

Abstract
PURPOSE:
Nephrolithiasis (NL) affects 1 in 11 individuals worldwide, leading to significant patient morbidity. NL is associated with nephrocalcinosis (NC), a risk factor for chronic kidney disease. Causative genetic variants are detected in 11–28% of NL and/or NC, suggesting additional NL/NC-associated genetic loci await discovery. Therefore, we employed genomic approaches to discover novel genetic forms of NL/NC.
METHODS:
Exome sequencing and directed sequencing of the OXGR1 locus were performed in a worldwide NL/NC cohort. Putatively deleterious rare OXGR1 variants were functionally characterized.
RESULTS:
Exome sequencing revealed a heterozygous OXGR1 missense variant (c.371T>G, p.L124R) co-segregating with calcium oxalate NL and/or NC disease in an autosomal dominant inheritance pattern within a multi-generational family with five affected individuals. OXGR1 encodes 2-oxoglutarate (α-ketoglutarate) receptor 1 in the distal nephron. In response to its ligand α-ketoglutarate (AKG), OXGR1 stimulates the chloride-bicarbonate exchanger Pendrin, which also regulates transepithelial calcium transport in cortical connecting tubules. Strong amino acid conservation in orthologues and paralogues, severe in silico prediction scores, and extreme rarity in exome population databases suggested the variant was deleterious. Interrogation of the OXGR1 locus in 1107 additional NL/NC families identified five additional deleterious dominant variants in five families with calcium oxalate NL/NC. Rare, potentially deleterious OXGR1 variants were enriched in NL/NC subjects relative to ExAC controls (Χ2=7.117, p=0.0076). Wildtype OXGR1-expressing Xenopus oocytes exhibited AKG-responsive Ca2+ uptake. Four of five NL/NC-associated missense variants revealed impaired AKG-dependent Ca2+ uptake, demonstrating loss-of-function.
CONCLUSION:
Rare, dominant loss-of-function OXGR1 variants are associated with recurrent calcium oxalate NL/NC disease.
Keywords: alpha-ketoglutarate, calcium oxalate, nephrolithiasis, genetics
INTRODUCTION
Nephrolithiasis (NL) affects nearly 10% of people worldwide, leading to significant patient morbidity1,2. NL is often associated with nephrocalcinosis (NC), a risk factor for chronic kidney disease3. Causative genetic variants are detected in 14–28% of NL/NC, suggesting novel NL/NC-associated genes await discovery4–6.
OXGR1 encodes 2-oxoglutarate (α-ketoglutarate) receptor 1, a G protein-coupled receptor (GPCR) expressed in cortical connecting tubule and collecting duct Type B intercalated cells7–9 (Figure S1). In response to its ligand α-ketoglutarate (AKG), OXGR1 mediates cellular Ca2+ uptake, an established second messenger of G-protein coupled receptors10,11. AKG is generated and renally excreted upon intake of the established calcium NL treatment citrate12,13. Mouse model and perfused connecting tubule studies suggest that, upon AKG binding, OXGR1 signals through Protein Kinase C (PKC) to promote the chloride-bicarbonate exchanger Pendrin7–9,14. Oxgr1−/− mice and Slc26a4/Pendrin−/− mice exhibited impaired urine alkalinization7–9. This parallels the impaired secretion of bicarbonate as well as reabsorption of sodium and chloride in perfused cortical connecting tubules from Oxgr1−/− mice in response to AKG7. In addition, Pendrin knockout mice showed urinary calcium wasting8,9, a risk factor for calcium stone formation13, in association with reduced expression of the apical TRPV5 and basolateral sodium-calcium exchanger (NCX1) in the cortical connecting tubule.
To discover a novel monogenic etiology of NL/NC disease, we performed exome sequencing (ES) in index family B1467 with five affected individuals exhibiting calcium oxalate NL and/or NC in an autosomal dominant inheritance pattern. We detected a rare heterozygous OXGR1 missense variant that co-segregated with NL/NC. Interrogation of the OXGR1 locus in 1107 additional NL/NC families revealed five dominant alleles in five families with calcium oxalate NL/NC disease. All were rare variants with multiple severe prediction scores. Rare, potentially deleterious OXGR1 variants were enriched in NL/NC subjects relative to ExAC controls. Wildtype OXGR1-expressing Xenopus oocytes demonstrated AKG-responsive Ca2+ uptake, whereas four of five NL/NC-associated OXGR1 missense variants exhibited impaired ligand-dependent Ca2+ uptake at pH 5 and/or 7.4. We propose rare dominant OXGR1 variants as a novel candidate etiology of NL/NC.
METHODS
Human Subjects Recruitment and Ethics Declaration
Subjects with pediatric and adult NL/NC disease were recruited with informed consent according to site-specific ethics committees and practices as discussed in the Supplementary Methods section. Subjects with potential secondary medical causes of NL/NC were excluded.
Molecular Genetics and Genomics Methods
Exome sequencing15, targeted next-generation sequencing16,17(p99), Sanger sequencing, homozygosity mapping18,19,20, variant calling15, and single cell mRNA sequencing and analysis21 were performed as previously described. (See Supplemental Methods for details and for cDNAs, cell lines, antibodies and reagents, and statistical analysis methods).
Xenopus oocyte methods
All Xenopus experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. Reagents and solutions used for cRNA transcription and expression in Xenopus laevis oocytes, and methods for measurement of unidirectional 45Ca2+ influx into oocytes, whole mount confocal immunofluorescence microscopy of oocytes, immunoblots of oocyte lysates, and statistical analysis of the oocyte data are described in detail in Supplemental Methods.
RESULTS
Exome Sequencing of Index Family B1467
Index case B1467_22, an Egyptian female born from a consanguineous union, developed recurrent nephrolithiasis at age 6 years (Figure 1A; Table 1, S1). Hypertension on physical examination was accompanied by microscopic hematuria and calcium oxalate crystalluria. Stone analysis demonstrated calcium oxalate monohydrate. Laboratory studies demonstrated reduced kidney function (BUN 21 mg/dL, serum creatinine 0.9 mg/dL) and metabolic acidemia (pH 7.32, HCO3 19 mmol/L). Serum electrolytes were otherwise normal. Urine metabolite analysis showed mild hyperoxaluria (50 mg/24hr/1.73 m^2) and hypocitraturia (373 mg/g Cr). Renal ultrasound revealed bilateral nephrocalcinosis (Figure 1B). 99Tc-dimercaptosuccinic acid (DMSA) scan showed differential function of 18% from the left kidney and 82% from the right kidney. Nephrocalcinosis was also found in the mother and three siblings, but the father’s ultrasound was normal (Table 1, S1). Clinical phenotyping of the three siblings revealed shared findings with the proband: renal dysfunction in one sibling (B1467_21); reduced serum bicarbonate levels in one sibling (B1467_23); calcium oxalate crystalluria in all siblings; and hypocitraturia and mild hyperoxaluria in all siblings (Table S1).
Figure 1. Exome and targeted sequencing in nephrolithiasis (NL)/nephrocalcinosis (NC) subjects revealed dominant OXGR1 variants, which impair Ca2+ uptake in response to ligand.
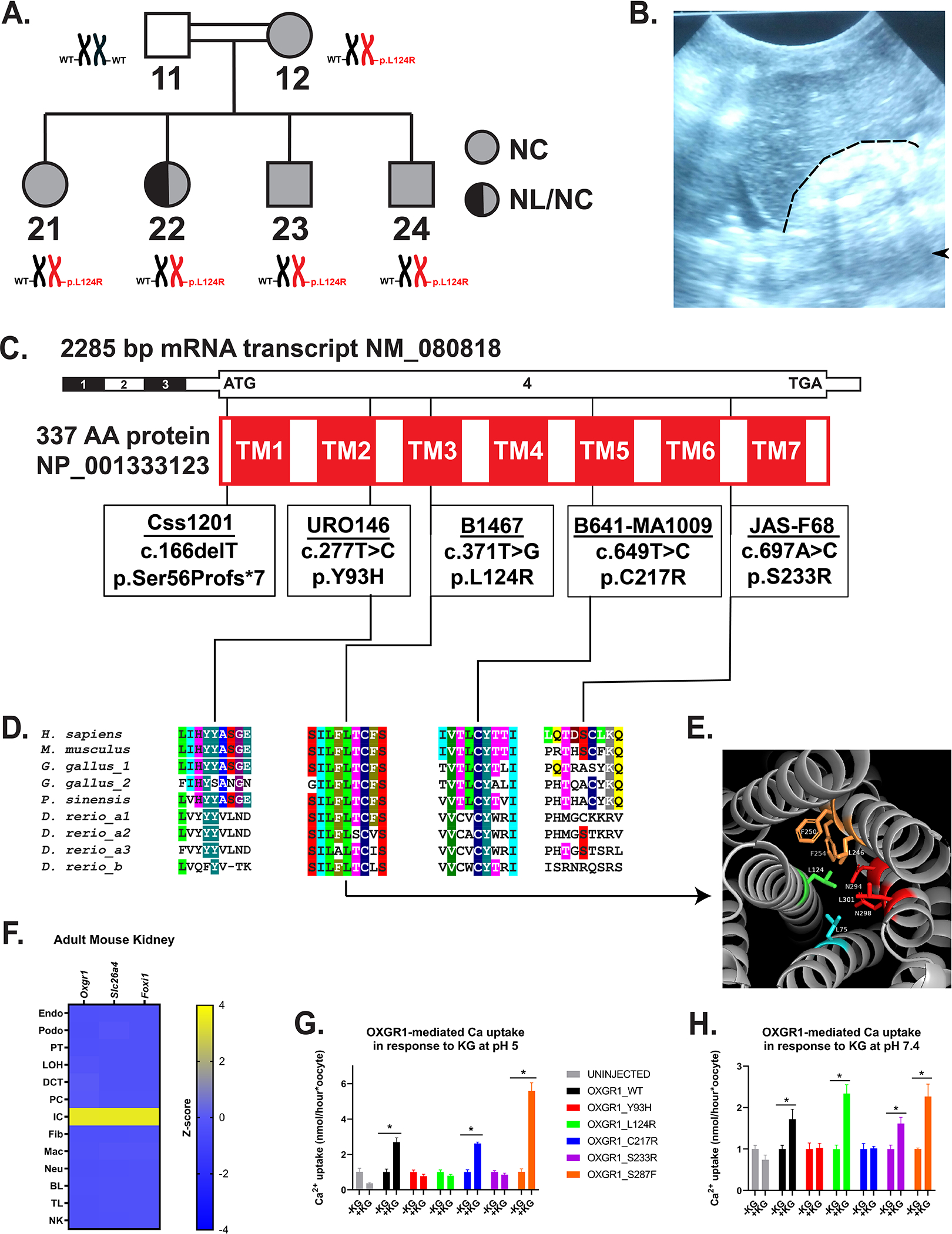
(A) Pedigree of Family B1467 with individual identifiers below symbols indicating absence or presence of NL and/or NC disease. Adjacent to each individual symbol is OXGR1 genotype.
(B) Renal ultrasound from subject B1467_21 shows increased echogenicity of right kidney (to right of and below dashed line) relative to adjacent liver with post-acoustic shadowing (arrowhead).
(C) Schematics of exon (black-white) and protein domain (red-white) structures of OXGR1. Black vertical lines indicate positions of variants (in boxes).
(D) Evolutionary conservation of amino acid sequence for each amino acid position impacted by OXGR1 missense variants. Protein sequences of putative orthologues from rodents (Mus musculus), birds (Gallus gallus), reptiles (Pelodiscus sinensis), and fish (Danio rerio) were used. Invertebrate orthologues have not been identified.
(E) Predicted human OXGR1 structure shown. Leucine 124 (stick model) present on the central transmembrane domain 3 (TM3, green) and protrudes into the core of the 7-transmembrane domain structure. It is within 4Å of seven amino acids, of which five are hydrophobic.
(F) Oxgr1 mRNA (z-score) was predominantly expressed in the intercalated cell (IC) cluster from single-cell mRNA sequencing data from adult mouse kidneys. Oxgr1 expression was highest in the IC cluster marked by expression of Slc26a4 and Foxi1, relative to other nephron tubular segment clusters to the left and interstitial or hematologic cell types to the right. Endo, endothelial cell; Podo, podocyte; PT, proximal tubular cell; LOH, loop of Henle cell; DCT, distal convoluted tubule; PC, principal cell; IC, intercalated cell; Fib, fibroblast; Mac, macrophage; Neu, neutrophil; BL, B lymphocyte; TL, T lymphocyte; NK, natural killer cell.
(G) Ca2+ uptake into Xenopus oocytes injected 48–72 hours previously with water or with cRNA (40 ng) encoding wild type human OXGR1 or the indicated variants. Uptake was measured at bath pH 5.0 for 30 min in the absence (−KG) or presence (+KG) of bath alpha-ketoglutarate (1 mM). Uptake measurements (nmol/hour) were normalized to the mean of the −KG group for each construct. *p<0.05 by Student’s t-test.
(H) Ca2+ uptake was measured as in (G) at bath pH 7.4. Uptake measurements were normalized to the mean of the −KG group for each construct. *p<0.05 by Student’s t-test.
Table 1.
Heterozygous variants in the gene OXGR1 (NM_080818.3) in 5 families with nephrolithiasis and/or nephrocalcinosis
| Family_Individual | Nucleotide / AA Change | In silico Severity Scores | Conservation through | ACMG/AMP Classification (Criteria) | ExAC; gnomAD (H, h, Tot) | SEX | Country of Origin / Ethnicity | Age of onset (yrs) | Stone Disease and Risk Factors |
|---|---|---|---|---|---|---|---|---|---|
| Css1201 | c.166delT / p.Ser56Profs*7 | - | - | LP (PVS1, PM2) | NP NP |
M | United Kingdom | 14 | Rec CaOx NL |
| URO 146 _21 _11 |
c.277T>C / p.Tyr93His | PP2 PD SIFT DL CADD 24.6 |
D. rerio | LP (PS3, PP1, PP3) | 0/1/121312 0/2/246250 |
M M |
Pakistan | 65 27 |
Rec NL Rec NL |
| B1476 _21 _22 _23 _12 |
c.371T>G / p.Leu124Arg | PP2 PD SIFT DL CADD 27.9 |
D. rerio | LP (PS3, PM1, PP1, PP3) | 0/1/121264 NP |
F F M M F |
Egypt | ND 6 ND ND ND |
NC, HOx, hCit Rec CaOx NL / NC, HOx, hCit NC, HOx, hCit NC, HOx, hCit NC |
| B641_MA1009 | c.649T>C / p.Cys217Arg | PP2 PD SIFT DL CADD 26.6 |
D. rerio | LP (PS3, PM2, PP3) | NP NP |
F | United States / Mixed European | 16 | Rec CaOx NL, HOx |
| JAS-F68_21 | c.697A>C / p.Ser233Arg | PP2 B SIFT DL CADD 21.5 |
D. rerio | VUS(PS3, PP3) | 0/3/121406 0/9/277214 |
M | United Kingdom | 50 | Rec CaOx NL / NC |
Abbreviations: AA, amino acid; B, benign; CADD, Combined Annotation Dependent Depletion; CaOx, calcium oxalate; DL, deleterious; ExAC, Exome Aggregation Consortium; F, female; gnomAD Genome Aggregation database; H, homozygous individuals in database; h, heterozygous alleles of particular variant in database; hCit, hypocitraturia; het, heterozygous; HOx, hyperoxaluria; LP, likely pathogenic; M, male; NA, not available; NC, nephrocalcinosis; NL nephrolithiasis; NP, not present in variant database; PD, probably damaging; PP2, PolyPhen-2 prediction score; Rec, recurrent; Seg, segregation; SIFT, Sorting Tolerant From Intolerant prediction score; Tot, total alleles at position in database; VUS, variant of unknown significance; yrs, years.
No causative variants in 30 established NL/NC disease genes4–6 were detected by ES. Vertical inheritance of the NL/NC trait suggested a rare autosomal dominant variant as cause of the disease. Evaluation of exome data revealed two co-segregating potentially deleterious heterozygous variants.
A heterozygous missense variant in OXGR1 (chr13:97639643A>C; NM_080818:c.371T>G, p.Leu124Arg) co-segregated with NL/NC (Figure 1C–D, S3; Table 1). It was deemed deleterious by multiple criteria: (i) it was extremely rare (one heterozygote among 121264 alleles) in the ExAC database and absent from the gnomAD database (Table 1); (ii) strong PolyPhen2, SIFT and CADD in silico prediction scores indicated a deleterious amino acid substitution (Table 1); (iii) Leu124 is conserved across vertebrate orthologues (Figure 1D) and in 216/300 (72%) of paralogous human GPCRs (Figure S4D).
Based on structural modeling, Leu124 resides in the third transmembrane helix of OXGR1, its side chain protruding into the receptor’s central core and surrounded within 4Å by predominantly hydrophobic (five of seven) amino acid side chains (Figures 1E, S4A–B). The L124R substitution is predicted to reduce OXGR1 stability (predicted pseudoΔΔG = −2.7) (Figure S4C, S6E).
A rare heterozygous co-segregating LMX1B variant (chr9:129455511; NM_001174147: c.650G>A, p.Arg217Gln) was deemed non-pathogenic in the absence of Nail-Patella syndrome features (proteinuria, skeletal abnormalities or ungual findings) in affected family members (Table S2). ES data from this consanguineous family was also screened for recessive variants within regions of homozygosity based on an autosomal homozygous recessive hypothesis (Figure S2). A rare homozygous GAS6 variant (chr13:114542707G>C; NM_000820:c.460G>C, p.Asp154His) was deemed non-pathogenic as the variant did not co-segregate with affected family members in recessive or recessive/dominant manners, and the residue was only partially conserved across vertebrates (Table S2).
Based on these analyses, we identified a candidate dominant variant in OXGR1 that was associated with the NL/NC trait in family B1467.
OXGR1 variants discovered in NL/NC families
We hypothesized that dominant OXGR1 loss-of-function variants may cause calcium NL/NC, based on the index family B1467 and the known expression and role of OXGR1 in intercalated cells (Figure 1F). We therefore examined the OXGR1 locus in 1107 additional NL/NC families through interrogation of exome data or by targeted sequencing (Figure S5A). OXGR1 variants with allele frequency <1% in dbSNP147 were further evaluated for (i) rare prevalence in ExAC and gnomAD databases and (ii) deleterious in silico prediction scores by at least two of three algorithms (SIFT, PolyPhen 2.0, and CADD) (Figure S5).
This approach identified five additional dominant variants in five families (Figure 1C–D, S3; Tables 1, S1, S3): one loss-of-function allele in family Css1201 (c.166delT; p.Ser56Profs*7), and four missense variants distributed throughout the protein (Figure 1C; Tables 1 and S3; Movie S1). Three of the four missense variants impact amino acid residues conserved across vertebrate orthologues (Figure 1D; Tables 1 and S3). The variant identified in subject B641-MA1009 impacts Cys217, a residue conserved in 122/300 (41%) of paralogous human GPCRs (Figure S4E).
OXGR1 NL/NC-associated missense variants were further characterized by structural modeling (Figure S6). Y93 is modeled to reside within 4Å of predominantly (4/6) polar residues (Figure S6A), and the variant Y93H is predicted to reduce intramolecular stability (pseudoΔΔG = −1.42) (Figure S6E). The C217R sidechain is modeled to closely approach only one residue, and is predicted to reduce intramolecular stability more modestly (pseudoΔΔG = −0.73) (Figures S6B, S6E). Neither S233R nor S287F are predicted to reduce stability (Figures S6C–E).
ES or gene panel sequencing performed in four families identified no pathogenic variants in established NL/NC disease genes4–6 (Figure S5). (B641_MA1009 DNA failed exome sequencing.) A variant of unknown significance was identified in ADCY10 in JAS-F684, but this locus is not established as a monogenic NL/NC disease gene.
Thus, six potentially deleterious variants were identified in six of 1108 NL/NC families (0.54%). Application of the same variant filtering criteria to exome data from 60,706 ExAC controls identified 66 variants in 99 subjects (0.16%), demonstrating enrichment for rare deleterious heterozygous OXGR1 variants in NL/NC subjects (Χ2=7.117, p=0.0076) (Figure S5, Table S4).
OXGR1 variants identified in recurrent calcium oxalate NL
Deeper clinical phenotyping in the six NL/NC families with OXGR1 rare potentially deleterious variants was performed and revealed the following trends (Table 1, S1, S3, S5). All six families (7/11 individuals) had history of NL with recurrence reported in five of six families (6/11 individuals). Stone composition revealed predominantly calcium oxalate in five families (5/5 individuals where composition data available). All six index cases, moreover, had a positive family history of NL/NC. Nephrocalcinosis was noted in two families (6/11 individuals): B1467 and JAS-F68. Hyperoxaluria was noted in two families (5/6 individuals where oxalate excretion available): B1467 and B641_MA1009. Urine sodium excretion was elevated in two families (2/2 individuals where sodium excretion available): Css1201 and B641_MA1009. Hypocitraturia was noted only in family B1467 (1/3 where data available). Hypercalciuria was not observed (0/5 families where data available).
OXGR1 missense variant impact on ligand-dependent OXGR1-mediated Ca2+ uptake
We explored the impact of OXGR1 NL/NC-associated missense variants on OXGR1 expression and function. Expression of N-terminal MYC-tagged wildtype OXGR1 protein in HEK293T cells revealed a multi-band pattern consistent with multi-merization by immunoblotting (Figure S7A–E). Constructs reflecting patient-derived OXGR1 missense variants exhibited similar patterns by immunoblotting (Figure S7G).
Wildtype and NL/NC-associated mutant proteins were comparably expressed in Xenopus oocyte lysates from injected cRNA (Figure S7H) and exhibited similar surface localization by confocal microscopy (Figure S8).
We next evaluated OXGR1-mediated cellular Ca2+ uptake as a measure of its GPCR activity11. Studies were conducted in response to AKG in oocytes at both normal and acidic bath pH, mimicking near-maximal collecting duct urinary acidification. Ca2+ uptake into oocytes expressing wildtype OXGR1 was stimulated by 1 mM AKG at both pH 5 and pH 7.4 (Figure 1G–H; Figure S9). In contrast, four of five NL/NC-derived missense variants (p.Tyr93His, p.Leu124Arg, p.Cys217Arg, p.Ser233Arg) demonstrated impaired AKG-dependent Ca2+ uptake at pH 5 and/or 7.4 (Figures 1G–H, S9).
These studies provide evidence of loss-of-function of four NL/NC-associated OXGR1-missense variants (Figure S5). As the p.Ser287Phe variant identified in subject B431_21 did not modify OXGR1-mediated Ca2+ uptake, it remains for now a variant of unknown significance (Table S3).
DISCUSSION
We have identified rare, dominant OXGR1 variants in human calcium kidney stone disease. These variants were enriched in NL/NC subjects relative to the ExAC cohort and further validated for loss-of-function in response to ligand. These findings suggest new pathogenic mechanisms and potential therapeutic targets in calcium oxalate nephrolithiasis.
Slc26a4/Pendrin knockout mice exhibit impaired transepithelial calcium transport in the cortical connecting tubule in association with altered apical and basal calcium transporter expression8,9. We confirmed that AKG-stimulated OXGR1 causes increased extracellular calcium uptake—an established second messenger of GPCR signaling10,11—and employed calcium uptake in Xenopus oocytes as a measure of receptor function (Figure 1G–H). However, it remains unclear if OXGR1 and acute AKG-mediated signaling regulate transepithelial calcium transport and urinary calcium excretion in vivo. Future studies assessing calcium excretion in Oxgr1 deficient mice and calcium transport in Oxgr1 deficient isolated connecting tubules will be critical to address these possibilities.
We posited that OXGR1 deficiency would lead to hypercalciuria and calcium nephrolithiasis in humans because OXGR1 positively regulates Pendrin activity7,8,14 and Pendrin deficiency causes urinary calcium wasting in mice9. While dominant loss-of-function OXGR1 variants are associated with calcium oxalate nephrolithiasis (Tables 1, S1), calcium excretion was normal in all subjects where data was available (0/5 families, Tables S1, S5). It remains possible that only severe OXGR1 deficiency caused by recessive variants, analogous to the homozygous null Pendrin mice8,9, would alter transepithelial calcium transport. An alternative pathogenic mechanism is that OXGR1 haploinsufficiency promotes calcium oxalate nephrolithiasis through direct effects on crystal formation. These possibilities should be evaluated in Oxgr1+/− and Oxgr1−/− mice through urine metabolite testing and oxalate calculi models22.
Hyperoxaluria and hypocitraturia were noted in a subset of OXGR1-variant associated NL/NC patients (2/3 and 1/3 families, respectively, where data is available). Discovery of additional NC/NC patients with OXGR1 variants is warranted to determine if these findings are truly associated or were incidentally observed due to other genetic or environmental factors. Moreover, there are no established mechanisms for how OXGR1/Pendrin signaling would alter oxalate or citrate excretion. Studies assessing excretion of these metabolites in Oxgr1 deficient mice are, therefore, warranted.
Citrate has been proposed to impair calcium stone formation by calcium chelation and inhibition of crystal formation13. Additionally, exogenous citrate is metabolized to alpha-ketoglutarate and renally excreted12. This suggests that citrate may impact stone formation in part through AKG-dependent mechanisms such as OXGR1 signaling7,10. In clinical phenotyping of the NL/NC patients with OXGR1 variants, citrate was only employed in one subject (Table S1). Therefore, we could not draw conclusions about the role of OXGR1 in the mechanism of action of citrate, but future controlled studies (e.g. in Oxgr1 deficient mice) are warranted to investigate this question.
A limitation of the study was that only probands were able to be recruited for four of six family members although all had a family history of NL/NC. In the future, it will be important to determine whether any of the affected family members share the identified OXGR1 variant.
Of note, we observed potentially deleterious OXGR1 variants in 0.16% of ExAC subjects, who are selected based on the absence of pediatric disease. However, this control cohort has the limitation that individuals may have subclinical or underreported NL/NC disease. Our Ca2+ uptake studies in Xenopus oocytes (Figure 1G–H) suggest that ExAC OXGR1 variants also warrant functional characterization and that environmental factors altering urinary pH (e.g. diet) may modify the functional impact of OXGR1 variants.
In summary, rare, dominant loss-of-function OXGR1 variants are associated with recurrent calcium oxalate NL/NC disease. These findings suggest new avenues of research into the pathogenesis and treatment of calcium oxalate nephrolithiasis.
Supplementary Material
ACKNOWLEDGEMENTS
F.H. is the William E. Harmon Professor of Pediatrics. This research is supported by a grant from the NIH to F.H. (DK068306-17). A.J.M. was supported by the NIH (T32DK-007726, 5K12HD052896-13, 1K08DK125768-01A1), Harvard Stem Cell Institute (F-KP-0003-17-00), American Society of Nephrology (Jared J. Grantham Research Fellowship 81471, Norman Siegel Research Scholar Career Grant 81542), and Manton Center for Orphan Disease Research (Junior Faculty Award). E.W. was supported by the Leopoldina Fellowship Program, German National Academy of Sciences Leopoldina (LPDS 2015-07). S.L.A. received research funds from Quest Diagnostics. The Yale Center for Mendelian Genomics (UM1HG006504) is funded by the National Human Genome Research Institute. The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. C.-H.W.W. was supported by funding from the American College of Medical Genetics and Genomics Foundation (ACMG/Takeda Next-Generation Biochemical Genetics Award) and National Institutes of Health (grant T32-GM007748). J.A.S. is supported by the Medical Research Council, the Northern Counties Kidney Research Fund and Kidney Research UK (Paed_RP_001_20180925). The NIHR Newcastle Biomedical Research Centre is a partnership between Newcastle Hospitals NHS Foundation Trust and Newcastle University, funded by the National Institute for Health and Care Research (NIHR). J.H. receives funding from the German Research Foundation (DFG, HA 6908/3-1 and HA 6908/4-1). F.B. was supported by a fellowship grant (404527522) from the German Research Foundation (DFG). T.J.S. received funding by the IZKF Erlangen (J70), the German Research Foundation (DFG, 281319475) and the Else Kröner-Fresenius Stiftung (RECORD program).
Footnotes
CONFLICT OF INTEREST STATEMENT
F.H. is a co-founder of Goldfinch Biopharma Inc. S.L.A. is consultant to and received funding from Quest Diagnostics, Inc. The other authors declare that they have no competing financial interests. No part of this manuscript has been previously published.
ETHICS DECLARATION STATEMENT
For this study, subjects with pediatric and adult NL/NC disease were recruited with informed consent according to site-specific ethics committees and practices as discussed in the Methods and Supplementary Methods sections.
SUPPLEMENTARY MATERIALS
Supplementary Methods (PDF)
Supplementary Table 1 (PDF)
Supplementary Table 2 (PDF)
Supplementary Table 3 (PDF)
Supplementary Table 4 (PDF)
Supplementary Table 5 (PDF)
Supplementary Figure 1 (PDF)
Supplementary Figure 2 (PDF)
Supplementary Figure 3 (PDF)
Supplementary Figure 4 (PDF)
Supplementary Figure 5 (PDF)
Supplementary Figure 6 (PDF)
Supplementary Figure 7 (PDF)
Supplementary Figure 8 (PDF)
Supplementary Figure 9 (PDF)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The genetics datasets supporting the current study have not been deposited in a public repository due to restriction by patient consent, but are available from the corresponding author on request.
REFERENCES
- 1.Scales CD, Smith AC, Hanley JM, Saigal CS. Prevalence of kidney stones in the United States. European Urology. 2012;62(1):160–165. doi: 10.1016/j.eururo.2012.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rule AD, Bergstralh EJ, Melton LJ, Li X, Weaver AL, Lieske JC. Kidney Stones and the Risk for Chronic Kidney Disease. Clinical Journal of the American Society of Nephrology. 2009;4(4):804–811. doi: 10.2215/CJN.05811108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shavit L, Jaeger P, Unwin RJ. What is nephrocalcinosis? Kidney International. 2015;88(1):35–43. doi: 10.1038/ki.2015.76 [DOI] [PubMed] [Google Scholar]
- 4.Halbritter J, Baum M, Hynes AM, et al. Fourteen Monogenic Genes Account for 15% of Nephrolithiasis/Nephrocalcinosis. Journal of the American Society of Nephrology. 2015;26(3):543–551. doi: 10.1681/ASN.2014040388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braun DA, Lawson JA, Gee HY, et al. Prevalence of Monogenic Causes in Pediatric Patients with Nephrolithiasis or Nephrocalcinosis. Clinical Journal of the American Society of Nephrology. 2016;11(4):664–672. doi: 10.2215/CJN.07540715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daga A, Majmundar AJ, Braun DA, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney International. Published online October 12, 2017. doi: 10.1016/j.kint.2017.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokonami N, Morla L, Centeno G, et al. α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest. 2013;123(7):3166–3171. doi: 10.1172/JCI67562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amlal H, Petrovic S, Xu J, et al. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl−/HCO3− exchanger activity and impairs bicarbonate secretion in kidney collecting duct. American Journal of Physiology-Cell Physiology. 2010;299(1):C33–C41. doi: 10.1152/ajpcell.00033.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barone S, Amlal H, Xu J, Soleimani M. Deletion of the Cl−/HCO3− exchanger pendrin downregulates calcium-absorbing proteins in the kidney and causes calcium wasting. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27(4):1368–1379. doi: 10.1093/ndt/gfr505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He W, Miao FJP, Lin DCH, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188. [DOI] [PubMed] [Google Scholar]
- 11.Dhyani V, Gare S, Gupta RK, Swain S, Venkatesh KV, Giri L. GPCR mediated control of calcium dynamics: A systems perspective. Cellular Signalling. 2020;74:109717. doi: 10.1016/j.cellsig.2020.109717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krebs HA, Salvin E, Johnson WA. The formation of citric and α-ketoglutaric acids in the mammalian body. Biochemical Journal. 1938;32(1):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coe FL, Worcester EM, Evan AP. Idiopathic hypercalciuria and formation of calcium renal stones. Nature Reviews Nephrology. 2016;12:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazo-Fernandez Y, Welling PA, Wall SM. α-Ketoglutarate stimulates pendrin-dependent Cl− absorption in the mouse CCD through protein kinase C. American Journal of Physiology-Renal Physiology. 2018;315(1):F7–F15. doi: 10.1152/ajprenal.00576.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connaughton DM, Kennedy C, Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney International. 2019;95(4):914–928. doi: 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halbritter J, Diaz K, Chaki M, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet. 2012;49(12):756. doi: 10.1136/jmedgenet-2012-100973 [DOI] [PubMed] [Google Scholar]
- 17.Halbritter J, Porath JD, Diaz KA, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Human genetics. 2013;132(8):865–884. doi: 10.1007/s00439-013-1297-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Research. 2009;37(suppl_2):W593–W599. doi: 10.1093/nar/gkp369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hildebrandt F, Heeringa SF, Rüschendorf F, et al. A Systematic Approach to Mapping Recessive Disease Genes in Individuals from Outbred Populations. PLOS Genetics. 2009;5(1):e1000353. doi: 10.1371/journal.pgen.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayer JA, Otto EA, O’Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nature Genetics. 2006;38:674. [DOI] [PubMed] [Google Scholar]
- 21.Park J, Shrestha R, Qiu C, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360(6390):758. doi: 10.1126/science.aar2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada A, Nomura S, Higashibata Y, et al. Successful formation of calcium oxalate crystal deposition in mouse kidney by intraabdominal glyoxylate injection. Urological Research. 2007;35(2):89–99. doi: 10.1007/s00240-007-0082-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The genetics datasets supporting the current study have not been deposited in a public repository due to restriction by patient consent, but are available from the corresponding author on request.