

Abstract
Cancer Stem-like Cells (CSCs) are often the root cause of refractive relapse due to their inherent resistance to most therapies and ability to rapidly self-propagate. Recently the antigen CD133 has been identified as a CSC marker on several cancer types and αCD133 therapies have demonstrated selective targeting against CSCs with minimal off-target toxicity. Theoretically, by selectively eliminating CSCs the sensitivity to bulk tumor targeting therapies should be enhanced. Previously, our laboratory has developed bispecific chemically self-assembled nanorings (CSANs) that successfully induced T-cell eradication of EpCAM positive tumors. We reasoned that targeting both CSCs (CD133+) and the bulk tumor (EpCAM+) simultaneously using our CSAN platform should produce a synergistic effect. We evaluated αCD133/αCD3 CSANs as both a single agent and in combination with αEpCAM/αCD3 CSANs to treat triple negative breast cancer (TNBC) cells which express a subpopulation of CD133+ cancer stem cells and EpCAM+ bulk tumor cells. Furthermore, an orthotopic breast cancer model validated the ability of αCD133 and αEpCAM targeting to combine synergistically in elimination of TNBC MDA-MB-231cells. Complete tumor eradication only occurred when EpCAM and CD133 were targeted simultaneously and lead to full remission in 80% of the test mice. Importantly, the depletion and enrichment of CD133 TNBCs highlighted the role of CD133 positive cancer cells in regulating tumor growth and progression. Collectively, our results demonstrate that dual targeting with bispecific CSANs can be effective against heterogenous tumor cell populations and that elimination of primary and CD133+ CSCs maybe necessary for eradication of at least a sub-set of TNBC.
Keywords: Immunotherapy, Nanotechnology, Bispecific, anti-cancer, T cell, CD133, EpCAM, Tumor Initiating Cells, Dual Targeting
INTRODUCTION
Recent response rates to targeted cancer therapies have been encouraging, yielding excellent growth inhibition and occasionally the complete clearance of primary tumors. Nevertheless, long-term remission remains a challenging endeavor to achieve. The past decade has successfully confirmed the existence of cancer like stem cells (CSCs), which can compose anywhere from 0.1% to 20% of the bulk tumor cells (1). CSCs, commonly referred to as tumor-initiating cells or tumor-propagating cells, are subpopulations of highly tumorigenic, self-propagating, and undifferentiated cells typically responsible for promoting tumor initiation, and differentiation into the various tumor cell entities of a carcinoma (2–4). Furthermore, CSCs are often the root cause of refractive relapse due to their resistance of chemotherapy and radiation treatment (5,6). CD133 is one recently identified CSC biomarker and has been reported to present on the surface of various tumor types, most notably: brain, breast, colon, pancreatic, and prostate, liver, ovarian and lung (7–12).
Previously, our laboratory has explored the use of chemically self-assembled nanoring (CSANs) as an alternative to current bispecific formats. CSANs have been formed by fusing a binding ligand such as a single-chain variable fragment (scFv) to two E. Coli dihydrofolate reductase (DHFR2) molecules that spontaneously assemble into octomeric nanorings upon the addition of a chemical dimerizer, bis-methotrexate (bisMTX) (Figure 1) (13). The self-assembly of an αCD3 fusion protein with a tumor targeting fusion protein results in the formation of bispecific, multivalent, CSANs able to selectively target T-cells to tumor cells and initiate tumor cell killing (14–16). We have previously incorporated a variety of targeting moieties (e.g. scFvs, fibronectins, and small peptide sequences) onto the C-termini of our DHFR2 proteins (e.g. scFv-DHFR2), thereby enabling the CSANs to selectively target cell surface receptors (17,18). A key feature of our approach is the ability to disassemble the CSANs in the presence of the FDA approved antibiotic, trimethoprim (13,14,19). CSANs displaying targeting moieties against EpCAM and EGFR receptors, αEpCAM/αCD3 and αEGFR/αCD3 CSANs respectively, demonstrated stable binding to T cells (14,16). In addition, when in the presence of the target antigen, CSAN directed T-cells demonstrated rapid activation with the upregulation of both activation markers (CD69 and CD25) and the production of interleukin-2 (IL-2) and interferon-γ (IFN-γ), followed by subsequent clearance of tumor cells, both in vitro and in vivo (14). Furthermore, our results demonstrated a significantly lower rate of naïve T-cell activation compared to memory cells, thus limiting the potential for induced naïve T-cell anergy. Due to the success of CSAN directed T-cells against solid tumors, we hypothesized that CSANs incorporating αCD133 might be applied to the selective elimination of CD133+ tumor initiating cells.
Figure 1. αEpCAM and αCD133 Chemically Self-Assembled Nanorings (CSANs).
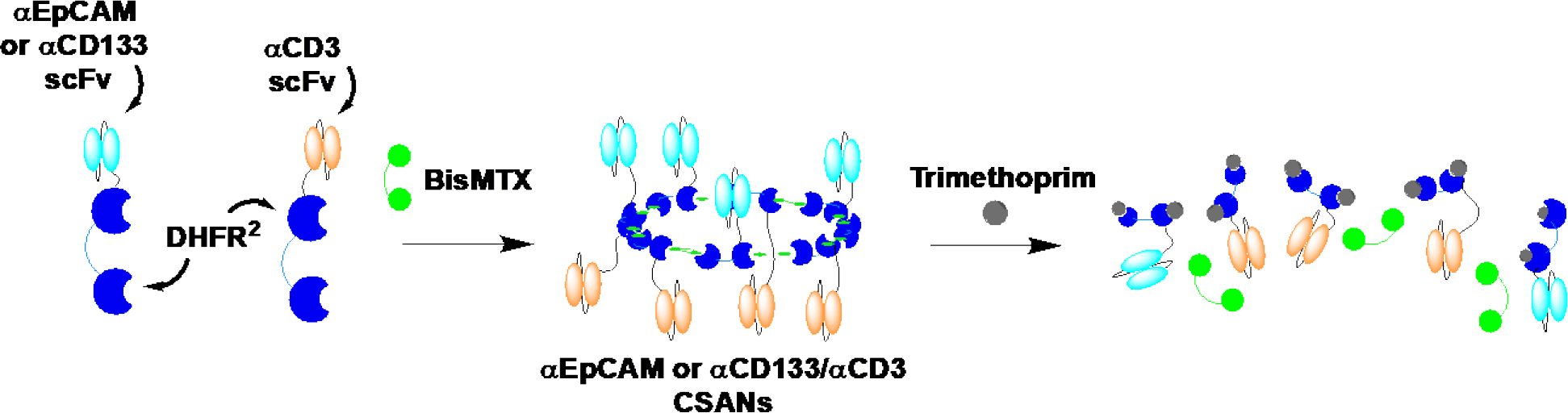
Monomers composed of either αEpCAM or αCD133 scFv and dihydrofolate reductase-dihydrofolate reductase (DHFR2) fusion proteins (DHFR2-αEpCAM, DHFR2-αCD3 and DHFR2-αCD3) were incubated with BisMTX resulting in their self-assembly into αEpCAM or αCD133/αCD3 CSANs. Treatment with the FDA approved antibiotic and competitive inhibitor, trimethoprim, results in disassembly of the CSANs.
Due to our previous success in eliminating EpCAM+ breast cancer tumors, we chose to target both EpCAM and CD133 in TNBC tumors using αCD133-DHFR2, capable of binding both glycosylated and non-glycosylated forms of CD133, and αEpCAM-DHFR2 (Figure 1) (20). Previously, expression of CD133 was confirmed on 2–4% of breast cancer cell lines generated from Brca1Δexon11/p53+/− mouse mammary tumors (21). Specifically, these CD133+ cells have been confirmed to display stem cell like characteristics; including the expression of stem cell genes, the ability to form spheroids and the in vivo reconstitution of tumors with as few as 100 cells in an immunocompromised mouse (21). TNBC represents a unique subgroup of breast cancer with a specific molecular profile, aggressive behavior pattern, lack of effective therapies and relatively poor prognosis (22). Furthermore, TNBC has been shown to have a CD133+ CSCs population that is responsible for tumor vasculogenesis (23,24).
In this report, we have characterized the ability of bispecific CSANs targeting EpCAM and CD133 separately and in combination to induce T-cell eradication of TNBC tumors and to assess the importance of CD133 expressing cells on tumor growth.
MATERIALS AND METHODS
Cell Lines and Blood Samples
HT-29 and MDA-MB-231 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and were monolayer cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and L-glutamine at 37°C in 5% CO2. Cell lines were validated via STR fingerprinting by Cytogenetics and Cell Authentication Core at MD Anderson Cancer Center (December 2019) and all studies are performed with validated stocks and kept under 5 passages. PCR Mycoplasma Detection Kit ((Applied Biological Materials Inc., Cat. # G238) was used to check for mycoplasma contamination (last confirmed January 2021). MDA-MB-231 cells were grown in 3D culture by 96-well hanging drop plates (Perfecta 3D, 3D Biomatrix, MI). 2.4×103 cells were seeded per well in 30 μl with complete medium for 48 hours before aggregates were collected by gentle PBS flushing into a 50 ml conical tube where the supernatant was aspirated, and spheroids were collected. Human PBMCs were isolated from buffy coats of healthy donor blood samples (obtained from Memorial Blood Centers, St. Paul, MN) by Ficoll density gradient centrifugation. PBMCs were cultured in complete RPMI 1640 medium (Lonza) supplemented with 10% (v/v) fetal bovine serum, L-glutamine (final concentration of 2mM), Penicillin (100 units/mL), and Streptomycin (100 μg/mL) in a humidified incubator with 5% CO2 at 37 °C.
T-cell Cytotoxicity Assays
Cell lysis was evaluated by measuring LDH (lactate dehydrogenase) release per the conditions provided by non-radioactive cytotoxicity assay (CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega). The day prior to the experiment 5×103 target cells, colon carcinoma (HT-29), TN Breast cancer (MDA-MB-231) cells or Glioblastoma cells (U87-MG) were seeded into a 96-well plate in 200 μL of RPMI-1640 media per well. The following day resting PBMCs, at a 10:1 effector (E) to target (T) cell (E:T) ratio, were counted and incubated with 0 to 200 nM of αEpCAM/αCD3 bispecific CSANs, αCD133/αCD3 bispecific CSANs or both for one hour at 37°C with 5% C02. Following the initial incubation, PBMCs were washed and resuspended in RPMI before addition to 96-well plate containing target cells and incubated for 24 hours under standard conditions.
Immunostaining and Cytokine Analysis
IL-2 and IFN-γ measurements in the cytotoxicity assay supernatants were analyzed using ELISA per the conditions provided by IFN-γ ELISA kit (Invitrogen) and IL-2 ELISA kit (Invitrogen). In brief, following the incubation period the supernatant was removed and 10-fold diluted into ELISA buffer (provided in the kit) prior to placing 50 μL into the respective well of the included ELISA 96-well plate. IFN-γ and IL-2 production of experimental wells was determined through a standard curve generated from known control sample concentrations.
Orthotopic TN Breast Cancer (MDA-MB-231) Model
All animal experiments were approved by and performed according to the guidelines of the Institutional Animal Care Committee of the University of Minnesota according to the guidelines specified by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals (reprint 2015) available from the Office of Laboratory Animal Welfare. Six- to 8-wk-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were injected unilaterally into the fourth mammary fat pad with 1.0×106 MDA-MB-231 cells while the mouse was under 2% isoflurane. The injection was performed in 50 μL of 50:50 Matrigel/PBS directly through the nipple. Once tumors were ~50 mm3 mice were randomized into cohorts of 5 mice, outliers were removed from the study, and those treatment groups receiving PBMC based therapy were IV infused with 20 million peripheral Blood Mononuclear Cells (PBMCs) for the engraftment of a human immune system. Treatments were initiated 4 days later by IV tail vein injection, including: PBS, PBMC only (this experimental group acquired treatments of PBS only following the initial PBMC engraftment), 1 mg/kg αCD3 monospecific CSANs, 1 mg/kg αCD133/αCD3 bispecific CSANs, 1 mg/kg αEpCAM/αCD3 bispecific CSANs, and both αCD133/αCD3 and αEpCAM/αCD3 bispecific CSANs at .5 mg/kg each. Treatments were administered every 2 days for a total of 6 treatments. Body weight was monitored daily, and tumor growth was monitored daily by caliper to measure the height x width x length and recorded as mm3.
A second cohort of mice were sacrificed 1 day after the final treatment and their tumors were isolated and minced. 10–15ml of warm tumor Collagenase solution was added tubes were shaken on rotary shaker for 2–3 hours until all larger tissue fragments were digested. HBSS with 2% FBS was added to the 50 mL tube and each sample was passed through a 40 uM cell filter. Samples were spun down, washed in HBSS and counted prior to flow cytometry analysis. Cells were stained with αCD133-PE, αHuman-BV421-IgG, αHuman-CD3-BV605, αEpCAM-AF647 and ghost red viable dye before analysis with an LSRFortessa H0081. Cells positive for ghost red viability, negative αHuman-BV421-IgG, and positive for αHuman-CD3-BV605 were removed from analysis by gating. All remaining αHuman positive cells were characterized for expression levels of CD133 and EpCAM. Data was collected for each sample until a total of ~10,000 ± 200 alive, human, and CD3 negative cells were obtained for analysis (i.e. alive tumor cells).
CD133 Depletion Assays
Non-coated NH2-Dynabeads were functionalized with an excess of Biotin-PEG4-NHS ester (per manufacturer recommended instructions). Bispecific CSANs comprised of αCD133-DHFR2 and Monovalent Streptavidin (mSA) functionalized DHFR2 (mSA-DHFR2) were generated at 100 nM (αCD133/mSA CSANs) and .1 mg of total protein was coated onto 500 μl of Biotin functionalized beads. Beads were washed with PBS x3 prior to incubation with 100 million MDA-MB-231 cells for 30 minutes at 4°C before magnet bead isolation. The supernatant, CD133- population, was removed and the beads were washed 3x with PBS to acquire pure CD133+ cells. The beads were incubated with 200 μM Trimethoprim, in PBS, for 30 minutes at room temp to disassemble the αCD133/mSA CSANs and release pure CD133+ cells into the supernatant. Isolated CD133 positive cells were added to cultured MDA-MB-231 cells for an enriched population. To ensure non-specific binding didn’t occur, isolation controls were generated by combining non-targeted DHFR2 (1DD) and mSA-DHFR2 to generate CSANs with only mSA binding potential (1DD/mSA CSANs). Isolations were validated by flow cytometry with αCD133-PE mAB.
8- to 10-week-old female NSG Mice were injected with either 106 standard MDA-MB-231 cells, Enriched CD133+ cells or depleted CD133− cells in the mammary fat pad through the 4th nipple. Injection was performed in 50 μL 50:50 Matrigel/PBS while the mouse was under general anesthesia. Once tumors reached 1000mm3 mice were euthanized and their tumors were isolated and minced as stated in the procedure above. Cells were stained with αCD133-PE, αHuman-BV421-IgG, αEpCAM-AF647 and ghost red viable dye before analysis with an LSRFortessa H0081. Cells positive for ghost red viability and negative for αHuman-BV421-IgG were removed from analysis by gating. All remaining αHuman positive cells were characterized for expression levels of CD133 and EpCAM. Data was collected for each sample until a total of ~10,000 ± 150 alive and human cells were obtained for analysis (i.e. alive tumor cells).
Statistical analysis
Data was analyzed with GraphPad Prism 5.0 software using two-tailed unpaired Student’s t-test for comparison of treatment to control groups. P < 0.05 was considered statistically significant and values are denoted with asterisks as follows: ns P > 0.05 not significant; * P < 0.05; ** P < 0.01; *** P < 0.001 or value is indicated within the figure and legend.
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.
RESULTS
In Vitro Analysis of αCD133/αCD3 Bispecific CSANs
To determine the potential for CSAN combination therapy, we decided to test the efficacy of bispecific CSANs against CD133 and EpCAM positive solid tumor tissues, targeting either antigen separately or in combination. Consequently, we prepared both αEpCAM/αCD3 and αCD133/αCD3 CSANs to characterize their ability to bind and activate T-cells in the presence of EpCAM and CD133 expressing tumor cells. The development, preparation and characterization of αEpCAM-DHFR2 and αCD3-DHFR2 proteins have been previously described.(13,14,19) Due to the importance of targeting both glycosylated and non-glycosylated forms of CD133, we chose to incorporate the scFv designed by Ohlfest and coworkers (derived from the αCD133 targeting clone 7 monoclonal antibody (mAb)) into our DHFR2 construct as a fusion protein.(25) The αCD133 sequence was generously obtained from Dr. Dan Vallera and subsequently ligated onto the N-terminus of our DHFR2 construct with a standard 13 amino acid linker (Supplemental Figure 1A). All plasmid constructs were verified by classic sanger sequencing before transformation into BL21 E. coli expression cells and expressed as an insoluble inclusion body. The isolated yield for the purified αCD133-DHFR2 was found to be 2 mg/L of culture (Supplemental Figure 1B).
Following the production and purification of the αCD133-DHFR2 monomer, we incubated the fusion protein with αCD3-DHFR2 and bisMTX dimerizer (1:1:2 equivalents) for 30 minutes to form αCD133/αCD3 CSANs, which were characterized by size exclusion chromatography (Figure 1 and Supplemental Figure 2A). As with previous octameric CSAN constructs, the rings are randomly generated with combinations of the two targeting elements and calculated to be statistically greater than ninety-nine percent bispecific in makeup. As anticipated, the αCD133/αCD3 based CSANs where consistent with previous scFv based constructs and eluted at ~19 minutes with nearly 100% oligomerization, whereas the smaller αCD133-DHFR2 monomer proteins eluted at 28.5 minutes. Importantly, the self-assembled αCD133/αCD3 CSANs exhibited the same size dimensions, and thus polyvalency (7–10), found for previous bispecific CSANs in the laboratory. This was further reinforced by dynamic light scattering (DLS) indicating the lack of aggregation and extremely tight size distribution characteristic of CSANs (Supplemental Figure 2B), as previously observed for other CSANs constructs.(14,16,18)
Binding studies were performed using flow cytometry, and the confirmed CSANs, to ensure the functionality of the CD133 targeting scFv. We utilized a colorectal carcinoma cell line (HT-29) for initial binding studies as they universally express CD133 and simplified early evaluations. Monospecific αCD133 CSANs were generated and incubated with HT-29 cells in addition to co-staining with αFLAG-PE to probe for the FLAG tag present on αCD133-DHFR2. As seen in supplemental figure 3 binding of the αCD133 CSANs to HT-29 cells was observed and comparable to binding seen from a commercially available αCD133 mAb. Interestingly, only the αCD133 constructs with the scFv fused to the N-terminus, opposed to C-terminus, of DHFR2 were capable of binding, most likely due to unforeseen steric hindrances.
Initial cytotoxicity studies were carried out with HT-29 cells due to both their universal expression levels of CD133 as well as high receptor density of 1.1×106 receptors per cell (supplemental figure 3). Unactivated T-cells, treated with increasing αCD133/αCD3 CSAN concentrations, were incubated with target HT-29 cells for 24 hours at an effector-to-target ratio of 10:1 (Figure 2A). Maximal cytotoxicity, 59% target cell lysis, was observed for cells incubated with 100 nM of the αCD133/αCD3 CSANs, with significant cytotoxicity observed at concentrations as low as 10 nM. These results were found to be consistent to HT-29 cells treated with αEpCAM/αCD3 CSANs alone or αCD133/αCD3 CSANs in addition to αEpCAM/αCD3 CSANs, which is not surprising as HT-29 cells are also EpCAM+ (7.0×106 EpCAM/cell) (Figure 2B, 2C and Supplemental Figure S4). Additionally, the relative killing potential was similar to previously seen scFv targeted CSANs (14–16). In fact, 60 – 70% is the maximum cell killing we would expect in this type of in vitro study, without Matrigel, due to limited T-cell motility. When targeted against U87-MG control cells which express neither EpCAM or CD133 we saw no cytotoxicity from either αEpCAM/αCD3 CSANs, αCD133/αCD3 CSANs or a combination (Supplemental Figure 5). Additionally, replacing αCD3-DHFR2 with non-targeted DHFR2 (1DD) eliminated all cell killing, whereas CSANS formed with 1DD replacing a tumor targeting moiety exhibited maximum cell killing of 19% (figure 2). This number is consistent with previous results indicating CD3 binding alone primes T-cells to a low level in vitro (14,15).
Figure 2. αCD133/αCD3 and αEpCAM/αCD3 CSAN labeled T-cells selectively activate and kill target CD133+ and EpCAM+ cells, respectively.
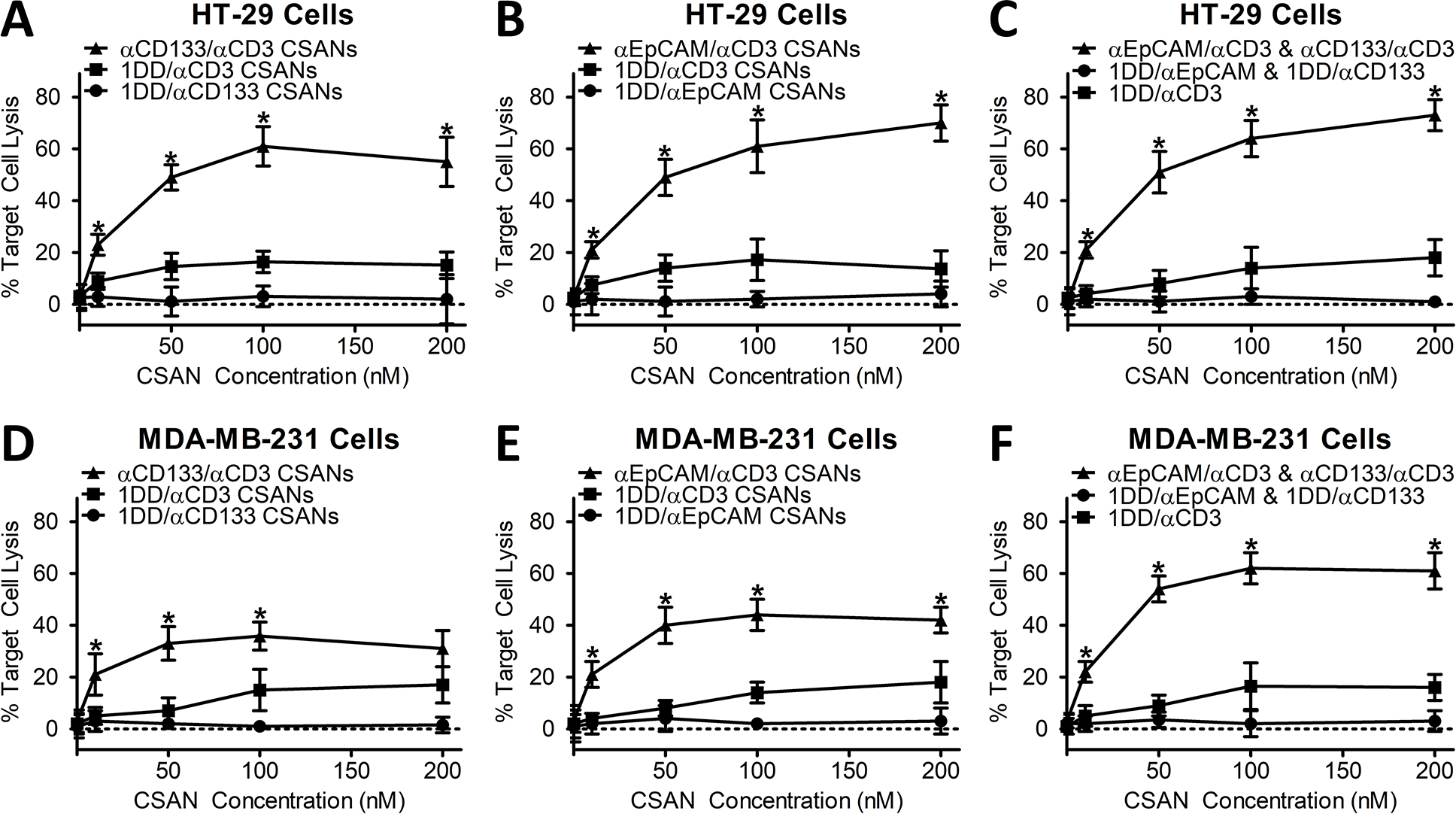
Target HT-29 cell (CD133highEpCAMhigh colon carcinoma) lysis, by unactivated PBMCs, was evaluated at a set 10:1 E:T ratio with increasing concentrations of (A) αCD133/αCD3 CSANs, (B) αEpCAM/αCD3 CSANs or (C) both αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs in combination. Target MDA-MB-231 cell (CD133lowEpCAMlow TNBC cells) lysis, by unactivated PBMCs, was evaluated at a set 10:1 E:T ratio with increasing concentrations of (D) αCD133/αCD3 CSANs and (E) αEpCAM/αCD3 CSANs, as well as in (F) combination. When treated in combination, both αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs were used at a 1:1 ratio. CSAN controls were generated by combining a targeted DHFR2 with a non-targeted DHFR2 (1DD). Data shown was obtained from one donor (n=3), but representative of three donors. *P<0.05 with respect to targeted CSAN therapy and non-targeted CD3 control, by 2-tailed Student’s t test.
Following confirmation that αCD133/αCD3 CSANS were capable of both target antigen binding and target cancer cell lysis, we chose to evaluate a triple negative breast cancer (TNBC) model to address the specific hypothesis that targeting T-cells to a primary tumor marker (such as EpCAM) as well as a CSC marker (such as CD133) are necessary for TNBC eradication. Approximately, 10–20% of MDA-MB-231 cells (a TNBC cell line) have been demonstrated as positive for CD133 (8,21,23,26). Consistent with literature values, when grown in spheroid, we found that MDA-MB-231 cells exhibited the following heterogeneity: 81.2±1.5% EpCAM+CD133−, 11.3±2.0% EpCAM+CD133+, 6.9±1.7% EpCAMlowCD133+ and 1.1±0.5% EpCAMlowCD133− (Supplemental Figure 6).
As expected, when MDA-MB-231 cells were treated with αCD133/αCD3 CSANs the maximal target cell killing was achieved at a much lower CSAN concentration, 50 nM, due to the low percentage of total MDA-MB-231 cells expressing CD133 (Figure 2D). In fact, the hook effect (decreased levels of cell killing at concentrations above saturation) was seen at concentrations immediately above 50 nM. Furthermore, a similar effect was seen with αEpCAM/αCD3 CSANs due to low receptor count of EpCAM seen on individual MDA-MB-231 cells (5.2×104 EpCAM receptors/cell), in contrast to HT-29 cells (Figure 2E).(18) However, when both αEpCAM and αCD133 CSANs were used simultaneously, we observed a significantly higher levels of target cell lysis (62%) compared to either αEpCAM (43%) or αCD133 (38%) CSANs treated individually (Figure 2F). Of note, a hook effect was not observed at concentrations of ≤ 200 nM when αEpCAM CSANs and αCD133 were used in combination, which is likely in response to the dilution of targeting elements when two CSAN constructs are present on the same cell surface. As observed with αEpCAM/αCD3 CSANs, full activation of the T-cells by αCD133/αCD3 CSANs and the combination of αCD133/αCD3 CSANs with αEpCAM/αCD3 CSANs was dependent on the presence of antigen-positive tumor cells.
We’ve previously performed extensive evaluations on how CSANs affect T-cell signaling and activation and decided to track IL-2 and IFNγ production to monitor if combination therapy exhibited any differences (14). When PBMCs were treated with αEpCAM/αCD3 CSANs in the presence of EpCAM+ expressing MCF-7 and HT-29 cells the production of IL-2 (Figure 3A) was enhanced by 12.0- and 12.7-fold, respectively, when compared to EpCAM− U87-MG control cells. However, in the presence of MDA-MB-231 cells, which express 74-fold less EpCAM receptors (5.2×104) per cell than MCF-7 cells (3.8×106), the enhancement was only 5.7-fold (18). Likely, the higher receptor density of EpCAM on MCF-7 and HT-29 cells lead to a more robust engagement between the cell surface of PBMCs and the target cell, thus leading to a stronger activation signal. Additionally, αCD133/αCD3 CSAN treated PBMCs showed a very similar level of IL-2 production (~10-fold increase) when targeting CD133 expressing HT-29 cells, compared to non-expressing U87-MG and MCF-7 control groups. While HT-29 cells express both CD133 and EpCAM, the receptor density of CD133 (1.1×106 per cell) is lower than that of EpCAM (7.0×106 per cell), leading to a moderately lower level of activation induced by αCD133/αCD3 CSANs relative to that of αEpCAM/αCD3 CSANs. Interestingly, when αCD133/αCD3 CSANs were incubated with MDA-MB-231 cells, which only express CD133 within a ~20% subpopulation, there was a 50% reduction in IL-2 production compared to HT-29 cells which are 100% CD133+ (Figure 3A). Importantly, while only 20% of the MDA-MB-231 cells express CD133, each individual CD133+ cell expresses CD133 receptor levels to a similar density of those of HT-29 cells (Figure S4). However, if the baseline (i.e., the non-CD133 expressing target cells) is removed this reduction correlates almost exactly to the 20% subpopulation of CD133 positive MDA-MB-231 cells. Lastly, while αCD133/αCD3 CSAN and αEpCAM/αCD3 CSAN targeting of MDA-MB-231 cells (which exhibit low EpCAM receptor density and a 20% CD133 positive subpopulation) individually produced less IL-2 when compared to targeting HT-29 cells (high EpCAM density), when combined they were able to produce a far greater response against MDA-MB-231 cells (10.9-fold)
Figure 3. Synergistic αEpCAM/αCD133 Cytokine Release.
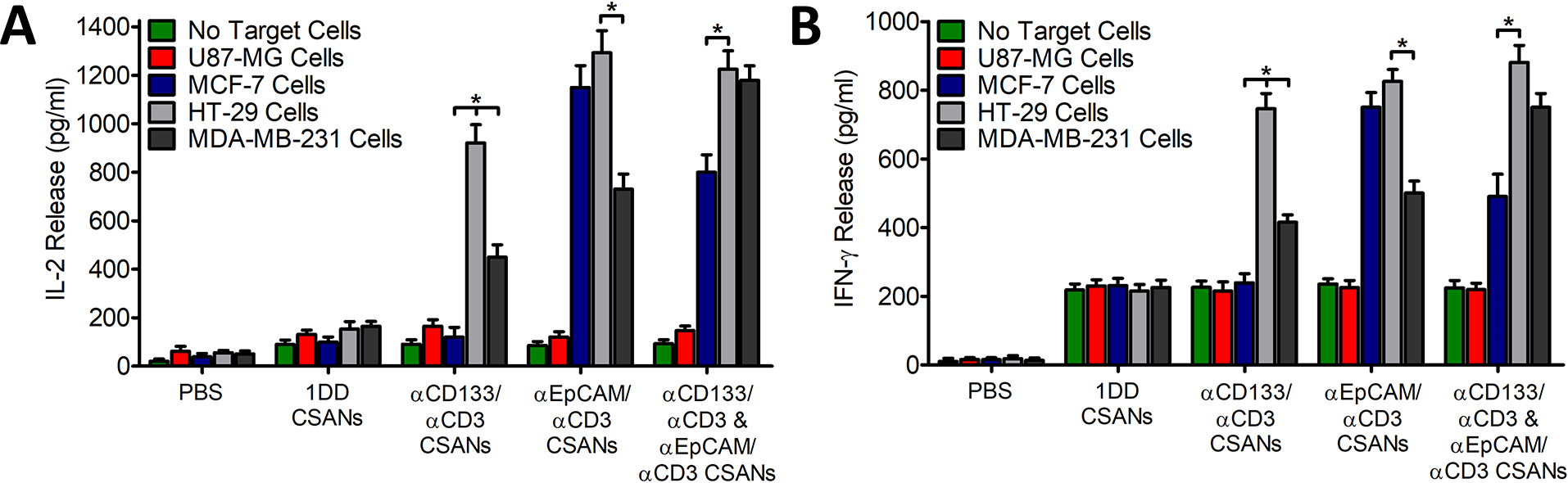
Unactivated PBMCs were co-cultured with media, 100 nM 1DD/αCD3 CSANS, 100 nM αCD133/αCD3 CSANs, 100 nM αEpCAM/αCD3 CSANs, or both 50 nM αCD133/αCD3 and 50 nM αEpCAM/αCD3 CSANs to generate non-labeled PBMCS, non-targeted CSANs, αCD133 CSANs, αEpCAM CSANs or dual targeting αCD133/αEpCAM CSANs, respectively. Generated constructs were incubated in the presence of no target cells, U87-MG cells (EpCAMnegCD133neg), MCF-7 cells (EpCAMhighCD133neg), HT-29 cells (EpCAMhighCD133pos), or MDA-MB-231 cells (EpCAMlowCD133partial pos). Following the 24-hour incubation, the media was analyzed for (A) IL-2 and (B) IFN-γ. Data shown was obtained from one donor (n=3), but representative of three donors. *P<0.05, by 2-tailed Student’s t test and one-way ANOVA.
In comparison to IL-2 production, the relative amounts of IFNγ bear a striking resemblance across all the treatment groups (Figure 3B). As observed previously, a modest amount of IFNγ is produced when αCD3 based CSANs are added to PBMCs, regardless of whether they are bispecific (14,15). A dramatic increase occurs once the bispecific CSANs engage with antigen expressing cells that is dependent on the amount of EpCAM and CD133 expression; also observed for IL-2 (Figure 3B). Thus, T-cell activation by αCD3 based CSANs appears to correlate with IL-2 and IFNγ production, with a modest level of IFNγ produced which is not indicative of CSAN directed activation.
In Vivo αEpCAM/αCD133 CSAN Anti-Tumor Activity
To establish a baseline efficacy for αCD133/αCD3 CSAN T-cells, we utilized a murine HT-29 xenograft model to ensure all tumor cells expressed CD133 antigen. To accomplish this, 1 million HT-29 cells were unilaterally injected into the flank of NOD.Cg-Prkdcscid Il2rγtm1Wjl/SzJ (NSG) mice. Once tumors reached 80 mm3, the mice were randomized into cohorts and inoculated with 20 million PBMCs. Mice were treated 4 days later by IV tail vein injection with either PBS, monospecific αCD3 CSANs (1 mg/kg), monospecific αCD133 CSANs (1 mg/kg) or bispecific αEpCAM/αCD3 CSANs (1 mg/kg) and compared to the non-treatment control. A two-day dosing schedule was chosen based on previous results demonstrating that CSANs are stable for up to 4 days on the surface of T-cells and result in potent in vivo anti-tumor activity (14). Over the course of the twelve days full tumor eradication was observed for the αCD133/αCD3 CSANs group (Figure 4A) compared to the rapid growth seen in non-targeted control groups. Additionally, the response was durable, with only a single tumor regrowth observable 30 days post treatment (Figure 4B). Similar levels of anti-tumor activity were found for NSG orthotopic xenographs of the breast cancer cell line, MCF-7 (EpCAM+) treated with αEpCAM/αCD3 CSANs (14).
Figure 4. In Vivo efficacy targeting CD133+ HT-29 colon cancer tumors with αCD133/αCD3 CSANs.
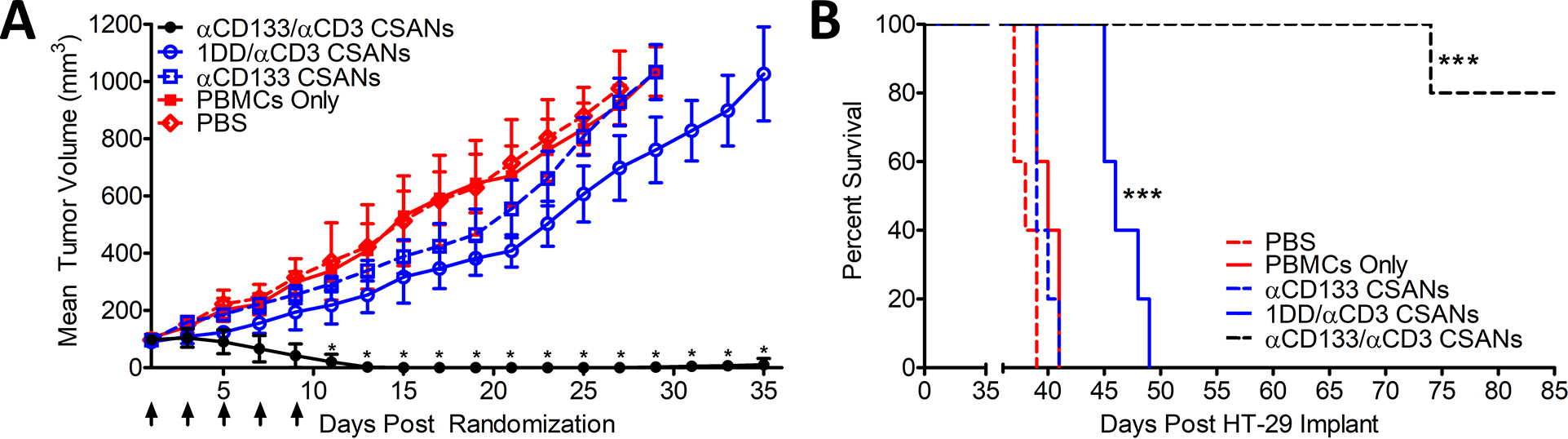
NSG mice were inoculated in the flank with 1.0×106 HT-29 cells. Cohorts were randomized when tumors were ~80 mm3 and IV inoculated with 20 million PBMCs. Treatments were initiated 4 days later, including: PBS, PBMC only (this experimental group acquired treatments of PBS only following the initial PBMC engraftment), 1 mg/kg αCD3 monospecific CSANs, 1 mg/kg αCD133 monospecific CSANs, and 1 mg/kg αEpCAM/αCD3 bispecific CSANs (n=5). Treatments were administered every 2 days for a total of 5 treatments. (A) Tumor growth was monitored every by caliper and recorded as mm3. *P<0.05 with respect to readings statistically significant from the 1DD/αCD3 CSAN control group, by 2-tailed Student’s t test. (B) Survival was monitored out to 85 days when graft vs host (GVH) disease symptoms were no longer manageable. Kaplan-Mier Survival Plot. *P<0.0001 with respect to PBS control, by log rank test. All in vivo experiments were performed independently and at least twice.
To characterize the in vivo anti-tumor activity of the αEpCAM/αCD3 CSANs and αCD133/αCD3 CSANs in combination, we utilized an orthotopic TNBC mouse model. MDA-MB-231 cells were unilaterally injected into the fourth mammary fat pad of NSG mice. Once tumors reached 50 mm3 mice were randomized in cohorts of 5 and treatment groups including T-cells were IV infused with 20 million PBMCs and allowed to engraft in NSG mice over 4 days. Following the engraftment period, mice were IV treated with either PBS, αCD3 CSANs (1 mg/kg), αEpCAM/αCD3 CSANs (1 mg/kg), αCD133/αCD3 CSANs (1 mg/kg), or a 1:1 mixture of αCD133/αCD3 and αEpCAM/αCD3 CSANs (1 mg/kg total) every two days, for 6 total treatments and compared to the non-treatment control group.
When αCD133/αCD3 or αEpCAM/αCD3 CSANs were dosed in mice bearing MDA-MB-231 tumors a partial tumor reduction response was observed (Figure 5A). αEpCAM/αCD3 CSANs rapidly induced a small decrease in tumor mass, however following completion of the dosing schedule the tumor volumes quickly rebounded to growth rates like those observed for the PBS and non-treatment control. As we previously demonstrated αEpCAM/αCD3 CSANs could eliminate MCF-7 (high EpCAM expression) breast cancer tumors, we reasoned that the inability to decrease MDA-MB-231 tumor mass further was due to the relatively low expression of EpCAM previously discussed. In contrast, αCD133/αCD3 CSANs demonstrated no initial anti-tumor activity. However, 8 days following the initial treatment, tumor growth plateaued and remained static for over 8 days following the final treatment. The delayed growth rate suggests that the smaller fraction of CD133+ MDA-MB-231 cells, relative to EpCAM+/CD133− cells, appreciably impacted tumor growth. By selectively targeting the CD133+ population, we likely reduced the most tumorigenic population. Indeed, when tumors were isolated and analyzed for EpCAM and CD133 expression we observed that αCD133/αCD3 CSANs were capable of preferentially eliminating EpCAM+CD133+ EpCAM−CD133+ cells (Figure 5C and S7). In contrast, αEpCAM/αCD3 CSANs selectively reduced the percentage EpCAM+CD133− cells, while the percentage of EpCAM+CD133+ and EpCAM−CD133+ cells increased by 50% and 63%, respectively. Given the increased resistance to apoptosis exhibited by CD133+ cells, this phenomenon may reflect the resistance of the CD133+ MDA-MB-231 cells to T-cell induced cytotoxicity, as recently observed for γδT-cells, unless CD133 is directly engaged (5,6,8). Thus, the CD133+ tumor cell population appears to be essential for tumor growth under conditions in which primary tumor cells are being targeted and sensitive to T-cell induced cytoxicity that directly targets CD133.
Figure 5. In Vivo αCD133 and αEpCAM CSAN Combination Therapy in an orthotopic TNBC Mouse Model.
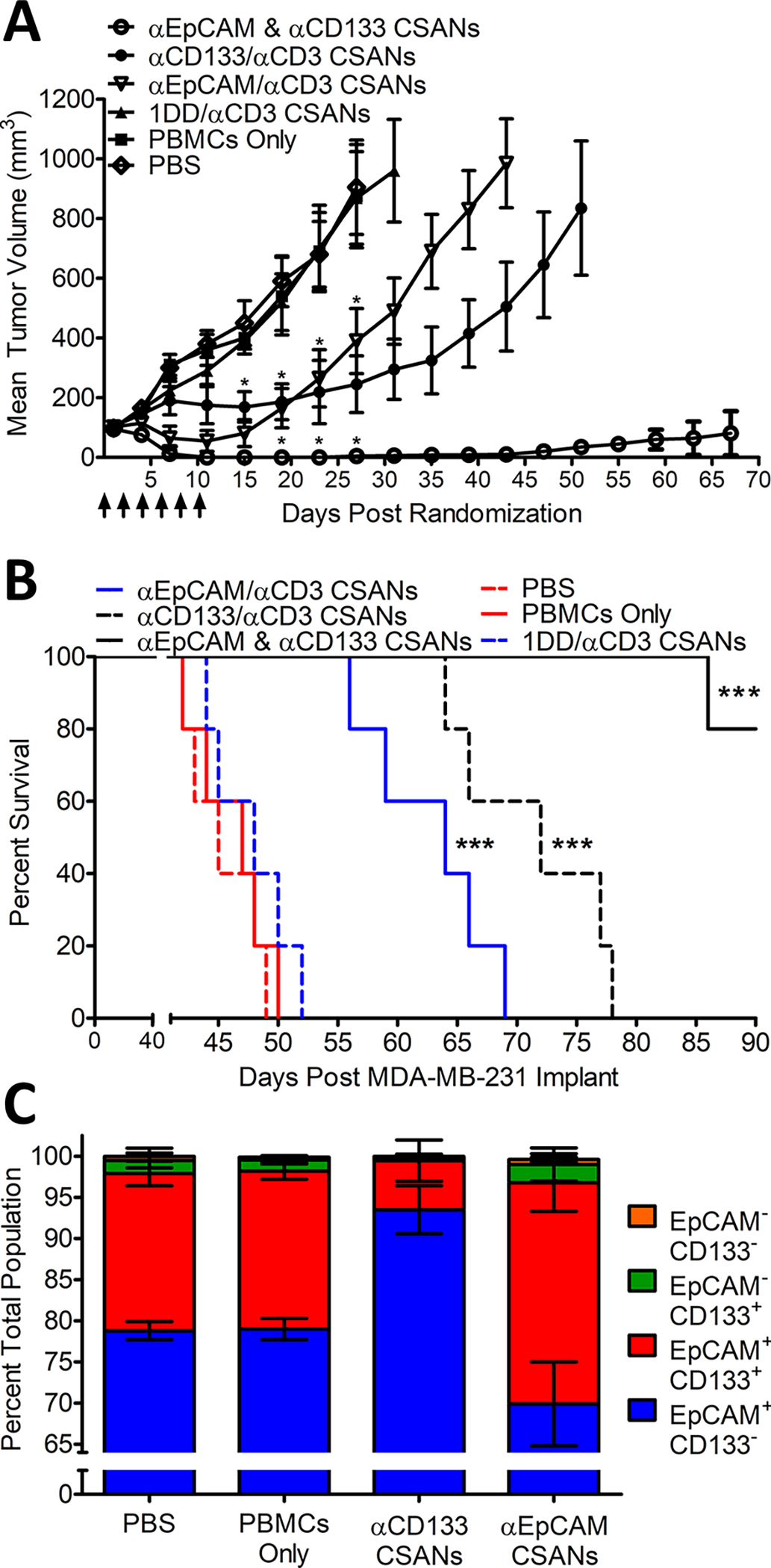
NSG mice were inoculated in the mammary fat pad with 1.0×106 MDA-MB-231 cells. Cohorts were randomized when tumors were ~80 mm3 and IV inoculated with 20 million PBMCs. Treatments were initiated 4 days later, including: PBS, PBMC only (this experimental group acquired treatments of PBS only following the initial PBMC engraftment), 1 mg/kg αCD3 monospecific CSANs, 1 mg/kg αCD133/αCD3 bispecific CSANs, 1 mg/kg αEpCAM/αCD3 bispecific CSANs, and both αCD133/αCD3 and αEpCAM/αCD3 bispecific CSANs at .5 mg/kg each (n=5). Treatments were administered every 2 days for a total of 6 treatments. (A) Tumor growth was monitored every two days by caliper and recorded as mm3. *P<0.05 with respect to readings statistically significant from the PBS control group, by 2-tailed Student’s t test. (B) Survival was monitored out to 90 days when graft vs host (GVH) disease symptoms were no longer manageable. (C) An identical cohort treated in parallel (n=5) and euthanized 1 day following the final treatment where tumors were isolated and homogenized. Cells were stained with αCD133-PE, αHuman-BV421-IgG, αHuman-CD3-BV605, αEpCAM-AF647 and ghost red viable dye before analysis with an LSRFortessa H0081. Cells positive for ghost red viability, positive for CD3 expression and negative for αHuman-BV421-IgG were removed from analysis by gating. The remaining αHuman positive cells were analyzed until a total cell count of 10,000 ± 200 was achieved and subsequently analyzed for expression levels of CD133 and EpCAM (A representative Flow Cytometry Diagram can be seen in supplemental Figure S6). Kaplan-Mier Survival Plot. *P<0.0001 with respect to PBS control, by log rank test. FACs data is presented as the mean ± standard deviation of three independent trials. All in vivo experiments were performed independently and at least twice.
Remarkably, when tumor bearing mice were treated with both αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs in combination, we observed full MDA-MB-231 tumor regression (Figure 5A). No tumor regrowth was evident, and a durable response was observed for the combination treatment with only a single remission out to 90 days (Figure 5B). Importantly, the total protein concentration of the CSANs used in the dual targeting application was the same as the individual αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs, half the amount of each, implying a significant synergism between the αEpCAM/αCD3 and αCD133/αCD3 CSANs when used to simultaneously direct T-cells to MDA-MB-231 orthotopic tumors.
Upon initiation of therapeutic treatment, a transient decrease in weight was observed for mice dosed with αCD133/αCD3 CSANs, αEpCAM/αCD3 CSANs or a combination. As the decrease was transient and less than 5% of the total mouse weight the likely cause was tumor cell lysis and transient immune response. Additionally, a small decrease was observed for the control animals, likely due to the initiation of graft vs host disease and tumor burden (27) (Supplemental Figure 8).
Importance of CD133 in Tumorigenicity
To further evaluate the importance of the stem-like properties exhibited from CD133+ cells, in MDA-MB-231 tumors, we developed a method for isolating CD133+ cells with the same αCD133 scFv used in our CSANs platform. Previously, we demonstrated that bispecific CSANs containing a monovalent streptavidin (mSA) targeting arm bind to biotinylated surfaces with high avidity and stability (28). Consequently, we generated mSA/αCD133 CSANs from mSA-DHFR2 and αCD133-DHFR2 and demonstrated that NH2-Dynabeads functionalized with Biotin-PEG4-NHS ester were able to rapidly and stably bind to both mSA/αCD133 CSANs and monovalent control mSA CSANs (Supplemental Figure 9). Beads were incubated with cell populations expressing CD133 and subsequently isolated by magnetic separation. As CSANs are rapidly disassembled in the presence of trimethoprim, the CD133+ cells were able to be quickly and efficiently released from the Dynabeads for further use and analysis.(14,15) CD133+ cells were validated with the commercially available αCD133 mAb. By using bead isolation, we decreased MDA-MD-231 CD133+ cell population by approximately 11-fold (Supplemental Figure 9). However, this number is likely a low estimate of the CD133+ cell depletion as all currently available commercial αCD133 mAbs only detects the glycosylated, but not non-glycosylated, forms of CD133.
Culturing of the CD133 depleted MDA-MB-231 cells for two weeks resulted in an increase by 8- fold in the percentage of CD133+ cells or 70% of the original non-depleted MDA-MB-231 control cells. In contrast, when MDA-MB-231 cells were enriched by the addition of 10% more CD133+ cells, no significant increase in the percentage of the CD133+ cells were observed. Consistent with the in vitro results, when implanted into NSG mice, the CD133 depleted MDA-MB-231 cells exhibited a significantly reduced initial rate of proliferation. While MDA-MB-231 cells enriched with CD133+ cells exhibited a significantly increased initial rate of proliferation, relative to the normal MDA-MB-231 control (Figure 6A). Nevertheless, after 4-weeks, no significant difference in the tumor growth rate for each set of conditions was observable. When tumors reached 1000 cm3 in size, the animals were euthanized, and tumors were isolated. Interestingly, no significant difference in the final percentage of CD133+ cells were found between the tumors generated from CD133 enriched or CD133 depleted MDA-MB-231 cells (Figure 6B). In addition, the final percentage of CD133+ cells found within the isolated tumors was similar to the percentages observed for cultured MDA-MB-231 spheroids. Consequently, our results indicate that tumor proliferation rates are potentially affected by the CD133+ cell population both in vitro and in vivo. However, regardless of the starting point (i.e., whether CD133 depleted, enriched or unaltered) the percentage of CD133+ cells required to maintain normal tumor growth appears to reach a homeostatic population.
Figure 6. Effects of CD133 Depletion and Enrichment on Orthotopic TN Breast Cancer Model.
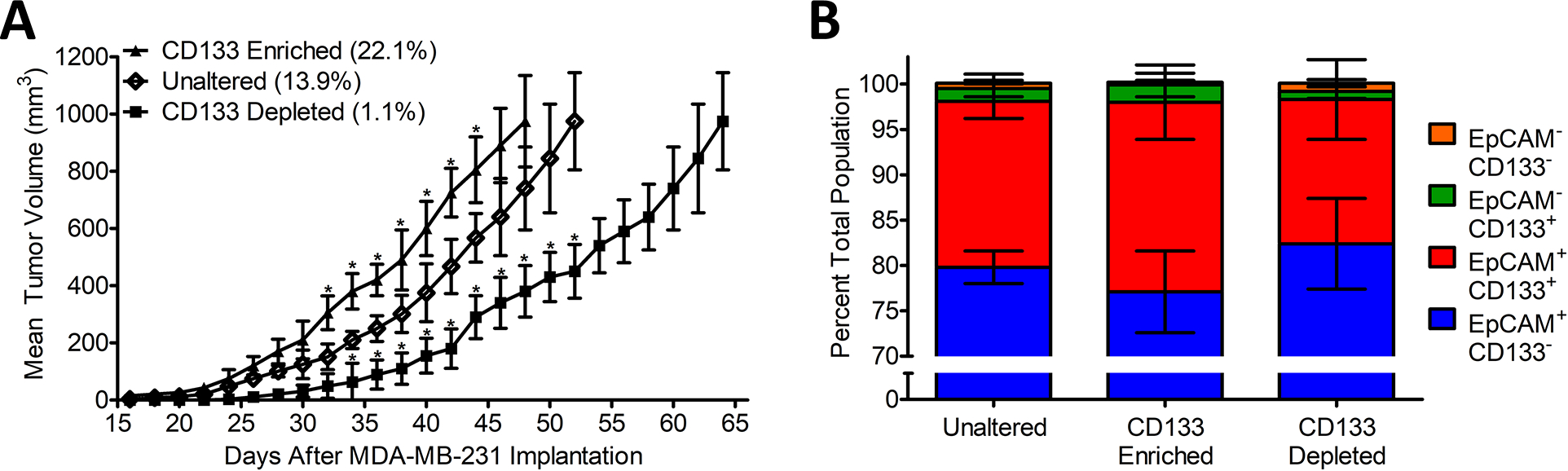
8- to 10-week-old female NSG Mice were injected with either 106 standard MDA-MB-231 cells (13.9% ± 1.1 CD133+), Enriched CD133+ cells (22.1% ± 1.3 CD133+), or depleted CD133− cells (1.1% ± 0.4 CD133+), in the mammary fat pad through the 4th nipple. CD133 Cells were either enriched or depleted by magnetic bead isolation as described in Supplemental Figure S7. Injection was performed in 50 μL 50:50 Matrigel/PBS while the mouse was under general anesthesia. (A) Tumor growth was monitored every two days by caliper and recorded as mm3. *P<0.05 with respect to readings statistically significant from the Unaltered control group, by 2-tailed Student’s t test. (B) Animals were euthanized, and their tumors were isolated once reaching 1000mm3. Tumor tissue was homogenized, and cells were stained with αCD133-PE, αHuman-BV421-IgG, αEpCAM-AF647 and ghost red viable dye before analysis with an LSRFortessa H0081. Cells positive for ghost red viability and negative for αHuman dyes were removed from analysis. All remaining αHuman positive cells were characterized for expression levels of CD133 and EpCAM. Data was collected for each sample until a total of ~10,000 ± 150 alive and human cells were obtained for analysis (i.e., alive tumor cells). A representative Flow Cytometry Diagram can be seen in supplemental Figure S7. The data is presented as the mean ± standard deviation of three independent FACs trials.
DISCUSSION
As previously mentioned, refractory relapse remains a concern for cancer therapy, and cancer stem cells (CSCs) have been linked to post-treatment tumor re-emergence. While CD133 alone as a CSC marker remains controversial, there is significant evidence supporting the antigen’s prognostic value association with stem-like properties (11). Moreover, CD133+ positive cells have been shown to display greater colony-forming efficiency, a higher proliferative rate, and a greater ability to form human xenograft tumors in NSG mice (29). Consequently, we chose to determine if bispecific CSANs capable of targeting T-cells to CD133+ stem-like cells and EpCAM+ primary tumor cells within TNBC would potentially lead to an increased overall therapeutic effect.
Both αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs demonstrated impressive T-cell targeting anti-tumor activity both in vitro and in vivo when utilized as single agents, particularly against nearly homogeneous antigen expressing cells such as CD133+ HT-29 cells and EpCAM+ MCF-7 cells. Additionally, both bispecific CSANs remained effective when targeting MDA-MB-231 cells, which are composed of a more heterogenous, and lower, antigen expression profile. However, treatment of MDA-MB-231 cells with both αCD133/αCD3 CSANs and αEpCAM/αCD3 CSANs resulted in a more profound anti-tumor response than treatment with either CSAN alone (Figure 2 and 5).
Interestingly, while αEpCAM/αCD3 CSANs demonstrated a more robust initial response in tumor clearance (Figure 5), αCD133/αCD3 CSANs provided a more sustained tumor reduction with a significantly greater median survival than αEpCAM/αCD3, indicating that both EpCAM+/CD133+ and EpCAM−/CD133+ cells may play a role in tumor survival and progression (Figure 5B). Furthermore, characterization of tumors following treatment confirmed that the observed decrease in tumor progression resulted from each bispecific CSANs selectively targeting and eliminating tumor cells expressing their respective target antigens. The increased percentage of EpCAM+CD133+ cells observed after treatment with αEpCAM/αCD3 CSANs maybe due to the enhanced ability of CD133+ cancer cells to resist apoptosis through autophagy and expression of anti-apoptotic factors (5,30). Alternatively, the phenomenon may result from a significantly lower level of EpCAM expression on the EpCAM+CD133+ cells MDA-MB-231 cells, relative to EpCAM+CD133− cells, thus reducing the effectiveness of the αEpCAM/αCD3 CSANs to induce targeted T-cell cytotoxicity.
To further probe the importance of CD133+ cells on TNBC tumor progression, using mSA/CD133 CSANs, we selectively removed or enriched CD133 expressing cells by magnetic bead isolation. When inoculated into NSG mice, CD133 depleted MDA-MB-231 cells displayed a significantly slower tumor growth rate than the non-depleted control cells. In comparison, a significantly faster rate of growth, compared to control cells, was observed for tumors resulting from CD133 enriched MDA-MB-231 cells. Nevertheless, after two weeks, no significant difference between the rate of growth of either the CD133 enriched, depleted tumors or control tumors was observed. In addition, regardless of the starting point, at the end of the study the amount of CD133+ cells converged to approximately the same percentage. Previously, MDA-MB-231 cells that were found to be low or non-expressors of the CSC biomarker aldehyde dehydrogenase, CD44+ and CD24+ exhibited a highly reduced ability to proliferate in vitro and in vivo relative to ALDHhi, CD44+CD24− cells (31,32). Recent more in depth studies utilizing a panel of antibodies that took in consideration the glycosylation state of CD133 demonstrated that approximately 3–4% of MDA-MB-231 cells grown in 2D cell culture express CD133 (26). Similar to the increase in CD133 expression found in spheroids of other tumor cell line types, we found that the percentage of CD133+ MDA-MB-231 cells grown in 3D cell culture rises to approximately 19% (21,23). Surprisingly, whether grown as spheroids or as implanted tumors, there appears to be a stable homeostatic population of CD133+ cells necessary to maintain tumor proliferation. It is not clear whether the origin of the expansion of the CD133+ cells is due to either the self-renewal of an undetectable population of remaining CD133+ cells or cellular plasticity. The apparent lack of a necessary stromal cell dependence could provide an explanation for the significant metastatic potential associated with CD133+ TNBC cells (8,31). The results of ongoing studies to address this question, as well as, the rationale for the apparent CD133+ cellular homeostasis will hopefully provide insights into this phenomenon. Nevertheless, utilizing T-cells to eliminate the MDA-MB-231 tumors by focusing on both the primary and CSC cell populations was found to be highly effective, which may in part result from greater overall T-cell infiltration and sensitivity of CD133+ cells to T-cell cytotoxicity when directly engaging CD133.
While the potential toxicity of targeting CD133 remains a concern, we did not observe any observable toxicities unrelated to graft vs host disease typically observed with NSG mice and implanted with human PBMCs, despite the cross reactivity of the aCD133 scFv with murine and human CD133. However, CD133 is commonly found as a marker of somatic stem cells, ranging from hematopoietic, neural, prostate, kidney, liver, and pancreas (33). Thus, targeting of CD133+ bears the potential risks for on target, off tumor, side effects. However, prior work performed by the Vallera lab, using the same CD133 scFv used in this study, evaluated the outcome of a targeted toxin in various progenitor assays including long-term culture and colony-forming assays (34). In each of their assays, including long term progenitor assays, minimal effects were found indicating that normal progenitors were not affected. Additionally, a recent phase 1 clinical trial was performed with a CD133 targeted chimeric antigen receptor T-cell therapy for advanced metastatic malignancies. The study reported overall acceptable toxicities and moderate off-target toxicities for hematopoietic stem cells, likely as result of the persistent nature of CAR therapy. Only a single significant toxicity was reported, and the suspected cause was bile duct stenosis (35). Consequently, αCD133 CSANs may have the potential to be employed as both a probe of the role of CD133 cells in cancer progression and metastasis, as well as a novel complementary immunotherapeutic approach. Nevertheless, future testing for long-term toxicities will be crucial for ongoing development.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NCI R01-CA247681(CRW), NCI R21-CA185627(CRW) and Tychon Bioscience, LLC. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.
Footnotes
Competing interests: Carston R. Wagner is the Chief Scientific Officer, a founder and shareholder in Tychon Bioscience, Inc, which has licensed technology related to this research.
REFERENCES
- 1.Cho RW, Clarke MF. Recent advances in cancer stem cells. Curr Opin Genet Dev 2008;18(1):48–53 doi 10.1016/j.gde.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene 2004;23(43):7274–82 doi 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 3.Boman BM, Wicha MS. Cancer stem cells: a step toward the cure. J Clin Oncol 2008;26(17):2795–9 doi 10.1200/JCO.2008.17.7436. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414(6859):105–11 doi 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008;26(17):2839–45 doi 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Germano S, O’Driscoll L. Breast cancer: understanding sensitivity and resistance to chemotherapy and targeted therapies to aid in personalised medicine. Curr Cancer Drug Targets 2009;9(3):398–418. [DOI] [PubMed] [Google Scholar]
- 7.Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets 2009;13(7):823–37 doi 10.1517/14728220903005616. [DOI] [PubMed] [Google Scholar]
- 8.Zhao P, Lu Y, Jiang X, Li X. Clinicopathological significance and prognostic value of CD133 expression in triple-negative breast carcinoma. Cancer Sci 2011;102(5):1107–11 doi 10.1111/j.1349-7006.2011.01894.x. [DOI] [PubMed] [Google Scholar]
- 9.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005;65(23):10946–51 doi 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 10.Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A 2008;105(36):13427–32 doi 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 12.Ferrandina G, Bonanno G, Pierelli L, Perillo A, Procoli A, Mariotti A, et al. Expression of CD133–1 and CD133–2 in ovarian cancer. Int J Gynecol Cancer 2008;18(3):506–14 doi 10.1111/j.1525-1438.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, So CR, Fegan A, Cody V, Sarikaya M, Vallera DA, et al. Chemically self-assembled antibody nanorings (CSANs): design and characterization of an anti-CD3 IgM biomimetic. J Am Chem Soc 2010;132(48):17247–57 doi 10.1021/ja107153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersburg JR, Shen J, Csizmar CM, Murphy KA, Spanier J, Gabrielse K, et al. Eradication of Established Tumors by Chemically Self-Assembled Nanoring Labeled T Cells. ACS Nano 2018;12(7):6563–76 doi 10.1021/acsnano.8b01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen J, Vallera DA, Wagner CR. Prosthetic Antigen Receptors. J Am Chem Soc 2015;137(32):10108–11 doi 10.1021/jacs.5b06166. [DOI] [PubMed] [Google Scholar]
- 16.Kilic O, Matos de Souza MR, Almotlak AA, Wang Y, Siegfried JM, Distefano MD, et al. Anti-EGFR Fibronectin Bispecific Chemically Self-Assembling Nanorings (CSANs) Induce Potent T Cell-Mediated Antitumor Responses and Downregulation of EGFR Signaling and PD-1/PD-L1 Expression. J Med Chem 2020;63(18):10235–45 doi 10.1021/acs.jmedchem.0c00489. [DOI] [PubMed] [Google Scholar]
- 17.Shah R, Petersburg J, Gangar AC, Fegan A, Wagner CR, Kumarapperuma SC. In Vivo Evaluation of Site-Specifically PEGylated Chemically Self-Assembled Protein Nanostructures. Mol Pharm 2016;13(7):2193–203 doi 10.1021/acs.molpharmaceut.6b00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csizmar CM, Petersburg J, Perry TJ, Rozumalski L, Hackel BJ, Wagner CR. Multivalent Ligand Binding to Cell Membrane Antigens: Defining the Interplay of Affinity, Valency, and Expression Density. J Am Chem Soc 2018. doi 10.1021/jacs.8b09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabrielse K, Gangar A, Kumar N, Lee JC, Fegan A, Shen JJ, et al. Reversible re-programing of cell-cell interactions. Angew Chem Int Ed Engl 2014;53(20):5112–6 doi 10.1002/anie.201310645. [DOI] [PubMed] [Google Scholar]
- 20.Swaminathan SK, Olin MR, Forster CL, Cruz KS, Panyam J, Ohlfest JR. Identification of a novel monoclonal antibody recognizing CD133. J Immunol Methods 2010;361(1–2):110–5 doi 10.1016/j.jim.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res 2008;10(1):R10 doi 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dean M Cancer stem cells: redefining the paradigm of cancer treatment strategies. Mol Interv 2006;6(3):140–8 doi 10.1124/mi.6.3.5. [DOI] [PubMed] [Google Scholar]
- 23.Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013;32(5):544–53 doi 10.1038/onc.2012.85. [DOI] [PubMed] [Google Scholar]
- 24.Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N, et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology 2010;51(2):545–56 doi 10.1002/hep.23311. [DOI] [PubMed] [Google Scholar]
- 25.Swaminathan SK, Niu L, Waldron N, Kalscheuer S, Zellmer DM, Olin MR, et al. Identification and characterization of a novel scFv recognizing human and mouse CD133. Drug Deliv Transl Res 2013;3(2):143–51 doi 10.1007/s13346-012-0099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohlfest JR, Zellmer DM, Panyam J, Swaminathan SK, Oh S, Waldron NN, et al. Immunotoxin targeting CD133(+) breast carcinoma cells. Drug Deliv Transl Res 2013;3(2):195–204 doi 10.1007/s13346-012-0066-2. [DOI] [PubMed] [Google Scholar]
- 27.Norelli M, Camisa B, Bondanza A. Modeling Human Graft-Versus-Host Disease in Immunocompromised Mice. Methods Mol Biol 2016;1393:127–32 doi 10.1007/978-1-4939-3338-9_12. [DOI] [PubMed] [Google Scholar]
- 28.Csizmar CM, Petersburg JR, Hendricks A, Stern LA, Hackel BJ, Wagner CR. Engineering Reversible Cell-Cell Interactions with Lipid Anchored Prosthetic Receptors. Bioconjug Chem 2018;29(4):1291–301 doi 10.1021/acs.bioconjchem.8b00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432(7015):396–401 doi 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 30.Zobalova R, McDermott L, Stantic M, Prokopova K, Dong LF, Neuzil J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys Res Commun 2008;373(4):567–71 doi 10.1016/j.bbrc.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 31.Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep 2017;7(1):13856 doi 10.1038/s41598-017-14364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 2009;13(8B):2236–52 doi 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, Wu PY. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev 2009;18(8):1127–34 doi 10.1089/scd.2008.0338. [DOI] [PubMed] [Google Scholar]
- 34.Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med (Berl) 2008;86(9):1025–32 doi 10.1007/s00109-008-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018;7(7):e1440169 doi 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.