

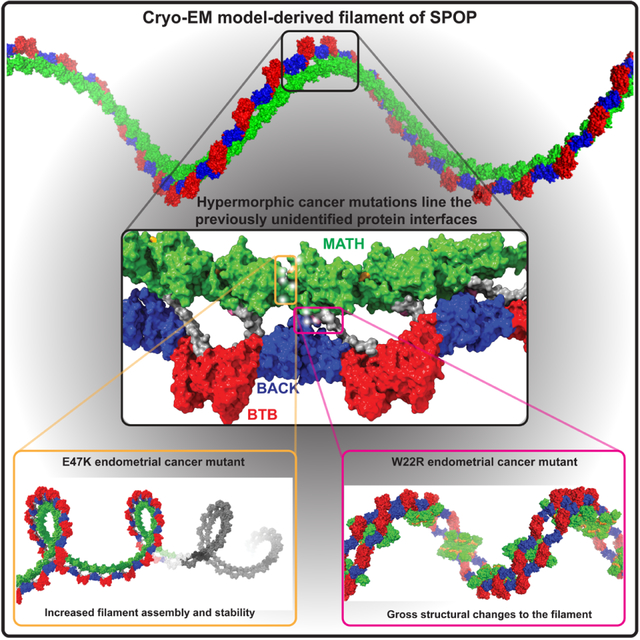
Summary
The speckle-type POZ protein (SPOP) functions in Cullin3-RING ubiquitin ligases (CRL3) as a receptor for the recognition of substrates involved in cell growth, survival and signaling. SPOP mutations have been attributed to the development of many types of cancers, including prostate and endometrial cancers. Prostate cancer mutations localize in the substrate-binding site of the substrate recognition (MATH) domain and reduce or prevent binding. However, most endometrial cancer mutations are dispersed in seemingly inconspicuous solvent-exposed regions of SPOP, offering no clear basis for their cancer-causing and peculiar gain-of-function properties. Herein, we present the first structure of SPOP in its oligomeric form, uncovering several new interfaces important for SPOP self-assembly and normal function. Given that many previously unaccounted-for cancer mutations are localized in these newly identified interfaces, we uncover molecular mechanisms underlying dysregulation of SPOP function, with effects ranging from gross structural changes to enhanced self-association, and heightened stability and activity.
Keywords: cryo-EM, structure, higher-order oligomer, filament, endometrial cancer, tumor suppressor, oncogene, prostate cancer
In Brief
The mechanism underlying endometrial cancer mutations of SPOP are incompletely understood – they seem solvent-exposed in available structures. Cuneo, O’Flynn et al. report the cryo-EM structure of SPOP, which reveals that these mutations are in previously uncharacterized interfaces. Mutations enhance assembly or alter quaternary structure, contributing to superphysiological function.
Graphical Abstract
Introduction
Ubiquitin ligases play critical roles in the maintenance of cellular proteostasis. In the modular Cullin-RING ubiquitin ligases (CRLs), substrate receptors recruit specific proteins, which are then marked by ubiquitination for proteasomal degradation.1 Hence, mutations in substrate receptors result in altered levels of their specific substrates and can result in disease processes. The speckle-type POZ protein (SPOP) is a substrate receptor of the Cullin3-RING ubiquitin ligase (CRL3). SPOP substrates include proto-oncogenes such as androgen, estrogen, and progesterone receptor, MYC, the Hedgehog pathway transcriptional regulators Gli2 and Gli3, the bromodomain and extraterminal domain (BET) family of proteins BRD2, BRD3 and BRD4, the apoptosis regulator death domain associated-protein (DAXX), and the DNA-damage response protein 53BP1.2–11 Dysregulation of SPOP activity alters the levels of these proteins6 and can result in oncogenesis in susceptible cell types. Indeed, SPOP is the most frequently mutated gene in prostate cancer and is also often mutated in endometrial, breast, colon, and other solid tumors.12,13 SPOP is thus regarded as an important tumor suppressor across cancers14 and it is important to understand how it functions at a molecular level.
High-resolution structures of each of SPOP domains are available, either in isolation or in combinations.15,16 A large number of these structures show the interaction of substrate peptides with the N-terminal, substrate-binding meprin and TRAF homology (MATH) domain. While many CRL substrate receptors form dimers, SPOP forms linear higher-order oligomers through the synergistic function of its two dimerization domains, a BTB (broad-complex, tramtrack and bric-a-brac) domain and a BACK (BTB and C-terminal Kelch) domain (Figure 1A).17 By superposition of known structures, a model of SPOP oligomers can be generated in which the position and orientation of domains relative to each other are uniquely defined (Figure 1B). In this structural model, homodimers of BTB and BACK domains alternate and give rise to a helical SPOP oligomer. MATH domains dimerize in a head-to-head orientation, and those dimers are connected to the helical filament via flexible linkers, facilitating their engagement with substrates.15 Each SPOP monomer can recruit Cullin3 (Cul3)18, the scaffold component of the CRL3. Cul3 in turn forms tight complexes with the RING domain protein RING-box domain protein 1 (Rbx1) and recruits the E2 conjugating enzyme to facilitate transfer of ubiquitin onto substrates (Figure 1A). Oligomeric SPOP is more enzymatically active than SPOP variants bearing mutations in the dimerization interfaces that are constitutively monomeric or dimeric.17,19 Mirroring the multivalency of SPOP, most of its known substrates contain multiple weakly interacting SPOP-binding motifs in flexible, disordered regions.20,21 SPOP oligomers can engage these multivalent substrates with high avidity forming complexes with longer lifetimes than those of single MATH:peptide complexes. This enables poly-ubiquitination of substrates via multiple rounds of enzymatic turnover and contributes to the higher activity of SPOP oligomers.19 The structural model and functional data therefore both assert that the substrate adaptor SPOP functions as an oligomer in the active CRL3SPOP ubiquitin ligase complex.
Figure 1. SPOP oligomer model cannot explain the molecular basis of cancer mutant dysfunction.
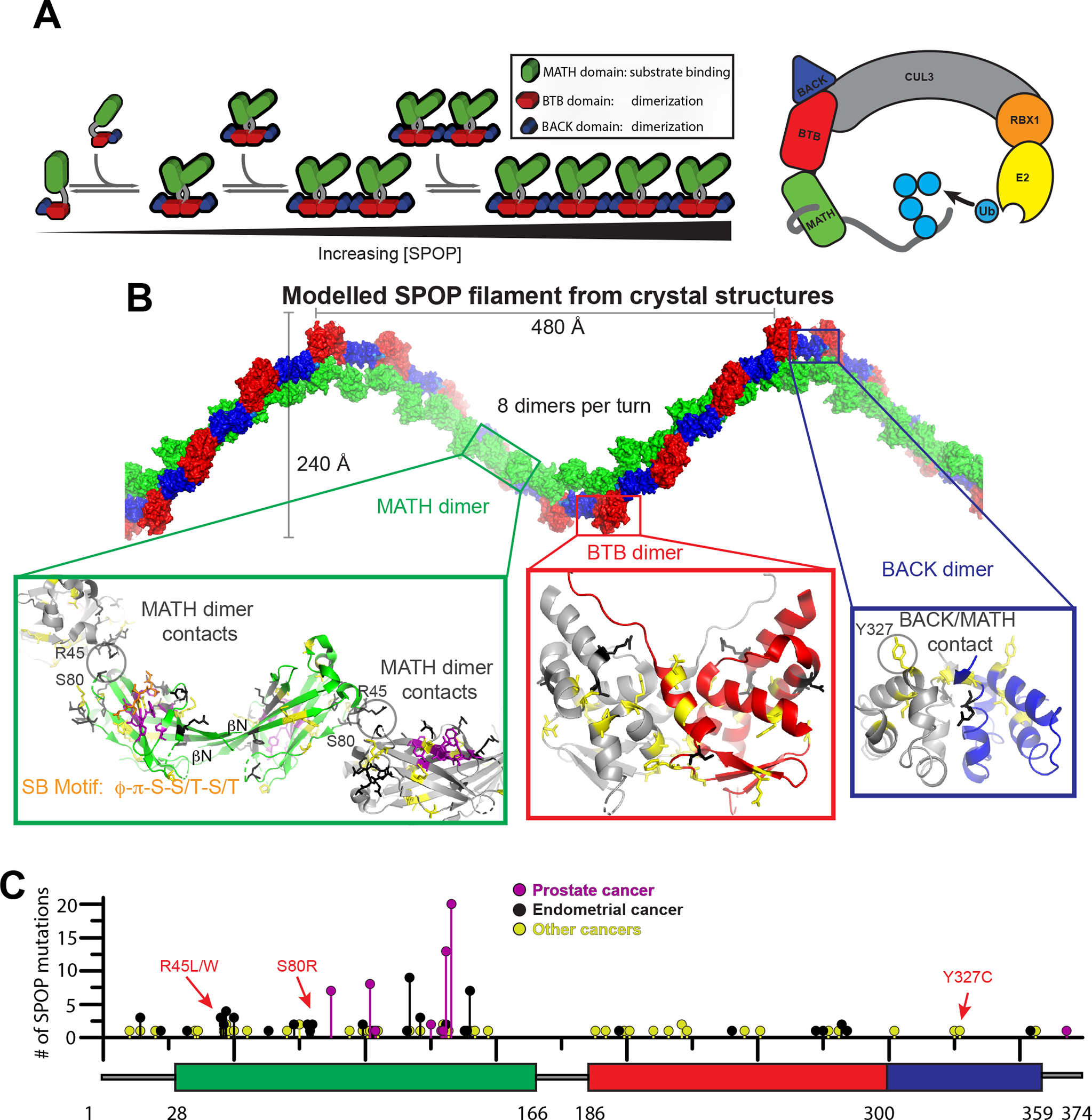
(A) Left, cartoon schematic of concentration dependent SPOP self-association. SPOP dimerizes strongly through the BTB domain. The resulting BTB dimers further dimerize through the BACK domain resulting in linear higher-order oligomers with defined size distribution.17 A SPOP octamer is shown as example.
(B) Model of a SPOP left-handed helical filament created through superposition of SPOP crystal structures (BTB dimer, PDB 3HQI15; BACK dimer, PDB 4HS216). The molecular effect of cancer mutations located in the BTB/BTB (red box) and BACK/BACK (blue box) interfaces are readily rationalized in the structural model. In the MATH domain (green box), all but one prostate cancer mutations (purple) are localized to the substrate binding cleft and are known to reduce substrate binding.22 Most endometrial and other cancer mutations are outside of the substrate binding cleft in regions where no previous function has been identified. Only a few mutations can be rationalized in terms of MATH head-to-head dimerization or tail-to-tail interactions (grey circles).
(C) The lollipop plot shows mutations identified in cancer patients in all three domains of SPOP (represented as a box plot). Prostate cancer mutations (magenta) are relatively frequent, occur in the substrate binding cleft and interfere with substrate binding. Intermediate-frequency endometrial cancer mutations (black) are clustered in regions of the MATH domain with no previously known function nor structural explanation by the filament model. Mutation data collated from cBioPortal.31,32
The available structural data offer some insight into the basis for dysfunction of disease mutants. Prostate cancer mutations are located in the substrate-binding groove of SPOP’s MATH domain (Figures 1B,C, magenta sticks) and severely weaken substrate binding.10,22 As a result, SPOP substrates are not effectively ubiquitinated and degraded, and their levels rise, transforming susceptible cell types. On the other hand, endometrial cancer mutations in the MATH domain are located outside the substrate-binding groove (Figures 1B,C, black sticks).5,23 These mutants are hypermorphic, promoting stronger ubiquitination of some substrates, such as BRD2, BRD3 and BRD4.5 The mechanisms underlying gain of function in these mutants are unclear: they do not display higher affinity toward substrates, compared to WT SPOP.24
The inability of current structural models of SPOP to explain the molecular basis of gain-of-function cancer mutations suggest that they do not fully capture the structure of the oligomer and relevant interactions remain to be identified. Here we present the first experimental, high-resolution structure of full-length SPOP oligomers. We observe three new intermolecular interfaces that have not previously been observed in SPOP structures and structural models. These include MATH:MATH interactions in a head-to-tail orientation that enable a continuous chain of MATH domains. Another interface, formed by three monomers, locks down the MATH domains in a repetitive, regular assembly on top of the BTB-BACK filament layer. Clustered in these newly observed interfaces are many so-far unexplained cancer mutations. We characterized several of those mutations and probed the structural and functional consequence of disrupting these interfaces. One mutation resulted in gross structural changes to SPOP assemblies. Another mutant structure was similar to the WT but the mutations in that interface enhanced self-assembly. The two distinct mechanisms of dysregulation both result in longer half-lives in cells and contribute to enhanced substrate ubiquitination. The new structural information we present here enables a deeper molecular understanding of function and cancer-related dysregulation of the SPOP tumor suppressor, while providing insights into one molecular basis of hypermorphic mutations.
Results
Structure of SPOP oligomers
We sought to determine an experimental structure of SPOP oligomers to close the current bottlenecks in our mechanistic understanding of SPOP. Cryo-electron microscopy (cryo-EM) images collected on a Titan Krios microscope showed linear oligomers with a variety of different lengths and curvatures (Figure S1A) but regular structure over short length scales (i.e., several dimers) as revealed by 2D classes (Figures 2A and S1B). We thus did not enforce regular symmetry along the full filament but instead optimized the box size that captured the largest regular unit of a continuous filament (see Figure S2 for picks along filaments).25
Figure 2. The cryo-EM structure of WT SPOP shows three previously unidentified protein-protein interfaces.
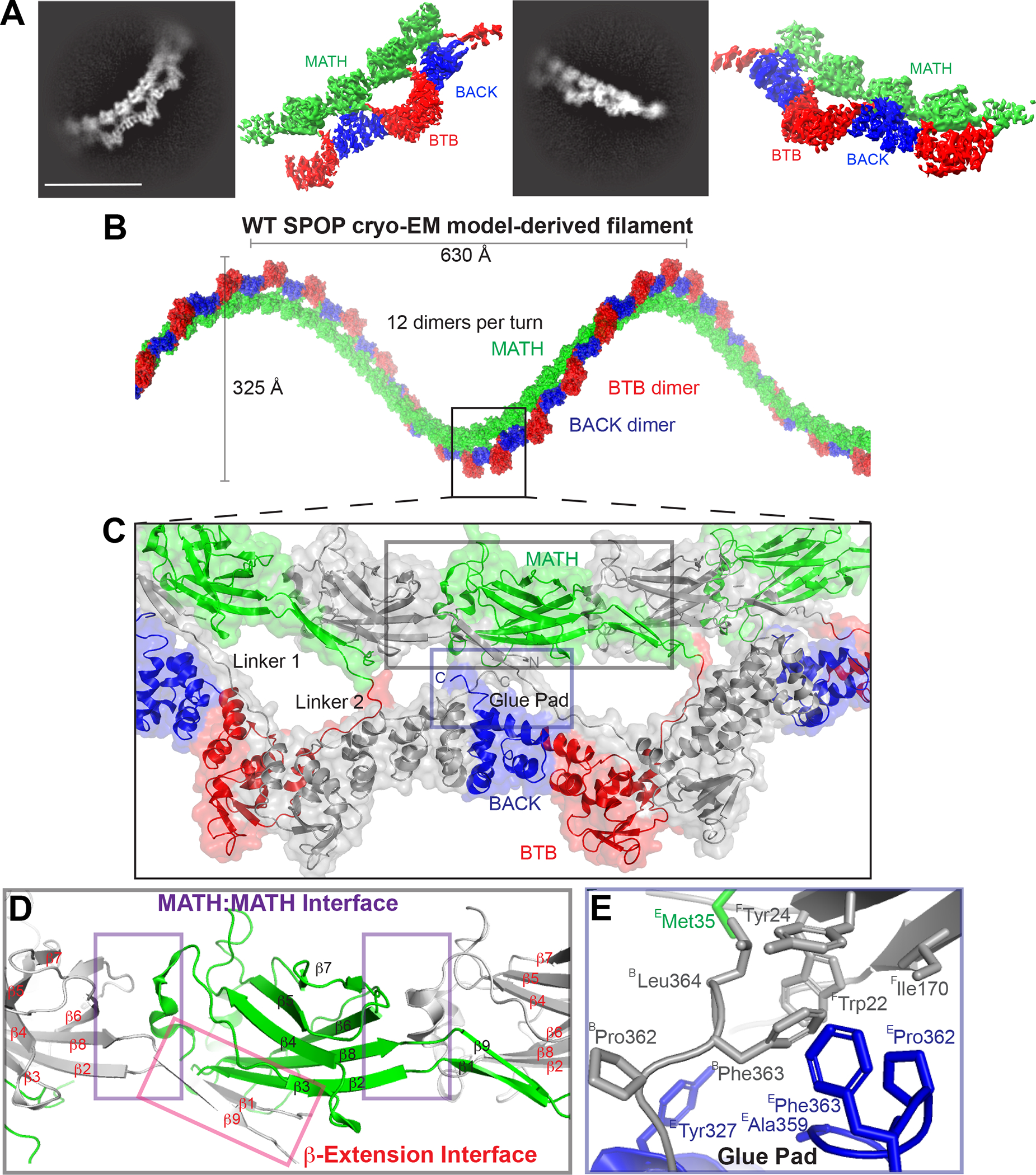
(A) Representative 2D classes and sharpened cryo-EM maps of WT-SPOP oligomers in similar orientation (see also Figure S1).
(B) The cryo-EM model of SPOP oligomers can be used to construct a left-handed helical filament by concatenation of tetramers.
(C) The structure reveals three previously unidentified protein interfaces that mediate extensive MATH:MATH interactions (grey inset box) and “glue” the MATH domains and the C-terminus into the repeating unit of the filament (blue inset box). The linker connecting the MATH domains with the oligomerization domains appears in two distinct repeating conformations; one interacts specifically with the BTB and BACK domains of an adjacent monomer (Linker 1), whereas the other interacts only with an adjacent BTB domain (Linker 2). Alternating monomers are colored grey for clarity.
(D) The MATH domains take on a continuous head-to-tail orientation along the filament with a repeating MATH:MATH interface (purple box). The β-extension interface (red box) mediates additional MATH:MATH self-association. Here, the N-terminus of SPOP, which was lacking from earlier studies, forms a well-ordered β-strand (β1) and a continuous β-sheet with the adjacent MATH domain as well as with what was in earlier crystal structures a disordered linker (now β9) following the structured MATH domain.
(E) Close-up view of the glue pad, which anchors the MATH domain into the repeating oligomerization domains through a hydrophobic interface formed by three polypeptide chains. (Superscripts on residue names indicate the protein chains.)
The local resolution decreases past a tetramer but is of sufficient quality to show that the repeating unit of the SPOP oligomer structure is a SPOP dimer: the two monomers in the dimer differ in their connectivity to the BTB-BACK layer (see Figure 2C, linker 1 vs linker 2, where monomers are shown in alternating grey and colored representation). Linking the tetramers into higher-order oligomers results in a helical filament with 630 Å pitch, 325 Å width, and 12 dimers per turn (Figure 2B). Of note, the different curvatures observed between and within filaments reveal flexibility in inter-subunit orientation, and the helical dimensions are therefore idealized parameters extrapolated from the local structure.
The filament has two layers. The α-helical layer is formed by alternating dimers of BTB and BACK domains; the β-rich layer is formed by evenly arranged MATH domains connected to and bound to the BTB-BACK layer. The local resolution peaks in the BTB-BACK layer at ~3.8 Å (Figures S1C–S1D). The MATH domain layer has a somewhat lower resolution due to the non-discrete dynamic nature of the MATH domain which could not be resolved with 3D variability analysis of the cryo-EM data. The BTB and BACK domain orientations in the filament are virtually identical to the previous SPOP oligomer model, while the MATH domain layer shows substantial differences, as described below.
Three intermonomer interfaces
Our structure reveals three new intermonomer interfaces (Figures 2C–2E) that together enforce a regular assembly of MATH domains on top of the BTB-BACK domains. These are the MATH:MATH interface, the β-extension interface (Figure 2D), and an area we refer to as the glue pad, which “glues” every second MATH domain onto the BTB-BACK filament layer via hydrophobic interactions (Figures 2C, 2E and S1E).
The MATH domains assemble in a regular head-to-tail orientation, which means that all MATH dimers are equivalent to each other (Figures 2B–2D). This is in sharp contrast to the previous SPOP structural model, in which two consecutive MATH domains formed head-to-head dimers, with little interaction in the tail-to-tail orientation; a small contact with little buried surface area involved residues R45 and S80 (Figures 1B–1C), which were found to be mutated in several endometrial cancer patients and therefore gave some support to this orientation. In contrast, the newly revealed head-to-tail interactions are stabilized by an extensive interface corresponding to a buried surface area of ~400 Å2 (Figure 2D). These interfaces seem to be identical between all pairs of MATH domains (grey to green and green to grey in Figure 2D), but local resolution limits the interpretation of detailed interactions.
The second new interface involves interactions by the N- and C-terminal extensions of the MATH domain; they form two additional β-strands (β1 and β9) and extend the β-sheet of the subsequent MATH domain by adding on to β3 intermolecularly (Figure 2D); we thus call this the β-extension interface. Notably, while the N-terminal 27 amino acids were lacking from constructs used in earlier structural studies, this region provides 735 Å2 of buried surface area with strand β3 of the adjacent MATH domain. Sequence inspection reveals that strand β3 does not have the characteristics of an edge strand, which usually contain charged residues. This is a natural negative design mechanism that prevents the association of additional β-strands and ultimately β-aggregation.26 β3 lacks charged residues and thus seems primed for accepting interactions with β1/ β9.
The MATH domains hold one another in place by continuous head-to-tail interactions (see above) and are furthermore glued in place on the BTB-BACK filament layer by the third interface (Figure 2C), the glue pad. The fact that the BTB-BACK domains form alternate head-to-head and tail-to-tail interactions, but the MATH domains form continuous head-to-tail interactions, results in a breaking of the symmetry between MATH domains. Only every second MATH domain is anchored to the BTB-BACK layer via the glue pad, and the linker conformations of alternating MATH domains therefore differ (see Figure 2C). Each second MATH domain forms an extended hydrophobic interface involving W22, Y24, and I170 from monomer F, M35, A359, P362 and F363 from monomer E, and P362, F363 and L364 from monomer B (Figure 2E). The glue pads facilitate the organization into a regular double-layer assembly of the α-helical BTB-BACK layer and the β-sheet MATH domain layer. These intermolecular interactions agree with contacts predicted from the covariation of SPOP ortholog sequences27 which were unaccounted for in the SPOP structural model. Notably, covariation does not predict inter-monomer contacts stemming from the tail-to-tail orientation observed in the crystal structure.
Previously unexplained cancer mutations map to new interfaces
When we mapped mutations found in endometrial and other types of cancers onto the experimental SPOP oligomer structure, we realized that many of the so-far unexplained mutations localize to the newly described interfaces (Figure 3), including to the glue pad and to the MATH:MATH interface. R45 and S80, the only contact in the previously supported tail-to-tail orientation of the MATH domains (as shown in Figure 1B), are involved in more extensive contacts in the experimental structure (Figure 3). In fact, a ridge of mutation hot spots extends along the MATH domain layer of the filament (Figure 3). Pathogenic mutations are enriched in interfaces as compared to other parts of protein structures, in particular in homomeric protein complexes.28 We thus hypothesized that the experimental SPOP structure has the potential to explain disease mechanisms and by extension function. We further hypothesized that disrupting the glue pad would liberate the MATH domains from their regular arrangement in the double-layer filament and might alter function. The WT structure revealed key buried residues in the glue pad including W22 and Y327. W22R is a mutation of unknown significance found in an endometrial cancer patient, and Y327F and Y327C were found in a lung adenocarcinoma patient and a hepatocellular carcinoma patient, respectively, supporting the idea that disruption of the glue pad may be detrimental. We decided to interrogate the effect of the W22R mutation.
Figure 3. SPOP cancer mutations line novel interfaces in the SPOP cryo-EM structure.
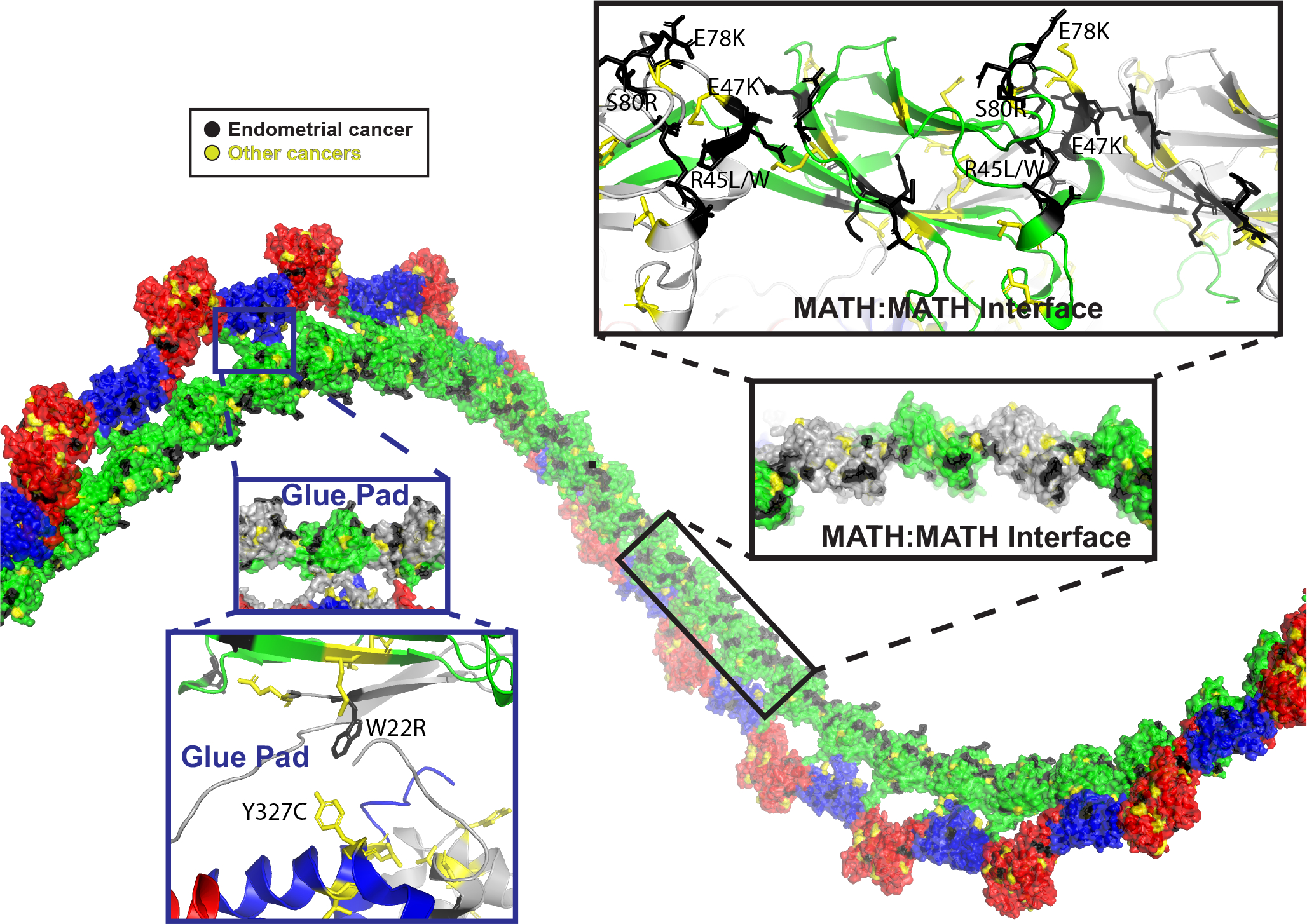
Previously unexplainable mutations are found in the MATH:MATH and β-extension interfaces, and in the glue pad. A ridge of highly clustered mutations follows the face of the MATH domain that is positioned towards the interior of the filament (black callout box). This mutation ridge contains the cluster of charge-altering mutations that span the MATH:MATH interface (black and yellow). An additional cluster of cancer mutations is found in the glue pad (blue callout box), including the endometrial cancer mutation W22R and Y327C/Y327F in hepatocellular carcinoma and lung adenocarcinoma, respectively.
Mutation W22R leads to gross alterations of the SPOP assembly architecture
We generated protein samples of SPOP W22R and visualized them by cryo-EM. The filaments were thicker than WT filaments (Figures S3 and S4), and 2D class averages showed different fine structure but nonetheless regular appearance. 3D classification revealed two different types of assemblies with populations of 62 vs 29%; the remaining 8% particles were discarded (Figures 4A, 4D and S5). Both contained double SPOP filaments, i.e., two filaments that run parallel to each other and intercalate via MATH-MATH interactions (Figure 4B,E); no contacts are formed between the BTB-BACK layers of the two filaments. In population 1, the repeating unit is likely an octamer, but the map density for two MATH domains was insufficient for modeling (Figures S6A and S6F). In population 2, the repeating unit is a tetramer (Figure S6B); the lower resolution (~6.0 Å) is sufficient to model its general assembly.
Figure 4. SPOP W22R forms dramatically altered filaments.
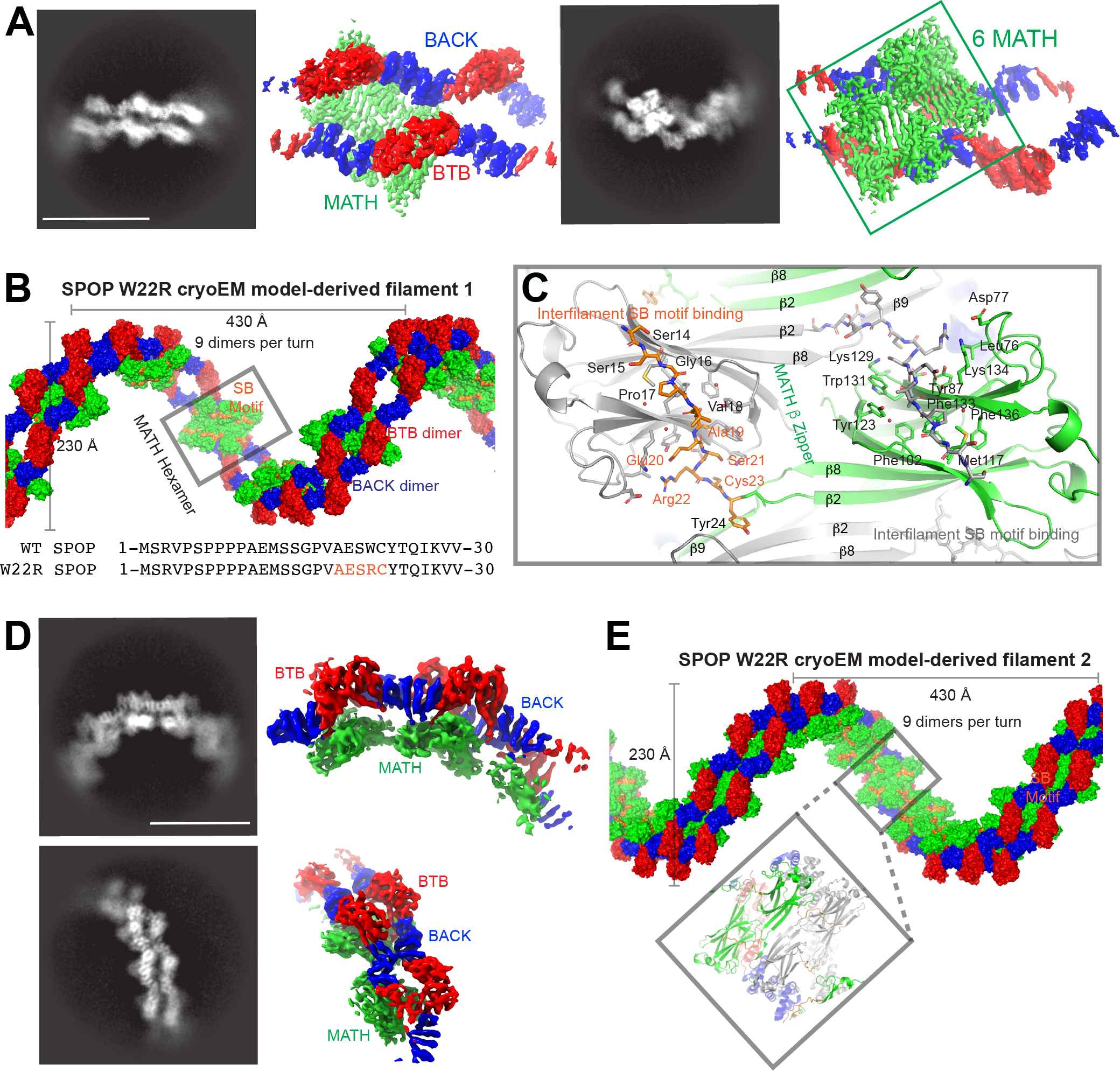
(A) Representative 2D classes (left) and sharpened cryo-EM maps in similar orientation (right) of SPOP W22R double filaments with MATH hexamer assemblies, i.e., population 1 (see also Figure S3).
(B) The cryo-EM model of population 1 of SPOP W22R can be used to construct a left-handed helical filament. Six ordered MATH domains (green surface, grey box) form a continuous assembly that link two individual SPOP filaments. The assembly is further held together by inter-monomer and inter-filament interactions of the substrate binding cleft in each MATH domain with a pseudo-SB motif sequence generated by mutation W22R (orange surface representation). Bottom, sequence alignment of N-terminal 30 amino acids of SPOP WT and W22R. Pseudo-SB motif in orange.
(C) Close up view of MATH domains. The β2 strands intercalate across the double filament in a zipper-like fashion (β-zipper) and link together two SPOP filaments. The R22-containing pseudo-SB motif binds to adjacent monomers and further links the two filaments together. MATH monomers from one filament are colored grey, whereas MATH monomers from the other filament are colored green; the N-terminus bound in the SB binding cleft is colored orange. See also Figure S6 for representations of the connectivity.
(D) Representative 2D classes (left) and sharpened cryo-EM maps in similar orientation (right) of SPOP W22R assemblies with MATH tetramer assemblies, i.e., population 2 (see also Figure S4).
(E) Population 2 of SPOP W22R particles have an alternative arrangement with a tetramer repeating unit. MATH tetramers link together two filaments. Helical parameters are similar to the population 1 of the SPOP W22R double filament.
In population 1, we clearly observe six MATH domains that intercalate with each other and hold the two filaments together (Figure S6A). The positions of the MATH domains are well determined, and they have a high local resolution of ~3.0 Å (Figure 4 and S3). We were hence able to visualize the interactions that lead to the intertwined MATH domains and double filament structure in detail. The hexamer of MATH domains has two-fold symmetry, hence can be described as two MATH domain trimers. Two of the three MATH domains are bound through extensive contacts between β2 in a head-to-head orientation (Figure S6C), in a manner similar to the MATH:MATH interactions in the SPOP filament model based on previous structures (in particular the SPOP28-337 dimer, PDB 3HQI, Figure 1). This arrangement is also present in population 2. The remaining monomer packs against an adjacent MATH domain in a different head-to-head orientation as well as forming interactions with strand β9 (Figure S6D). The two-fold symmetry of this hexamer means that the trimers from both filaments attach to each other via a zipper-like chain of β-strands (Figure 4C, contributions from the two filaments shown in green and grey). This β-zipper is less extensive in population 2, likely leading to the observed large decrease in resolution as a consequence of increased local dynamics.
Strikingly, the disruption of the glue pad liberates the mutated residue R22 from the β-extension and glue-pad interfaces of the wild-type structure. This region now binds within the substrate binding cleft of adjacent MATH domains and further stabilizes intermolecular MATH interactions (Figures 4B–4C and S6D–S6E). Indeed, the sequence AESR22C acts as a suboptimal SPOP-binding motif and binds an adjacent MATH domain binding site in a canonical conformation despite the deviations from the consensus motif. A total of 550 Å2 of solvent-accessible surface area is buried upon binding of the region encompassing each single suboptimal motif to each MATH domain of the other filament. Interestingly, the substrate binding grooves in the three consecutive MATH domains in population 1 are lined up in a manner that a multivalent substrate may be able to bind with high avidity (Figure 4B). The remaining two MATH domains, which we infer based on the presence of additional BTB-BACK domains in the assembly (Figure S6F), are presumably located on opposite sides of this hexamer, and therefore lack the interconnectivity of the interior six MATH domains. This may result in a relatively independent ability to move and may explain our inability to reliably localize them and infer their positions based on diffuse excess density.
In conclusion, the W22R mutation results in disruption of the glue pad interface found in the WT protein, which enables de novo interactions and results in two altered assembly stoichiometries, and potentially accounts for the hypermorphic phenotype previously found for endometrial cancer mutants.5
The E47K mutation leaves the structure largely unperturbed
While prostate cancer mutations localize to the substrate binding cleft, we find that other types of cancer mutations are enriched in the newly revealed MATH:MATH interface, including endometrial mutations R45L/W, E47K, E78K and S80R (Figure 3); these mutations involve removal or addition of charged residues, and we wondered whether altered self-association might explain their previously observed gain-of-function towards ubiquitination of certain substrates.5 We hence generated and purified SPOP E47K because its gain of function was previously characterized. Cryo-electron micrographs showed regular filaments (Figure S7A), and 2D class averages showed detailed, regular structures (Figures 5A and S7B). We determined the oligomer structure, which is nearly indistinguishable in its overall characteristics and specific conformations from the WT structure. The repeating unit of SPOP E47K is also a dimer. Concatenation of the experimental tetramer results in a single helical filament with 660 Å pitch, 380 Å width, and 12 monomers per turn, i.e., similar to the WT filament. The local resolution around mutated residue K47 is limited, making an interpretation of details such as small changes to the hydrogen-bonding network in the interface difficult (Figures S7C, S7E and S7F). Given the lack of overt structural changes, we considered whether the high occurrence of mutations in this newly revealed interface points to alterations of self-association, a typical consequence of disease-associated mutations in interfaces.29
Figure 5. The E47K mutation leaves the structure of SPOP relatively unperturbed.
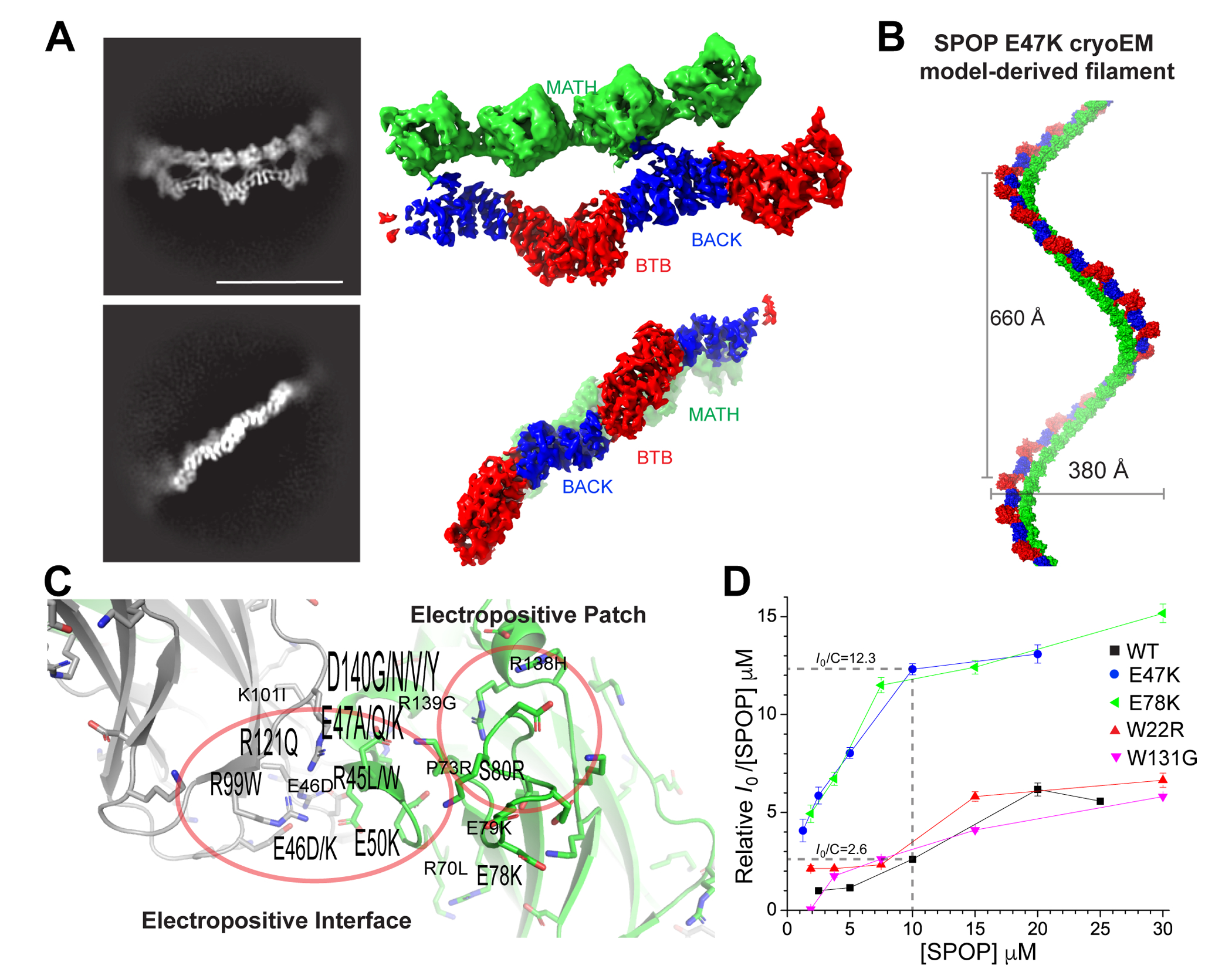
(A) Representative 2D classes (left) and sharpened cryo-EM maps in similar orientation (right) of SPOP E47K oligomers (see also Figure S7).
(B) Left-handed helical filament constructed from the cryo-EM model of SPOP E47K; the helical parameters are similar to the WT filament, and the domains also have highly similar orientations relative to each other.
(C) Ribbon representation of the MATH:MATH interface; monomer 1 is colored grey, monomer 2 green. A cluster of mutations that alter charge are found in the interface. The height of the labels indicates the number of identified mutations at the given position (e.g., K101I was found once, and D140G/N/V/Y was found 8 times).
(D) Self-assembly of MATH:MATH interface mutants is enhanced. Relative molecular mass of WT SPOP and SPOP mutants determined from small-angle X-ray scattering intensity at zero scattering angle (Io). All data are normalized relative to WT SPOP at the lowest concentration. Lines are drawn to connect individual concentration measurements and guide the eye. Error bars indicate the error in P(r)-based I0 determination with Gnom.33 Dashed lines represent the 4.7-fold difference in molecular mass between WT and E47K at 10 μM. (For full SAXS curves see Figure S8.)
We characterized the self-association characteristics of WT and mutants by small-angle X-ray scattering (SAXS), which probes the size and shape of assemblies. The scattering intensity at zero angle is a direct indicator of molecular mass. At equal concentrations, the scattering intensity at zero angle of SPOP E47K was higher than that of the WT (Figure 5D and S8A). We found that this was true over all examined concentrations. We probed the behavior of an additional endometrial mutant from the MATH:MATH interface, E78K, and observed that it also assembled into larger structures than WT at all tested concentrations (Figure 5D), suggesting that stabilization of the filament may be one of the molecular mechanisms underlying the dysfunction of endometrial cancer mutants. Indeed at a concentration of 10 μM, the MATH:MATH mutants of SPOP formed filaments ~five times larger than the wild-type. The size of SPOP W22R assemblies was not enhanced compared to the WT, suggesting that its altered function is the result of its gross structural perturbation. The prostate cancer mutant SPOP W131G had nearly identical self-association behavior as the WT, reflecting that its loss-of-function phenotype is caused by inability to bind substrate, not by changes in self-association. In conclusion, the interrogated endometrial MATH:MATH interface mutants formed structurally similar but larger filaments than WT SPOP.
Interface mutations enhance SPOP activity towards BRD3
Given the importance of SPOP self-association for its function, we next asked whether the enhanced self-association of E47K and E78K increased its ubiquitination activity. In in vitro ubiquitination assays with neddylated CRL3SPOP, we used BRD3 as a substrate because its ubiquitination has been shown to be enhanced by SPOP endometrial cancer mutants.5 We observed robust poly-ubiquitination within tens of minutes (Figure 6A). E47K and E78K resulted in slightly faster appearance of poly-ubiquitinated BRD3, but the differences were small. In contrast, SPOP W22R, the glue pad mutant, had lower intrinsic activity than the WT. The same trends were observed when the CRL3:SPOP molar ratio was less than one (Figure S8B). Given that these results did not recapitulate the previously reported enhanced ubiquitination of certain substrates by SPOP endometrial mutants, we probed their function in an in-cell ubiquitination assay. We used inducible SPOP constructs to limit the expression of SPOP to close to endogenous levels. This strategy in turn also limited the expression of the substrate BRD3. We immuno-precipitated His-tagged ubiquitin and monitored BRD3 in the fraction of ubiquitinated proteins by Western blotting. The SPOP mutants W22R, E47K and E78K resulted in strongly increased levels of poly-ubiquitinated BRD3 (as evidenced by a more pronounced smear to higher molecular weights) as compared to WT SPOP (Figure 6B), in agreement with previous observations that endometrial mutants increase BRD3 ubiquitination.5 The enhanced activity of the mutants therefore was not intrinsic but manifested in cells.
Figure 6. Endometrial cancer mutations enhance protein stability and therefore substrate turnover.
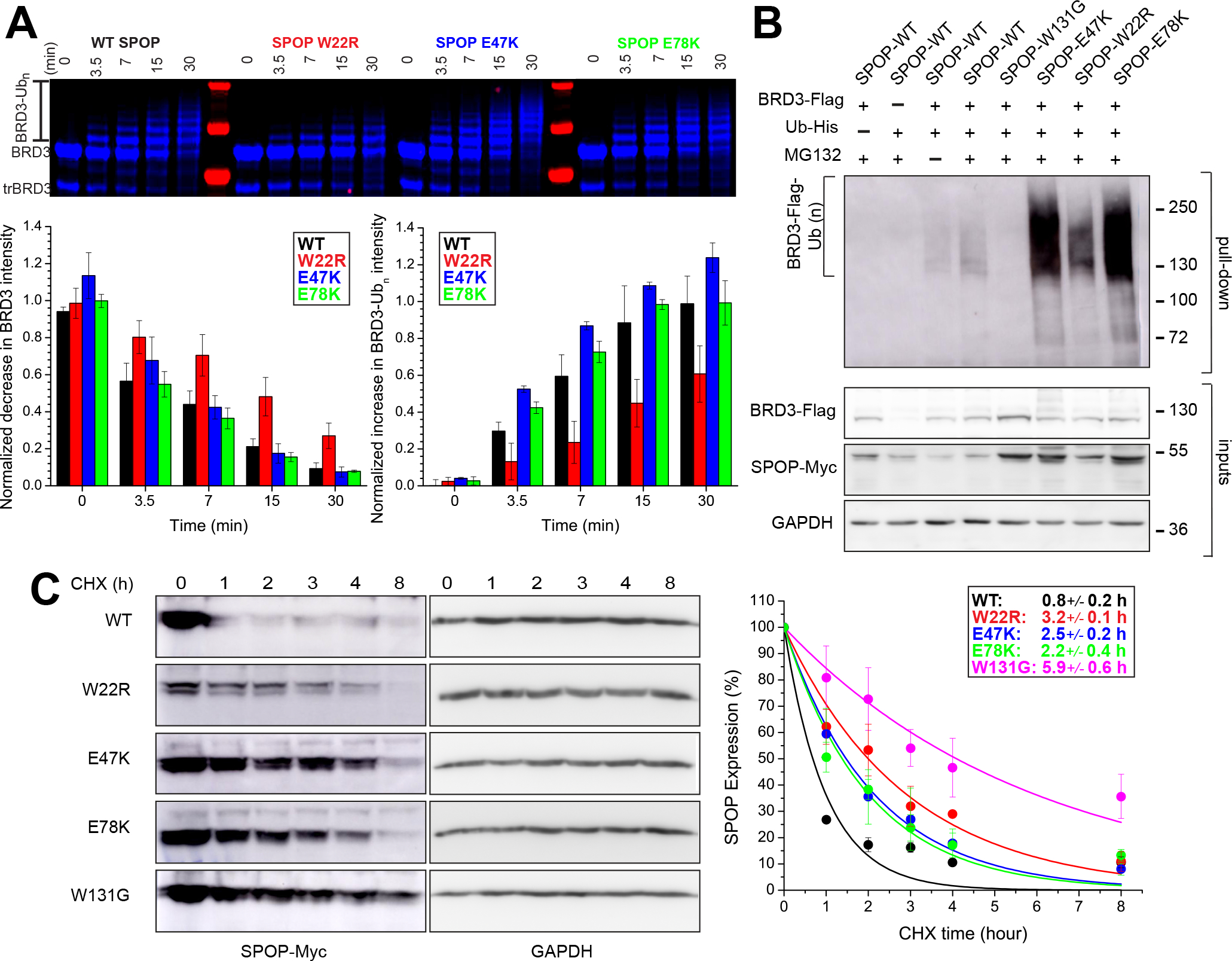
(A) E47K and E78K SPOP marginally increase ubiquitination activity in vitro, W22R does not. In vitro ubiquitination assays with CRL3SPOP were performed as described previously (Zhuang et al, 2009) using fluorescently labeled BRD3 as substrate, and WT SPOP or one of the characterized endometrial cancer mutants W22R, E47K and E78K SPOP. Top, ubiquitination efficiency was monitored by SDS–PAGE and fluorescent imaging. Bottom left and right, quantification of the decrease of unmodified BRD3 and of the increase of ubiquitinated BRD3 (Ubn-BRD3), respectively, as a function of time, in 3 independent assays. The mean value ± the S.D. are reported. (For additional conditions see Figure S8.)
(B) SPOP endometrial cancer mutants enhance BRD3 ubiquitination in cells. Immunoblots showing BRD3-Flag ubiquitination in T-REX-293 cells transfected with the indicated constructs. 24 h post-transfection, cells were incubated with MG132 or DMSO at 20 μM for 4 hours. Cell lysates were subjected to His-Ub pulldown using nickel-NTA beads under denaturing conditions, followed by SDS–PAGE, and immunoblotting with anti-FLAG antibody. Input materials were subjected to immunoblotting using antibodies for Flag, MYC, and GAPDH (loading control).
(C) SPOP endometrial cancer mutants increase the half-life of SPOP as determined by cycloheximide (CHX) chase assay in T-REX-293 cells. Left, a representative immunoblot analysis of CHX-treated T-REX-293 cells. Cells were transfected with the indicated SPOP constructs for 16 h and supplemented with 100 μg/ml of CHX for the indicated time periods. Right, the SPOP-Myc level at each time point relative to the level at time zero is the mean from three biological replicates and fit (solid line) to determine t1/2 values (inset box). Error bars are ± S.D.
This raises the question what the factors are that lead to high activity in cells but are not recapitulated in vitro. We observed that the cellular levels of SPOP mutants were higher than of the WT (Figure 6B), pointing to the possibility that their turnover was decreased rather than their activity enhanced. We thus dampened translation with cycloheximide to probe protein stability. We found that all mutants had longer half-lives than the WT, but to different extents. Levels of the prostate cancer mutant W131G fell most slowly, presumably due to its inactivity and consequential lack of autoregulation. W22R, E47K and E78K were also retained at higher levels than WT, in order of decreasing effect (Figure 6C). In conclusion, our data show that gain of function due to SPOP endometrial cancer mutations is likely the result of two different molecular mechanisms that enhance protein stability without sacrificing activity: Glue pad mutations form structurally altered assemblies, and MATH:MATH interface mutations strengthen self-association. Our structural analysis has thus revealed molecular mechanisms that may underlie hypermorphic mutations of the SPOP tumor suppressor.
Discussion
Here we report the first structure of full-length SPOP oligomers. It recapitulates several features of previous partial structures and the filament model built from them; it is also in agreement with biophysical data that shows SPOP undergoes linear oligomerization. However, it reveals new structural insights that are critical to understanding SPOP function and dysfunction. We observed three new interfaces that facilitate the formation of extended intermonomer β-sheets and lock the MATH domains in place on top of the BTB-BACK filament layer. Notably, mutations found in cancer patients line these new interfaces and point to their functional importance. While most of the mutations are currently classified as variants of unknown significance, we show here for two distinct interfaces that mutations enhance BRD3 ubiquitination. Our results agree with a previous report that shows a hypermorphic character of endometrial cancer mutants towards several substrates including BRD2, BRD3 and BRD4.5 In addition, here we were able to identify two distinct molecular mechanisms underlying this hypermorphic character, exemplified by two types of mutations: (1) Mutation W22R disrupts the glue pad and results in gross structural changes involving rewiring of inter-monomer MATH:MATH interactions. (2) Mutations E47K and E78K are examples of mutations in a charged patch between MATH domains, leading to enhanced oligomer assembly. Both mutations result in enhanced ubiquitination capacity in cells, and our data indicate that this is largely driven by the increased half-lives of the SPOP mutants. By extending our understanding of SPOP oligomer structure, our results provide new insights into the mechanisms that can lead to SPOP gain-of-function mutations; a contrast to the well-understood loss-of-function prostate cancer mutations, which disrupt substrate binding. Previous analyses had proposed that SPOP has characteristics of an oncogene30, and our results reveal the potential underlying molecular mechanisms.
Among the totality of possible mutations, gain-of-function mutations are uncommon. Most single amino acid mutations reduce either protein stability, which results in degradation, or they reduce enzymatic activity or partner binding. This is also the case for SPOP prostate cancer mutations, which weaken substrate binding and ubiquitination. However, it was previously recognized that endometrial cancer mutations lead to gain of function, and we have shown here that two distinct molecular mechanisms enhance the stability of SPOP mutants in the cell. Is this the sole underlying mechanism of gain-of-function? Given the slightly enhanced activity of E47K and E78K in vitro, we propose that intrinsic activity and protein stability synergize in their gain of function. W22R showed a somewhat decreased ubiquitination activity in vitro, but the structures reveal interesting features that may enhance activity towards specific substrates. The substrate binding sites in MATH domain tetramers and hexamers are aligned (Figures 4A, 4E and 7C), which we expect to favor binding of multivalent substrates with spacing of SPOP-binding motifs that match the distances in the assemblies. The occupancy through pseudo-SB motifs from adjacent MATH domains may counteract the stimulating effect somewhat. In contrast, the substrate binding clefts in WT filaments are placed parallel to each other, likely requiring relatively large spacing between SB motifs for optimal multivalent engagement. The two structures may therefore explain why endometrial cancer mutants enhance ubiquitination of some substrates but not others as previously shown.5
Figure 7. SPOP filament structure provides insights into functional CRL assembly.
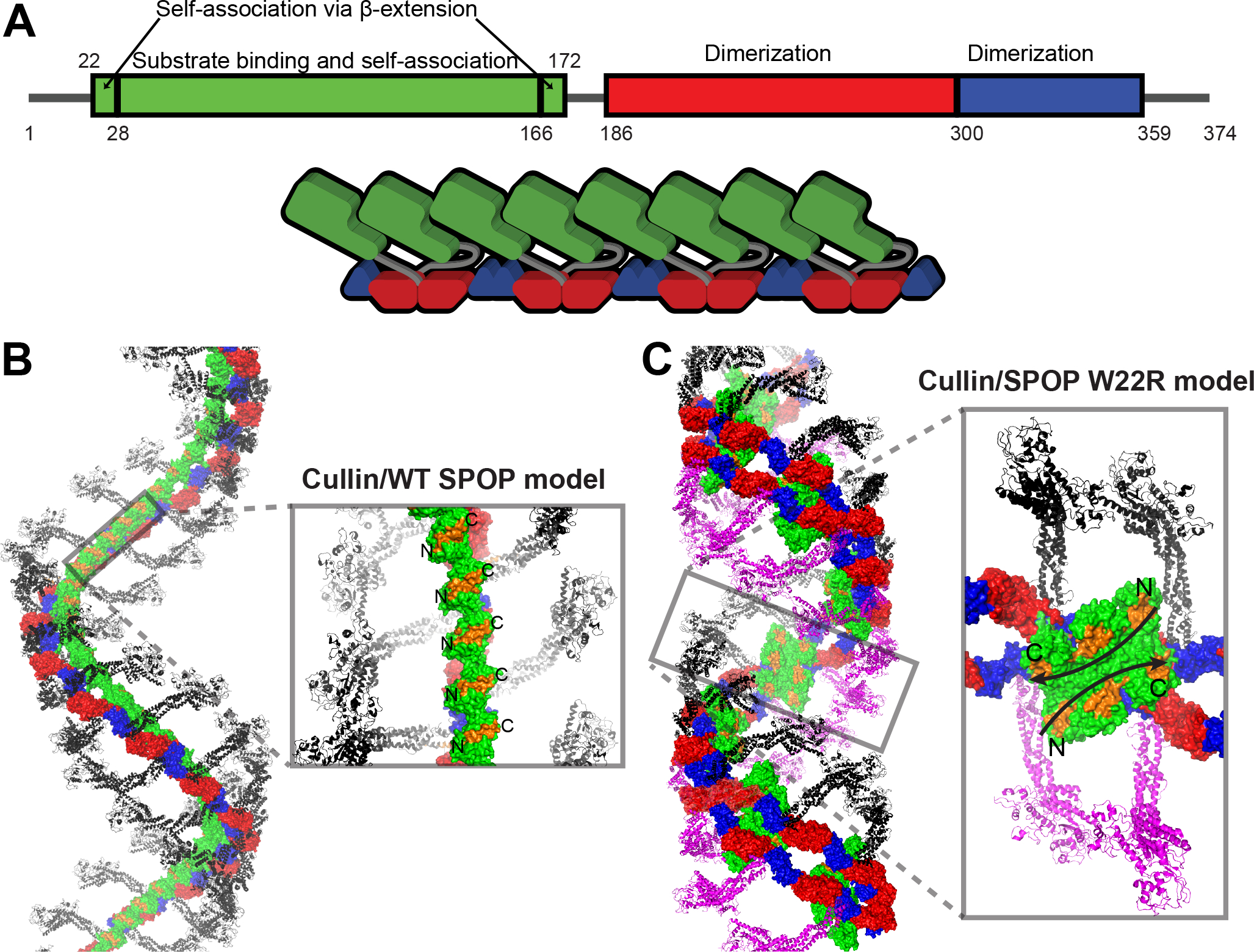
(A) Updated box-plot of SPOP domains (top). Updated schematic of SPOP domain structure and SPOP filament highlighting the head-to-tail assembly of MATH domains and that every second MATH domain is locked into place on the BTB-BACK filament via the glue pad (bottom). As a consequence, alternate linkers have different conformations.
(B) Model of the complete CRL3SPOP filament generated by superposition of X-ray crystal structures onto the cryoEM structure of the SPOP filament. The Cul3 N-terminal domain/SPOP complex (PDB ID 4EOZ) was used to orient the structure of the Cul1-Rbx1-Skp1-F-box-Skp2 SCF ubiquitin ligase complex. Each SPOP monomer can bind one Cullin without steric clashes. The substrate binding sites (orange surfaces) in the MATH domain are in a parallel orientation.
(C) Model of the SPOP W22R filament in complex with Cullin, constructed as in panel (B). Binding of one Cullin per SPOP monomer creates steric clashes (grey inset); each SPOP dimer can bind one Cullin. The central hexameric MATH assembly creates two large anti-parallel continuous substrate binding sites (orange surface with black arrow showing direction of substrate chain), which may favor binding of multivalent substrates with closely spaced SPOP-binding motifs.
The W22R mutation results in the disruption of the glue pad and leads to extensive MATH:MATH interactions not observed in the WT. Interestingly, individual dimers within the hexamer and tetramer assemblies of populations 1 and 2, respectively, resemble the head-to-head assembly of MATH domains in a previously resolved SPOP dimer.15 Of note, this SPOP dimer lacked the N-terminus, BACK domain and C-terminus and was hence neither able to form higher-order oligomers beyond the dimers nor the glue pad. The observed interactions in the dimer thus mirror the interactions of the W22R mutant (Figure S6C).
SPOP mutations in patients are typically heterozygous. Our structures show that some mutants adopt highly similar structures as the WT and can likely co-assemble with WT protein, such as E47K and probably E78K. Mixtures of SPOP WT and W22R may not co-assemble but rather form separate assemblies due to their highly different assembly forms. These differences expected between mutants may provide a possible explanation for a spectrum of phenotypes from dominant-negative to intermediate.
The WT SPOP filament modeled from the cryo-EM structure can accommodate binding of one Cul3 molecule per SPOP monomer (Figures 7A–7B). Such an arrangement likely enables effective poly-ubiquitination assuming that several Cul3 scaffold molecules may be able to contribute ubiquitin molecules to the growing ubiquitin chain with its changing distances for ubiquitin transfer. In contrast, the W22R double filaments can only accommodate one Cullin molecule per SPOP dimer; additional Cullins result in steric clashes (Figure 7C), at least if we assume conformational rigidity. This may explain the slightly lower intrinsic activity of W22R SPOP in vitro, although we found no defect in ubiquitination activity when the ratio of CRL3:SPOP was 0.5 (Figure S8B).
The helical dimensions of the idealized filament models for WT and E47K are somewhat different, but these should not be seen as absolute values given that the curvature of filaments is variable in cryo-EM micrographs. This variability is evidence of dynamic equilibria in the SPOP assemblies and may impact how well the MATH domains are “glued” on the BTB-BACK filament via the glue pad interfaces. Indeed, the lower local resolution of the MATH domains suggests continuous motions of the MATH domains, rather than discrete sub-states. This leads us to wonder whether the MATH domains can dissociate from the filament and explore conformational space via long disordered linkers as previously described for SPOP dimers.15 Such conformational freedom would enable recognition of a greater variety of SPOP substrates with variable SB motif spacing. However, the loss of considerable buried surface area, hydrogen bonding and the required disentanglement of a set of consecutive interfaces suggests that this is thermodynamically unfavorable and may only be relevant for substrates with several high-affinity SB motifs but unfavorable spacing. Such substrates have not been identified to our knowledge.
SPOP differs from other Cul3 substrate receptors in its ability to make higher-order oligomers, and this is important for its activity.17,18 Our work reveals that the extent of oligomerization has a direct and powerful effect on activity and therefore must be under tight control. One factor that controls SPOP oligomerization which has been previously described is the CRL3 substrate receptor SPOP-like (SPOPL). SPOPL can dimerize with SPOP through its BTB domain but has a loop insertion in its BACK domain that prevents BACK-BACK association and therefore caps SPOP oligomer size.18 The SPOP levels in the cell must be another direct factor in determining SPOP oligomer size and must therefore be kept under tight control to prevent activity fluctuations. How SPOP levels are controlled warrants future characterization. The molecular mechanism underlying the enhanced half-live of SPOP endometrial cancer mutants is unclear, but it seems to pertain to their enhanced or altered assembly. We speculate that SPOP turnover occurs from the accessible ends of the SPOP oligomer, in which case the assembly into fewer, longer filaments would reduce turnover. Future investigation of this hypothesis is required.
The detailed structural architecture of the tumor suppressor SPOP we reveal here provides new insight into molecular mechanisms underlying so far unexplained cancer mutations. The structural insights may reveal mutant-specific strategies for interfering with SPOP self-association as a cancer therapeutic and caution at the same time against simplified assumptions regarding expected effects in proteins with dual tumor suppressor/oncogene properties.
Limitations of the Study
Our work identifies enhanced or altered assembly of endometrial SPOP cancer mutants as one molecular mechanism underlying their hypermorphic function towards BRD3. Janouskova et al. have reported such effects of SPOP endometrial cancer mutants towards several substrates including also BRD2, BRD4 and DEK.5 Interestingly, they found that the same mutants had hypomorphic effects towards ubiquitination of several other substrates. The molecular basis for these differences is still unknown and will require further investigation. We suspect that the spacing of SPOP-binding motifs in these substrates may be unfavorable for engaging SPOP.
STAR Methods
RESOURCE AVAILABILITY
Lead contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tanja Mittag (tanja.mittag@stjude.org).
Materials availability:
Plasmids generated in this study have been deposited to Addgene.
Data and code availability:
Cryo-EM structures have been deposited at PDB and EMDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH antibody | Cell Signaling | Cat# 2118; RRID:AB_561053 |
| Mouse monoclonal anti-Flag antibody | Sigma | Cat# F1804; RRID:AB_262044 |
| Mouse monoclonal anti-Flag antibody | Cell Signaling | Cat# 2276; RRID:AB_331783 |
| Bacterial and Virus Strains | ||
| BL21-RIPL cells | BL21-RIPL cells | Cat# 230280 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ubiquitin | Boston Biochem | Cat# U-100H-10M |
| MG132 | Selleck Chemicals | Cat# S2619 |
| cycloheximide | Cell Signaling | Cat# 2112 |
| Effectene Transfection Reagent | Qiagen | Cat# 301425 |
| Imidazole | Sigma | Cat# I2399 |
| Deposited Data | ||
| Cryo-EM structure of wild-type SPOP | This study | PDB: 8DWV; EMDB: EMD-27761 |
| Cryo-EM structure of E47K SPOP | This study | PDB: 8DWS; EMDB: EMD-27758 |
| Cryo-EM structure of W22R SPOP form 1 | This study | PDB: 8DWU; EMDB: EMD-27760 |
| Cryo-EM structure of W22R SPOP form 2 | This study | PDB: 8DWT; EMDB: EMD-265983 |
| Other data | This study | Mendeley: DOI: 10.17632/kn52rzcckn.1 |
| Experimental Models: Cell Lines | ||
| Human: T-REx-293 | Thermo Fisher Scientific | Cat# R71007 |
| Recombinant DNA | ||
| His-SUMO-SPOP1-374 | Marzahn et al.45 | N/A |
| His-SUMO-SPOP1-374 W22R | This study | N/A |
| His-SUMO-SPOP1-374 E47K | This study | N/A |
| His-SUMO-SPOP1-374 E78K | This study | N/A |
| His-BRD3 pET28a | This study | N/A |
| pET-DUET-1-His-Cul3/Rbx1 | Small et al.46 | N/A |
| GST pGEX4T1 GST-ThrombinNEDD8 | Walden et al.47 | N/A |
| pGEX4T1 GST-Thrombin- UBCH5 | Huang et al.48 | N/A |
| pGEX4T1 GST-Thrombin-UBC12 | Duda et al.40 | N/A |
| pcDNA4/TO-SPOP-Myc wild-type | This study | N/A |
| pcDNA4/TO-SPOP-Myc W131G | This study | N/A |
| pcDNA4/TO-SPOP-Myc W22R | This study | N/A |
| pcDNA4/TO-SPOP-Myc E78K | This study | N/A |
| pcDNA4/TO-SPOP-Myc E47K | This study | N/A |
| pcDNA3-Myc-Cul3 | Ohta et al.49 | Addgene #19893 |
| pcDNA3-HA-Rbx1 | Ohta et al.49 | Addgene #19897 |
| Ub-His | Wang et al.50 | |
| BRD3-Flag | Wai et al.51 | |
| His-SUMO-SPOP1-374 W131G | This study | N/A |
| Software and Algorithms | ||
| cryoSPARC | Pujani et al.52 | https://cryosparc.com/ |
| Coot | Emsley et al.53 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Afonine et al.54 | https://www.phenix-online.org |
| MolProbity | Chen et al.55 | http://molprobity.biochem.duke.edu |
| Chimera | Pettersen at al.56 | https://www.cgl.ucsf.edu/chimera |
| PyMOL | Schrodinger | http://www.pymol.org |
| Other | ||
| C-flat Holey Carbon Grid Gold 1.2 μm hole 1.3 μm space 400 mesh | Electron Microscopy Sciences | Cat# CF413-50-Au |
| C-flat Holey Carbon Grid 2.0 μm hole 2.0 μm space 300 Mesh | Electron Microscopy Sciences | Cat# CF-223C-50 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines:
Human T-REX-293 cells (RRID:CVCL_D585, female origin) were obtained from Thermo Fisher and tested negative for mycoplasma contamination using Universal Mycoplasma Detection kit from ATCC.
METHOD DETAILS
Plasmids:
The open reading frame of mouse SPOP (Uniport: Q6ZWS8-1) and its corresponding mutants were cloned into pcDNA4/TO/myc-His vector (Thermo Fisher) to produce C-terminally Myc-tagged proteins. pcDNA3-Myc-Cullin3, and pcDNA3-HA-Rbx1 were obtained from Addgene (#19893 and #19897, respectively).34 Ub-His and BRD3-Flag plasmids were kind gifts from Wenyi Wei (Harvard Medical School) and Joel Mackay (University of Sydney), respectively. The full-length human SPOP gene encoding residues 1-374 (His-SUMO-SPOP1-374) was cloned into a pEXP vector containing an N-terminal hexa-histidine-SUMO tag for bacterial expression and affinity purification. Rolling circle mutagenesis of this vector backbone was used to create all SPOP point mutants used in this study. A synthetic gene for BRD3, codon-optimized for E. coli, was ordered from Genscript in a pET28a vector. A hexa-histidine tag followed by a TEV protease cleavage site was placed at the N-terminus of the open reading frame.
Protein expression and purification:
His-SUMO-SPOP1-374 was transformed into BL21-RIPL cells and expressed in auto-induction media.35 Cells were harvested, lysed, and cell debris was pelleted by centrifugation. The clarified supernatant was applied to a gravity Ni Sepharose resin equilibrated in resuspension buffer (30 mM imidazole, 1 M NaCl, pH 7.8). After washing with wash buffer (75 mM imidazole, 1 M NaCl, pH 7.8), the protein was eluted with a buffer containing 1 M imidazole, 1 M NaCl, pH 7.8. The eluted protein solution was diluted prior to loading onto a Fast Flow Q-column that was equilibrated with 20 mM HEPES pH 7.8, 150 mM NaCl and was subsequently eluted using a step NaCl gradient. TEV protease was added to the eluted protein and the reaction was left at 4 °C overnight. The cleaved protein was then re-purified using a Q-column as above and concentrated to 30–100 μM prior to dialysis in 20 mM HEPES pH 7.5, 400 mM NaCl and 5 mM dithiothreitol (DTT) before flash freezing and storage at −80 °C.
The BRD3 plasmid was transformed into BL21-RIPL cells, cultures were grown in LB medium, and expression initiated by addition of IPTG. Cells were lysed in 30 mM imidazole pH 7.8, 1 M NaCl with a sonicator. The clarified lysate was loaded onto a 5 mL Fast Flow Chelating Sepharose gravity column and washed with the resuspension buffer. Protein was eluted in 300 mM imidazole pH 7.8, 300 mM NaCl and subsequently diluted three-fold before loading onto a HiTrap Heparin column, attached to a HiTrap Q column for the removal of bound nucleic acid. The protein was eluted with a NaCl gradient in 20 mM HEPES pH 7.5 and concentrated to 100 μM. Protein was then dialyzed into 20 mM Tris pH 7.8, 150 mM NaCl, 5 mM DTT and flash frozen and stored at −80 °C.
E2 and E3 were purified as described in the following. UBCH7 was expressed in E. coli BL21 Gold (DE3) cells as a GST-fusion protein and purified as previously described.36,37 First, the protein was purified by glutathione affinity chromatography in 20 mM Tris pH 7.8, 300 mM NaCl, 5 mM DTT, and subsequently subjected to thrombin cleavage. It was then diluted with a low salt buffer and further purified by ion-exchange chromatography, and size exclusion chromatography (SEC). NEDD8 was expressed in E. coli BL21 Gold (DE3) cells as GST-fusion protein, and purified in the same was as UBCH7 by glutathione affinity, thrombin cleavage, glutathione pass-back, and SEC, as previously described.38 Cul3/Rbx1 were co-expressed in E. coli BL21 Gold (DE3) and purified by Ni chromatography in 20 mM Tris pH 7.5, 500 mM NaCl, 2 mM β-mercaptoethanol. The protein complex was subjected to thrombin cleavage and polished by SEC, as previously described.39 All SEC steps above were performed in 20 mM Tris pH 7.8, 150 mM NaCl, 5 mM DTT, and the protein samples concentrated to the intended concentration, flash frozen and stored at −80 °C. Neddylation of 12 μM Cul3/Rbx1 was accomplished by incubating with 1 μM UBC12, 0.1 μM APPBP1-UBA3, and 20 μM NEDD8 with 5 mM ATP and 10 mM MgCl2 for several hours as previously described and monitoring the reaction by SDS-PAGE.40
Cryo-EM grid preparation and data collection:
3 μl of SPOP1-374 (concentration 0.5 – 0.8 mg/ml) was applied to a freshly glow-discharged grid (WT, C-Flat R1.2/1.3-4Au; W22R and E47K, C-Flat R2/2). An FEI Vitrobot plunge freezer, with humidity in the chamber at 100% and temperature at 4 °C, was used for plunge freezing after a 20 s blotting time.
Electron micrographs were recorded on an FEI Titan Krios with a Gatan Summit K3 electron detector (data collection parameters shown in Table 1). 60 frames were collected and aligned with the MotionCorr2 software using a dose filter.
Table 1.
Cryo-EM data collection, refinement and validation.
| WT | W22R Population 1 | W22R Population 2 | E47K | |
|---|---|---|---|---|
| EMDB – 27761 | EMDB – 27760 | EMDB – 27759 | EMDB – 27758 | |
| PDB – 8DWV | PDB – 8DWU | PDB – 8DWT | PDB – 8DWS | |
|
| ||||
| Data collection and processing | ||||
| Magnification | 81,000 | 130,000 | 130,000 | |
| Voltage (kV) | 300 | 300 | 300 | |
| Electron exposure (e−Å2) | 69.6 | 69.2 | 65.0 | |
| Defocus range (μm) | −0.6/−1.8 | −0.6/−1.8 | −0.6/−1.6 | |
| Pixel size (Å) | 1.057 | 0.6485 | 0.6485 | |
| Symmetry imposed | C1 | C1 | C1 | |
| Initial particle images (no.) | 2.3M | 8.3M | 6.5M | |
| Final particle images (no.) | 572K | 164K | 121K | 350K |
| Map resolution (Å) | 3.2 | 3.4 | 6.0 | 3.6 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 3–6 | 3–6 | 6–9 | 3–6 |
|
| ||||
| Refinement | ||||
| Initial model used (PDB code) | manually built | manually built | manually built | manually built |
| Model resolution (Å) | ||||
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 |
| Model resolution Range (Å) | 3–6 | 3–6 | 6–9 | 3–6 |
| Map sharpening B factor (Å2) | −164.6 | −102.4 | −501.5 | −148.0 |
| Model composition | ||||
| Non-hydrogen atoms | 16480 | 21100 | 32810 | 17229 |
| Protein residues | 2084 | 2668 | 4176 | 2182 |
| B factors (Å2) | 51.2 | 41.0 | 102.6 | 41.0 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.004 | 0.005 | 0.005 | 0.004 |
| Bond lengths (°) | 0.745 | 1.258 | 0.880 | 0.805 |
| Validation | ||||
| MolProbity score | 1.9 | 1.76 | 1.91 | 1.88 |
| Clashscore | 6.82 | 5.35 | 6.88 | 6.75 |
| Poor rotamers (%) | 0 | 0.05 | 0.03 | 0.3 |
| Ramachandran plot | ||||
| Favored (%) | 90.5 | 92.3 | 90.7 | 91.2 |
| Allowed (%) | 8.9 | 7.2 | 8.8 | 8.2 |
| Disallowed (%) | 0.6 | 0.5 | 0.5 | 0.6 |
An initial subset of frames, typically 500–2000 movies, were used for blob particle picking and 2D-classification in CryoSPARC.41 Given that the curvature of the filaments was variable, we did not enforce helical symmetry along the filament but instead optimized the box size that captured the largest regular unit of a continuous filament (also used by 25). The resulting 2D classes were filtered and used to generate maps, the classes from which were used as templates for a second round of picking using the entire dataset. The particles used for reconstruction lie, as expected along filaments (see Figure S2). Particles selected for W22R via blob picking were not improved with template picking, so blob picking was used on the entire dataset. Neural network picking also did not improve particle picking. Subsequent 2D, 3D, and non-uniform refinement and polishing steps led to the generation of the final processed maps (Figure S5).
Molecular models were built using Coot, where domain fragments from previous crystal structures (MATH and BTB domains, PDB ID: 3HQI; BACK domain, PDB ID: 4HS2) and manual building of previously un-modeled regions were used to construct a full-length SPOP model. The full-length molecular model was then superimposed onto maps using Chimera, rebuilt where necessary in Coot, and then subsequently refined in Phenix (Table 1).42 All images of molecular models and maps were generated with either Pymol or ChimeraX.43
Small-angle X-ray scattering:
Small-angle X-ray scattering (SAXS) experiments were performed at the LIX-beamline (16-ID) of the National Synchrotron Light Source II (Upton, NY).44 Data were collected at a wavelength of 1.0 Å, yielding an accessible scattering angle range of 0.006 < q < 3.2 Å−1, where q is the momentum transfer, defined as q = 4π sin(θ)/λ, where λ is the X-ray wavelength and 2θ is the scattering angle. Data with q < 0.2 Å−1 were used for all analyses. Prior to data collection, wild-type SPOP1-374 and mutants were dialyzed into 20 mM HEPES pH 7.5, 400 mM NaCl and 5 mM DTT. Samples were loaded into a 1 mm capillary for ten 1 s X-ray exposures. Data were reduced at the beamline using the Python package py4xs and intensity at zero scattering angle (Io) values were determined using GNOM.33
Cell lines, cell culture, transfection, and treatments
Cells were grown at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% GlutaMAX, and 100 U/mL Penicillin-Streptomycin (Thermo Fisher). Transfections were carried out using the Effectene Transfection Reagent from Qiagen. Tetracycline (Sigma) was added to the culture media to induce the expression of pcDNA4/TO/myc-SPOP. MG132 and cycloheximide (CHX) were purchased from Selleck Chemicals and Cell Signaling, respectively.
Immunoblotting
Immunoblotting assays were conducted using primary antibodies for Flag (Santa Cruz), Myc (Cell Signaling), and GAPDH (Cell Signaling). The ECL and ECL Select reagents (GE Healthcare) were used to develop immunoblots. Images were quantified with the ImageJ software (National Institutes of Health).
Cycloheximide assays
16 hr after transfection, T-REX-293 cells were treated with 100 μg/ml of cycloheximide for the indicated time points. Collected cells were lysed directly in the SDS loading dye and analyzed by Western blotting. SPOP levels were normalized relative to GAPDH levels.
In-cell ubiquitination assays
T-REX-293 cells were transfected with 400 ng of plasmids for SPOP-Myc, Ub-His, Myc-Cullin3, HA-Rbx1, and Flag-tagged BRD3. Tetracycline was added to the culture media to induce SPOP-Myc expression. At 24 hr after transfection, MG132 was added to the cells at a final concentration of 20 μM for another 4–5 hr. Cells were lysed in buffer A (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4 pH 8.0, 10 mM imidazole). The lysates were sonicated, cleared, and incubated with Ni-NTA agarose (Qiagen) for His-tag pull down. The beads were washed twice with buffer A, twice with A/T buffer composed of one volume of buffer A and three volumes of buffer T (25 mM Tris pH 8.0, 20 mM imidazole), and twice with buffer T. Beads were incubated in SDS-PAGE loading dye containing 300 mM imidazole for 15 min and boiled for 5 min to elute protein.
In vitro ubiquitination assays
BRD3 ubiquitination was carried out in 50 mM Tris (pH 7.5), 250 mM NaCl, 10 mM MgCl2, 5 mM ATP and 1 mM DTT at room temperature at time points from 0 to 30 min. The reaction mixture contained ubiquitinating enzymes at final concentrations of 0.25 μM UBA1 (E1), 8 μM UbcH5B (E2), NEDD8-CUL3-Rbx1 (at 1 or 5 μM) (E3), SPOP (2.0 μM), 75 μM ubiquitin (BostonBiochem) and 10 μM BRD3. The reactions were quenched by addition of SDS-PAGE loading buffer with 4 M urea, loaded on SDS-PAGE gels for visualization and visualized on a Amershaem I600 imager (GE Healthcare). ImageJ was used for determination of band intensities.
QUANTIFICATION AND STATISTICAL ANALYSIS
The in vitro and in-cell ubiquitination assays and cycloheximide assays were performed three times as independent biological replicates. The immunoblots and gel images show representative results. The quantitative analyses report the mean ± S.D. The error bars on the SAXS data represent the S.D. from reduction of 2D to 1D data.
Supplementary Material
Highlights.
The structure of the oligomeric tumor suppressor SPOP reveals three new interfaces.
These interfaces are populated with previously not understood cancer mutations.
One mutation promotes changes to the quaternary structure.
Other interface mutants promote assembly and increase SPOP half-life in cells.
Acknowledgements
We thank Israel Fernandez for his guidance in cryo-EM data analysis, Shaker Krit for IT support for the cryo-EM facility at SJCRH, Zhaowen “Norman” Luo for help with figures, and Richard Kriwacki and members of the Mittag lab for fruitful discussions. T.M. acknowledges funding by NIH grant R01GM112846, and by the American Lebanese Syrian Associated Charities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The LiX beamline is part of the Center for BioMolecular Structure (CBMS), which is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). LiX also received additional support from NIH Grant S10 OD012331. As part of NSLS-II, a national user facility at Brookhaven National Laboratory, work performed at the CBMS is supported in part by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Program under contract number DE-SC0012704.
Footnotes
Declaration of Interests Statement
T.M. was a consultant for Faze Medicines, Inc. and is a member of the journal’s advisory board.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
VIDEOS
Figure3_360.mp4; related to Figure 3: SPOP cancer mutations line interfaces revealed by the SPOP cryo-EM structure
Figure4_360.mp4; related to Figure 4: Architecture of the SPOP W22R filament
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baek K, Scott DC, and Schulman BA (2021). NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr Opin Struct Biol 67, 101–109. 10.1016/j.sbi.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao K, Jin X, Tang Y, Ma J, Peng J, Yu L, Zhang P, and Wang C (2015). Tumor suppressor SPOP mediates the proteasomal degradation of progesterone receptors (PRs) in breast cancer cells. Am J Cancer Res 5, 3210–3220. [PMC free article] [PubMed] [Google Scholar]
- 3.Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J, Foley C, Dong B, Zhang L, Kwon OJ, et al. (2017). SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 36, 4767–4777. 10.1038/onc.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. (2013). Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 110, 6997–7002. 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al. (2017). Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 23, 1046–1054. 10.1038/nm.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, et al. (2014). Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 346, 85–89. 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Ma J, Botuyan MV, Cui G, Yan Y, Ding D, Zhou Y, Krueger EW, Pei J, Wu X, et al. (2021). ATM-phosphorylated SPOP contributes to 53BP1 exclusion from chromatin during DNA replication. Sci Adv 7. 10.1126/sciadv.abd9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, et al. (2017). Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med 23, 1055–1062. 10.1038/nm.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 23, 1063–1071. 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An J, Wang C, Deng Y, Yu L, and Huang H (2014). Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 6, 657–669. 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, et al. (2014). Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 74, 5631–5643. 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark A, and Burleson M (2020). SPOP and cancer: a systematic review. Am J Cancer Res 10, 704–726. [PMC free article] [PubMed] [Google Scholar]
- 13.Song Y, Xu Y, Pan C, Yan L, Wang ZW, and Zhu X (2020). The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer 19, 2. 10.1186/s12943-019-1124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. (2009). Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 36, 39–50. 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Geersdaele LK, Stead MA, Harrison CM, Carr SB, Close HJ, Rosbrook GO, Connell SD, and Wright SC (2013). Structural basis of high-order oligomerization of the cullin-3 adaptor SPOP. Acta Crystallogr D Biol Crystallogr 69, 1677–1684. 10.1107/S0907444913012687. [DOI] [PubMed] [Google Scholar]
- 17.Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H, Ben-Nissan G, Kolaitis RM, Peters JL, Pounds S, et al. (2016). Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J 35, 1254–1275. 10.15252/embj.201593169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Errington WJ, Khan MQ, Bueler SA, Rubinstein JL, Chakrabartty A, and Prive GG (2012). Adaptor protein self-assembly drives the control of a cullin-RING ubiquitin ligase. Structure 20, 1141–1153. 10.1016/j.str.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Pierce WK, Grace CR, Lee J, Nourse A, Marzahn MR, Watson ER, High AA, Peng J, Schulman BA, and Mittag T (2016). Multiple Weak Linear Motifs Enhance Recruitment and Processivity in SPOP-Mediated Substrate Ubiquitination. J Mol Biol 428, 1256–1271. 10.1016/j.jmb.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuneo MJ, and Mittag T (2019). The ubiquitin ligase adaptor SPOP in cancer. FEBS J 286, 3946–3958. 10.1111/febs.15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, and Jiang J (2009). Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci U S A 106, 21191–21196. 10.1073/pnas.0912008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. (2012). Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44, 685–689. 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et al. (2012). Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 44, 1310–1315. 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostertag MS, Hutwelker W, Plettenburg O, Sattler M, and Popowicz GM (2019). Structural Insights into BET Client Recognition of Endometrial and Prostate Cancer-Associated SPOP Mutants. J Mol Biol 431, 2213–2221. 10.1016/j.jmb.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Hansen JM, Horowitz A, Lynch EM, Farrell DP, Quispe J, DiMaio F, and Kollman JM (2021). Cryo-EM structures of CTP synthase filaments reveal mechanism of pH-sensitive assembly during budding yeast starvation. Elife 10. 10.7554/eLife.73368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson JS, and Richardson DC (2002). Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A 99, 2754–2759. 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, et al. (2018). Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol Cell 72, 19–36 e18. 10.1016/j.molcel.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livesey BJ, and Marsh JA (2022). The properties of human disease mutations at protein interfaces. PLoS Comput Biol 18, e1009858. 10.1371/journal.pcbi.1009858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerasimavicius L, Livesey BJ, and Marsh JA (2022). Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure: implications for variant effect prediction. bioRxiv, 2021.2010.2023.465554. 10.1101/2021.10.23.465554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svergun D (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of applied crystallography 25, 495–503. [Google Scholar]
- 34.Ohta T, Michel JJ, and Xiong Y (1999). Association with cullin partners protects ROC proteins from proteasome-dependent degradation. Oncogene 18, 6758–6766. [DOI] [PubMed] [Google Scholar]
- 35.Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 36.Huang DT, Zhuang M, Ayrault O, and Schulman BA (2008). Identification of conjugation specificity determinants unmasks vestigial preference for ubiquitin within the NEDD8 E2. Nat Struct Mol Biol 15, 280–287. 10.1038/nsmb.1387. [DOI] [PubMed] [Google Scholar]
- 37.Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA, de Jong A, Ovaa H, Alpi AF, Harper JW, and Schulman BA (2016). Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell 166, 1198–1214 e1124. 10.1016/j.cell.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, and Schulman BA (2014). Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell 157, 1671–1684. 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small E, Eggler A, and Mesecar AD (2010). Development of an efficient E. coli expression and purification system for a catalytically active, human Cullin3-RINGBox1 protein complex and elucidation of its quaternary structure with Keap1. Biochem Biophys Res Commun 400, 471–475. 10.1016/j.bbrc.2010.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, and Schulman BA (2008). Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006. 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 42.Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, and McCoy AJ (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D: Structural Biology 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiFabio J, Chodankar S, Pjerov S, Jakoncic J, Lucas M, Krywka C, Graziano V, and Yang L (2016). The life science x-ray scattering beamline at NSLS-II. In 1. (AIP Publishing LLC; ), pp. 030049. [Google Scholar]
- 45.Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H, Ben-Nissan G, Kolaitis RM, Peters JL, and Pounds S (2016). Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. The EMBO journal 35, 1254–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Small E, Eggler A, and Mesecar AD (2010). Development of an efficient E. coli expression and purification system for a catalytically active, human Cullin3–RINGBox1 protein complex and elucidation of its quaternary structure with Keap1. Biochemical and biophysical research communications 400, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walden H, Podgorski MS, and Schulman BA (2003). Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature 422, 330–334. [DOI] [PubMed] [Google Scholar]
- 48.Huang DT, Zhuang M, Ayrault O, and Schulman BA (2008). Identification of conjugation specificity determinants unmasks vestigial preference for ubiquitin within the NEDD8 E2. Nature structural & molecular biology 15, 280–287. [DOI] [PubMed] [Google Scholar]
- 49.Ohta T, Michel JJ, Schottelius AJ, and Xiong Y (1999). ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Molecular cell 3, 535–541. [DOI] [PubMed] [Google Scholar]
- 50.Wang B, Jie Z, Joo D, Ordureau A, Liu P, Gan W, Guo J, Zhang J, North BJ, and Dai X (2017). TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 545, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wai DC, Szyszka TN, Campbell AE, Kwong C, Wilkinson-White LE, Silva AP, Low JK, Kwan AH, Gamsjaeger R, and Chalmers JD (2018). The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators. Journal of Biological Chemistry 293, 7160–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallographica Section D: Biological Crystallography 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallographica Section D: Structural Biology 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM structures have been deposited at PDB and EMDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH antibody | Cell Signaling | Cat# 2118; RRID:AB_561053 |
| Mouse monoclonal anti-Flag antibody | Sigma | Cat# F1804; RRID:AB_262044 |
| Mouse monoclonal anti-Flag antibody | Cell Signaling | Cat# 2276; RRID:AB_331783 |
| Bacterial and Virus Strains | ||
| BL21-RIPL cells | BL21-RIPL cells | Cat# 230280 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ubiquitin | Boston Biochem | Cat# U-100H-10M |
| MG132 | Selleck Chemicals | Cat# S2619 |
| cycloheximide | Cell Signaling | Cat# 2112 |
| Effectene Transfection Reagent | Qiagen | Cat# 301425 |
| Imidazole | Sigma | Cat# I2399 |
| Deposited Data | ||
| Cryo-EM structure of wild-type SPOP | This study | PDB: 8DWV; EMDB: EMD-27761 |
| Cryo-EM structure of E47K SPOP | This study | PDB: 8DWS; EMDB: EMD-27758 |
| Cryo-EM structure of W22R SPOP form 1 | This study | PDB: 8DWU; EMDB: EMD-27760 |
| Cryo-EM structure of W22R SPOP form 2 | This study | PDB: 8DWT; EMDB: EMD-265983 |
| Other data | This study | Mendeley: DOI: 10.17632/kn52rzcckn.1 |
| Experimental Models: Cell Lines | ||
| Human: T-REx-293 | Thermo Fisher Scientific | Cat# R71007 |
| Recombinant DNA | ||
| His-SUMO-SPOP1-374 | Marzahn et al.45 | N/A |
| His-SUMO-SPOP1-374 W22R | This study | N/A |
| His-SUMO-SPOP1-374 E47K | This study | N/A |
| His-SUMO-SPOP1-374 E78K | This study | N/A |
| His-BRD3 pET28a | This study | N/A |
| pET-DUET-1-His-Cul3/Rbx1 | Small et al.46 | N/A |
| GST pGEX4T1 GST-ThrombinNEDD8 | Walden et al.47 | N/A |
| pGEX4T1 GST-Thrombin- UBCH5 | Huang et al.48 | N/A |
| pGEX4T1 GST-Thrombin-UBC12 | Duda et al.40 | N/A |
| pcDNA4/TO-SPOP-Myc wild-type | This study | N/A |
| pcDNA4/TO-SPOP-Myc W131G | This study | N/A |
| pcDNA4/TO-SPOP-Myc W22R | This study | N/A |
| pcDNA4/TO-SPOP-Myc E78K | This study | N/A |
| pcDNA4/TO-SPOP-Myc E47K | This study | N/A |
| pcDNA3-Myc-Cul3 | Ohta et al.49 | Addgene #19893 |
| pcDNA3-HA-Rbx1 | Ohta et al.49 | Addgene #19897 |
| Ub-His | Wang et al.50 | |
| BRD3-Flag | Wai et al.51 | |
| His-SUMO-SPOP1-374 W131G | This study | N/A |
| Software and Algorithms | ||
| cryoSPARC | Pujani et al.52 | https://cryosparc.com/ |
| Coot | Emsley et al.53 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Afonine et al.54 | https://www.phenix-online.org |
| MolProbity | Chen et al.55 | http://molprobity.biochem.duke.edu |
| Chimera | Pettersen at al.56 | https://www.cgl.ucsf.edu/chimera |
| PyMOL | Schrodinger | http://www.pymol.org |
| Other | ||
| C-flat Holey Carbon Grid Gold 1.2 μm hole 1.3 μm space 400 mesh | Electron Microscopy Sciences | Cat# CF413-50-Au |
| C-flat Holey Carbon Grid 2.0 μm hole 2.0 μm space 300 Mesh | Electron Microscopy Sciences | Cat# CF-223C-50 |