

Abstract
Enzyme function requires conformational changes to achieve substrate binding, domain rearrangements, and interactions with partner proteins, but these movements are difficult to observe. Small-angle X-ray scattering (SAXS) is a versatile structural technique that can probe such conformational changes under solution conditions that are physiologically relevant. Although it is generally considered a low-resolution structural technique, when used to study conformational changes as a function of time, ligand binding, or protein interactions, SAXS can provide rich insight into enzyme behavior, including subtle domain movements. In this perspective, we highlight recent uses of SAXS in to probe structural enzyme changes upon ligand and partner-protein binding and discuss tools for signal deconvolution of complex protein solutions.
Graphical Abstract
Introduction to SAXS
In a biological SAXS experiment, a monochromatic collimated X-ray beam is incident upon a solution sample, and the scattering is measured by a downstream detector (Figure 1A).[1] A solution of randomly oriented protein produces scattering that is symmetric about the beam center. The 2D image is therefore integrated azimuthally to obtain a 1D profile. Once background scattering is subtracted, we obtain the scattering profile, I(q) vs. q, where I is the net intensity, and q is the momentum transfer variable, defined as q = (4π sin θ)/λ. Here, 2θ is the scattering angle, λ is the incident X-ray wavelength, and the unit of q is Å−1.
Figure 1. SAXS overview.
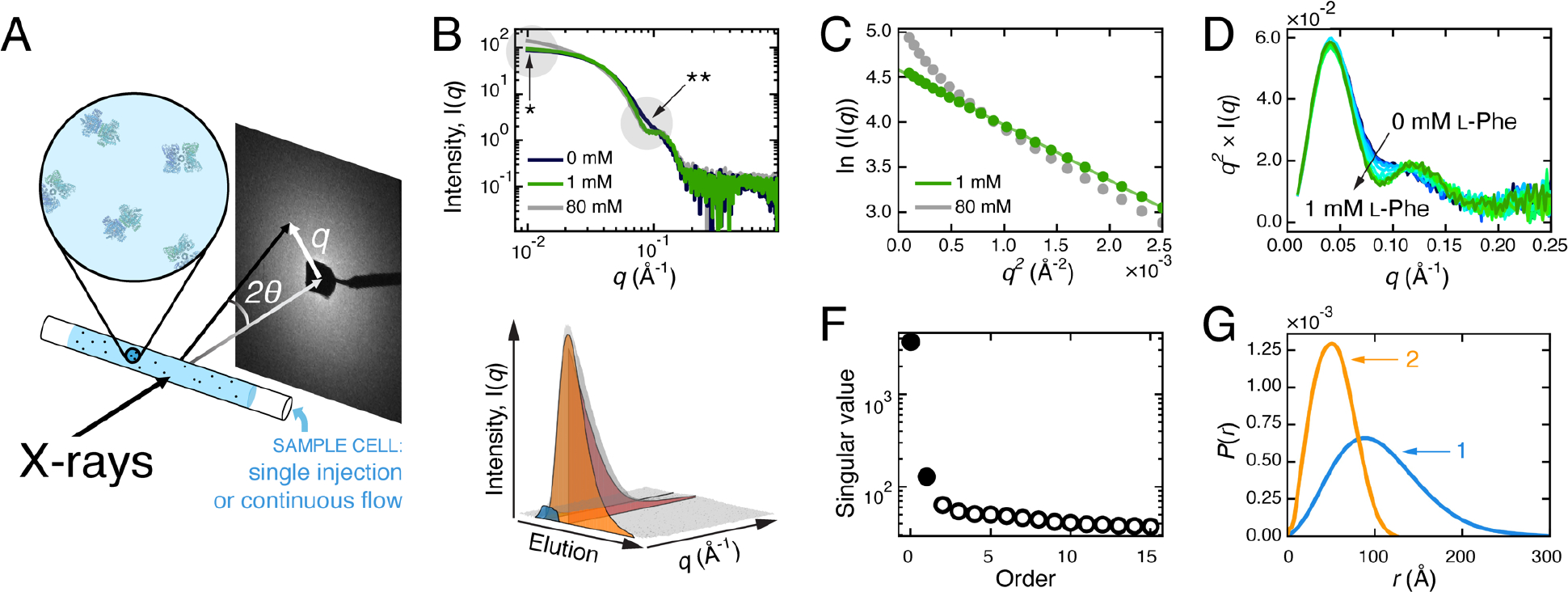
A) SAXS experimental set-up. B-G) SAXS analysis example illustrating the effect of L-phenylalanine (l-Phe) on mammalian phenylalanine hydroxylase (PheH) [4]. B) Scattering profiles of PheH with different L-Phe concentrations (0 mM, dark blue; 1 mM, green; 80 mM, grey). Changes in the low- (*) and mid-q regions (**) can be further examined by Guinier and Kratky analyses. C) The Guinier plot of PheH at 1 mM L-Phe (green) is linear, indicating that the Rg is well-defined, while at 80 mM L-Phe (grey), the upturn at low q indicates aggregation. D) The Kratky plot of PheH at several concentrations of L-Phe from 0 mM (dark blue) to 1 mM (green) clearly shows changes in mid-q region. E) SEC-SAXS dataset collected on ligand-free PheH (grey surface) shows elution and scattering angle on two separate axes. To recover the scattering and elution profiles of the two over-lapping species (blue and orange surfaces), EFA or REGALS can be used. F) Prior to deconvolution, SVD analysis of the SEC-SAXS dataset (grey surface in panel E) revealed two significant components in the dataset (filled circles). There is a third significant value in this dataset because of a buffer component that eluted after the protein (not shown in panel E). G) P(r) plot shows how the shape and Dmax of the two conformationally distinct species (labeled 1, 2) differ.
Because of the reciprocal relationship between q and length scale, the low-q region of a scattering profile reports on the overall size and shape of a protein, while higher-q regions report on smaller structural features (Figure 1B). Guinier analysis (Figure 1C) utilizes the low-q region to produce I(0), the forward scattering (a function of molecular weight), and Rg, the radius of gyration (a function of spatial size), while Kratky analysis (Figure 1D) highlights structural features observed in the mid-q range, such as domain arrangements. For monodisperse samples, SAXS data can also be assessed in real space using the pair-distance distribution function, P(r), which is obtained by an indirect Fourier transform of I(q) (Figure 1G). P(r) can be understood as a histogram of all electron pair distances, r, within a protein, and therefore provides an estimate of maximum dimension, Dmax, and another means to calculate I(0) and Rg. Further details on these basic analysis methods can be found elsewhere [1,2].
A useful feature of SAXS is that the signal is roughly additive at protein concentrations typically used, and therefore, the scattering from a mixture can be treated as a linear sum of the individual contributions. When the structures of the individual components are known, the sum of their theoretical scattering can be fit to SAXS data to estimate the concentration of each species [3]. However, in recent years, model-independent methods have appeared, in part due to the introduction of size-exclusion chromatography-coupled SAXS (SEC-SAXS). Because SAXS is extremely sensitive to aggregation, mismatched buffer, and other issues with sample quality, SEC-SAXS is now a common mode of data collection at most beamlines. Scattering is measured from samples immediately as they elute from a column, resulting in a 3D dataset with scattering intensity as a function of both q and elution volume (Figure 1E). If there is no change in the scaled scattering across multiple frames, the profiles can be averaged to increase signal-to-noise. However, in practice, chromatography is often unable to adequately separate protein mixtures. Protein complexes with relatively high dissociation constants or a species that is interconverting rapidly with another species (e.g., a dynamic monomer-dimer equilibrium), may not elute as sharp, isolated peaks. Singular value decomposition (SVD) of SEC-SAXS data often reveals that there are multiple species even within single peaks (Figure 1F).
Evolving Factor Analysis (EFA) was developed to utilize iterative SVD to recover scattering profiles and elution peaks of overlapping species from SEC-SAXS data in a model-independent way (Figure 1E,F,G) [4]. Recently a more general method, known as regularized alternating least-squares (REGALS), was developed for deconvoluting complex SAXS data without imposing prior structural or physiochemical knowledge [5]. These deconvolution methods can be applied to datasets that capture changes in populations, and thus SAXS is especially powerful for the study of conformational changes.
To take full advantages of SAXS, it is also important to be aware of the limitations. SAXS data have very subtle features, and distinct structures may produce nearly indistinguishable scattering profiles.[6] Moreover, SVD can only discriminate species with scattering profiles that are distinguishable above the noise. Having biochemical knowledge of the sample is therefore advantageous for designing SAXS experiments that yield interpretable results. Experimental considerations include maintaining structural stability (e.g., samples can dilute by an order of magnitude in SEC) under solution conditions that are biochemically relevant. It is also important to note that techniques like EFA or REGALS should not be used in place of preparing the highest quality samples possible. In ideal cases, data decomposition with EFA and REGALS leads to stable solutions where the species of interest are dominant, and the extracted scattering profiles do not significantly change over a wide range of parameters. Confidence in data interpretation is demonstrated by consistency among the various types of SAXS analyses as well as consistency with other biochemical and biophysical data.
Using SAXS to Assess Compaction and Changes in Flexibility
Changes in the extendedness or flexibility of a protein can be readily assessed by SAXS [7–14]. Below, we highlight two case studies [15,16]. In both, Kratky and P(r) analyses were used to probe domain movements in response to allosteric activation, and SEC-SAXS was used to obtain the highest quality data possible, which is necessary to make robust conclusions about subtle changes.
Allosteric enzymes play an important role in controlling the flux of biosynthetic pathways. In the shikimate pathway, there are two major points for controlling the biosynthesis of aromatic amino acids: the entry point and branch point. The pathway begins with the condensation of phosphoenolpyruvate (PEP) and erythrose 4-phosphate (E4P) and ends with the production of chorismate. The enzyme 3-deoxy-D-arabino heptulosonate-7-phosphate synthase (DAH7PS) performs the first step, while chorismate mutase (CM) serves as the branch point for the synthesis of Tyr and Phe by converting chorismate to prephenate. In Prevotella nigrescens, CM is fused to the C-terminus of DAH7PS to form one bienzyme DAH7PS-CM. Bai and Parker use SAXS to examine an unusual form of allostery where the product of CM acts as a feedback inhibitor for DAH7PS, and the substrates of DAH7PS upregulate the activity of CM [15]. Scattering data were collected with an SEC-SAXS setup at the Australian Synchrotron (SAXS/WAXS beamline) with various ligand combinations. Addition of DAH7PS substrates led to a reduction in Rg of the bienzyme concomitant with a sharpening of the Kratky peak (Figure 2A) and a flattening of the plateau region in the Porod-Debye plot (Figure 2B) – all consistent with compaction of the bienzyme. In contrast, in the presence of prephenate, the bienzyme adopts a compact form with or without DAH7PS substrates (Figure 2C,D). These results suggested that when prephenate levels are low, the DAH7PS substrates activate CM by inducing a compact conformation (Figure 2E). By upregulating CM, prephenate would begin to accumulate, and with the bienzyme already in a compact configuration, only a subtle conformational change would be needed to occlude the active sites of DAH7PS and in turn, shut down the pathway.
Figure 2. Pn DAH7PS-CM enzyme compaction in response to substrate and product binding.
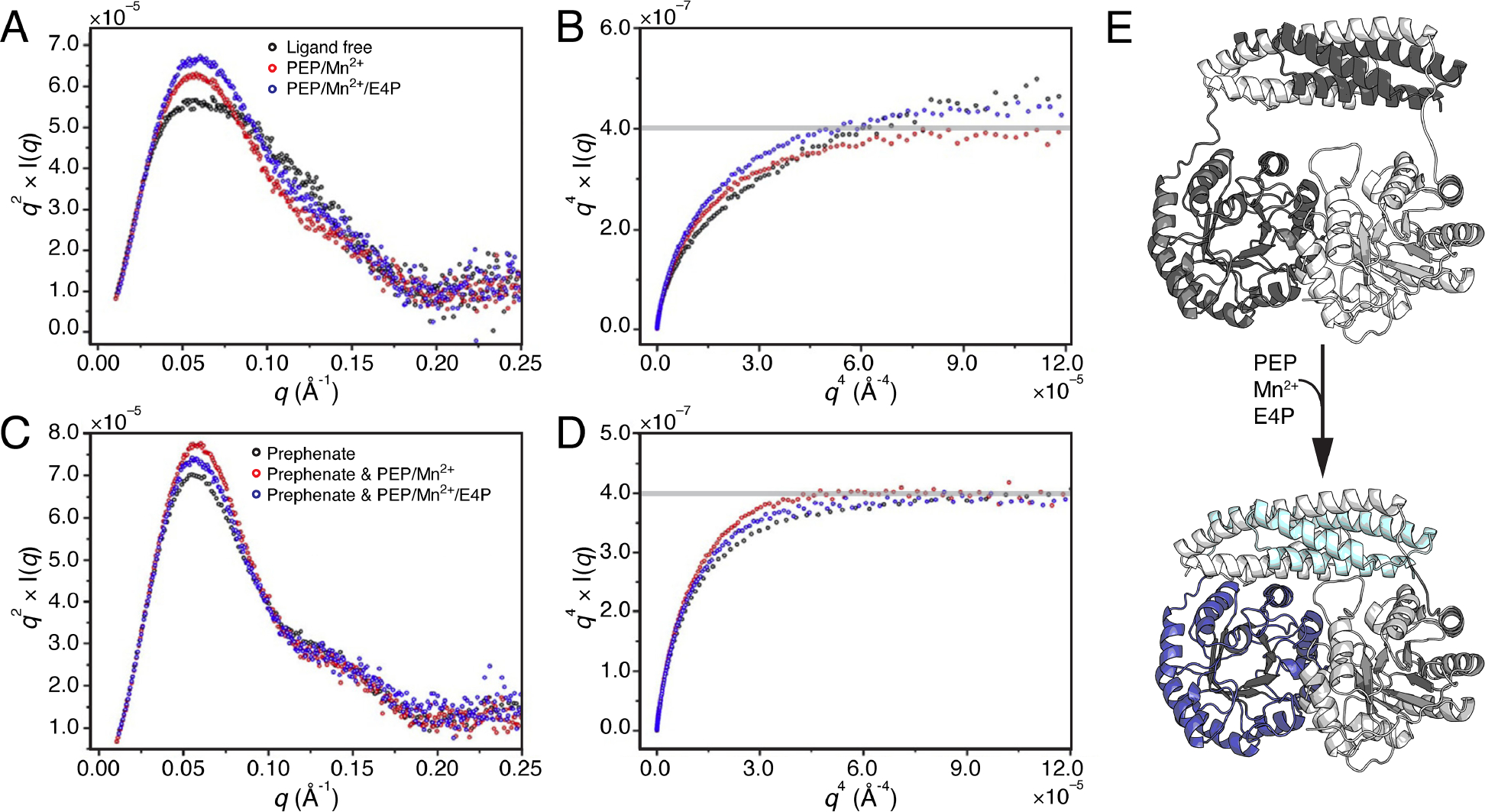
Top panels compare Pn DAH7PS-CM SAXS analysis for ligand-free state (black) with addition of DAH7PS substrates/cofactor, PEP/Mn2+ (red) or PEP/Mn2+/E4P (blue), via A) Kratky and B) Porod-Debye plots. The bottom panels demonstrate lack of change when CM product prephenate is bound alone (black), in the presence of PEP/Mn2+ (red), or PEP/Mn2+/E4P (blue), via C) Kratky and D) Porod-Debye plots. Panels A–D: adapted under permission (copyright Elsevier, 2021).[15] E) AlphaFold2 models of Pn DAH7PS-CM illustrating compaction expected upon DAH7PS substrate binding (not fit to original data).
Similar SAXS analyses were used by Sieng and co-workers to study allosteric activation of a phospholipase [16]. Phospholipase Cε (PLCε) hydrolyzes lipids from the cellular membrane to produce signaling molecules, and the enzyme is activated by Rap1A, a small Ras-GTPase. From a structural standpoint, PLCε is interesting as it consists of several domains: an N-terminal guanine-exchange domain, multiple core domains, and C-terminal Ras-binding domains. The authors first used truncated constructs of Rattus norvegicus PLCε to determine which domains were necessary for activation by a constitutively active Rap1A variant (Rap1AG12V). While it was expected that the C-terminal domains would be required, the authors found that core domains that are N-terminal to the catalytic domain were also necessary. SEC-SAXS and EFA were performed at the Advanced Photon Source (BioCAT beamline) using a PLCε construct consisting of the core domains and C-terminal Ras-binding domains only (PH-COOH). Addition of Rap1AG12V led to a slight reduction in Rg, accompanied by subtle sharpening of both P(r) and the Kratky peak. Notably, P(r) of PH-COOH on its own is skewed, indicating that the enzyme is somewhat extended. When complexed with Rap1AG12V, P(r) is more symmetric, indicating that the complex is more compact. Combined, these results suggest that PLCε is flexible and that Rap1A-dependent activation entails recruitment of multiple domains, likely both N- and C-terminal to the catalytic domain, to form a compact state. In the full-length enzyme, this conformational change, in turn, may also serve to bring the tethered guanine-exchange domain closer to the bound GTPase.
SAXS to Build Models of Protein Complexes and Assemblies
Because the scattering intensity scales as the square of the molecular weight, SAXS is particularly sensitive to changes in oligomerization state. As a result, SAXS is a useful tool for understanding how proteins associate to form complexes and assemblies [11,12,17–27]. Similarly, SAXS can also assist in the design of de novo protein assemblies [28,29].
SAXS can also be combined with prior knowledge from other methods to define the overall architecture of protein complexes and assemblies that are otherwise challenging to visualize. The trans-membrane assembly involved in bacterial chemotaxis is one such system. To adjust flagella rotation in response to the environment, bacteria like Escherichia coli (Ec) and Thermotoga maritima use a complex chemoreceptor array consisting of methyl-accepting chemotaxis proteins (MCPs), a coupling protein CheW, and a protein kinase CheA to relay chemical signals from the cell surface to the cytoplasm. In a study by Muok and co-workers [30], SAXS was combined with multiple biophysical and biochemical methods to piece together how these proteins interact. Engineered complexes were stabilized by disuccinimidyl sulfoxide crosslinking and examined by SEC-SAXS at the Cornell High Energy Synchrotron Source (formerly G1 line, currently known as ID7A). The Kratky plot of the EFA-separated profile displayed a sharp peak indicating that the complex is well-packed, compared to the apparent flexibility of CheA alone [31]. However, it also displayed a shoulder that could be a result of an open structure between some subunits, as observed in the 2D classes from cryo-EM. Because a 3D reconstruction could not be obtained by cryo-EM, the complex was modeled computationally by incorporating restraints from cross-linking mass spectrometry analysis, pulse-dipolar electron-spin resonance spectroscopy (PDS), and subunit crystal structures. Multiple models were generated, in which flexible linkages were conformationally sampled in Rosetta, and the final working model was selected based on agreement with SAXS data.
Structure determination of complex systems, like the above example, remain challenging because they may exhibit conformational and/or compositional heterogeneity. A major advantage of using SEC-SAXS with EFA is that they can enable the characterization of compositionally pure states. Although flexibility poses a challenge to structure determination, the structures of folded domains and subunits can now be more easily predicted by methods like AlphaFold [32] and RoseTTA Fold [33]. By combining partial models with distance restraints from other biophysical methods (e.g., PDS, single-molecule Förster resonance electron transfer (FRET), protein cross-linking analysis), models of the full assembly can be generated for validation against SAXS data. We anticipate that this approach will be especially useful for investigating the assembly of protein complexes and of highly flexible multi-domain enzymes.
Detailed SAXS Analysis of Conformational Transitions
One of the most powerful uses of SAXS is to assess conformational transitions upon ligand or protein-partner binding [8,9,34]. A notable example is a study by Horst and co-workers, in which cryo-EM and SAXS were used to study the activation of soluble guanylate cyclase (sGC) by nitric oxide (NO) [35]. In mammals, sGC plays an important role in the NO signaling pathways of cardiovascular and neurological systems. Structurally, sGC consists of two subunits (α and β), each forming a dumbbell wherein regulatory domains are connected to a catalytic domain at the other end by a coiled-coil domain. The α and β subunits dimerize in parallel orientation such that the catalytic domains form an active site at the dimer interface. The regulatory lobe of the enzyme is asymmetric in that only the β subunit contains the heme cofactor needed for NO binding. To investigate structural changes upon activation of Manduca sexta sGC, the authors performed SEC-SAXS at the Advanced Light Source (SIBLYS beamline) using diethylamine NONOate to introduce NO into the solution. Of the analyses presented, the Kratky plots showed the clearest and most dramatic change. In response to NO binding, the shoulder in the Kratky plot becomes significantly more pronounced, indicating that activation leads to an opening of the structure. Using cryo-EM and a small-molecule stimulator, the authors investigated the transition to an even more active form and found that ligand-binding in the regulatory lobe leads to rotational movement of the coiled-coil domains that transmits to the catalytic lobe, ultimately opening a pocket for the substrate.
SAXS can also be used to characterize conformational transitions and guide structure determination of stable states. The work by Thomas and co-workers on ribonucleotide reductase (RNR) is an exemplar of this type of application [36]. RNRs catalyze a reaction essential to DNA-based life, the conversion of ribonucleotides to deoxyribonucleotides. Class I RNRs require two subunits for turnover: a catalytic α-dimer (α2) where nucleotide reduction occurs and a β-dimer (β2) where electron transfer is initiated. Many RNRs possess an N-terminal ATP-cone domain on the α subunit that, in response to dATP binding, induces higher order oligomerization which inhibits electron transfer between α2 and β2. Activity regulation was thought to not occur in class Ib RNRs due to a characteristic truncation of the ATP-cone. However, work on the Bacillus subtilis (Bs) class Ib RNR showed that it is inhibited by dATP and that inhibition is enhanced by dAMP binding the truncated ATP-cone [37]. In this study, anion-exchange chromatography-coupled SAXS (AEX-SAXS) and REGALS were critical in demonstrating that the α subunit is a monomer in the nucleotide-free state and an elongated dimer with a non-canonical interface when bound to dAMP (Figure 3D).
Figure 3. Deconvolution of Bs RNR conformational transition observed by SAXS.
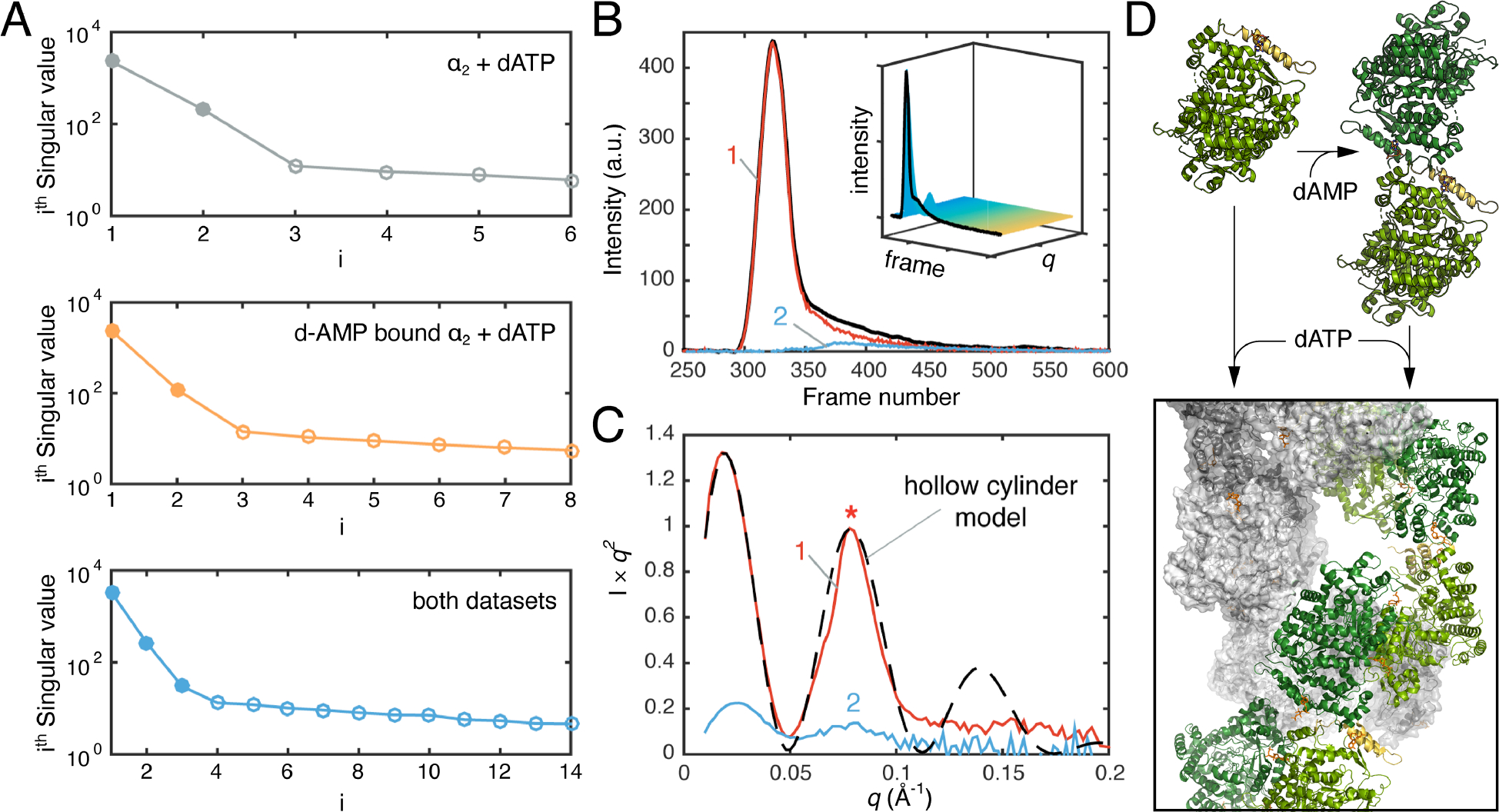
A) The singular values (filled in circles) derived from SVD analysis of the titration series of the ligand-free α subunit of Bs RNR with dATP (grey), the dAMP-bound non-canonical α2 with dATP (orange), and a combination of both datasets (blue). B) SEC-SAXS was performed on the dAMP-bound non-canonical α2 with a saturating concentration of dATP in the presence of substrate (inset). Two scattering components were deconvoluted by EFA, denoted as 1 & 2 (main plot). C) The major species, 1, shows features of a hollow cylinder model. Panels A–C: adapted under Creative Commons Attribution 4.0 International License.[36] D) Structures illustrating conversion of RNR α monomer (PDB: 6MT9) and dAMP-bound non-canonical α2 (PDB: 6CGL) to inactive filament (PDB: 6MYX) in the presence of dATP.
Building from this work, Thomas, et al. performed SAXS titrations on both the nucleotide-free α and the dAMP-bound non-canonical α2 [36]. For both, addition of dATP led to a dramatic increase in the Rg, indicating that Bs RNR undergoes higher order oligomerization, much like an RNR with a full ATP-cone. SVD on each of the titration series yielded two significant singular values, corresponding to a starting and final species, while the SVD of the two series combined surprisingly yielded three, indicating that the two titrations lead to the same end point (Figure 3A). This result suggested that the non-canonical α2 must be a component of the final structure. By performing SEC-SAXS and EFA (Figure 3B), the authors isolated the scattering from the dATP-bound α and found that the Kratky plot could be described by a hollow cylinder (Figure 3C). Using solution conditions determined by SAXS, Thomas, et al. then determined the cryo-EM structure and found that indeed, dATP binds two different allosteric sites to promote the assembly of the α subunit into helical structures, which in turn dimerize into a hollow, double helical filament (Figure 3D). In this study, titrations and SEC-SAXS performed at the Cornell High Energy Synchrotron Source (beamline G1/ID7A) were used extensively to map six oligomerization states of Bs RNR and guide structure determination by cryo-EM and crystallography.
Capturing Transient States with Time-Resolved SAXS
Finally, a key application of SAXS is to study transient structural states. Because of the relatively simple requirements for a SAXS setup, many different types of experiments can be performed. Conformational changes can be probed in a time-resolved manner using various mixing methods (e.g., stopped flow, laminar flow, chaotic flow) as well as pump-probe methods.
Josts and co-workers used two different experimental setups to probe the ATP-induced dimerization of the nucleotide-binding domains (NBDs) of a bacterial lipid flippase (Ec MsbA) and their subsequent dissociation following ATP hydrolysis [38,39]. In the first study, the authors used a continuous flow setup at the European Synchrotron Radiation Facility (beamline ID09) to capture early time points in this reaction by using photo-dissociating ATP (i.e., 1-(2-nitrophenyl)-caged ATP) to release ATP into the into reaction mixture in < 10 ms [38]. SAXS data were collected at several time points from 50 ms to 1.4 s. As is common with pump-probe SAXS experiments, a scattering profile collected in the dark was subtracted from the dataset to produce difference profiles. SVD performed on the difference dataset yielded two significant singular values, one major and one minor. In a difference dataset, a two-state transition yields one significant singular value. The presence of a second significant singular value thus led to the conclusion that the dataset captured an early intermediate in addition to a monomer-to-dimer transition. In the second study, a stop-flow mixer at PETRA III (beamline P12) was used to examine the time points between 175 ms to 120 s [39]. SVD analysis performed on a buffer-subtracted dataset led to two significant singular values that corresponded to monomer and dimer. (Figure 4A) Each scattering curve was then fit to linear combinations of the theoretical profiles of the two species generated from their reported crystal structures. This analysis revealed that introduction of ATP leads to dimerization of a fraction of the NBDs during the first 10 s, followed by their slow dissociation (Figure 4A, D).
Figure 4. Time-resolved SAXS of the nucleotide-binding domain (NBD) of Ec MsbA.
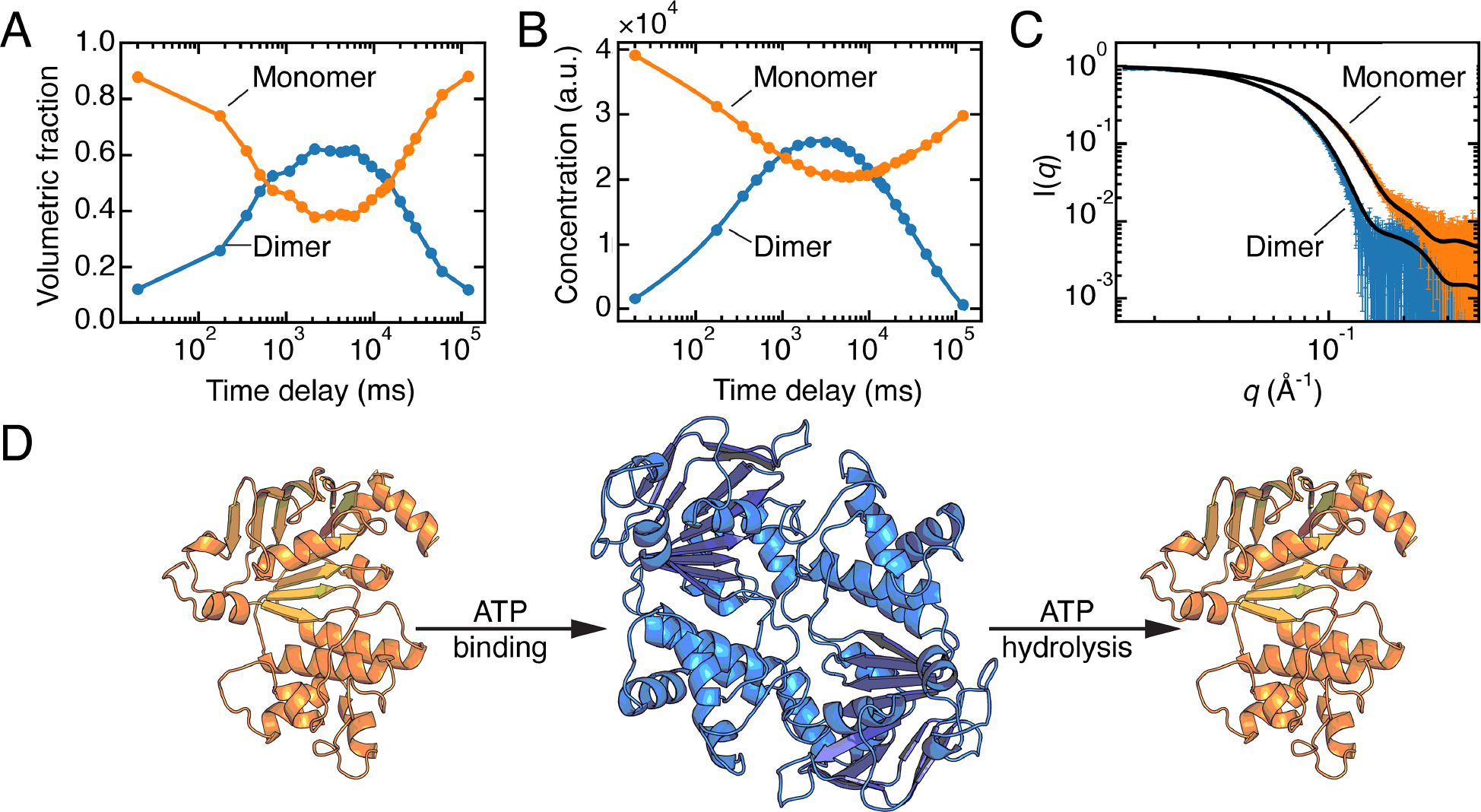
A) Concentrations of individual species obtained by fitting linear combinations of the theoretical SAXS profiles calculated from models of the monomer and dimer derived from a crystal structure (PDB: 3B60).[39] Figure was reproduced from the original data (SASDB entry: SASDGV5) following the methods in the original paper. B) Concentrations derived from REGALS are in agreement with panel A. C) SAXS profiles of the monomer and dimer of Ec MsbA-NBD extracted from the time-resolved experiment by REGALS agree with CRYSOL predictions from the models described in panel A. Panels B, C: adapted under Attribution 4.0 International (CC BY 4.0) license [5]. D) Illustration of the transition between monomer and dimer during ATP binding and hydrolysis, generated via AlphaFold2 from the sequence of Ec MsbA-NBD (residues 337–582).
Although the transient MsbA-NBD dimer could be quantified because of the availability of crystal structures, high-resolution structures may not always be available or may not accurately represent solution structures. To address this issue, Meisburger, et al. developed REGALS and applied it to the MsbA-NBD stopped-flow dataset [5]. The REGALS analysis incorporated basic assumptions that: (1) the transition involved only two species, as determined by SVD; (2) scattering profiles should be smooth; (3) dimers had completely dissociated at the final time point; and (4) monomer and dimer have maximum dimensions in solution. By applying alternating least-square refinement between observed and calculated scattering intensities under the constraint of these assumptions, REGALS was able to decompose time-resolved SAXS data without using prior structural knowledge. The recovered concentration curves agreed well with the results of the original paper (Figure 4B), and the extracted scattering profiles of the MsbA-NBD monomer and dimer agree with theoretical profiles calculated from models based on crystal structures (Figure 4C,D). While EFA can be used for SEC-SAXS data because of the assumption that eluting species have zero concentration at the start and end of their elution, REGALS was developed for more complex datasets, such as anion-exchange chromatography-coupled SAXS (where the salt gradient adds a changing background signal), ligand titrations (where the structures and/or the concentrations of the interconverting species are unknown), and time-resolved data (where the goal is to capture unknown transient intermediates).
Conclusions
In addition to the approaches discussed above, new SAXS methods are constantly being developed that will continue to expand our toolbox for the study of enzyme function. At the Cornell High Energy Synchrotron Source, for example, enzymes from extremophiles can be studied at variable pressures and temperatures [40,41]. In addition, anoxic SAXS has recently become available for the general user, enabling both flow-cell and SEC-SAXS of oxygen-sensitive samples, including metalloenzymes. At the Advanced Photon Source, time-resolved SAXS, a technique that was once only available to expert users, is available for studying enzyme kinetics over a wide range of timescales [42,43]. Finally, in terms of biochemical problems that remain challenging to solve, enzymes that exhibit extreme flexibility are at the top of the list – not just for SAXS but for all structural techniques. Such enzymes include assembly-line enzymes like non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), which may contain many domains and disordered regions. With the recent advances in structure prediction, it will be interesting to see how these computational methods may be interfaced with experimental methods to tackle such challenging problems.
Acknowledgments
The authors thank Da Xu (Ando Lab) for helping with Figure 4A. This work is supported by National Institutes of Health grants F32GM140763 (to A.S.B.) and GM124847 (to N.A.).
List of Abbreviations
- SAXS
Small-angle X-ray scattering
- SEC-SAXS
Size-exclusion chromatography-coupled SAXS
- AEX-SAXS
Anion-exchange chromatography-coupled SAXS
- SVD
Singular value decomposition
- EFA
Evolving factor analysis
- REGALS
Regularized alternating least squares
- l-Phe
l-phenylalanine
- PheH
Phenylalanine hydroxylase
- PEP
Phosphoenolpyruvate
- E4P
Erythrose 4-phosphate
- DAH7PS
3-deoxy-d-arabino heptulosonate-7-phosphate synthase
- CM
Chorismate mutase
- PLCε
Phospholipase Cε
- MCP
Methyl-accepting chemotaxis proteins
- PDS
Pulse-dipolar electron-spin resonance spectroscopy
- FRET
Förster resonance energy transfer (microscopy)
- sGC
Soluble guanylate cyclase
- RNR
Ribonucleotide reductase
- dAMP
Deoxyadenosine monophosphate
- dATP
Deoxyadenosine triphosphate
- ATP
Adenosine triphosphate
- PETRA III
Positron-Elektron-Tandem-Ring-Anlage III
- NBD
Nucleotide-binding domain
References
- 1.Skou S, Gillilan RE, Ando N: Synchrotron-based small-angle X-ray scattering of proteins in solution. Nature Protocols 2014, 9:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacques DA, Trewhella J: Small-angle scattering for structural biology--expanding the frontier while avoiding the pitfalls. Protein Sci 2010, 19:642–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI: PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography 2003, 36:1277–1282. [Google Scholar]
- 4.Meisburger SP, Taylor AB, Khan CA, Zhang S, Fitzpatrick PF, Ando N: Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J Am Chem Soc 2016, 138:6506–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meisburger SP, Xu D, Ando N: REGALS: a general method to deconvolve X-ray scattering data from evolving mixtures. IUCrJ 2021, 8:225–237. * This paper describes the development of a method for deconvoluting complex SAXS data with minimal prior knowledge. The method is demonstrated with data from ligand titration, ion exchange chromatography, and time-resolved SAXS experiments.
- 6.Volkov VV, Svergun DI: Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 2003, 36:860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickramaratne AC, Li L, Hopkins JB, Joachimiak LA, Green CB: The Disordered Amino Terminus of the Circadian Enzyme Nocturnin Modulates Its NADP(H) Phosphatase Activity by Changing Protein Dynamics. Biochemistry 2022, doi: 10.1021/acs.biochem.2c00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Léger C, Pitard I, Sadi M, Carvalho N, Brier S, Mechaly A, Raoux-Barbot D, Davi M, Hoos S, Weber P, et al. : Dynamics and structural changes of calmodulin upon interaction with the antagonist calmidazolium. BMC Biol 2022, 20:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMahon SA, Zhu W, Graham S, Rambo R, White MF, Gloster TM: Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat Commun 2020, 11:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunnak W, Winter AJ, Lazarus CM, Crump MP, Race PR, Wattana-Amorn P: SAXS reveals highly flexible interdomain linkers of tandem acyl carrier protein-thioesterase domains from a fungal nonreducing polyketide synthase. FEBS Lett 2021, 595:133–144. [DOI] [PubMed] [Google Scholar]
- 11.Klaus M, Rossini E, Linden A, Paithankar KS, Zeug M, Ignatova Z, Urlaub H, Khosla C, Köfinger J, Hummer G, et al. : Solution Structure and Conformational Flexibility of a Polyketide Synthase Module. JACS Au 2021, 1:2162–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Gillilan R, Abbaspourrad A: Tuning C-Phycocyanin Photoactivity via pH-Mediated Assembly-Disassembly. Biomacromolecules 2021, 22:5128–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Brien DP, Brier S, Ladant D, Durand D, Chenal A, Vachette P: SEC-SAXS and HDX-MS: A powerful combination. The case of the calcium-binding domain of a bacterial toxin. Biotechnol Appl Biochem 2018, 65:62–68. [DOI] [PubMed] [Google Scholar]
- 14.Tikhonova E, Mariasina S, Arkova O, Maksimenko O, Georgiev P, Bonchuk A: Dimerization Activity of a Disordered N-Terminal Domain from Drosophila CLAMP Protein. Int J Mol Sci 2022, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai Y, Parker EJ: Reciprocal allostery arising from a bienzyme assembly controls aromatic amino acid biosynthesis in Prevotella nigrescens. J Biol Chem 2021, 297:101038. * Bai and Parker use SAXS to examine an bienzyme from the shikimate pathway where the substrates of one enzyme activates the other enzyme, and the product of the second enzyme inhibits the first enzyme.
- 16.Sieng M, Selvia AF, Garland-Kuntz EE, Hopkins JB, Fisher IJ, Marti AT, Lyon AM: Functional and structural characterization of allosteric activation of phospholipase Cε by Rap1A. J Biol Chem 2020, 295:16562–16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michael AK, Fribourgh JL, Chelliah Y, Sandate CR, Hura GL, Schneidman-Duhovny D, Tripathi SM, Takahashi JS, Partch CL: Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proc Natl Acad Sci U S A 2017, 114:1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perry-Hauser NA, Hopkins JB, Zhuo Y, Zheng C, Perez I, Schultz KM, Vishnivetskiy SA, Kaya AI, Sharma P, Dalby KN, et al. : The Two Non-Visual Arrestins Engage ERK2 Differently. J Mol Biol 2022, 434:167465. * In the study of a G-protein coupled receptor pathway, SAXS is used to examine the interaction of two enzymes (an extracellular signal-regulated kinase ERK1/2 and arrestin2/3).
- 19.Bláha J, Skálová T, Kalousková B, Skořepa O, Cmunt D, Grobárová V, Pazicky S, Poláchová E, Abreu C, Stránský J, et al. : Structure of the human NK cell NKR-P1:LLT1 receptor:ligand complex reveals clustering in the immune synapse. Nat Commun 2022, 13:5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golub M, Lokstein H, Soloviov D, Kuklin A, Wieland DCF, Pieper J: Light-Harvesting Complex II Adopts Different Quaternary Structures in Solution as Observed Using Small-Angle Scattering. J Phys Chem Lett 2022, 13:1258–1265. [DOI] [PubMed] [Google Scholar]
- 21.Xi Z, Ilina TV, Guerrero M, Fan L, Sluis-Cremer N, Wang Y-X, Ishima R: Relative domain orientation of the L289K HIV-1 reverse transcriptase monomer. Protein Sci 2022, 31:e4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta K, Wen Y, Ninan NS, Raimer AC, Sharp R, Spring AM, Sarachan KL, Johnson MC, Van Duyne GD, Matera AG: Assembly of higher-order SMN oligomers is essential for metazoan viability and requires an exposed structural motif present in the YG zipper dimer. Nucleic Acids Res 2021, 49:7644–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourhis J-M, Yabukarski F, Communie G, Schneider R, Volchkova VA, Frénéat M, Gérard FC, Ducournau C, Mas C, Tarbouriech N, et al. : Structural Dynamics of the C-terminal X Domain of Nipah and Hendra Viruses Controls the Attachment to the C-terminal Tail of the Nucleocapsid Protein. J Mol Biol 2022, 434:167551. [DOI] [PubMed] [Google Scholar]
- 24.Marchany-Rivera D, Estremera-Andújar RA, Nieves-Marrero C, Ruiz-Martínez CR, Bauer W, López-Garriga J: SAXS structure of homodimeric oxyHemoglobin III from bivalve Lucina pectinata. Biopolymers 2021, 112:e23427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petit GA, Hong Y, Djoko KY, Whitten AE, Furlong EJ, McCoy AJ, Gulbis JM, Totsika M, Martin JL, Halili MA: The suppressor of copper sensitivity protein C from Caulobacter crescentus is a trimeric disulfide isomerase that binds copper(I) with subpicomolar affinity. Acta Crystallogr D Struct Biol 2022, 78:337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sestok AE, Brown JB, Obi JO, O’Sullivan SM, Garcin ED, Deredge DJ, Smith AT: A fusion of the Bacteroides fragilis ferrous iron import proteins reveals a role for FeoA in stabilizing GTP-bound FeoB. J Biol Chem 2022, 298:101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamrakar A, Singh R, Kumar A, Makde RD, Ashish, Kodgire P: Biophysical characterization of the homodimers of HomA and HomB, outer membrane proteins of Helicobacter pylori. Sci Rep 2021, 11:24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Courbet A, Hansen J, Hsia Y, Bethel N, Park Y-J, Xu C, Moyer A, Boyken SE, Ueda G, Nattermann U, et al. : Computational design of mechanically coupled axle-rotor protein assemblies. Science 2022, 376:383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi TS, Tezcan FA: Overcoming universal restrictions on metal selectivity by protein design. Nature 2022, 603:522–527. * In this paper, Choi and Tezcan used SAXS to verify that metal-mediated structures of a designed metal-binding protein observed by crystallography were consistent in solution.
- 30.Muok AR, Chua TK, Srivastava M, Yang W, Maschmann Z, Borbat PP, Chong J, Zhang S, Freed JH, Briegel A, et al. : Engineered chemotaxis core signaling units indicate a constrained kinase-off state. Sci Signal 2020, 13:eabc1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenswag AR, Muok A, Li X, Crane BR: Conformational transitions that enable histidine kinase autophosphorylation and receptor array integration. J Mol Biol 2015, 427:3890–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. : Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. : Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu J-N, Chen J-S, Lin S-M, Hong J-Y, Chen Y-J, Jeng U-S, Luo S-Y, Hou M-H: Targeting the N-Terminus Domain of the Coronavirus Nucleocapsid Protein Induces Abnormal Oligomerization via Allosteric Modulation. Front Mol Biosci 2022, 9:871499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Horst BG, Yokom AL, Rosenberg DJ, Morris KL, Hammel M, Hurley JH, Marletta MA: Allosteric activation of the nitric oxide receptor soluble guanylate cyclase mapped by cryo-electron microscopy. Elife 2019, 8. ** Horst and co-workers use SAXS and cryo-EM to probe the complex regulation of the nitric oxide receptor soluble guanylate cyclase. This paper is especially interesting as a gas molecule induces a large conformational change.
- 36. Thomas WC, Brooks FP 3rd, Burnim AA, Bacik J-P, Stubbe J, Kaelber JT, Chen JZ, Ando N: Convergent allostery in ribonucleotide reductase. Nat Commun 2019, 10:2653. ** This paper demonstrates how SAXS can be used to comprehensively study the conformational landscape of a complex allosteric enzyme. SAXS titrations and SVD are used to characterize oligomeric transitions, and SEC-SAXS and EFA are used to produce high-quality scattering profiles for structural interpretation. SAXS results facilitated the determination of two cryo-EM structures and three crystal structures.
- 37. Parker MJ, Maggiolo AO, Thomas WC, Kim A, Meisburger SP, Ando N, Boal AK, Stubbe J: An endogenous dAMP ligand in Bacillus subtilis class Ib RNR promotes assembly of a noncanonical dimer for regulation by dATP. Proceedings of the National Academy of Sciences 2018, 115:E4594–E4603. ** Josts and co-workers examine the timing of domain association and dissocitaion in an ABC lipid transporter, using time-resolved techniques to capture ms conformational changes.
- 38.Josts I, Niebling S, Gao Y, Levantino M, Tidow H, Monteiro D: Photocage-initiated time-resolved solution X-ray scattering investigation of protein dimerization. IUCrJ 2018, 5:667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Josts I, Gao Y, Monteiro DCF, Niebling S, Nitsche J, Veith K, Gräwert TW, Blanchet CE, Schroer MA, Huse N, et al. : Structural Kinetics of MsbA Investigated by Stopped-Flow Time-Resolved Small-Angle X-Ray Scattering. Structure 2020, 28:348–354.e3. [DOI] [PubMed] [Google Scholar]
- 40.Rai DK, Gillilan RE, Huang Q, Miller R, Ting E, Lazarev A, Tate MW, Gruner SM: High-pressure small-angle X-ray scattering cell for biological solutions and soft materials. J Appl Crystallogr 2021, 54:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller R, Cummings C, Huang Q, Ando N, Gillilan RE: Inline SAXS-coupled chromatography under extreme hydrostatic pressure. bioRxiv 2022, doi: 10.1101/2022.08.15.503920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin EW, Harmon TS, Hopkins JB, Chakravarthy S, Incicco JJ, Schuck P, Soranno A, Mittag T: A multi-step nucleation process determines the kinetics of prion-like domain phase separation. Nat Commun 2021, 12:4513. ** In this paper, a chaotic mixer was used to obtain SAXS data as early as 69 μs after initiating a reaction.
- 43.Pabit SA, Sutton JL, Chen H, Pollack L: Role of ion valence in the submillisecond collapse and folding of a small RNA domain. Biochemistry 2013, 52:1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]