

Editor summary:
Matters arising regarding the lipidation form of plasma APOC3 that induces an alternative NLRP3 activation pathway.
Increased levels of apolipoprotein C3 (APOC3) in the blood predict cardiovascular disease risk in people with and without diabetes1, 2, 3, 4. Conversely, APOC3 loss-of-function variants are associated with cardioprotection in humans5, 6, 7. The mechanisms whereby APOC3 promotes atherosclerotic cardiovascular disease risk have been the topic of intense studies. It is known that APOC3 slows the clearance of triglyceride-rich lipoproteins (TRLs) and their remnant lipoprotein particles (RLPs) by inhibiting the activity of lipoprotein lipase and by preventing hepatic uptake of TRLs and RLPs, leading to increased levels of TRLs and their remnants in circulation8. The atherogenic effects of APOC3 are likely mediated, at least in part, by its effects on TRL metabolism, but recent findings that APOC3 predicts future coronary artery disease events in individuals with type 1 diabetes (T1D), who often have normal plasma lipid profiles1, 2 suggest that other mechanisms might be at play. As cardiovascular disease is accompanied by heightened inflammation, and the recent Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) highlighted a causal role for the cytokine interleukin-1β (IL-1β) in incident cardiovascular disease9, a possible pro-inflammatory role for APOC3 has generated much interest.
Zewinger and colleagues elegantly demonstrated that de-lipidated APOC3 induces IL-1β release from human monocytes without requiring priming by lipopolysaccharide (LPS)10. The mechanism involved APOC3-induced dimerization of toll-like receptor (TLR) 2 and TLR4, Ca2+-dependent superoxide production and activation of caspase 8 via an alternative NLRP3 inflammasome activation pathway. The ability of native and de-lipidated very low-density lipoprotein (VLDL, a TRL lipoprotein) to mimic the effects of APOC3 on IL-1β release led to the conclusion that APOC3 bound to VLDL activates the NLRP3 inflammasome. However, we find that the ability of APOC3 to induce IL-1β release from human and mouse monocytes is dependent on the lipidation state of APOC3, and that only de-lipidated APOC3 is able to induce IL-1β release.
We show that both de-lipidated APOC3 (4 mg/dL) and ultrapure LPS induce a striking increase in IL-1β release from human blood monocytes without prior priming of the NLRP3 inflammasome, consistent with the study by Zewinger and colleagues (Figure 1a). However, this effect was not mimicked by human VLDL, which contained sufficient APOC3 (0.35 mg/dL) to induce IL-1β release (Extended Data Figure 1a). To investigate whether the APOC3-induced alternative inflammasome pathway is unique to human monocytes, we isolated mouse bone marrow monocytes, using an immunomagnetic negative selection method. Like in the human monocytes, de-lipidated APOC3 (0.004 – 4 mg/dL) induced significant increases in IL-1β release from mouse monocytes (Figure 1b, Extended Data Figure 1a–b). Again, VLDL was unable to induce IL-1β release from these cells.
Figure 1. De-lipidated APOC3, but not lipid-bound APOC3, induces IL-1β release from human and mouse monocytes.
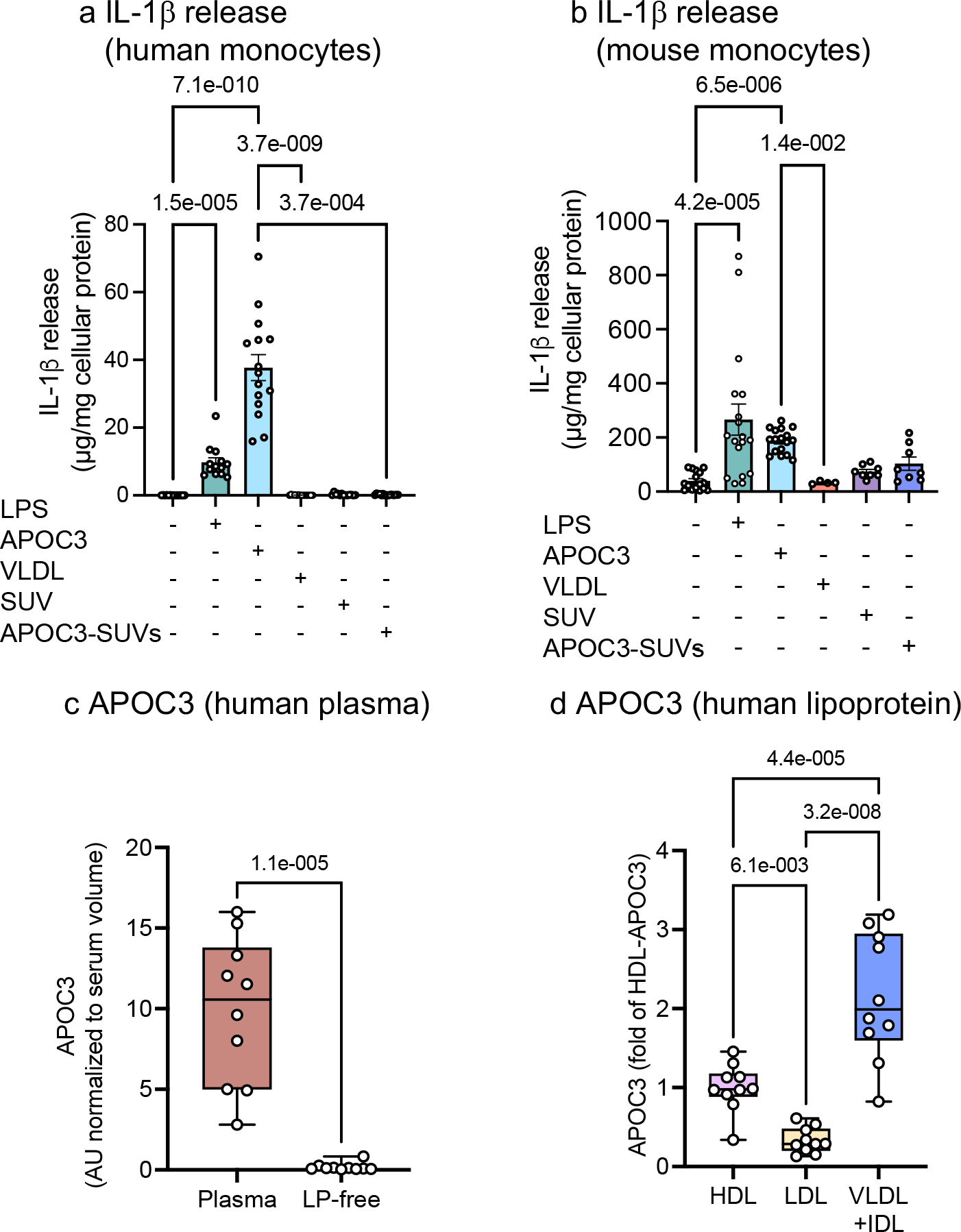
a, IL-1β release from human blood monocytes incubated with or without ultrapure LPS (10 ng/mL), small unilamellar vesicles (SUVs) with an average diameter of 27 nm (70.5 mg/dL), de-lipidated human APOC3 (4 mg/dL), APOC3-SUVs complex (4 mg/dL APOC3 and 70.5 mg/dL SUVs), or human VLDL (4 mg/dL) for 16 hr. b, IL-1β release from mouse bone marrow-derived monocytes incubated as in a. IL-1β measurements were conducted by ELISA and the data were normalized to total cellular protein determined by a BCA protein assay. Data show means ± SEM (n=14, 13, 15, 13, 10, 15 replicates/group, respectively, of pooled samples isolated from 7 subjects in a and n=16, 18, 17, 8, 8, 4 individual mice/group in b). c-d, Serum samples from 10 individuals with T1D were used to investigate total serum levels and relative levels of APOC3 in different lipoprotein fractions and the lipoprotein-free (LP-free) fraction. Lipoprotein fractions were isolated by density ultracentrifugation and APOC3 levels were measured by targeted mass spectrometry (DALS peptide). c, APOC3 levels in whole serum versus the LP-free fraction (d>1.021 g/mL). d, APOC3 levels in HDL, LDL and VLDL+IDL. The data are shown in box-and-whisker plots (the bound of the box extends from the 25th to 75th percentiles and the whisker covers the minima and maxima) with the median marked by a center line. Statistical analyses were performed by Kruskal-Wallis test and Dunn’s multiple comparisons tests in a and b, two-sided, Mann-Whitney test in c, and one-way ANOVA followed by Holm-Šídák’s multiple comparisons test in d. Normality test was performed by D’Agostino & Pearson test. Outliers were removed based on robust regression and outlier removal method with Q=0.1%. (0, 0, 1, 2, 0, 0) datapoints were removed in a; no datapoint was removed in b, c, or d.
APOC3 is an amphipathic lipid-binding apolipoprotein11. We therefore investigated whether the IL-1β stimulating effect of de-lipidated APOC3 is also observed when APOC3 is lipid-bound. We generated lipidated APOC3 by synthesizing small unilamellar vesicles (SUVs), approximately 27 nm in size (Extended Data Figure 1c–e), with 1-palmitoyl-2-oleoyl-glycerol-3-phosphocholine (POPC)12 and bound de-lipidated APOC3 to these vesicles. Lipidated APOC3 was co-incubated with isolated human and mouse monocytes as above. Strikingly, lipidated APOC3 at the same concentration as the de-lipidated APOC3 failed to induce IL-1β release from either human or mouse monocytes (Figure 1a–b). These results are consistent with the interpretation that the ability of APOC3 to induce IL-1β release in monocytes is dependent on the lipidation state of APOC3. The lack of IL-1β-inducing effect of lipidated APOC3 is consistent with the lack of VLDL-induced IL-1β release from monocytes observed in our study. The discrepancy in the ability of VLDL to induce IL-1β release in our study and that of Zewinger et al.10 could be due to differences in VLDL preparations or endotoxin levels.
De-lipidated APOC3 failed to induce significant IL-1β release from differentiated mouse bone marrow-derived macrophages (vehicle = 0.50±0.50 pg/mL; APOC3 = 2.39±1.27 pg/mL; mean±SEM; n=3; p=0.4 by Mann-Whitney test), consistent with the findings of Zewinger et al10 and with the ability of TLR activation alone to induce release IL-1β in monocytes, but not in macrophages,13 suggesting that the ability of de-lipidated APOC3 to induce inflammasome activation is lost with monocyte to macrophage maturation.
APOC3 predicts incident coronary artery events in individuals with T1D1, 2, 3. The lack of VLDL-induced IL-1β release from monocytes prompted us to question whether free APOC3 exists at sufficiently high levels to activate the inflammasome under physiological conditions in diabetes. We collected serum samples from individuals with T1D and measured the amount of APOC3 within each lipoprotein fraction (VLDL + intermediate-density lipoprotein [IDL], LDL, and HDL) and in the lipoprotein-free fraction by targeted mass spectrometry. We observed APOC3 in all the lipoprotein fractions, but the lipid-free fraction contained little APOC3 (0.00024 ±0.000007 mg/dL, mean±SEM, n=10, Figure 1c–d). These results show that APOC3 exists primarily in a lipid-bound state in circulation. Concentrations of APOC3 mimicking those free in circulation were unable to induce IL-1β release (Extended Data Figure 1a). However, we cannot exclude the possibility that sufficient free APOC3 exists physiologically in tissues at concentrations high enough to induce inflammasome activation in vivo14. Investigation of whether this is the case in humans or animal models will be challenging, although novel approaches, such as single-cell transcriptomics and spatial proteomics and metabolomics in conjunction with artificial intelligence technology provide strides in the right direction15, 16.
To investigate the in vivo effects of APOC3 reductions on inflammasome activity, we evaluated two mouse model systems in which we used antisense oligonucleotide (ASO) or small interfering RNAs (siRNA) approaches to reduce APOC3. In the first, we silenced hepatic APOC3 production using a liver-targeted APOC3 GalNAc-conjugated ASO in non-diabetic and diabetic mice (Figure 2a). Controls were treated with a control GalNAc ASO. After four weeks, diabetic mice exhibited elevated blood glucose, plasma triglycerides, APOC3, and IL-18 levels (Figure 2b–e). IL-18, like IL-1β, is released through the inflammasome pathway. It should be noted that IL-18 is bound to IL-18 binding proteins in plasma, and therefore the total concentrations of IL-18 do not reflect its biological activity17. Silencing of hepatic APOC3 markedly reduced plasma APOC3 levels in diabetic mice, however, the elevated plasma IL-18 remained unaffected. Similarly, although plasma IL-1β levels were approximately 100 times lower than those of IL-18, APOC3 ASO treatment did not significantly reduce plasma IL-1β levels in diabetic mice (control ASO = 3.8 ± 1.2 pg/mL, APOC3 ASO = 1.7 ± 0.8 pg/mL, mean±SEM; n=12–14; p=0.17 by Mann-Whitney test). These results indicate that endogenous APOC3 does not contribute significantly to inflammasome activation in diabetic mice in vivo.
Figure 2. Endogenous APOC3 does not alter plasma IL-18.
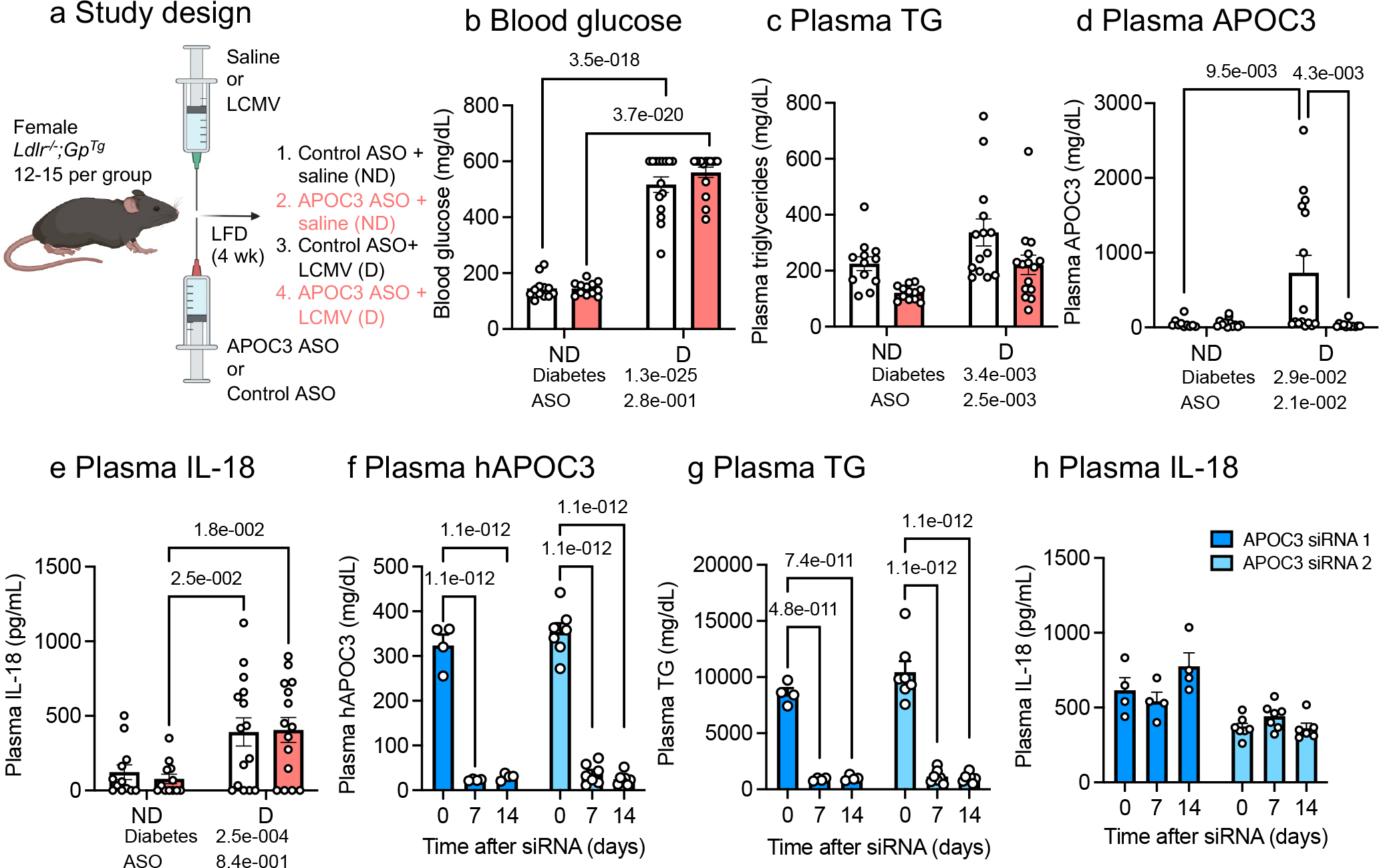
Female low-density lipoprotein receptor-deficient mice with a virus glycoprotein transgene (Ldlr−/−;GPTg) were rendered diabetic (D) with lymphocytic choriomeningitis virus (LCMV). Saline was used as a control (non-diabetic, ND). At the onset of diabetes, the animals were switched to a low-fat, semipurified diet (LFD) and maintained for 4 weeks. Animals received weekly i.p. injections of 10 mg/kg control or APOC3 GalNAc ASOs. a, Schematic of study design for panel b-e. b, Blood glucose levels. c, Plasma triglyceride (TG) levels. d, Plasma APOC3. e, Plasma IL-18. Blood glucose was measured via a glucose meter. Plasma triglyceride levels were measured via colorimetric assay. Plasma APOC3 and IL-18 levels were measured via ELISA. f-h, Plasma samples collected from hAPOC3 transgenic mice treated with two different human APOC3 siRNAs were measured via ELISA. f, Plasma hAPOC3. g, Plasma TGs. h, Plasma IL-18. Control GalNAc ASO did not affect plasma TG, APOC3, or IL-18 levels. Data show means ± SEM (n=12, 12, 15, 15 individual mice in b and e; n=12, 12, 14, 15 in c; n=10, 15, 12, 12 in d) and (n=4 and n=7 individual mice for APOC3 siRNA 1 and siRNA 2 in f-h, except n=6 for t=14 for siRNA 2 in h); two-way ANOVA followed by Tukey multiple comparisons tests vs day 0. Outliers were removed based on robust regression and outlier removal method with Q=0.1%; (0, 0; 1, 0) datapoint was removed in c; (2, 0; 0, 3) datapoints in d; and (0,0,0; 0,0,1) datapoints in h. Figure 2a was created with BioRender.com.
In a second mouse model, we evaluated mice with transgenic expression of human APOC3 to strengthen the conclusion that plasma APOC3 does not impact inflammasome activity. We measured plasma levels of IL-1β and IL-18 from human APOC3 transgenic mice treated with saline or two different human APOC3 GalNAc-conjugated siRNAs. Despite the highly elevated plasma APOC3 and triglycerides, which were markedly reduced by the APOC3 siRNA treatment, levels of IL-18 were unchanged (Figure 2f–h). It should be noted that these mice exhibited 62-times higher plasma APOC3 concentrations versus the free APOC3 used in our in vitro assay and 22-times higher plasma APOC3 levels versus that observed in individuals with diabetes2, 18. Human APOC3 transgenic mice had plasma IL-1β levels below the lowest standard (<0.3 pg/mL, n=4–7). These results strongly support the conclusion that APOC3 does not lead to elevated levels of circulating IL-1β and IL-18 in vivo in mice.
Finally, Zewinger and colleagues demonstrated that subjects with elevated plasma levels of APOC3 also had elevated levels of high sensitivity C-reactive protein (hsCRP; a marker of systemic inflammation) when adjusted for age and sex10. If this observed positive correlation is reflective of a causal relationship between APOC3 and systemic inflammation, a reasonable expectation might be that genetically-mediated reductions in APOC3 would result in reduced levels of hsCRP. We utilized data from the UK Biobank to explore whether this hypothesis can be supported by analyses in carriers of APOC3 loss-of-function variants (Extended Data Figure 2 and Extended Data Table 1). We identified 1,928 individuals carrying rs138326449 (IVS2 + 1G-A), a rare (MAF = 0.002) APOC3 loss-of-function variant that results in ~50% reduced circulating APOC3 in heterozygotes5. We confirmed that APOC3 loss-of-function is significantly associated with decreased serum triglycerides and nominally associated with decreased risk of myocardial infarction. However, paradoxically, APOC3 loss-of-function is suggestively associated with modestly increased levels of serum hsCRP, rather than reduced levels as would be expected if APOC3 promoted inflammatory processes. The association between rs138326449 and elevated hsCRP was robust to adjustment for potential confounders present at the time of serum sample ascertainment, including the use of lipid-lowering medications and diabetes (Extended Data Table 2). Overall, the data from the UK Biobank support the hypothesis that the atherosclerotic cardiovascular disease protection conferred by APOC3 loss-of-function is mediated by triglyceride lowering rather than by reduced inflammation.
In summary, de-lipidated APOC3 stimulates IL-1β release from human and mouse monocytes, consistent with the results by Zewinger and colleagues10, but we provide strong evidence from an array of different analyses – including in vitro, in vivo, and genomic data - demonstrating that this effect is lost when APOC3 is bound to a lipid particle. The very low levels of free APOC3 in serum samples from individuals with T1D, the inability of APOC3 silencing or overexpression to alter plasma levels of IL-18 and IL-1β in mice and the analyses of APOC3 loss-of-function variant carriers in the UK Biobank also support the conclusion that endogenous lipid-bound APOC3 does not induce inflammasome activation in vivo. Overall, our data suggest that the atherogenic effects of APOC3 are principally mediated by its effects on TRL/RLP metabolism rather than by increased inflammasome activation. Future studies should determine whether lipid-free APOC3 exists in vascular tissue at sufficient levels to elicit atherogenic inflammatory responses.
Methods
APOC3 and other reagents
Ultrapure LPS from Escherichia coli was purchased from List Lab (catalog#421). Human VLDL was purchased from Kalen Biomedical, LLC (catalog#770100–7). The VLDL preparations used for this study contained endotoxin levels less than 0.1 EU/mL, as measured by the amebocyte lysate assay (Pierce™ Chromogenic Endotoxin Quant Kit, ThermoFisher Scientific, catelog#A39552). De-lipidated human APOC3 was purchased from Academy Bio-medical Company, Inc. (catalog#33P-102). We used the same source and range of concentrations of de-lipidated APOC3 as Zewinger et al.10 Endotoxin levels in the batches of de-lipidated APOC3 available from the company for our study were higher than those reported by Zewinger et al (<0.03 EU/mL). Endotoxin levels in several batches of purchased de-lipidated APOC3, measured in the linear range (dilution in endotoxin-free water) by the amebocyte lysate assay (Pierce™ Chromogenic Endotoxin Quant Kit, ThermoFisher Scientific, catelog#A39552) ranged between <0.01 to 1.7 EU/mL in the concentrations of APOC3 used (0.0004 – 4 mg/dL) as shown by Extended Data Figure 1a–b. These findings suggest that de-lipidated APOC3 is capable of inducing IL-1β release even when endotoxin levels are very low or undetectable. Human APOC3 ELISA and mouse APOC3 ELISA were purchased from Abcam (Human, catalog#ab154131; mouse, catalog#ab217777).
Mice.
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Washington (protocol 3154–01) or by the Ionis Institutional Animal Care and Use Committee, and were conducted in conformity with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Adult (8–12 weeks old) female and male LDL receptor-deficient (Ldlr−/−) mice with or without a lymphocytic choriomeningitis virus (LCMV) glycoprotein transgene (GPTg) under control of the rat insulin promoter on a C57BL/J6 background were used for part of this study19. All mice were housed in a specific pathogen-free facility and kept in a temperature-controlled room set to a light and dark cycle of 12 hours each. The mice had ad libitum access to standard mouse chow (LabDiet, catalog#5053), or a low-fat diet19, and water during the study. Ldlr−/− mice without the GPTg were used for monocyte experiments in vitro. Females with the GPTg were used for the type 1 diabetes study. Female mice were injected with LCMV to induce diabetes. After the onset of diabetes, diabetic and non-diabetic mice were injected i.p. with 10 mg/kg/week GalNac control or APOC3 ASOs. Mice carrying a human APOC3 transgene were purchased from The Jackson Laboratory (B6;CBA-Tg(APOC3)3707Bres/J; JAX stock #006907). Chow-fed male hAPOC3 mice with plasma triglycerides greater than ~4,000 mg/dL were selected for this study.
Mouse monocyte experiments
Mouse bone marrow cells were harvested from the femur and tibia with 8 seconds centrifugation at 11,000xg in the presence of sterile ACK lysing buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA). Bone marrow cells were further enriched for monocytes using an immunomagnetic negative selection kit from STEMCELL Technologies (catalog#19861) as per manufacturer’s instructions. 0.2×106 freshly isolated monocytes per well were cultured in 96-well tissue-culture-treated flat-bottom plates (Corning, catalog#353072) with 0.22 μm filtered (Millipore, catalog#S2GPU05RE) complete medium (RPMI with 10% fetal bovine serum and 1% penicillin/streptomycin) in a water-jacket incubator for 1 hour at 37°C with 5% CO2. Non-adhered cells were removed by aspiration and cultured with serum-free medium (RPMI with 1% penicillin/streptomycin) with or without treatments for 16 hours at 37°C with 5% CO2. Conditioned media were harvested for IL-1β release measurements via ELISA (ThermoFisher Scientific, catalog# 88701388). Cells were lysed with RIPA lysis buffer (ThermoFisher Scientific, catalog#89901) with protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, catalog#78430). Total protein concentration was measured with the BCA protein assay (ThermoFisher Scientific, catalog#23227). Measurements were taken from distinct samples.
Mouse bone-marrow-derived macrophage experiments
Mouse bone marrow cells were harvested as described above in the murine monocyte experiments. The bone marrow cells were cultured in complete medium (RPMI, 30% L-conditioned medium, 7% fetal bovine serum, 1% penicillin/streptomycin) in a water-jacket incubator for 7 days at 37°C with 5% CO2. Cultured macrophages were incubated with or without treatments for 16 hours at 37°C with 5% CO2. For LPS priming, cells were stimulated with 10 ng/mL ultrapure LPS for 4 hours before treatments. Conditioned media were harvested for IL-1β measurements via ELISA (ThermoFisher Scientific, catalog# 88701388). Cells were lysed with RIPA lysis buffer (ThermoFisher Scientific, catalog#89901) with a protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, catalog#78430). Total protein concentration was measured with the BCA protein assay (ThermoFisher Scientific, catalog#23227). Measurements were taken from distinct samples.
Mouse models with APOC3 ASO or APOC3 siRNA treatment
Type 1 diabetes mouse model:
8- to 12-weeks old female Ldlr−/−;GPTg mice were injected with saline or LCMV. Diabetic (defined by blood glucose levels > 250 mg/dl) and non-diabetic mice were fed a low-fat, semi-purified diet without added cholesterol, as previously described19. The diabetic mice received insulin pellets (LinShin Canada Inc.) to provide baseline insulin and were treated with liquid insulin (Lantus; Sanofi) as needed to prevent ketonuria and extensive weight loss. Blood glucose was measured via a glucose meter. Values above 600 mg/dL are shown as 600 mg/dL due to the maximal range of the glucometer. After the onset of diabetes, diabetic and non-diabetic mice were injected i.p. with 10 mg/kg/week liver-targeted GalNAC control or APOC3 ASOs. Liver-specific delivery was achieved by targeting the hepatocyte-specific asialoglycoprotein receptor via triantennary GalNAc conjugation to the 5’ end of the sense strand20. The APOC3 ASO (CCAGCTTTATTAGGGACAGC) and control ASO (CCTTCCCTGAAGGTTCCTCC) were produced by Ionis Pharmaceuticals. The animals were maintained for 4 weeks before the termination of the study.
Human APOC3 transgenic mouse model:
Chow-fed 8- to 12-weeks old male hAPOC3 mice (JAX stock #006907)21 were injected with saline or 1 mg/kg of a siRNA targeting hAPOC3. The siRNAs consisted of a chemically modified antisense strand with sequence TCACTGAGAATACTGTCCCTTTT (hAPOC3 siRNA 1) or TCACTGAGAATACTGTCCCTT (hAPOC3 siRNA 2), hybridized with a chemically modified sense strand of sequence AAGGGACAGTATTCTCAGTGA (hAPOC3 siRNA 1) or AAGGGACAGTATTCTCAGTGA (hAPOC3 siRNA 2). Liver-specific delivery was achieved by targeting the hepatocyte-specific asialoglycoprotein receptor via triantennary GalNAc conjugation to the 5’ end of the sense strand20.
Plasma IL-18 and IL-1β levels were measured by ELISA (ThermoFisher Scientific, catalog#BMS618–3; BD Biosciences, catalog# 562278). Plasma human APOC3 levels were measured by an immunoturbidimetric automated assay using the Olympus/Beckman Coulter AU400 Chemistry Analyzer and ApoC3 K-Assay (Kamiya Biomedical). Measurements were taken from distinct samples but different analytes were measured in the same samples for different figure panels.
Human monocyte isolation
Experiments were approved by the Institutional Review Board at the University of Washington (IRB# STUDY00008441) and were performed according to local ethics regulations and NIH guidelines. Informed consent was obtained from each participant and they received donuts or similar snacks and juice or water and $15 for participating in the study. Human peripheral blood mononuclear cells were isolated from the peripheral blood of healthy subjects (4 male and 3 female participants between the ages of 21 and 55) and enriched for monocytes with an immunodensity negative selection cocktail from STEMCELL Technologies (catalog#15068) as per the manufacturer’s instructions. 1×106 enriched freshly isolated human monocytes per well were cultured in 96-well tissue-culture-treated flat-bottom plates (Corning, catalog#353072) with 0.22 μm filtered (Millipore, catalog#S2GPU05RE) complete medium (RPMI, 1xGlutaMax, 1% penicillin/streptomycin, 10% fetal bovine serum, and 10 mM HEPES pH 7.4) in a water-jacket incubator for 1 hour at 37°C with 5% CO2. Non-adhered cells were removed by aspiration and were then incubated in serum-free medium (RPMI, 1xGlutaMax, 1% penicillin/streptomycin, and 10 mM HEPES pH 7.4) with or without treatments for 16 hours at 37°C with 5% CO2. Conditioned media were harvested for IL-1β measurements via ELISA (ThermoFisher Scientific, catalog# BMS224–2). Cells were lysed with RIPA lysis buffer (ThermoFisher Scientific, catalog#89901) with protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, catalog#78430). Total protein concentration was measured with the BCA protein assay (ThermoFisher Scientific, catalog#23227). Measurements were taken from distinct samples.
Small unilamellar vesicles and lipidated APOC3 synthesis
1-palmitoyl-2-oleoyl-glycerol-3-phosphocholine (POPC) was purchased from Avanti Polar Lipids (catalog#850457). The chloroform from the POPC stock was completely removed by evaporation under nitrogen gas for 24 hours on ice. 3 mL of sterile PBS (pH 7.4, ThermoFisher Scientific, catalog#10010023) was added and the samples were vortexed until the lipid was in suspension. The samples were pulse-sonicated on ice for 1 hour with a 1-second on and 1-second off-duty cycle at 75% of the probe’s maximum setting (Fisher Biosciences probe). The vesicles were centrifuged at 300×g for 20 minutes at 4°C to remove unreacted lipids and titanium residues. Reacted vesicles were applied to a Superose 6 gel filtration column (Amersham Biosciences) and SUV fractions 16–17, corresponding to an average SUV size of 27 nm were collected (Extended Data Figure 1). The SUV fractions were concentrated with a 3 kDa molecular cutoff filter (Millipore, catalog#UFC5003) and the phospholipid content was quantified (Sigma-Aldrich, catalog#MAK122). Purified and concentrated SUVs were analyzed with ion mobility analysis on differential mobility for size distribution as previously described22. Homogenized samples were used for the lipidation of APOC3. De-lipidated APOC3 was incubated with SUVs at a 1:10 molecular ratio for 24 hours at 4°C. Samples were filtered and concentrated with a 100 kDa molecular cutoff filter (Millipore, catalog#UFC5100) to remove free APOC3. SUVs and lipidated APOC3 were sterile filtered with a 0.45 μm filter (Corning, catalog#8162) before applying to monocytes. The SUV preparations contained no detectable endotoxin (Pierce™ Chromogenic Endotoxin Quant Kit, ThermoFisher Scientific, catelog#A39552).
Isolation of HDL, LDL, VLDL + IDL, and lipoprotein-free serum from subjects with type 1 diabetes
HDL (density 1.063–1.21 g/mL), LDL (1.019–1.063 g/mL), VLDL + IDL (<1.019 g/mL), and lipoprotein-free serum (>1.21 g/mL) were isolated by density gradient ultracentrifugation from rapidly thawed previously collected serum from 10 subjects with T1D in the Coronary Artery Calcification in Type 1 Diabetes (CACTI)1 study cohort as previously reported23. For HDL: 50 μL serum was adjusted to a density of 1.21 g/mL using potassium bromide solution (KBr) in a 230 μL centrifuge tube. The samples were spun in a TLA 100 rotor (Beckman Coulter) at 436,000×g for 5.5 hours at 5°C. The top 57.5 μL was collected with a pipette for HDL isolation and the bottom fraction was used for the isolation of the lipoprotein-free fraction. The top fraction was adjusted to a density of 1.063 g/mL and the samples were spun in a TLA 100 rotor at 436,000×g for 5.5 hours at 5°C. The bottom 58 μL was collected with a Hamilton syringe as the HDL fraction. For LDL: 100 μL serum was adjusted to a density of 1.063 g/mL using KBr in a 230 μL centrifuge tube. The samples were spun in a TLA 100 rotor at 436,000×g for 5.5 hours at 5°C. The top 57.5 μL was collected with a pipette and was adjusted to a density of 1.019 g/mL using KBr Then the samples were spun in a TLA 100 rotor at 436,000×g for 5.5 hours at 5°C. The bottom 58 μL was collected with a Hamilton syringe as the LDL fraction. For VLDL and IDL: 300 μL serum was adjusted to a density of 1.019 g/mL using KBr in a 500 μL centrifuge tube and the samples were spun in a TLA 120 rotor (Beckman Coulter) at 627,000×g for 12 hours at 5°C. The top 125 μL was collected with a pipette as the VLDL and IDL fraction. For the lipoprotein-free fraction: the remaining bottom fraction from the HDL isolation spin (above) was adjusted to a density of 1.21 g/mL using KBr and spun in a TLA 100 rotor at 436,000×g for 5.5 hours at 5°C. The bottom 58 μL was collected with a Hamilton syringe as the lipid-free fraction. All fractions were dialyzed against 20 mM potassium phosphate (pH 7.0) with 100 μM DTPA before digestion and mass spectrometry analysis of APOC3.
Digestion of HDL, LDL, VLDL + IDL, and lipoprotein-free serum
Following the addition of freshly prepared methionine (5 mM), 10 μg of HDL, LDL, VLDL + IDL, or lipid-free serum was reduced with dithiothreitol and then alkylated with iodoacetamide in 3% sodium deoxycholate (SDC) and 100 mM ammonium bicarbonate (NH4HCO3). The HDL, LDL, VLDL+IDL, or lipoprotein free serum proteins were then incubated overnight (18 hours) at 37°C with 20:1 (w/w, protein/enzyme) of sequencing grade modified trypsin. An appropriate amount of 20% trifluoroacetic acid was added to the reaction mixture (to pH 2–3) to stop the digestion and precipitate the SDC. The SDC precipitation was spun down in a microcentrifuge (Eppendorf® 5424R) at 19,745×g for 20 minutes. The supernatant of the digested samples was desalted and cleaned up by solid-phase extraction using Waters HLB 1cc (30 mg) extraction cartridges. The digested peptides were eluted from the cartridges with 60% of acetonitrile containing 0.3% of trifluoroacetic acid, and the eluted samples were dried and stored at −80°C until mass spectrometry analysis.
Liquid chromatography-electrospray ionization tandem mass spectrometric (LC-ESI-MS/MS) analysis of APOC3 in HDL, LDL, VLDL +IDL, and lipoprotein-free serum by parallel reaction monitoring (PRM)
We used targeted proteomics with PRM to quantify APOC3 in HDL, LDL, VLDL+IDL, and lipoprotein-free serum23. Briefly, LC-ESI-MS/MS analyses were performed in the positive ion mode with an ultrahigh-resolution accurate mass Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to a nano-flow UHPLC system (EASY-nLC™ 1200 System, Thermo Fisher Scientific). Peptide digests (equivalent to 0.3 μg of protein) were loaded onto a C-18 trap column (0.1 × 40 mm) at a flow rate of 2.5 μL/min for 4 minutes and desalted for 10 minutes using 0.1% formic acid in water (solvent A). They were then separated at a flow rate of 0.3 μL/min, using a C-18 analytical column (0.1 × 200 mm). A multistep gradient of solvent A and 0.1% formic acid in 90% acetonitrile (solvent B) was used for the separation. The trap and analytical columns were packed in-house with Magic C-18 reverse-phase resin (5 μm; 100 Å; Michrom Bioresources). The columns were kept at room temperature, and the peptides were separated using a multistep gradient as follows: 1% to 8% solvent B for 1 min; 8% to 28% solvent B for 24 min; 28% to 40% solvent B for 6 min; and 40% to 90% solvent B for 5 min. The column was subsequently washed for 3 min in 90% B and re-equilibrated in 1% B for 12 min. The mass spectrometer was operated in the data-independent acquisition PRM mode.
Quantifying APOC3 in HDL, LDL, VLDL + IDL, and lipoprotein-free serum
The PRM data were analyzed with Skyline (version 21.1.0.278), an open-source program24. The peptide DALSSVQESQVAQQAR from APOC3 was monitored for quantifying APOC3. To get the total peak area for the peptide, the peak areas of all the transitions of the peptide detected by PRM analysis were summed but the transitions with interferences were deleted. Because the intensity of the doubly charged and triply charged peptides was comparable, we measured the peak areas of the peptide at both charge states and their areas were added. In order to compare the relative intensity of APOC3 in different fractions, the sum peak area of that peptide from each sample was normalized to the average peak area of that peptide in HDL in the control group. Thus, the average level of APOC3 in HDL in the control group was defined as an arbitrary unit of 1 and the relative levels of APOC3 in other lipoprotein fractions and lipoprotein-free serum were expressed as the percentage of HDL levels. Measurements were taken from distinct samples.
Analysis of association between an APOC3 loss-of-function variant, triglycerides, high sensitivity C-reactive protein and myocardial infarction in the UK Biobank
The UK Biobank is a prospective cohort of approximately 500,000 individuals in the UK aged 40–69 at the time of recruitment between 2006 and 201025. Genotyping was performed on one of two genotyping arrays: the UK BiLEVE Axiom Array (49,950 participants) or the UK Biobank Axiom Array (438,427 participants). To obtain a more comprehensive set of genetic variants that were not directly genotyped on the arrays, we performed imputation utilizing the TOPMed reference panel via the TOPMed imputation server26, 27. We focused our analyses on 452,401 individuals of broadly European ancestry and on rs138326449 (IVS2 + 1G-A), a rare (MAF = 0.002) APOC3 splice donor variant that results in APOC3 loss-of-function and for which carriers exhibit ~50% reduced circulating APOC35. rs138326449 was well imputed using the TOPMed reference panel (R2~0.9). We identified 1,928 carriers of the rs138326449 alternative (A) allele, of which 1,926 are heterozygotes and 2 are homozygotes.
We performed association analyses under an additive model using GloWGR, a distributed version of the REGENIE whole-genome regression method28. Analyses were adjusted for participant age, genetic sex, genotyping batch, assessment center, and the first ten principal components of genetic ancestry. We evaluated associations between rs138326449 and three phenotypes: serum triglycerides, serum high sensitivity C-reactive protein (hsCRP) and myocardial infarction (MI). Serum triglyceride (n=430,833) and hsCRP (n=426,774) measures were derived from the blood biochemistry assays (https://biobank.ctsu.ox.ac.uk/ukb/ukb/docs/serum_biochemistry.pdf) generated by UK Biobank by first removing outlier values ± 5 standard deviations from the mean and then log10 transforming remaining values prior to analysis. MI cases (n=20,851) and controls (n=318,164) were defined using composite criteria from linked longitudinal health records, death registry data and self-reported information from the UK Biobank in-person evaluations. Briefly, cases were defined as individuals with codes that capture incident or prevalent MI (ICD-10 codes: I21*, I22*, I23*; UK Biobank fields: 20002_1075, 6150_1) and controls were defined as individuals without any codes that capture incident or prevalent ischemic heart disease more broadly (ICD-10 codes: I20*, I21*, I22*, I23*, I24*, I25*, Z95.1, Z95.5; OPCS-4 Codes: K40*, K45*, K49*, K50*, K75*; UK Biobank fields: 20002_1074, 20002_1075, 6150_1, 6150_2, 20004_1070, 20004_1095, 20004_1523).
In order to evaluate whether the observed association between rs138326449 and elevated hsCRP was robust to adjustment for potential confounders present at the time of serum sample ascertainment, these analyses were repeated with the addition of relevant covariates, including use of lipid-lowering medications (captured by UK Biobank fields (limited to Instance ‘0’): 20003_1140861958, 20003_1141146234, 20003_1141146138, 20003_1141192410, 20003_1141192736, 20003_1140888648, 20003_1141192740, 20003_1140861954, 20003_1141188146, 20003_1140861924, 20003_1141192414, 20003_1140888594, 20003_1141162544, 20003_1140862026, 20003_1140861928, 20003_1141172214, 20003_1140881748, 20003_1140861926, 20003_1140861970, 20003_1141201306, 20003_1140862028, 20003_1141157260, 20003_1140861856, 20003_1140864592, 20003_1141171548, 6177_1, 6153_1) and diabetes (captured by UK Biobank fields (limited to Instance ‘0’): 20002_1220, 20002_1222, 20002_1223, 2443_1, 20003_1140884600, 20003_1140883066, 20003_1140874744, 20003_1141171646, 20003_1141177600, 20003_1141152590, 20003_1140874686, 20003_1141189090, 20003_1141189094, 20003_1140874718, 20003_1140874646, 20003_1141171652, 20003_1141168660, 20003_1140910566, 20003_1141177606, 20003_1140874746, 20003_1140874674, 20003_1140868902, 20003_1141156984, 20003_1141173882).
Statistical analysis
Power analyses were performed to determine the number of mice needed for experiments when sufficient preliminary information on response variables was available. For example, one critical parameter was the plasma IL-18 in mice. Preliminary studies indicated a mean value of 124 pg/mL in non-diabetic mice and a mean value of 257 pg/mL in diabetic mice with a standard deviation of 93 pg/mL. Power analysis showed that n=11 or n= 13 was needed to reject the null hypothesis with 90% or 95% probability and 5% type 1 error in a two-sided test. Statistical analyses were performed using GraphPad Prism 9.4.0 (GraphPad Software). Samples were assigned to various groups by block randomization with a block size of 2–4 (depending on the experiment). Data collection and analysis were performed blind to the conditions of the experiments. Normality tests were performed by the D’Agostino & Pearson test and outliers were removed based on the robust regression and outlier removal (ROUT) method with Q=0.1%. Data are displayed as mean ± SEM. Based on the normality of the data, they were analyzed with two-sided Mann-Whitney test, one-way ANOVA followed by Holm-Šídák’s multiple comparisons test, Kruskal-Wallis test and Dunn’s multiple comparisons tests, or two-way ANOVA followed by Tukey multiple comparisons test. A p-value of less than 0.05 was considered statistically significant.
Extended Data
Extended Data Fig. 1. Characterization of small unilamellar vesicles (SUVs) and APOC3.

a, IL-1β release from mouse bone marrow-derived monocytes incubated in indicated APOC3 concentrations. IL-1β measurements were conducted by ELISA and the data were normalized to total cellular protein determined by a BCA protein assay. Data show means ± SEM (n=5, 5, 5, 5, 5, 8 replicates/group, respectively). b, Endotoxin levels in the final concentrations of APOC3 used in panel a, measured by the amebocyte lysate assay (Pierce™ Chromogenic Endotoxin Quant Kit). Data show means ± SEM (n=3 replicates/group). In a and b, statistical analyses were performed by Kruskal-Wallis tests and Dunn’s multiple comparisons tests. c, 1-palmitoyl-2-oleoyl-glycerol-3-phosphocholine (POPC) in PBS was sonicated on ice to allow the formation of small unilamellar vesicles (SUVs). The SUVs were purified on a Superose 6 column before the incorporation of APOC3. SUV-APOC3 was isolated with various centrifugation columns and was sterile filtered. The molar concentrations of APOC3 and SUVs were measured by ELISA, BCA protein assay, and phospholipid assay. d, FPLC profile of synthesized SUVs and fractions 16 and 17 (shown in red) were purified and used for the lipidation of APOC3. e, Ion mobility analysis on differential mobility analyzer (DMA) of SUVs from fractions 16 and 17, demonstrating a homogenized particle population around 27 nm. Extended Data Figure 1c was created with BioRender.com.
Extended Data Fig. 2. Distribution and density of serum triglyceride and high sensitivity C-reactive protein (hsCRP) values by APOC3 rs138326449 genotype in the UK Biobank.
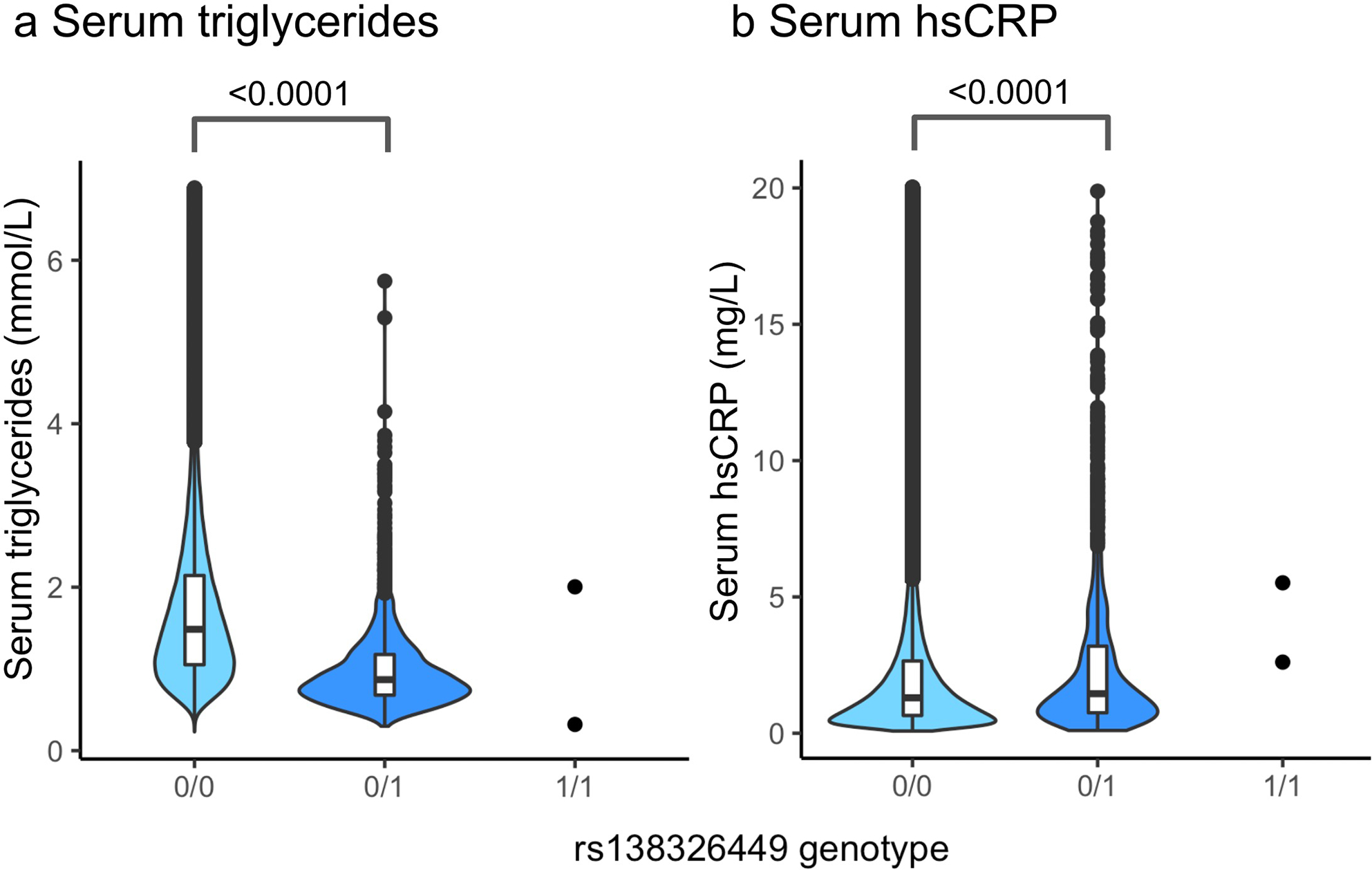
Unadjusted serum levels of a, triglycerides (n=430,833) and b, hsCRP (n=426,774) among UK Biobank carriers of 0, 1 or 2 alternative (A) alleles of the APOC3 loss-of-function variant rs138326449 (IVS2 + 1G-A). Specifically, 0/0 indicates homozygosity of the reference (G) allele, 0/1 indicates heterozygosity and 1/1 indicates homozygosity of the alternative (A) allele. The data are shown in box-and-whisker plots (the bound of the box extends from the 25th to 75th percentiles and the whisker covers the minima and maxima) with the median marked by a center line, remaining distribution and kernel density estimations of each trait (outlier values ± 5 standard deviations from the mean were removed). Compared with rs138326449-G homozygotes, rs138326449-A heterozygotes demonstrate a 0.62 mmol/L decrease in median triglycerides (p = <2.00 × 10−16 by one-way ANOVA) and a 0.16 mg/L increase in median hsCRP (p = 1.54 × 10−5 by one-way ANOVA).
Extended Data Table. 1. Associations between APOC3 rs138326449 and serum triglycerides, high-sensitivity C-reactive protein, and myocardial infarction were evaluated in UK Biobank participants.
Analyses were performed under an additive model in GloWGR and adjusted for participant age, genetic sex, genotyping batch, assessment center, and the first ten principal components of genetic ancestry. A linear regression model was used to test associations between rs138326449 and quantitative traits (serum triglycerides and serum high-sensitivity C-reactive protein) and a logistic regression model was used to test the association between rs138326449 and the binary trait (myocardial infarction). The p value for triglycerides (<4.90E-324) represents the system minimum floating-point value, indicating that the true p value is lower than this value. The p values presented in the table have not been adjusted for multiple comparisons.
| rsID | Consequence | MAF | Serum Triglycerides (n=430,833) | Serum High-Sensitivity C-Reactive Protein (n=426,774) | Myocardial Infarction (n=20,851) | |||
|---|---|---|---|---|---|---|---|---|
| p value | beta (95% CI) | p value | beta (95% CI) | p value | OR (95% CI) | |||
| rs138326449 | APOC3 Splice Donor Variant (IVS2 + 1G-A) | 0.002 | <4.90E-324 | −0.224 (−0.234, −0.215) | 1.75E-07 | 0.051(0.032, 0.070) | 0.025 | 0.765 (0.602, 0.971) |
Extended Data Table. 2. Conditioned APOC3 rs138326449 high-sensitivity C-reactive protein (hsCRP) associations in the UK Biobank.
Associations between rs138326449 and hsCRP with and without adjustment for potential confounders present at the time of serum sample ascertainment. All analyses were performed under an additive linear regression model in GloWGR and adjusted for participant age, genetic sex, genotyping batch, assessment center, and the first ten principal components of genetic ancestry (Standard Covariates), with the addition of a covariate indicating either the use of lipid-lowering medication (Lipid Rx) or diabetes (D). The p-values presented in the table have not been adjusted for multiple comparisons.
| rsID | Consequence | MAF | Serum hsCRP [Standard Covariates] (n=426,774) | Serum hsCRP [Standard Covariates + Lipid Rx] (n=426,774) | Serum hsCRP [Standard Covariates + D] (n=426,774) | |||
|---|---|---|---|---|---|---|---|---|
| p value | beta (95% CI) | p value | beta (95% CI) | p value | beta (95% CI) | |||
| rs138326449 | APOC3 Splice Donor Variant (IVS2 + 1G-A) | 0.002 | 1.75E-07 | 0.051 (0.032, 0.070) | 8.29E-08 | 0.052 (0.033, 0.071) | 1.25E-07 | 0.051 (0.032, 0.070) |
Supplementary Material
Acknowledgments
This study was supported by NIH grants R35HL150754, P01HL151328 and R01HL161829 (KEB) and a Predoctoral Fellowship Award from the American Heart Association #828090 (CCH). UK Biobank analyses were conducted using the UK Biobank Resource under Application Number 34229.
Footnotes
Competing interests
A.E.M. is employed by Ionis Pharmaceuticals. O.G., S.B., and G.M. are employed by Empirico Inc.. K.E.B. serves on the scientific advisory board of Esperion Therapeutics, Inc. J.L.W receives royalties from patents on oxidation-specific antibodies and biomarkers related to oxidized lipoproteins held by UCSD. J.L.W. is a co-founder of Oxitope, Inc. and Kleanthi Diagnostics, LLC, and is a consultant for Ionis Pharmaceuticals. The remaining authors declare no competing interests.
Peer Review Information:
Nature Immunology thanks Marit Westerterp and Laurent Yvan-Charvet for their contribution to the peer review of this work.
Online content. Nature Research reporting summaries and supplementary information are available in the online supplements.
Data Availability.
All original data are available from the corresponding author upon request.
References
- 1.Kanter JE et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest 129, 4165–4179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu A et al. Serum apolipoproteins and apolipoprotein-defined lipoprotein subclasses: a hypothesis-generating prospective study of cardiovascular events in T1D. J Lipid Res 60, 1432–1439 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansson Sigfrids F et al. Apolipoprotein C-III predicts cardiovascular events and mortality in individuals with type 1 diabetes and albuminuria. J Intern Med 291, 338–349 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheffer PG et al. Increased plasma apolipoprotein C-III concentration independently predicts cardiovascular mortality: the Hoorn Study. Clin Chem 54, 1325–1330 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Tg et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 371, 22–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG & Tybjaerg-Hansen A Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 371, 32–41 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Pollin TI et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boren J, Packard CJ & Taskinen MR The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front Endocrinol (Lausanne) 11, 474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zewinger S et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol 21, 30–41 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Segrest JP et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res 33, 141–166 (1992). [PubMed] [Google Scholar]
- 12.Davidson WS et al. The biotin-capture lipid affinity assay: a rapid method for determining lipid binding parameters for apolipoproteins. J Lipid Res 47, 440–449 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Netea MG et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlotter F et al. ApoC-III is a novel inducer of calcification in human aortic valves. J Biol Chem 296, 100193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z & Zhang Z Mapping cell types across human tissues. Science 376, 695–696 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Mund A, Brunner AD & Mann M Unbiased spatial proteomics with single-cell resolution in tissues. Mol Cell 82, 2335–2349 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA, Novick D, Kim S & Kaplanski G Interleukin-18 and IL-18 binding protein. Front Immunol 4, 289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qamar A et al. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Arterioscler Thromb Vasc Biol 35, 1880–1888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 19.Renard CB et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest 114, 659–668 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prakash TP et al. Comprehensive Structure-Activity Relationship of Triantennary N-Acetylgalactosamine Conjugated Antisense Oligonucleotides for Targeted Delivery to Hepatocytes. J Med Chem 59, 2718–2733 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Reaven GM, Mondon CE, Chen YD & Breslow JL Hypertriglyceridemic mice transgenic for the human apolipoprotein C-III gene are neither insulin resistant nor hyperinsulinemic. J Lipid Res 35, 820–824 (1994). [PubMed] [Google Scholar]
- 22.Vaisar T et al. High Concentration of Medium-Sized HDL Particles and Enrichment in HDL Paraoxonase 1 Associate With Protection From Vascular Complications in People With Long-standing Type 1 Diabetes. Diabetes Care 43, 178–186 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao B et al. Pulmonary surfactant protein B carried by HDL predicts incident CVD in patients with type 1 diabetes. J Lipid Res 63, 100196 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacLean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mbatchou J et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet 53, 1097–1103 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data are available from the corresponding author upon request.