic pancreatic cancer harboring germline BRCA1/2 mutation after first-line platinum-based chemotherapy. | Olaparib 300mg BID vs placebo. | Anemia 10% vs 2%, fatigue 5% vs 1%, decreased appetite 3% vs 0%, any 40% vs 23%. | Median PFS 7.4 vs 3.8 mo (HR 0.53, 95% CI 0.35–0.82; P=0.004). Median OS 18.9 mo vs 18.1 mo (HR 0.91, 95% CI 0.56–1.46; P=0.68). | Led to FDA approval of olaparib as first-line maintenance treatment of germline BRCA-mutated metastatic pancreatic cancer |
| PROfound; 6 | III (randomized treatment vs SOC) | Metastatic castrate-resistant, HRR mutant, prostate cancer with disease progression on a hormonal agent. | Olaparib 300mg BID vs physician's choice of enzalutamide or abiraterone. | Anemia 21% vs 3%, fatigue 3% vs 5%, any 51% vs 38% | BRCA1/2 or ATM-mutant; 7.4 mo vs 3.6 mo (HR 0.34, 95% CI 0.25–0.47; P<0.001). ORR 33% vs 2%, (odds of OR 20.86; 95% CI, 4.18 to 379; P<0.001). | Led to FDA approval of olaparib as first-line maintenance treatment of HRR gene-mutated metastatic castration-resistant prostate cancer, adding ATM mutation as an indication. |
| TRITON2, 7 | II (non-randomized, treatment only) | Metastatic castrate-resistant prostate cancer harboring PARP-sensitive DDR deficiency, having progressed on 1–2 lines of androgen receptor-directed and one taxane-based therapy. | Rucaparib 600mg BID. | Anemia 25%, thrombocytopenia 10%, fatigue 9%, ALT/AST increase 5%. | Median PFS 9.0 mo, 95% CI 8.3 to 13.5 mo. ORR in 27/62 (43.5%), 95% CI, 31%-56.7% with DOR not reached. Stable disease or better in 88.7%. | Led to FDA approval of rucaparib monotherapy as treatment for patients with BRCA1/2 mutant, metastatic castrate-resistance prostate cancer. |
| OlympiA; 8 | III (randomized, placebo-controlled) | Localized HER2-breast cancer with germline BRCA1/2 mutation following definitive local treatment and neoadjuvant or adjuvant chemotherapy. | Olaparib 300mg BID vs placebo. | Anemia 9% vs 0%, neutropenia 5% vs 1%, leukopenia 3% vs 0%, fatigue 2% vs 0%, any 24% vs 11%. | Median 3-year disease-free survival 86% vs 77% (HR 0.58, 95% CI 0.41–0.82; P<0.001). Median 3-year distant disease-free survival 88% vs 80% (HR 0.57, 95% CI 0.39–0.83; P<0.001). Median 3-year OS 92% vs 88% (HR 0.68, 99% CI 0.44–1.05; P=0.02). | Led to FDA approval of olaparib as adjuvant therapy for patients with germline BRCA-mutated HER2-negative high-risk early breast cancer. |
Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G., Scott, C., Meier, W., Shapira-Frommer, R., Safra, T., et al. (2012). Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366, 1382–1392. 10.1056/NEJMoa1105535.
Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G., Scott, C.L., Meier, W., Shapira-Frommer, R., Safra, T., et al. (2014). Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15, 852–861. 10.1016/S1470-2045(14)70228-1.
Coleman, R.L., Oza, A.M., Lorusso, D., Aghajanian, C., Oaknin, A., Dean, A., Colombo, N., Weberpals, J.I., Clamp, A., Scambia, G., et al. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961. 10.1016/S0140-6736(17)32440-6.
Robson, M., Im, S.A., Senkus, E., Xu, B., Domchek, S.M., Masuda, N., Delaloge, S., Li, W., Tung, N., Armstrong, A., et al. (2017). Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 377, 523–533. 10.1056/NEJMoa1706450.
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M.J., Park, J.O., Hochhauser, D., Arnold, D., Oh, D.Y., et al. (2019). Maintenance Olaparib for Germline. N Engl J Med 381, 317–327. 10.1056/NEJMoa1903387.
de Bono, J., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S., Chi, K.N., Sartor, O., Agarwal, N., Olmos, D., et al. (2020). Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 382, 2091–2102. 10.1056/NEJMoa1911440.
Abida, W., Patnaik, A., Campbell, D., Shapiro, J., Bryce, A.H., McDermott, R., Sautois, B., Vogelzang, N.J., Bambury, R.M., Voog, E., et al. (2020). Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a. J Clin Oncol 38, 3763–3772. 10.1200/JCO.20.01035.
Tutt, A.N.J., Garber, J.E., Kaufman, B., Viale, G., Fumagalli, D., Rastogi, P., Gelber, R.D., de Azambuja, E., Fielding, A., Balmaña, J., et al. (2021). Adjuvant Olaparib for Patients with. N Engl J Med 384, 2394–2405. 10.1056/NEJMoa2105215.
Despite a conserved mechanism of action, the ability of different PARP inhibitors to promote PARP-trapping varies considerably. Talazoparib (the most potent PARP trapper) demonstrates nanomolar cytotoxicity while veliparib, which has the least PARP trapping, is not active at 100mM 33. Clinically, the maximum tolerated dose of these medications mirrors their PARP-trapping capability. This realization has important clinical implications and should be considered when studying combination treatments43,44.
Acquired resistance mechanisms to PARPi are actively being evaluated, but three mechanisms are generally implicated: 1. Drug target-related effects like upregulation of efflux pumps or mutations in PARP genes. 2. Restoration of HR via a PARP-independent mechanism. 3. Loss of DNA end-protection and/or restoration of replication fork stability45. Multiple models have demonstrated upregulation of the ABCB1 drug efflux transporter upon development of PARPi resistance as a putative resistance mechanism. While the clinical relevance of this finding is unclear, resistance to PARPi could be theoretically reversed with the addition of an ABCB1 inhibitor46. Since all PARPis target the catalytic domain of PARP enzymes by competing with NAD+, resistance can arise from PARP mutations that reduce PARPi affinity, preserve enzymatic activity despite PARPi binding, or result in ineffective PARP trapping47. As synthetic lethality depends on insufficient HR, cell survival is facilitated by restoration of HR via BRCA-mediated (secondary activating mutations or loss of promoter hypermethylation) or BRCA-independent mechanisms.
Mismatch Repair Deficiency (MMRd):
Following the ground-breaking discovery that BRCA inactivation sensitizes tumors to PARP inhibition, subsequent studies have searched for other PARP inhibitor sensitizing genetic defects in the DNA repair machinery. These efforts helped discover that tumors demonstrating mismatch repair deficiency (MMRd) are sensitive to PARP inhibition48. Several groups have showed that genetic defects in key mismatch repair genes MLH1, PMS2, MSH2, and MSH6 all confer MMRd. Here, loss of MSH2 or MSH6 prevents recognition of base-base mismatches and most insertion-deletion loops, thus preventing MMR initialization and completion of MMR8,49. Functionally, decreased MMR increases somatic mutation accumulation. MMRd also increases expansion and contraction of microsatellite sequences in a phenomenon known as microsatellite instability (MSI). These known impacts of MMRd did not immediately explain why MMRd tumors are sensitive to PARP inhibition. Recent studies have uncovered that MLH1, PMS2, MSH2, and MSH6 also play key roles in repair of interstrand DNA crosslinks50. Koto et al. demonstrated that these proteins help recognize and repair interstrand DNA crosslinks (ICLs) in a non-replication-based mechanism, distinguishing it from Fanconi-Anemia mediated ICL repair. This discovery is significant as interstrand crosslinks are potent inhibitors of DNA replication that induce double-strand break accumulation if not properly repaired. Here, MMRd tumors are similar to BRCA deficient tumors where a genetic defect predisposes them to double stranded DNA break accumulation. This predisposition can thereby be exacerbated by PARP inhibition, which also forces double-stranded break accumulation, ultimately promoting tumor cell death.
ERCC1:
ERCC1 plays important roles in both nucleotide excision repair and interstrand crosslink repair. For NER, ERCC1 forms a heterodimer with the endonuclease XPF and controls 5’ incision, the first major step of NER following damage verification7. NER is the main cellular mechanism for repairing bulky DNA lesions that block DNA replication. In the absence of timely repair, unrepaired bulky lesions promote replication fork collapse and formation of double-stranded DNA breaks. ERCC1 loss also promotes double-strand break formation through impaired interstrand crosslink repair. Studies have shown that the ERCC1:XPF heterodimer is an important mediator of DNA incisions that are crucial for repair of interstrand crosslinks 10. Much like BRCA deficiency and MMRd, ERCC1 loss synergizes with PARP inhibition through toxic double-stranded DNA break accumulation51,52. This is significant as inactivating mutations in ERCC1 represent the most frequent DDR defect in non-small cell lung cancer (NSCLC), occurring in 30%–50% of cases 53.
DNA Repair-Related Genes:
Recently, several studies have demonstrated that genetic defects in DNA repair-related genes also sensitize tumors to targeted PARP inhibition. Two prominent examples are discussed below.
PBRM1:
Polybromo 1 (PBRM1) encodes the BAF180 protein and is a key member of the SWI/SNF-B (PBAF) chromatin remodeling complex. PBRM1 is one of the most frequently inactivated genes across cancers, highlighted by ~70% loss of expression in renal clear cell carcinoma 54. In addition to its role in chromatin remodeling, PBRM1 plays an important role in regulating transcriptional silencing in regions adjacent to double-stranded DNA breaks55. In the absence of efficient transcriptional silencing near double-stranded breaks, cells accumulate transcription-replication conflicts, marked by R-loop accumulation, DNA damage foci, and replication stress. Studies have demonstrated that increased transcription-replication conflicts promote double-stranded break formation56. Given this, it fits well that PBRM1 inactivation has been shown to confer sensitivity to PARP inhibition. Here, PBRM1 inactivation increases development of double-strand break formation which is only further magnified by PARP inhibition57.
ARID1A:
AT-Rich Domain 1A (ARID1A) is a key component of the SWI/SNF complex and is also highly mutated across cancers. To this end, nearly 50% of endometrial cancers as well as 30% of gastric and bladder cancers carry inactivating mutations in ARID1A58,59. Beyond chromatin remodeling, ARID1A plays an important role in facilitating DNA repair. Shen et al. found that ARID1A is recruited to double-strand break sites along with the DNA damage kinase ATR. At these sites, ARID1A plays a key role in facilitating double-strand break resection and sustaining ATR activity60. Importantly, both end resection and ATR signaling are required for proper repair of double-strand breaks. Thus, ARID1A deficiency decreases double-strand break repair. As with BRCA deficiency, MMRd, ERCC1 inactivation, and PBRM1 loss, ARID1A deficiency sensitizes cells to PARP inhibition through cumulative accumulation of intrinsic and therapeutically induced toxic double-stranded DNA breaks. Early phase clinical trials in relapsed, ARID1A-deficient gynecological cancers are testing the efficacy of PARP and ATR inhibitor combinations61.
Genetic Alterations Sensitizing to Non-PARPi DDRi Strategies:
The promise of PARP inhibition has prompted the development of several other targeted DDRi agents, including inhibitors blocking DNA damage signaling effectors ATR and CHK1. Below, we discuss genetic alterations that predispose tumors to increased sensitivity to DDR inhibitors targeting ATR or CHK1.
ATM Loss Sensitizes to ATRi:
Ataxia Telangiectasia Mutated (ATM) is an essential component of the cellular DNA damage sensing and checkpoint machinery. In replicating cells, ATM is recruited to the sites of double-strand DNA breaks through interaction with the MRE11-RAD50-NBS1 (MRN) complex62. Once bound, ATM helps catalyze the DNA end resection activity of CtIP and MRE11, promoting resected end formation required for HR. Studies have also shown that ATM helps facilitate HR repair completion downstream of initial resection. Following activation, ATM also initiates a signaling cascade, including CHK2 kinase activation, that helps coordinate cell cycle arrest and DNA damage transcriptional responses. ATM is frequently dysregulated by inactivating mutations, homozygous deletions, and genomic fusion events across tumor types resulting in decreased HR-mediated double-strand break repair and cellular damage signaling1. ATM deficient cells become increasingly reliant on other mechanisms to repair double-stranded breaks. Furthermore, ATM deficient cells become hyper dependent on the ATR-CHK1 signaling pathway. Several preclinical studies have demonstrated that targeted ATR inhibition represents a unique therapeutic vulnerability in ATM deficient tumors, explained by further loss of cell cycle checkpoint activity63,64. By blocking ATR signaling, ATM deficient cells lose the last predominant cellular mechanism that coordinates cell cycle arrest and facilitates DNA repair. Collectively, loss of cell cycle checkpoint activity promotes unscheduled cell cycle entry into mitosis. Here, unrepaired DNA damage and under-replicated DNA become substrates for chromosome missegregation and help promote mitotic catastrophe, or damage-induced cell death during mitosis65.
ERCC1 Loss Sensitizes to CHK1i:
In addition to conferring PARP inhibitor sensitivity, ERCC1 loss also creates a unique vulnerability to CHK1 inhibition. CHK1 is a key mediator of the cellular DNA damage response that is activated by ATR. Functionally, CHK1 coordinates several essential cellular processes to allow cells to cope with increased replication stress. These processes include coordination of cell cycle arrest, replication fork stabilization, and suppression of replication origin firing. CHK1 coordinates cell cycle arrest by directing phosphorylating members of the CDC25 family of phosphatases. These CDC25 phosphatases promote cell cycle progression by removing inhibitory phosphorylation marks on cyclin dependent kinases (CDKs), thereby increasing their activity18. Once activated, CHK1 phosphorylates CDC25A, preventing CDK2:CCNE or CDK2:CCNA activity required for S/G2 progression. Additionally, CHK1 phosphorylates CDC25C to prevent CDK1:CCNB1 activity and mitotic entry, allowing time for DNA repair coordination prior to cell cycle progression. CHK1 also directly phosphorylates and inactivates EXO1, the main exonuclease responsible for resection of stalled replication forks. By inactivating EXO1, CHK1 promotes stalled fork stability and prevents double-strand DNA break formation. In the context of ERCC1 loss, cells have decreased capacity for NER and interstrand crosslink repair resulting in replication stress and heightened dependence on the ATR-CHK1 pathway. Much like with ATR inhibition in the context of ATM deficiency, inhibition of CHK1 in cells with ERCC1 loss promotes double-strand break accumulation, driving cancer cell death66.
Fanconi Anemia (FA) Loss Sensitizes to ATRi and CHK1i:
As with ERCC1 loss, loss of various FA pathway members sensitizes cells to CHK1 inhibition. The FA pathway is responsible for repair of interstrand DNA crosslinks encountered during replication. Additionally, studies have shown that the FA pathway is essential for resolving transcription-replication conflicts at sites of replication stress67. Despite this essentiality, various FA pathway members are inactivated by mutation or deep deletions across bladder, ovarian, breast, skin, uterine, head and neck, prostate, pancreatic, and colorectal cancers1. Of these members, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCG, FANCI, FANCL, and FANCP/SLX4 are the most commonly inactivated effectors68. Loss of these genes impairs interstrand crosslink recognition and repair as well as R-loop resolution. This loss of function is significant as unresolved interstrand crosslinks and R-loops stall the replicative machinery, increasing replication stress. Under these conditions, cells with an impaired FA pathway are hyper dependent on the ATR-CHK1 pathway to coordinate cell cycle arrest and replication fork stability. Studies have demonstrated that CHK1 inhibition is lethal to cells with impaired FA69. By impairing CHK1 activity, FA deficient cells accumulate chromosomal breaks, unresolved DNA damage, and undergo widespread cell death.
While PARPi is currently the only widely adopted DDRi strategy in the clinic, there are several clinical trials underway evaluating other DDR inhibitors, many of which target ATR or CHK170. Early phase trials evaluating ATR inhibitors have been conducted as monotherapy and in conjunction with chemotherapy, DDRi, and immunotherapy across many solid tumor contexts including melanoma, NSCLC, ovarian, and solid-tumor basket trials. These trials demonstrated an acceptable tolerability with predominantly hematologic side effects from bone marrow toxicity. Response rates were modest and favored patients with genomic alterations like BRCA mutations, other DNA repair defects, and elevated tumor mutational burden (TMB)71–74. Several CHK1 monotherapy trials have been conducted with little success, likely due to inadequate patient selection as these trials did not necessitate existing susceptibilities to DDRi and were evaluated in the treatment refractory setting. CHK1 monotherapy demonstrated high levels (frequently above 70%) of grade 4 neutropenia which warrants particularly thoughtful patient selection in future studies75–77. Moving forward, defining the underlying causes of DDRi sensitivity will be required to extend benefit of these agents to new cohorts of patients in the clinic.
DDRi Promotes an Anti-Tumor Immune Response:
In addition to tumor intrinsic effects, targeted DDRi has been shown to stimulate anti-tumor immune responses. This increased immune system engagement occurs from both dying and surviving cancer cell populations treated with DDRi through distinct mechanisms. Mechanisms of DDRi-directed immune engagement are discussed below.
Cancer Cell Death: Danger Associated Molecular Patterns (DAMPs) and Neoantigen Presentation Awaken the Immune System:
As outlined above, targeted DDRi induces cancer cell death by overwhelming or disarming key DNA repair pathways, but also provides an avenue for stimulating anti-tumor immune responses (Figure 2). Much research has shown that cancer cell death can occur through “programmed” or “immunogenic” death mechanisms78. Traditionally, apoptosis has been classified as a “programmed” or non-immunogenic cell death mechanism. This is largely because cell membrane integrity is maintained during apoptosis, whereas cell membrane rupture is a defining feature of immunogenic cell death mechanisms like necroptosis and pyroptosis. This is significant as cell membrane rupture releases DAMPs and neoantigens into the tumor microenvironment. DAMPs—such as calreticulin, ATP, and HMGB1—are taken up by tissue-resident dendritic cells, stimulating their activity and maturation, ultimately promoting neoantigen processing and presentation79. This neoantigen presentation activity is essential for generating tumor specific CD4 and CD8 T-cell responses, which are ultimately required for durable anti-tumor immune control. Dendritic cells are also capable of presenting tumor-specific neoantigens following phagocytosis of apoptotic tumor cells80. Thus, DDRi directed cancer cell death plays an essential role in potentiating anti-tumor adaptive immune responses.
Figure 2: DDRi-induced cancer cell death activates dendritic cells and promotes T-cell mediated anti-tumor responses.
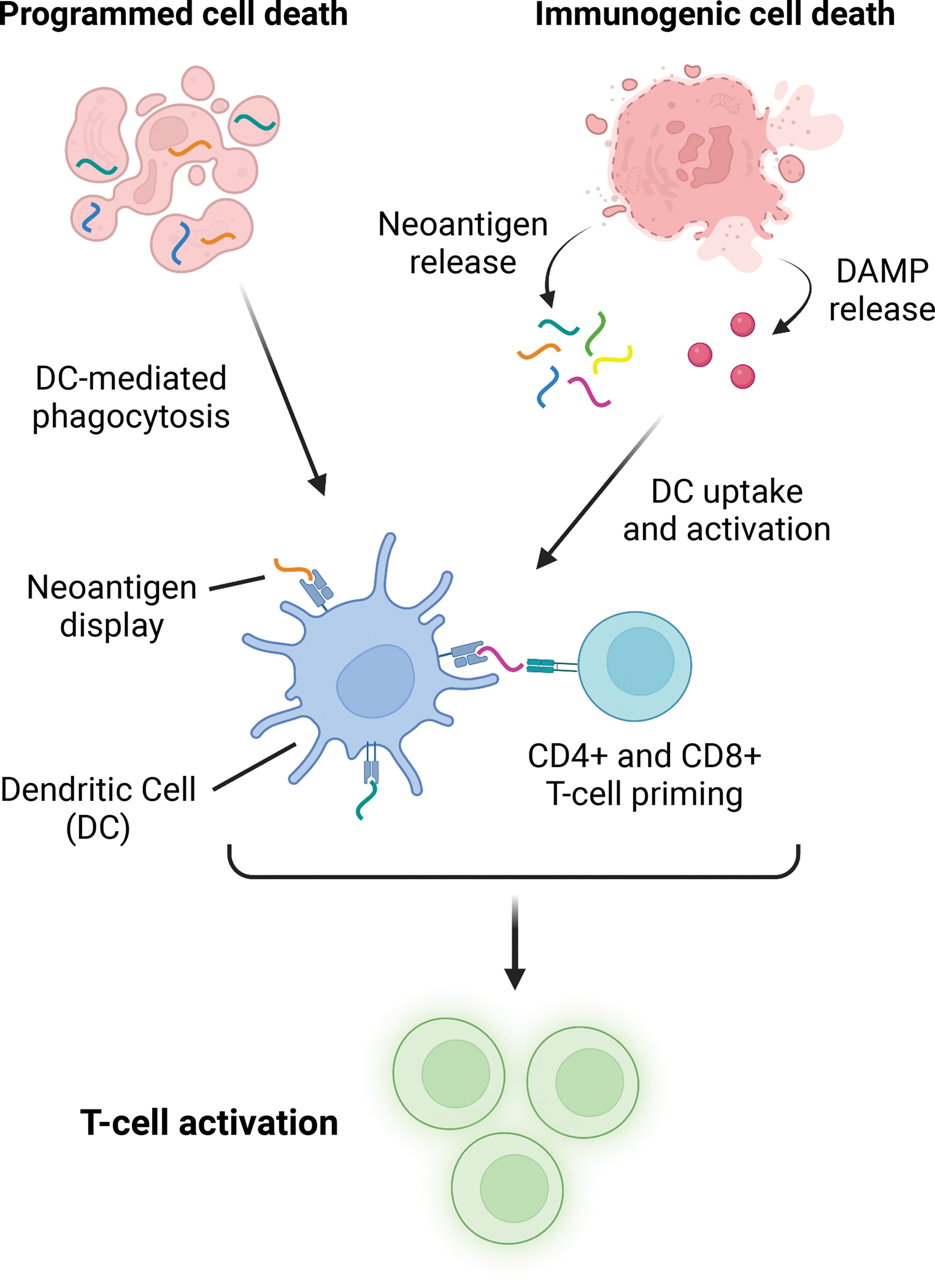
DDRi induces cancer cell death. While cancer cells can die through multiple cellular mechanisms, cell death is mainly characterized as “programmed” or “immunogenic.” Here, programmed cell death refers to a process where cells die, but cell membranes remain intact. Immunogenic cell death, on the other hand, occurs when cells die and their membranes rupture, spilling cellular contents into the tumor microenvironment. During this process, tumor neoantigens and danger associated molecular patterns (DAMPs) are released. Tumor resident dendritic cells are in turn activated by DAMP release and take up released neoantigens. Once activated, dendritic cells present tumor neoantigens to CD4 and CD8 T-cells, generating tumor-specific adaptive immune responses. Importantly, programmed cell death can also generate T-cell responses via dendritic cell activation. Here, dead cells are phagocytosed by dendritic cells. These dendritic cells then display tumor neoantigens to CD4 and CD8 T-cells, promoting their activation.
cGAS-STING Activation and Type I Interferon Responses:
Targeted DDRi also increases immune system engagement by activating cGAS-STING signaling in surviving cancer cells (Figure 3). The cGAS-STING pathway originally evolved as a mechanism for helping mammalian cells detect viral infection through sensing of cytosolic DNA, as the cytosol is a DNA-free compartment in non-diseased states. CGAS-STING signaling is initiated following binding of dsDNA by cyclic-GMP-AMP synthase (cGAS) (Figure 4). Once activated, cGAS catalyzes the conversion of ATP and GTP into 2’,3’-cyclic GMP-AMP (cGAMP)81. CGAMP functions as a second messenger to activate the Stimulator of Interferon Genes (STING) protein located in the endoplasmic reticulum (ER)82,83. Upon activation, STING then migrates to the ER-Golgi intermediate compartments where it recruits TANK Binding Kinase 1 (TBK1), a serine/threonine kinase that phosphorylates Interferon Regulatory Factor 3 (IRF3)84–86. IRF3 in-turn localizes to the nucleus where it mediates the expression and secretion of type 1 interferons. STING also stimulates IKK-alpha/beta, triggering transcriptional activation of Nuclear Factor Kappa-light-chain Enhancer of Activated B cells (NF-kB) which also mediates transcription of interferons (IFN) and interleukin-687. Once secreted into the microenvironment, these cytokines help activate tissue-resident dendritic cells and ultimately promote the generation of CD4 and CD8-mediated immune responses (Figure 3, Figure 4A). In addition to this direct activation, cGAMP generated by tumoral cGAS can diffuse into the microenvironment where it acts as a second messenger for neighboring myeloid and lymphoid populations, further stimulating interferon production88,89.
Figure 3: DDRi-induced DNA damage generates cytosolic DNA and activates the cGAS/STING pathway.
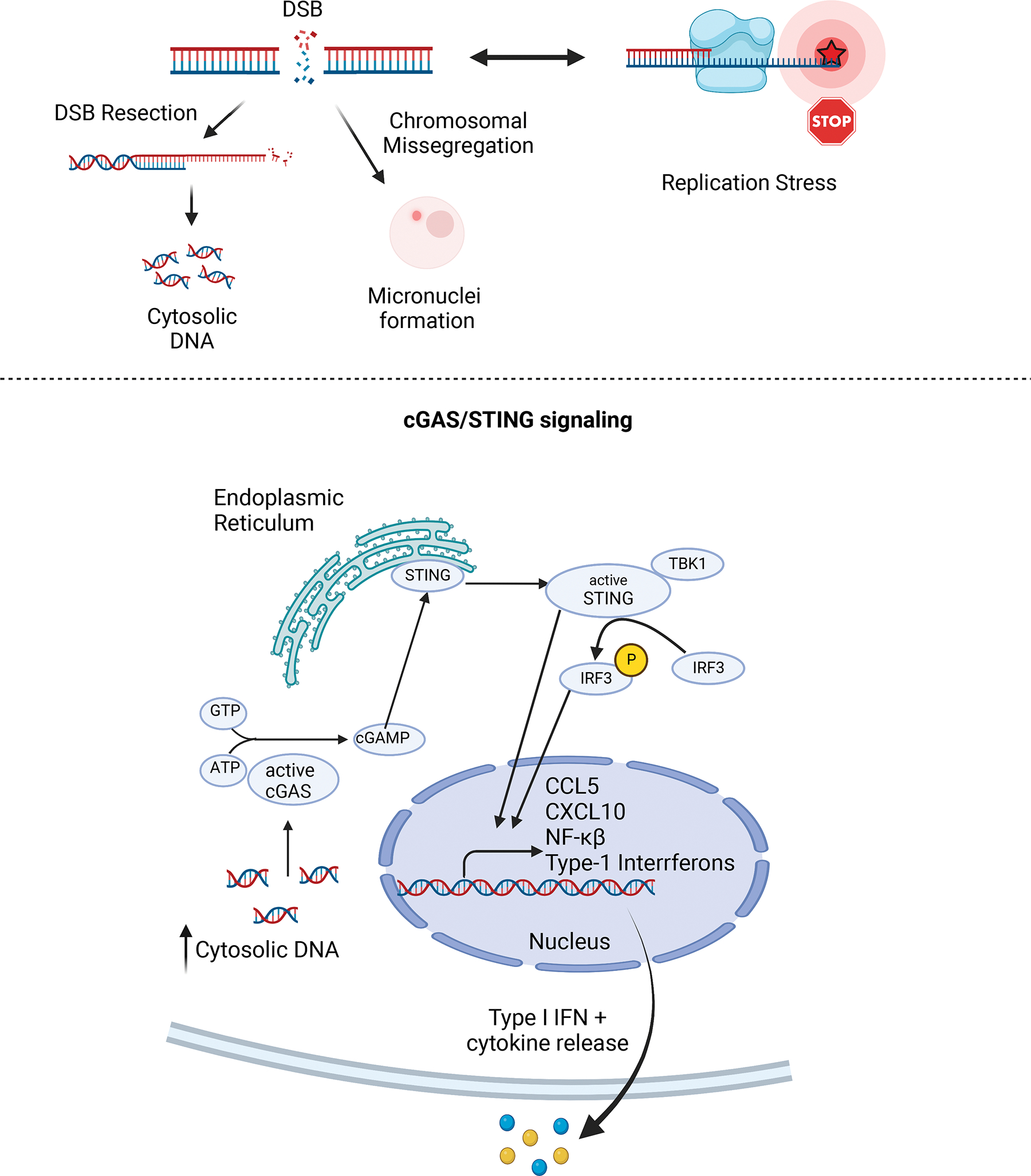
A) DDRi agents promote DNA damage accumulation in surviving cancer cell populations. Specifically, DDRi promotes double-stranded DNA break accumulation and replication stress. Once formed, double-stranded DNA breaks are often resected in order to initiate repair. Increased DNA resection activity generates excessive double-stranded DNA production and cytosolic DNA accumulation. Furthermore, double-strand breaks that are poorly repaired can promote micronuclei accumulation via chromosomal missegregation. When present, micronuclei also activate the cGAS-STING pathway.
B) Cytosolic DNA is sensed by the cGAS-STING pathway. Once bound to dsDNA, cGAS becomes activated and produces cGAMP. cGAMP production in turn activates STING. Activated STING initiates an inflammatory transcriptional program marked by production of Type I IFNs and other cytokines. These cytokines are then secreted into the tumor microenvironment.
Figure 4: DDRi promotes cGAS/STING signaling, engaging both adaptive and innate anti-tumor immune responses.
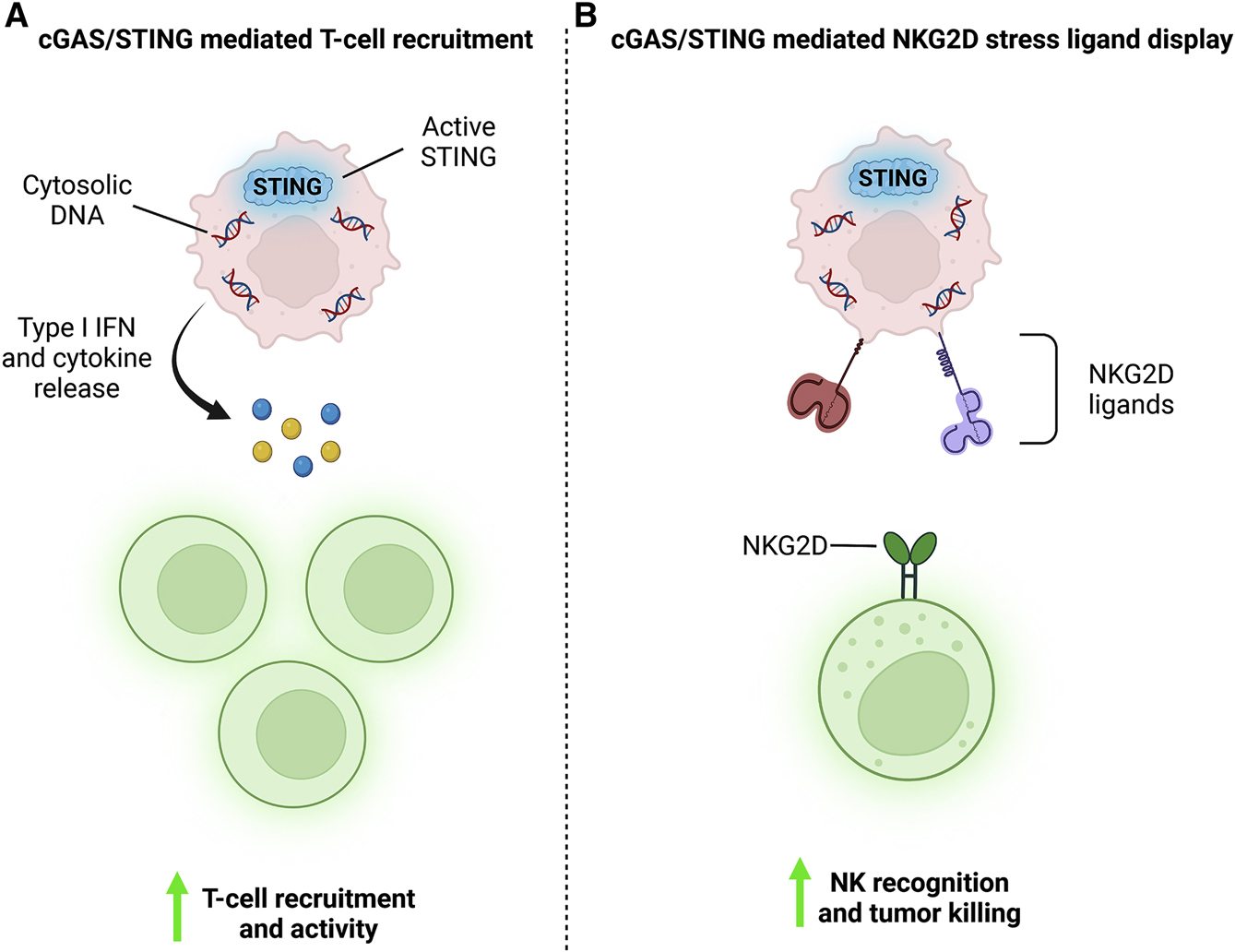
A) DDRi induced cytosolic DNA activates the cGAS/STING pathway. Activate STING initiates transcription of Type I IFNs and other inflammatory cytokines. These cytokines are secreted into the tumor microenvironment where they help recruit and activate anti-tumor T-cells. B) Additionally, cGAS-STING signaling drives NKG2D stress ligand display on the surface of cancer cells. When displayed, these ligands help tumor resident NK cells bind to and kill cancer cells. Thus, cGAS-STING signaling helps engage both adaptive and intrinsic anti-tumor immune responses.
Mechanistically, DDRi activates cGAS-STING signaling through double-stranded DNA break generation (Figure 3). Repair of double-stranded DNA breaks often involves resection which produces small DNA fragments that accumulate in the cytosol, where they are bound by cGAS and activate cGAS-STING signaling90. Additionally, error-prone repair of double-stranded DNA breaks can cause chromosomal missegregation and micronuclei formation. Micronuclei can then rupture, introducing tumor DNA into the cytosol, promoting cGAS activation87,91. Studies have demonstrated that cGAS can enter micronuclei and initiate cGAS-STING signaling even in the absence of micronuclei rupture 83. DDRi forces surviving cancer cells to generate these second messengers responsible for recruiting adaptive anti-tumor immune responses.
DNA Damage Induces NKG2D Ligand Display for NK Cell Recognition:
Lastly, seminal studies have demonstrated that DNA damage accumulation promotes anti-tumor NK cell responses by inducing tumor cell display of NKG2D ligands MICA, MICB, and ULBP4/RAET1 (Figure 4B) 92. When present, these ligands bind the NKG2D receptor present on NK cells and promote tumor recognition and NK-mediated cytotoxicity93,94. Interestingly, studies have shown that cGAS-STING pathway signaling downstream of DNA damage accumulation is required for NKG2D ligand display95. These data demonstrate that DDRi directed DNA damage accumulation engages both the innate immune response (NK cells) and the adaptive immune response (T-cells) to further potentiate anti-tumor immune responses.
Two is Better Than One: Leveraging DDR Deficiencies to Potentiate Immunotherapy Responses
The observation that DNA damage accumulation, produced by genetic DDR compromise or targeted DDRi therapy, results in increased IFN signaling responses invites the question: does DDRi demonstrate synergy when combined with immune-mediated therapy? Below we discuss data from pre-clinical studies and clinical trials supporting DDRi and immunotherapy combination strategies.
Preclinical Data Leveraging DDR Deficiencies and IO:
Many groups have studied how DDRi-mediated DNA damage accumulation and cGAS-STING activation synergize with immunotherapy in preclinical models. BRCA mutations were the first highly studied genetic predisposition to immunotherapy. Acutely after BRCA2 knockdown in ovarian cancer cell lines, genes related to cell cycle progression, DNA replication, and DNA repair are downregulated, supporting cell cycle arrest in G1 of the cell cycle. However, several weeks after BRCA2 knockdown the cells upregulate interferon-stimulated genes and the cGAS-STING-STAT pathway becomes activated. Patient data from TCGA corroborated this association between BRCA2 deficiencies and increased immune response genes using mRNA expression96. This mechanism was also shown in breast cancer syngeneic immunocompetent mouse models. Niraparib-responsive BRCA-deficient tumors were treated with niraparib and exhibited increased RNA expression of immune-related genes like the inflammatory response, TNFA signaling, interferon gamma, and interferon alpha. These genetic signatures correlated with increased tumor infiltrating lymphocytes (TILs). This prompted evaluation of combination PARPi-PD-L1 treatment in humanized mouse models harboring BRCA-deficient tumors, which did indeed demonstrate synergistic killing97.
It was initially demonstrated in BRCA-deficient, triple-negative breast cancer (TNBC; breast cancer cells lacking the estrogen, progesterone, and HER2 receptors), that the PARP inhibitor olaparib induced CD8+ T-cell infiltration and activation in vivo, supporting T-cell activation as a predominant mechanism of cytotoxicity. CRISPR mediated knockout of STING prevented olaparib-induced proinflammatory signaling and the associated T-cell infiltration. Modulation of the T-cell response by treating immunodeficient SCID mice with PARPi or treating immunocompetent mice with anti-CD8 antibodies in combination with PARPi, significantly reduced median survival when compared to immunocompetent PARPi monotherapy98.
DDRi-IO synergy was also demonstrated in BRCA-deficient ovarian cancers treated with olaparib and PD-L1 inhibition which demonstrated significant synergistic cytotoxic effect through innate and adaptive immune responses. The impressive response observed in BRCA-deficient ovarian cancer treated with olaparib was abrogated when such cells were orthotopically injected into STING deficient mice as compared to wild-type99. Further work was done evaluating PARPi in colorectal and ovarian tumors confirmed to have no mutations in genes involved in the HR repair pathway. Syngeneic immunocompetent colon and ovarian cell line-derived mouse models were treated with PARPi in athymic (immunodeficient) or thymus intact C57BL/6 mice. PARP inhibition had no effect on overall survival in athymic mice but demonstrated a significant improvement in immunocompetent mice. Further work with in the same syngeneic model demonstrated that neither PARPi, anti-PD-L1, or the combination had any effect on tumor volume in athymic mice, but all showed decreased tumor size in the immunocompetent model, with the combination of PARPi and anti-PD-L1 therapy showing significant synergy100. Collectively, these data strongly support that an intact immune system is necessary for a PARP-mediated antitumoral effect.
Landmark Clinical Data Leveraging Immunotherapy in DDR Deficient Tumors:
The precedent for combining DDRi and immunotherapy in the clinic was set years prior to the first PARP inhibitor trials with the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) antibody ipilimumab. Ipiliumab became FDA approved in 2011 following a landmark study demonstrating that previously treated metastatic melanoma patients receiving ipilimumab had a 23.5% 2-year survival compared to 13.7% among those who received a cancer-associated peptide vaccine101. This trial also reported a remarkable complete response in 6% of patients with a median duration of response of at least 59 months at 7-year follow-up102. Melanoma represents the solid tumor with the highest rate of TMB, due to related UV damage from sun exposure, which sensitizes these cancers to immunotherapy. The discovery that CTLA-4 inhibition produces cures in a universally fatal malignancy prompted the awarding of a Nobel Prize to Drs. James Allison and Tasuko Honju, and a paradigm shift toward developing immune checkpoint blockade (ICB) strategies across all malignancies.
CTLA-4 inhibition was quickly followed by additional ICB strategies like programmed death-1 (PD-1, on T-cells) and PD-1 ligand (PD-L1, on antigen-presenting cells) targeting monoclonal antibodies. Upon PD-1 activation, CD8+ T cells demonstrate reduced proliferation and decreased interleukin and interferon-gamma secretion103,104. Upregulation of PD-L1 is commonly seen as an immune-surveillance escape mechanism among many tumor types. Inhibition of PD-1 and its ligand has been a widely successful strategy in promoting immune-mediated tumoral killing. This was first successfully exploited in the Keynote 001 study using the anti-PD-L1 antibody pembrolizumab against three groups of previously treated tumor types: metastatic melanomas, NSCLC, and other solid tumors. The melanoma data was the first to publish, demonstrating stable or improved disease in over half of the patients treated, despite these patients having progressed on ipilimumab. The median survival at 18 months was not reached at any dose evaluated in the study105. Updated data from the Keynote 001 trial demonstrated a 5-year OS of 34% with an observed plateau suggestive of long-term benefit and possibly cure, at 48 months of treatment106. The metastatic NSCLC arm data was published shortly thereafter demonstrating the scope of this strategy, but also the importance of PD-L1 as a predictive biomarker in certain cancer histology types. Median survival was not reached at 28 months among patients with ≥50% PD-L1 proportion score but was just over eight months among those with PD-L1 proportion scores of 1–49%. 5-year OS data exceeded 25% among patients with a PD-L1 proportion score of≥50%107.
While ICB represents an overwhelming success among a subset of patients with cancer, many do not derive benefit. One important study supporting the connection between mutational burden and subsequent immune activation was Keynote-158. Published in 2020, this was the first tissue-agnostic trial to exclusively enroll both microsatellite instable (MSI-High) and MMRd tumors to be treated with ICB via the PD-1 targeting antibody pembrolizumab. In this study, endometrial cancer demonstrated an impressive overall response rate of 57.1% with 8/49 participated obtaining a complete response. In contrast, patients with brain or pancreatic tumors had ORR of 0% and 2.1% respectively, despite also having microsatellite instability (MSI) or MMRd. Those who responded in the trial frequently demonstrated durable responses, with the median duration of response not reached after two years108. These variable response rates further demonstrate that no single biomarker has been universally predictive of response across tumor types.
The maximum therapeutic potential of biomarker-directed treatment is only realized under the ideal patient selection criteria. A recent study evaluating twelve patients with MMRd, locally advanced, rectal adenocarcinoma treated with single-agent anti-PD-1 therapy demonstrated this. Immunotherapy was intended to be followed by standard of care chemoradiotherapy and surgery. However, every patient in the trial demonstrated a clinical complete response after the immunotherapy, so none required additional chemotherapy, radiation, or surgery109. The standard of care treatment for these tumors frequently requires a highly morbid operation resulting in a permanent ostomy bag. A paradigm shift toward patients not even requiring chemotherapy or radiation has appropriately generated excitement sounding through the echo chambers of the lay media.
Immune-mediated strategies are nothing short of miraculous for certain patient subgroups but dramatic variability in treatment responsiveness is seen across and within tumor types. IO-associated benefit has led to improved overall mortality in cancers like NSCLC, which were previously driven by varying rates of tobacco use110. A cross-sectional analysis from 2011 to 2018 of publicly available data from the American Cancer Society and FDA approved indications for drug use evaluated checkpoint inhibitor indications and expected response rates. Six checkpoint inhibitor drugs were approved over this time period resulting in an increase in patient eligibility for ICB from 1.54% to 43.6%; however, the percentage of patients estimated to respond only increased from 0.14% to only 12.5% across all indications. It is unlikely that all patients eligible for ICB received ICB, so the percentage of patients who derived benefit from ICB over that time period was likely lower111.
Combination DDRi Strategies:
Borrowing lessons learned from kinase inhibition, investigators have attempted to simultaneously inhibit multiple DDR proteins to maximize benefit. An early phase trial evaluating an ATRi-PARPi combination in advanced DDR-deficient cancers yielded only an 8.3% response rate, but two patients harboring ATM mutations had complete responses with myelosuppression-related dose reductions was seen in 20% of patients112. The ATRi-PARPi combination was further evaluated in the CAPRI trail which included 12 patients evaluable for response (three with BRCA mutations). There were no objective responses in these platinum-resistant patients, but stable disease was achieved in 9/12 patients. Benefit was still driven by BRCA mutation status with a median PFS of 8.2 and 4.2 months for BRCA mutant vs wildtype tumors respectively113.
PARPi-CHK1i combination therapy was evaluated in twenty-nine PARPi refractory, BRCA-mutant, predominantly high-grade serous ovarian cancer or fallopian tube cancer patients. The study included a 7-day olaparib alone lead-in to avoid profound myelosuppression as prexasertib (CHK1i) monotherapy previously demonstrated a 73.3% rate of neutropenia and anemia. The trial was designed with prexasertib dose-escalation but was limited to the initial dose due to prolonged grade 4 neutropenia. Continuous olaparib dosing was also transitioned to intermittent dosing due to toxicity. Grade 4 neutropenia was seen in 20/29 patients, with similar rates of less severe anemia and thrombocytopenia. Of the BRCA+ ovarian or fallopian tube patients, 10/18 remained on study for 4+ cycles, 4/18 achieved a PR, and three patients enjoyed PRs for greater than nine cycles114.These studies demonstrate the potential benefits and toxicities while highlighting the importance of strict enrollment criteria when investigating combination DDRi treatment strategies.
Complementary DDRi strategies may promote even greater cGAS/STING activation and further potential ICB efficacy. This principle was supported by the results of a genome-wide CRISPR-CAS9 KO screen applied to PRE1-hTERT cells subsequently treated with combination ATRi/PARPi. RNASEH2B, an RNAse which functions to degrade the RNA associated with RNA:DNA hybrids, was identified as a top hit. This corroborated previous studies describing PARPi and ATRi sensitivity in RNAse H2-deficient cells producing elevated level of DNA damage and loss of RNASEH2 demonstrating synthetic lethality with ATR inhibitors115,116.
Extending DDR/IO Benefit Beyond DDR Inactivating Genetic Alterations:
While many associate PARPi efficacy with predisposing BRCA deficiencies, it is well characterized that BRCA-proficient tumors may also be targeted in this way. Ovarian and colon syngeneic mouse models have demonstrated sensitivity to PARPi without known HR repair pathway deficiencies. PARPi-mediated increases in cytosolic DNA activated cGAS/STING to promote a type I interferon response in the absence of the traditional synthetic lethal context. This effect was not observed in an immunodeficient system harboring the same xenograft, suggesting the necessity of an intact immune system for PARPi efficacy. Anti-PD-L1 therapy further potentiated the effect of PARPi in the immune-intact system yielding longer survival times and decreased tumor growth. The PARPi-IO combination yielded greater CCL5 levels, CXCL10 levels, p-IRF3 and STING expression, and ultimately greater CD8+ T-cell infiltration, further supporting immune-mediated cytotoxicity in this model100.
Our group and others discovered that CHK1 and PARP inhibitors induce genomic instability and increase the generation of micronuclei, a potent activator of the cGAS-STING pathway, in small-cell lung cancer (SCLC) cell lines. DDRi universally and significantly increased T-cell infiltration and abrogated T-cell exhaustion in vivo, despite increasing PD-L1 expression. After treating with CHK1i-anti-PD-L1 combination therapy, immunocompetent SCLC mouse models yielded 6/10 complete responses compared to no response with IBC alone117. This is consistent with the poor response seen clinically with ICB in SCLC. Our group observed similar results with PARPi-ICB combination therapy. DDRi-IO also shows promise across other tissue types. Immunocompetent C57BL/6 mice harboring bladder, colon, or pancreatic flank tumors treated with CHK1i-anti-PD-L1 therapy demonstrated tumor growth inhibition greater than either agent given alone118. Similar results were seen with syngeneic prostate cancer models treated with combination ATR-anti-PD-L1; with single agents demonstrating modest effects but combination treatment producing near complete responses119.
A phase-2 trial (N=20) evaluated the durvalumab-olaparib combination in previously treated ES-SCLC patients. Most had progressive disease (12/19 evaluable patients), but four patients achieved stable disease and one had a CR. Grade 3 or higher side effects were seen in 9/20 patients, predominantly lymphopenia. Pre-treatment tumor analysis identified that PD-L1 was required, but not sufficient, to generate a pre-treatment inflamed-phenotype. Conversely, none of the PD-L1 negative tumors had pre-treatment tumor infiltrating lymphocytes (TILs). Post-treatment biopsies demonstrated that responding tumors had dense TILs with increased PD-L1 expression. Non-responding tumors lacked PD-L1 and displayed minimal or no TILs. These data support the observation that immunologically “cold” tumors, like SCLC, may respond to T-cell mediated cytotoxicity under the appropriate microenvironmental conditions120. These data may be confounded by treatment resistance obtained during prior chemotherapy or prior PARPi administration. DDRi+IO therapy shows promise, even in highly recalcitrant tumors where any benefit warrants further investigation. Future studies will require careful patient selection, further emphasis on the timing of treatment, and intentional evaluation of potential resistance mechanisms.
One strategy to maximize short-term ICB benefit is to evaluate efficacy in the neoadjuvant setting (given before surgery) by evaluating surgical outcomes. The phase-II I-SPY2 trial included a comparison of neoadjuvant durvalumab (anti-PDL1), olaparib (PARPi), and paclitaxel (DOP) vs a paclitaxel-alone control group in patients with HER-2 negative breast cancer121. The population that received the DOP regimen had higher rates of pathologic complete responses (pCR) compared to chemotherapy alone group across all breast cancer subtypes, with TNBC increasing from 27% to 47%. Despite patients receiving the DOP combination experiencing increased rates of grade 3–4 neutropenia compared to paclitaxel alone (12.3% vs 9%), rates of early discontinuation were similar across intervention and control arms. Furthermore, 0% of the DOP group demonstrated progression/lack of response before surgery compared to 11% in the control group. These results were similar to prior trials evaluating ICB-chemotherapy neoadjuvant combinations lacking DDRi, suggesting DNA damage accumulation is the primary driver of clinical benefit in this setting. Predictive biomarker analysis using a 13-gene panel, 10 of which representing immune signatures, demonstrated a positive relationship between gene expression and pCR in both arms, while the DNA repair deficiency (PARPi7) and mitotic signatures correlated positively with pCR only in the DOP arm122. Determining whether chemotherapy can be de-escalated in the setting of ICB-DDRi without sacrificing efficacy is an area of great and growing interest.
The MEDIOLA single-arm basket trial evaluated the durvalumab-olaparib combination across multiple tumor types including BRCA-mutated metastatic breast cancer, metastatic ovarian cancer, metastatic gastric cancer, and relapsed SCLC. To-date, the breast cancer cohort (N=34) is the only group published and demonstrates grade 3 or worse toxicity in 32% of patients with anemia (12%), neutropenia (9%), and pancreatitis (6%) being the most common. Notably, 24/30 patients eligible for drug-activity analysis had disease control at 12 weeks, suggesting further studies are warranted. RNA-seq analysis using patient samples from this trial identified that olaparib treatment increased STING and IFN-1 pathway activity in 5/6 patients analyzed, and the patient who did not have elevated STING/IFN-1 pathway activity progressed quickly. The JAK-STAT pathway showed parallel modulation following olaparib treatment further suggesting pathway activation is a resistance mechanism to immune cell-mediated therapies123,124. This group also evaluated the olaparib-ICB combination in BRCA-deficient syngeneic mouse models. The combination not only demonstrated significant synergy, but also promoted the development immune memory cells. Combination treatment yielded more CRs (5/30 with IO alone vs 19/30 with PARPi-IO), and mice with CRs were then re-challenged with tumor cell implantation. All re-challenged animals showed initial tumor growth followed by complete and durable tumor rejection without additional treatments125.
DDRi side effects are generally tolerable; however, DDRi-associated lymphopenia predisposes patients to potentially life-threatening infections and white blood cell counts must be monitored while on therapy. T-cells undergo rigorous selection in the thymus, both positive (confirming they function) and negative (to prevent auto-immunity). This delicate process is susceptible to external perturbations, including PARPi, as thymocytes are dependent on PARP-2 function for normal development126. Models of individual PARP-1 or PARP-2 deficiency do not dramatically change peripheral T-cell numbers, but dual deficiency causes dramatic decrease in CD4+ and CD8+ T-cells, likely due to synthetic lethality. T-cells are susceptible to this insult as they proliferate in response to antigenic stimuli127. Patients treated with PARP inhibitors frequently harbor DDRi sensitizing germline mutations which also predisposes to toxicity.
CAR-Ts, TCR-Ts, and BiTEs: New Immunotherapy Strategies May Synergize with DDRi
Newer strategies to directly target tumor cell surface proteins using Bi-specific T-Cell Engager compounds (BiTEs) and cellular therapy approaches show significant promise (Figure 5). Most cellular therapies fall within two categories: Chimeric antigen receptor T-cells (CAR)-Ts and T-cell receptor engineered cells (TCR)-Ts. BiTE compounds serve as a highly specific T-cell honing mechanism, bringing together T-cells with cancer cells. They are composed of two antibody single chain variable fragments, one directed toward an antigen and the other toward a T-cell surface protein (typically CD3), connected by a linker protein128. Blinatumumab, a BiTE colocalizing CD19+ B-cells and CD3+ T-cells, has demonstrated remarkable efficacy in targeting B-cell leukemias/lymphomas resulting in FDA approval in 2018129. To date, there have been no subsequent BiTE approvals, suggesting that further optimization of this immunologic strategy may be needed. Areas of potential improvement include improved target selection, BiTE engineering, or combinations with ICB, STING pathway agonism, tumor-directed radiation, or other novel immune infiltration strategies. It should be noted that cellular therapies are also particularly efficacious in leukemias/lymphomas. An overall response rate of 71% has been estimated across hematologic malignancies treated with engineered T-cell therapies as compared to 26% across solid tumors. As approximately 90% of cancer-related mortality is driven by solid tumors, improving this discrepancy is critical to improving cancer outcomes130.
Figure 5: Existing and Novel Immuno-Oncology Strategies.
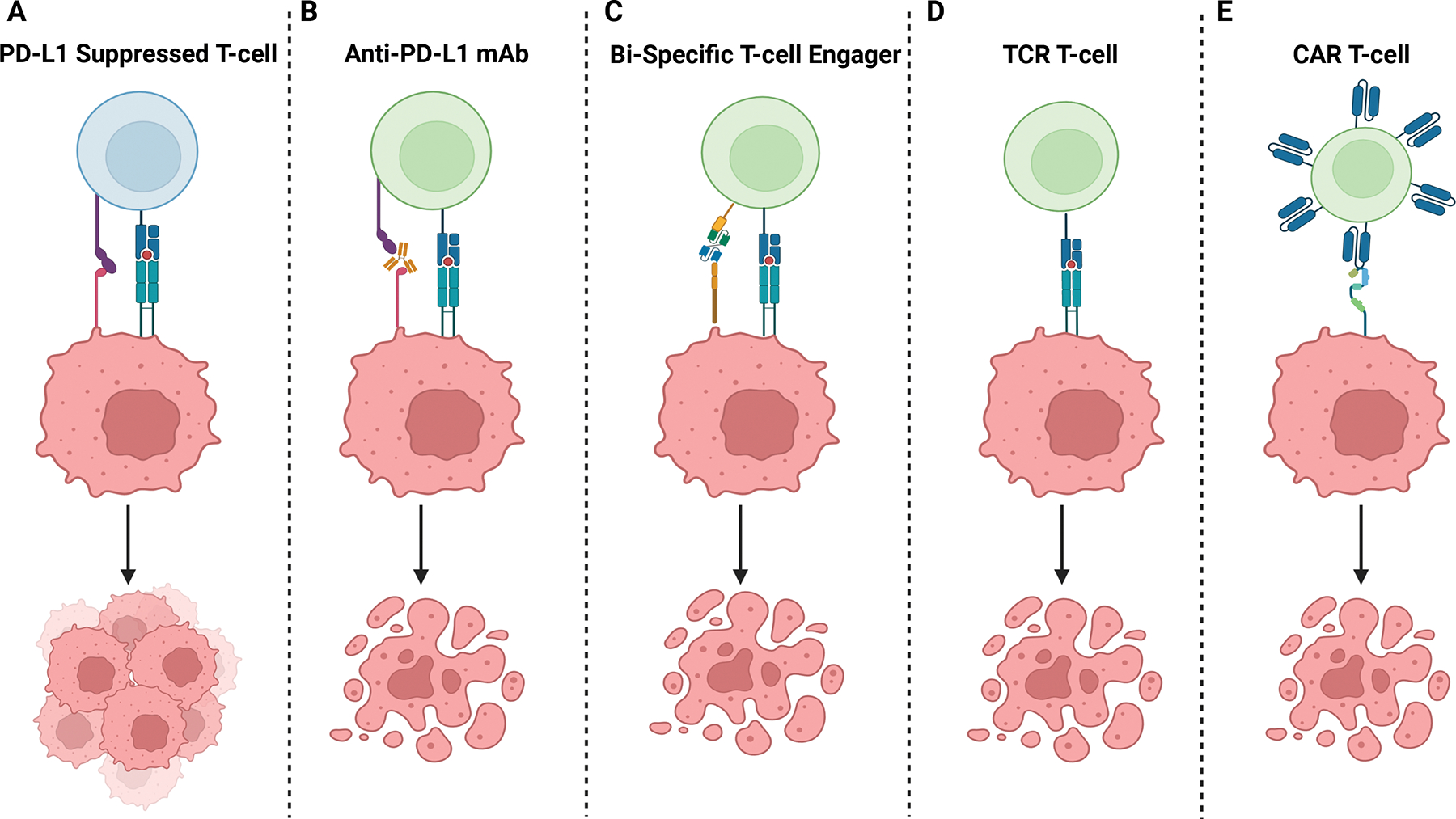
A) Tumors cells often upregulate inhibitor ligands like PD-L1 to suppress T-cell killing. B) Inhibitory ligand receptor-blocking antibodies, like anti-PD-L1 antibodies, promote immune cell activation and cell death. C) Bi-specific T-cell engager molecules (BiTEs) simultaneously bind a target (CD19 on B-cells for example) and effector cells (typically CD3 on T-cells) promoting co-localization and subsequent activation of tumor-targeting T-cells. D) Cancer-specific T-cells, either harvested and expanded or engineered (TCR T-cells), target tumor-associated antigens using a natural T-cell receptor reacting to antigenic peptides presented on MHC molecules. E) Chimeric antigen receptor (CAR) T-cells contain extracellular protein-specific (CD19 and BCMA for example) antibody-based extracellular domains which activate intracellular T-cell domains upon ligand binding.
Data supporting methods to potentiate cellular therapy is limited but do support DDRi or direct STING pathway agonism. Combining PARPi with anti-CD70 CAR T-cell therapy yielded 100% complete responses in mouse renal cell carcinoma (RCC) xenografts compared to 60% complete responses when treating with the CAR T-cell therapy alone. The combination PARPi-CAR-T yielded higher serum levels of interferons and CAR T-cell infiltration than either treatment alone; a cytokine effect that was abrogated with shRNA KO of STING in the RCC cells. Interestingly, CAR T-cells remained in circulation 15 days post combination PARPi-CAR T-cell treatment but were absent in the CART-cell alone group131. Similar work was done evaluating EGFR-targeting B57BL/6 mouse-derived CAR T-cells in EGFR+ breast cancer cells in vivo. Tumors treated with PARPi-CAR T-cell therapy resulted in significantly decreased tumor volumes, greater tumor infiltration of CAR T-cells, CD4+, and CD8+ cells, and higher CAR T-cell DNA copy-number in residual tumors after 10 days, as compared to PARPi or CAR T-cell therapy alone. PARPi also decreased recruitment of myeloid-derived suppressor cells, possibly through downregulation of the SDF1alpha/CXCR4 axis132. Neither of these studies reported severe toxicities associated with the combination, consistent with their non-overlapping toxicity profiles.
STINGa-CAR T-cell co-treatment is another area of novel development. Immunocompetent syngeneic mice with breast cancer treated with combination STINGa-CAR T-cell demonstrated decreased myeloid-derived suppressor cells, increased T-cell infiltration, an increase in immune-potentiating M1-like macrophages, and a decreased in immunosuppressive M2-like macrophage infiltration, as compared to single agent treatment. The addition of STINGa was also associated with increased persisting CAR T-cells at 7 days post treatment. Tumor growth was significantly reduced, and survival significantly improved with combination therapy compared to either treatment alone133. This mechanism is contracted by other models, including a BRCA1 deficient breast cancer syngeneic mouse model, which demonstrated that tumor-associated macrophages (TAMs) blunt PARPi efficacy in vivo and in vitro via pro-tumor polarization of TAMS, suppression of DNA damage, and impairment of STING-dependent anti-tumor immunity134. These data suggest that STING-mediated promotion of immune infiltration, by DDRi or STINGa, serves as a promising avenue of additional investigation to potentiate cellular therapies, but additional histology-specific studies are needed.
Emerging Biomarkers for DDRi/IO Combinations: SLFN11, “POLEness,” and STK11
There are several immune-related factors that positively correlate with ICB sensitivity. Those most widely studied include PD-L1 expression, gamma-interferon expression, MMRd, T-cell receptor diversity, neoantigen load, and TMB135,136. These factors contribute to enhanced novel peptide detection and subsequent immune cell activation in response to malignancy associated genomic alterations, but do not universally correlate with enhanced ICB activity. TMB has generated significant interest across tumor types as an independent determinant of IO efficacy with a 117 trial meta-analysis finding significantly increased response rates in TMB-high tumors (n=12,450) treated with ICB as compared to TMB-low137. However, this association only holds true for certain cancers—namely those in which neoantigen load and CD8+ T-cell infiltration is positively correlated like melanoma, lung, and bladder cancers. Breast cancer, prostate cancer, and glioma do not demonstrate improved ICB responses with increasing TMB, demonstrating that additional markers of response are needed138.
SLFN11:
Schlafen-11 (SLFN11; in German Schlafen means “sleeping”) has been identified as an important mediator of cellular fate and the immune-response when DNA-damage is present. While the ATR/CHK1 pathway works to bind damaged DNA-associated replication protein A (RPA), as a means of promoting DNA repair, SLFN11 binds to RPA and promotes cellular death139. Once bound to the stalled replication fork’s RPA tag, SLFN11 interacts with the replicative helicases minichromosome maintenance complex component 3 (MCM3) and DExH-box helicase 9 (DHX9). Upon binding, SLFN11 inhibits replication fork progression by opening chromatin ahead of the replication initiation sites, thus preventing replication fork progression and promoting cellular death. In the absence of SLFN11, stalled replication fork RPA will undergo DNA-repair via ATR/CHK1140. In addition to acting as a master determinant of DNA-damage-associated cellular fate, SLFN11 has been identified as one of the interferon (IFN)-stimulated genes promoting innate and adaptive immunity141.
Cell line and PDX models across multiple tumor types have demonstrated that SLFN11 expression predicts sensitivity to many conventional DNA-damaging agents (platinum agents, topoisomerase inhibitors, alkylating agents, and antimetabolites) and PARP inhibitors. SLFN11 also predicts treatment response across multiple tumor types, including hepatocellular carcinoma, colorectal cancer, breast cancer, and prostate cancer. High SLFN11 strongly correlates with improved response from DNA-damaging agents and PARP inhibitors, and is associated with increased TILs142–146. The ongoing SWOG1929 trial was subsequently opened to prospectively evaluate SLFN11 as a predictive marker conferring benefit of maintenance DDRi with standard of care maintenance immunotherapy. ES-SCLC patients whose tumors are positive for SLFN11 on IHC are randomized to receive maintenance atezolizumab alone per standard of care, or atezolizumab with the PARPi talazoparib.
Clinical associations between SLFN11 expression and improved overall treatment response have also been demonstrated in hepatocellular carcinoma, colorectal cancer, breast cancer, and prostate cancer. SLFN11 expression is predictive of treatment responses in these contexts, but typically is not independently prognostic of outcome147–149. A notable exception is that high SLFN11 was as an independent prognostic factor in patients with localized esophageal cancer treated with curative chemoradiation. However, overall numbers were low (N=73) and additional studies are needed150. In addition to tumor IHC staining for SLFN11, blood-based markers like cfDNA methylation and circulating tumor cell (CTC) analysis are actively being evaluated151.
”POLEness:”
Despite tremendous advancements in DNA sequencing techniques, there has been a paucity of genomic markers of ICB efficacy until recently. It has been known that MMRd tumors demonstrate increased sensitivity to IO-based treatments but specific genomic lesions predictive of responsiveness had not been elucidated until recently. Whole-genome sequencing on 22 MMRd tumors undergoing ICB therapy identified that increasing “POLEness” scores (tumors sharing mutation signatures associated with POLE-mutant cancers) were associated with increased responsiveness to ICB. Interestingly, no such associate was seen with “MMRdness,” although this was likely due to all patients in the cohort being MMRd, thus complicating the statistical analysis. This was the first mutational signature distinguishing ICB responders from non-responders and demonstrates that further investigation may be fruitful with eventual transition into peripheral-blood based assays152. A larger study evaluated 458 patients across numerous tumor types who harbored POLE mutations, with 121 patients having received a form of ICB. Patients who had received an ICB-only regimen (N=82) and had pathogenic POLE mutations demonstrated an improved median PFS of 15.1 vs 2.5 months (p<0.001) among those with benign POLE mutations. Additionally, those with pathogenic mutations had a median OS of 29.5 vs 6.8 months (P<0.001) among those with benign POLE mutations. The number of co-mutations was not associated with improved response to ICB, suggesting that a factor intrinsic to the POLE mutational profile sensitizes tumors to immunotherapy153.
STK11:
Many tumors demonstrate a high TMB phenotype yet often respond poorly to IO treatment strategies. For example, KRAS-mutant NSCLC commonly contains loss-of-function mutations in STK11 (also called LKB1), termed KL-tumors. The loss of STK11/LKB1 is strongly associated loss of PD-L1 and downregulation of STING through hyperactivation of DNMT1 and EZH2 with subsequent epigenetic silencing. This leads to diminished cytosolic dsDNA sensing and diminished immune activation through pIRF3 signaling; an effect which is abrogated upon LKB1 reconstitution. This work suggests that decreased STING expression through loss-of-function mutations in STK11/LKB1 is a putative resistance mechanism to IO therapies154. The growing appreciation for STING’s importance in immune-mediated therapies has led to the development of multiple STING agonists which are currently being evaluated in clinical trials155.
Summary and Future Directions:
Targeted DDRi drives cancer cell death by overwhelming cellular DNA damage responses. DDRi agents have shown greatest efficacy in a pan-cancer class of tumors harboring inactivating genetic alterations in DNA repair genes. New studies are extending these seminal discoveries by demonstrating that targeted DDRi agents have activity in tumors without known DNA repair defects. Beyond promoting tumor intrinsic cell death, extensive studies have shown that DDRi generates robust anti-tumor immune responses by engaging both the innate and adaptive immune system. These data have laid the groundwork for numerous ongoing clinical trials combining DDRi agents with immune checkpoint inhibitors targeting PD-1, PD-L1, and CTLA-4. Next generation therapies such as BiTEs, CAR-Ts, and TCR-Ts are generating promising results and offer new avenues to combine DDRi and immunotherapy for increased tumor control. Moving forward, rationally designed DDRi and immunotherapy combinations, along with application of emerging biomarkers, will help improve outcomes for patients with aggressive cancers.
There remain many limitations to DDRi-IO strategies and much is unknown regarding resistance mechanisms to these combinations. As novel therapies continue to progress into clinical trials, it is imperative that the field of cancer medicine adapts to subsequent novel resistance mechanisms. This will require maximally leveraging our current analytic modalities like whole-exome sequencing, single-cell RNAseq, tumor micro-environment profiling, and other tools; but it will also require re-evaluating the use of existing adjuvant modalities like radiation therapy in this new context. For example, ATR inhibition has been shown to significantly potentiate the STING pathway agonism caused by ionizing radiation through cGAS/STING in cell line models47. Novel delivery methods like antibody-conjugated radionucleotides are another example of this concept being well executed. Most trials that set the standards of radiation therapy preceded the immuno-oncology era, and it is well know that radiation induces immune responses, warranting further analysis. Several questions still exist about how to best leverage and build upon our existing knowledge (Box 1) and we are optimistic that future studies will continue to be fruitful.
Box 1. Outstanding Questions.
Why do many tumors that demonstrate increased genomic instability respond poorly to IO or targeted DDRi?
Newer DDRi agents are being tested/under development (POLQi, etc.), do they work in the clinic? How can we use them to pair with IO? Do they have a similar or different mechanism of action for engaging the immune system?
With numerous options for IO strategies (PD-L1/PD-1, CTLA-4, LAG-3/TIGIT, CAR T-cells, TCR T-cells, BiTEs, etc.), how do we prioritize clinical trials?
What are tumor-specific and tumor-agnostic non-germline genetic biomarkers (mutational signatures, copy number signatures) of response for DDRi or IO strategies?
What effect may the microbiome have on DDRi/IO sensitivity?
How should we integrate radiation therapy with these strategies?
The predecessors of immunotherapy, including chemotherapy and targeted therapy, were founded on initial groundbreaking discoveries followed by gradual incremental successes. Immunotherapy has followed this paradigm as well, but there remains a seemingly endless number of potential adaptations to existing and novel immuno-oncologic strategies. Harnessing the immunologic effects of increased DNA damage demonstrates one promising peak amidst a cascade of what will surely be greater peaks to come.
Special Acknowledgement:
Jordan Pietz – graphic designer assisting with Figures 1–4.
Dr. Lauren Byers serves as a consultant or in an advisory role for Merck Sharp & Dohme Corp., Arrowhead Pharmaceuticals, Chugai Pharma, AstraZeneca, Genentech Inc., AbbVie, BeiGene, and Jazz Pharmaceuticals, and receives research funding from AstraZeneca, Amgen, and Jazz Pharmaceuticals.
Dr. Carl Gay serves as a consultant or in an advisory role for G1 Therapeutics, AstraZeneca, BeiGene, Bristol Myers Squibb, Jazz Pharmaceuticals, and MonteRosa, and receives research funding from AstraZeneca.
Funding:
BBM is a TRIUMPH Fellow in the CPRIT Research Training Program (RP210028).
Footnotes
Dr. Kyle Concannon has no competing interests to declare.
Dr. Benjamin Morris has no competing interests to declare.
REFERENCES
- 1.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254 e236. 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 4.Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weller M, Tabatabai G, Kastner B, Felsberg J, Steinbach JP, Wick A, Schnell O, Hau P, Herrlinger U, Sabel MC, et al. (2015). MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin Cancer Res 21, 2057–2064. 10.1158/1078-0432.CCR-14-2737. [DOI] [PubMed] [Google Scholar]
- 6.Krokan HE, and Bjoras M (2013). Base excision repair. Cold Spring Harb Perspect Biol 5, a012583. 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scharer OD (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5, a012609. 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiricny J (2006). The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7, 335–346. 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 9.Yi C, and He C (2013). DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol 5, a012575. 10.1101/cshperspect.a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17, 337–349. 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 11.Black SJ, Ozdemir AY, Kashkina E, Kent T, Rusanov T, Ristic D, Shin Y, Suma A, Hoang T, Chandramouly G, et al. (2019). Molecular basis of microhomology-mediated end-joining by purified full-length Poltheta. Nat Commun 10, 4423. 10.1038/s41467-019-12272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krejci L, Altmannova V, Spirek M, and Zhao X (2012). Homologous recombination and its regulation. Nucleic Acids Res 40, 5795–5818. 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stinson BM, and Loparo JJ (2021). Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu Rev Biochem 90, 137–164. 10.1146/annurev-biochem-080320-110356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceccaldi R, Rondinelli B, and D’Andrea AD (2016). Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol 26, 52–64. 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ottaviani D, LeCain M, and Sheer D (2014). The role of microhomology in genomic structural variation. Trends Genet 30, 85–94. 10.1016/j.tig.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Yang W, and Gao Y (2018). Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem 87, 239–261. 10.1146/annurev-biochem-062917-012405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith J, Tho LM, Xu N, and Gillespie DA (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 108, 73–112. 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 18.Lanz MC, Dibitetto D, and Smolka MB (2019). DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J 38, e101801. 10.15252/embj.2019101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, and et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71. 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 20.Turner N, Tutt A, and Ashworth A (2004). Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4, 814–819. 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 21.Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, et al. (2013). Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 31, 1748–1757. 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, Foulkes WD, Armel S, Eisen A, Neuhausen SL, et al. (2012). The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 107, 2005–2009. 10.1038/bjc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O’Brien L, Sawyer E, et al. (2011). BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer 105, 1230–1234. 10.1038/bjc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, et al. (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317, 2402–2416. 10.1001/jama.2017.7112. [DOI] [PubMed] [Google Scholar]
- 25.Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, et al. (2013). Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 105, 812–822. 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- 26.Prakash R, Zhang Y, Feng W, and Jasin M (2015). Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7, a016600. 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. (2016). Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res 22, 3764–3773. 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, et al. (2020). Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 383, 2207–2218. 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 30.Ashworth A (2008). A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 26, 3785–3790. 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 31.Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18, 610–621. 10.1038/nrm.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murai J, and Pommier Y (2019). PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annual Review of Cancer Biology 3, 131–150. 10.1146/annurev-cancerbio-030518-055914. [DOI] [Google Scholar]
- 33.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599. 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33, 244–250. 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961. 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. (2019). Maintenance Olaparib for Germline. N Engl J Med 381, 317–327. 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. (2019). Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 381, 2391–2402. 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 38.Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D, et al. (2020). Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 383, 2345–2357. 10.1056/NEJMoa2022485. [DOI] [PubMed] [Google Scholar]
- 39.LaFargue CJ, Dal Molin GZ, Sood AK, and Coleman RL (2019). Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol 20, e15–e28. 10.1016/S1470-2045(18)30786-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. (2014). Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15, 852–861. 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 41.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, et al. (2018). Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 379, 2495–2505. 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 42.Poveda A, Floquet A, Ledermann JA, Asher R, Penson RT, Oza AM, Korach J, Huzarski T, Pignata S, Friedlander M, et al. (2021). Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 22, 620–631. 10.1016/S1470-2045(21)00073-5. [DOI] [PubMed] [Google Scholar]
- 43.Carney B, Kossatz S, Lok BH, Schneeberger V, Gangangari KK, Pillarsetty NVK, Weber WA, Rudin CM, Poirier JT, and Reiner T (2018). Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat Commun 9, 176. 10.1038/s41467-017-02096-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown JS, Kaye SB, and Yap TA (2016). PARP inhibitors: the race is on. Br J Cancer 114, 713–715. 10.1038/bjc.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liontos M, Terpos E, Markellos C, Zagouri F, Briasoulis A, Katsiana I, Skafida E, Fiste O, Kunadis E, Andrikopoulou A, et al. (2021). Immunological Response to COVID-19 Vaccination in Ovarian Cancer Patients Receiving PARP Inhibitors. Vaccines (Basel) 9. 10.3390/vaccines9101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaspers JE, Sol W, Kersbergen A, Schlicker A, Guyader C, Xu G, Wessels L, Borst P, Jonkers J, and Rottenberg S (2015). BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res 75, 732–741. 10.1158/0008-5472.CAN-14-0839. [DOI] [PubMed] [Google Scholar]
- 47.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, et al. (2018). Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 9, 1849. 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao H, Thienpont B, Yesilyurt BT, Moisse M, Reumers J, Coenegrachts L, Sagaert X, Schrauwen S, Smeets D, Matthijs G, et al. (2014). Mismatch repair deficiency endows tumors with a unique mutation signature and sensitivity to DNA double-strand breaks. Elife 3, e02725. 10.7554/eLife.02725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richman S (2015). Deficient mismatch repair: Read all about it (Review). Int J Oncol 47, 1189–1202. 10.3892/ijo.2015.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kato N, Kawasoe Y, Williams H, Coates E, Roy U, Shi Y, Beese LS, Scharer OD, Yan H, Gottesman ME, et al. (2017). Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway. Cell Rep 21, 1375–1385. 10.1016/j.celrep.2017.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng H, Zhang Z, Borczuk A, Powell CA, Balajee AS, Lieberman HB, and Halmos B (2013). PARP inhibition selectively increases sensitivity to cisplatin in ERCC1-low non-small cell lung cancer cells. Carcinogenesis 34, 739–749. 10.1093/carcin/bgs393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Henon C, Dorvault N, Rouanne M, et al. (2019). PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest 129, 1211–1228. 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Postel-Vinay S, Vanhecke E, Olaussen KA, Lord CJ, Ashworth A, and Soria JC (2012). The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat Rev Clin Oncol 9, 144–155. 10.1038/nrclinonc.2012.3. [DOI] [PubMed] [Google Scholar]
- 54.Pawlowski R, Muhl SM, Sulser T, Krek W, Moch H, and Schraml P (2013). Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int J Cancer 132, E11–17. 10.1002/ijc.27822. [DOI] [PubMed] [Google Scholar]
- 55.Kakarougkas A, Ismail A, Chambers AL, Riballo E, Herbert AD, Kunzel J, Lobrich M, Jeggo PA, and Downs JA (2014). Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell 55, 723–732. 10.1016/j.molcel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lalonde M, Trauner M, Werner M, and Hamperl S (2021). Consequences and Resolution of Transcription-Replication Conflicts. Life (Basel) 11. 10.3390/life11070637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, Garrido M, Meisenberg C, Lamb A, Ngo C, et al. (2021). PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res 81, 2888–2902. 10.1158/0008-5472.CAN-21-0628. [DOI] [PubMed] [Google Scholar]
- 58.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. (2011). Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43, 875–878. 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al. (2012). Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res 22, 2120–2129. 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, et al. (2015). ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov 5, 752–767. 10.1158/2159-8290.CD-14-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banerjee S, Stewart J, Porta N, Toms C, Leary A, Lheureux S, Khalique S, Tai J, Attygalle A, Vroobel K, et al. (2021). ATARI trial: ATR inhibitor in combination with olaparib in gynecological cancers with ARID1A loss or no loss (ENGOT/GYN1/NCRI). Int J Gynecol Cancer 31, 1471–1475. 10.1136/ijgc-2021-002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rafiei S, Fitzpatrick K, Liu D, Cai MY, Elmarakeby HA, Park J, Ricker C, Kochupurakkal BS, Choudhury AD, Hahn WC, et al. (2020). ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res 80, 2094–2100. 10.1158/0008-5472.CAN-19-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, et al. (2016). ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 127, 582–595. 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 65.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, and Kroemer G (2004). Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837. 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 66.Mohni KN, Kavanaugh GM, and Cortez D (2014). ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res 74, 2835–2845. 10.1158/0008-5472.CAN-13-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell 60, 351–361. 10.1016/j.molcel.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niraj J, Farkkila A, and D’Andrea AD (2019). The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478. 10.1146/annurev-cancerbio-030617-050422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen CC, Kennedy RD, Sidi S, Look AT, and D’Andrea A (2009). CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol Cancer 8, 24. 10.1186/1476-4598-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barnieh FM, Loadman PM, and Falconer RA (2021). Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr Res Pharmacol Drug Discov 2, 100017. 10.1016/j.crphar.2021.100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim R, Kwon M, An M, Kim ST, Smith SA, Loembé AB, Mortimer PGS, Armenia J, Lukashchuk N, Shah N, et al. (2022). Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann Oncol 33, 193–203. 10.1016/j.annonc.2021.10.009. [DOI] [PubMed] [Google Scholar]
- 72.Middleton MR, Dean E, Evans TRJ, Shapiro GI, Pollard J, Hendriks BS, Falk M, Diaz-Padilla I, and Plummer R (2021). Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br J Cancer 125, 510–519. 10.1038/s41416-021-01405-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plummer R, Dean E, Arkenau HT, Redfern C, Spira AI, Melear JM, Chung KY, Ferrer-Playan J, Goddemeier T, Locatelli G, et al. (2022). A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 163, 19–26. 10.1016/j.lungcan.2021.11.011. [DOI] [PubMed] [Google Scholar]
- 74.Thomas A, Redon CE, Sciuto L, Padiernos E, Ji J, Lee MJ, Yuno A, Lee S, Zhang Y, Tran L, et al. (2018). Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J Clin Oncol 36, 1594–1602. 10.1200/JCO.2017.76.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, et al. (2016). Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol 34, 1764–1771. 10.1200/JCO.2015.64.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong DS, Moore K, Patel M, Grant SC, Burris HA 3rd, William WN Jr., Jones S, Meric-Bernstam F, Infante J, Golden L, et al. (2018). Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin Cancer Res 24, 3263–3272. 10.1158/1078-0432.CCR-17-3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iwasa S, Yamamoto N, Shitara K, Tamura K, Matsubara N, Tajimi M, Lin AB, Asou H, Cai Z, Inoue K, et al. (2018). Dose-finding study of the checkpoint kinase 1 inhibitor, prexasertib, in Japanese patients with advanced solid tumors. Cancer Sci 109, 3216–3223. 10.1111/cas.13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Green DR, Ferguson T, Zitvogel L, and Kroemer G (2009). Immunogenic and tolerogenic cell death. Nat Rev Immunol 9, 353–363. 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kroemer G, Galluzzi L, Kepp O, and Zitvogel L (2013). Immunogenic cell death in cancer therapy. Annu Rev Immunol 31, 51–72. 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 80.Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, and Bhardwaj N (1998). Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med 188, 1359–1368. 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hopfner KP, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21, 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 82.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kwon J, and Bakhoum SF (2020). The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 10, 26–39. 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, and Taguchi T (2016). Activation of STING requires palmitoylation at the Golgi. Nat Commun 7, 11932. 10.1038/ncomms11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang C, Shang G, Gui X, Zhang X, Bai XC, and Chen ZJ (2019). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. 10.1038/s41586-019-1000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reisländer T, Groelly FJ, and Tarsounas M (2020). DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell 80, 21–28. 10.1016/j.molcel.2020.07.026. [DOI] [PubMed] [Google Scholar]
- 88.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763.e754. 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, Bolt MJ, Arina A, Parry R, Kron SJ, et al. (2019). STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res 79, 1465–1479. 10.1158/0008-5472.CAN-18-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Erdal E, Haider S, Rehwinkel J, Harris AL, and McHugh PJ (2017). A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev 31, 353–369. 10.1101/gad.289769.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299. 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gasser S, Orsulic S, Brown EJ, and Raulet DH (2005). The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190. 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, and Raulet DH (2002). The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 17, 19–29. 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 94.Nausch N, and Cerwenka A (2008). NKG2D ligands in tumor immunity. Oncogene 27, 5944–5958. 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 95.Lam AR, Bert NL, Ho SS, Shen YJ, Tang LF, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, et al. (2014). RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 74, 2193–2203. 10.1158/0008-5472.CAN-13-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reisländer T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, Wright B, Lockstone H, Biroccio A, Harris A, et al. (2019). BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun 10, 3143. 10.1038/s41467-019-11048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, Feng N, Vellano CP, Federico L, Marszalek JR, et al. (2019). Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep 9, 1853. 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP Inhibitor Efficacy Depends on CD8. Cancer Discov 9, 722–737. 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. (2018). PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 25, 2972–2980.e2975. 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, and Peng G (2019). PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 79, 311–319. 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711–723. 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, and Wolchok JD (2015). Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 33, 1889–1894. 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O’Shea MA, and Fauci AS (2008). The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol 181, 6738–6746. 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 104.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF (2013). Upregulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5, 200ra116. 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, et al. (2014). Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384, 1109–1117. 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 106.Hamid O, Puzanov I, Dummer R, Schachter J, Daud A, Schadendorf D, Blank C, Cranmer LD, Robert C, Pavlick AC, et al. (2017). Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer 86, 37–45. 10.1016/j.ejca.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 107.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018–2028. 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 108.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, Geva R, Gottfried M, Penel N, Hansen AR, et al. (2020). Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 38, 1–10. 10.1200/JCO.19.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, El Dika IH, Segal N, Shcherba M, Sugarman R, et al. (2022). PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N Engl J Med 386, 2363–2376. 10.1056/NEJMoa2201445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, Mariotto AB, Lowy DR, and Feuer EJ (2020). The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N Engl J Med 383, 640–649. 10.1056/NEJMoa1916623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haslam A, and Prasad V (2019). Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535. 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, LoRusso P, Jurgensmeier J, Avedissian M, Sklar J, et al. (2021). Ceralasertib-Mediated ATR Inhibition Combined With Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis Oncol 5. 10.1200/PO.20.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP, Morgan M, Burger RA, Haggerty A, Zarrin H, et al. (2021). Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol 163, 246–253. 10.1016/j.ygyno.2021.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A, Quinn N, Gannon C, Vuong L, Parmar K, et al. (2021). Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin Cancer Res 27, 4710–4716. 10.1158/1078-0432.CCR-21-1279. [DOI] [PubMed] [Google Scholar]
- 115.Zimmermann M, Bernier C, Kaiser B, Fournier S, Li L, Desjardins J, Skeldon A, Rimkunas V, Veloso A, Young JTF, et al. (2022). Guiding ATR and PARP inhibitor combinationswith chemogenomic screens. Cell Rep 40, 111081. 10.1016/j.celrep.2022.111081. [DOI] [PubMed] [Google Scholar]
- 116.Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al. (2019). Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38, 2451–2463. 10.1038/s41388-018-0606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. (2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 9, 646–661. 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, Gay CM, Stewart CA, Fan Y, Shen L, et al. (2019). Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J Thorac Oncol 14, 2152–2163. 10.1016/j.jtho.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tang Z, Pilié PG, Geng C, Manyam GC, Yang G, Park S, Wang D, Peng S, Wu C, Peng G, et al. (2021). ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin Cancer Res 27, 4898–4909. 10.1158/1078-0432.CCR-21-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thomas A, Vilimas R, Trindade C, Erwin-Cohen R, Roper N, Xi L, Krishnasamy V, Levy E, Mammen A, Nichols S, et al. (2019). Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J Thorac Oncol 14, 1447–1457. 10.1016/j.jtho.2019.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, String-Reasor E, Boughey JC, Chien AJ, Elias AD, et al. (2021). Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e985. 10.1016/j.ccell.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, Chien AJ, Forero-Torres A, Ellis E, Han H, et al. (2020). Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol 6, 676–684. 10.1001/jamaoncol.2019.6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, Alexandre J, You B, Bastian S, Krebs MG, et al. (2020). Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol 21, 1155–1164. 10.1016/S1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 124.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 7, 188–201. 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Staniszewska AD, Armenia J, King M, Michaloglou C, Reddy A, Singh M, San Martin M, Prickett L, Wilson Z, Proia T, et al. (2022). PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. Oncoimmunology 11, 2083755. 10.1080/2162402X.2022.2083755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nicolas L, Martinez C, Baro C, Rodriguez M, Baroja-Mazo A, Sole F, Flores JM, Ampurdanes C, Dantzer F, Martin-Caballero J, et al. (2010). Loss of poly(ADP-ribose) polymerase-2 leads to rapid development of spontaneous T-cell lymphomas in p53-deficient mice. Oncogene 29, 2877–2883. 10.1038/onc.2010.11. [DOI] [PubMed] [Google Scholar]
- 127.Navarro J, Gozalbo-Lopez B, Mendez AC, Dantzer F, Schreiber V, Martinez C, Arana DM, Farres J, Revilla-Nuin B, Bueno MF, et al. (2017). PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci Rep 7, 41962. 10.1038/srep41962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goebeler ME, and Bargou RC (2020). T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol 17, 418–434. 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 129.Gökbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, Diedrich H, Topp MS, Brüggemann M, Horst HA, et al. (2018). Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 131, 1522–1531. 10.1182/blood-2017-08-798322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Britten CM, Shalabi A, and Hoos A (2021). Industrializing engineered autologous T cells as medicines for solid tumours. Nat Rev Drug Discov 20, 476–488. 10.1038/s41573-021-00175-8. [DOI] [PubMed] [Google Scholar]
- 131.Ji F, Zhang F, Zhang M, Long K, Xia M, Lu F, Li E, Chen J, Li J, Chen Z, et al. (2021). Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J Hematol Oncol 14, 152. 10.1186/s13045-021-01168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sun R, Luo H, Su J, Di S, Zhou M, Shi B, Sun Y, Du G, Zhang H, Jiang H, and Li Z (2021). Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol Ther 29, 60–74. 10.1016/j.ymthe.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, Willis C, Dotti G, Vincent BG, Restifo NP, and Serody JS (2021). STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 218. 10.1084/jem.20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Q, Bergholz JS, Ding L, Lin Z, Kabraji SK, Hughes ME, He X, Xie S, Jiang T, Wang W, et al. (2022). STING agonism reprograms tumor-associated macrophages and overcomes resistance to PARP inhibition in BRCA1-deficient models of breast cancer. Nat Commun 13, 3022. 10.1038/s41467-022-30568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li X, Song W, Shao C, Shi Y, and Han W (2019). Emerging predictors of the response to the blockade of immune checkpoints in cancer therapy. Cell Mol Immunol 16, 28–39. 10.1038/s41423-018-0086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Osipov A, Lim SJ, Popovic A, Azad NS, Laheru DA, Zheng L, Jaffee EM, Wang H, and Yarchoan M (2020). Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-regression Analysis. Clin Cancer Res 26, 4842–4851. 10.1158/1078-0432.CCR-20-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, Jonasch E, Khasraw M, Heimberger AB, Lim B, et al. (2021). High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol 32, 661–672. 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang B, Ramkumar K, Cardnell RJ, Gay CM, Stewart CA, Wang WL, Fujimoto J, Wistuba II, and Byers LA (2021). A wake-up call for cancer DNA damage: the role of Schlafen 11 (SLFN11) across multiple cancers. Br J Cancer 125, 1333–1340. 10.1038/s41416-021-01476-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, Al Abo M, Rajapakse VN, Nakamura E, Jenkins LMM, et al. (2018). SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol Cell 69, 371–384.e376. 10.1016/j.molcel.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mavrommatis E, Fish EN, and Platanias LC (2013). The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res 33, 206–210. 10.1089/jir.2012.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Byers LA, Bentsion D, Gans S, Penkov K, Son C, Sibille A, Owonikoko TK, Groen HJM, Gay CM, Fujimoto J, et al. (2021). Veliparib in Combination with Carboplatin and Etoposide in Patients with Treatment-Naïve Extensive-Stage Small Cell Lung Cancer: A Phase 2 Randomized Study. Clin Cancer Res 27, 3884–3895. 10.1158/1078-0432.CCR-20-4259. [DOI] [PubMed] [Google Scholar]
- 143.Deng Y, Cai Y, Huang Y, Yang Z, Bai Y, Liu Y, Deng X, and Wang J (2015). High SLFN11 expression predicts better survival for patients with KRAS exon 2 wild type colorectal cancer after treated with adjuvant oxaliplatin-based treatment. BMC Cancer 15, 833. 10.1186/s12885-015-1840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Isnaldi E, Ferraioli D, Ferrando L, Brohée S, Ferrando F, Fregatti P, Bedognetti D, Ballestrero A, and Zoppoli G (2019). Schlafen-11 expression is associated with immune signatures and basal-like phenotype in breast cancer. Breast Cancer Res Treat 177, 335–343. 10.1007/s10549-019-05313-w. [DOI] [PubMed] [Google Scholar]
- 145.Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, Owonikoko TK, Woo KM, Cardnell RJ, Fujimoto J, et al. (2018). Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol 36, 2386–2394. 10.1200/JCO.2018.77.7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Winkler C, Armenia J, Jones GN, Tobalina L, Sale MJ, Petreus T, Baird T, Serra V, Wang AT, Lau A, et al. (2021). SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br J Cancer 124, 951–962. 10.1038/s41416-020-01199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Conteduca V, Ku SY, Puca L, Slade M, Fernandez L, Hess J, Bareja R, Vlachostergios PJ, Sigouros M, Mosquera JM, et al. (2020). SLFN11 Expression in Advanced Prostate Cancer and Response to Platinum-based Chemotherapy. Mol Cancer Ther 19, 1157–1164. 10.1158/1535-7163.MCT-19-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coussy F, El-Botty R, Château-Joubert S, Dahmani A, Montaudon E, Leboucher S, Morisset L, Painsec P, Sourd L, Huguet L, et al. (2020). BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci Transl Med 12. 10.1126/scitranslmed.aax2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.He T, Zhang M, Zheng R, Zheng S, Linghu E, Herman JG, and Guo M (2017). Methylation of SLFN11 is a marker of poor prognosis and cisplatin resistance in colorectal cancer. Epigenomics 9, 849–862. 10.2217/epi-2017-0019. [DOI] [PubMed] [Google Scholar]
- 150.Kagami T, Yamade M, Suzuki T, Uotani T, Tani S, Hamaya Y, Iwaizumi M, Osawa S, Sugimoto K, Miyajima H, et al. (2020). The first evidence for SLFN11 expression as an independent prognostic factor for patients with esophageal cancer after chemoradiotherapy. BMC Cancer 20, 1123. 10.1186/s12885-020-07574-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tserpeli V, Stergiopoulou D, Londra D, Giannopoulou L, Buderath P, Balgkouranidou I, Xenidis N, Grech C, Obermayr E, Zeillinger R, et al. (2021). Prognostic Significance of. Cancers (Basel) 14. 10.3390/cancers14010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chung J, Maruvka YE, Sudhaman S, Kelly J, Haradhvala NJ, Bianchi V, Edwards M, Forster VJ, Nunes NM, Galati MA, et al. (2021). DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov 11, 1176–1191. 10.1158/2159-8290.CD-20-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Garmezy B, Gheeya J, Lin HY, Huang Y, Kim T, Jiang X, Thein KZ, Pilié PG, Zeineddine F, Wang W, et al. (2022). Clinical and Molecular Characterization of. JCO Precis Oncol 6, e2100267. 10.1200/PO.21.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, Tange S, Mitsuishi Y, Thai TC, Masuda S, et al. (2019). Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 9, 34–45. 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, and Kroemer G (2020). Trial watch: STING agonists in cancer therapy. Oncoimmunology 9, 1777624. 10.1080/2162402X.2020.1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]