

Summary
Kinetochores control eukaryotic chromosome segregation by connecting chromosomal centromeres to spindle microtubules. Duplication of centromeric DNA necessitates kinetochore disassembly and subsequent reassembly on the nascent sisters. To search for a regulatory mechanism that controls the earliest steps of this process, we studied Mif2/CENP-C, an essential basal component of the kinetochore. We found that phosphorylation of a central region of Mif2 (Mif2-PEST) enhances inner kinetochore assembly. Eliminating Mif2-PEST phosphorylation sites progressively impairs cellular fitness. The most severe Mif2-PEST mutations are lethal in cells lacking otherwise non-essential inner kinetochore factors. These data show that multi-site phosphorylation of Mif2/CENP-C controls inner kinetochore assembly.
eTOC Blurb
Kinetochores, which are the chromosomal attachment points for spindle microtubules, are dynamic protein assemblies that must change shape to accommodate the cell cycle. Hinshaw et al. find that multi-site phosphorylation of a basal kinetochore component by cell cycle kinases controls kinetochore assembly.
Graphical Abstract
Introduction
Kinetochores, which connect centromeres to the mitotic spindle, supply and respond to the kinase signals that drive the cell cycle. They change shape and composition to accommodate DNA replication, tension sensing during metaphase, and chromosome movement during anaphase. While kinase activities required for progression from metaphase through anaphase have been identified, those that control kinetochore assembly during the preceding stages of the cell cycle have not.
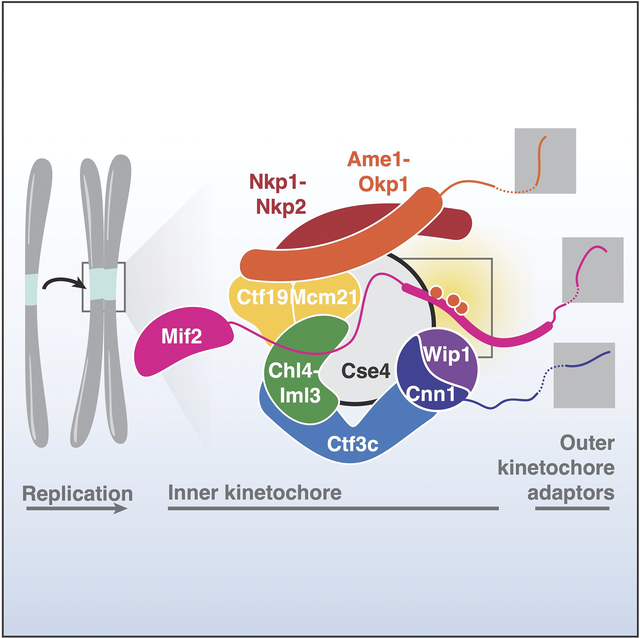
Kinetochores comprise modular protein complexes classified as inner or outer components.1 Outer kinetochores contact microtubules. Inner kinetochores recognize centromeric DNA and can be further subdivided into two groups: the Ctf19 complex (Ctf19c) and a second complex containing the centromeric nucleosome and its essential adaptor protein, Mif2/CENP-C (hereafter Mif2).2–5 In vertebrates, the Ctf19c is called the CCAN (Constitutive Centromere Associated Network). At least seven of the 13 Ctf19c proteins are phosphorylated in vivo (Okp1/CENP-Q, Ame1/CENP-U, Ctf19/CENP-P, Mcm21/CENP-O, Nkp1, Chl4/CENP-N, and Cnn1/CENP-T).6 Mif2 and the centromeric histone Cse4/CENP-A are also phosphorylated.7,8 Whether phosphorylation of these factors influences kinetochore assembly is an important open question.
We and others have reported structures of inner kinetochore protein assemblies.9–13 With one exception (discussed below),14 these structures show fixed contacts and do not address regulated inner kinetochore assembly. Divergent models for Cse4/CENP-A recognition by the Ctf19c have been proposed.9,12,15–18 Identifying regulatory mechanisms is a necessary step towards evaluating these models.
Kinetochore assembly requires Mif2/CENP-C in yeast and vertebrate cells.19,20 A conserved Mif2/CENP-C motif selectively binds Cse4/CENP-A versus histone H3.21 Human CENP-C has functionally redundant CENP-A binding motifs,22–24 and nearby CDK1 phosphorylation enhances CENP-A binding by promoting CENP-C folding.14,24 Similarly, Mif2-Cse4 binding is thought to alter Mif2 folding, which enables outer kinetochore assembly.25 Although multiple Mif2 phosphorylation sites have been identified,8,26 their functions are unknown, and the vertebrate CDK1 site is not conserved.
Though generally thought to be a constitutive feature of centromeres, Ctf19c/CCAN assembly is probably regulated during S phase, when the underlying centromeric DNA is replicated. Reports of structural rearrangements and regulated subunit recruitment support this view.2,14,24,27,28 Likewise, improved methods for kinetochore reconstitution from yeast cell extracts indicate that cell cycle stage influences yeast inner kinetochore assembly or stability in vitro.29 What kinase activities control inner kinetochore assembly, how might they influence its structure, and how does this relate to cell cycle progression?
To address these questions, we focused on the conserved and essential inner kinetochore protein, Mif2. We report that Mif2 phosphorylation stabilizes the inner kinetochore. This and associated findings connect cell cycle regulation to the functional plasticity of the inner kinetochore.
Results
Genetic dissection of Mif2 and its interactions with Ctf19 complex factors
We used genetic complementation to identify the minimal Mif2 protein required for cell division.30 To do so, we expressed MIF2 or its mutants from a plasmid and tested their ability to rescue cell growth upon auxin-mediated depletion of chromosomally encoded Mif2-AID (Auxin-Inducible Degron; Figure 1A). Mif2 lacking amino acids 1–200 (mif2-Δ200) supported viability, but a version lacking residues 1–255 (mif2-Δ255) did not.
Figure 1 – Mif2-PEST region phosphorylation supports kinetochore function.
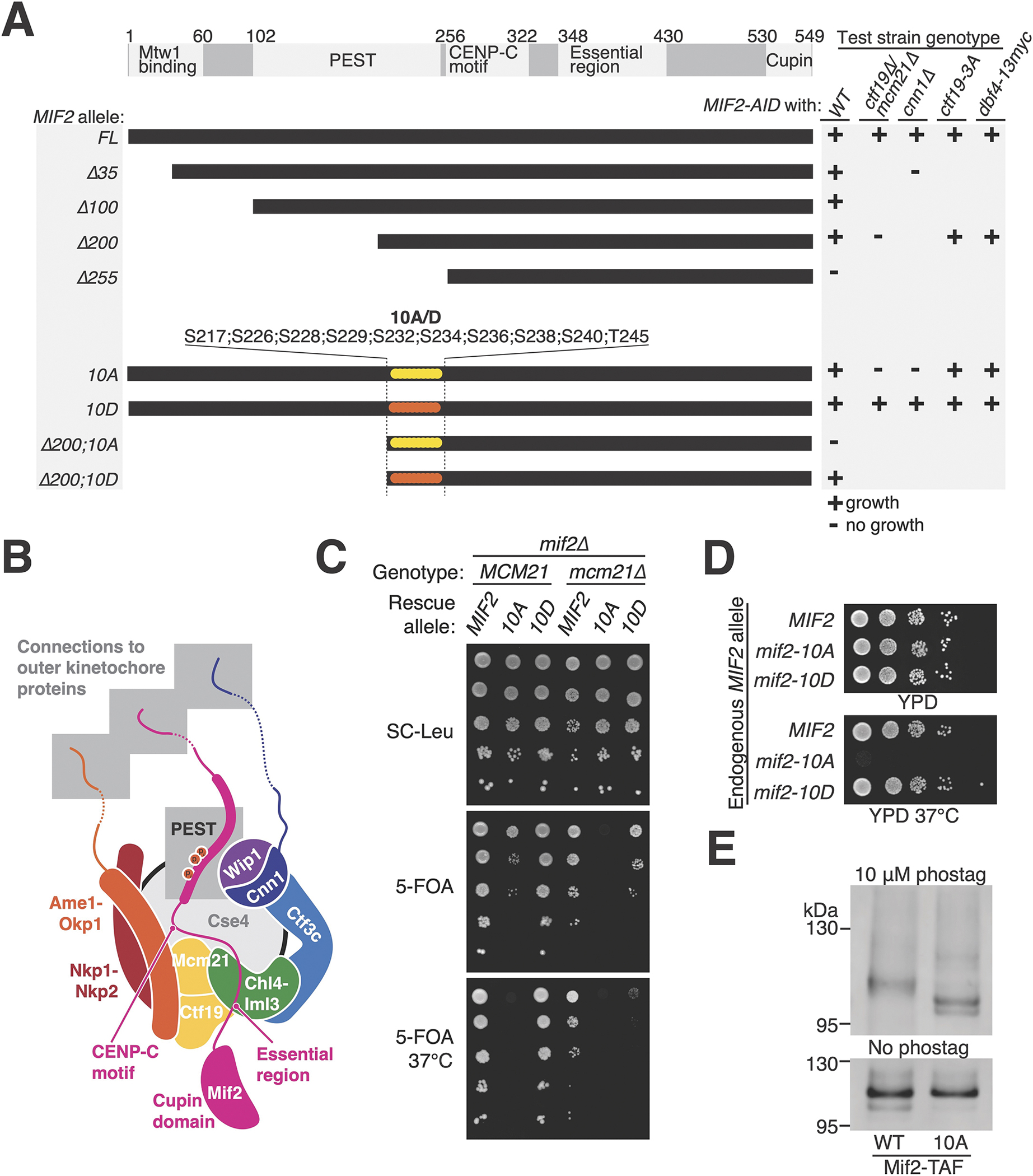
(A) MIF2 genetic complementation experiments. Rescue plasmids coding for MIF2 or its mutants (CEN/ARS-LEU2 MIF2; listed at left) were tested for their ability to complement depletion of endogenous Mif2-AID protein in strains with the indicated genotypes (listed at right). See also Figure S1 and S2. (B) Illustration of the inner kinetochore. Shaded boxes show three peptides that recruit outer kinetochore proteins (top) and the phosphorylated Mif2-PEST (middle). (C) Synthetic lethal interaction between mcm21Δ and mif2-10A. mif2Δ (left) or mif2Δ mcm21Δ (right) cells carried the indicated MIF2 rescue allele on a test plasmid (CEN/ARS-LEU2 with MIF2, mif2-10A, or mif2-10D). A second complementing plasmid (CEN/ARS-URA3 with MIF2 or both MCM21 and MIF2) was ejected by growth on 5-FOA (middle and bottom), leaving only the test plasmid. (D) Heat stress (37 °C) kills mif2-10A mutant cells. MIF2, mif2-10A, or mif2-10D were integrated into the native MIF2 locus in otherwise wild type cells. (E) Mif2-TAF was purified from asynchronous cultures, resolved by SDS-PAGE with 10 μM (top) or 0 μM (bottom) Phos-tag acrylamide, and detected by anti-protein A Western blot (TAF: 6xHIS-3xFLAG-ProteinA tag). See also Figure S3A and Tables S1–2.
We used a second genetic complementation system for a finer MIF2 deletion analysis covering Mif2 residues 1–240 (Figure S1). In these experiments, mif2Δ cells were propagated with a complementing plasmid carrying MIF2 and a test plasmid carrying MIF2 or its mutants. Removal of the complementing plasmid revealed the phenotype conferred by the test plasmid. We identified a mif2 internal deletion mutant that was sensitive to high temperature, hydroxyurea (HU), and benomyl (mif2-Δ181-240; Figure S1). This part of Mif2 was previously named the PEST region (for Proline-, Glutamate (E)-, Serine-, and Threonine-rich; Mif2-PEST). Temperature-sensitivity of mif2-ΔPEST cells is known.30
The Mif2-PEST region is well-situated to regulate inner kinetochore assembly: it is immediately N-terminal to the Cse4-binding motif, it is positioned near Ctf19c proteins in the assembled kinetochore, and its human counterpart binds CCAN factors (CENP-H/I and CENP-N; Figure 1B).31 We used the Mif2-AID complementation system described above to search for Mif2-PEST phosphorylation sites that might influence Ctf19c function. Cells lacking Mif2-N depend on the Ctf19c for viability.25,32 Accordingly, we searched for serine/threonine-to-alanine missense mutations that were selectively lethal when introduced into mif2-Δ200. We identified ten Mif2-PEST serine/threonine residues that, when converted to alanine, were tolerated in full-length MIF2 but lethal in mif2-Δ200 (Figure 1 and Figure S2A). Conversion of the same residues to aspartate did not produce a growth defect.
To test the idea that the mif2-10A allele weakens the inner kinetochore, we combined this mutation with Ctf19c mutations. ctf19Δ or cnn1Δ strains have partially defective inner kinetochores, and mif2-10A failed to support the viability of either Ctf19c deletion strain (ctf19Δ or cnn1Δ; Figure 1A and Figure S2B). In addition to its function in kinetochore assembly, CTF19 ensures timely centromeric cohesin recruitment and early replication of centromeric DNA.33,34 To test whether these CTF19 functions are dispensable in mif2-10A cells, we used the ctf19-3A and dbf4-13myc alleles to specifically inactivate centromeric cohesin recruitment and early centromere replication, respectively. mif2-10A supported growth in both mutant backgrounds, indicating that neither function is required for cell growth in the absence of Mif2-PEST phosphorylation.
Extragenic mif2-10A caused a dominant growth defect in cells with Ctf19c mutations. Specifically, mcm21Δ, ctf19Δ, or cnn1Δ cells carrying both MIF2 and mif2-10A were sick (Figure S2B). To bypass this dominance, we created a complementing plasmid carrying both MIF2 and MCM21. Ejection of this plasmid in mif2Δ mcm21Δ cells enabled simultaneous deletion of MIF2 and MCM21, revealing the phenotype associated with MIF2 or its mutants provided on a separate test plasmid (Figure 1C). This experiment confirmed that mif2-10A does not support viability in mcm21Δ cells. MIF2 complementation was incomplete on selective medium, consistent with defective plasmid propagation in mcm21Δ cells. To eliminate the need for plasmid complementation, we replaced chromosomal MIF2 by mif2-10A or -10D in otherwise wild type cells. Replacement by mif2-10A prevented growth at elevated temperature (Figure 1D) and made cells hypersensitive to the microtubule poison, benomyl (Figure S1D).
To determine whether the Mif2-PEST region is phosphorylated, we used Phos-tag acrylamide gel electrophoresis (Figure 1E).35 Mif2-WT purified from asynchronous cultures migrated more slowly than Mif2-10A. Phosphatase treatment converted these bands to a single fast migrating species, confirming Mif2 phosphorylation at the mutated Mif2-PEST sites (Figure S3A).
DDK, Cdc5, and Ipl1 phosphorylate Mif2 in vitro
The Mif2-PEST resembles known DDK and Cdc5 substrates.36,37 We therefore tested whether DDK and Cdc5 could phosphorylate Mif2 in vitro. We also included Ipl1, a kinase known to phosphorylate residues outside the Mif2-PEST.8 All three kinases robustly phosphorylated full length Mif2-WT (Figure 2A). Mif2-10A was a poor substrate for both Cdc5 and DDK (Figure 2B). Ipl1 phosphorylated Mif2-WT, -10A, and -10D equally, whereas further mutation of eight known or mapped Ipl1 sites to make Mif2-18D prevented Mif2 phosphorylation by Ipl1 (Figure S3B). Cdc5 and DDK can act cooperatively,38 and a mixture of Cdc5 and DDK phosphorylated Mif2 more than would be expected were both kinases working individually (Figure 2B). The double kinase treatment maintained the preference for Mif2-WT over Mif2-10A. Therefore, Cdc5 and DDK can cooperatively phosphorylate the Mif2-PEST region in vitro, and these and other kinases may phosphorylate the Mif2-PEST region in vivo.
Figure 2 – DDK and Cdc5 phosphorylate the Mif2 PEST region in vitro.
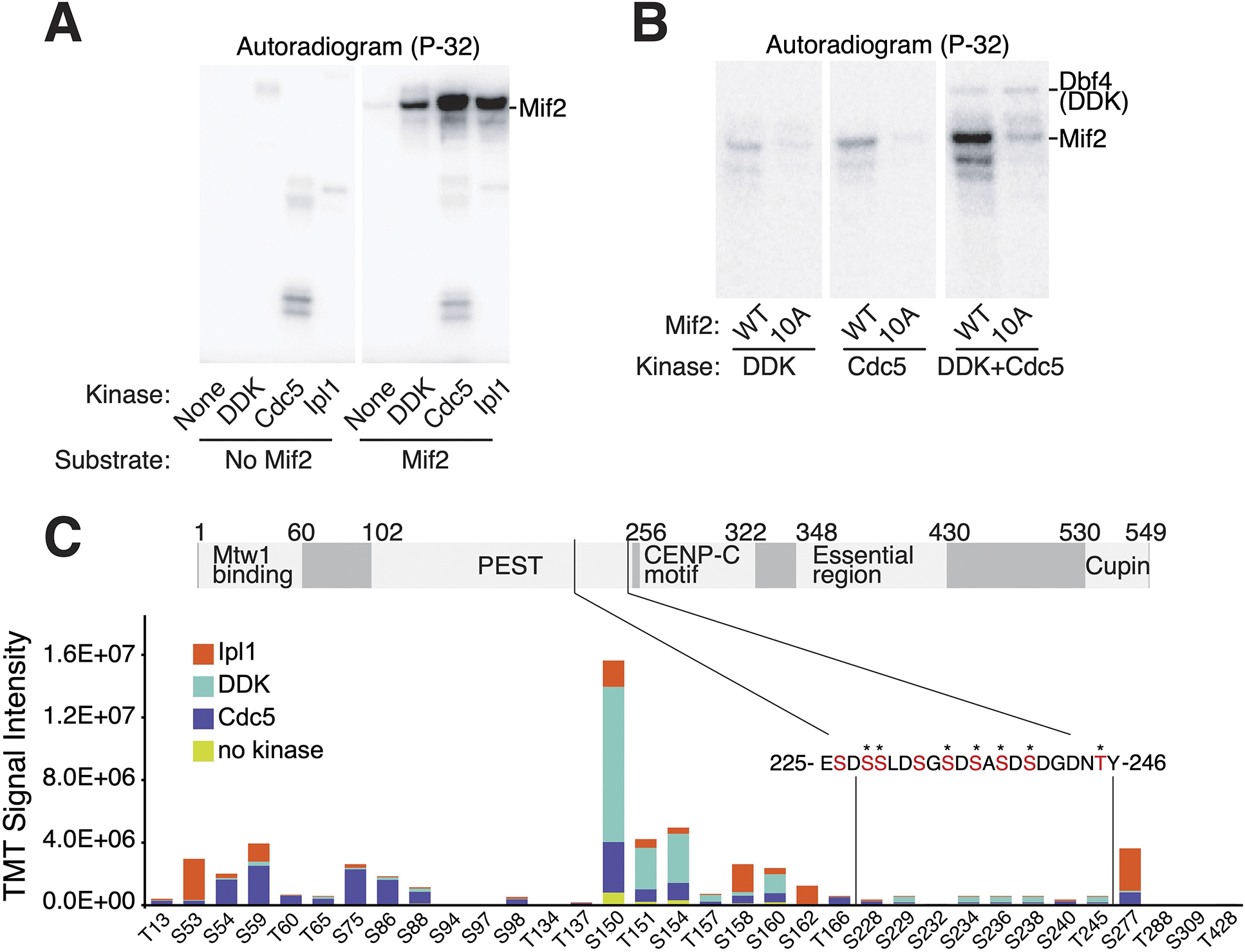
(A) DDK, Cdc5, and Ipl1 phosphorylate Mif2 in vitro. Purified full length Mif2 was incubated with the indicated kinases, and phosphate transfer from [γ-32P]ATP was detected by autoradiography. See also figure S3. (B) Kinase assays were performed as in panel A with the indicated substrates and enzymes (all panels from a single exposure). (C) Phosphorylation reactions with Mif2 and Cdc5, DDK, or Ipl1 were subjected to TMT-MS analysis (see Methods). Relative intensities of the same peptide treated with different kinases and unique TMT labels provide an accurate assessment of the relative contribution of each kinase to the abundance of a given phosphopeptide. Total signal intensities for different peptides primarily reflect unequal ionization efficiencies. The sequence of Mif2 residues 225–246 is shown, and residues that could be conclusively identified as phosphorylated are marked by stars. See also Data S1 and Table S1.
We used Tandem Mass Tag (TMT) labeling and mass spectrometry (MS) to directly observe Mif2 phosphopeptides after in vitro kinase reactions with Ipl1, Cdc5, or DDK (Figure 2C and Data S1). A given phosphopeptide’s abundance, propensity for ionization, and level of phosphorylation determine its detectability by MS. These characteristics make multi-phosphorylated peptides difficult to detect, which includes the Mif2-PEST. Nevertheless, we mapped several phosphopeptides to the Mif2-PEST region after Mif2 treatment with Cdc5 and DDK. The Ipl1-, Cdc5-, and DDK-modified Mif2 sites match the known consensus motifs of these kinases.36,37,39 MS analysis of Mif2-WT purified from yeast identified matching phosphopeptides (Data S1), although Mif2-PEST phosphopeptides were not detected due to the lower abundance of endogenous Mif2 protein and for the technical reasons stated above.
Mif2 phosphorylation enhances inner kinetochore assembly in vitro
To test the effects of Mif2-PEST mutations on inner kinetochore assembly, we reconstituted this process in vitro and subjected the resulting complexes to gel filtration chromatography. For these experiments, purified Ctf19c and Mif2 proteins were first dephosphorylated. In the absence of kinase activity, Mif2 and the Cse4 nucleosome associated weakly with the intact Ctf19c (Figure 3A, middle panel). Substitution of Mif2 by Mif2-10D enhanced complex formation (Figure 3A, bottom panel), suggesting that mimicking Mif2 PEST phosphorylation facilitates inner kinetochore assembly.
Figure 3 – Biochemical reconstitution of inner kinetochore assembly.
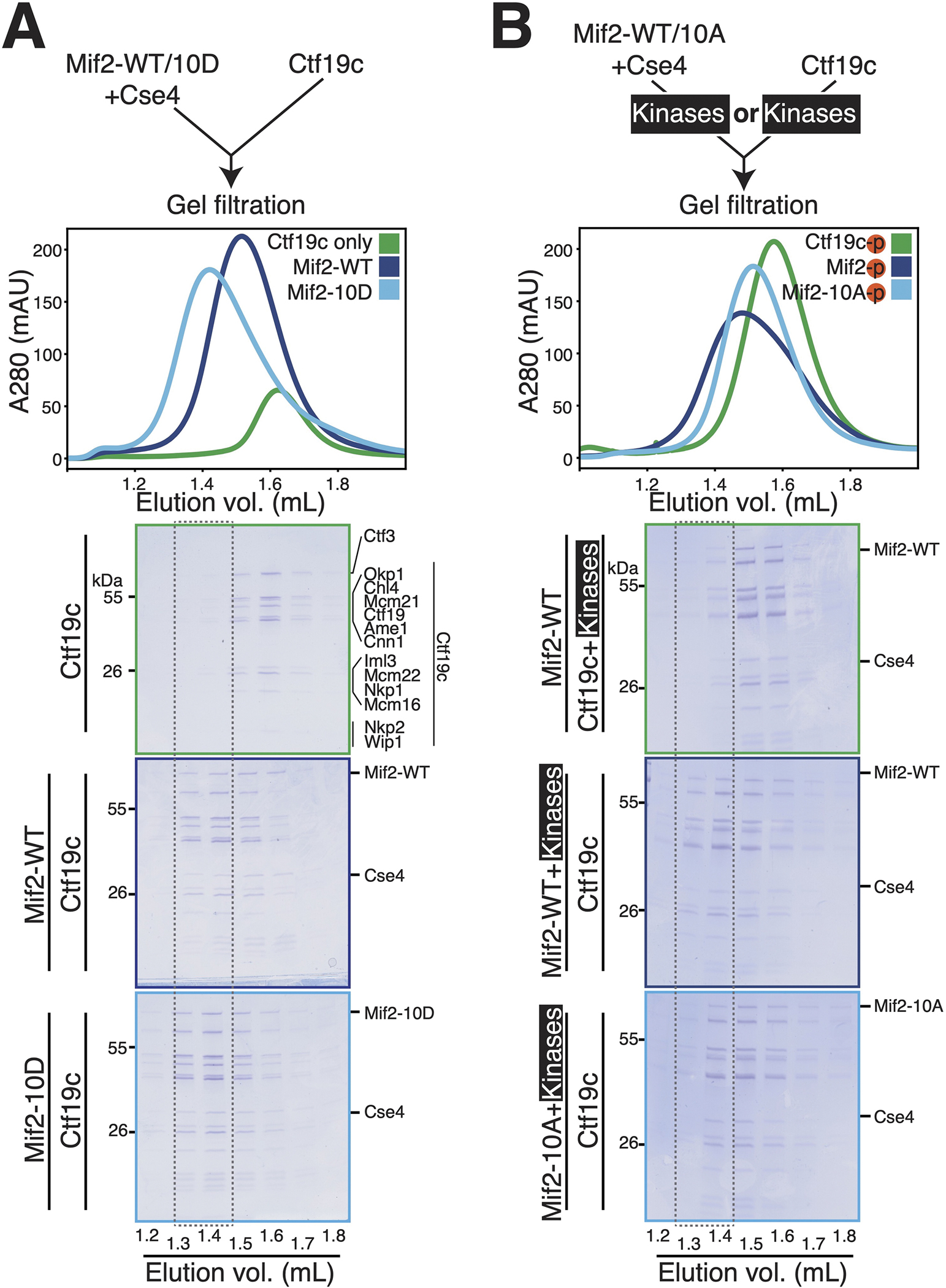
(A) Analysis of inner kinetochore assembly in the absence of kinase activity. The diagram shows the experimental setup. Chromatograms show the gel filtration elution profiles for the indicated samples. The dotted outline is a visual aid to highlight equivalent fractions for each experiment. The curves show the following: green – Ctf19c only; dark blue – Mif2-WT, Cse4 nucleosome, and Ctf19c; light blue – Mif2-10D, Cse4 nucleosome, and Ctf19c. (B) Mif2-Cse4 nucleosome or the Ctf19c was phosphorylated before complex formation and gel filtration. The diagram shows the experimental setup (top). The curves show the following: green – Mif2-Cse4 nucleosome complex with phosphorylated Ctf19c; dark blue – phosphorylated Mif2-Cse4 nucleosome complex with Ctf19c; light blue – phosphorylated Mif2-10A-Cse4 nucleosome complex with Ctf19c. See also Figure S3 and Table S1.
We next assessed the effects of Mif2 phosphorylation using the same system. We treated either the Mif2-nucleosome complex or the Ctf19c with Cdc5 and DDK before a final incubation step in which both complexes were combined (Figure 3B). Treatment of the Mif2-nucleosome complex with kinases enhanced its interaction with the Ctf19c (Figure 3B, middle panel). Inclusion of Mif2-10A in place of Mif2-WT largely prevented complex stabilization by kinases (Figure 3B, bottom panel). Therefore, Mif2 phosphorylation promotes inner kinetochore assembly. A comparison of complexes formed with unmodified and phosphorylated Mif2 can be found in Figure S3D.
To determine whether the foregoing observations could trivially be explained by stronger DNA binding by phosphorylated Mif2, we performed EMSA experiments. Mif2 phosphorylation by Cdc5 and DDK slightly weakened its association with the Cse4 nucleosome (Figure S3E). Consistent with this, Mif2-10A has a higher affinity than Mif2-10D for the DNA used in these experiments (Figure S3F). Therefore, enhanced inner kinetochore assembly upon Mif2 phosphorylation is not caused by tighter Mif2-DNA or -nucleosome contact. Instead, Mif2 phosphorylation facilitates Ctf19c recruitment.
Mif2 phosphorylation enhances stable Mif2-Cse4 association in vivo
To determine whether Mif2-PEST phosphorylation influences inner kinetochore assembly in vivo, we performed chromatin immunoprecipitation and quantitative PCR experiments (ChIP-qPCR) to examine Mif2 localization. Mif2 localized to CEN3 but not to a non-centromeric locus (Figure 4A). Mif2-10A was partially defective in its localization, and Mif2-10D retained centromere localization. Therefore, Mif2-PEST phosphorylation is required for normal centromere recruitment, and constitutively mimicking phosphorylation with acidic residues bypasses this requirement. That Mif2 recruitment does not strictly reflect the relative DNA binding strengths of purified Mif2-10A and Mif2-10D (Figure S3F) indicates that defective centromere-Mif2-10A contact in vivo is a secondary effect of a poorly assembled Ctf19c. Indeed, Ctf19c assembly is required for robust Mif2-centromere contact (see Discussion).40
Figure 4 – Mif2-PEST mutation interferes with Mif2-Cse4 association in vivo.
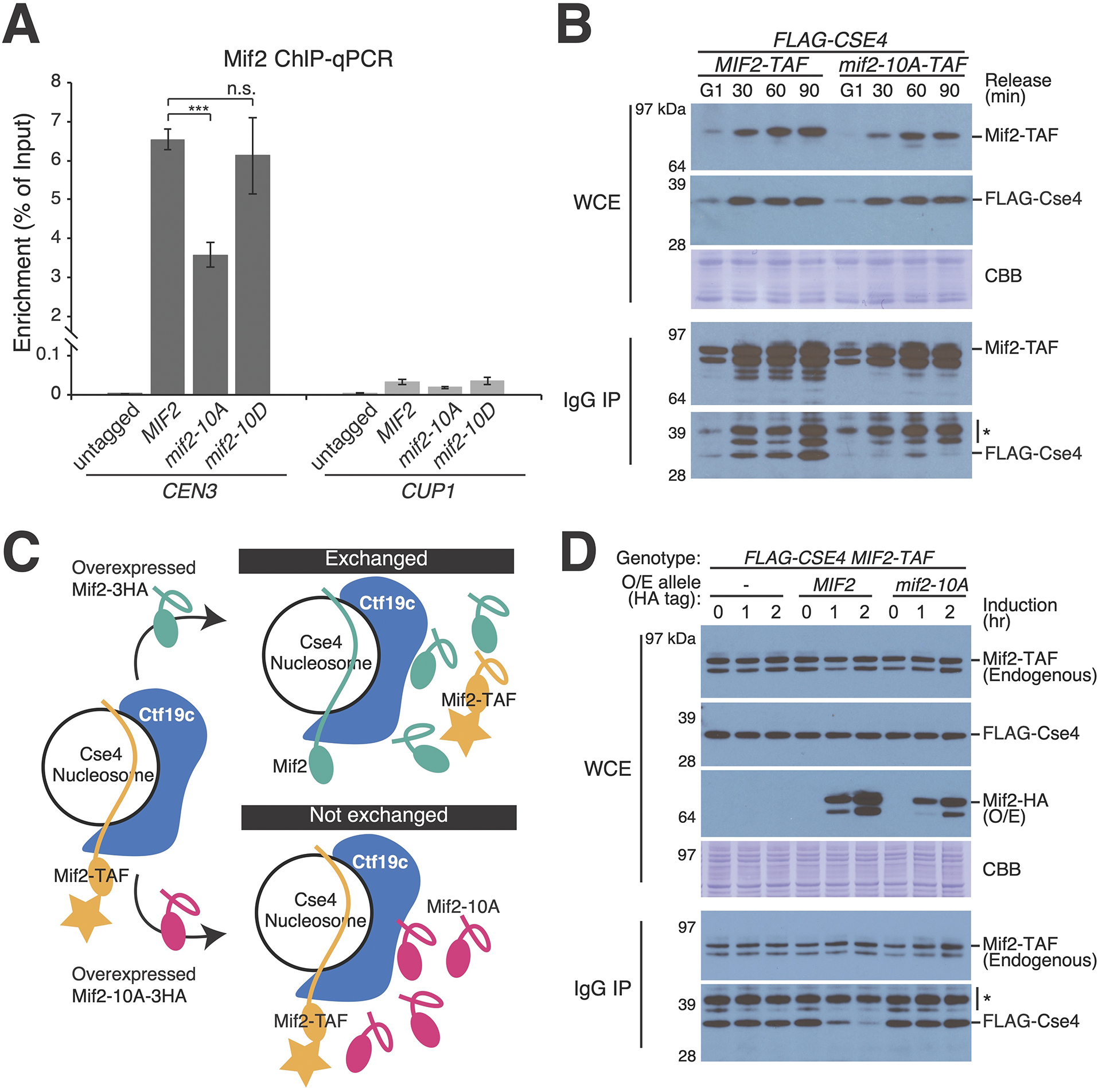
(A) Measurement of Mif2-CEN3 association in asynchronous cells with the indicated MIF2 alleles coding for Mif2-TAF proteins. ChIP-qPCR for Mif2-TAF is shown. Mif2 association with a non-centromeric locus (CUP1) is shown at right (*** p < 0.001, Student’s t-test). (B) Co-purification of Mif2 and Cse4 analyzed during cell cycle progression. Cells were arrested in G1 with alpha factor and released into the cell cycle for the indicated amounts of time (top). Mif2-TAF was immunopurified. Co-purifying FLAG-Cse4 was detected by Western blot (WCE – whole cell extract; IgG IP – Mif2-TAF immunoprecipitation; CBB – Coomassie brilliant blue for total protein; * – background from partially degraded Mif2 protein A tag). (C) Schematic showing the experimental design for competition pull-down experiments (panel D). Upon induction of Mif2-HA expression from an ectopic locus, endogenous Mif2 (Mif2-TAF) was affinity-purified, and copurifying Cse4 was detected by Western blot. See also Figure S4A. (D) Overexpressed Mif2 but not Mif2-10A competes with endogenous Mif2 for Cse4 binding. Pulldown experiments were performed as in panel A. Time elapsed after induction of extragenic Mif2-WT or Mif2-10A is shown (O/E – overexpression; * – background band). See also Figure S4B and Tables S1–2.
To further evaluate Mif2-Cse4 association in vivo, we measured their interaction by co-immunoprecipitation. Cse4 co-purified with Mif2-WT, and less Cse4 co-purified with Mif2-10A (Figure 4B). Mif2-Cse4 binding was weakest in G1, and it increased as cells progressed through the cell cycle. To directly compare Cse4 association with Mif2-WT and Mif2-10A, we performed competitive binding experiments by overexpressing a second copy of MIF2 (Figure 4C). Overexpressed Mif2-WT but not Mif2-10A interfered with the ability of endogenous Mif2-WT to bind Cse4 (Figure 4D). Cells lacking Mcm21 show the same pattern (Figure S4). Therefore, Mif2-10A is defective in its association with Cse4 in vivo.
Mif2 phosphorylation and Ctf19c assembly cooperate to ensure kinetochore stability
As a qualitative measure of kinetochore function in vivo, we assessed tolerance to a dicentric plasmid (Figure 5A). Mitotic cells establish biorientation for each centromere independently. A single dicentric chromatid can attach to opposite spindle pole bodies, mimicking the merotelic attachments seen in organisms with multivalent centromeres.41 Budding yeast have no mechanism to correct this error, and the result is a severe viability defect.42 We confirmed MCM21 deletion bypasses the lethality of a dicentric plasmid.43 mif2-10A cells carrying the dicentric plasmid were also viable, indicating that these cells have hypomorphic kinetochores. In contrast, cells bearing the mif2-10D mutation did not tolerate the dicentric plasmid.
Figure 5 – Genetic analysis of Mif2-PEST phosphorylation sites.
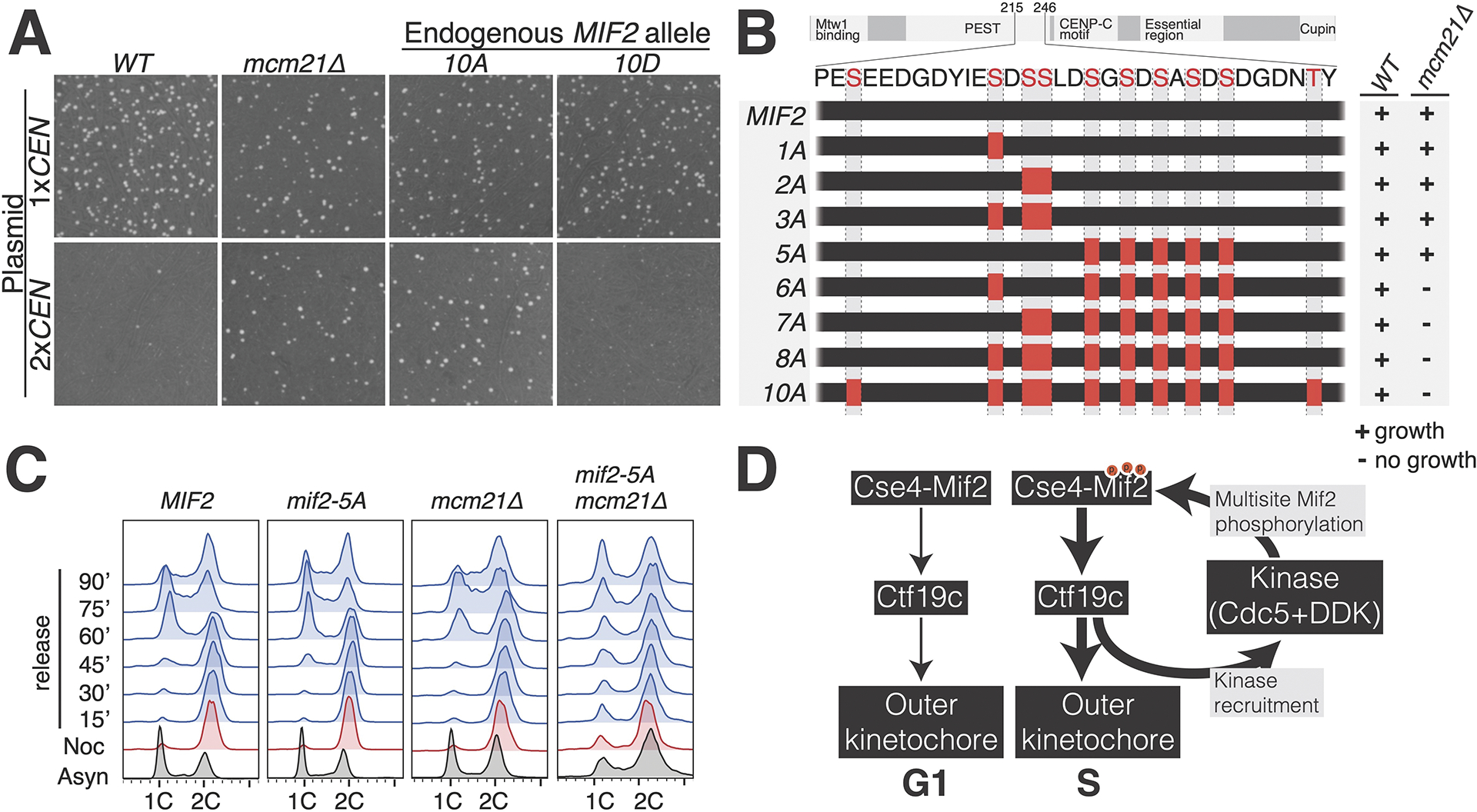
(A) The indicated strains were propagated with a monocentric or dicentric plasmid (1xCEN and 2xCEN, respectively). Viability was assessed by colony formation under selection for the plasmid. Plate images are shown. (B) Diagram showing Mif2 residues 215–246. Alanine mutations corresponding to the indicated MIF2 alleles are shown as red boxes. Viability of the corresponding genotypes was determined by sporulation analysis. The results are summarized at right. See also Figure S5. (C) Recovery from mitotic arrest was analyzed by flow cytometry in the indicated strains. Cells were arrested in G2/M before release into fresh medium. In MIF2 cells, the reappearance (60 minutes) and subsequent disappearance (90 minutes) of the 1C peak shows synchronous cell cycle progression. (D) Model showing kinase regulation of inner kinetochore assembly. Cdc5 and DDK are listed as the likely kinases, and other kinases may contribute at this step (Cdc28/CDK1) 44. Cdc5 and DDK are recruited by inner kinetochore proteins at the G1/S transition 33,34, and this stabilizes inner kinetochore assembly via phosphorylation of Mif2 and possibly other targets. See also Tables S1–2.
The PEST region is a universal feature of CENP-C proteins. Mif2-PEST and homologous peptides contain abundant acid- and proline-directed putative phosphorylation sites, but their individual positions are not fixed, even in closely related species. This and the absence of predicted secondary structure elements suggests that function depends on the combined actions of multiple phosphorylated residues and not on a single site. To test this idea, we created a series of MIF2 alleles encoding subsets of mutations (Figure 5B). Sporulation of heterozygous mcm21Δ mif2-10A cells confirmed that mif2-10A cells are viable but the double mutant cells are not (Figure S5A). Likewise, mif2-8A, -7A, and -6A mcm21Δ double mutant spores were inviable. In contrast, mif2-1A, -2A, -3A, and -5A mcm21Δ double mutant spores were viable, and these had increasing sensitivities to heat stress (37 °C), HU, and benomyl (Ben) (Figure S5B). The mif2-3A and mif2-5A mutations ablate non-overlapping subsets of phosphorylation sites, but they produced nearly identical growth defects in the absence of MCM21. Therefore, no single phosphorylation site can explain the viability defects observed in these mutants.
The results above indicate that the Mif2-PEST region works with the Ctf19c to ensure normal kinetochore function. We used mif2-5A, the most perturbative Mif2-PEST mutant viable in mcm21Δ cells, to examine the consequence of partial Mif2-PEST inactivation. To study mitosis in mcm21Δ mif2-5A double mutant cells, we released cells from metaphase arrest and tracked their entry into the following cell cycle (Figure 5C). Wild type, mcm21Δ, and mif2-5A cells synchronously re-entered the cell cycle upon release. In contrast, mcm21Δ mif2-5A double mutant cells displayed defective chromosome segregation upon release (Figure 5C, right column). At 90 minutes after release, the double mutant cultures were essentially asynchronous, indicating long and variable delays in the execution of mitosis. Consistently, G2/M cells accumulated in asynchronous mcm21Δ mif2-5A cultures (Figure 5C, bottom row). We also measured cell viability, normalized to the beginning of the procedure, of cells released from a nocodazole arrest (Figure S5C). Whereas wild type, mcm21Δ, and mif2-5A cells largely maintained their viability throughout the arrest, the viability of mcm21Δ mif2-5A cells dropped significantly.
Discussion
Kinetochores must accommodate the events of the cell cycle. These include replication of underlying centromeric DNA, attachment to spindle microtubules, establishment of tension-sensing microtubule attachments, and maintenance of the attachments through anaphase. Kinases drive the transitions between these states. We found that phosphorylation of Mif2, a conserved structural component of kinetochores, enhances inner kinetochore assembly. Mif2 phosphorylation is essential in cells with weakened kinetochores.
How might Mif2-PEST phosphorylation prepare the inner kinetochore for mitosis? Hagemann et al. now report that Mif2-PEST phosphorylation strengthens Mif2-Okp1 binding tenfold.44 Direct contact between Mif2-PEST and Okp1 is not a foregone conclusion. Firstly, Mif2-10D enhances inner kinetochore assembly despite the chemical distinction between aspartate and phosphorylated serine. Secondly, proposed foldback autoinhibition of Mif2 suggests enhanced Mif2-Okp1 binding could be an indirect consequence of phosphorylation (see below).25 Tighter Mif2-Okp1 binding provides an explanation for mif2-10A cnn1Δ synthetic lethality; linkages with the centromere (via Okp1-Mif2) and with the outer kinetochore (via Cnn1) are severely compromised in this double-mutant. It bears mentioning that we cannot rule out the possibility that Mif2-PEST phosphorylation has consequences not recapitulated in reductive biochemical experiments.
In published structures, CENP-C folds back on itself at its contact point with CENP-A.14,15,23 In this conformation, phosphorylated Mif2-PEST would be in position to satisfy positively charged Mif2 DNA binding segments.21 In line with this idea, recombinant Mif2 used in published experiments showing foldback autoinhibition25 and Okp1 binding32 was almost certainly phosphorylated in the PEST region. Mif2 regulation by autoinhibition was originally suggested by Brown,45 and a related mechanism controls the DNA binding activity of the Ets-1 transcription factor.46,47 Vertebrate Haspin provides a mitotic parallel: CDK and PLK generate a Haspin phosphopeptide that sequesters an adjacent lysine-rich segment.48 Whether phosphorylation regulates Mif2 function via a similar mechanism remains to be seen. Answering this question will likely require structural studies of properly assembled Mif2-containing kinetochores.
We propose that feed-forward regulation progressively stabilizes key kinetochore interfaces. The inner kinetochore recruits DDK to the centromere,33,34 and we have shown here that the reverse is also true. The resulting positive feedback loop progressively stabilizes kinetochores (Figure 5D). Cooperative Mif2-PEST phosphorylation by Cdc5, DDK, and possibly CDK144 (Figure 2B) provides a compelling biochemical explanation for progressive stabilization. Conversely, any mutations that interrupt positive feedback should interfere with Mif2 and Ctf19c function. This is true: mif2-10A phenocopies mcm21Δ in the dicentric plasmid assay (Figure 5A), and defective Mif2/Cse4 centromere loading is a known consequence of MCM21 deletion.40 The observed synthetic lethality between mif2-10A and mcm21Δ may reflect a catastrophic disruption of the inner kinetochore caused by both mutants.
Why should inner kinetochore assembly be subject to kinase regulation? In yeast, DDK phosphorylates Ctf19 at the onset of S phase,33,34 and recruitment of DDK to the inner kinetochore specifies centromeric replication origins as the first to fire in S phase.34 These facts indicate that Mif2-PEST-dependent inner kinetochore strengthening probably coincides with firing of centromere-associated replication origins, thus preventing premature inner kinetochore stabilization before replication of the underlying chromosome. The foregoing findings motivate further experiments to test this model.
STAR Methods
RESOURCE AVAILABILITY
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Stephen M. Hinshaw (hinshaw@stanford.edu).
Materials availability
Plasmids and yeast strains are listed in the Key Resources Table, Table S1, and Table S2. Requests will be fulfilled by the lead contact. Where indicated, plasmids are available from Addgene.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-FLAG-HRP (mouse monoclonal) | Sigma | A8592 |
| anti-FLAG (mouse monoclonal) | Sigma | F3165 |
| anti-HA | Sigma | 3F10 |
| anti-ProtA | Sigma | P3775 |
| anti-FLAG magnetic beads | Sigma | M8823 |
| Protein G Dynabeads | ThermoFisher | 10003D |
| Bacterial and virus strains | ||
| Rosetta 2(DE3)pLysS; E. coli | EMD Millipore | 71403 |
| Chemicals, peptides, and recombinant proteins | ||
| Lambda phosphatase | Hinshaw stock | N/A |
| IAA (3-Indoleacetic acid) | Sigma | I2886 |
| Benzonase | EMD Millipore | 70746 |
| [γ-32P]ATP | Perkin Elmer | BLU502A |
| SYTOX Green Nucleic Acid Stain | ThermoFisher | S7020 |
| SYBR Gold Nucleic Acid Gel Stain | ThermoFisher | S11494 |
| SYBR Green 2× Master Mix | ThermoFisher | A46012 |
| 1-Napthaleneacetic acid | Sigma | N0640 |
| Phostag acrylamide | APExBIO | F4002 |
| Experimental models: Cell lines | ||
| High Five cells; Trichoplusia ni | ThermoFisher | B85502 |
| Sf9 cells; Spodoptera frugiperda | ThermoFisher | 11496015 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae strains used in this study | See Table S2 | N/A |
| Oligonucleotides | ||
| ATCGAGAATCCCGGTGCC | IDT | oSMH1950 |
| ATCGGATGTATATATCTGACACGTGC | IDT | oSMH1951 |
| ATCAGCGCCAAACAATATGGAAAA | IDT | CEN3_fwd |
| GAGCAAAACTTCCACCAGTAAACG | IDT | CEN3_rev |
| AACTTCCAAAATGAAGGTCA | IDT | CUP1_fwd |
| GCATGACTTCTTGGTTTCTT | IDT | CUP1_rev |
| Recombinant DNA | ||
| Plasmids used in this study | See Table S1 | N/A |
| Software and algorithms | ||
| COMET | Seattle Proteome Center | https://uwpr.github.io/Comet/ |
| Graphpad Prism v9 (graphpad.com) | Dotmatics | N/A |
Data and code availability
There was no large dataset generated and deposited along with this paper.
This paper does not report original code.
The data reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmid and strain construction
Yeast strains used in this study are listed in Table S1.
The MIF2 locus including flanking sequence (see pSMH1409 and pHZE1904 in Table S1) was cloned into CEN/ARS plasmids via conventional restriction cloning (pSMH1409; XhoI and NotI) or by yeast recombination repair (pHZE1904). To generate MIF2-TAF:HisMX6 (where TAF indicates a 6xHIS-3xFLAG-ProteinA tag), the Mif2-expressing plasmid was linearized by SphI, and a PCR product of TAF:HisMX6 was fused to the MIF2 C-terminus via yeast recombination. Deletion and point mutations of Mif2 were then introduced in these plasmids.
For modification of the chromosomal MIF2 locus (besides MIF2-AID; described below), restriction fragments of plasmids containing MIF2 or its mutants were integrated into the chromosomal MIF2 locus. The parental strain genotype was mif2Δ, and this deficiency was complemented by a CEN/ARS plasmid with the URA3 marker (pHZE1905). His-positive colonies were replicated on plates containing 5-fluoroorotic acid (5-FOA) to evict the complementing MIF2:URA3 plasmid. Successful integration was confirmed by nourseothricin-sensitivity and PCR tests. Transformations for all other genomic modifications were done by standard methods.49 All strains were grown at 30 °C in yeast extract peptone dextrose medium (YPD) unless otherwise noted. For growth assays with strains harboring modifications at the endogenous MIF2 locus, cells were grown and plated on YPD with the indicated additives.
MIF2-AID complementation experiments
PCR integration was used to introduce a C-terminal auxin-inducible degron (AID) tag50 at the endogenous MIF2 locus in a strain constitutively expressing TIR1 (pADH1-OsTIR1-9xMyc). Where indicated, further genetic manipulations (cnn1Δ, for example) were done in this strain background. Complementing MIF2 and the specified mutant alleles were supplied on a CEN6-containing plasmid (pCEN/ARS-LEU2) that included ~500 bp flanking genomic sequence on either side of the MIF2 gene. Plasmid transformants were selected on synthetic complete medium without leucine (Sc -leu). Transformants were picked, streaked, and grown overnight in liquid culture (SC -leu) before plating a five-fold dilution series on Sc -leu with or without 1-Napthaleneacetic acid (auxin). All plates were grown for two days at 30 °C unless otherwise indicated.
MIF2 plasmid shuffling experiments using URA3/5-FOA
Two strains, mif2Δ pCEN/ARS-URA3-MIF2 (HZY366) and mif2Δmcm21Δ pCEN/ARS-URA3-MIF2-MCM21 (HZY1247), were transformed with the indicated plasmids bearing MIF2, mif2-10A and mif2-10D mutants. Successful transformants were grown up in Sc -Leu media to an OD600 ~1, normalized based on cell density, and then spotted on the indicated plates following 5-fold serial dilutions. 5-fluoroorotic acid (Sc supplemented with 0.1% 5-FOA) treatment removes the complementing MIF2 plasmid in HZY366, or the MCM21 and MIF2 co-expression plasmid in HZY1247, exposing the phenotypes of mif2 mutants in WT MCM21 cells or mcm21Δ cells. Plates were incubated at 30 °C for 2 or 3 days before taking images using a BioRad ChemiDoc MP imaging system.
METHOD DETAILS
Cell viability upon release from a nocodazole arrest
Ten clones of each MIF2, mif2-5A, mcm21Δ, and mif2-5A mcm21Δ cells were grown in YPD media supplemented with 1% DMSO to early logarithmic. Cell cultures were arrest in G2/M phase by the addition of 15 μg/mL Nocodazole for 3 hours. The same volume of cell culture was taken before and after nocodazole arrest, followed by dilution and plating on YPD plates. Plates were incubated at 30 °C for 2 or 3 days. Cell viability is calculated by dividing the colony number of pre-treated cells to that of nocodazole treated.
Mif2-FLAG pull-down, phosphatase treatment, and Phos-tag gel experiments
Asynchronously dividing cultures were grown in YPD and harvested by centrifugation. Cell pellets were washed once in PBS, and dried cell pellets were frozen at −80 °C. To break cells, pellets were thawed on ice by addition of 450 μL buffer L (25 mM HEPES, pH 7.5; 2 mM MgCl2; 0.1 mM EDTA; 0.5 mM EGTA; 0.1 % NP-40; 175 mM potassium glutamate; 15 % glycerol by volume; 2 μg/ml aprotinin, pepstatin, and leupeptin; 1 mM PMSF and benzamidine) with phosphatase inhibitors added: 80 mM NaF; 20 mM NaVO4; 1x complete EDTA-free Protease Inhibitor Cocktail (Roche), 1x PhosSTOP (Roche), and Phosphatase Inhibitor Cocktail 2 (Sigma; 1% by volume). Cells were lysed by bead beating via vortex with 0.5 mM borosilicate glass balls (6 × 30 sec cycles with cooling on ice). Cleared supernatants were mixed with anti-FLAG magnetic beads (Sigma) and rotated at 4 °C for one hour. Beads were washed four times with lysis buffer containing phosphatase and protease inhibitors (all except Phosphatase Inhibitor Cocktail 2) before elution by boiling in SDS-PAGE loading buffer. For phosphatase treatment, beads were washed four more times with phosphatase wash buffer (50 mM HEPES, pH 7.5; 0.2 mM MnCl2; 100 mM NaCl; 5% glycerol; 2 μg/ml aprotinin, pepstatin, and leupeptin; 2 mM β-mercaptoethanol). Each pulldown sample was resuspended in 50 μL phosphatase buffer and split into equivalent tubes before addition of 4 μL buffer or purified lambda phosphatase. Beads were incubated 30 min at 37 °C before removal of unbound material. Bound material was eluted by boiling in SDS-PAGE loading buffer.
To resolve eluted proteins, a neutral pH Phos-tag acrylamide electrophoresis system was used essentially as described 35. 0.75 mm Phos-tag acrylamide gels were prepared with 10% acrylamide/bis-acrylamide and 350 mM BIS-TRIS HCl, pH 6.8. For Phos-tag gels, 20 μM ZnCl2 and 10 μM Phos-tag reagent (APExBIO) were added before polymerization from 10 mM aqueous and 5 mM methanol stock solutions, respectively. Gels were run in neutral running buffer: 0.1 M Tris base; 0.1 M MOPS; 0.1 % (w/v) SDS; 5 mM sodium bisulfite at 30 mA for 120 min. Before transferring to PVDF membrane, gels were washed three times ten minutes in Tris-glycine buffer. 10 mM EDTA was included in the first two washes. Standard wet tank transfer to PVDF (1 hour at 370 mA) was carried out before probing with an anti-Protein A antibody.
Cell cycle analysis by flow cytometry
Liquid cell culture at each desired time point was fixed with 70 % ethanol (final concentration). All samples were left in ethanol at −20 °C overnight. The next day, fixed cells were collected by centrifugation, resuspended in 50 mM sodium citrate pH 7.0 with RNaseA (250 μg per sample) and Proteinase K (250 μg per sample), and incubated overnight at 37 °C. On the third day, the cells were collected by centrifugation, resuspended in 50mM sodium citrate with 1 μM Sytox Green (ThermoFisher), and sonicated at 80 kHz for 3–5 seconds. After incubation in a dark room for an hour, the samples were analyzed, and data was collected with the BD LSRFortessa X-20 cytometer.
Recombinant protein expression and purification
Ipl1 kinase was expressed in E. coli Rosetta 2(DE3) cells (EMD Millipore). A polycistronic co-expression vector coding for Sli15-580-698 and Ipl1-AS651 was constructed by ligation independent cloning (LIC) and overlapping PCR. Both proteins had N-terminal 6-His tags. Cdc5 was cloned by LIC into a vector coding for an N-terminal 6-His-MBP tag. Histone proteins were cloned by LIC and overlapping PCR to create co-expression plasmids coding for all four histone proteins on a single transcript. Histone H2A and codon-optimized histone H4 carried N-terminal 6-His tags. DDK (Dbf4 and Cdc7-AS3)52 was cloned into a single baculovirus transfer vector by LIC.53 Both proteins had N-terminal 6-His tags. Ctf19c components were cloned sequentially into BioBrick-enabled LIC vectors 53 to create two coexpression plasmids coding for eleven of the thirteen components (Ame1-Okp1-Nkp1-Nkp2-Ctf19-Mcm21 and Ctf3-Mcm16-Mcm22-Cnn1-Wip1). The remaining two Ctf19c components, Chl4 and Iml3, were purified from E. coli transformed with pSMH104.54 Mif2 and associated mutants were cloned into a baculovirus transfer vector using LIC.
For protein expression in E. coli, cells were grown at 37 °C to OD ~1 before induction with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were incubated overnight at 18 °C before harvesting by centrifugation and resuspension in buffer D800 (20 mM HEPES, pH 7.5; 10 mM imidazole, pH 8.0; 150 mM NaCl; 10 % glycerol by volume; 2 mM β-mercaptoethanol) supplemented with 2 μg/ml aprotinin, leupeptin, and pepstatin and 1 mM PMSF and benzamidine. Cells were stored at −80 °C until purification. For protein expression in insect cells, High Five cells (T. ni, ThermoFisher) were grown in EX-CELL 405 medium (Sigma) and infected with P3 virus at a cell density of ~1 million/mL. 72 hours after infection, cells were harvested by centrifugation, resuspended in buffer B50 (B800 with 50 mM NaCl) supplemented with protease inhibitors and stored at −80 °C until purification. Baculovirus stocks were generated according to the manufacturer’s recommendation in Sf9 cells (S. frugiperda, ThermoFisher).
For protein purification from both E. coli and insect cells, lysis was done by sonication, and cell extracts were clarified by centrifugation at 43549.6 ×g for 60 minutes. The NaCl concentration was adjusted to 800 mM or 2 M before lysis for insect cell or histone octamer expressions, respectively. 6xHis-tagged proteins were purified by Co2+ affinity and eluted in C150 (D800 with 400 mM imidazole and 150 mM NaCl; Cdc5), C100 (D800 with 100 mM NaCl and 400 mM imidazole; DDK, Ipl1-Sli15, Ctf19c components, Mif2 and mutants), or C1000 (D800 with 1M NaCl, 400 mM imidazole, and 10 mM EDTA; histone octamers).
For kinases, the following further purifications were carried out. Ipl1-Sli15 and DDK were applied to a 5 mL cation exchange column (HiTrap SP HP; GE) and eluted with a linear gradient from B50 (Ipl1-Sli15) or B100 (DDK) to D800. Peak fractions were pooled, concentrated by ultrafiltration, and applied to an S200 column (S200 10/300; GE) equilibrated with GF150 (20 mM Tris-HCl, pH 8.5; 150 mM NaCl; 1 mM TCEP). Cdc5 was concentrated by ultrafiltration before injection onto an S200 column equilibrated with GF150. The pooled eluate was concentrated by ultrafiltration, glycerol was added to a final concentration of 5% by volume, and protein was stored at −80 °C until use.
For Ctf19c reconstitution, the Ame1-Okp1-Nkp1-Nkp2-Ctf19-Mcm21 and Ctf3-Mcm16-Mcm22-Cnn1-Wip1 complexes were purified separately as described above. Eluates from the metal affinity step were applied to a 5 mL cation exchange column (HiTrap SP HP; GE). Peak fractions were pooled, concentrated, and mixed at an equimolar ratio at a final volume of ~1 mL, to which a molar excess of TEV-cleaved Chl4-Iml3 was added. Lambda phosphatase, MnCl2 (final concentration 1 mM), and benzonase (~10 U/mL final concentration) were added, and the reaction was incubated at 30 °C for one hour before application to an S200 column equilibrated with GF150. The pooled eluate was concentrated and stored as described above.
As for Ctf19c preparations, all recombinant Mif2 proteins were dephosphorylated before the final purification step. Specifically, Mif2 and its variants were purified exactly as for Ctf19c factors with the following exceptions. A 5 mL anion exchange column (HiTrap Q HP; GE) was used. Peak fractions were treated with lambda phosphatase but not benzonase. Concentrated and dephosphorylated Mif2 was applied to an S200 column equilibrated in GF500 (GF150 with 500 mM NaCl) before concentration and freezing as described above.
Histone octamers were further purified by concentration and application to an S200 column equilibrated in GF1000 (GF150 with 1000 mM NaCl). Recombinant nucleosome core particles were reconstituted on 147 bp 601 DNA by salt gradient dialysis. 601 DNA was amplified by PCR from a plasmid template using the following oligonucleotides at large scale:
oSMH1950 – ATCGAGAATCCCGGTGCC
oSMH1951 – ATCGGATGTATATATCTGACACGTGC.
Reaction products were pooled, diluted with water and 100 mM HEPES, pH 7.5, 10 mM EDTA, and 400 mM NaCl (final concentrations) before purification by anion exchange chromatography (HiTrap Q HP; GE). Peak fractions were precipitated with 70 % ethanol by volume (final) at −20 °C, pelleted, reconstituted in 50 mM Tris-HCl, pH 8.5, and stored at −20 °C until use. For nucleosome wrapping reactions, a 1.2 molar ratio of histone octamer to 601 DNA was mixed in NCP-hi (1000 mM NaCl, 10 mM Tris-HCl pH 7, 1 mM EDTA, 1 mM dithiothreitol (DTT)), and overnight gradient dialysis was carried out at room temperature into NCP-lo (NCP-hi with 10 mM NaCl). After a final ~2 h dialysis in NCP-lo, the sample was analyzed by native gel electrophoresis as described below. Nucleosome particles were used for EMSA or reconstitution experiments within 48 hours of gradient dialysis.
In vitro kinase assays
The indicated kinases and substrates were mixed before addition of an equal volume of 2x ATP with [γ-32P]-ATP tracer mix (0.2 mM ATP-MgCl2, 0.5 μC/μL [γ-32P]ATP) in 2x kinase buffer (100 mM HEPES, pH 7.5; 10 mM Mg(OAc)2, 20% (v/v) glycerol, 400 mM potassium glutamate, 2 mM EDTA, 0.02% (v/v) NP-40 substitute; 4 mM NaoVO4; 40 mM NaF; 2 mM β-mercaptoethanol) to start the reaction for a final concentration 1x tracer mix and 1x kinase buffer. Mif2 was included at a final concentration of 600 nM, MBP-Cdc5 at 2 μM, and DDK at 400 nM. Reactions were incubated at 30 °C for one hour before boiling with SDS-PAGE sample buffer. Reaction products were separated by SDS-PAGE, and dried gels were imaged using autoradiography.
TMT-mass spectrometry to measure phosphopeptide abundance
To analyze the purified and kinase-treated Mif2 proteins, we applied Tandem Mass Tag (TMT) labeling system (ThermoFisher) to identify and quantify phospho-peptides within each kinase-treated sample (Ipl1, DDK, Cdc5, or none). Briefly, 15 μg purified Mif2 proteins, with or without kinase treatment, were first dissolved in 50 μL of 6 M urea with 50 mM NH4HCO3. Then, each sample was treated with 10 mM DTT to disrupt the disulfide bonds and subsequently alkylated with 30 mM iodoacetamide. 1 μL beta-mercaptoethanol was added after 30 minutes to quench the alkylation reaction. 5 μL of the quenched samples were diluted in 25 μL of 50 mM NH4HCO3 and digested by trypsin or chymotrypsin for 2 hours at 37 °C. To end the digestion, trifluoroacetic acid (TFA) was added to a final concentration of 0.2 % in each sample. Each set of samples, digested by trypsin or chymotrypsin, was combined, desalted by C18 columns, and dried completely before resuspension in 20 μL of 50 mM KH2PO4 (pH 8.0). Each sample was treated with 8 μL of TMT labeling reagent at room temperature overnight. The next day, 1 μL of 1 M Tris-HCl, pH 8.0 was added to quench any unincorporated labeling reagent. After labeling, all samples were combined, desalted by C18 columns, and dried under vacuum. The dried sample was resuspended in 0.5 % acetic acid and processed for analysis using a ThermoFisher Orbitrap Fusion LUMOS Tribrid mass spectrometer as described.55
To identify the in vivo phosphorylation sites of Mif2, 1 liter culture of Mif2-TAF cells grown in YPD at OD600 ~1.0 were collected and lysed with a PBS buffer containing protease inhibitors, PMSF, 0.5 M NaF, 0.5 M beta-glycerophosphate, 20 mM EDTA, and 1 μM Okadaic acid. Cells were lysed by glass beads beating at 4 °C for up to 2 hours (1 minute breaking, 2 minutes cooling program). After centrifugation, the clarified lysate was collected and incubated with 100 μL of anti-FLAG M2 beads (Sigma) at 4 °C overnight. The next day, anti-FLAG beads were washed with 1 mL of ice-cold lysis buffer for 5 times and eluted in 2 quick steps by 300 μL 0.1 M Glycine-HCl (pH 2.0). The total elution volume was 600 uL and each elution was completed within 15 seconds. After evaluation of Mif2 purification by Western blot (anti-Protein A), the samples were neutralized, reduced, alkylated, digested with trypsin, acidified, and desalted using the protocol described above for the in vitro phosphorylated samples.
To enrich phosphopeptides, the treated sample was purified with an IMAC (immobilized metal affinity chromatography) column as described 56. Briefly, to make fresh IMAC columns, the beads were recovered from a Qiagen Ni-NTA spin column (Cat No. 31014, one spin column is enough to make ~70 μL IMAC beads) and Ni2+ were stripped by gently shaking in 50 mM EDTA, 1 M NaCl for an hour, then washed with ddH2O, 0.6 % acetic acid, and recharged with Fe3+ by gently rotating in 50 mL of 0.1 M FeCl3 in 0.3 % acetic acid for an hour. Beads were washed once with 0.6 % acetic acid, then twice with 0.1 % acetic acid. Once prepared, beads can be stored in 0.1 % acetic acid for up to a week at 4 °C. To purify phosphopeptides, we packed 10 μL of IMAC beads in a gel loading tip, resuspended dried peptides in 40 μl of 1 % acetic acid (pH 3–4), then loaded the peptide sample slowly into the IMAC column by slightly applying syringe pressure. The IMAC column was washed twice with one bed volume of 0.6% acetic acid and once with a half bed volume of ddH2O. Phosphopeptides were eluted with three bed volume of 6 % NH4OH, dried, and resuspended in 5 μL of 0.6 % acetic acid for MS analysis, as described above. COMET (Seattle Proteome Center: Trans Proteomic Pipeline) software package was used for database searching. A static mass modification of 57.021464 Da for cysteine residues and a differential modification of 79.966331 Da for Ser/Thr phosphorylation were used.
Inner kinetochore reconstitution experiments
The indicated components were mixed at a final volume of 50 μL in GF150. The Mif2-Cse4 nucleosome complex was pre-formed on ice for 60 minutes before use. For kinase treatments, the relevant complexes were mixed with purified kinases (MBP-Cdc5 at 800 nM and DDK at 1 μM) and ATP-MgCl2 (1 mM final concentration) and incubated for 1 hour at 30 °C. Buffer and ATP-MgCl2 were added to unphosphorylated reactions, which were also incubated at 30 °C. Subsequent inner kinetochore assembly reactions were initiated by mixing Ctf19c and Mif2-Cse4 complexes on ice for one hour in the presence of 2 mM ADP (final concentration). Concentrations during the final incubation step were Ctf19c at 1.5 μM and Mif2-Cse4 nucleosome at 2 μM. Separation was carried out on a Superose 6 column (5/150 GL; 0.1 mL per min pump setting) equilibrated in GF150-HEPES (GF150 with HEPES, pH 8.6 replacing Tris-HCl). Fractions were taken every 30 sec, and identical fractions at 1 min spacing from each injection were analyzed by SDS-PAGE.
Gel shift assay (EMSA)
Recombinant Mif2 and nucleosome samples were mixed in a final volume of 5 μL and incubated on ice for one hour before separation by native gel electrophoresis. SDS-free sample buffer was added, and 1 % acrylamide gels equilibrated in chilled 0.5 × TBE buffer were used to separate the indicated reaction products. DNA was visualized with SYBR Gold Nucleic Acid Gel Stain (ThermoFisher) according to the manufacturer’s instructions. Nucleosome concentration was estimated using absorbance at 260 nM. Titrations were carried out to determine the minimal concentration that could be visualized using SYBR Gold stain, and this was ~100 nM for all experiments shown. Mif2 kinase treatments were carried out as described for inner kinetochore reconstitution experiments, and total NaCl concentration was normalized to 108 mM for every binding reaction.
ChIP-qPCR assay
To evaluate the localization of Mif2, Mif2-10A, and Mif2-10D to the centromere, ChIP was performed 55,57. Briefly, yeast cultures (150 mL, enough for three immunoprecipitation experiments) were grown to an OD600 of 0.8 and cross-linked for 15 min with 1 % formaldehyde at room temperature. Whole-cell lysates were prepared in 0.8 ml ChIP lysis buffer (50 mM Hepes, pH 7.6; 140 mM NaCl; 1 mM EDTA; 1 % Triton, and 0.1% sodium deoxycholate) supplemented with protein inhibitors by glass bead beating and sonication to shear the genomic DNA to an average size of 300–500 bp. Immunoprecipitation was performed using 50 μL Dynabeads (Protein G; ThermoFisher) and 3 μL anti-Flag antibody M2 (Sigma; F3165). After binding, beads were washed as follows: once in 1 mL lysis buffer with 5 min incubation, twice in 1 mL washing buffer (100 mM Tris-Cl, pH 8.0; 250 mM LiCl; 0.5% NP-40; 0.5% deoxycholate; and 1 mM EDTA) with 5 min incubations, and once with 1 mL TE buffer (10 mM Tris-Cl, pH 8.0, and 1 mM EDTA) with 1 min incubation. After the washes, the samples were first eluted in 40 μL of TE buffer with 1% SDS at 65 °C for 10 min, and this was saved as elution 1. 162 μL of DNA extraction buffer (135 μL of TE, 15 μL of 10% SDS, 12 μL of 5 M NaCl) was then added to the beads with 1.5 μL of RNaseA and incubated at 37 °C for 30min, and this was elution 2. Both eluates were mixed and incubated at 37 °C for another 30 min. The inputs were treated with 162 μL of DNA extraction buffer and 1.5 μL of RNaseA with 1 h incubation at 37 °C.
The input and immunoprecipitated DNA were incubated in the same buffer with the addition of 20 μg Protease K at 65 °C overnight to reverse crosslinks before purification using a QIAquick PCR Purification kit (QIAGEN). Before qPCR analysis, the input DNA was diluted 1:100, and immuno-purified DNA was diluted 1:10 by volume. qPCR was done using SYBR Green 2x master mix (KAPA Biosystems) on a Roche LightCycler 480 system. Three independent immunoprecipitation experiments were performed. qPCR primer sequences were:
CEN3_fwd: 5′-ATCAGCGCCAAACAATATGGAAAA-3′
CEN3_rev: 5′-GAGCAAAACTTCCACCAGTAAACG-3′
CUP1_fwd: 5′-AACTTCCAAAATGAAGGTCA-3′
CUP1_rev: 5′-GCATGACTTCTTGGTTTCTT-3′.
Co-immunoprecipitation and Mif2 competition experiments
To detect the in vivo association of endogenous Mif2-WT/10A with the Cse4 nucleosome, we used strains expressing Mif2-TAF variants and tagged Cse4 (internal 3xFLAG) from their respective chromosomal loci. The 3xFLAG tag was inserted at the XbaI site of Cse4, similar to a previous report.58 Mif2-TAF/FLAG-Cse4 or mif2-10A-TAF/FLAG-Cse4 cells were harvested by centrifugation and washed (PBSN, 1 mM EDTA, 1 mM NaF, 1 mM beta-glycerophosphate, PI and PMSF). For each IP, 80 mL of OD600 1 cells were resuspended in 4x cell pellet volumes of lysis buffer (~0.8 mL PBSN, 0.2% NP-40) and frozen dropwise in liquid nitrogen before lysis using a freezer mill. The resulting powder was thawed at 4 °C before clarification by centrifugation, and the protein concentration of each cell extract was normalized to ~10 mg/mL. 0.8 mL of clarified lysate was incubated with 30 μL of IgG beads (human IgG, ~3 mg Protein A per mL capacity) at 4 °C overnight with rotation. The beads were then washed with 1mL of ice-cold lysis buffer four times before elution of bound material via a 5 min incubation at 98 °C with 30 μL of 2x LDS buffer (Invitrogen). About 25 μL of eluate was collected by centrifugation. 2 μL of 1 M DTT was added to each sample before heating at 98 °C for 2 min to reduce the protein samples.
To evaluate the stability of the pre-existing Mif2-Cse4 binding against competition by newly synthesized Mif2-WT, Mif2-10A and Mif2-10D proteins, we transformed the Mif2-TAF/FLAG-Cse4 strain with high-copy 2-micron pRS425 plasmids coding for galactose-inducible Mif2-3xHA, Mif2-10A-3xHA, or Mif2-10D-3xHA. The Mif2-3xHA variants were induced by addition of 2% galactose to log phase cultures grown in SC-leu/raf (SC-leu with 2 % raffinose instead of glucose). For these experiments, cells at three time points were collected: pre-induction, one hour after induction, and two hours after induction. The co-IP method described above was used to evaluate endogenous Mif2-Cse4 binding in the presence of Mif2 competitors.
Input and eluted samples were loaded onto a NuPAGE gradient gel (4–12 %; ThermoFisher) and run in MOPS-SDS buffer (ThermoFisher) at 200 V for 70 min. After transferring at 100V for 100 min to a PVDF membrane, the membrane was blotted with anti-Protein A (1:10,000, Sigma), anti-HA (1:2000, 3F10, Sigma), and anti-FLAG (1:2000, Sigma) primary antibodies to detect Mif2-TAF, Mif2-3xHA, and FLAG-Cse4, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were done using Graphpad Prism v9. Phosphopeptide quantification was carried out as described above using COMET (Seattle Proteome Center). Statistical tests used are indicated in figure legends.
Supplementary Material
Highlights.
Multi-site phosphorylation controls kinetochore activity
Multiple kinases can regulate the chromosomal anchor of the kinetochore
Model for cell cycle-dependent kinetochore strengthening
Acknowledgments
We thank Stephen Harrison and Kevin Corbett for discussions and for comments on the manuscript. We thank Phong Lee for advice on nucleosome reconstitutions. This work was supported by NIH GM116897, OD023498, and University of California CRCC faculty seed grant to H.Z. S.M.H. was supported in part by funding from HHMI and the Helen Hay Whitney Foundation.
Footnotes
Declaration of interests
The authors declare no competing interests.
Supplemental items
Data S1 – Peptide tables from Mif2 mass spectrometry experiments. Related to Figure 2.
(A) Phospho-peptides observed in mass spectrometry experiments listed with the indicated phosphorylations. In vivo phosphorylations from Mif2 immunoprecipitations are shown. (B) Mif2 phosphorylations from in vitro experiments are shown with absolute and relative TMT intensities for each peptide plotted. (C) Venn diagram that compares the results of in vitro and in vivo (endogenous) phosphorylation mapping experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Biggins S (2013). The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194, 817–846. 10.1534/genetics.112.145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hara M, and Fukagawa T (2020). Dynamics of kinetochore structure and its regulations during mitotic progression. Cell Mol Life Sci 77, 2981–2995. 10.1007/s00018-020-03472-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinshaw SM, and Harrison SC (2018). Kinetochore Function from the Bottom Up. Trends Cell Biol 28, 22–33. 10.1016/j.tcb.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 4.McKinley KL, and Cheeseman IM (2016). The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17, 16–29. 10.1038/nrm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musacchio A, and Desai A (2017). A Molecular View of Kinetochore Assembly and Function. Biology (Basel) 6. 10.3390/biology6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohm M, Killinger K, Dudziak A, Pant P, Janen K, Hohoff S, Mechtler K, Ord M, Loog M, Sanchez-Garcia E, and Westermann S (2021). Cdc4 phospho-degrons allow differential regulation of Ame1(CENP-U) protein stability across the cell cycle. Elife 10. 10.7554/eLife.67390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boeckmann L, Takahashi Y, Au WC, Mishra PK, Choy JS, Dawson AR, Szeto MY, Waybright TJ, Heger C, McAndrew C, et al. (2013). Phosphorylation of centromeric histone H3 variant regulates chromosome segregation in Saccharomyces cerevisiae. Mol Biol Cell 24, 2034–2044. 10.1091/mbc.E12-12-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westermann S, Cheeseman IM, Anderson S, Yates JR 3rd, Drubin DG, and Barnes G (2003). Architecture of the budding yeast kinetochore reveals a conserved molecular core. J Cell Biol 163, 215–222. 10.1083/jcb.200305100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinshaw SM, and Harrison SC (2019). The structure of the Ctf19c/CCAN from budding yeast. Elife 8. 10.7554/eLife.44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kixmoeller K, Allu PK, and Black BE (2020). The centromere comes into focus: from CENP-A nucleosomes to kinetochore connections with the spindle. Open Biol 10, 200051. 10.1098/rsob.200051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pesenti ME, Raisch T, Conti D, Walstein K, Hoffmann I, Vogt D, Prumbaum D, Vetter IR, Raunser S, and Musacchio A (2022). Structure of the human inner kinetochore CCAN complex and its significance for human centromere organization. Mol Cell. 10.1016/j.molcel.2022.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan K, Yang J, Zhang Z, McLaughlin SH, Chang L, Fasci D, Ehrenhofer-Murray AE, Heck AJR, and Barford D (2019). Structure of the inner kinetochore CCAN complex assembled onto a centromeric nucleosome. Nature 574, 278–282. 10.1038/s41586-019-1609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yatskevich S, Muir KW, Bellini D, Zhang Z, Yang J, Tischer T, Predin M, Dendooven T, McLaughlin SH, and Barford D (2022). Structure of the human inner kinetochore bound to a centromeric CENP-A nucleosome. Science 376, 844–852. 10.1126/science.abn3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ariyoshi M, Makino F, Watanabe R, Nakagawa R, Kato T, Namba K, Arimura Y, Fujita R, Kurumizaka H, Okumura EI, et al. (2021). Cryo-EM structure of the CENP-A nucleosome in complex with phosphorylated CENP-C. EMBO J, e105671. 10.15252/embj.2020105671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allu PK, Dawicki-McKenna JM, Van Eeuwen T, Slavin M, Braitbard M, Xu C, Kalisman N, Murakami K, and Black BE (2019). Structure of the Human Core Centromeric Nucleosome Complex. Curr Biol 29, 2625–2639 e2625. 10.1016/j.cub.2019.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chittori S, Hong J, Saunders H, Feng H, Ghirlando R, Kelly AE, Bai Y, and Subramaniam S (2018). Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science 359, 339–343. 10.1126/science.aar2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pentakota S, Zhou K, Smith C, Maffini S, Petrovic A, Morgan GP, Weir JR, Vetter IR, Musacchio A, and Luger K (2017). Decoding the centromeric nucleosome through CENP-N. Elife 6. 10.7554/eLife.33442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou K, Gebala M, Woods D, Sundararajan K, Edwards G, Krzizike D, Wereszczynski J, Straight AF, and Luger K (2022). CENP-N promotes the compaction of centromeric chromatin. Nat Struct Mol Biol 29, 403–413. 10.1038/s41594-022-00758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown MT, Goetsch L, and Hartwell LH (1993). MIF2 is required for mitotic spindle integrity during anaphase spindle elongation in Saccharomyces cerevisiae. J Cell Biol 123, 387–403. 10.1083/jcb.123.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon MS, Hori T, Okada M, and Fukagawa T (2007). CENP-C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol Biol Cell 18, 2155–2168. 10.1091/mbc.e07-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao H, Wang F, Wisniewski J, Shaytan AK, Ghirlando R, FitzGerald PC, Huang Y, Wei D, Li S, Landsman D, et al. (2017). Molecular basis of CENP-C association with the CENP-A nucleosome at yeast centromeres. Genes Dev 31, 1958–1972. 10.1101/gad.304782.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carroll CW, Milks KJ, and Straight AF (2010). Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 189, 1143–1155. 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, and Bai Y (2013). A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science 340, 1110–1113. 10.1126/science.1235532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe R, Hara M, Okumura EI, Herve S, Fachinetti D, Ariyoshi M, and Fukagawa T (2019). CDK1-mediated CENP-C phosphorylation modulates CENP-A binding and mitotic kinetochore localization. J Cell Biol 218, 4042–4062. 10.1083/jcb.201907006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Killinger K, Bohm M, Steinbach P, Hagemann G, Bluggel M, Janen K, Hohoff S, Bayer P, Herzog F, and Westermann S (2020). Auto-inhibition of Mif2/CENP-C ensures centromere-dependent kinetochore assembly in budding yeast. EMBO J 39, e102938. 10.15252/embj.2019102938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Y, Wong CC, Nakajima Y, Tyers RG, Sarkeshik AS, Yates J 3rd, Drubin DG, and Barnes G (2011). Overlapping kinetochore targets of CK2 and Aurora B kinases in mitotic regulation. Mol Biol Cell 22, 2680–2689. 10.1091/mbc.E10-11-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinshaw SM, and Harrison SC (2020). The Structural Basis for Kinetochore Stabilization by Cnn1/CENP-T. Curr Biol 30, 3425–3431 e3423. 10.1016/j.cub.2020.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Nagpal H, Hori T, Furukawa A, Sugase K, Osakabe A, Kurumizaka H, and Fukagawa T (2015). Dynamic changes in CCAN organization through CENP-C during cell-cycle progression. Mol Biol Cell 26, 3768–3776. 10.1091/mbc.E15-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang J, Barber A, and Biggins S (2018). An assay for de novo kinetochore assembly reveals a key role for the CENP-T pathway in budding yeast. Elife 7. 10.7554/eLife.37819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen RL, Espelin CW, De Wulf P, Sorger PK, Harrison SC, and Simons KT (2008). Structural and functional dissection of Mif2p, a conserved DNA-binding kinetochore protein. Mol Biol Cell 19, 4480–4491. 10.1091/mbc.E08-03-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klare K, Weir JR, Basilico F, Zimniak T, Massimiliano L, Ludwigs N, Herzog F, and Musacchio A (2015). CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. J Cell Biol 210, 11–22. 10.1083/jcb.201412028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hornung P, Troc P, Malvezzi F, Maier M, Demianova Z, Zimniak T, Litos G, Lampert F, Schleiffer A, Brunner M, et al. (2014). A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP-A. J Cell Biol 206, 509–524. 10.1083/jcb.201403081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinshaw SM, Makrantoni V, Harrison SC, and Marston AL (2017). The Kinetochore Receptor for the Cohesin Loading Complex. Cell 171, 72–84 e13. 10.1016/j.cell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Natsume T, Muller CA, Katou Y, Retkute R, Gierlinski M, Araki H, Blow JJ, Shirahige K, Nieduszynski CA, and Tanaka TU (2013). Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol Cell 50, 661–674. 10.1016/j.molcel.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagy Z, Comer S, and Smolenski A (2018). Analysis of Protein Phosphorylation Using Phos-Tag Gels. Curr Protoc Protein Sci 93, e64. 10.1002/cpps.64. [DOI] [PubMed] [Google Scholar]
- 36.Botchkarev VV Jr., and Haber JE (2018). Functions and regulation of the Polo-like kinase Cdc5 in the absence and presence of DNA damage. Curr Genet 64, 87–96. 10.1007/s00294-017-0727-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheu YJ, and Stillman B (2006). Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol Cell 24, 101–113. 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Princz LN, Wild P, Bittmann J, Aguado FJ, Blanco MG, Matos J, and Pfander B (2017). Dbf4-dependent kinase and the Rtt107 scaffold promote Mus81-Mms4 resolvase activation during mitosis. EMBO J 36, 664–678. 10.15252/embj.201694831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR 3rd, Chan CS, Drubin DG, and Barnes G (2002). Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell 111, 163–172. 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- 40.Borek WE, Vincenten N, Duro E, Makrantoni V, Spanos C, Sarangapani KK, de Lima Alves F, Kelly DA, Asbury CL, Rappsilber J, and Marston AL (2021). The Proteomic Landscape of Centromeric Chromatin Reveals an Essential Role for the Ctf19(CCAN) Complex in Meiotic Kinetochore Assembly. Curr Biol 31, 283–296 e287. 10.1016/j.cub.2020.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewar H, Tanaka K, Nasmyth K, and Tanaka TU (2004). Tension between two kinetochores suffices for their bi-orientation on the mitotic spindle. Nature 428, 93–97. 10.1038/nature02328. [DOI] [PubMed] [Google Scholar]
- 42.Koshland D, Rutledge L, Fitzgerald-Hayes M, and Hartwell LH (1987). A genetic analysis of dicentric minichromosomes in Saccharomyces cerevisiae. Cell 48, 801–812. 10.1016/0092-8674(87)90077-8. [DOI] [PubMed] [Google Scholar]
- 43.Poddar A, Roy N, and Sinha P (1999). MCM21 and MCM22, two novel genes of the yeast Saccharomyces cerevisiae are required for chromosome transmission. Mol Microbiol 31, 349–360. 10.1046/j.1365-2958.1999.01179.x. [DOI] [PubMed] [Google Scholar]
- 44.Hagemann G, Solis-Mezarino V, Singh S, Potocnjak M, Kumar C, and Herzog F (2022). Quantitative crosslinking and mass spectrometry determine binding interfaces and affinities mediating kinetochore stabilization. bioRxiv. 10.1101/2022.03.31.486303. [DOI] [Google Scholar]
- 45.Brown MT (1995). Sequence similarities between the yeast chromosome segregation protein Mif2 and the mammalian centromere protein CENP-C. Gene 160, 111–116. 10.1016/0378-1119(95)00163-z. [DOI] [PubMed] [Google Scholar]
- 46.Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, McIntosh LP, and Graves BJ (2005). Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145. 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 47.Serber Z, and Ferrell JE Jr. (2007). Tuning bulk electrostatics to regulate protein function. Cell 128, 441–444. 10.1016/j.cell.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 48.Ghenoiu C, Wheelock MS, and Funabiki H (2013). Autoinhibition and Polo-dependent multisite phosphorylation restrict activity of the histone H3 kinase Haspin to mitosis. Mol Cell 52, 734–745. 10.1016/j.molcel.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. . [DOI] [PubMed] [Google Scholar]
- 50.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922. 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 51.Kung C, Kenski DM, Dickerson SH, Howson RW, Kuyper LF, Madhani HD, and Shokat KM (2005). Chemical genomic profiling to identify intracellular targets of a multiplex kinase inhibitor. Proc Natl Acad Sci U S A 102, 3587–3592. 10.1073/pnas.0407170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wan L, Zhang C, Shokat KM, and Hollingsworth NM (2006). Chemical inactivation of cdc7 kinase in budding yeast results in a reversible arrest that allows efficient cell synchronization prior to meiotic recombination. Genetics 174, 1767–1774. 10.1534/genetics.106.064303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gradia SD, Ishida JP, Tsai MS, Jeans C, Tainer JA, and Fuss JO (2017). MacroBac: New Technologies for Robust and Efficient Large-Scale Production of Recombinant Multiprotein Complexes. Methods Enzymol 592, 1–26. 10.1016/bs.mie.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinshaw SM, and Harrison SC (2013). An Iml3-Chl4 heterodimer links the core centromere to factors required for accurate chromosome segregation. Cell Rep 5, 29–36. 10.1016/j.celrep.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suhandynata RT, Quan Y, Yang Y, Yuan WT, Albuquerque CP, and Zhou H (2019). Recruitment of the Ulp2 protease to the inner kinetochore prevents its hyper-sumoylation to ensure accurate chromosome segregation. PLoS Genet 15, e1008477. 10.1371/journal.pgen.1008477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suhandynata R, Liang J, Albuquerque CP, Zhou H, and Hollingsworth NM (2014). A method for sporulating budding yeast cells that allows for unbiased identification of kinase substrates using stable isotope labeling by amino acids in cell culture. G3 (Bethesda) 4, 2125–2135. 10.1534/g3.114.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meluh PB, and Koshland D (1997). Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev 11, 3401–3412. 10.1101/gad.11.24.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wisniewski J, Hajj B, Chen J, Mizuguchi G, Xiao H, Wei D, Dahan M, and Wu C (2014). Imaging the fate of histone Cse4 reveals de novo replacement in S phase and subsequent stable residence at centromeres. Elife 3, e02203. 10.7554/eLife.02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There was no large dataset generated and deposited along with this paper.
This paper does not report original code.
The data reported in this paper will be shared by the lead contact upon request.