

Abstract
Myocardial hypoxia is the low oxygen tension in the heart tissue implicated in many diseases, including ischemia, cardiac dysfunction, or after heart procurement for transplantation. Oxygen-generating microparticles have recently emerged as a potential strategy for supplying oxygen to sustain cell survival, growth, and tissue functionality in hypoxia. Here, we prepared oxygen-generating microparticles with poly D, L-lactic-co-glycolic acid, and calcium peroxide (CPO), which yielded a continuous morphology capable of sustained oxygen release for up to 24 hrs. We demonstrated that CPO-microparticles increased primary rat cardiomyocyte metabolic activity while not affecting cell viability during hypoxia. Moreover, hypoxia-inducible factor (HIF)-1α, which is upregulated during hypoxia, can be downregulated by delivering oxygen using CPO microparticles. Single-cell traction force microscopy data demonstrated that the reduced energy generated by the hypoxic cells could be restored using CPO microparticles. We engineered cardiac tissues that showed higher contractility in the presence of CPO microparticles compared to hypoxic cells. Finally, we observed reduced myocardial injuries in ex vivo rabbit hearts treated with CPO microparticles. In contrast, an acute early myocardial injury was observed for the hearts treated with control saline solution in hypoxia. In conclusion, CPO microparticles improve cell and tissue contractility and gene expression while reducing hypoxia-induced myocardial injuries in the heart.
Keywords: Oxygen releasing microparticles, hypoxia, Traction force microscopy, organ-on-a-chip, heart transplant
Graphical Abstract
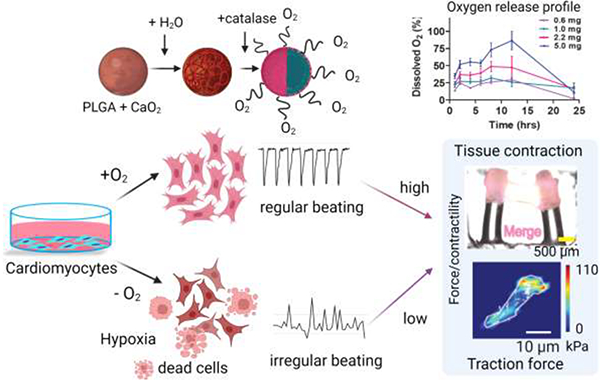
1. Introduction:
Hypoxia is one of the major reasons for tissue damage caused by the limited oxygen (O2) supply resulting from vascular network destruction. Recent studies have demonstrated the critical role of hypoxia in many diseases, including myocardial ischemia, pulmonary hypertension, or cardiomyopathy.[1, 2] The treatment of ischemic heart disease remains largely elusive.[1] Besides, hypoxia has been a significant challenge for engineering tissue constructs, especially in large tissue scaffolds (>1 mm) that create a hypoxic core when implanted in vivo.[3–5] Moreover, before heart transplantation, the procured heart undergoes ischemia if the time window is more than 4 to 6 hrs. The ischemic reperfusion injuries of heart allografts contribute to short and long-term adverse effects that lead to primary graft dysfunction and rejection.[6–8] Heart preservation time can be prolonged by using a perfusion machine that supplies oxygen and removes toxic metabolites to reduce ischemic injuries to the donor’s heart.[9] As a result, machine perfusion improves ventricular function and minimizes the risk of cardiac allograft vasculopathy after transplantation. However, conventional methods cannot supply an adequate amount of oxygen as the solubility of gaseous O2 in the cold (4 – 8 °C) organ preservation solution is low. Even with the maximum infusion rate, the supplied oxygen amount was 8% of the total amount of oxygen required for an adult.[2, 10]
Ischemia/reperfusion injuries exhibit functional dysregulation at the single-cell level, which is characterized by myocardial apoptosis, sudden generation of reactive oxygen species (ROS), or adenosine triphosphate (ATP) depletion.[11–14] For example, hypoxia induces cell cycle arrest in postnatal cardiomyocytes (CMs) by increasing mitochondrial oxidative phosphorylation.[1, 15] Mechanistically, the production of free radicals increases with low oxygen levels leading to an increase in DNA damage.[15] As a result of hypoxia, many signaling pathways are altered, including the hypoxia-inducible factor (HIF) 1 and 2, which regulates the oxygen homeostasis in the cell.[16] In normoxia, HIF-1α is unstable and degrades quickly by hydroxylation, acetylation, and post-transcriptional activity. On the contrary, hypoxia decreases the degradation of HIF-1α, resulting in its accumulation, increasing its stability, and regulating its transcriptional activity.[17] In hypoxia, HIF-1α is upregulated and decreases apoptosis by reducing ROS generation leading to a decrease in DNA damage.[18] Cardiac troponin (cTn) level in the blood is another biomarker of cardiac tissue damage, and cTn is elevated after MI.[16, 19] In addition, cTn increases after orthotopic heart transplantation and is used as a biomarker to assess myocardial cell damage.[20] Liu et al. used a newborn pig model and showed that cTn levels increased 6 hrs after the onset of hypoxic insult in hypoxic-ischemic encephalopathy (HIE), which occurs due to a severe lack of oxygen during birth.[21]
Severe hypoxia also affects cell contractility. It has been previously shown that hypoxia reduces tissue contractility and adaption of skeletal muscle (i.e., increase in muscle fiber diameter), which occurs in hypertrophy.[22] Mechanical regulation in the heart is vital for proper beating, and cellular contractility.[12, 23] In many cardiac diseases, including cardiac ischemia and hypertrophy, cardiac contractility and beating rhythms are often dysregulated, which affects the heart’s ability to pump blood efficiently.[24, 25] Using neonatal rat CMs, Kumuro and Yazaki et al. showed that a short-term stretch of cardiac tissue could improve cardiac contractile function.[24, 26, 27] Moreover, this mechanical stretch induces genomic reprogramming leading to activation of stressed-activated protein kinase, protein kinase C, and mitogen-activated protein, which restore cardiac function.[27] The cardiac tissue partially recovers its contractile function by providing antioxidants that act as superoxide scavengers.[22] However, the mechanism is not fully understood and remains to be investigated. Traditionally, 2D models have been used to assess functional phenotypes of cardiac cells; however, recently, more sophisticated 3D microphysiological models that better mimic the in vivo physiology have been increasingly used to assess cellular contractility.[28–31]
In an earlier study, free oxygen gas was infused into the human bloodstream, resulting in pulmonary emboli.[2] Moreover, there is a serious limitation in supplying a sufficient amount of gaseous oxygen, as it increases perfusion pressure.[32] To address these problems, in recent years, many oxygen-releasing carriers have emerged to deliver oxygen in the bloodstream and improve tissue functionality, showing potential use in vivo.[5, 33, 34]. We previously reported oxygen-generating 3D hydrogels that improved cardiac cell function in hypoxia by incorporating free CPO in cell-laden gelatin methacryloyl (GelMA) hydrogels.[35] Different compounds have been used as oxygen-releasing agents, including calcium peroxide (CaO2), hydrogen peroxide (H2O2), magnesium peroxide (MgO2), and sodium percarbonate (Na2CO3).[36, 37] These compounds are encapsulated in biodegradable polymers or lipids to form microparticles for sustained O2 release to improve tissue regeneration capacity, stem cell differentiation, or tissue functionality in implanted tissues, such as hearts or lungs.[37–39] For example, Poly D,L-lactic-co-Glycolic Acid (PLGA), poly-caprolactone (PCL), and poly(N-vinylpyrrolidone) (PVP) are used for encapsulation due to their biodegradable and biocompatible properties.[40] By varying the polymer composition, molecular weight, and the amount of O2-releasing agent, O2 release can be maintained for up to 3 to 20 days.[33, 41] CaO2 is the most commonly used O2-releasing agent due to its sustained release profile when encapsulated; however, it also increases the pH of the aqueous solvent in which it is dissolved.[37, 42] Recently, lipid-based O2 carriers have been used to load O2 gas and release O2 in the blood under a hypoxic condition.[43] However, the fragility of lipid membranes raises the risk of breakage. Thus, it may result in a sudden release of O2, which is cytotoxic.[44] Therefore, biomaterials used for delivering O2 should be carefully selected depending on their biocompatibility, biodegradability, and, most importantly, the biological application to release the desired amount of O2 with a targeted spatiotemporal profile.[45]
In this work, we prepared O2-releasing microparticles using PLGA as the biopolymer and CaO2 as the O2-releasing agent to achieve the sustained release of O2 that shows a potential application in reducing ischemic injury during heart transplantation. Neonatal rat CMs were subjected to hypoxia to model the ischemic injury and to study the effect of CPO microparticles on single cells and tissue-level functions. The alteration of the HIF-1α signaling pathway when cells are subjected to hypoxic insult and after O2 treatment was tested by performing western blot and quantitative real-time (qRT)-PCR and immunostaining. We showed the effect of CPO microparticles on cell contractility using traction force microscopy and micropillar deflection assays, potentially paving the way to improve cardiac functional activity, contractility, and force generation in hypoxia. Finally, we investigated the effect of a modified organ preservation solution formulation with CPO microparticles versus standard saline control using rabbit heart to model ex vivo cardiac ischemic injury after procurement before heart transplantation.
2. Materials and Methods
2.1. Oxygen-releasing microparticle synthesis
Microparticles were synthesized using CaO2 (75%, −200 mesh, Mw-72.08 (Sigma-Aldrich)) as an O2-releasing agent. Three types of microparticles were formulated using CaO2 concentration of 9% (CPO1), 20% (CPO2), or 33% (CPO3) (w/w) in PLGA (Resomer® RG 502 H, Mw- 7000 – 17000 (Sigma-Aldrich)). In addition, PLGA microparticles were synthesized in similar ways without adding CaO2. An emulsion-based method was used to prepare CPO microparticles comprised of two steps. In the first step, an oil bath was prepared using 2% (w/v) span80 (Sigma Aldrich) in 100 ml cottonseed oil (Sigma Aldrich) by stirring the mixture at 400 rpm for 1 hr. at room temperature (RT).[39] [46] In the second step, the respective amount of CaO2 (for CPO1-100 mg, CPO2-250 mg, or CPO3-500 mg) and PLGA (1 g) were dissolved in an 8 ml acetonitrile (Sigma-Aldrich) solution. Subsequently, the solution was probe sonicated (QSonica Sconicator, Microtip Probes, for Q500/Q700 Sconicators) at an intensity of 50% and with 30 seconds on and 5 seconds off cycle, or constant sonication for 10 mins. The solution was immediately added to the oil bath dropwise. The resultant solution was stirred using a magnetic stirring plate for another 20 mins. Then 100 ml of a hexane solution was added and stirred for 1 hr to generate white microparticles. Lastly, microparticles were purified by centrifugation (Beckman Coulter) using hexane for 5 mins at 800 rpm and dried using a vacuum desiccator for 2 days before use.
2.2. Characterization of CPO microparticles
SEM imaging:
The morphology of the different formulations of CPO microparticles was characterized by Scanning Electron Microscopy (ZEISS Supra 40VP S.E.M., Germany). Dry CPO microparticles were attached on a silver stub using a conducting tape, followed by a gold coating using a sputter coater. Particle size distributions were obtained using Image J analysis. The internal structure of microparticles was investigated in two different ways. Microparticles were paraffinized and sectioned. Sectioned slices were mounted on the sliver stab using conducting tape. Second, microparticles were frozen in liquid nitrogen and broken. Broken microparticles were mounted a similar way on the sliver stub and prepared for SEM.[46]
2.3. Zeta potential Measurement
Particles were dissolved in water at a 1 mg/ml concentration. Zeta potential was measured using Malvern Analytical Zetasizer.
2.4. Determining CaO2 encapsulation:
2.4.1. Alizarin Red S
Particles were stained with 2% Alizarin Red S in Di water, and pH was adjusted to 4.2. After 5 mins of incubation, particles were imaged under the microscope (Keyence). The percentage of calcium loaded in the microparticles was characterized using Alizarin Red S (Sigma-Aldrich) staining. Briefly, we dissolved 1 mg of microparticles in 1 ml ethyl acetate (Sigma Aldrich) for 30 mins. Next, the microparticles were sonicated for 5 mins at 50 °C following 3 mins of vortexing. The solution was centrifuged at 13,000 rpm for 30 mins, and the microparticle pellet was dried after removing the supernatant. Afterward, 2% (w/v) Alizarin Red S solution was used to stain the microparticles for 10 mins at RT in the dark. Finally, we collected the precipitation, followed by washing and dissolving steps, as described before.[41, 47] The absorbance was measured (405 nm and 570nm) using a plate reader (Thermo Scientific, VARIOSKAN LUX). The amount of calcium was quantified using a standard curve obtained from the absorbance of CPO powders with known concentrations (0.1 mg to 1 mg/ml). The efficiency of encapsulated CaO2 in CPO microparticles was determined using the following:
2.4.2. ICP-MS
The amount of calcium encapsulated in microparticles was measured by Inductively coupled plasma mass spectrometry (ICP-MS). First, 1 mg of microparticles were digested with 200 μl of aqua regia for 4 hrs. Next, each digested sample was diluted to 1 ml with Millipore water, and the resultant solution was subjected to ICP-MS analysis. Each sample was tested in triplicate measurements with background correction.
2.5. Characterization of Oxygen release profile
The oxygen release profiles were obtained for all CPO microparticles (CPO1-3) formulation for 24 hrs., using the oxygen probe ISO-OXY-2 Oxygen Sensor-2mm (World Precision Instruments). Solutions with CPO microparticles were prepared in 1X PBS (Gibco ™) with 0.05% (w/v) catalase from bovine liver (Sigma Aldrich). 0.6, 1, 2.2, and 5 mg/ml were used to obtain the O2 release profile. A hypoxic chamber (Billups Rothenberg, Inc) was used to maintain the microenvironment in hypoxia containing 1% O2, 5% CO2, and 94% nitrogen (N2). Voltage values for the sensor were obtained for known oxygen values dissolved in PBS for calibration.
2.5. Rat cardiomyocyte isolation:
Primary CMs were isolated freshly from Charles River Sprague Dawley neonatal rat pups (24–48-hrs old). Neonatal pups were collected and then euthanized by decapitation without anesthesia. The hearts were excised with scissors, and ventricles were collected and rinsed briefly in ADS buffer. Using an enzyme digestion solution containing 0.02% w/v collagenase II (Worthington) and 0.06% (w/v) pancreatin (Sigma-Aldrich), the harvested hearts were digested. Next, neonatal calf Serum (NSC) was added to the enzyme solution to deactivate the enzymatic activity. Then cells were then centrifuged for 3 mins at 2200 rpm. The above process was repeated 4X times to ensure the complete extraction of cells, subsequently suspended in Percoll (Pharmacia) gradients to separate myocytes.
2.6. Cell culture
Initially, primary CMs were cultured in collagen type I (corning) -coated plates at 37°C and 5% CO2 in RPMI 1640 (Cytiva) with 10% (v/v) Fetal Bovine Serum (FBS) (Life Technologies), 1% (v/v) penicillin-streptomycin (Caisson Labs), 1% (v/v) B-27 supplement (Thermo Scientific), and 1% (v/v) insulin (Corning). HUVEC cells were maintained in Endothelial Cell Growth Medium, including basal medium and Supplement Mix (Promo Cell). For all cell experiments, cells were cultured for 48 hrs at 37°C and 5% CO2 before transferring into the hypoxic chamber filled with 1% O2, 5% CO2, and 94% N2 for 48 hrs. Dissolved O2 in the media was removed by purging with nitrogen gas before adding it to cells and incubated in a hypoxic chamber for 48 hrs. Next, CPO microparticles were added into cells using Transwell inserts with fresh media containing 0.2 mM HEPES buffer and catalase (0.1%). Cells were incubated overnight with CPO microparticles, and separate experiments were performed. Since a hypoxic chamber was used and oxygen was removed from the medium before adding it to cells, the O2 available to cells is due to CPO microparticles when added to the media. And the amount of O2 can be obtained at any time from the O2 release kinetics experiment.
CMs were plated at 2 × 105 cells/well in 24 well plates for cytotoxicity experiments. The control dish was left in hypoxia, and CPO microparticles of concentration 1 or 2 mg/ml were added.
2.7. Cardiomyocyte beating assay
CMs were cultured on collagen-coated 6 well plates (Corning) at a density of 1.5 ×106 cells/well to create a monolayer for 2D tissue beating assays for 48 hrs. Similarly, followed by hypoxic treatments for 48 hrs, CPO microparticles of concentration 1 mg/ml were added into the wells and incubated overnight. Times lapse images were acquired to capture the beating of the monolayer at different positions.
2.8. Cell viability and metabolic activity assay
Cells were cultured in well plates washed with PBS and then stained with the LIVE/DEAD viability/cytotoxicity kit (Life Technologies) by incubating with 2 μM ethidium homodimer-1 and 2 μM calcein-AM (diluted in PBS) for 45 mins at RT in the dark. Images of cells were acquired using a Keyence fluorescence microscope (BZ-X700 Series) with 4X or 10X objectives. A metabolic activity assay was performed using the PrestoBlue cell viability reagent (Molecular Probes, Invitrogen, reference). Briefly, the old culture medium was removed and washed with PBS for each well. A fresh complete culture medium was prepared with 10% (v/v) of PrestoBlue added to the well, followed by a 60-min incubation at 37 °C and 5% CO2. The PrestoBlue-containing media were transferred into 96-well plates for fluorescence measurement via a plate reader (excitation at 560 nm and emission at 590 nm).
2.9. Immunostaining
Cells were grown on a glass coverslip for immunostaining and then provided with hypoxic treatment or the addition of CPO microparticles, as described in the cell culture section. Paraformaldehyde (PFA) (Sigma Aldrich) 4% in PBS for 15 mins was used to fix the cells, followed by permeabilization with 0.05% (v/v) Triton X-100 and blocking with 1% (w/v) BSA (Thermo Scientific) in PBS for 30 mins, all at RT. Next, fixed cells were stained with primary antibodies: Cardiac Troponin T Monoclonal Antibody (13-11), Mouse / IgG1 (Thermo Scientific), and hypoxia-inducible factor 1 (HIF-1α) antibody (H1Alpha67) (Novus Biologicals) for 1 hr. at RT. Following PBS washes, samples were incubated with phalloidin, DAPI, and goat anti-mouse secondary antibodies conjugated (Thermo Scientific) with either Alexa 594 or Alexa 488 (as applicable) for HIF-1α or cTn for 1 hr at RT. After washing with PBS, coverslips were mounted, and fluorescent images were acquired using a Keyence microscope with complementary Image software.
2.10. Microscopy and image analysis:
Cells were imaged either with an inverted fluorescence microscope (Keyence) or a confocal microscope (Carl Zeiss) equipped with 4X, 10X, 20X, and 40X objectives. The lasers Ar and HeNe were used with excitation 458, 488, and 514 nm; 543 and 633 nm. The beating of cardiac monolayers was captured via video microscopy at 29 frames per sec for 60 secs using a 10X objective lens (Keyence fluorescence microscope). The intensity profile of the region of interest (ROI) was obtained using Image J, from which beating frequencies were determined. Phase and bead images were acquired for traction force measurement using the Keyence microscope with a 40X objective. Timelapse images were processed for analysis using ImageJ, and images for traction force microscopy were processed using MATLAB software (R2021a) for calculation.
2.11. Western blot:
Primary CMs were cultured as discussed in the cell culture section. Cells were lysed using RIPA lysis buffer (Cell Signaling) with protease and phosphatase cocktail inhibitor (Thermo Scientific). Cells were assayed for immunoblotting using an ani-tubulin antibody (Cell Signaling) and anti-HIF-1α antibody (Novus Biologicals). HIF-1α expressions were normalized against the expression levels of tubulin.
2.12. RNA isolation and qRT-PCR
In the analytical evolution of the RT-qPCR assay for the targeted gene, we first extracted total RNA from the cells as mentioned in the cell culture section using the RNeasy Mini kit (QIAGEN). cDNA was synthesized and served as a template for the targeted gene amplification using QuantiTect Reverse Transcription Kit (QIAGEN). Concentrations of RNA and cDNA were quantified using NanoDrop Technologies. SYBR Green Master mix (Applied Biosystems) was used for targeted gene amplifications. All experiments were performed as mentioned in the manufacturer’s protocol. Primers were designed using Primer blast (National Center for Biotechnology Information) and purchased from Integrated DNA Technologies. Following primer sequences for targeted genes were used:
HIF-1α Forward Primer CAAAGACAATAGCTTTGCAGAATG,
HIF-1α Reverse Primer ACGGTCACCTGGTTGCTG
GAPDH Forward primer TCTCTGCTCCTCCCTGTTCTA
GAPDH Reverse primer ATGAAGGGGTCGTTGATGGC
The delta-delta Ct (2−ΔΔCt) method was used to calculate the relative expression of gene fold change. GAPDH was used to normalize obtained HIF-1α values.
2.13. Fabrication of micropillars and cardiac tissue contractility assay
A computer-aided design (CAD) of the device was created using CorelDraw2020 (64-bit, -Corel Corp, Ottawa, ON) for PMMA (Fisher Scientific) master mold. Polymethylmethacrylate (PMMA, ACRYLIC) (Goodfellows) was used to create pillars that were 3.17 mm in height and 0.8 mm in diameter, separated by a space with 3.2-mm in the center-to-center distance using a laser cutting system (VLS 2.30 Desktop Laser, Universal Laser Systems Inc.). The laser-cut pieces of PMMA were assembled to create a master mold for PDMS casting. The device was prepared with PDMS 20:1 (w/w) and was placed in the oven at 80 °C for 2 hrs.
The cardiac tissue construct was prepared by adding 10% of M199 media (Wisent Bioproducts), sodium bicarbonate 2 mg/ml, 10 mM sodium hydroxide (Sigma Aldrich), 28% deionized water, 50% collagen type I (3.8 mg/ml), and primary rat CMs at a concentration of 60 × 106 cells/ml. The desired mixture containing 1.7 million cells was added to the fabricated posts to create cardiac tissue-on-a-chip. Cell mixture on chips was incubated for 30 mins to create a hydrogel construct and incubated with the complete culture media for 48 hrs., followed by hypoxic treatment for 48 hrs. before adding CPO microparticles as discussed in the cell culture section.
The contractile force was calculated from a small deflection of posts based on the cantilever beam theory:
| (1) |
Where F is the force at a single micro-post, k is the bending stiffness of the post, and δ is its deflection measured by displacement. Here, each micro-post is modeled as a cantilever beam. The cantilevers were fixed at one end and underwent small deflections at the other. The stiffness can be calculated using the beam bending theory:
| (2) |
In the equation, E is the Young’s modulus, with the elasticity of PDMS being approximately 1.1 MPa for a 1:20 mixture of PDMS mixture. The diameter D and length L of the microposts were obtained from side-view images of the device and cross-referenced with the original CAD.
2.14. Traction force microscopy measurement
Cell traction forces were measured on polyacrylamide (PAA) (Bio-Rad) hydrogel substrates with a stiffness of 10 kPa. Pre-gel mixtures were prepared as following the previous protocol.[48–50] Briefly, 8% acrylamide and 0.1% bis-acrylamide (Bio-Rad), 2% N-Hydroxy succinimide (dissolved in Dimethyl Sulfoxide (DMSO)) (Sigma Aldrich), 0.75 % 3-aminopropylsilyl (Thermo Fisher Scientific), and 0.25% tetramethyl ethylenediamine (Millipore Sigma) were added to water to prepare the pre-gel mixture. In addition, 1% of 200-nm fluorescently labeled green beads (2% solid, Thermo Fisher Scientific) were added to the gel mix. Petri dishes were treated with 1M NaOH for 1 hr followed by (3-aminopropyl) triethoxysilane and glutaraldehyde for 1 and 2 hrs, respectively. The gel mix was added to the treated dish for gelation and left for 30 to 45 mins for polymerization at RT. Gel surface was treated with Sulfo-SANPAH (Bio Vision) under UV for 3 mins.[50] Rat tail collagen I at a 100 μg/ml concentration was used for laminating the gel. Cells were grown for 48 hrs for complete spreading and attachment, followed by 48 hrs in hypoxia before treatment with CPO microparticles (see cell culture section). Brightfield images of cells and fluorescence images of the nanobeads were captured before (stressed) and after (relaxed), removing the cells to relieve traction stress. A custom-built MATLAB code was used for traction force microscopy (TFM) analysis,.[48, 50] Briefly, stressed, and relaxed images of the beads were used to obtain the displacement field. The image correlation calculation used a 32×32-pixel region of interest (ROI). The cell traction field was obtained from the displacement field by using constrained Fourier transform traction microscopy (FTTM).
2.15. Procurement of rabbit hearts, histology pathology
We have used an ex vivo ischemic injury model using hearts procured from rabbits. Freshly harvested rabbit hearts (from Sierra For Medical Sciences, Inc) were immersed in 25 ml of regular saline (0.9% NaCl) and media solution adapted from Celsior recipes with and without CPO microparticles for 3 to 4 hrs. at RT.[51] The formulated medium composition contained 10% mannitol, 28% lactobionic acid, 3% glutamic acid, 4.5 % histidine, 0.03 % calcium chloride, 1.1% potassium chloride, 4% sodium hydroxide, 0.5% catalase, with and without 1 mg/ml CPO microparticles in MilliQ water (all chemicals purchased from Sigma-Aldrich). We added catalase and HEPES in the solution at the same concentration as in vitro experiments. After incubation for 3 to 4 hrs, the hearts were dissected and cryopreserved in optimal cutting temperature (OCT) for cryosectioning or fixed in 4% formalin to make paraffin blocks. Formalin-fixed tissues were dehydrated (Thermo Electron Corporation) and embedded in paraffin. Paraffin blocks were sectioned (4 μm thickness) using (Leica, RM2255) and mounted on slides for hematoxylin and eosin (H&E) staining and histopathology analyses (H&E staining Kit Abcam).
Statistical Analysis:
Data are presented as mean value ± standard error of the mean. Each experiment has been performed a minimum of two to five times (duplicate and triplicate samples were used as well), as stated in the figure legend. The two-way ANOVA, one-way ANOVA, or unpaired Student’s t-test with one tail unequal variance were used to determine statistical significance as appropriate using GraphPad Prism 9 or in excel. A statistical significance level of 0.05 was used. Denotations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p < 0.001; ns, p > 0.05.
3. Results and Discussion:
3.1. Synthesis and characterization of CPO microparticles
Regardless of the methods used to prepare microparticles (i.e., flow lithography or emulsion), the solubility of CaO2 is a major concern. Therefore, incorporating CaO2 into polymers to fabricate appropriate microparticles capable of sustained O2 release is challenging.[42, 52] The use of hydrophobic polymers, such as PLGA, is critical because of their high surface area and miscibility with organic solvents. Moreover, CaO2 has a low affinity to hydrophobic polymers, leading to the heterogeneous distribution of CaO2 in the matrix.[53] Among all other inorganic peroxides, CaO2 shows slower O2 release as the decomposition of CaO2 into O2 is a two-step process. However, the process is pH-dependent.[54] We prepared microparticles with PLGA and CaO2 by the emulsion method (Figure 1A). We used span80, a surfactant that acts as an emulsifier and facilitates microparticle coalescence in an oil bath. Homogeneous encapsulation of CaO2 within PLGA particles was improved by reducing the surface tension using span80, leading to a lowering in the size of the microparticles.[55, 56] Unlike the microfluidic method, the emulsion method allows the straightforward synthesis of microparticles in a time-efficient manner, and most importantly, its purification steps do not involve contact with water.[57] Since any interaction of CaO2 with H2O would start the decomposition of CaO2 at the microparticle preparation stage, H2O was avoided in the emulsion method by using an organic solvent.
Figure 1:
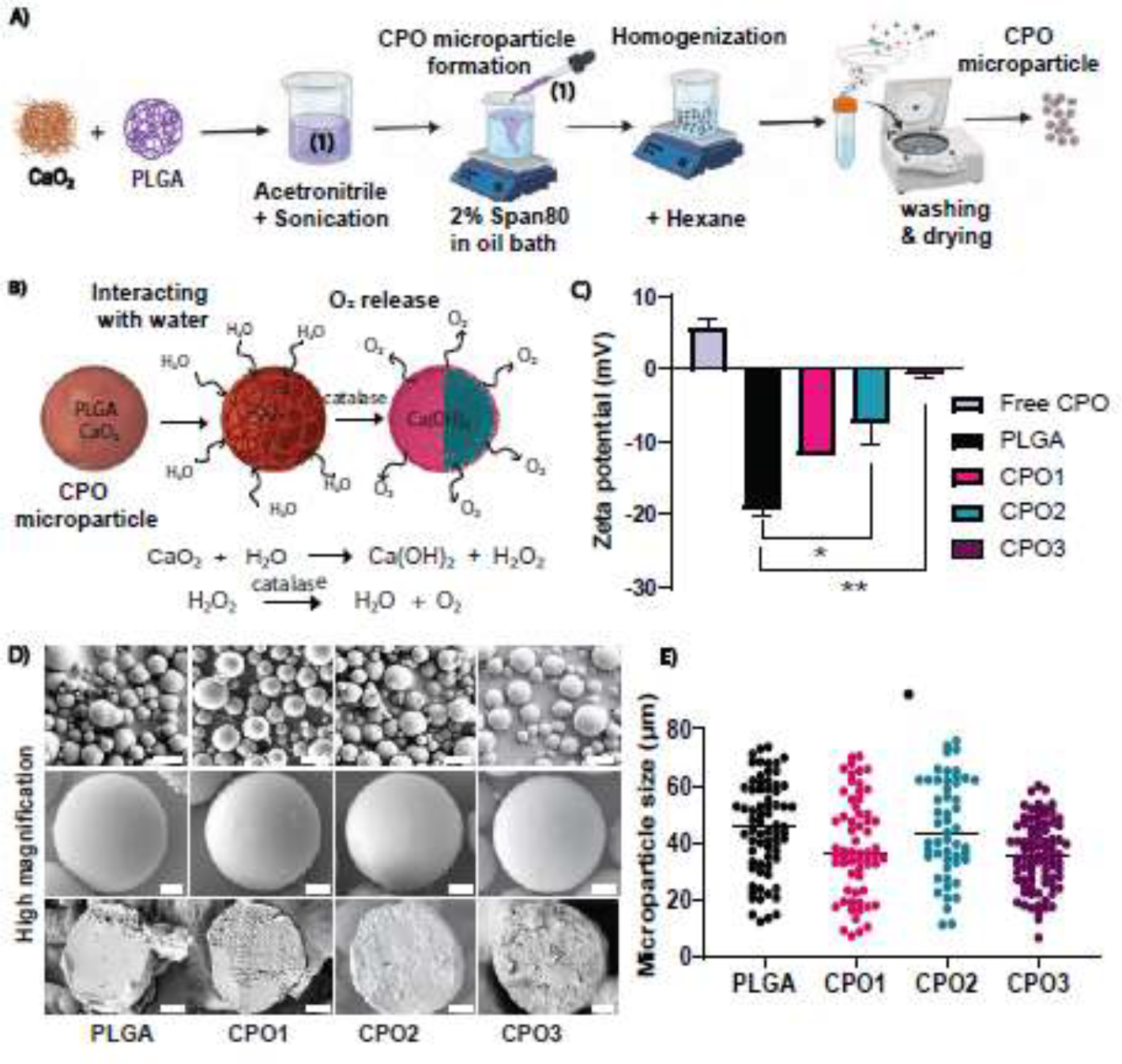
Microparticle preparation and morphological characterization. (A) Schematic representation of the formulation strategy used to synthesize PLGA-based oxygen-releasing microparticles using CaO2 as an O2-releasing agent. An acetonitrile phase containing PLGA and CaO2 was prepared and homogenized. An oil phase was prepared with Span80 as an emulsifier. As the acetonitrile phase diffused into an oil bath, the nonsolvent PLGA/CaO2 phase was separated to form a droplet. The acetonitrile solvent in the presence of Span80 resulted in the spontaneous formation of continuous polylithic CaO2 /PLGA microparticles followed by washing and drying. (B) Schematic representation of the oxygen-release mechanism from the fabricated microparticles with different compositions of CaO2 encapsulated in PLGA coalesced during fabrication to yield a continuous morphology. (C) Zeta potential distribution of CPO microparticles and free CPO were measured. Three different PLGA to CaO2 ratios were used to prepare the microparticles: 1:10, 1:4, and 1:2. CPO microparticles showed an increase in charge with higher CaO2 loading (n=3). (D) SEM images of CPO microparticles: (i) PLGA, (ii) CPO1, (iii) CPO2, and (iv) CPO3. Scale bar = 50 μm (upper panel). A cross-sectional view of microparticles obtained from a paraffin block revealed the internal structures of the microparticles. Scale bar = 10 μm (middle and lower panel) for the respective microparticle composition. (E) CPO microparticle size distributions show the average sizes of three different CPO microparticle formulations. (Number of particles, n = 71, 62, 51, and 83 for PLGA, CPO1, CPO2, and CPO3, respectively). One-way ANOVA was used for the significance test (* p < 0.05).
We synthesized three different CPO microparticles with varying ratios (w/w) of PLGA to CaO2: 10:1, 4:1, and 2:1 for CPO1, CPO2, and CPO3, respectively (See Materials and Methods). The concentrations of CaO2 encapsulated in CPO1, CPO2, and CPO3 were 9% (w/w), 20% (w/w), and 33% (w/w), respectively. Figure 1B depicts the release pathway of O2 from CaO2. During the hydrolytic decomposition of CPO microparticles, CaO2 breaks into hydrogen peroxide (H2O2), which further decomposes into O2 in the presence of the catalase enzyme. To investigate the stability of the fabricated CPO microparticles, we measured the surface charge of the microparticles by zeta potential (ζ). The negative charge of PLGA microparticles without CaO2 was consistent with previously reported data, with an average ζ value of around − 20 mV. In contrast, free CPO had a positive charge with an average ζ value of around + 6 mV.[58] Thus, increasing the ratio of CaO2 in the formulations resulted in increasing surface charge ranging between −20 mV to −0.6 mV (Figure 1C), confirming the stability of the microparticles.
The morphology of the microparticles was characterized for all formulations by SEM (Figure 1D). The images indicated heterogeneous size distributions of the microparticles (Figure 1D, Supplementary Figure S1A–D). The SEM results showed an average diameter of around 40 ± 5 μm for all CPO-microparticle formulations with statistically significant differences (Figure 1E). The internal structure of the fractured microparticles was obtained using SEM after sectioning. A continuous polylithic morphology was exhibited by all microparticles with a few pores (Figure 1D). However, the freeze-drying process did not fracture the particles, indicating their structural stability (Supplementary Figure S1E). A smooth cross-section was observed in the case of microparticles with PLGA alone. In the case of CPO microparticles, increased surface roughness was observed, which indicates an encapsulation of CaO2 within the PLGA polymer (Figure 1D).
To verify the successful encapsulation of CaO2 in PLGA, we measured the encapsulation efficiency of CPO microparticles by staining calcium with Alizarin Red S.[4] Fluorescent images showing calcium staining of CPO particles confirmed the encapsulation of CaO2 (Figures 2A). In addition, we extracted the encapsulated CaO2 by dissolving PLGA. The absorbance of obtained CaO2 was measured using Alizarin Red S staining. We determined the normalized absorbance with respect to PLGA microparticles. A significant increase in the absorbance values was observed as the CaO2 amount increased in PLGA, with CPO3 showing the highest intensity compared to CPO1, CPO2, and PLGA (Figure 2B). We further quantified the encapsulation efficiency showing nearly 23%, 45%, and 83% (w/w) encapsulation of CaO2 in CPO1, CPO2, and CPO3 microparticles, respectively (Figure 2C). The amount of CaO2 encapsulated in CPO2 microparticles was also calculated using ICP-MS; we found that 50% w/w CaO2 was encapsulated in the CPO2 microparticles (Supplementary Figure S1F&G). Altogether, the measured encapsulation efficiency of CaO2 with different concentrations varied between ~ 20% to 80% (w/w) depending upon the initial amount and the technique that was used for the measurement.
Figure 2:
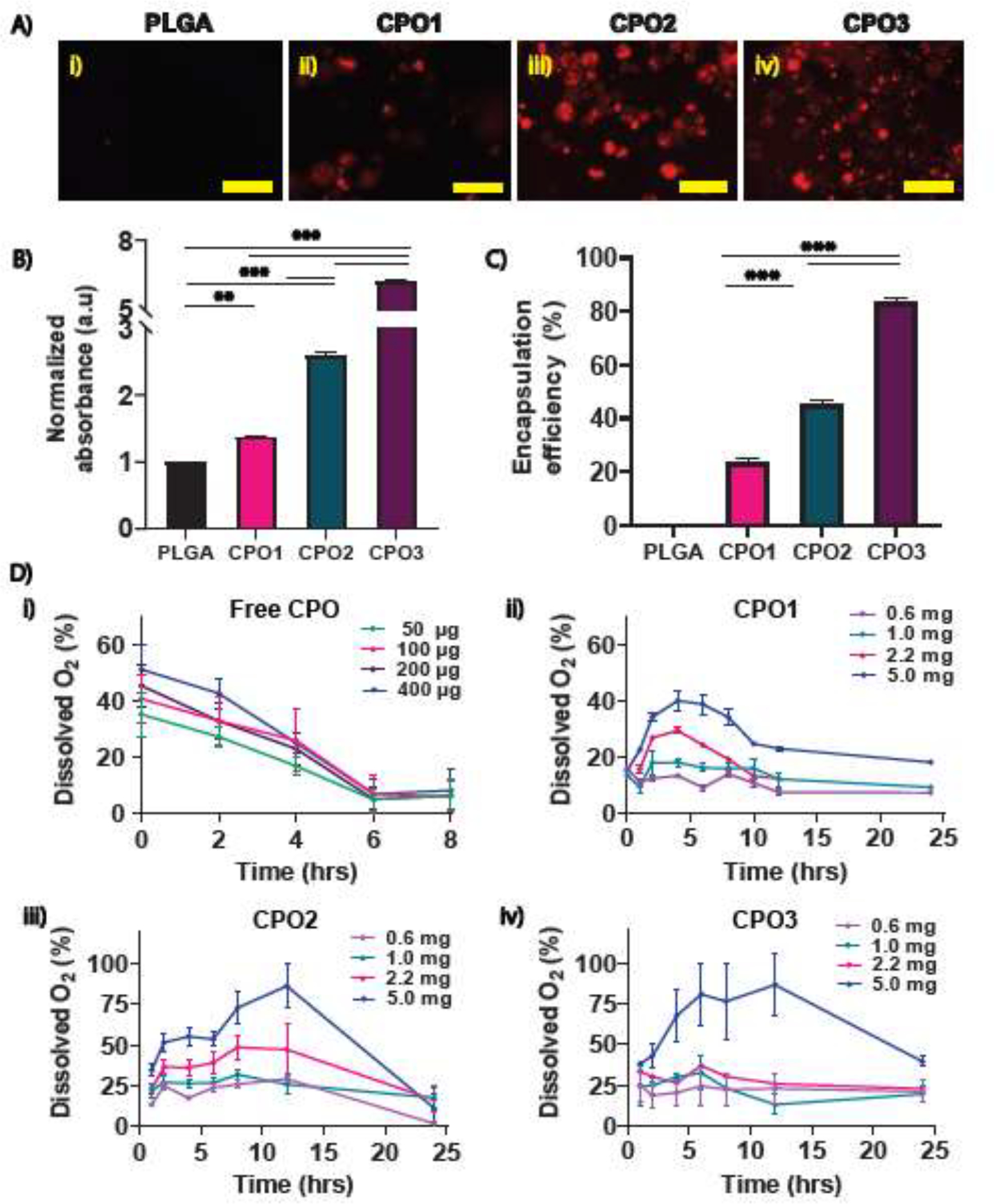
Evaluation of CaO2 encapsulation into microparticles and oxygen release kinetics. (A) Alizarin Red S staining was obtained by fluorescence imaging for the different CPO microparticle formulations revealing CaO2 encapsulation for (i) PLGA, (ii) CPO1, (iii) CPO2, and (iv) CPO3 microparticles (red). Scale bar = 150 μm. (B) Normalized absorbance of CaO2 extracted from the CPO microparticles stained with Alizarin Red S. (C) Encapsulation efficiency of CPO microparticles in the PLGA was obtained. (n=2 with triplicate). (D) The oxygen release profiles of (i) free CPO (powder), (ii) CPO1, (iii) CPO2, and (iv) CPO3 microparticles up to 24 hrs in hypoxia were measured. (n=3 to 4 for each concentration of all microparticles). Student’s t-test was performed for the significance tests (* p ≤ 0.05, ** p ≤ 0.01, *** p< 0.001).
To evaluate the O2-releasing kinetics, different concentrations of CPO microparticles were used (0.6, 1, 2.2, and 5 mg/mL in hypoxic PBS). Microparticles were dissolved in catalase-containing PBS and placed inside a hypoxic chamber (Supplementary Figure S1H). We removed the dissolved oxygen by flowing nitrogen gas from PBS before adding the CPO particles to the solution to estimate oxygen release from CPO microparticles accurately. We prepared different concentrations of free CPO (50, 100, 200, and 500 μg/ml) dissolved in deoxygenated PBS. Microparticles started releasing gas bubbles upon hydration. The size of the bubbles from different formulations increased with increasing CaO2 concentrations, as observed and shown in (Supplementary Figure S1H). The Ruthenium sensor probe measured the oxygen-releasing kinetics of CPO microparticles and free CPO (Figure 2D). The total amount of dissolved O2 was plotted as a function of time.
Figure 2D shows the total amount of dissolved oxygen for free CPO, CPO1, CPO2, and CPO3 with different microparticle concentrations plotted over time. Free CPO showed a burst release of O2 immediately after adding hypoxic PBS containing catalase, followed by a decrease in the release profile as depicted in Figure 2D (i). A complete release was observed within 6 hrs with a declining release profile over time. The data demonstrated a lower release profile for CPO1 than CPO2 and CPO3 (Figure 2D (ii), (iii), and (iv)). Unlike free CPO, dissolved oxygen showed a sustained release of oxygen for up to 24 hrs before showing a decrease in release profile, confirming the controlled release of CaO2. The initial increase in O2 release is due to the existence of CaO2 at the surface of microparticles. Microparticle concentrations of 0.6, 1, and 2.2 mg/mL showed a nearly constant amount of O2 release over the 24 hrs period, whereas 5 mg/ml displayed a noticeable increase in its release profile. A large amount of dissolved O2 was released in case of using a particle concentration of 5 mg/ml reaching saturation for both CPO2 and CPO3 (Figure 2D). Thus, an optimum concentration of particles was required to achieve a sustained release profile for a given amount of PBS. However, the increased CaO2 loading in CPO3 compared to CPO2 did not significantly change O2 release profiles for a given concentration, as observed in Figure 2D (iii & iv). Further optimization using higher molecular weights or varying the backbone of PLGA will likely lead to sustained release of oxygen over several days.[39] Moreover, the size of the microparticles, which influences the O2 release kinetics, can be controlled by using a microfluidics-based microparticle fabrication or by filtration for the desired size range to improve the release profile further.[59] In our microparticle distribution, we have various sizes of microparticles (Figure 1.D & E) that have a cumulative release (Figure 2.D) profile. However, in all the experimental conditions, we have a similar distribution of particles, leading to the same cumulative release profiles. Thus, we assume we have the same percentage of oxygen in all conditions. However, particle sizes can be improved. Since CPO2 at a concentration of 1mg/ml released O2 at the physiological level, we selected CPO2 for further cell studies.
3.2. Biocompatibility and signaling pathways:
Next, we studied the cytotoxicity of CPO microparticles on primary rat CMs. We assessed cell viability and metabolic activity under hypoxic conditions with and without CPO microparticles. During hydrolytic decomposition, CPO microparticles generate free radicals that change the pH of the media, which is cytotoxic to cells. This can be mitigated by adding a HEPES buffer in the media. We first measured the pH of cell culture media containing CPO microparticles with and without adding HEPES buffer. Data showed that adding 0.2 mM and 0.3 mM of HEPES reduced the pH values from 8 to 7.4 (Supplementary Figure S2A). We added catalase to facilitate the decomposition of H2O2 into O2. It is worth noting that excess O2 is also cytotoxic to cells.[44, 60] Therefore, we chose CPO2 microparticles, which demonstrated the most physiologically relevant release profile with the optimum CaO2 encapsulation required for maximum cell and tissue survival. Moreover, CPO2 showed sustained release for up to 24 hrs when used at 1 mg/ml (Figure 2D (iii)). Thus, CPO2 was chosen for the rest of the studies in this work.
We seeded primary rat CMs at 5 × 105 cells/ml with and without CPO microparticles at 1 mg/ml and 2 mg/ml for cell viability assay. We performed live (green) and dead (read) assays and found an increase in cell viability after the addition of CPO microparticles (Figure 3B). The percentage of dead cells decreased significantly when CPO microparticles were added to hypoxic cells (Figure 3C). To determine the effect of CPO microparticles on cell metabolic activity, we performed a PrestoBlue cell viability assay for CMs. Data showed that cell metabolic activity significantly increased after the treatment with 1 mg/ml CPO microparticles (Figure 3D). However, with the increased particle concentration from 1 mg/ml to 2 mg/ml, metabolic activity increased, confirming the effect of excessive O2 and the need for an optimum amount of O2 in a cellular microenvironment with low O2 tension. In addition to CMs, we also measured cell metabolic activity of 3T3 fibroblasts (Supplementary Figure S2B) and human umbilical vein endothelial cells (HUVECs). Metabolic activity of HUVECs increased with an increasing cell concentration from 1 × 105 to 2 × 105 cells/well (Supplementary Figure S2 C) when 1 mg/ml CPO microparticles were used. These results were similar to those obtained with CMs showing increased metabolic activity upon the addition of CPO microparticles in hypoxia (Figure 3E). Moreover, we observed that cell metabolic activity depended upon the microparticle concentration for a given number of HUVEC and 3T3 fibroblast cells, which agreed well with the results obtained for CMs.
Figure 3:
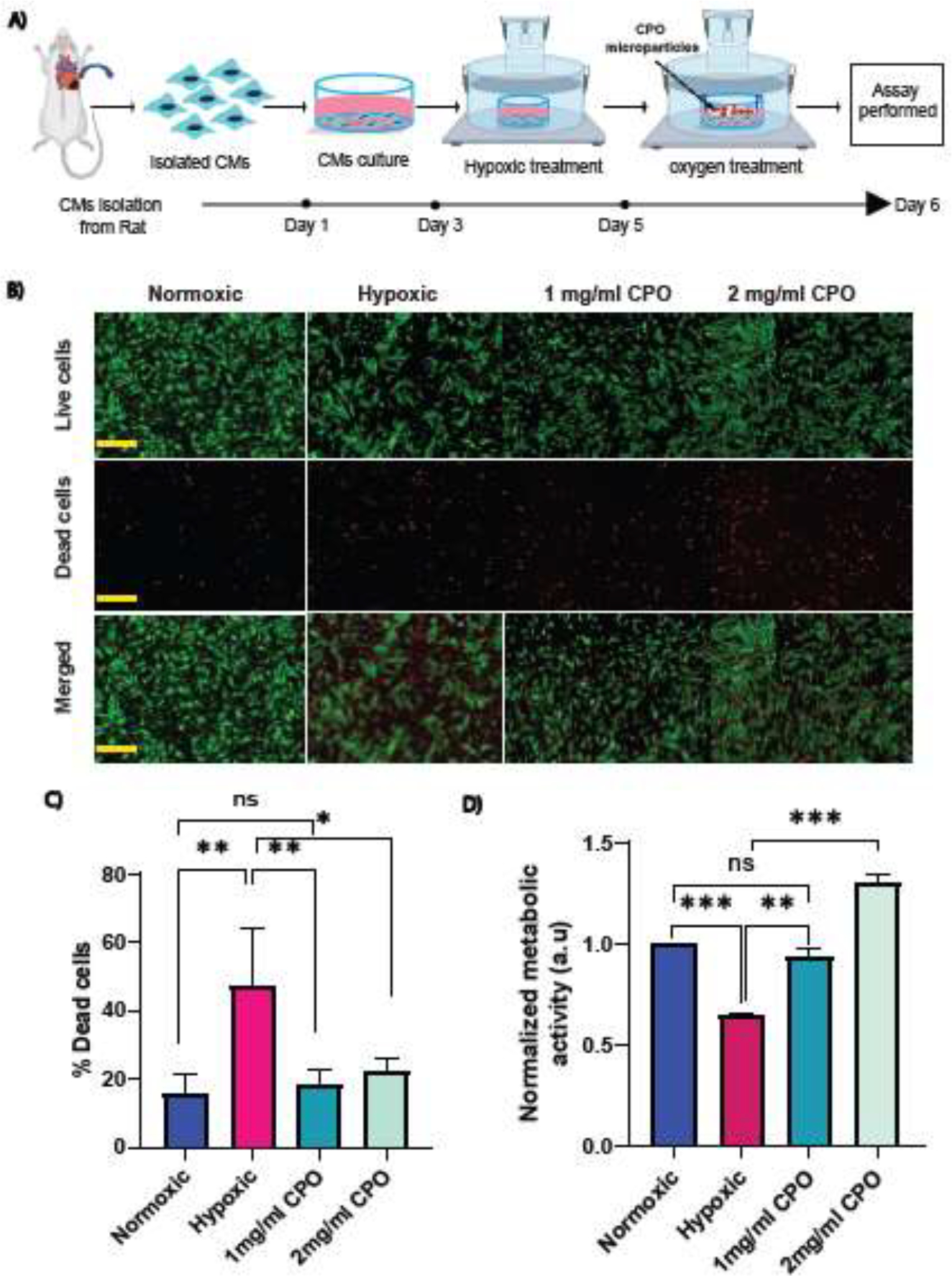
Cell viability and metabolic activity assays in the presence of CPO2 microparticles. (A) Schematic of the experimental setup, where rat CMs were cultured in a hypoxic chamber with or without microparticles, which were loaded in cell inserts and dipped into cell culture media. (B) Live (green) and dead (red) staining with calcein AM and ethidium homodimer-1 for CMs with and without microparticles at a concentration of 1 mg/ml or 2 mg/ml in hypoxia. Scale bar = 150 μm. (C) Quantification of the percentage of dead cells with respect to living cells. (n=4 to 6 images/ conditions) (D) CM metabolic activity during hypoxia with and without CPO microparticles of 1 mg/ml or 2 mg/ml using PrestoBlue. Student’s t-test was performed for the significance test (* p ≤ 0.05, ** p ≤ 0.01, *** p< 0.001).
One of the possible mechanisms of action for CPO microparticles to enhance cellular function is through the alteration of HIF-1α, which is a master regulator translocated into the nucleus during the adaptation of a cell to hypoxia. Moreover, HIF-1α and cTn are both used as biomarkers to monitor many cardiac diseases. To better understand the effects of hypoxia and CPO microparticles at the subcellular level, rat CMs were stained for their subcellular structures such as actin, nucleus, and HIF-1α. The subcellular structures were evaluated by immunofluorescence microscopy. The results showed that HIF-1α expression increased due to hypoxia compared to normoxic cells (Figure 4A & B). Furthermore, cells incubated with CPO microparticles significantly reduced HIF-1α intensity levels under hypoxic conditions. To quantify further the HIF-1α protein expression level, we performed western blot experiments, which showed a significant difference when quantifying the blot intensity between hypoxic and O2 treated cells (Figure 4D & E). We also performed qRT-PCR to assess genomic reprogramming of HIF-1α in response to O2 tension or CPO treatment. We observed a significantly higher genomic expression level of HIF-1α in cells under hypoxia compared to normoxic cells, and upon addition of CPO microparticles, there was a downregulation (Figure 4E). We also investigated the cTn expression, one of the important biomarkers of ischemic injuries. We observed a significant increase in the fluorescence intensity level of cTn-1 (Figure 4F & G). The higher fluorescence intensity of cTn-1 may be due to the fractional increase of free cTn-1 from the irreversibly bound pool in the cytoplasm before being released from the cells. However, no structural difference was observed in the actin network between the hypoxic and O2-treated groups (Supplementary Figure S2F). In conclusion, the current study shows that CPO microparticles could modulate HIF-1α expression.
Figure 4:
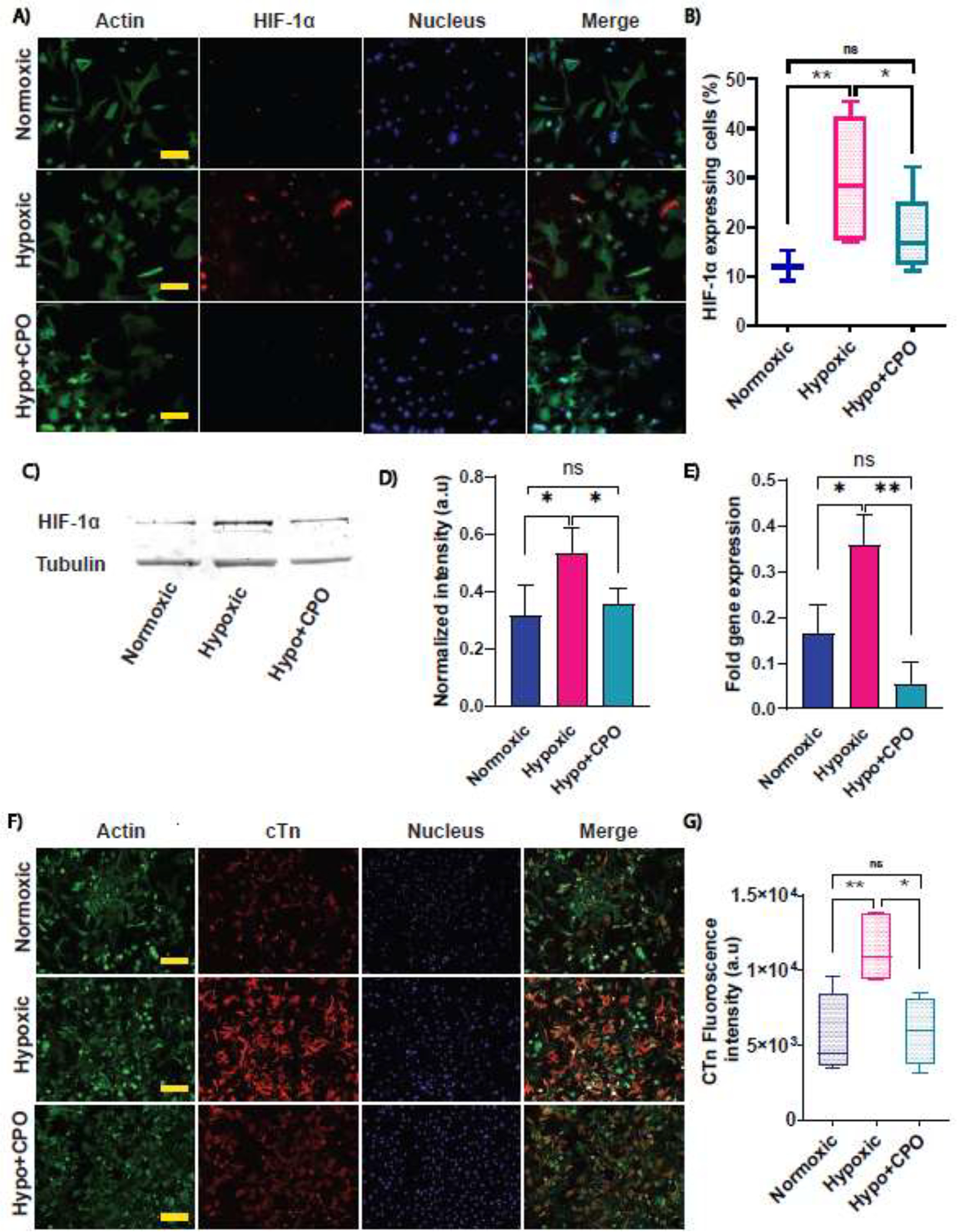
Effects of hypoxia and CPO microparticles on HIF-1α and cTnI expression. CMs were cultured for a total of 6 days in normoxia, followed by hypoxia (control), and with CPO microparticles in hypoxia. (A) Immunofluorescence images of HIF-1α staining (red), actin (green), nucleus (blue), and merged (4th column). Scale bar = 50 μm. (B) Quantification of HIF-1α expression in normoxic, hypoxic (control), and hypo+CPO. (Number of cells n= 148,142 and 158 for normoxic, hypoxic, and hypo+CPO, respectively, from 3 to 6 images/condition). (C) The HIF-1α expression after hypoxic and oxygen treatment was revealed by a western blot experiment. (D) The quantification of HIF-1α protein expressions for hypoxic and oxygen-treated cells from the blot normalized by the loading control, tubulin. (n=3) (E) Genomic expression of HIF-1α was obtained by performing qRT-PCR. Data are normalized by GAPDH. The delta-delta method was used for quantification (n=2 with triplicate). (F) Immunofluorescence images of actin (green), nucleus (blue), cTn I (red), and merged (4th column). Scale bar = 100 μm. (G) Quantifying cTn I expression in normoxic, hypoxic (control), and hypoxic with CPO microparticles. (Number of images n= 4, 6, and 4 for normoxic, hypoxic, and hypo+CPO, respectively). Student’s t-test was performed for the significance test (* p ≤ 0.05, ** p ≤ 0.01, *** p< 0.001). Error bars represent the standard error of the mean with respect to the number of samples mentioned in each plot.
3.3. Contractility of single cells and engineered cardiac tissues in hypoxia
This study aimed to evaluate CPO treatment on cell contractility during hypoxia, which is commonly observed in ischemic injuries. Mechanical tension and forces generated by single cells, 2D cardiac monolayers, and 3D engineered tissues were measured using cell traction force microscopy and engineered micropillar assay in normoxia, hypoxia with or without our CPO microparticles. Neonatal rat CMs are sensitive to changes in their environmental conditions, as shown in the previous section (Figure 3C) by metabolic activity assay and HIF-1α expression level. Therefore, we measured cellular contractility in response to low O2 tension and CPO microparticles.
3.3.1. Single-cell traction force measurement:
For single-cell force measurement, primary rat CMs were cultured on a soft (E = 10 kPa) hydrogel substrate laminated with collagen to facilitate CMs adhesion to the substrate (Figure 5A).[48, 49] Hydrogels embedded with fluorescent nanoparticles (200 nm in diameter) were displaced due to induced deformation on the substrate generated by the cell. Brightfield images of individual cells and stressed and relaxed images of the fluorescent nanoparticles were acquired for the force calculation (Figure 5B). Briefly, the displacement field of the nanoparticles was obtained using a stressed and relaxed image pair using particle image velocimetry.[61, 62]
Figure 5:
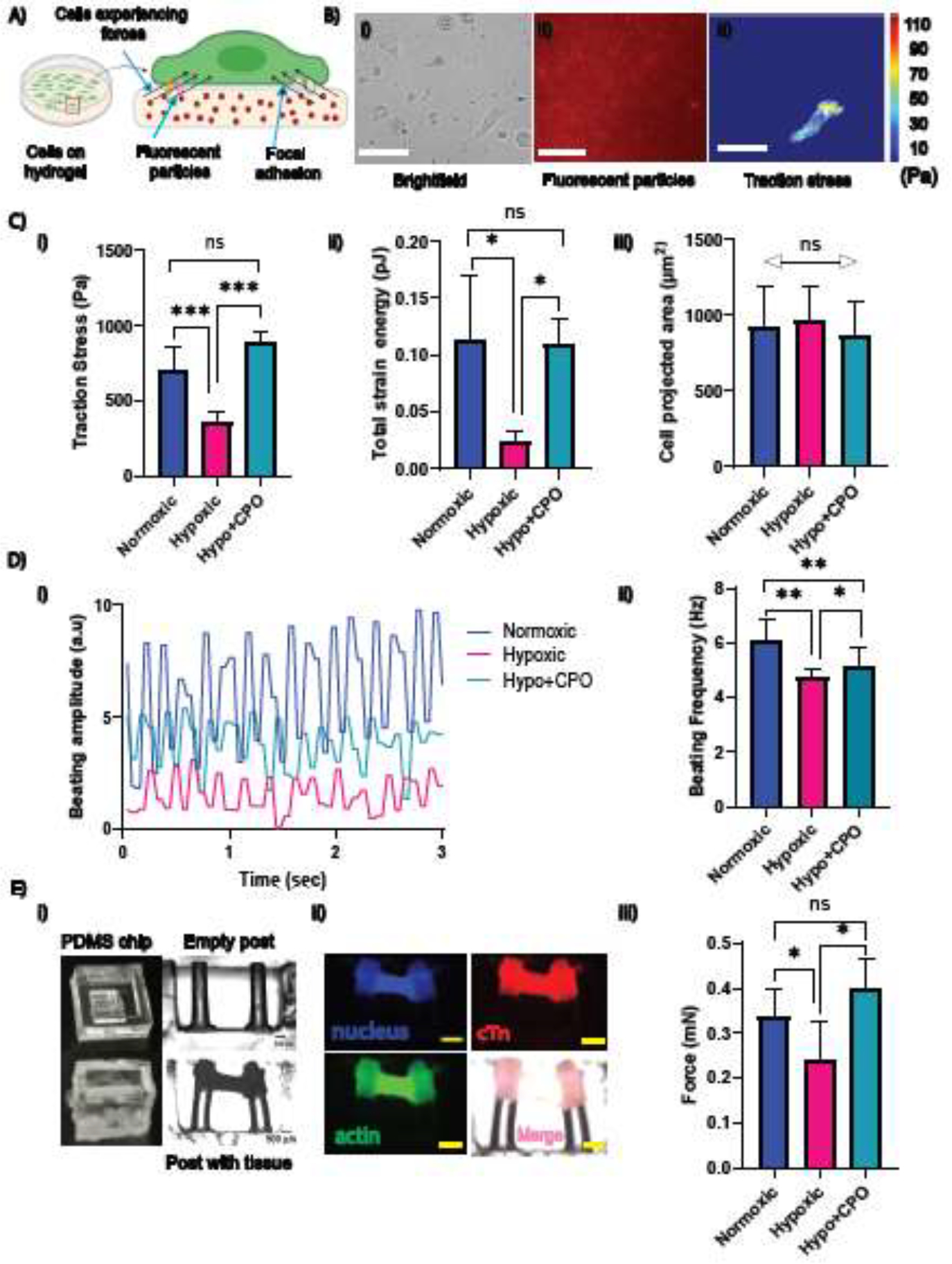
Restoring contractile mechanics of single CMs and cardiac tissue using CPO microparticles. (A) Schematic illustration depicting the cell traction force measurement of cells on the substrate. (B) Cells (left) cultured on a 10 kPa polyacrylamide-based 2D soft substrate with 200 nm fluorescent microparticles embedded within: (i) a brightfield image of cells, (ii) fluorescent microparticles, and (iii) a cell traction stress map obtained from the microparticles’ displacement field. Scale bar = 30 μm. (C) (i) traction force, (ii) contractile energy, and (iii) cell projected area of CMs with and without CPO microparticles in a hypoxic condition (n = 13 cells for each condition). (D) CMs created a monolayer for beating in (i) normoxia and hypoxia (ii) with and (iii) without microparticles showing higher amplitude with CPO particles. (iv) Average beating frequency in normoxic, hypoxic, and hypoxic with CPO microparticles (n = 8 movies for each). (E) Force generated by cardiac tissue measured by micro-post assay: (i) images captured for empty PDMS chip, micropillars without tissue, and brightfield image of cardiac tissue engineered between a pair of parallel micropillars. Displacement of the pillars due to tissue contraction is visible in the brightfield image of tissue construction. (Side view) (ii) Immunostaining images of cardiac tissue between micropillars are shown: actin (green), nucleus (blue), cTn (red), and merged all (pink). Scale bar = 500 μm (all images were captured with the same magnification). (iii) Forces in normoxia (p=0.055), hypoxia with and without CPO microparticles generated by the engineered tissue. The number of posts, n = 7, 4, and 4 normoxic, hypoxic and hypoxic with CPO microparticles, respectively. The student’s t-test was performed for significance. (* p ≤ 0.05, **, p ≤ 0.01; *** p < 0.001, ns p > 0.05.).
Traction forces were obtained using the integral equation
| (3) |
where,
| (4) |
In the above equations, Gij = Green’s Tensor,ui = displacement, fj = force, υ = Poisson’s ratio, E = elasticity of the substrate, and (x, y) = the coordinate of the beads. A Fast Fourier Traction Cytometry was used to measure traction forces.[48, 50] A cell traction stress map was obtained from the displacement field (Figure 5B). Cell traction stress increased significantly for cells with CPO microparticles compared to hypoxic cells (Figure 5C (i)). We calculated strain energy generated due to elastic deformation. This measured contractile strength significantly increased when CPO microparticles were added (Figure 5C (ii)). However, there was no significant difference between the projected area of the cells in both hypoxic control and CPO microparticle treatment groups. It has been previously shown that traction force and cell area are correlated.[63] Therefore, it is important to ensure that the observed effect on cell traction force is not confounded by the projected cell area. [63] Thus our non-significant area differences confirm that the differences in forces are solely due to oxygen tension.
3.3.2. CM beating assay:
We observed spontaneous CM beating after 48 hrs of culture. CMs were cultured for 3 days to form a tissue-like structure that displayed synchronous beating. We recorded the beating kinetics of CMs in hypoxia with and without CPO microparticles at a high temporal resolution. Signals generated by contractile CM monolayers were obtained (Figure 5D (i)). Contraction/relaxation frequencies were obtained from the signals, showing a significant decrease in hypoxia and an increase with CPO microparticles (Figure 5D (ii)). Our data showed that adding CPO microparticles in hypoxia could improve the cardiac beating rate.
3.3.3. Engineered 3D cardiac tissue contractility assay:
We fabricated PDMS-based chips, each containing a small chamber within (Supplementary Figure S3A). Two micropillars were fabricated within the chamber, separated by a space to measure traction forces generated by cardiac tissue (Figure 5E (i)). We cultured 3D engineered cardiac tissue constructs in a collagen gel between two micropillars for tissue contractility assay. As the tissue becomes mature, tension is generated between the pillars leading to a change in the deflection of the pillars. To confirm CM encapsulation and gel formation, we stained for actin, nucleus, and cTn, which showed a cardiac tissue band formed between the micropillars (Figure 5E(ii), Supplementary Figure S3B). Traction forces generated by the tissue can be obtained from the displacement of the pillars using Euler-Bernoulli’s theory, as mentioned in eq. (2). The change in deflection for each micropillar was quantified by a series of image analyses of blank chips and chips with tissues. We obtained mechanical tension generated by cardiac tissue in hypoxia with and without CPO microparticles showing significantly higher displacement and hence the force for those with CPO particles (Figure 5F (iii)). Our data suggested that CPO microparticles improve cardiac tissue contractility and force generation capacity.
It is worthwhile to note that previous studies reported 10 kPa as an optimum matrix stiffness for cardiac tissue and single-cell functionality, where reduced beating rates were observed when the cells were cultured on substrates having a matrix stiffness exceeding 10 kPa matrix.[64] Thus, the force and energy obtained in TFM were possibly the maximum values. The contraction stress was also improved in engineered cardiac tissue by adding CPO microparticles. It was previously shown that the contractile function of the rat diaphragm muscle strip is depressed when exposed to hypoxia.[65] In another study, it was demonstrated that antioxidants partially protect the contractile function in severe hypoxia.[66] These prior findings are aligned with and support our results. The possible mechanism is that the oxidants inhibit glycolysis and oxidative phosphorylation in the heart during ischemia. In contrast, the addition of antioxidants preserves C-reactive protein (CrP) levels in the rat heart muscle at the end of ischemic exposure resulting in the preservation of contractile function.[67]
3.4. Reducing ischemic injury in heart tissue:
Storage of the heart in a cold preservation solution after donations remains the standard.[68] The success of heart transplantation depends upon the development of graft vasculature for oxygen supply. [32] The imbalance of O2 supply-demand raises the risk of MI or may lead to the rejection of allografts. Currently, ischemic time is still 4–6 hrs using the cold static storage. Before transplantation, machine perfusions are used for oxygen supply and removal of metabolites, increasing preservation time by up to 10 hrs. However, with a higher perfusion pressure and flow, there is a high risk of hemolysis.[68] This study aimed to demonstrate a proof of concept for using CPO microparticles during perfusion to reduce ischemic injury. Thus, an ex vivo model of freshly harvested rabbit hearts (Supplementary Figure S3C) was used and treated with four different solutions: saline as a control group (grp I), formulated solution adapted from Celsior organ preservation media composition (grp II), saline with CPO microparticles (grp III), and formulated media (grp IV), prepared as discussed in the Materials and Methods section.
H&E staining was performed for all four groups to study ischemic injury during the first 3 to 4 hrs as observed during heart transplantation (Supplementary Figure S3D). As part of the innate immune system, neutrophils infiltrate tissue after acute inflammation.[69] Coagulative necrosis is another hallmark of MI characterized by irreversible, permanent damage to tissue. Our H&E showed no sign of neutrophilic infiltration or coagulative necrosis in all four groups after a hypoxic insult. However, some of the heart samples in all four groups presented signs of early acute myocardial injury (Figure 6). The percentage of early acute myocardial injury was defined by the appearance of wavy myocardial fibers and focal edema. This was different in each group, as listed in Table 1. Multiple patches of wavy patterns of myocytes in H&E-stained tissue sections appeared for the control groups with higher percentages of acute myocardial injury (44.4 % and 34.2 %) compared to solutions with CPO microparticles (10.8 % and 9.3 %) for saline and media, respectively (Figure 6), respectively, reflecting time course protection of the rabbit hearts by CPO microparticles during the ischemic time.
Figure 6:
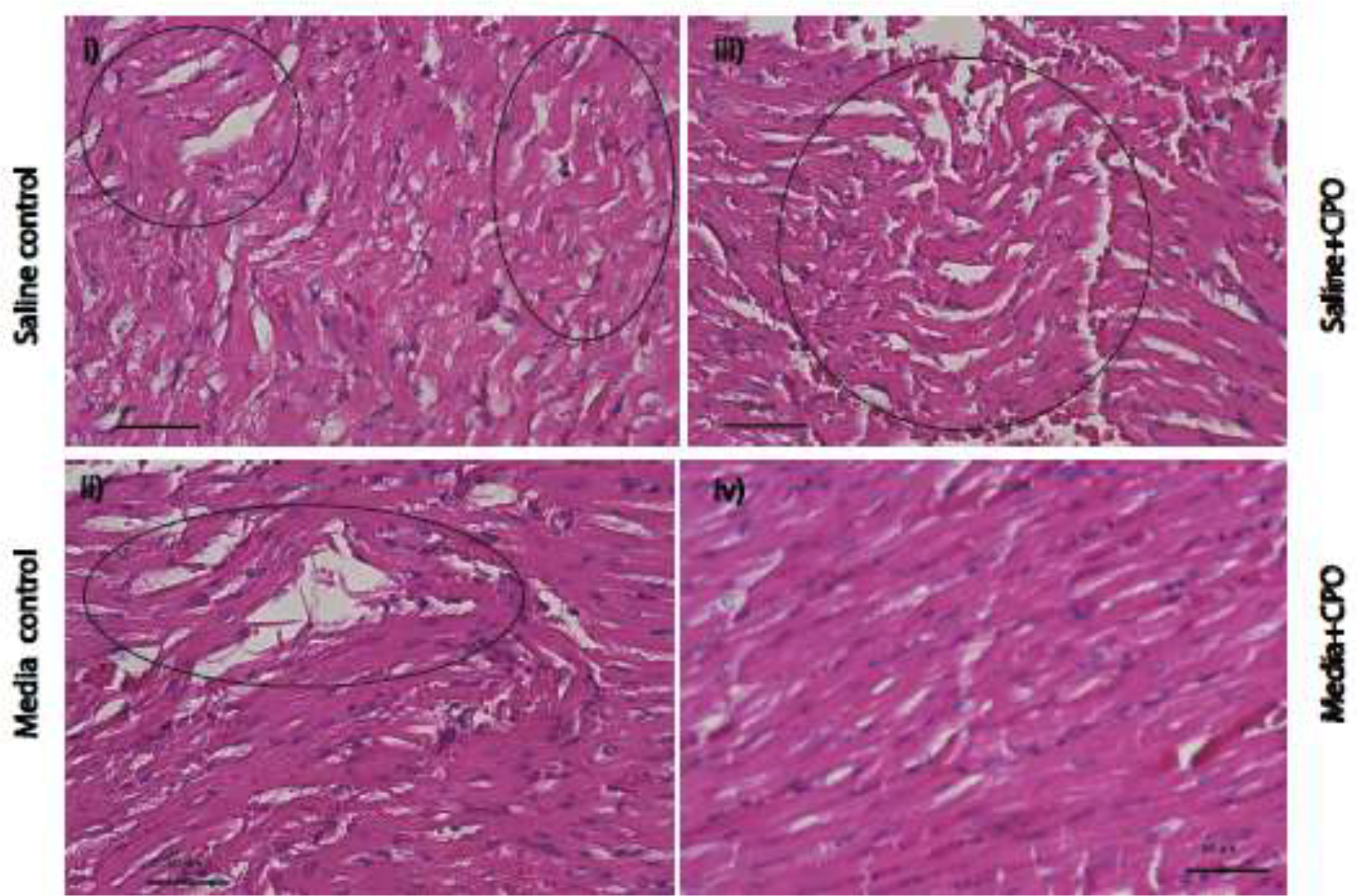
Rabbit heart histology displaying acute myocardial injuries with myofiber waviness and focal edema after 4 hrs. of heart procurement with and without CPO microparticles. Myocardial injuries were observed in rabbit hearts incubated in saline control (grp I), formulated media control (grp II), without (grp III), and with (grp IV) CPO microparticles for 4 hrs. Changes induced in all groups are marked by circles. All sections were stained with the H&E. Scale bar = 50 μm. All images were acquired with the same magnification.
Table 1.
Histology analysis for four condition groups: (i) saline control (grp I) (hypoxic), (ii) formulated media control (grp II) (hypoxic), (iii) saline with CPO (grp III), and (iv) formulated media with CPO (grp IV). Percentage calculated from 30–50 images per condition.
| Groups | Coagulative Necrosis Changes (Neutrophilic Infiltration) | Coagulative Necrosis Changes (Myocyte Nuclear Changes) | Early Signs of Ischemic Changes (Wavy Patterns of Myocytes) |
|---|---|---|---|
|
| |||
| Saline Control (grp. I) | Negative | Negative | 44.4 % |
| Media Control (grp. II) | Negative | Negative | 34.2% |
| Saline with CPO (grp. III) | Negative | Negative | 10.8 % |
| Media with CPO (grp. IV) | Negative | Negative | 9.3% |
MI is a leading cause of death and is associated with significant tissue damage and subsequent heart dysfunction. It is previously reported that MI can occur due to spontaneous coronary artery dissection or embolization, known as MI type 2.[6]. Contraction bands (eosinophilic bands) of myocytes appear due to the clustering of hypercontracted contractile proteins in the cytoplasm of the cells after early ischemia.[6] This may be the possible mechanism of wavy pattern appearance in the control group. Previously reported histopathological data showed predominant infiltration of eosinophils in MI after 12 to 24 hrs. of ischemic insult. However, there is an ongoing controversy about whether the eosinophil infiltration is attributed to either pathophysiological or vascular injury.[7] Delivering oxygen using CPO microparticles during transplantation may reduces myocardial injuries after heart transplantation.
4. Conclusion:
We demonstrated the fabrication of O2-releasing microparticles with PLGA and CaO2 as an O2-releasing agent that exhibits a sustained O2-release profile for up to 24 hrs or more. In addition, the optimization of CaO2 concentration in PLGA for required O2 release revealed the best fit for CPO2 with a PLGA to CaO2 ratio of 4:1. As the generation of free radicals during CaO2 decomposition increases the pH of cell culture media, we demonstrated that the HEPES buffer could be used to control the pH alteration in the media, maintaining cell viability. Moreover, CM metabolic activity and cell viability could be protected using CPO microparticles in hypoxia. Released oxygen from 1 mg/ml CPO microparticles was sufficient for 5 × 105 primary rat CMs to be protected from hypoxia. Our data suggest that an optimized CPO microparticle concentration must be used for a given cell density and type. Contractility is the central function of CMs. Our force data from TFM and micropillar assay proved that the contractile functions could be preserved in hypoxia by adding our O2-releasing microparticles. Likewise, compared to hypoxic control cells, single CMs generated higher strain energy on a soft substrate of stiffness 10 kPa when CPO microparticles were added. The contraction stress was also improved in engineered cardiac tissue by adding CPO microparticles. Another possible mechanism of action for CPO microparticles is through the alteration of the HIF-1α signaling pathway. In fact, in the current study, CPO microparticles downregulated HIF-1α expression, inversely correlated with force generation.
Our study suggests that CPO microparticles can potentially be used for many biomedical applications in which a supply of oxygen is required, including tissue engineering, wound healing, cardiac patch implant, and cardiac ischemic injury during heart transplantation to improve patient outcomes. These CPO microparticles can potentially be used to increase heart transplantation widow by reducing ischemic injury. Indeed, we demonstrated that myocardial injuries could be reduced by using CPO microparticles for 4 hours following heart procurements in ex vivo heart models. In addition, we believe that future studies incorporating CPO microparticles in machine perfusion are needed to validate the application and will allow the successful clinical translation of CPO microparticles as a preventative/treatment strategy for reducing ischemic injury.
Supplementary Material
Statement of significance.
Oxygen-releasing microparticles can reduce myocardial ischemia, allograft rejection, or irregular heartbeats after heart transplantation. Here we present biodegradable oxygen-releasing microparticles that are capable of sustained oxygen release for more than 24hrs. We then studied the application of sustained release of oxygen-releasing microparticles for cardiomyocytes’ cell and tissue function. Previous studies have not measured cardiac tissue or cell mechanics, which is important for understanding proper cardiac function and beating. Using traction force microscopy and an engineered tissue-on-a-chip, we demonstrated that our oxygen-releasing microparticles improve cell and tissue contractility during hypoxia while downregulating the HIF-1α expression level. Finally, using the microparticles, we showed reduced myocardial injuries in rabbit heart tissue, confirming the potential of the particles for organ transplantation or tissue engineering.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health (R01AR074234, R01GM126571, and U01CA214411) and the American Heart Association Transformational Project Award (18TPA34230036). Thanks to NRVM heart core for providing us with primary rat cardiomyocytes. The authors thank Dr. Hanjun Kim, Dr. Alireza Hassani, Dr. Saurabh Sharma, and Dr. Menekse Ermis Sen for their valuable discussion.
Footnotes
Conflict of Interest: Authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Muz B, de la Puente P, Azab F, Azab AK, The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy, Hypoxia (Auckland, N.Z.) 3 (2015) 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mik EG, Johannes T, Zuurbier CJ, Heinen A, Houben-Weerts JH, Balestra GM, Stap J, Beek JF, Ince C, In vivo mitochondrial oxygen tension measured by a delayed fluorescence lifetime technique, Biophys J 95(8) (2008) 3977–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].You JO, Rafat M, Ye GJ, Auguste DT, Nanoengineering the Heart: Conductive Scaffolds Enhance Connexin 43 Expression, Nano Lett. 11 (2011) 3643. [DOI] [PubMed] [Google Scholar]
- [4].Gholipourmalekabadi M, Zhao S, Harrison BS, Mozafari M, Seifalian AM, Oxygen-Generating Biomaterials: A New, Viable Paradigm for Tissue Engineering?, Trends in Biotechnology 34(12) (2016) 1010–1021. [DOI] [PubMed] [Google Scholar]
- [5].Liang J-P, Accolla RP, Soundirarajan M, Emerson A, Coronel MM, Stabler CL, Engineering a macroporous oxygen-generating scaffold for enhancing islet cell transplantation within an extrahepatic site, Acta Biomaterialia 130 (2021) 268–280. [DOI] [PubMed] [Google Scholar]
- [6].Michaud K, Basso C, d’Amati G, Giordano C, Kholová I, Preston SD, Rizzo S, Sabatasso S, Sheppard MN, Vink A, van der Wal AC, P. Association for European Cardiovascular, Diagnosis of myocardial infarction at autopsy: AECVP reappraisal in the light of the current clinical classification, Virchows Arch 476(2) (2020) 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hayes SN, Kim ESH, Saw J, Adlam D, Arslanian-Engoren C, Economy KE, Ganesh SK, Gulati R, Lindsay ME, Mieres JH, Naderi S, Shah S, Thaler DE, Tweet MS, Wood MJ, Spontaneous Coronary Artery Dissection: Current State of the Science: A Scientific Statement From the American Heart Association, Circulation 137(19) (2018) e523–e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pagliaro P, Gattullo D, Rastaldo R, Losano G, Ischemic preconditioning: from the first to the second window of protection, Life Sci 69(1) (2001) 1–15. [DOI] [PubMed] [Google Scholar]
- [9].Radisic M, Yang L, Boublik J, Cohen RJ, Langer R, Freed LE, Vunjak-Novakovic G, Medium Perfusion Enables Engineering of Compact and Contractile Cardiac Tissue, Am. J. Physiol. 286 (2004) H507. [DOI] [PubMed] [Google Scholar]
- [10].Tunnicliffe FW, Stebbing GF, THE INTRAVENOUS INJECTION OF OXYGEN GAS AS A THERAPEUTIC MEASURE, The Lancet 188(4851) (1916) 321–323. [Google Scholar]
- [11].Tanumihardja E, Slaats RH, van der Meer AD, Passier R, Olthuis W, van den Berg A, Measuring Both pH and O2 with a Single On-Chip Sensor in Cultures of Human Pluripotent Stem Cell-Derived Cardiomyocytes to Track Induced Changes in Cellular Metabolism, ACS Sensors 6(1) (2021) 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brady AJ, Mechanical properties of isolated cardiac myocytes, Physiol Rev 71(2) (1991) 413–28. [DOI] [PubMed] [Google Scholar]
- [13].Ye L, Qiu L, Feng B, Jiang C, Huang Y, Zhang H, Zhang H, Hong H, Liu J, Role of Blood Oxygen Saturation During Post-Natal Human Cardiomyocyte Cell Cycle Activities, JACC: Basic to Translational Science 5(5) (2020) 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xin T, Lv W, Liu D, Jing Y, Hu F, Opa1 Reduces Hypoxia-Induced Cardiomyocyte Death by Improving Mitochondrial Quality Control, Frontiers in Cell and Developmental Biology 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [15].Puente Bao N., W. Kimura, Shalini A. Muralidhar, J. Moon, James F. Amatruda, Kate L. Phelps, D. Grinsfelder, Beverly A. Rothermel, R. Chen, Joseph A. Garcia, Celio X. Santos, S. Thet, E. Mori, Michael T. Kinter, Paul M. Rindler, S. Zacchigna, S. Mukherjee, David J. Chen, Ahmed I. Mahmoud, M. Giacca, Peter S. Rabinovitch, A. Asaithamby, Ajay M. Shah, Luke I. Szweda, Hesham A. Sadek, The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response, Cell 157(3) (2014) 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li F, Hu Y, Nie J, Fu FH, Effects of acute, intermittent exercise in hypoxic environments on the release of cardiac troponin, Scand J Med Sci Sports 26(4) (2016) 397–403. [DOI] [PubMed] [Google Scholar]
- [17].Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW, Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions, Exp Mol Med 36(1) (2004) 1–12. [DOI] [PubMed] [Google Scholar]
- [18].Datta Chaudhuri R, Banik A, Mandal B, Sarkar S, Cardiac-specific overexpression of HIF-1α during acute myocardial infarction ameliorates cardiomyocyte apoptosis via differential regulation of hypoxia-inducible pro-apoptotic and anti-oxidative genes, Biochemical and Biophysical Research Communications 537 (2021) 100–108. [DOI] [PubMed] [Google Scholar]
- [19].Madan S, Saeed O, Shin J, Sims D, Goldstein D, Piña I, Jorde U, Patel SR, Donor Troponin and Survival After Cardiac Transplantation, Circulation: Heart Failure 9(6) (2016) e002909. [DOI] [PubMed] [Google Scholar]
- [20].Zimmermann R, Baki S, Dengler TJ, Ring GH, Remppis A, Lange R, Hagl S, Kübler W, Katus HA, Troponin T release after heart transplantation, Br Heart J 69(5) (1993) 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu X, Tooley J, Løberg EM, Suleiman MS, Thoresen M, Immediate hypothermia reduces cardiac troponin I after hypoxic-ischemic encephalopathy in newborn pigs, Pediatr Res 70(4) (2011) 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chaillou T, Skeletal Muscle Fiber Type in Hypoxia: Adaptation to High-Altitude Exposure and Under Conditions of Pathological Hypoxia, Front Physiol 9 (2018) 1450–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Terracio L, Miller B, Borg TK, Effects of cyclic mechanical stimulation of the cellular components of the heart: in vitro, In Vitro Cell Dev Biol 24(1) (1988) 53–8. [DOI] [PubMed] [Google Scholar]
- [24].Yamazaki T, Komuro I, Kudoh S, Zou Y, Nagai R, Aikawa R, Uozumi H, Yazaki Y, Role of ion channels and exchangers in mechanical stretch-induced cardiomyocyte hypertrophy, Circ Res 82(4) (1998) 430–7. [DOI] [PubMed] [Google Scholar]
- [25].Sadeghi AH, Shin SR, Deddens JC, Fratta G, Mandla S, Yazdi IK, Prakash G, Antona S, Demarchi D, Buijsrogge MP, Sluijter JPG, Hjortnaes J, Khademhosseini A, Engineered 3D Cardiac Fibrotic Tissue to Study Fibrotic Remodeling, Adv Healthc Mater 6(11) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fink C, Ergün S, Kralisch D, Remmers U, Weil J, Eschenhagen T, Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement, Faseb j 14(5) (2000) 669–79. [DOI] [PubMed] [Google Scholar]
- [27].Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, Takaku F, Yazaki Y, Stretching cardiac myocytes stimulates protooncogene expression, J Biol Chem 265(7) (1990) 3595–8. [PubMed] [Google Scholar]
- [28].Cooper G.t., Cardiocyte adaptation to chronically altered load, Annu Rev Physiol 49 (1987) 501–18. [DOI] [PubMed] [Google Scholar]
- [29].Annabi N, Selimovid Š, Acevedo Cox JP, Ribas J, Afshar Bakooshli M, Heintze D, Weiss AS, Cropek D, Khademhosseini A, Hydrogel-coated microfluidic channels for cardiomyocyte culture, Lab on a chip 13(18) (2013) 3569–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Camci-Unal G, Annabi N, Dokmeci MR, Liao R, Khademhosseini A, Hydrogels for cardiac tissue engineering, NPG Asia Materials 6(5) (2014) e99–e99. [Google Scholar]
- [31].Luo Z, Zhou X, Mandal K, He N, Wennerberg W, Qu M, Jiang X, Sun W, Khademhosseini A, Reconstructing the tumor architecture into organoids, Advanced Drug Delivery Reviews 176 (2021) 113839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Peltz M, Cobert ML, Rosenbaum DH, West LM, Jessen ME, Myocardial perfusion characteristics during machine perfusion for heart transplantation, Surgery 144(2) (2008) 225–32. [DOI] [PubMed] [Google Scholar]
- [33].Ashammakhi N, Darabi MA, Kehr NS, Erdem A, S.-k. Hu, M.R. Dokmeci, A.S. Nasr, A. Khademhosseini, Advances in Controlled Oxygen Generating Biomaterials for Tissue Engineering and Regenerative Therapy, Biomacromolecules 21(1) (2020) 56–72. [DOI] [PubMed] [Google Scholar]
- [34].Motealleh A, Schäfer AH, Fromm O, Kehr NS, 3D-Printed Oxygen-Carrying Nanocomposite Hydrogels for Enhanced Cell Viability under Hypoxic and Normoxic Conditions, Biomacromolecules 22(11) (2021) 4758–4769. [DOI] [PubMed] [Google Scholar]
- [35].Alemdar N, Leijten J, Camci-Unal G, Hjortnaes J, Ribas J, Paul A, Mostafalu P, Gaharwar AK, Qiu Y, Sonkusale S, Liao R, Khademhosseini A, Oxygen-Generating Photo-Cross-Linkable Hydrogels Support Cardiac Progenitor Cell Survival by Reducing Hypoxia-Induced Necrosis, ACS Biomater. Sci. Eng. 3 (2017) 1964. [DOI] [PubMed] [Google Scholar]
- [36].Hsieh T-E, Lin S-J, Chen L-C, Chen C-C, Lai P-L, Huang C-C, Optimizing an Injectable Composite Oxygen-Generating System for Relieving Tissue Hypoxia, Frontiers in Bioengineering and Biotechnology 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khorshidi S, Karkhaneh A, Bonakdar S, Fabrication of amine-decorated nonspherical microparticles with calcium peroxide cargo for controlled release of oxygen, J Biomed Mater Res A 108(1) (2020) 136–147. [DOI] [PubMed] [Google Scholar]
- [38].Erdem A, Haghniaz R, Ertas YN, Sangabathuni SK, Nasr AS, Swieszkowski W, Ashammakhi N, Methods for fabricating oxygen releasing biomaterials, Journal of Drug Targeting 30(2) (2022) 188–199. [DOI] [PubMed] [Google Scholar]
- [39].Mohseni-Vadeghani E, Karimi-Soflou R, Khorshidi S, Karkhaneh A, Fabrication of oxygen and calcium releasing microcarriers with different internal structures for bone tissue engineering: Solid filled versus hollow microparticles, Colloids and Surfaces B: Biointerfaces 197 (2021) 111376. [DOI] [PubMed] [Google Scholar]
- [40].Essa D, Kondiah PPD, Choonara YE, Pillay V, The Design of Poly(lactide-co-glycolide) Nanocarriers for Medical Applications, Front Bioeng Biotechnol 8 (2020) 48–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shiekh PA, Singh A, Kumar A, Oxygen-Releasing Antioxidant Cryogel Scaffolds with Sustained Oxygen Delivery for Tissue Engineering Applications, ACS Applied Materials & Interfaces 10(22) (2018) 18458–18469. [DOI] [PubMed] [Google Scholar]
- [42].Morais AI, Wang X, Vieira EG, Viana BC, Silva-Filho EC, Osajima JA, Afewerki S, Corat MA, Silva HS, Marciano FR, Ruiz-Esparza GU, Stocco TD, de Paula MM, Lobo AO, Electrospraying Oxygen-Generating Microparticles for Tissue Engineering Applications, Int J Nanomedicine 15 (2020) 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Seekell RP, Lock AT, Peng Y, Cole AR, Perry DA, Kheir JN, Polizzotti BD, Oxygen delivery using engineered microparticles, Proceedings of the National Academy of Sciences 113(44) (2016) 12380–12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ma C, Beyer AM, Durand M, Clough AV, Zhu D, Norwood Toro L, Terashvili M, Ebben JD, Hill RB, Audi SH, Medhora M, Jacobs ER, Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins, Arteriosclerosis, Thrombosis, and Vascular Biology 38(3) (2018) 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Suszynski TM, Rizzari MD, Scott WE, Eckman PM, Fonger JD, John R, Chronos N, Tempelman LA, Sutherland DE, Papas KK, Persufflation (gaseous oxygen perfusion) as a method of heart preservation, Journal of Cardiothoracic Surgery 8(1) (2013) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Abulateefeh SR, Alkilany AM, Synthesis and Characterization of PLGA Shell Microcapsules Containing Aqueous Cores Prepared by Internal Phase Separation, AAPS PharmSciTech 17(4) (2016) 891–897. [DOI] [PubMed] [Google Scholar]
- [47].Wang J, Zhu Y, Bawa HK, Ng G, Wu Y, Libera M, van der Mei HC, Busscher HJ, Yu X, Oxygen-Generating Nanofiber Cell Scaffolds with Antimicrobial Properties, ACS Appl. Mater. Interfaces 3 (2011) 67. [DOI] [PubMed] [Google Scholar]
- [48].Mandal K, Wang I, Vitiello E, Orellana LAC, Balland M, Cell dipole behaviour revealed by ECM sub-cellular geometry, Nature Communications 5(1) (2014) 5749. [DOI] [PubMed] [Google Scholar]
- [49].Mandal K, Raz-Ben Aroush D, Graber ZT, Wu B, Park CY, Fredberg JJ, Guo W, Baumgart T, Janmey PA, Soft Hyaluronic Gels Promote Cell Spreading, Stress Fibers, Focal Adhesion, and Membrane Tension by Phosphoinositide Signaling, Not Traction Force, ACS Nano 13(1) (2019) 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mandal K, Gong Z, Rylander A, Shenoy VB, Janmey PA, Opposite responses of normal hepatocytes and hepatocellular carcinoma cells to substrate viscoelasticity, Biomaterials Science 8(5) (2020) 1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. https://wtrs.com/celsior-cold-storage-solution/
- [52].Gauvin R, Chen YC, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A, Microfabrication of Complex Porous Tissue Engineering Scaffolds Using 3D Projection Stereolithography, Biomaterials 33 (2012) 3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kim H-W, Lee H-H, Knowles JC , Electrospinning biomedical nanocomposite fibers of hydroxyapatite/poly(lactic acid) for bone regeneration, Journal of Biomedical Materials Research Part A 79A(3) (2006) 643–649. [DOI] [PubMed] [Google Scholar]
- [54].Rastinfard A, Nazarpak MH, Moztarzadeh F, Controlled chemical synthesis of CaO2 particles coated with polyethylene glycol: characterization of crystallite size and oxygen release kinetics, RSC Advances 8 (2018) 91–101. [Google Scholar]
- [55].Pachuau L, Mazumder B, A study on the effects of different surfactants on Ethylcellulose microspheres, 2009. [Google Scholar]
- [56].Jelvehgari M, Nokhodchi A, Rezapour M, Valizadeh H, Effect of formulation and processing variables on the characteristics of tolmetin microspheres prepared by double emulsion solvent diffusion method, Indian J Pharm Sci 72(1) (2010) 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li W, Zhang L, Ge X, Xu B, Zhang W, Qu L, Choi C-H, Xu J, Zhang A, Lee H, Weitz DA, Microfluidic fabrication of microparticles for biomedical applications, Chem Soc Rev 47(15) (2018) 5646–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stromberg ZR, Lisa Phipps M, Magurudeniya HD, Pedersen CA, Rajale T, Sheehan CJ, Courtney SJ, Bradfute SB, Hraber P, Rush MN, Kubicek-Sutherland JZ, Martinez JS, Formulation of stabilizer-free, nontoxic PLGA and elastin-PLGA nanoparticle delivery systems, International Journal of Pharmaceutics 597 (2021) 120340. [DOI] [PubMed] [Google Scholar]
- [59].Lagreca E, Onesto V, Di Natale C, La Manna S, Netti PA, Vecchione R, Recent advances in the formulation of PLGA microparticles for controlled drug delivery, Progress in Biomaterials 9(4) (2020) 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tretter V, Zach M-L, Böhme S, Ullrich R, Markstaller K, Klein KU, Investigating Disturbances of Oxygen Homeostasis: From Cellular Mechanisms to the Clinical Practice, Front Physiol 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Butler JP, Tolić-Nørrelykke IM, Fabry B, Fredberg JJ, Traction fields, moments, and strain energy that cells exert on their surroundings, Am J Physiol Cell Physiol 282(3) (2002) C595–605. [DOI] [PubMed] [Google Scholar]
- [62].Sabass B, Gardel ML, Waterman CM, Schwarz US, High resolution traction force microscopy based on experimental and computational advances, Biophysical journal 94(1) (2008) 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Califano JP, Reinhart-King CA, Substrate Stiffness and Cell Area Predict Cellular Traction Stresses in Single Cells and Cells in Contact, Cell Mol Bioeng 3(1) (2010) 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang H-Y, Speicher DW, Sanger JW, Sanger JM, Discher DE, Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating, J Cell Sci 121(Pt 22) (2008) 3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gölgeli A, Coşkun A, Ozesmi C, Rat diaphragm muscle contraction with and without oxygen enrichment, Res Commun Mol Pathol Pharmacol 90(1) (1995) 87–95. [PubMed] [Google Scholar]
- [66].Wright VP, Klawitter PF, Iscru DF, Merola AJ, Clanton TL, Superoxide scavengers augment contractile but not energetic responses to hypoxia in rat diaphragm, Journal of Applied Physiology 98(5) (2005) 1753–1760. [DOI] [PubMed] [Google Scholar]
- [67].Depré C, Vanoverschelde JL, Goudemant JF, Mottet I, Hue L, Protection against ischemic injury by nonvasoactive concentrations of nitric oxide synthase inhibitors in the perfused rabbit heart, Circulation 92(7) (1995) 1911–8. [DOI] [PubMed] [Google Scholar]
- [68].Qin G, Jernryd V, Sjöberg T, Steen S, Nilsson J, Machine Perfusion for Human Heart Preservation: A Systematic Review, Transpl Int 35 (2022) 10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li J, Conrad C, Mills TW, Berg NK, Kim B, Ruan W, Lee JW, Zhang X, Yuan X, Eltzschig HK, PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling, Journal of Experimental Medicine 218(6) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.