

Abstract
Hereditary cancer syndromes (HCSs) are arguably the most frequent category of Mendelian genetic diseases, as at least 2% of presumably healthy subjects carry highly-penetrant tumor-predisposing pathogenic variants (PVs). Hereditary breast-ovarian cancer and Lynch syndrome make the highest contribution to cancer morbidity; in addition, there are several dozen less frequent types of familial tumors. The development of the majority albeit not all hereditary malignancies involves two-hit mechanism, i.e. the somatic inactivation of the remaining copy of the affected gene. Earlier studies on cancer families suggested nearly fatal penetrance for the majority of HCS genes; however, population-based investigations and especially large-scale next-generation sequencing data sets demonstrate that the presence of some highly-penetrant PVs is often compatible with healthy status. Hereditary cancer research initially focused mainly on cancer detection and prevention. Recent studies identified multiple HCS-specific drug vulnerabilities, which translated into the development of highly efficient therapeutic options.
Keywords: Hereditary cancer syndromes, Germline pathogenic variants, Cancer predisposition, Cancer treatment, Next-generation sequencing
Core Tip: There are many reviews describing particular types of hereditary cancer syndromes (HCSs) (e.g., hereditary breast-ovarian cancer, Lynch syndrome, Li-Fraumeni syndrome, etc.). However, for the last 15-20 years there were no publications providing a general overview on familial cancers. Our paper describes mechanisms underlying genetic cancer predisposition, lists major types of HCSs, and comments on therapeutic advances in the management of hereditary tumors.
INTRODUCTION
Hereditary cancer syndromes (HCSs) are a heterogeneous group of genetic diseases, which are associated with significantly increased risk of tumor development. There is a number of severe inborn disorders characterized by profound multiorgan failures, where cancer susceptibility constitutes only a part of clinical presentation of the disease (e.g., Bloom syndrome, Fanconi anemia, Nijmegen breakage syndrome, ataxia-telangiectasia, etc.). Most of these syndromes involve biallelic inactivation of genes involved in DNA repair and are characterized by severe immune deficiency[1,2]. Subjects affected by “genuine” HCSs usually do not have any detectable phenotypic malfunctions, they differ from truly healthy people only by a highly elevated propensity to develop malignant disease in certain organs.
Hereditary cancers apparently represent the most common category of vertically transmitted disorders. Indeed, while the occurrence of the best known genetic diseases, e.g., cystic fibrosis or phenylketonuria, usually falls below 1:10000, the population frequency of BRCA1/2-associated hereditary breast-ovarian cancer (HBOC) or MLH1/MSH2-linked Lynch syndrome is about 25-30 times higher and approaches approximately 1:300–1:400[3-6]. Collectively, at least 2% of presumably healthy subjects carry germline PVs associated with highly increased and often a nearly-fatal risk of a certain cancer type, and these estimates can be significantly higher in populations with pronounced founder effect[5,7].
Earlier studies on HCSs usually assumed that almost all carriers of pathogenic alleles are destined to develop cancer, i.e. they considered mainly families and genes with almost 100% disease penetrance. The development of genetic technologies and the availability of large collections of cancer patients and healthy subjects resulted in the discovery of genes, whose alteration is associated with less pronounced but still medically relevant (2-3-fold) increase of cancer risk. These moderately penetrant alleles rarely cause familial clustering of malignancies and present a challenge for defining disease-preventive strategies. Furthermore, unbiased case-control studies revealed that earlier family-based HCS investigations overestimated disease risks for the majority of cancer genes; in fact, seemingly none of the well-established HCS genes has a complete penetrance, with the most of estimates falling within 40%–80% probability of tumor development for germline pathogenic variant (PV) carriers[4-6,8,9].
Virtually all HCSs are more or less organ-specific, i.e. they mainly manifest by cancers arising in particular anatomic sites or tissues. However, the development of hereditary cancer registries and large data sets led to the understanding that many HCSs are associated with a wider spectrum of cancers than was initially suggested, although most of the newly added tumor types are characterized only by a marginal increase of their lifetime risk. For example, BRCA1 and BRCA2 were discovered as breast-ovarian cancer genes. Recent data indicate that carriers of BRCA1/2 PVs may have a borderline elevation of the probability of development for almost all major cancer types[10-16].
MECHANISMS OF HEREDITARY CANCER PREDISPOSITION
The acquisition of a single mutation in oncogene or suppressor gene is usually fully tolerable for a human cell due to the existence of multiple cancer-protecting biological mechanisms. The process of malignant transformation ultimately requires accumulation of several cancer-driving events in the same cell clone. Consequently, when a single cancer-associated PV is inherited from the parents, its carrier remains phenotypically healthy despite the presence of the pathogenic allele in every cell of the body. However, the number of additional events necessary for cancer manifestation decreases by one, therefore the probability of tumor development in this subject is manifold higher as compared to general population (Figure 1).
Figure 1.
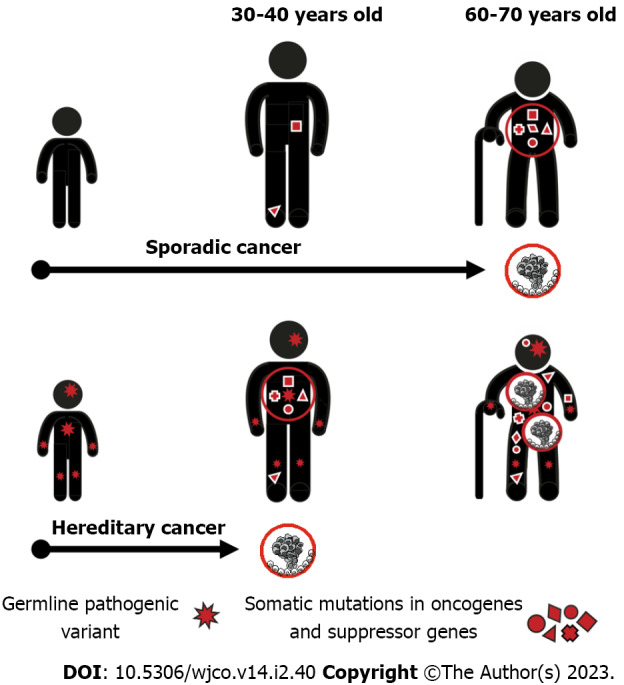
Mechanisms of hereditary cancer predisposition. Single cancer-driving mutation is usually fully compensated, therefore carriers of germline pathogenic variants may remain healthy during a prolonged period of time. However, since every cell in the target organ already contains one alteration in cancer gene, the probability of accumulation of a critical mass of additional oncogenic mutations in any given cell clone is high, and cancer manifestation often occurs at a relatively young age.
The majority of known HCS genes are suppressor genes, which require biallelic inactivation to exert their action. When inactivating PV in a single allele is inherited, the remaining copy of the gene retains its function and the normal health status is preserved. The process of malignant transformation is usually triggered by the “second hit”, i.e. by a somatic inactivation of the remaining allele occurring in any cell located within the target organ. This mechanism is highly characteristic for the best known HCS genes, e.g., RB1, BRCA1, BRCA2, MLH1, MSH2, etc[4,17-19]. There are examples of mutated suppressor genes, which contribute to the development of hereditary cancers without mandatory inactivation of the remaining gene copy. It is suggested that the reduced gene dosage, so-called haploinsufficiency, is a primary cause of malignant transformation in these situations. Interestingly, some genes, e.g., PALB2 and CHEK2, may utilize both mechanisms: Indeed, instances of both monoallelic and biallelic inactivation of these genes in human tumors have been described in the literature, and there are clear biological differences between carcinomas associated with haploinsufficiency vs second-hit loss-of-function of the above genes[20,21].
A few human cancers are caused by the inheritance of activated oncogene. The best known example is the syndrome of multiple endocrine neoplasia (MEN) type 2A and 2B (now sometimes classified as MEN2 and MEN3, respectively), which is associated with gain-of-function PVs in RET receptor tyrosine kinase[22].
HCSs have a Mendelian mode of inheritance. Most of currently described hereditary cancers are transmitted by autosomal-dominant mechanisms. Recessive inheritance of cancer predisposition is more difficult to study, especially for common tumor types, therefore only a few examples of biallelic cancer-predisposing gene defects have been identified so far[23,24]. There are also reports describing instances of oligogenic inheritance, i.e. the combination of genetic variants resulting in significant increase of cancer risks[25-28].
Hereditary cancers usually have peculiar phenotypic characteristics attributed to their mechanisms of development[29]. Most of HCSs arising in adults manifest after the peak of reproductive activity, so cancer predisposition is transmitted through generations virtually without negative selection and HCS patients often describe multiple instances of the same disease in their relatives. Presence of the first cancer-predisposing mutation in every cell of the human organism ensures highly increased risk of cancer disease as long as target organs or their parts remain in the body. Consequently, HCSs often manifest by multiple primary malignancies[30]. Furthermore, given that the cancer development in PV carriers requires less additional somatic events as compared to genetically healthy subjects, hereditary cancers commonly demonstrate younger age at onset. The development of HCS usually involves gene-specific pathways, therefore these cancers are often distinguished by predetermined molecular portrait and histological appearance. For example, BRCA1-associated breast carcinomas are usually triple-negative, chromosomally unstable and carry somatic mutation in the TP53 suppressor gene[31-34]. All these features, i.e., family cancer history, presence of multiple primary tumors, young age at onset, and especial phenotypic characteristics, represent well-recognized clinical signs of HCSs[29].
MAJOR TYPES OF HCSs
Breast and ovarian carcinomas
It is difficult to discuss hereditary breast cancer (BC) and hereditary ovarian cancer (OC) as two separate disease entities, because the best known and the most frequent genetic causes for these diseases are represented by PVs located within the same genes, BRCA1 and BRCA2 (Figure 2). Nevertheless, there are essential differences between BC and OC, which may critically affect genetic investigations of these diseases. The lifetime risk for BC in Western countries is around 1:8, therefore about 1 out of 60-70 mother-daughter or sister-sister pairs would share this disease just by chance[35,36]. OC is significantly less common, with population occurrence approaching close to 1:60–1:70; therefore, the probability of “random” co-occurrence of OC in two female first-degree relatives is very low, falling within 1:3500–1:5000[36,37]. Furthermore, while two-thirds of OC cases belong to its major histological entity, i.e. high-grade serous ovarian carcinoma, breast carcinomas are characterized by significant biological diversity manifested by differences in their receptor status and other essential tumor features[38,39]. It appears that hereditary BC research has more confounding factors as compared to the analysis of OC familial clustering.
Figure 2.
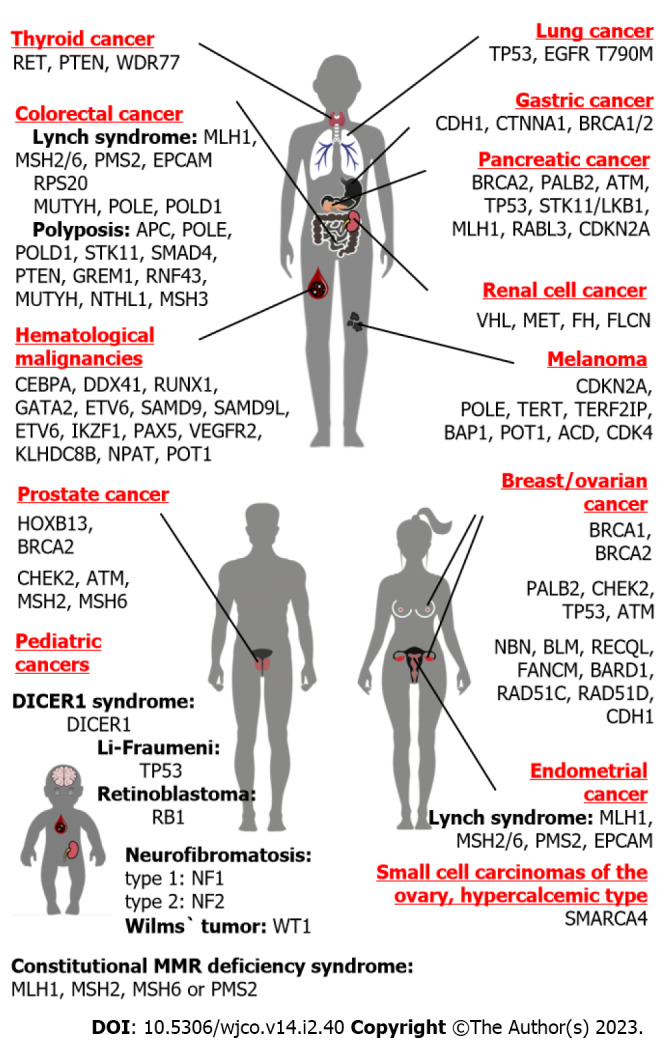
Main hereditary cancer genes and organs at risk. This figure illustrates major hereditary cancer types observed in females, males, adults of both genders, and children.
The causes of HBOC syndrome are considerably better understood than the genetic basis of hereditary BC alone. There are two major contributors to BC and OC predisposition, BRCA1 and BRCA2 (Table 1). Both these genes are involved in double-strand DNA repair by homologous recombination. BRCA2-associated cancers tend to have older age at onset as compared to BRCA1-driven malignancies. PVs in both BRCA1 and BRCA2 genes confer approximately 70% lifetime risk for BC; the cumulative risk for OC is estimated to be 44% and 17% for BRCA1 and BRCA2 genes, respectively[40]. Importantly, these collective calculations may somehow be misleading, because some PVs located within these genes predispose preferentially to BC, while others are associated with more pronounced OC risk; in fact, there are so-called BC and OC cluster regions located within these genes[41]. There are multiple genetic and non-genetic factors, which modify the risk of cancer disease in BRCA1/2 PV carriers[42]. BRCA1/2 make significant contribution to cancer morbidity: These PVs are observed in approximately 2%–5% of BC patients and up to 25%–30% of women diagnosed with high-grade serous OC[5,6,43-46]. In addition to BRCA1 and BRCA2, some RAD51 paralogs, namely RAD51C and RAD51D, predispose both to BC and OC[5,47,48]. Recent data also suggest the involvement of RAD51B germline PVs in breast- OC susceptibility[49]. The occurrence of PVs in newly described HBOC genes is an order of magnitude lower as compared to BRCA1/2[5,47].
Table 1.
Health impact of major hereditary cancer genes: Frequency of pathogenic variants in non-selected subjects and oncological patients
|
Gene
|
Frequency of pathogenic variants in population
|
Contribution in cancer morbidity
|
Ref.
|
| BRCA1 | Approximately 0.1%; > 1% in some founder populations | Breast cancer: 1%-3%; High-grade serous ovarian cancer: 15%-30% | [5,6,45,230-233] |
| BRCA2 | Approximately 0.3%; > 1% in some founder populations | Breast cancer: 1%-3%; High-grade serous ovarian cancer: 7%-12%; Prostate cancer: 2%-4%; Pancreatic cancer: 2%-3% | [5,6,45,99,102,112,232,233] |
| PALB2 | Approximately 0.1% | Breast cancer: Approximately 0.5%-1% | [5,6,45] |
| CHEK2 | 0.5%-0.7% | Breast cancer: 0.5%-2%; Moderately elevated frequencies across several cancer types | [5,6,25,113,234,235] |
| ATM | 0.3%-0.5% | Breast cancer: 0.5%-0.8%; Moderately elevated frequencies across several cancer types | [5,6,45,99,102,113] |
| MLH1, MSH2, MSH6, PMS2, EPCAM | 0.02%-0.05% for MLH1, MSH2, MSH6, EPCAM each; approximately 0.1% for PMS2 | Colorectal cancer: 1%-6%; Endometrial cancer: 2%-6% | [5,6,76,236-238] |
| CDH1 | < 0.005% | Diffuse gastric cancer: 7%; Lobular breast cancer: 0.3% | [5,6,92] |
| TP53 | < 0.01% | Breast cancer in women < 30 years old: 2%-6%; Pediatric cancers: 8%; Osteosarcoma: 4% | [161,239,240] |
| HOXB13 | 0.2%-0.4% | Prostate cancer: Approximately 1% | [112,117,241] |
PALB2 is the third most important BC-predisposing gene after BRCA1 and BRCA2[50]. Its penetrance towards BC is similar to BRCA2, while the data regarding the role of PALB2 PVs in OC predisposition are conflicting[47,51]. There are two middle-penetrance genes, ATM and CHEK2, which are associated with 2-3-fold elevation of the risk of BC development but are unlikely to contribute to increased OC susceptibility[47]. Moderate BC predisposing roles were also suggested for NBN (NBS1), BLM, RECQL, FANCM, BARD1 and several other genes, but, contrary to the evidence obtained for ATM and CHEK2, these observations have not been uniformly reproduced across distinct data sets[5,6,47,52-54]. BRIP1 is the only known gene, which is associated with hereditary OC but not with hereditary BC[47]. There are no mechanistic explanations, why some genes predispose to BC, others to OC, and a few to both BC and OC.
Many “novel” BC/OC-predisposing loci were discovered by candidate gene approach, where genes with similar to BRCA1/2 functions, i.e., the participants of DNA repair pathways, were selected for DNA testing in case-control studies. These functional considerations also influenced the interpretation of whole-exome studies, i.e., the priority was given to genes involved in the maintenance of cellular genome[55,56]. Overall, exome sequencing studies largely failed to reveal novel BC predisposing genes whose contribution to BC morbidity is comparable with the impact of BRCA1/2, PALB2 or CHEK2 germline PVs[53,57,58].
BC may arise as a part of multiorgan cancer syndrome. Germline TP53 PVs predispose to Li-Fraumeni syndrome, which is manifested by a wide spectrum of tumors. TP53 PVs are particularly common in very young patients with BC[59]. Recent large-scale next-generation sequencing (NGS) studies suggest that mutated TP53 can be found in non-selected BC patients, which do not have personal or family history of non-breast tumors[60-63]. A rare BC subtype, lobular BC, is associated with CDH1 germline PVs predisposing to diffuse stomach cancer[47,64].
There are convincing data indicating that patients with Lynch syndrome, i.e., hereditary predisposition to colorectal and endometrial cancer, develop OC more often than in general population[46,65-69]. Unlike BRCA1/2-driven tumors, Lynch syndrome associated OCs often have non-serous histology[68]. Several other multiorgan cancer syndromes also render marginally increased OC risk[46,70].
Exome sequencing studies of OC families identified several promising OC-predisposing candidates, e.g., ANKRD11 and POLE genes[71]. Some data indicate that protein-truncating germline PVs in the ERCC3 gene may confer increased OC risk[72]. Validation of these findings is complicated due to rarity of BRCA1/2-independent familial OC clustering.
Small cell carcinomas of the ovary, hypercalcemic type (SCCOHTs) constitute a rare variety of OC. SCCOHTs are associated with germline PV in the SMARCA4 gene, which plays a role in chromatin remodeling[70].
Colorectal tumors
The accumulation of multiple cases of colorectal cancer (CRC) in pedigrees was systematically described in 1967 by Lynch et al[73]. Lynch syndrome, also called hereditary non-polyposis colorectal cancer (HNPCC), is the best-known genetic cause of CRC predisposition. HNPCC is associated with heterozygous germline inactivation of genes involved in DNA mismatch repair (MMR), namely MLH1, MSH2, MSH6 or PMS2 (Table 1). In addition, some Lynch syndrome patients carry deletion of the last portion of epithelial cell adhesion molecule (EPCAM), a gene located upstream to the MSH2 genomic segment. This deletion results in the loss of transcription of the termination polyadenylation signal at the end of EPCAM and consequent emergence of the read-through EPCAM-MSH2 fusion RNA message; furthermore, cells expressing the EPCAM-MSH2 chimera demonstrate methylation of the MSH2 promoter and failure to produce functional MSH2 protein[74]. The genetic causes of Lynch syndrome are apparently limited to the germline inactivation of MLH1, MSH2, MSH6 or PMS2 genes, as attempts to link this disease with PVs in other participants of MMR were unsuccessful[4]. The lifetime risk of CRC for the carriers of pathogenic alleles falls within 40%–70% for MLH1 and MSH2 genes, however it reaches only 10%–20% for MSH6 and PMS2 heterozygous individuals. Lynch syndrome contributes approximately to 3% of CRC morbidity in Western countries, however this estimate is significantly lower in some other populations[3,4,75-79]. In addition to CRC, Lynch syndrome is associated with a highly elevated risk of endometrial cancer as well as increased susceptibility to gastric, small bowel, biliary, urothelial, ovarian, brain, and some other malignancies. The spectrum and the risk of extracolonic and extraendometrial cancers varies depending on the gene involved[4,77,80]. The development of tumors in Lynch syndrome patients involves somatic second-hit inactivation of the remaining copy of the disease-causing gene[4].
Malfunction of MMR in HNPCC-associated tumors results in a high tumor mutation burden (TMB). Short repetitive sequences, so-called microsatellites, are particularly prone to MMR defects. Consequently, Lynch syndrome tumors have high-level microsatellite instability (MSI-H) diagnosed by electrophoretic detection of multiple changes in the length of mononucleotide repeats. Electrophoretic equipment is not a component of the standard morphological laboratory; therefore, many hospitals chose to use immunohistochemical (IHC) detection of MMR deficiency (MMR-D). Indeed, tumors arising in carriers of MLH1 PVs lack the expression of MLH1 and PMS2 proteins, while MSH2-related CRCs show concomitant loss of MSH2 and MSH6 staining. Germline heterozygosity for MSH6 or PMS2 genes is accompanied by tumor-specific IHC negativity for MSH6 or PMS2, respectively[77,81]. Importantly, only a minority of tumors with MSI-H/MMR-D phenotype are hereditary cancers. MSI-H/MMR-D is also highly characteristic for sporadic colorectal, gastric and endometrial carcinomas, especially for malignancies occurring in elderly patients. Inactivation of MMR in sporadic tumors is usually attributed to the down-regulation of the MLH1 gene via promoter hypermethylation[81]. For the time being, MSI-H/MMR-D screening is recommended for all patients with CRC[82]. The selection of patients with MSI-H/MMR-D phenotype for subsequent germline testing may include consideration of age, family history of cancer, tumor location, and, in some instances, molecular characteristics of cancer cells. For example, Lynch syndrome related CRCs usually do not have mutation in the BRAF oncogene and demonstrate lack of methylation in the MLH1 gene promoter[81]. Increasing availability of NGS is likely to result in the acceptance of uniform germline testing for all patients with microsatellite unstable colorectal and endometrial cancer, therefore the significance of procedures applied for the patient selection may diminish in the near future.
CRC familial clustering commonly occurs irrespective of MSI-H/MMR-D and Lynch syndrome. Surprisingly, the attempts to identify other than Lynch syndrome hereditary CRC genes were largely unsuccessful. Besides MLH1, MSH2, MSH6, PMS2 and EPCAM, there is only one hereditary CRC gene with proven significance, RPS20. However, RPS20 is altered only in a minority of multi-case CRC families and its impact is limited to a few selected populations[4,76,79].
Some germline PVs predispose to polyposis of gastrointestinal tract and increased risk of malignant transformation. There is a number of polyposis-related genes, which are associated with several scenarios of the disease development, e.g., either the emergence of CRC in combination with the presence of multiple polyps, or, alternatively, the appearance of CRC in the absence of benign colon lesions. Some polyposis syndromes are transmitted by autosomal-dominant mode (APC, POLE, POLD1, STK11, SMAD4, BMPR1A, PTEN, GREM1, RNF43), while others involve recessive inheritance and biallelic gene inactivation in affected patients (MUTYH, NTHL1, MSH3, MBD4)[23,24,83].
The most known polyposis gene, adenomatous polyposis coli (APC), is associated with very severe impairment of gastrointestinal tract, although some hypomorphic APC variants cause an attenuated form of this disease. APC is a tumor suppressor gene, its inactivation results in up-regulation of the WNT signaling pathway. The incidence of APC is around 1:10000, and approximately 30% of detected APC PVs are de novo mutations. In addition to colon polyposis and CRC, there are some common extracolonic features of this disease, in particular, duodenal polyps and carcinomas, stomach polyps, osteomas, desmoid tumors and congenital hypertrophy of the retinal pigmented epithelium[84].
MUTYH-associated polyposis (MAP) has a somewhat lower incidence than APC, with estimates approaching approximately 1:20000. MUTYH gene is involved in base excision repair (BER), therefore its biallelic deficiency is associated with increased risk of accumulation of oncogenic mutations. MAP is usually characterized by a moderate number of polyps and relatively late disease onset. However, the probability of CRC development in MAP patients is high and approaches approximately 80%. MUTYH-driven CRCs often contain KRAS G12C substitution. Approximately 5% of patients with KRAS G12C-mutated CRC are biallelic carriers of MUTYH pathogenic alleles, therefore somatic KRAS status may be used as an indicator for MAP screening in CRC patients. Extracolonic manifestations of MAP are relatively uncommon, with the exception of highly increased risk for kidney cancer[83]. Most patients of European ancestry with genetic MAP diagnosis are homozygotes or compound heterozygotes for founder MUTYH alleles, Y165C and/or G382D[3,84-86].
NTHL1-related polyposis is similar to MAP, as it is caused by germline biallelic inactivation of the gene involved in BER. It is exceptionally rare, with estimated incidence falling below 1:100000. Various extracolonic tumors are highly characteristic for this syndrome, with a particularly elevated risk for BC[24]. A recent study identified MBD4, another participant of BER pathway, as a genetic cause of polyposis and multiorgan cancer predisposition[83].
Heterozygous germline PVs in POLE and POLD1 genes predispose to gastrointestinal polyposis, CRC, endometrial carcinomas and some other malignancies. Inactivation of these genes results in failure of proofreading activities of DNA polymerases, therefore tumors arising in carriers of POLE and POLD1 pathogenic alleles contain ultrahigh number of somatic mutations[24,76,85,87].
Gastric cancer
Gastric cancer (GC) is among the most common malignancies worldwide. Its incidence is highly influenced by environmental and behavioral factors: GC risk is significantly associated with Helicobacter pylori infection, low hygienic standard, high consumption of salt, “Northern” diet, alcohol abuse, etc.[88]. Consequently, family clustering of GC is not necessarily attributed to genetic factors, but may also be observed due to sharing of some GC-predisposing attitudes.
Strong evidence for the role of heredity is obtained only for diffuse GC, a histological variety of GC characterized by poor differentiation and presence of signet-ring cells[9,89]. The causative gene, CDH1, was initially discovered in New Zealand Maori families characterized by an exceptionally high incidence of diffuse GC[90]. CDH1 encodes E-cadherin, a protein involved in cell adhesion. CDH1 germline PVs are uncommon in the majority of analyzed populations, with the frequency being around 1:5000–1:20000[5,6,91], while the proportion of CDH1 heterozygotes in consecutive series of GC patients approaches approximately 7% for diffuse GC and 2% for non-selected GC[92]. A few hundred CDH1-related GC pedigrees have been described worldwide. Presence of CDH1 germline PVs is also associated with high risk of lobular BC, a peculiar and relatively uncommon variety of BC disease. Family studies estimated penetrance of CDH1 PVs to be around 70% for GC and 40% for BC[9]. Unbiased NGS data sets revealed instances of CDH1 germline PVs unrelated to clinically diagnosed diffuse GC, therefore, there are yet unknown factors modifying phenotypic consequences of CDH1 heterozygosity[5,6,91]. Genetic analysis of CDH1 PV-negative diffuse GC families led to the identification of subjects with inactivating PVs in CTNNA1 gene, which encodes alpha-catenin and interacts with beta-catenin and E-cadherin[9].
There are studies suggesting the role of PVs in double-strand DNA repair genes in GC predisposition. For example, contribution of PALB2 PVs has been suggested in some investigations[93,94], however the analysis of PALB2-related families did not confirm these findings[95]. GC is likely to be a part of BRCA1/2 syndrome, as some GCs arise on BRCA1/2-mutated background and demonstrate somatic loss of the remaining allele of the involved gene[13,96,97]. Lynch syndrome and some hereditary polyposis syndromes may involve malignant transformation of stomach epithelia. The lifetime GC risk in carriers of MLH1 or MSH2 PVs approaches 7%–8%. Specific nucleotide substitutions located in the promoter 1B region of the APC gene cause a condition, which is called gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS). GAPPS is attributed to down-regulation of APC transcription in gastric mucosa; interestingly GAPPS patients do not have extensive involvement of the colon because APC expression in colonic epithelium is regulated by the promoter 1A[9,23,24,84,98].
Pancreatic cancer
Predisposition to pancreatic cancer (PanCa) is usually inherited as a part of multi-organ HCS. BRCA2 is the best-established PanCa-predisposing gene (Table 1). PVs in BRCA2 confer approximately 5%–10% lifetime risk of developing PanCa, which is an order of magnitude higher than in general population[99-102]. In contrast to BRCA2, the data on the contribution of BRCA1 in PanCa morbidity are controversial[103]. It is safe to state that if BRCA1 indeed plays a role in PanCa susceptibility, its penetrance towards this cancer type is significantly lower as compared to BRCA2[99-101].
The association of the PALB2 gene with familial PanCa was initially demonstrated by exome sequencing analysis of a PanCa patient whose sister also suffered from this disease[104]. Family-based studies of PALB2-related pedigrees have confirmed this association, although the risk of PanCa associated with PALB2 PVs is moderate[95]. Moderate-to-high elevation of PanCa risk is also characteristic for ATM heterozygotes[99,105-108].
PanCa may emerge as a part of Li-Fraumeni syndrome, a disease caused by TP53 germline PVs, as well as a manifestation of Lynch syndrome[99,101,107]. Peutz-Jeghers syndrome (PJS) (attributed to PVs in STK11/LKB1) and CDKN2A-driven familial melanoma syndrome are associated with 20%–25% lifetime risk of PanCa[101,107,109].
Whole-genome sequencing study of a PanCa family revealed segregation of this disease with RABL3 truncating PV[110]. RABL3 is involved in the prenylation of KRAS protein. However, PVs in the RABL3 gene appear to be exceptionally rare and are unlikely to significantly contribute to overall PanCa morbidity[111].
Prostate cancer
PVs in two genes, HOXB13 and BRCA2, are associated with more than 5-fold elevation of prostate cancer (PrCa) risk, and, therefore, with almost 1:2 probability of developing this disease during lifetime. HOXB13 is the only known gene specifically associated with PrCa (Table 1). It encodes a prostate-specific homeobox transcription factor. Its PVs are represented by several ethnicity-specific missense mutations, which affect the interaction between HOXB13 protein and MEIS homeobox cofactor. HOXB13 PVs contribute to approximately 1% of PrCa incidence[112-114].
BRCA2 is apparently the most frequent cause of hereditary PrCa. Its penetrance towards PrCa in men is comparable to the risk estimates observed for BC in female BRCA2 PV carriers[103,112,114,115]. Similar to pancreatic cancer, evidences regarding the contribution of BRCA1 in PrCa morbidity are controversial, and associated risks are at best low-to-moderate[103,115,116]. The role of ATM PVs in PrCa predisposition is well established; ATM-heterozygous men have an approximately 2-fold elevation of the probability of PrCa development[112,114,117]. The impact of PALB2 PVs has been suggested in some studies, although systematic investigations failed to validate these findings[95]. Lynch syndrome associated with PVs in MSH2 and MSH6 genes may also render an increased PrCa risk[118].
Renal cell cancer
Next-generation sequencing of DNA obtained from renal cell carcinoma (RCC) patients revealed an unexpectedly high frequency of germline PVs: Pathogenic or likely pathogenic alleles were detected in 41/254 (16%) analyzed subjects[119]. Approximately 5% of RCC incidence is associated with RCC-predisposing syndromes[119]. Von-Hippel-Lindau syndrome caused by germline PVs in the von Hippel-Lindau (VHL) gene renders approximately 30%–40% lifetime risk of RCC and is also associated with the development of pancreatic neuroendocrine tumors, pheochromocytomas and hemangioblastomas. PVs in the fumarate hydratase (FH) gene are responsible for hereditary leiomyomatosis and renal cell cancer. Germline PVs in MET receptor tyrosine kinase confer a fatal risk of papillary RCC. RCC is also characteristic for Birt-Hogg-Dubé syndrome, a disease caused by PVs in the FLCN gene and associated with slowly progressing renal lesions, skin fibrofolliculomas and lung cysts[120]. The risk of various types of RCC is increased in patients with tuberous sclerosis syndrome[121].
Lung cancer
Genuine hereditary lung cancer (LC) is an exceptionally rare disease. The best-described cause of familial LC is the inheritance of the epidermal growth factor receptor (EGFR) T790M variant[122,123]. EGFR T790M was initially discovered as a secondary somatic mutation acquired during the course of therapy by EGFR inhibitors[124,125]. Subsequent studies demonstrated that some subjects carry this missense substitution in germline. Inborn EGFR T790M allele is associated with the development of lung tumors, which contain tyrosine kinase inhibitor sensitizing mutations in exons 19 and 21 of the EGFR gene[126]. Only a few dozen subjects carrying germline EGFR T790M allele have been described worldwide[123]. The frequency of the EGFR T790M allele in consecutive LC series is vanishingly low[127,128]. In addition to EGFR T790M, a few unique LC families with other germline pathogenic EGFR variants have been described[123,128]. LC may also arise as a part of Li-Fraumeni syndrome, being attributed to germline TP53 pathogenic allele[8,129].
Melanoma
Germline PVs in the CDKN2A gene have been detected in 20%–40% of families with multiple instances of cutaneous melanoma. CDKN2A PV carriers are at risk of development of other tumor types, particularly pancreatic cancer[130,131]. CDKN2A pathogenic alleles are associated with a more aggressive superficial spreading subtype, however there are controversial data with regard to their impact on melanoma-specific survival[132]. There are several described pedigrees where melanoma incidence is segregated with pathogenic alleles in CDK4, POT1 or TERT genes[133].
Multiple endocrine neoplasia
Multiple endocrine neoplasia (MEN) type 1 affects parathyroid glands, pancreatic islet cells and the anterior pituitary. It is caused by heterozygous inactivation of the MEN1 tumor suppressor gene, which encodes menin, a protein involved in regulation of a spectrum of biological processes. The prevalence of MEN1 syndrome is approximately 1:30000[22], although the population frequency of MEN1 PVs may be slightly higher[5]. Most of MEN1 patients demonstrate primary hyperparathyroidism caused by parathyroid hyperplasia. This condition is accompanied by hypercalcemia with varying degrees of its consequences. Duodeno-pancreatic neuroendocrine tumors of pancreas are represented by gastrinomas, non-functioning tumors, insulinomas, glucagonomas and vasoactive intestinal peptide producing tumors. Anterior pituitary neoplasms include prolactinomas as well as somatropin-, adrenocorticotropic hormone- and gonadotropin-secreting adenomas. In addition to the above three organs, MEN1 may manifest by adrenocortical, bronchopulmonary and thymic neuroendocrine tumors as well as by a number of non-endocrine neoplasms[134]. Unexpectedly, a strong association between MEN1 heterozygosity and highly increased risk of acute pancreatitis has been demonstrated in a recent study[108]. Some patients, who have MEN1-related phenotype, but lack PVs in the MEN1 gene, carry CDNK1B pathogenic alleles. CDNK1B-related MEN is now classified as MEN4 syndrome[22].
MEN2A (MEN2) and MEN2B (MEN3) syndromes are caused by activating PVs in RET receptor tyrosine kinase. Both these conditions are strongly associated with the development of medullary thyroid carcinoma (MTC). MTC is a relatively rare subtype of thyroid cancer, however germline RET pathogenic alleles make a very significant contribution to the incidence of this disease being detected in about a quarter of MTC patients. Besides MTC, approximately half of subjects with MEN2A syndrome develop pheochromocytomas, and up to a third of MEN2A cases are characterized by hyperparathyroidism. The prevalence of MEN2A is similar to the one for MEN1. MEN2A is caused by RET PVs in codon 634, or less, frequently, in codons 609, 611, 618, 620 or 630. These PVs, being located in the extracellular domain and resulting in replacements of cysteines, induce conformational changes in RET protein, which facilitate dimerization and cross-phosphorylation of this receptor. There are some other point mutations, which do not affect cysteines and generally cause a milder disease phenotype, i.e. the development of MTC in the absence of other endocrine tumors; isolated MTC may also be associated with cysteine mutations involving other than 634 codons of the RET oncogene. MEN2B (MEN3), being an order of magnitude less common than MEN2A, is a significantly more aggressive disease manifested in the first or second decade of life with highly metastatic and potentially fatal MTC. Patients with MEN2B also often develop pheochromocytomas as well as some non-endocrine features, e.g., neuromas and musculoskeletal abnormalities. MEN2B is usually caused by RET M918T allele or, in less than 5% of cases, A883F substitution. These amino acid substitutions are located in the kinase domain and render dimerization-independent activation of RET receptor. Overall, the distinction between familial MTC, MEN2A and MEN2B may look counter-intuitive, as these maladies are all related to RET activating alleles and differ from each other mainly by the disease severity but not by underlying biological mechanisms[22,135,136].
Carney complex manifests with adrenocortical disease, pituitary adenomas, gonadal and thyroid tumors, spotty skin pigmentation, cardiac and cutaneous myxomas, and some other non-endocrine neoplasms. This condition is caused by PRKAR1A germline PVs[137]. There is a number of genes, associated with isolated endocrine cancers. Germline PVs in the WDR77 gene have been recently shown to predispose to papillary subtype of thyroid cancer. WDR77 is a component of a transmethylase complex responsible for posttranslational modification of histone H4[138]. Genetic susceptibility to pheochromocytoma and/or paraganglioma may be rendered by PVs affecting SDHAF2, SDHB, SDHC, SDHD, MAX, TMEM127 or some other genes[139]. There are instances of familial pituitary adenoma associated with AIP germline PVs[140,141].
Li-Fraumeni syndrome
Li-Fraumeni syndrome is caused by PVs in the TP53 gene. TP53 is apparently the best-studied tumor suppressor gene, which is involved in the regulation of DNA damage response, programmed cell death, cell cycle and several other biological processes. Population occurrence of TP53 germline heterozygosity is well below 1:10000, although some communities demonstrate a noticeable frequency of founder hypomorphic TP53 variants[5,6,142]. Earlier family-based studies suggested nearly-fatal penetrance for TP53 germline PVs, although recent data indicate that some carriers of TP53 pathogenic alleles manage to achieve late adulthood without being affected by cancer disease[8].
TP53 PVs render a highly increased risk of childhood cancers. Li-Fraumeni syndrome-associated pediatric malignancies include adrenal cortical carcinomas, choroid plexus carcinomas, rhabdomyosarcomas and medulloblastomas. Adult cancers are mainly represented by very-young-onset BC in females as well as lung carcinomas, osteosarcomas, soft-tissue sarcomas and brain tumors[8,63,143]. Breast carcinomas arising in TP53 PV carriers frequently carry HER2 amplification[144]. Li-Fraumeni syndrome related lung carcinomas are characterized by an exceptionally high frequency of EGFR somatic mutations[129,145]. Carriers of TP53 PVs also have highly elevated risk of hematological malignancies[146]. The analysis of specific groups of consecutive patients revealed that Li-Fraumeni syndrome is a significant contributor to the incidence of pediatric cancers, very-young-onset breast carcinomas and osteosarcomas[142,146-150].
PTEN hamartoma tumor syndrome
PTEN hamartoma tumor syndrome (PHTS) is manifested by multiple benign and malignant tumors affecting breast, thyroid, endometrium, skin, kidney, colon and some other organs[151-153]. It is caused by heterozygous inactivating PVs in the PTEN gene, which is involved in the negative regulation of phosphatidylinositol 3-kinase/AKT/mechanistic target of rapamycin (mTOR) pathway and plays a role in the regulation of cell survival, proliferation, apoptosis and various metabolic processes[152,154]. PTEN-related syndrome is commonly known as Cowden syndrome, however the PHTS is a more preferable definition as it includes some other PTEN-associated maladies, e.g., Bannayan-Riley-Ruvalcaba syndrome and Lhermitte–Duclos disease[151,152]. Patients with PHTS often have a wide range of skin and mucosal manifestations and frequently present with macrocephaly[151]. Based on clinical considerations, the reported frequency of PHTS is approximately 1:200000[154], although unbiased NGS studies suggest that approximately 1:10000 healthy people are PTEN heterozygotes[5,6]. Activating germline PVs in the WWP1 gene, which encodes E3 ubiquitin ligase and negatively regulates PTEN, were detected in some PTEN-wild-type patients with PHTS-associated tumors[155].
PJS
PJS manifests via characteristic mucocutaneous pigmentations and various polyp-related complications. Multiple gastrointestinal hamartomatous polyps in the affected patients are located mainly in the small bowel. The disease is caused by heterozygous inactivating PVs in tumor suppressor kinase STK11/LKB1. STK11/LKB1 is involved in the regulation of cell cycle, apoptosis and cell metabolism. Population occurrence of PJS is estimated to be within 1:50000–1:200000, however as many as 1 out of 10000 apparently healthy subjects may carry STK11/LKB1 PVs[5,156]. STK11/LKB1 is a highly-penetrant cancer-predisposing gene. This genetic condition is associated with highly elevated risk of breast, colon, stomach, pancreatic and some other malignancies[156]. In addition, there are rare tumor subtypes specifically linked to PJS, e.g., so-called sex cord tumors with annular tubules affecting ovaries[157]. Clinical presentation of PJS may depend on the type of STK11/LKB1 PVs[158].
Gorlin syndrome
Gorlin syndrome [nevoid basal cell carcinoma (BCC) syndrome] is characterized by the appearance of BCCs and the development of odontogenic keratocysts. This disease is also associated with increased risk of medulloblastoma. In addition, various developmental abnormalities are frequently seen in patients with this condition. Gorlin syndrome is a rare disease, being observed in approximately 1:30000–1:300000 subjects. The most frequent cause of Gorlin syndrome is a heterozygous inactivating PV in the PTCH1 gene. SUFU or PTCH2 pathogenic alleles have been identified in the affected subjects, who are mutation-negative for PTCH1. Tumor development in Gorlin syndrome patients involves upregulation of the Hedgehog signaling pathway due to loss of its negative regulation by PTCH1, SUFU or PTCH2[159]. BCC predisposition may also be rendered by heterozygous inactivating PVs in the PTPN14 tumor suppressor gene[160].
Pediatric cancers
It is difficult to draw a strict distinction between “pediatric” and “adult” hereditary cancers because many HCSs may present with various manifestations both in childhood and in the middle of life. Relevant examples include Li-Fraumeni syndrome, Cowden syndrome, PJS, neurofibromatosis, RET-related malignancies, etc. Expectedly, NGS analysis of non-selected patients with pediatric cancers revealed elevated frequency of PVs in known cancer-predisposing genes[161,162].
Retinoblastoma was the first pediatric tumor for which the genetic origin was convincingly established and the causative gene was identified. Hereditary retinoblastoma is caused by germline inactivation of the RB1 gene. RB1, being the first cloned tumor suppressor gene, is implicated in the negative regulation of the cell cycle[19]. RB1 germline alterations are observed in all patients with familial and/or bilateral retinoblastoma as well as in 14% of subjects with sporadic unilateral appearance of this disease[163]. Retinoblastoma survivors are at high risk of developing other neoplasms, particularly sarcomas[164]. Spliceosome dysfunction has been recently shown to underlie the emergence of bone malignancies in RB1 heterozygotes[165].
Wilms` tumor (nephroblastoma, WT) is a relatively common pediatric cancer. The most frequent genetic cause of WT is a mutation in the WT1 gene, which can be associated either with isolated WT, or with its combination with aniridia, nephrotic syndrome and/or abnormal genitalia. WT can also be a part of so-called overgrowth syndromes (Beckwith-Wiedemann syndrome, Sotos syndrome, Simpson–Golabi–Behmel syndrome, Perlman syndrome) or several syndromes associated with a wide spectrum of cancers (Li-Fraumeni syndrome, Bloom syndrome, Fanconi anemia, etc.)[166].
Neurofibromatosis type 1 is caused by inactivating heterozygous PVs in the NF1 gene. NF1 is a negative regulator of the RAS signaling pathway. NF1 heterozygosity is estimated to occur in 1:3500 newborns and is manifested by cafe au lait spots, axillary freckles, Lisch nodules and neurofibromas. This syndrome is associated with a high risk of development of gliomas, hematological malignancies, pheochromocytomas and some other tumors. Neurofibromatosis type 2 is ten times less common than the type 1 disease. The NF2 gene encodes merlin, its inactivation is associated with the development of schwannomas and meningiomas in adolescence or adulthood[167].
DICER1 syndrome has been described relatively recently[168]. It is associated with heterozygous germline inactivation of the DICER1 gene. DICER1, a ribonuclease III family enzyme, is responsible for the maturation of microRNA. The pathogenesis of DICER1-related malignancies usually involves somatic alteration of the remaining gene allele. DICER1 PVs are characterized by incomplete penetrance. Carriers of DICER1 PVs are at risk of developing pleuropulmonary blastomas, gynandroblastomas, sarcomas, Sertoli-Leydig cell tumors and some other neoplasms[169,170].
PVs in the SMARC family genes, which regulate chromatin remodeling, are responsible for the rhabdoid tumor predisposition syndrome[171]. SMARCB1 pathogenic alleles are associated with the development of malignant rhabdoid tumors of the central nervous system and kidneys. Hypomorphic SMARCB1 PVs are also implicated in familial schwannomatosis where the development of schwannomas involves concomitant down-regulation of both SMARCB1 and NF2 genes[172]. SMARCE1 PVs predispose to the development of meningiomas. SMARCA4 pathogenic alleles are associated with rhabdoid tumors as well as small-cell OC, hypercalcemic type[171].
Constitutional mismatch repair deficiency syndrome (CMMRD) is an autosomal-recessive disorder caused by biallelic inactivation of MMR genes[4]. This condition has characteristic cutaneous manifestations and renders a high probability of developing brain, gastrointestinal and hematological malignancies at a young age[173].
Hematological malignancies
Hematological malignancies often manifest as a part of a syndromic condition. Various abnormalities of hematopoiesis resulting in the depletion of some cell lineages are frequently accompanied by myeloid-derived neoplasms. Immune deficiencies render an increased risk of development of lymphomas[174]. Familial clustering of acute myeloid leukemia may be attributed to germline PVs in CEBPA, DDX41, RUNX1, GATA2, ETV6, SAMD9, SAMD9L and some other genes. Hereditary acute lymphoblastic leukemia is related to germline PVs in ETV6, IKZF1 or PAX5 genes and may as well be a part of clinical manifestation of Li-Fraumeni syndrome[175]. Alterations in the KDR (vascular endothelial growth factor 2) receptor tyrosine kinase are the most frequent cause of hereditary Hodgkin lymphoma; high risk of this disease may also be rendered by germline PVs located in KLHDC8B, NPAT or POT1 genes[176].
MANAGEMENT OF HEREDITARY TUMORS
Cancer detection and prevention
The research on HCSs was initially viewed mainly as a part of prophylactic medicine. Indeed, there is a strong emphasis on the identification of yet healthy people, who are carriers of tumor-predisposing PVs and may significantly benefit from early cancer detection and prevention (Figure 3). Diagnostic surveillance strategies have been articulated for all major cancer syndromes. For example, female carriers of BRCA1, BRCA2 and some other pathogenic alleles are advised to start breast self-examination from 18 years old; regular clinical breast examination and magnetic resonance imaging are usually added beginning from 25 years, and they are supplemented by annual mammography in women aged 30–75 years. OC screening includes annual transvaginal ultrasound examination and CA-125 serum marker measurement starting at 30–35 years[84]. Clinical efficacy of surveillance is considerably higher in patients with Lynch syndrome. The adherence to colonoscopy performed every 1–2 years beginning from 20–25 years of age, upper endoscopy every 3–5 years starting at 30–35 years as well as endometrial cancer screening, significantly reduces individual risk of cancer death[84]. Effective surveillance is more complicated in subjects with multiorgan cancer predisposition. In particular, carriers of TP53 germline PVs are advised to begin cancer screening in early childhood and, wherever possible, to abstain from potentially mutagenic diagnostic procedures, e.g., X-ray examination[146]. The development of screening recommendations for subjects with HCSs is a continuous process, which is usually coordinated by international and national healthcare professional societies or initiative groups, involves interaction of a high number of experts working in different areas of medicine, requires significant research efforts aimed at collection of real-world data and is a subject of regular updates[84,146,177,178]. There is a multitude of published guidelines, which generally suggest similar diagnostic algorithms but differ from each other in many nuances. The detailed discussion on existing recommendations is beyond the scope of this review.
Figure 3.

Management of hereditary cancer syndromes. PARPi: Poly (ADP-ribose) polymerase inhibitors; TMB: Tumor mutation burden; HIF-2α: Hypoxia inducible factor-2α; VHL: von Hippel-Lindau; mTOR: Mechanistic target of rapamycin; MAPK: Mitogen-activated protein kinase signaling pathway; MEK: Mitogen-activated protein kinase; VEGFR: Vascular endothelial growth factor receptor; SMO: Smoothened; CMMRD: Constitutional mismatch repair deficiency syndrome.
Prophylactic risk-reducing surgery has become a standard medical intervention, being particularly well investigated in subjects with the HBOC syndrome, hereditary diffuse GC, hereditary medullary thyroid cancer, etc.[9,22,146,179-181]. It is self-explanatory that surgical removal of the organ(s) at-risk may be applied only in situations when this procedure is not associated with life-threatening adverse effects or disproportional decrease of the quality of life, and only for syndromes with insufficient reliability of early cancer diagnosis. Carriers of highly-penetrant BC-predisposing PVs (BRCA1, BRCA2, PALB2, TP53, etc.) are encouraged to undergo risk-reducing breast surgery, given that even high compliance with diagnostic check-ups does not fully warrant cancer detection at early stage or good treatment outcome[182]. BRCA1/2 heterozygous women are strongly recommended to opt for prophylactic salpingo-oophorectomy at the age of 35–45 years (or after the completion of childbearing)[177,178,183]. This procedure is justified by the poor clinical efficacy of OC screening and dispensability of ovaries for women entering their second half of life. Prophylactic gastrectomy in CDH1 PV carriers is associated with severe impairment of the quality of life, however the abstinence from this procedure is associated with a significant risk of death due to diffuse GC[9]. Risk-reducing thyroidectomy followed by hormone replacement therapy is a standard option for carriers of RET high-risk PVs. This surgery is usually performed in childhood, and the recommended age for intervention varies depending on the type of RET PV[184,185].
Benefit from risk-reducing surgeries has been confirmed by real-world data, however this experience is mainly limited to healthy relatives of cancer patients, who were found to be heterozygous for a highly-penetrant pathogenic allele[184,186,187]. Recent large-scale genetic investigations have identified some carriers of tumor-predisposing variants, who do not have a family history of cancers associated with their genetic findings[5,6]. Apparently, these individuals should be advised to undergo full-scale diagnostic surveillance, whereas great caution must be taken while considering prophylactic surgical interventions in subjects with favorable pedigree data[23].
Advances in cytotoxic and targeted therapy
Despite substantial advances in early detection and prevention of malignant diseases, cancer genetics remained an “exotic” discipline for many practicing oncologists until the second decade of this century. This was due to relative rarity of familial tumors and limited impact of germline DNA testing on the treatment strategies. Several discoveries, which were made within the past 10–15 years and resulted in the recognition of specific drug vulnerabilities in hereditary cancers, have moved familial cancer studies to the frontline of medical oncology[188,189].
BRCA1/2-driven breast and ovarian carcinomas arise due to somatic inactivation of the remaining allele of the involved gene (Figure 3 and Table 2). Consequently, these tumors are deficient in DNA double-strand break repair and demonstrate pronounced sensitivity to platinum compounds, mitomycin C, bifunctional alkylating agents and poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi). Several clinical studies involving cisplatin or carboplatin suggested that platinum-based regimens are highly effective in women with breast or ovarian BRCA1/2-associated cancer[190-192]. Combined administration of cisplatin and mitomycin C resulted in a remarkable improvement of treatment outcomes in patients with BRCA1-mutated carcinomas[193,194]. There are a number of successful clinical investigations, which resulted in the approval of PARPi for the treatment of hereditary breast, ovarian, pancreatic and prostate malignancies[195]. Interestingly, non-breast/ovarian carcinomas arising in BRCA1/2 PV carriers often retain the second BRCA1/2 allele and therefore do not have this drug vulnerability. Findings obtained on BRCA1/2 PV carriers may or may not be applicable to other genes involved in homologous recombination, as not all of the latter trigger tumor development by the two-hit mechanism, and even biallelic defects in some genes, e.g., ATM or CHEK2, are not necessarily associated with platinum or PARPi sensitivity[21,196-198].
Table 2.
Cytotoxic and targeted therapy for tumors arising in carriers of cancer-predisposing alleles
|
Tumor type
|
Target
|
Drugs
|
Ref.
|
| BRCA1/2-driven carcinomas and their phenocopies | BRCA1/2 inactivation resulting in the deficiency of DNA repair by homologous recombination | Platinum derivatives, Mitomycin C, Bifunctional alkylating agents, PARPi | [190-193,195] |
| Hypermutated cancers (Lynch syndrome associated microsatellite unstable tumors; POLD1/POLE-deficient cancers; MUTYH-associated colorectal carcinomas; tumors in patients with CMMRD syndrome) | High tumor mutation burden resulting in excessive number of neoantigens | Immune checkpoint inhibitors | [199-206] |
| RET-associated malignancies | RET tyrosine kinase | RET inhibitors | [207-209] |
| Neurofibromatosis, type 1 | Upregulation of RAS/RAF/MEK pathway due to NF1 inactivation | MEK inhibitors | [210,211] |
| Basal cell carcinomas in patients with Gorlin syndrome | Hedgehog pathway | SMO inhibitors | [213] |
| Tumors arising in patients with tuberous sclerosis | mTOR pathway | mTOR inhibitors | [214,215] |
| Renal cell carcinomas associated with von Hippel-Lindau syndrome | Up-regulation of HIF-2α due to VHL gene inactivation | HIF-2α inhibitors | [216] |
HIF-2α: Hypoxia inducible factor-2α; PARPi: Poly (ADP-ribose) polymerase inhibitors; CMMRD: Constitutional mismatch repair deficiency syndrome; MEK: Mitogen-activated protein kinase; SMO: Smoothened; mTOR: Mechanistic target of rapamycin; VHL: von Hippel-Lindau.
Microsatellite-unstable cancers, including tumors arising due to Lynch syndrome, are characterized by an excessive number of somatic mutations, and, consequently, high tumor antigenicity. These malignancies can be managed by the administration of so-called immune checkpoint inhibitors, the drugs which antagonize immune suppressor molecules and restore proper antitumor immunity[199]. Clinical studies on microsatellite-unstable cancers involved both patients with Lynch syndrome and subjects with sporadic carcinomas. Pembrolizumab has been approved for the treatment of MSI-H tumors irrespective of their organ localization[200]. Interestingly, a small study comparing hereditary vs sporadic microsatellite-unstable endometrial carcinomas revealed that tumors associated with a germline pathogenic allele have higher TMB and are more responsive to this drug[201]. The results of available clinical trials support the use of pembrolizumab or a combination of nivolumab and ipilimumab in the first-line therapy of metastatic MSI-H CRC[199-202]. There are instances of successful utilization of immune checkpoint inhibitors for the treatment of POLE/POLD1- and MUTYH-related malignancies[203,204]. Several case studies reported clinical benefit from immune therapy in patients with CMMRD-associated tumors[205,206].
Some hereditary cancers are associated with the upregulation of specific signaling pathways. A multikinase inhibitor vandetanib, which has activity towards RET and several other tyrosine kinases, has demonstrated significant clinical activity in patients with hereditary MTCs[207]. Clinical studies on selective RET inhibitors, selpercatinib and pralsetinib, included subjects with both hereditary and sporadic RET-driven thyroid tumors, and demonstrated remarkable benefit from these drugs[136,208,209].
Tumors arising in patients with neurofibromatosis type 1 are characterized by inactivation of NF1 gene, which is a negative regulator of RAS/RAF/MEK pathway. Consequently, these malignancies are potentially sensitive to MEK inhibition[210,211]. MEK inhibitor selumetinib has been evaluated in 25 children with recurrent, refractory, or progressive pediatric low-grade NF1-related gliomas, which failed at least one prior therapy. Objective response was documented in 10 (40%) cases, and 24 (96%) patients experienced no progression of the disease within 2 years[210]. Another study included children with NF1-associated symptomatic inoperable plexiform neurofibromas. Objective responses were observed in 37/50 (70%) patients, with 28 instances of response lasting more than 1 year[211]. Activating mutations in RAS/RAF/MEK pathway are also characteristic for hypermutated cancers arising in CMMRD patients. Pronounced efficacy of selumetinib or trametinib has been demonstrated in several patients with heavily pretreated CMMRD-related brain tumors[212].
Gorlin syndrome related BCCs can be managed by down-regulation of G-protein coupled receptor smoothened (SMO), which is involved in the activation of the Hedgehog pathway. Vismodegib, a selective SMO inhibitor, has been evaluated in placebo-controlled trial involving 46 patients, who had at least ten tumors each. All subjects receiving this drug experienced the decrease of existing tumor burden. Furthermore, the use of vismodegib slowed the emergence of new cancer lesions in patients with Gorlin syndrome[213].
Cancers associated with tuberous sclerosis are responsive to mTOR targeted drugs. Clinical efficacy of everolimus has been repeatedly demonstrated in angiomyolipomas and subependymal giant cell astrocytomas associated with this syndorme[214,215]. There are promising results of the treatment of VHL syndrome related tumors by hypoxia-inducible factor-2α inhibitor belzutifan[216]. FH-deficient RCCs often respond to the combination of anti-vascular endothelial growth factor therapy and mTOR antagonists or to multitargeted tyrosine kinase inhibitors[217,218].
Drug vulnerabilities detected in hereditary cancer often have clinical relevance to their sporadic phenocopies. For example, platinum/PARPi sensitivity was initially described in BRCA1/2-driven carcinomas, but subsequent research revealed that tumors with BRCA1/2-like (BRCAness) properties, e.g., a specific pattern of chromosomal instability, are also sensitive to these compounds[219,220].
CONCLUSION
Increasing involvement of healthy people in whole exome or multigene sequencing will certainly identify a huge number of subjects, who have a potentially severe disease according to a genetic test, but continue to remain unaffected until the elderly age. We are already witnessing that virtually all updated penetrance estimates are significantly lower than the ones observed by earlier studies, and, vice versa, the population frequency of some presumably “fatal” germline PVs is manifold higher than the observed incidence of corresponding genetic diseases[4-6,8,9]. The distinction between genetic health and disease is likely to be reconsidered in the near future.
Earlier cancer genetic studies produced rather straightforward gene-disease interactions, where all relevant genes and associated diseases could be easily presented in a table-like format. Systematic large-scale investigations carried out in the last decade revealed substantial promiscuity in genotype-phenotype interactions, thus complicating the clinical diagnosis of HCSs and interpretation of genetic findings[4,17,103,108,119,149,161,162,221]. The unbiased cataloging of patient data may help to account for the diversity of HCS manifestations.
Most of the known non-cancer genetic diseases are recessive, while most of the already identified cancer predisposition syndromes are dominant. This difference is unlikely to be related to genuine biological reasons, but is rather attributed to difficulties in the genetic studies of common cancer types. Virtually all “classic” genetic pathologies are orphan maladies (e.g., cystic fibrosis or phenylketonuria), so the appearance of even 2-3 patients with a unique phenotype in the same family/pedigree, or in the same neighborhood, is immediately recognizable by practicing physicians or clinical investigators. However, if we consider a recessive mechanism for say, conventional breast, lung, or colorectal carcinomas, i.e., the situation when both parents are asymptomatic heterozygous carriers of a recessive tumor-predisposing allele, and the disease is manifested only in subjects with biallelic gene involvement, there is little if any chance to distinguish these subjects from sporadic phenocopies[222]. Indeed, already known recessive tumor-predisposing syndromes include mainly rare diseases with very characteristic phenotypic manifestation, e.g., some hereditary polyposis syndromes[84]. Systematic germline sequencing of cancer patients and the analysis of accumulated “big data” may eventually identify some examples of recessive predisposition to common cancer types. Focus on large communities with pronounced founder effect may facilitate the research in this direction.
The critical mass of advances in clinical genetics, including studies on HCSs, has been achieved due to efforts of scientists working mainly in North America, Western Europe, Japan, and several other parts of the world distinguished by the combination of an exceptionally high level of technological development and strong dedication to biomedical research. Consequently, current knowledge on pathogenic alleles and corresponding familial diseases mainly reflects the genetic background of Western European populations and some Eastern Asian communities. It is self-explanatory that each ethnic group has its own ancestors, who have a unique composition of pathogenic gene variants. Consequently, the distribution of genetic diseases is a subject of major interethnic variations, with a number of maladies observed only in selected populations. It is important to encourage ethnicity-specific cataloging of pathogenic alleles and corresponding phenotypes in order to support proper practical implementation of gene-based tests. Furthermore, analysis of “novel” populations is likely to result in the discovery of new medically relevant genes and corresponding genetic diseases[36,223-226].
Most of cancer studies rely mainly on the identification of protein-truncating variants. The clarification of functional/pathogenic significance for missense mutations is complicated, and there is a need for robust bioinformatic and laboratory pipelines supporting the distinction between disease-causing and neutral amino acid substitutions[227,228]. Current research is mainly focused on the coding regions of the genome; however other genetic loci, to be studied by whole genome cataloging, are also very likely to be a source of disease-predisposing variations[229].
Identification of cancer-predisposing genes is an example of triumph of translational medicine. The development of methods of non-surgical prevention of tumor progression in carriers of disease-associated pathogenic alleles is an obvious priority for future studies in this field.
Footnotes
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: November 17, 2022
First decision: November 30, 2022
Article in press: February 14, 2023
Specialty type: Oncology
Country/Territory of origin: Russia
Peer-review report’s scientific quality classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Stabellini N, United States; Zhang X, China S-Editor: Fan JR L-Editor: A P-Editor: Fan JR
Contributor Information
Evgeny N Imyanitov, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Ekaterina S Kuligina, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Anna P Sokolenko, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Evgeny N Suspitsin, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia. evgeny@imyanitov.spb.ru.
Grigoriy A Yanus, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Aglaya G Iyevleva, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Alexandr O Ivantsov, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
Svetlana N Aleksakhina, Department of Tumor Growth Biology, N.N. Petrov Institute of Oncology, St.-Petersburg 197758, Russia; Department of Clinical Genetics, St.-Petersburg Pediatric Medical University, St.-Petersburg 194100, Russia.
References
- 1.Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, Molenaar JJ, Porter CC, Sandlund JT, Plon SE, Wang LL, Savage SA. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clin Cancer Res. 2017;23:e23–e31. doi: 10.1158/1078-0432.CCR-17-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma R, Lewis S, Wlodarski MW. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front Pediatr. 2020;8:570084. doi: 10.3389/fped.2020.570084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valle L, de Voer RM, Goldberg Y, Sjursen W, Försti A, Ruiz-Ponte C, Caldés T, Garré P, Olsen MF, Nordling M, Castellvi-Bel S, Hemminki K. Update on genetic predisposition to colorectal cancer and polyposis. Mol Aspects Med. 2019;69:10–26. doi: 10.1016/j.mam.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Peltomäki P, Olkinuora A, Nieminen TT. Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Rev Gastroenterol Hepatol. 2020;14:707–720. doi: 10.1080/17474124.2020.1782187. [DOI] [PubMed] [Google Scholar]
- 5.Breast Cancer Association Consortium. Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, Wahlström C, Pooley KA, Parsons MT, Fortuno C, Wang Q, Bolla MK, Dennis J, Keeman R, Alonso MR, Álvarez N, Herraez B, Fernandez V, Núñez-Torres R, Osorio A, Valcich J, Li M, Törngren T, Harrington PA, Baynes C, Conroy DM, Decker B, Fachal L, Mavaddat N, Ahearn T, Aittomäki K, Antonenkova NN, Arnold N, Arveux P, Ausems MGEM, Auvinen P, Becher H, Beckmann MW, Behrens S, Bermisheva M, Białkowska K, Blomqvist C, Bogdanova NV, Bogdanova-Markov N, Bojesen SE, Bonanni B, Børresen-Dale AL, Brauch H, Bremer M, Briceno I, Brüning T, Burwinkel B, Cameron DA, Camp NJ, Campbell A, Carracedo A, Castelao JE, Cessna MH, Chanock SJ, Christiansen H, Collée JM, Cordina-Duverger E, Cornelissen S, Czene K, Dörk T, Ekici AB, Engel C, Eriksson M, Fasching PA, Figueroa J, Flyger H, Försti A, Gabrielson M, Gago-Dominguez M, Georgoulias V, Gil F, Giles GG, Glendon G, Garcia EBG, Alnæs GIG, Guénel P, Hadjisavvas A, Haeberle L, Hahnen E, Hall P, Hamann U, Harkness EF, Hartikainen JM, Hartman M, He W, Heemskerk-Gerritsen BAM, Hillemanns P, Hogervorst FBL, Hollestelle A, Ho WK, Hooning MJ, Howell A, Humphreys K, Idris F, Jakubowska A, Jung A, Kapoor PM, Kerin MJ, Khusnutdinova E, Kim SW, Ko YD, Kosma VM, Kristensen VN, Kyriacou K, Lakeman IMM, Lee JW, Lee MH, Li J, Lindblom A, Lo WY, Loizidou MA, Lophatananon A, Lubiński J, MacInnis RJ, Madsen MJ, Mannermaa A, Manoochehri M, Manoukian S, Margolin S, Martinez ME, Maurer T, Mavroudis D, McLean C, Meindl A, Mensenkamp AR, Michailidou K, Miller N, Mohd Taib NA, Muir K, Mulligan AM, Nevanlinna H, Newman WG, Nordestgaard BG, Ng PS, Oosterwijk JC, Park SK, Park-Simon TW, Perez JIA, Peterlongo P, Porteous DJ, Prajzendanc K, Prokofyeva D, Radice P, Rashid MU, Rhenius V, Rookus MA, Rüdiger T, Saloustros E, Sawyer EJ, Schmutzler RK, Schneeweiss A, Schürmann P, Shah M, Sohn C, Southey MC, Surowy H, Suvanto M, Thanasitthichai S, Tomlinson I, Torres D, Truong T, Tzardi M, Valova Y, van Asperen CJ, Van Dam RM, van den Ouweland AMW, van der Kolk LE, van Veen EM, Wendt C, Williams JA, Yang XR, Yoon SY, Zamora MP, Evans DG, de la Hoya M, Simard J, Antoniou AC, Borg Å, Andrulis IL, Chang-Claude J, García-Closas M, Chenevix-Trench G, Milne RL, Pharoah PDP, Schmidt MK, Spurdle AB, Vreeswijk MPG, Benitez J, Dunning AM, Kvist A, Teo SH, Devilee P, Easton DF. Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med. 2021;384:428–439. doi: 10.1056/NEJMoa1913948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu C, Hart SN, Gnanaolivu R, Huang H, Lee KY, Na J, Gao C, Lilyquist J, Yadav S, Boddicker NJ, Samara R, Klebba J, Ambrosone CB, Anton-Culver H, Auer P, Bandera EV, Bernstein L, Bertrand KA, Burnside ES, Carter BD, Eliassen H, Gapstur SM, Gaudet M, Haiman C, Hodge JM, Hunter DJ, Jacobs EJ, John EM, Kooperberg C, Kurian AW, Le Marchand L, Lindstroem S, Lindstrom T, Ma H, Neuhausen S, Newcomb PA, O'Brien KM, Olson JE, Ong IM, Pal T, Palmer JR, Patel AV, Reid S, Rosenberg L, Sandler DP, Scott C, Tamimi R, Taylor JA, Trentham-Dietz A, Vachon CM, Weinberg C, Yao S, Ziogas A, Weitzel JN, Goldgar DE, Domchek SM, Nathanson KL, Kraft P, Polley EC, Couch FJ. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med. 2021;384:440–451. doi: 10.1056/NEJMoa2005936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14:185–187. doi: 10.1038/ng1096-185. [DOI] [PubMed] [Google Scholar]
- 8.Amadou A, Achatz MIW, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr Opin Oncol. 2018;30:23–29. doi: 10.1097/CCO.0000000000000423. [DOI] [PubMed] [Google Scholar]
- 9.van der Post RS, Oliveira C, Guilford P, Carneiro F. Hereditary gastric cancer: what's new? Fam Cancer. 2019;18:363–367. doi: 10.1007/s10689-019-00127-7. [DOI] [PubMed] [Google Scholar]
- 10.Reckamp KL, Behrendt CE, Slavin TP, Gray SW, Castillo DK, Koczywas M, Cristea MC, Babski KM, Stearns D, Marcum CA, Rodriguez YP, Hass AJ, Vecchio MM, Mora P, Cervantes AE, Sand SR, Mejia RM, Tsou TC, Salgia R, Weitzel JN. Germline mutations and age at onset of lung adenocarcinoma. Cancer. 2021;127:2801–2806. doi: 10.1002/cncr.33573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh M, McBride A, Yun S, Bhattacharjee S, Slack M, Martin JR, Jeter J, Abraham I. BRCA1 and BRCA2 Gene Mutations and Colorectal Cancer Risk: Systematic Review and Meta-analysis. J Natl Cancer Inst. 2018;110:1178–1189. doi: 10.1093/jnci/djy148. [DOI] [PubMed] [Google Scholar]
- 12.de Jonge MM, Ritterhouse LL, de Kroon CD, Vreeswijk MPG, Segal JP, Puranik R, Hollema H, Rookus MA, van Asperen CJ, van Leeuwen FE, Smit VTHBM, Howitt BE, Bosse T HEBON Group. Germline BRCA-Associated Endometrial Carcinoma Is a Distinct Clinicopathologic Entity. Clin Cancer Res. 2019;25:7517–7526. doi: 10.1158/1078-0432.CCR-19-0848. [DOI] [PubMed] [Google Scholar]
- 13.Avanesyan AA, Sokolenko AP, Ivantsov AO, Kleshchev MA, Maydin MA, Bizin IV, Raskin GA, Shelekhova KV, Gorodnova TV, Bessonov AA, Anisimova EI, Volynshchikova OA, Romanko AA, Ni VI, Broyde RV, Tkachenko OB, Whitehead AJ, Scherbakov AM, Imyanitov EN. Gastric Cancer in BRCA1 Germline Mutation Carriers: Results of Endoscopic Screening and Molecular Analysis of Tumor Tissues. Pathobiology. 2020;87:367–374. doi: 10.1159/000511323. [DOI] [PubMed] [Google Scholar]
- 14.Kim H, Choi DH, Park W, Im YH, Ahn JS, Park YH, Nam SJ, Kim SW, Lee JE, Yu JH, Lee SK, Jung BY. The association between non-breast and ovary cancers and BRCA mutation in first- and second-degree relatives of high-risk breast cancer patients: a large-scale study of Koreans. Hered Cancer Clin Pract. 2019;17:1. doi: 10.1186/s13053-018-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko JM, Ning L, Zhao XK, Chai AWY, Lei LC, Choi SSA, Tao L, Law S, Kwong A, Lee NP, Chan KT, Lo A, Song X, Chen PN, Chang YL, Wang LD, Lung ML. BRCA2 loss-of-function germline mutations are associated with esophageal squamous cell carcinoma risk in Chinese. Int J Cancer. 2020;146:1042–1051. doi: 10.1002/ijc.32619. [DOI] [PubMed] [Google Scholar]
- 16.Maccaroni E, Giampieri R, Lenci E, Scortichini L, Bianchi F, Belvederesi L, Brugiati C, Pagliaretta S, Ambrosini E, Berardi R. BRCA mutations and gastrointestinal cancers: When to expect the unexpected? World J Clin Oncol. 2021;12:565–580. doi: 10.5306/wjco.v12.i7.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, Rosen EY, Richards AL, Bouvier N, Selcuklu SD, Bielski CM, Abida W, Mandelker D, Birsoy O, Zhang L, Zehir A, Donoghue MTA, Baselga J, Offit K, Scher HI, O'Reilly EM, Stadler ZK, Schultz N, Socci ND, Viale A, Ladanyi M, Robson ME, Hyman DM, Berger MF, Solit DB, Taylor BS. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571:576–579. doi: 10.1038/s41586-019-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodgson D, Lai Z, Dearden S, Barrett JC, Harrington EA, Timms K, Lanchbury J, Wu W, Allen A, Senkus E, Domchek SM, Robson M. Analysis of mutation status and homologous recombination deficiency in tumors of patients with germline BRCA1 or BRCA2 mutations and metastatic breast cancer: OlympiAD. Ann Oncol. 2021;32:1582–1589. doi: 10.1016/j.annonc.2021.08.2154. [DOI] [PubMed] [Google Scholar]
- 19.Martínez-Sánchez M, Hernandez-Monge J, Rangel M, Olivares-Illana V. Retinoblastoma: from discovery to clinical management. FEBS J. 2022;289:4371–4382. doi: 10.1111/febs.16035. [DOI] [PubMed] [Google Scholar]
- 20.Preobrazhenskaya EV, Shleykina AU, Gorustovich OA, Martianov AS, Bizin IV, Anisimova EI, Sokolova TN, Chuinyshena SA, Kuligina ES, Togo AV, Belyaev AM, Ivantsov AO, Sokolenko AP, Imyanitov EN. Frequency and molecular characteristics of PALB2-associated cancers in Russian patients. Int J Cancer. 2021;148:203–210. doi: 10.1002/ijc.33317. [DOI] [PubMed] [Google Scholar]
- 21.Iyevleva AG, Aleksakhina SN, Sokolenko AP, Baskina SV, Venina AR, Anisimova EI, Bizin IV, Ivantsov AO, Belysheva YV, Chernyakova AP, Togo AV, Imyanitov EN. Somatic loss of the remaining allele occurs approximately in half of CHEK2-driven breast cancers and is accompanied by a border-line increase of chromosomal instability. Breast Cancer Res Treat. 2022;192:283–291. doi: 10.1007/s10549-022-06517-3. [DOI] [PubMed] [Google Scholar]
- 22.McDonnell JE, Gild ML, Clifton-Bligh RJ, Robinson BG. Multiple endocrine neoplasia: an update. Intern Med J. 2019;49:954–961. doi: 10.1111/imj.14394. [DOI] [PubMed] [Google Scholar]
- 23.Powers JM, Ebrahimzadeh JE, Katona BW. Genetic testing for hereditary gastrointestinal cancer syndromes: Interpreting results in today's practice. Curr Treat Options Gastroenterol. 2019;17:636–649. doi: 10.1007/s11938-019-00253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magrin L, Fanale D, Brando C, Fiorino A, Corsini LR, Sciacchitano R, Filorizzo C, Dimino A, Russo A, Bazan V. POLE, POLD1, and NTHL1: the last but not the least hereditary cancer-predisposing genes. Oncogene. 2021;40:5893–5901. doi: 10.1038/s41388-021-01984-2. [DOI] [PubMed] [Google Scholar]
- 25.Sokolenko AP, Bogdanova N, Kluzniak W, Preobrazhenskaya EV, Kuligina ES, Iyevleva AG, Aleksakhina SN, Mitiushkina NV, Gorodnova TV, Bessonov AA, Togo AV, Lubiński J, Cybulski C, Jakubowska A, Dörk T, Imyanitov EN. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res Treat. 2014;145:553–562. doi: 10.1007/s10549-014-2971-1. [DOI] [PubMed] [Google Scholar]
- 26.Hilbers FS, van 't Hof PJ, Meijers CM, Mei H, Michailidou K, Dennis J, Hogervorst FBL, Nederlof PM, van Asperen CJ, Devilee P. Clustering of known low and moderate risk alleles rather than a novel recessive high-risk gene in non-BRCA1/2 sib trios affected with breast cancer. Int J Cancer. 2020;147:2708–2716. doi: 10.1002/ijc.33039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schubert SA, Ruano D, Tiersma Y, Drost M, de Wind N, Nielsen M, van Hest LP, Morreau H, de Miranda NFCC, van Wezel T. Digenic inheritance of MSH6 and MUTYH variants in familial colorectal cancer. Genes Chromosomes Cancer. 2020;59:697–701. doi: 10.1002/gcc.22883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao C, Polley EC, Hart SN, Huang H, Hu C, Gnanaolivu R, Lilyquist J, Boddicker NJ, Na J, Ambrosone CB, Auer PL, Bernstein L, Burnside ES, Eliassen AH, Gaudet MM, Haiman C, Hunter DJ, Jacobs EJ, John EM, Lindström S, Ma H, Neuhausen SL, Newcomb PA, O'Brien KM, Olson JE, Ong IM, Patel AV, Palmer JR, Sandler DP, Tamimi R, Taylor JA, Teras LR, Trentham-Dietz A, Vachon CM, Weinberg CR, Yao S, Weitzel JN, Goldgar DE, Domchek SM, Nathanson KL, Couch FJ, Kraft P. Risk of Breast Cancer Among Carriers of Pathogenic Variants in Breast Cancer Predisposition Genes Varies by Polygenic Risk Score. J Clin Oncol. 2021;39:2564–2573. doi: 10.1200/JCO.20.01992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown GR, Simon M, Wentling C, Spencer DM, Parker AN, Rogers CA. A review of inherited cancer susceptibility syndromes. JAAPA. 2020;33:10–16. doi: 10.1097/01.JAA.0000721648.46099.2c. [DOI] [PubMed] [Google Scholar]
- 30.Whitworth J, Smith PS, Martin JE, West H, Luchetti A, Rodger F, Clark G, Carss K, Stephens J, Stirrups K, Penkett C, Mapeta R, Ashford S, Megy K, Shakeel H, Ahmed M, Adlard J, Barwell J, Brewer C, Casey RT, Armstrong R, Cole T, Evans DG, Fostira F, Greenhalgh L, Hanson H, Henderson A, Hoffman J, Izatt L, Kumar A, Kwong A, Lalloo F, Ong KR, Paterson J, Park SM, Chen-Shtoyerman R, Searle C, Side L, Skytte AB, Snape K, Woodward ER NIHR BioResource Rare Diseases Consortium, Tischkowitz MD, Maher ER. Comprehensive Cancer-Predisposition Gene Testing in an Adult Multiple Primary Tumor Series Shows a Broad Range of Deleterious Variants and Atypical Tumor Phenotypes. Am J Hum Genet. 2018;103:3–18. doi: 10.1016/j.ajhg.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res. 2009;69:3625–3633. doi: 10.1158/0008-5472.CAN-08-3426. [DOI] [PubMed] [Google Scholar]
- 32.Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D, Nevanlinna H, Ramus SJ, Spurdle A, Robson M, Sherman M, Mulligan AM, Couch FJ, Engel C, McGuffog L, Healey S, Sinilnikova OM, Southey MC, Terry MB, Goldgar D, O'Malley F, John EM, Janavicius R, Tihomirova L, Hansen TV, Nielsen FC, Osorio A, Stavropoulou A, Benítez J, Manoukian S, Peissel B, Barile M, Volorio S, Pasini B, Dolcetti R, Putignano AL, Ottini L, Radice P, Hamann U, Rashid MU, Hogervorst FB, Kriege M, van der Luijt RB HEBON, Peock S, Frost D, Evans DG, Brewer C, Walker L, Rogers MT, Side LE, Houghton C; EMBRACE, Weaver J, Godwin AK, Schmutzler RK, Wappenschmidt B, Meindl A, Kast K, Arnold N, Niederacher D, Sutter C, Deissler H, Gadzicki D, Preisler-Adams S, Varon-Mateeva R, Schönbuchner I, Gevensleben H, Stoppa-Lyonnet D, Belotti M, Barjhoux L; GEMO Study Collaborators, Isaacs C, Peshkin BN, Caldes T, de la Hoya M, Cañadas C, Heikkinen T, Heikkilä P, Aittomäki K, Blanco I, Lazaro C, Brunet J, Agnarsson BA, Arason A, Barkardottir RB, Dumont M, Simard J, Montagna M, Agata S, D'Andrea E, Yan M, Fox S; kConFab Investigators, Rebbeck TR, Rubinstein W, Tung N, Garber JE, Wang X, Fredericksen Z, Pankratz VS, Lindor NM, Szabo C, Offit K, Sakr R, Gaudet MM, Singer CF, Tea MK, Rappaport C, Mai PL, Greene MH, Sokolenko A, Imyanitov E, Toland AE, Senter L, Sweet K, Thomassen M, Gerdes AM, Kruse T, Caligo M, Aretini P, Rantala J, von Wachenfeld A, Henriksson K; SWE-BRCA Collaborators, Steele L, Neuhausen SL, Nussbaum R, Beattie M, Odunsi K, Sucheston L, Gayther SA, Nathanson K, Gross J, Walsh C, Karlan B, Chenevix-Trench G, Easton DF, Antoniou AC; Consortium of Investigators of Modifiers of BRCA1/2. Pathology of breast and OCs among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) Cancer Epidemiol Biomarkers Prev. 2012;21:134–147. [Google Scholar]
- 33.Hoyer J, Vasileiou G, Uebe S, Wunderle M, Kraus C, Fasching PA, Thiel CT, Hartmann A, Beckmann MW, Lux MP, Reis A. Addition of triple negativity of breast cancer as an indicator for germline mutations in predisposing genes increases sensitivity of clinical selection criteria. BMC Cancer. 2018;18:926. doi: 10.1186/s12885-018-4821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breast Cancer Association Consortium. Mavaddat N, Dorling L, Carvalho S, Allen J, González-Neira A, Keeman R, Bolla MK, Dennis J, Wang Q, Ahearn TU, Andrulis IL, Beckmann MW, Behrens S, Benitez J, Bermisheva M, Blomqvist C, Bogdanova NV, Bojesen SE, Briceno I, Brüning T, Camp NJ, Campbell A, Castelao JE, Chang-Claude J, Chanock SJ, Chenevix-Trench G, Christiansen H, Czene K, Dörk T, Eriksson M, Evans DG, Fasching PA, Figueroa JD, Flyger H, Gabrielson M, Gago-Dominguez M, Geisler J, Giles GG, Guénel P, Hadjisavvas A, Hahnen E, Hall P, Hamann U, Hartikainen JM, Hartman M, Hoppe R, Howell A, Jakubowska A, Jung A, Khusnutdinova EK, Kristensen VN, Li J, Lim SH, Lindblom A, Loizidou MA, Lophatananon A, Lubinski J, Madsen MJ, Mannermaa A, Manoochehri M, Margolin S, Mavroudis D, Milne RL, Mohd Taib NA, Morra A, Muir K, Obi N, Osorio A, Park-Simon TW, Peterlongo P, Radice P, Saloustros E, Sawyer EJ, Schmutzler RK, Shah M, Sim X, Southey MC, Thorne H, Tomlinson I, Torres D, Truong T, Yip CH, Spurdle AB, Vreeswijk MPG, Dunning AM, García-Closas M, Pharoah PDP, Kvist A, Muranen TA, Nevanlinna H, Teo SH, Devilee P, Schmidt MK, Easton DF. Pathology of Tumors Associated With Pathogenic Germline Variants in 9 Breast Cancer Susceptibility Genes. JAMA Oncol. 2022;8:e216744. doi: 10.1001/jamaoncol.2021.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321:288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 36.Fierheller CT, Alenezi WM, Tonin PN. The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers (Basel) 2021;13 doi: 10.3390/cancers13143406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 38.Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet. 2019;393:1240–1253. doi: 10.1016/S0140-6736(18)32552-2. [DOI] [PubMed] [Google Scholar]
- 39.Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397:1750–1769. doi: 10.1016/S0140-6736(20)32381-3. [DOI] [PubMed] [Google Scholar]
- 40.Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, Easton DF, Antoniou AC BRCA1 and BRCA2 Cohort Consortium, McGuffog L, Evans DG, Barrowdale D, Frost D, Adlard J, Ong KR, Izatt L, Tischkowitz M, Eeles R, Davidson R, Hodgson S, Ellis S, Nogues C, Lasset C, Stoppa-Lyonnet D, Fricker JP, Faivre L, Berthet P, Hooning MJ, van der Kolk LE, Kets CM, Adank MA, John EM, Chung WK, Andrulis IL, Southey M, Daly MB, Buys SS, Osorio A, Engel C, Kast K, Schmutzler RK, Caldes T, Jakubowska A, Simard J, Friedlander ML, McLachlan SA, Machackova E, Foretova L, Tan YY, Singer CF, Olah E, Gerdes AM, Arver B, Olsson H. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317:2402–2416. doi: 10.1001/jama.2017.7112. [DOI] [PubMed] [Google Scholar]
- 41.Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, Nathanson KL CIMBA Consortium, Laitman Y, Kushnir A, Paluch-Shimon S, Berger R, Zidan J, Friedman E, Ehrencrona H, Stenmark-Askmalm M, Einbeigi Z, Loman N, Harbst K, Rantala J, Melin B, Huo D, Olopade OI, Seldon J, Ganz PA, Nussbaum RL, Chan SB, Odunsi K, Gayther SA, Domchek SM, Arun BK, Lu KH, Mitchell G, Karlan BY, Walsh C, Lester J, Godwin AK, Pathak H, Ross E, Daly MB, Whittemore AS, John EM, Miron A, Terry MB, Chung WK, Goldgar DE, Buys SS, Janavicius R, Tihomirova L, Tung N, Dorfling CM, van Rensburg EJ, Steele L, Neuhausen SL, Ding YC, Ejlertsen B, Gerdes AM, Hansen Tv, Ramón y Cajal T, Osorio A, Benitez J, Godino J, Tejada MI, Duran M, Weitzel JN, Bobolis KA, Sand SR, Fontaine A, Savarese A, Pasini B, Peissel B, Bonanni B, Zaffaroni D, Vignolo-Lutati F, Scuvera G, Giannini G, Bernard L, Genuardi M, Radice P, Dolcetti R, Manoukian S, Pensotti V, Gismondi V, Yannoukakos D, Fostira F, Garber J, Torres D, Rashid MU, Hamann U, Peock S, Frost D, Platte R, Evans DG, Eeles R, Davidson R, Eccles D, Cole T, Cook J, Brewer C, Hodgson S, Morrison PJ, Walker L, Porteous ME, Kennedy MJ, Izatt L, Adlard J, Donaldson A, Ellis S, Sharma P, Schmutzler RK, Wappenschmidt B, Becker A, Rhiem K, Hahnen E, Engel C, Meindl A, Engert S, Ditsch N, Arnold N, Plendl HJ, Mundhenke C, Niederacher D, Fleisch M, Sutter C, Bartram CR, Dikow N, Wang-Gohrke S, Gadzicki D, Steinemann D, Kast K, Beer M, Varon-Mateeva R, Gehrig A, Weber BH, Stoppa-Lyonnet D, Sinilnikova OM, Mazoyer S, Houdayer C, Belotti M, Gauthier-Villars M, Damiola F, Boutry-Kryza N, Lasset C, Sobol H, Peyrat JP, Muller D, Fricker JP, Collonge-Rame MA, Mortemousque I, Nogues C, Rouleau E, Isaacs C, De Paepe A, Poppe B, Claes K, De Leeneer K, Piedmonte M, Rodriguez G, Wakely K, Boggess J, Blank SV, Basil J, Azodi M, Phillips KA, Caldes T, de la Hoya M, Romero A, Nevanlinna H, Aittomäki K, van der Hout AH, Hogervorst FB, Verhoef S, Collée JM, Seynaeve C, Oosterwijk JC, Gille JJ, Wijnen JT, Gómez Garcia EB, Kets CM, Ausems MG, Aalfs CM, Devilee P, Mensenkamp AR, Kwong A, Olah E, Papp J, Diez O, Lazaro C, Darder E, Blanco I, Salinas M, Jakubowska A, Lubinski J, Gronwald J, Jaworska-Bieniek K, Durda K, Sukiennicki G, Huzarski T, Byrski T, Cybulski C, Toloczko-Grabarek A, Złowocka-Perłowska E, Menkiszak J, Arason A, Barkardottir RB, Simard J, Laframboise R, Montagna M, Agata S, Alducci E, Peixoto A, Teixeira MR, Spurdle AB, Lee MH, Park SK, Kim SW, Friebel TM, Couch FJ, Lindor NM, Pankratz VS, Guidugli L, Wang X, Tischkowitz M, Foretova L, Vijai J, Offit K, Robson M, Rau-Murthy R, Kauff N, Fink-Retter A, Singer CF, Rappaport C, Gschwantler-Kaulich D, Pfeiler G, Tea MK, Berger A, Greene MH, Mai PL, Imyanitov EN, Toland AE, Senter L, Bojesen A, Pedersen IS, Skytte AB, Sunde L, Thomassen M, Moeller ST, Kruse TA, Jensen UB, Caligo MA, Aretini P, Teo SH, Selkirk CG, Hulick PJ, Andrulis I. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA. 2015;313:1347–1361. doi: 10.1001/jama.2014.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milne RL, Antoniou AC. Modifiers of breast and ovarian cancer risks for BRCA1 and BRCA2 mutation carriers. Endocr Relat Cancer. 2016;23:T69–T84. doi: 10.1530/ERC-16-0277. [DOI] [PubMed] [Google Scholar]
- 43.Sun J, Meng H, Yao L, Lv M, Bai J, Zhang J, Wang L, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Xie Y. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin Cancer Res. 2017;23:6113–6119. doi: 10.1158/1078-0432.CCR-16-3227. [DOI] [PubMed] [Google Scholar]
- 44.Gorodnova T, Sokolenko A, Ni V, Ivantsov A, Kotiv K, Petrik S, Amelina I, Berlev I, Imyanitov E. BRCA1-associated and sporadic ovarian carcinomas: outcomes of primary cytoreductive surgery or neoadjuvant chemotherapy. Int J Gynecol Cancer. 2019;29:779–786. doi: 10.1136/ijgc-2018-000175. [DOI] [PubMed] [Google Scholar]
- 45.Kurian AW, Ward KC, Howlader N, Deapen D, Hamilton AS, Mariotto A, Miller D, Penberthy LS, Katz SJ. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol. 2019;37:1305–1315. doi: 10.1200/JCO.18.01854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pietragalla A, Arcieri M, Marchetti C, Scambia G, Fagotti A. Ovarian cancer predisposition beyond BRCA1 and BRCA2 genes. Int J Gynecol Cancer. 2020;30:1803–1810. doi: 10.1136/ijgc-2020-001556. [DOI] [PubMed] [Google Scholar]
- 47.Colas C, Golmard L, de Pauw A, Caputo SM, Stoppa-Lyonnet D. "Decoding hereditary breast cancer" benefits and questions from multigene panel testing. Breast. 2019;45:29–35. doi: 10.1016/j.breast.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Song H, Leslie G, Engel C, Hahnen E, Auber B, Horváth J, Kast K, Niederacher D, Turnbull C, Houlston R, Hanson H, Loveday C, Dolinsky JS, LaDuca H, Ramus SJ, Menon U, Rosenthal AN, Jacobs I, Gayther SA, Dicks E, Nevanlinna H, Aittomäki K, Pelttari LM, Ehrencrona H, Borg Å, Kvist A, Rivera B, Hansen TVO, Djursby M, Lee A, Dennis J, Bowtell DD, Traficante N, Diez O, Balmaña J, Gruber SB, Chenevix-Trench G, Investigators K, Jensen A, Kjær SK, Høgdall E, Castéra L, Garber J, Janavicius R, Osorio A, Golmard L, Vega A, Couch FJ, Robson M, Gronwald J, Domchek SM, Culver JO, de la Hoya M, Easton DF, Foulkes WD, Tischkowitz M, Meindl A, Schmutzler RK, Pharoah PDP, Antoniou AC. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J Natl Cancer Inst. 2020;112:1242–1250. doi: 10.1093/jnci/djaa030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Setton J, Selenica P, Mukherjee S, Shah R, Pecorari I, McMillan B, Pei IX, Kemel Y, Ceyhan-Birsoy O, Sheehan M, Tkachuk K, Brown DN, Zhang L, Cadoo K, Powell S, Weigelt B, Robson M, Riaz N, Offit K, Reis-Filho JS, Mandelker D. Germline RAD51B variants confer susceptibility to breast and ovarian cancers deficient in homologous recombination. NPJ Breast Cancer. 2021;7:135. doi: 10.1038/s41523-021-00339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, Khan S, Aittomäki K, Moilanen JS, Turnbull C, Seal S, Mannermaa A, Kallioniemi A, Lindeman GJ, Buys SS, Andrulis IL, Radice P, Tondini C, Manoukian S, Toland AE, Miron P, Weitzel JN, Domchek SM, Poppe B, Claes KB, Yannoukakos D, Concannon P, Bernstein JL, James PA, Easton DF, Goldgar DE, Hopper JL, Rahman N, Peterlongo P, Nevanlinna H, King MC, Couch FJ, Southey MC, Winqvist R, Foulkes WD, Tischkowitz M. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song H, Dicks EM, Tyrer J, Intermaggio M, Chenevix-Trench G, Bowtell DD, Traficante N Group A; Brenton J, Goranova T, Hosking K, Piskorz A, van Oudenhove E, Doherty J, Harris HR, Rossing MA, Duerst M, Dork T, Bogdanova NV, Modugno F, Moysich K, Odunsi K, Ness R, Karlan BY, Lester J, Jensen A, Krüger Kjaer S, Høgdall E, Campbell IG, Lázaro C, Pujara MA, Cunningham J, Vierkant R, Winham SJ, Hildebrandt M, Huff C, Li D, Wu X, Yu Y, Permuth JB, Levine DA, Schildkraut JM, Riggan MJ, Berchuck A, Webb PM Group OS. Cybulski C, Gronwald J, Jakubowska A, Lubinski J, Alsop J, Harrington P, Chan I, Menon U, Pearce CL, Wu AH, de Fazio A, Kennedy CJ, Goode E, Ramus S, Gayther S, Pharoah P. Population-based targeted sequencing of 54 candidate genes identifies PALB2 as a susceptibility gene for high-grade serous ovarian cancer. J Med Genet. 2021;58:305–313. doi: 10.1136/jmedgenet-2019-106739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sokolenko AP, Iyevleva AG, Preobrazhenskaya EV, Mitiushkina NV, Abysheva SN, Suspitsin EN, Kuligina ESh, Gorodnova TV, Pfeifer W, Togo AV, Turkevich EA, Ivantsov AO, Voskresenskiy DV, Dolmatov GD, Bit-Sava EM, Matsko DE, Semiglazov VF, Fichtner I, Larionov AA, Kuznetsov SG, Antoniou AC, Imyanitov EN. High prevalence and breast cancer predisposing role of the BLM c.1642 C>T (Q548X) mutation in Russia. Int J Cancer. 2012;130:2867–2873. doi: 10.1002/ijc.26342. [DOI] [PubMed] [Google Scholar]
- 53.Cybulski C, Carrot-Zhang J, Kluźniak W, Rivera B, Kashyap A, Wokołorczyk D, Giroux S, Nadaf J, Hamel N, Zhang S, Huzarski T, Gronwald J, Byrski T, Szwiec M, Jakubowska A, Rudnicka H, Lener M, Masojć B, Tonin PN, Rousseau F, Górski B, Dębniak T, Majewski J, Lubiński J, Foulkes WD, Narod SA, Akbari MR. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet. 2015;47:643–646. doi: 10.1038/ng.3284. [DOI] [PubMed] [Google Scholar]
- 54.Neidhardt G, Hauke J, Ramser J, Groß E, Gehrig A, Müller CR, Kahlert AK, Hackmann K, Honisch E, Niederacher D, Heilmann-Heimbach S, Franke A, Lieb W, Thiele H, Altmüller J, Nürnberg P, Klaschik K, Ernst C, Ditsch N, Jessen F, Ramirez A, Wappenschmidt B, Engel C, Rhiem K, Meindl A, Schmutzler RK, Hahnen E. Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, Thorne H kConFab, Barnes DR, Li J, Ellul J, Philip GK, Antill YC, James PA, Trainer AH, Mitchell G, Campbell IG. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8:e1002894. doi: 10.1371/journal.pgen.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Felicio PS, Grasel RS, Campacci N, de Paula AE, Galvão HCR, Torrezan GT, Sabato CS, Fernandes GC, Souza CP, Michelli RD, Andrade CE, Barros BDF, Matsushita MM, Revil T, Ragoussis J, Couch FJ, Hart SN, Reis RM, Melendez ME, Tonin PN, Carraro DM, Palmero EI. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum Mutat. 2021;42:290–299. doi: 10.1002/humu.24158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuligina ES, Sokolenko AP, Bizin IV, Romanko AA, Zagorodnev KA, Anisimova MO, Krylova DD, Anisimova EI, Mantseva MA, Varma AK, Hasan SK, Ni VI, Koloskov AV, Suspitsin EN, Venina AR, Aleksakhina SN, Sokolova TN, Milanović AM, Schürmann P, Prokofyeva DS, Bermisheva MA, Khusnutdinova EK, Bogdanova N, Dörk T, Imyanitov EN. Exome sequencing study of Russian breast cancer patients suggests a predisposing role for USP39. Breast Cancer Res Treat. 2020;179:731–742. doi: 10.1007/s10549-019-05492-6. [DOI] [PubMed] [Google Scholar]
- 58.Koivuluoma S, Tervasmäki A, Kauppila S, Winqvist R, Kumpula T, Kuismin O, Moilanen J, Pylkäs K. Exome sequencing identifies a recurrent variant in SERPINA3 associating with hereditary susceptibility to breast cancer. Eur J Cancer. 2021;143:46–51. doi: 10.1016/j.ejca.2020.10.033. [DOI] [PubMed] [Google Scholar]
- 59.Shin SJ, Dodd-Eaton EB, Peng G, Bojadzieva J, Chen J, Amos CI, Frone MN, Khincha PP, Mai PL, Savage SA, Ballinger ML, Thomas DM, Yuan Y, Strong LC, Wang W. Penetrance of Different Cancer Types in Families with Li-Fraumeni Syndrome: A Validation Study Using Multicenter Cohorts. Cancer Res. 2020;80:354–360. doi: 10.1158/0008-5472.CAN-19-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lolas Hamameh S, Renbaum P, Kamal L, Dweik D, Salahat M, Jaraysa T, Abu Rayyan A, Casadei S, Mandell JB, Gulsuner S, Lee MK, Walsh T, King MC, Levy-Lahad E, Kanaan M. Genomic analysis of inherited breast cancer among Palestinian women: Genetic heterogeneity and a founder mutation in TP53. Int J Cancer. 2017;141:750–756. doi: 10.1002/ijc.30771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fortuno C, James PA, Spurdle AB. Current review of TP53 pathogenic germline variants in breast cancer patients outside Li-Fraumeni syndrome. Hum Mutat. 2018;39:1764–1773. doi: 10.1002/humu.23656. [DOI] [PubMed] [Google Scholar]
- 62.Rana HQ, Gelman R, LaDuca H, McFarland R, Dalton E, Thompson J, Speare V, Dolinsky JS, Chao EC, Garber JE. Differences in TP53 Mutation Carrier Phenotypes Emerge From Panel-Based Testing. J Natl Cancer Inst. 2018;110:863–870. doi: 10.1093/jnci/djy001. [DOI] [PubMed] [Google Scholar]
- 63.Kratz CP, Freycon C, Maxwell KN, Nichols KE, Schiffman JD, Evans DG, Achatz MI, Savage SA, Weitzel JN, Garber JE, Hainaut P, Malkin D. Analysis of the Li-Fraumeni Spectrum Based on an International Germline TP53 Variant Data Set: An International Agency for Research on Cancer TP53 Database Analysis. JAMA Oncol. 2021;7:1800–1805. doi: 10.1001/jamaoncol.2021.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corso G, Montagna G, Figueiredo J, La Vecchia C, Fumagalli Romario U, Fernandes MS, Seixas S, Roviello F, Trovato C, Guerini-Rocco E, Fusco N, Pravettoni G, Petrocchi S, Rotili A, Massari G, Magnoni F, De Lorenzi F, Bottoni M, Galimberti V, Sanches JM, Calvello M, Seruca R, Bonanni B. Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers (Basel) 2020;12 doi: 10.3390/cancers12061598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonadona V, Bonaïti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Noguès C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frébourg T, Sobol H, Lasset C, Bonaïti-Pellié C French Cancer Genetics Network. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305:2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- 66.Therkildsen C, Ladelund S, Smith-Hansen L, Lindberg LJ, Nilbert M. Towards gene- and gender-based risk estimates in Lynch syndrome; age-specific incidences for 13 extra-colorectal cancer types. Br J Cancer. 2017;117:1702–1710. doi: 10.1038/bjc.2017.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carnevali I, Riva C, Chiaravalli AM, Sahnane N, Di Lauro E, Viel A, Rovera F, Formenti G, Ghezzi F, Sessa F, Tibiletti MG. Inherited cancer syndromes in 220 Italian ovarian cancer patients. Cancer Genet. 2019;237:55–62. doi: 10.1016/j.cancergen.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer JP, Nakken S, Engel C, Aretz S, Jenkins MA, Sunde L, Bernstein I, Capella G, Balaguer F, Thomas H, Evans DG, Burn J, Greenblatt M, Hovig E, de Vos Tot Nederveen Cappel WH, Sijmons RH, Bertario L, Tibiletti MG, Cavestro GM, Lindblom A, Della Valle A, Lopez-Köstner F, Gluck N, Katz LH, Heinimann K, Vaccaro CA, Büttner R, Görgens H, Holinski-Feder E, Morak M, Holzapfel S, Hüneburg R, Knebel Doeberitz MV, Loeffler M, Rahner N, Schackert HK, Steinke-Lange V, Schmiegel W, Vangala D, Pylvänäinen K, Renkonen-Sinisalo L, Hopper JL, Win AK, Haile RW, Lindor NM, Gallinger S, Le Marchand L, Newcomb PA, Figueiredo JC, Thibodeau SN, Wadt K, Therkildsen C, Okkels H, Ketabi Z, Moreira L, Sánchez A, Serra-Burriel M, Pineda M, Navarro M, Blanco I, Green K, Lalloo F, Crosbie EJ, Hill J, Denton OG, Frayling IM, Rødland EA, Vasen H, Mints M, Neffa F, Esperon P, Alvarez K, Kariv R, Rosner G, Pinero TA, Gonzalez ML, Kalfayan P, Tjandra D, Winship IM, Macrae F, Möslein G, Mecklin JP, Nielsen M, Møller P. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22:15–25. doi: 10.1038/s41436-019-0596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lerner-Ellis J, Mighton C, Lazaro C, Watkins N, Di Gioacchino V, Wong A, Chang MC, Charames GS. Multigene panel testing for hereditary breast and ovarian cancer in the province of Ontario. J Cancer Res Clin Oncol. 2021;147:871–879. doi: 10.1007/s00432-020-03377-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shanbhogue KP, Prasad AS, Ucisik-Keser FE, Katabathina VS, Morani AC. Hereditary ovarian tumour syndromes: current update on genetics and imaging. Clin Radiol. 2021;76:313.e15–313.e26. doi: 10.1016/j.crad.2020.11.116. [DOI] [PubMed] [Google Scholar]
- 71.Zhu Q, Zhang J, Chen Y, Hu Q, Shen H, Huang RY, Liu Q, Kaur J, Long M, Battaglia S, Eng KH, Lele SB, Zsiros E, Villella J, Lugade A, Yao S, Liu S, Moysich K, Odunsi KO. Whole-exome sequencing of ovarian cancer families uncovers putative predisposition genes. Int J Cancer. 2020;146:2147–2155. doi: 10.1002/ijc.32545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stradella A, Del Valle J, Rofes P, Vargas-Parra G, Salinas M, González S, Montes E, López-Doriga A, Gómez C, de Cid R, Darder E, Teulé A, Solanes A, Munté E, Capellà G, Pineda M, Feliubadaló L, Brunet J, Lázaro C. ERCC3, a new ovarian cancer susceptibility gene? Eur J Cancer. 2020;141:1–8. doi: 10.1016/j.ejca.2020.09.023. [DOI] [PubMed] [Google Scholar]
- 73.Lynch HT, Krush AJ, Larsen AL. Heredity and multiple primary malignant neoplasms: six cancer families. Am J Med Sci. 1967;254:322–329. doi: 10.1097/00000441-196709000-00007. [DOI] [PubMed] [Google Scholar]
- 74.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 75.Yanus GA, Belyaeva AV, Ivantsov AO, Kuligina ESh, Suspitsin EN, Mitiushkina NV, Aleksakhina SN, Iyevleva AG, Zaitseva OA, Yatsuk OS, Gorodnova TV, Strelkova TN, Efremova SA, Lepenchuk AY, Ochir-Garyaev AN, Paneyah MB, Matsko DE, Togo AV, Imyanitov EN. Pattern of clinically relevant mutations in consecutive series of Russian colorectal cancer patients. Med Oncol. 2013;30:686. doi: 10.1007/s12032-013-0686-5. [DOI] [PubMed] [Google Scholar]
- 76.Valle L, Vilar E, Tavtigian SV, Stoffel EM. Genetic predisposition to colorectal cancer: syndromes, genes, classification of genetic variants and implications for precision medicine. J Pathol. 2019;247:574–588. doi: 10.1002/path.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cohen SA, Pritchard CC, Jarvik GP. Lynch Syndrome: From Screening to Diagnosis to Treatment in the Era of Modern Molecular Oncology. Annu Rev Genomics Hum Genet. 2019;20:293–307. doi: 10.1146/annurev-genom-083118-015406. [DOI] [PubMed] [Google Scholar]
- 78.Clark SK. Management of genetically determined colorectal cancer. Surgeon. 2019;17:165–171. doi: 10.1016/j.surge.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 79.Terradas M, Capellá G, Valle L. Dominantly Inherited Hereditary Nonpolyposis Colorectal Cancer Not Caused by MMR Genes. J Clin Med. 2020;9 doi: 10.3390/jcm9061954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, Middha S, Hechtman J, Zehir A, Dubard-Gault M, Tran C, Stewart C, Sheehan M, Penson A, DeLair D, Yaeger R, Vijai J, Mukherjee S, Galle J, Dickson MA, Janjigian Y, O'Reilly EM, Segal N, Saltz LB, Reidy-Lagunes D, Varghese AM, Bajorin D, Carlo MI, Cadoo K, Walsh MF, Weiser M, Aguilar JG, Klimstra DS, Diaz LA Jr, Baselga J, Zhang L, Ladanyi M, Hyman DM, Solit DB, Robson ME, Taylor BS, Offit K, Berger MF, Stadler ZK. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019;37:286–295. doi: 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–162. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cavazza A, Radia C, Harlow C, Monahan KJ. Experience of the implementation and outcomes of universal testing for Lynch syndrome in the United Kingdom. Colorectal Dis. 2019;21:760–766. doi: 10.1111/codi.14597. [DOI] [PubMed] [Google Scholar]
- 83.Palles C, West HD, Chew E, Galavotti S, Flensburg C, Grolleman JE, Jansen EAM, Curley H, Chegwidden L, Arbe-Barnes EH, Lander N, Truscott R, Pagan J, Bajel A, Sherwood K, Martin L, Thomas H, Georgiou D, Fostira F, Goldberg Y, Adams DJ, van der Biezen SAM, Christie M, Clendenning M, Thomas LE, Deltas C, Dimovski AJ, Dymerska D, Lubinski J, Mahmood K, van der Post RS, Sanders M, Weitz J, Taylor JC, Turnbull C, Vreede L, van Wezel T, Whalley C, Arnedo-Pac C, Caravagna G, Cross W, Chubb D, Frangou A, Gruber AJ, Kinnersley B, Noyvert B, Church D, Graham T, Houlston R, Lopez-Bigas N, Sottoriva A, Wedge D Genomics England Research Consortium; CORGI Consortium; WGS500 Consortium, Jenkins MA, Kuiper RP, Roberts AW, Cheadle JP, Ligtenberg MJL, Hoogerbrugge N, Koelzer VH, Rivas AD, Winship IM, Ponte CR, Buchanan DD, Power DG, Green A, Tomlinson IPM, Sampson JR, Majewski IJ, de Voer RM. Germline MBD4 deficiency causes a multi-tumor predisposition syndrome. Am J Hum Genet. 2022;109:953–960. doi: 10.1016/j.ajhg.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samadder NJ, Baffy N, Giridhar KV, Couch FJ, Riegert-Johnson D. Hereditary Cancer Syndromes-A Primer on Diagnosis and Management, Part 2: Gastrointestinal Cancer Syndromes. Mayo Clin Proc. 2019;94:1099–1116. doi: 10.1016/j.mayocp.2019.01.042. [DOI] [PubMed] [Google Scholar]
- 85.Daca Alvarez M, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The Inherited and Familial Component of Early-Onset Colorectal Cancer. Cells. 2021;10 doi: 10.3390/cells10030710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yanus GA, Akhapkina TA, Ivantsov AO, Preobrazhenskaya EV, Aleksakhina SN, Bizin IV, Sokolenko AP, Mitiushkina NV, Kuligina ES, Suspitsin EN, Venina AR, Holmatov MM, Zaitseva OA, Yatsuk OS, Pashkov DV, Belyaev AM, Togo AV, Imyanitov EN, Iyevleva AG. Spectrum of APC and MUTYH germ-line mutations in Russian patients with colorectal malignancies. Clin Genet. 2018;93:1015–1021. doi: 10.1111/cge.13228. [DOI] [PubMed] [Google Scholar]
- 87.Zarkavelis G, Boussios S, Papadaki A, Katsanos KH, Christodoulou DK, Pentheroudakis G. Current and future biomarkers in colorectal cancer. Ann Gastroenterol. 2017;30:613–621. doi: 10.20524/aog.2017.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–648. doi: 10.1016/S0140-6736(20)31288-5. [DOI] [PubMed] [Google Scholar]
- 89.Cosma LS, Schlosser S, Tews HC, Müller M, Kandulski A. Hereditary Diffuse Gastric Cancer: Molecular Genetics, Biological Mechanisms and Current Therapeutic Approaches. Int J Mol Sci. 2022;23 doi: 10.3390/ijms23147821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 91.Blair VR, McLeod M, Carneiro F, Coit DG, D'Addario JL, van Dieren JM, Harris KL, Hoogerbrugge N, Oliveira C, van der Post RS, Arnold J, Benusiglio PR, Bisseling TM, Boussioutas A, Cats A, Charlton A, Schreiber KEC, Davis JL, Pietro MD, Fitzgerald RC, Ford JM, Gamet K, Gullo I, Hardwick RH, Huntsman DG, Kaurah P, Kupfer SS, Latchford A, Mansfield PF, Nakajima T, Parry S, Rossaak J, Sugimura H, Svrcek M, Tischkowitz M, Ushijima T, Yamada H, Yang HK, Claydon A, Figueiredo J, Paringatai K, Seruca R, Bougen-Zhukov N, Brew T, Busija S, Carneiro P, DeGregorio L, Fisher H, Gardner E, Godwin TD, Holm KN, Humar B, Lintott CJ, Monroe EC, Muller MD, Norero E, Nouri Y, Paredes J, Sanches JM, Schulpen E, Ribeiro AS, Sporle A, Whitworth J, Zhang L, Reeve AE, Guilford P. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 2020;21:e386–e397. doi: 10.1016/S1470-2045(20)30219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Adib E, El Zarif T, Nassar AH, Akl EW, Abou Alaiwi S, Mouhieddine TH, Esplin ED, Hatchell K, Nielsen SM, Rana HQ, Choueiri TK, Kwiatkowski DJ, Sonpavde G. CDH1 germline variants are enriched in patients with colorectal cancer, gastric cancer, and breast cancer. Br J Cancer. 2022;126:797–803. doi: 10.1038/s41416-021-01673-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sahasrabudhe R, Lott P, Bohorquez M, Toal T, Estrada AP, Suarez JJ, Brea-Fernández A, Cameselle-Teijeiro J, Pinto C, Ramos I, Mantilla A, Prieto R, Corvalan A, Norero E, Alvarez C, Tapia T, Carvallo P, Gonzalez LM, Cock-Rada A, Solano A, Neffa F, Della Valle A, Yau C, Soares G, Borowsky A, Hu N, He LJ, Han XY Latin American Gastric Cancer Genetics Collaborative Group, Taylor PR, Goldstein AM, Torres J, Echeverry M, Ruiz-Ponte C, Teixeira MR, Carvajal-Carmona LG. Germline Mutations in PALB2, BRCA1, and RAD51C, Which Regulate DNA Recombination Repair, in Patients With Gastric Cancer. Gastroenterology. 2017;152:983–986.e6. doi: 10.1053/j.gastro.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fewings E, Larionov A, Redman J, Goldgraben MA, Scarth J, Richardson S, Brewer C, Davidson R, Ellis I, Evans DG, Halliday D, Izatt L, Marks P, McConnell V, Verbist L, Mayes R, Clark GR, Hadfield J, Chin SF, Teixeira MR, Giger OT, Hardwick R, di Pietro M, O'Donovan M, Pharoah P, Caldas C, Fitzgerald RC, Tischkowitz M. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol Hepatol. 2018;3:489–498. doi: 10.1016/S2468-1253(18)30079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, Dunning AM, Redman J, Scarth J, Plaskocinska I, Luccarini C, Shah M, Pooley K, Dorling L, Lee A, Adank MA, Adlard J, Aittomäki K, Andrulis IL, Ang P, Barwell J, Bernstein JL, Bobolis K, Borg Å, Blomqvist C, Claes KBM, Concannon P, Cuggia A, Culver JO, Damiola F, de Pauw A, Diez O, Dolinsky JS, Domchek SM, Engel C, Evans DG, Fostira F, Garber J, Golmard L, Goode EL, Gruber SB, Hahnen E, Hake C, Heikkinen T, Hurley JE, Janavicius R, Kleibl Z, Kleiblova P, Konstantopoulou I, Kvist A, Laduca H, Lee ASG, Lesueur F, Maher ER, Mannermaa A, Manoukian S, McFarland R, McKinnon W, Meindl A, Metcalfe K, Mohd Taib NA, Moilanen J, Nathanson KL, Neuhausen S, Ng PS, Nguyen-Dumont T, Nielsen SM, Obermair F, Offit K, Olopade OI, Ottini L, Penkert J, Pylkäs K, Radice P, Ramus SJ, Rudaitis V, Side L, Silva-Smith R, Silvestri V, Skytte AB, Slavin T, Soukupova J, Tondini C, Trainer AH, Unzeitig G, Usha L, van Overeem Hansen T, Whitworth J, Wood M, Yip CH, Yoon SY, Yussuf A, Zogopoulos G, Goldgar D, Hopper JL, Chenevix-Trench G, Pharoah P, George SHL, Balmaña J, Houdayer C, James P, El-Haffaf Z, Ehrencrona H, Janatova M, Peterlongo P, Nevanlinna H, Schmutzler R, Teo SH, Robson M, Pal T, Couch F, Weitzel JN, Elliott A, Southey M, Winqvist R, Easton DF, Foulkes WD, Antoniou AC, Tischkowitz M. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J Clin Oncol. 2020;38:674–685. doi: 10.1200/JCO.19.01907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moiseyenko VM, Volkov NM, Suspistin EN, Yanus GA, Iyevleva AG, Kuligina ESh, Togo AV, Kornilov AV, Ivantsov AO, Imyanitov EN. Evidence for predictive role of BRCA1 and bTUBIII in gastric cancer. Med Oncol. 2013;30:545. doi: 10.1007/s12032-013-0545-4. [DOI] [PubMed] [Google Scholar]
- 97.Li S, Silvestri V, Leslie G, Rebbeck TR, Neuhausen SL, Hopper JL, Nielsen HR, Lee A, Yang X, McGuffog L, Parsons MT, Andrulis IL, Arnold N, Belotti M, Borg Å, Buecher B, Buys SS, Caputo SM, Chung WK, Colas C, Colonna SV, Cook J, Daly MB, de la Hoya M, de Pauw A, Delhomelle H, Eason J, Engel C, Evans DG, Faust U, Fehm TN, Fostira F, Fountzilas G, Frone M, Garcia-Barberan V, Garre P, Gauthier-Villars M, Gehrig A, Glendon G, Goldgar DE, Golmard L, Greene MH, Hahnen E, Hamann U, Hanson H, Hassan T, Hentschel J, Horvath J, Izatt L, Janavicius R, Jiao Y, John EM, Karlan BY, Kim SW, Konstantopoulou I, Kwong A, Laugé A, Lee JW, Lesueur F, Mebirouk N, Meindl A, Mouret-Fourme E, Musgrave H, Ngeow Yuen Yie J, Niederacher D, Park SK, Pedersen IS, Ramser J, Ramus SJ, Rantala J, Rashid MU, Reichl F, Ritter J, Rump A, Santamariña M, Saule C, Schmidt G, Schmutzler RK, Senter L, Shariff S, Singer CF, Southey MC, Stoppa-Lyonnet D, Sutter C, Tan Y, Teo SH, Terry MB, Thomassen M, Tischkowitz M, Toland AE, Torres D, Vega A, Wagner SA, Wang-Gohrke S, Wappenschmidt B, Weber BHF, Yannoukakos D, Spurdle AB, Easton DF, Chenevix-Trench G, Ottini L, Antoniou AC. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J Clin Oncol. 2022;40:1529–1541. doi: 10.1200/JCO.21.02112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lerner BA, Llor X. Genetic Gastric Cancer Risk Syndromes. Curr Treat Options Gastroenterol. 2020;18:604–615. doi: 10.1007/s11938-020-00312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, Lilyquist J, Na J, Moore R, Antwi SO, Bamlet WR, Chaffee KG, DiCarlo J, Wu Z, Samara R, Kasi PM, McWilliams RR, Petersen GM, Couch FJ. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA. 2018;319:2401–2409. doi: 10.1001/jama.2018.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Llach J, Carballal S, Moreira L. Familial Pancreatic Cancer: Current Perspectives. Cancer Manag Res. 2020;12:743–758. doi: 10.2147/CMAR.S172421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abe K, Kitago M, Kitagawa Y, Hirasawa A. Hereditary pancreatic cancer. Int J Clin Oncol. 2021;26:1784–1792. doi: 10.1007/s10147-021-02015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Astiazaran-Symonds E, Goldstein AM. A systematic review of the prevalence of germline pathogenic variants in patients with pancreatic cancer. J Gastroenterol. 2021;56:713–721. doi: 10.1007/s00535-021-01806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–275. doi: 10.1002/cncr.29041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, Gallinger S, Schwartz AG, Syngal S, Cote ML, Axilbund J, Schulick R, Ali SZ, Eshleman JR, Velculescu VE, Goggins M, Vogelstein B, Papadopoulos N, Hruban RH, Kinzler KW, Klein AP. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hsu FC, Roberts NJ, Childs E, Porter N, Rabe KG, Borgida A, Ukaegbu C, Goggins MG, Hruban RH, Zogopoulos G, Syngal S, Gallinger S, Petersen GM, Klein AP. Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol. 2021;7:1664–1668. doi: 10.1001/jamaoncol.2021.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gardiner A, Kidd J, Elias MC, Young K, Mabey B, Taherian N, Cummings S, Malafa M, Rosenthal E, Permuth JB. Pancreatic Ductal Carcinoma Risk Associated With Hereditary Cancer-Risk Genes. J Natl Cancer Inst. 2022;114:996–1002. doi: 10.1093/jnci/djac069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zeng C, Bastarache LA, Tao R, Venner E, Hebbring S, Andujar JD, Bland ST, Crosslin DR, Pratap S, Cooley A, Pacheco JA, Christensen KD, Perez E, Zawatsky CLB, Witkowski L, Zouk H, Weng C, Leppig KA, Sleiman PMA, Hakonarson H, Williams MS, Luo Y, Jarvik GP, Green RC, Chung WK, Gharavi AG, Lennon NJ, Rehm HL, Gibbs RA, Peterson JF, Roden DM, Wiesner GL, Denny JC. Association of Pathogenic Variants in Hereditary Cancer Genes With Multiple Diseases. JAMA Oncol. 2022;8:835–844. doi: 10.1001/jamaoncol.2022.0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chan SH, Chiang J, Ngeow J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hered Cancer Clin Pract. 2021;19:21. doi: 10.1186/s13053-021-00178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nissim S, Leshchiner I, Mancias JD, Greenblatt MB, Maertens O, Cassa CA, Rosenfeld JA, Cox AG, Hedgepeth J, Wucherpfennig JI, Kim AJ, Henderson JE, Gonyo P, Brandt A, Lorimer E, Unger B, Prokop JW, Heidel JR, Wang XX, Ukaegbu CI, Jennings BC, Paulo JA, Gableske S, Fierke CA, Getz G, Sunyaev SR, Wade Harper J, Cichowski K, Kimmelman AC, Houvras Y, Syngal S, Williams C, Goessling W. Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat Genet. 2019;51:1308–1314. doi: 10.1038/s41588-019-0475-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roberts NJ, Grant RC, Gallinger S, Klein AP Familial Pancreatic Cancer Genome Sequencing Project. Germline sequence analysis of RABL3 in a large series of pancreatic ductal adenocarcinoma patients reveals no evidence of deleterious variants. Genes Chromosomes Cancer. 2021;60:559–564. doi: 10.1002/gcc.22947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nicolosi P, Ledet E, Yang S, Michalski S, Freschi B, O'Leary E, Esplin ED, Nussbaum RL, Sartor O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019;5:523–528. doi: 10.1001/jamaoncol.2018.6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bree KK, Hensley PJ, Pettaway CA. Germline Predisposition to Prostate Cancer in Diverse Populations. Urol Clin North Am. 2021;48:411–423. doi: 10.1016/j.ucl.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 114.Bhanji Y, Isaacs WB, Xu J, Cooney KA. Prostate Cancer Predisposition. Urol Clin North Am. 2021;48:283–296. doi: 10.1016/j.ucl.2021.03.001. [DOI] [PubMed] [Google Scholar]
- 115.Nyberg T, Tischkowitz M, Antoniou AC. BRCA1 and BRCA2 pathogenic variants and prostate cancer risk: systematic review and meta-analysis. Br J Cancer. 2022;126:1067–1081. doi: 10.1038/s41416-021-01675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Silvestri V, Leslie G, Barnes DR CIMBA Group, Agnarsson BA, Aittomäki K, Alducci E, Andrulis IL, Barkardottir RB, Barroso A, Barrowdale D, Benitez J, Bonanni B, Borg A, Buys SS, Caldés T, Caligo MA, Capalbo C, Campbell I, Chung WK, Claes KBM, Colonna SV, Cortesi L, Couch FJ, de la Hoya M, Diez O, Ding YC, Domchek S, Easton DF, Ejlertsen B, Engel C, Evans DG, Feliubadalò L, Foretova L, Fostira F, Géczi L, Gerdes AM, Glendon G, Godwin AK, Goldgar DE, Hahnen E, Hogervorst FBL, Hopper JL, Hulick PJ, Isaacs C, Izquierdo A, James PA, Janavicius R, Jensen UB, John EM, Joseph V, Konstantopoulou I, Kurian AW, Kwong A, Landucci E, Lesueur F, Loud JT, Machackova E, Mai PL, Majidzadeh-A K, Manoukian S, Montagna M, Moserle L, Mulligan AM, Nathanson KL, Nevanlinna H, Ngeow J, Nikitina-Zake L, Offit K, Olah E, Olopade OI, Osorio A, Papi L, Park SK, Pedersen IS, Perez-Segura P, Petersen AH, Pinto P, Porfirio B, Pujana MA, Radice P, Rantala J, Rashid MU, Rosenzweig B, Rossing M, Santamariña M, Schmutzler RK, Senter L, Simard J, Singer CF, Solano AR, Southey MC, Steele L, Steinsnyder Z, Stoppa-Lyonnet D, Tan YY, Teixeira MR, Teo SH, Terry MB, Thomassen M, Toland AE, Torres-Esquius S, Tung N, van Asperen CJ, Vega A, Viel A, Vierstraete J, Wappenschmidt B, Weitzel JN, Wieme G, Yoon SY, Zorn KK, McGuffog L, Parsons MT, Hamann U, Greene MH, Kirk JA, Neuhausen SL, Rebbeck TR, Tischkowitz M, Chenevix-Trench G, Antoniou AC, Friedman E, Ottini L. Characterization of the Cancer Spectrum in Men With Germline BRCA1 and BRCA2 Pathogenic Variants: Results From the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) JAMA Oncol. 2020;6:1218–1230. doi: 10.1001/jamaoncol.2020.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Momozawa Y, Iwasaki Y, Hirata M, Liu X, Kamatani Y, Takahashi A, Sugano K, Yoshida T, Murakami Y, Matsuda K, Nakagawa H, Spurdle AB, Kubo M. Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12 366 Controls. J Natl Cancer Inst. 2020;112:369–376. doi: 10.1093/jnci/djz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bancroft EK, Page EC, Brook MN, Thomas S, Taylor N, Pope J, McHugh J, Jones AB, Karlsson Q, Merson S, Ong KR, Hoffman J, Huber C, Maehle L, Grindedal EM, Stormorken A, Evans DG, Rothwell J, Lalloo F, Brady AF, Bartlett M, Snape K, Hanson H, James P, McKinley J, Mascarenhas L, Syngal S, Ukaegbu C, Side L, Thomas T, Barwell J, Teixeira MR, Izatt L, Suri M, Macrae FA, Poplawski N, Chen-Shtoyerman R, Ahmed M, Musgrave H, Nicolai N, Greenhalgh L, Brewer C, Pachter N, Spigelman AD, Azzabi A, Helfand BT, Halliday D, Buys S, Ramon Y Cajal T, Donaldson A, Cooney KA, Harris M, McGrath J, Davidson R, Taylor A, Cooke P, Myhill K, Hogben M, Aaronson NK, Ardern-Jones A, Bangma CH, Castro E, Dearnaley D, Dias A, Dudderidge T, Eccles DM, Green K, Eyfjord J, Falconer A, Foster CS, Gronberg H, Hamdy FC, Johannsson O, Khoo V, Lilja H, Lindeman GJ, Lubinski J, Axcrona K, Mikropoulos C, Mitra AV, Moynihan C, Ni Raghallaigh H, Rennert G, Collier R IMPACT Study Collaborators, Offman J, Kote-Jarai Z, Eeles RA. A prospective prostate cancer screening programme for men with pathogenic variants in mismatch repair genes (IMPACT): initial results from an international prospective study. Lancet Oncol. 2021;22:1618–1631. doi: 10.1016/S1470-2045(21)00522-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carlo MI, Mukherjee S, Mandelker D, Vijai J, Kemel Y, Zhang L, Knezevic A, Patil S, Ceyhan-Birsoy O, Huang KC, Redzematovic A, Coskey DT, Stewart C, Pradhan N, Arnold AG, Hakimi AA, Chen YB, Coleman JA, Hyman DM, Ladanyi M, Cadoo KA, Walsh MF, Stadler ZK, Lee CH, Feldman DR, Voss MH, Robson M, Motzer RJ, Offit K. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients With Advanced Renal Cell Carcinoma. JAMA Oncol. 2018;4:1228–1235. doi: 10.1001/jamaoncol.2018.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Al-Shinnag M, Marfan H, Susman R, Wakeling J, Gustafson S, Wood S, Mallett AJ. Birt-Hogg-Dubé Syndrome and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome: An Effective Multidisciplinary Approach to Hereditary Renal Cancer Predisposing Syndromes. Front Oncol. 2021;11:738822. doi: 10.3389/fonc.2021.738822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shuch B, Zhang J. Genetic Predisposition to Renal Cell Carcinoma: Implications for Counseling, Testing, Screening, and Management. J Clin Oncol. 2018:JCO2018792523. doi: 10.1200/JCO.2018.79.2523. [DOI] [PubMed] [Google Scholar]
- 122.de Alencar VTL, Formiga MN, de Lima VCC. Inherited lung cancer: a review. Ecancermedicalscience. 2020;14:1008. doi: 10.3332/ecancer.2020.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Benusiglio PR, Fallet V, Sanchis-Borja M, Coulet F, Cadranel J. Lung cancer is also a hereditary disease. Eur Respir Rev. 2021;30 doi: 10.1183/16000617.0045-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 126.Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, Sharma SV, Brannigan BW, Mohapatra G, Settleman J, Haber DA. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37:1315–1316. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 127.Lavdovskaia ED, Iyevleva AG, Sokolenko AP, Mitiushkina NV, Preobrazhenskaya EV, Tiurin VI, Ivantsov AO, Bizin IV, Stelmakh LV, Moiseyenko FV, Karaseva NA, Orlov SV, Moiseyenko VM, Korzhenevskaya MA, Zaitsev IA, Kozak AR, Chistyakov IV, Akopov AL, Volkov NM, Togo AV, Imyanitov EN. EGFR T790M Mutation in TKI-Naïve Clinical Samples: Frequency, Tissue Mosaicism, Predictive Value and Awareness on Artifacts. Oncol Res Treat. 2018;41:634–642. doi: 10.1159/000491441. [DOI] [PubMed] [Google Scholar]
- 128.Lu S, Yu Y, Li Z, Yu R, Wu X, Bao H, Ding Y, Shao YW, Jian H. EGFR and ERBB2 Germline Mutations in Chinese Lung Cancer Patients and Their Roles in Genetic Susceptibility to Cancer. J Thorac Oncol. 2019;14:732–736. doi: 10.1016/j.jtho.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 129.Mezquita L, Jové M, Nadal E, Kfoury M, Morán T, Ricordel C, Dhooge M, Tlemsani C, Léna H, Teulé A, Álvarez JV, Raimbourg J, Hiret S, Lacroix L, Menéndez M, Saldaña J, Brunet J, Lianes P, Coupier I, Auclin E, Recondo G, Friboulet L, Adam J, Green E, Planchard D, Frébourg T, Capellà G, Rouleau E, Lázaro C, Caron O, Besse B. High Prevalence of Somatic Oncogenic Driver Alterations in Patients With NSCLC and Li-Fraumeni Syndrome. J Thorac Oncol. 2020;15:1232–1239. doi: 10.1016/j.jtho.2020.03.005. [DOI] [PubMed] [Google Scholar]
- 130.Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87:809–811. [PubMed] [Google Scholar]
- 131.Potjer TP, van der Stoep N, Houwing-Duistermaat JJ, Konings IC, Aalfs CM, van den Akker PC, Ausems MG, Dommering CJ, van der Kolk LE, Maiburg MC, Spruijt L, Wagner A, Vasen HF, Hes FJ. Pancreatic cancer-associated gene polymorphisms in a nation-wide cohort of p16-Leiden germline mutation carriers; a case-control study. BMC Res Notes. 2015;8:264. doi: 10.1186/s13104-015-1235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Revythis A, Shah S, Kutka M, Moschetta M, Ozturk MA, Pappas-Gogos G, Ioannidou E, Sheriff M, Rassy E, Boussios S. Unraveling the Wide Spectrum of Melanoma Biomarkers. Diagnostics (Basel) 2021;11 doi: 10.3390/diagnostics11081341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rossi M, Pellegrini C, Cardelli L, Ciciarelli V, Di Nardo L, Fargnoli MC. Familial Melanoma: Diagnostic and Management Implications. Dermatol Pract Concept. 2019;9:10–16. doi: 10.5826/dpc.0901a03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Al-Salameh A, Cadiot G, Calender A, Goudet P, Chanson P. Clinical aspects of multiple endocrine neoplasia type 1. Nat Rev Endocrinol. 2021;17:207–224. doi: 10.1038/s41574-021-00468-3. [DOI] [PubMed] [Google Scholar]
- 135.Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15:150. doi: 10.1038/nrclinonc.2017.188. [DOI] [PubMed] [Google Scholar]
- 136.Subbiah V, Yang D, Velcheti V, Drilon A, Meric-Bernstam F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J Clin Oncol. 2020;38:1209–1221. doi: 10.1200/JCO.19.02551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bouys L, Bertherat J. MANAGEMENT OF ENDOCRINE DISEASE: Carney complex: clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur J Endocrinol. 2021;184:R99–R109. doi: 10.1530/EJE-20-1120. [DOI] [PubMed] [Google Scholar]
- 138.Zhao Y, Yu T, Sun J, Wang F, Cheng C, He S, Chen L, Xie D, Fu L, Guan X, Yan A, Li Y, Miao G, Zhu X. Germ-line mutations in WDR77 predispose to familial papillary thyroid cancer. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2026327118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Muth A, Crona J, Gimm O, Elmgren A, Filipsson K, Stenmark Askmalm M, Sandstedt J, Tengvar M, Tham E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med. 2019;285:187–204. doi: 10.1111/joim.12869. [DOI] [PubMed] [Google Scholar]
- 140.Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, Young J, Guiochon-Mantel A, Chanson P. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab. 2012;97:E663–E670. doi: 10.1210/jc.2011-2291. [DOI] [PubMed] [Google Scholar]
- 141.Chang M, Yang C, Bao X, Wang R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front Endocrinol (Lausanne) 2020;11:596554. doi: 10.3389/fendo.2020.596554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hahn EC, Bittar CM, Vianna FSL, Netto CBO, Biazús JV, Cericatto R, Cavalheiro JA, de Melo MP, Menke CH, Rabin E, Leistner-Segal S, Ashton-Prolla P. TP53 p.Arg337His germline mutation prevalence in Southern Brazil: Further evidence for mutation testing in young breast cancer patients. PLoS One. 2018;13:e0209934. doi: 10.1371/journal.pone.0209934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rocca V, Blandino G, D'Antona L, Iuliano R, Di Agostino S. Li-Fraumeni Syndrome: Mutation of TP53 Is a Biomarker of Hereditary Predisposition to Tumor: New Insights and Advances in the Treatment. Cancers (Basel) 2022;14 doi: 10.3390/cancers14153664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, Litton JK, Olopade OI, Hortobagyi GN, Strong LC, Arun BK. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer. 2012;118:908–913. doi: 10.1002/cncr.26377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sokolova TN, Breder VV, Shumskaya IS, Suspitsin EN, Aleksakhina SN, Yanus GA, Tiurin VI, Ivantsov AO, Vona B, Raskin GA, Gamajunov SV, Imyanitov EN. Revisiting multiple erroneous genetic testing results and clinical misinterpretations in a patient with Li-Fraumeni syndrome: lessons for translational medicine. Hered Cancer Clin Pract. 2021;19:2. doi: 10.1186/s13053-020-00157-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kratz CP, Achatz MI, Brugières L, Frebourg T, Garber JE, Greer MC, Hansford JR, Janeway KA, Kohlmann WK, McGee R, Mullighan CG, Onel K, Pajtler KW, Pfister SM, Savage SA, Schiffman JD, Schneider KA, Strong LC, Evans DGR, Wasserman JD, Villani A, Malkin D. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res. 2017;23:e38–e45. doi: 10.1158/1078-0432.CCR-17-0408. [DOI] [PubMed] [Google Scholar]
- 147.Lin YH, Jewell BE, Gingold J, Lu L, Zhao R, Wang LL, Lee DF. Osteosarcoma: Molecular Pathogenesis and iPSC Modeling. Trends Mol Med. 2017;23:737–755. doi: 10.1016/j.molmed.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Evans DG, Woodward ER, Bajalica-Lagercrantz S, Oliveira C, Frebourg T. Germline TP53 Testing in Breast Cancers: Why, When and How? Cancers (Basel) 2020;12 doi: 10.3390/cancers12123762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M, Gianferante M, Spector LG, Morton LM, Karyadi D, Robison LL, Armstrong GT, Bhatia S, Song L, Pankratz N, Pinheiro M, Gastier-Foster JM, Gorlick R, de Toledo SRC, Petrilli AS, Patino-Garcia A, Lecanda F, Gutierrez-Jimeno M, Serra M, Hattinger C, Picci P, Scotlandi K, Flanagan AM, Tirabosco R, Amary MF, Kurucu N, Ilhan IE, Ballinger ML, Thomas DM, Barkauskas DA, Mejia-Baltodano G, Valverde P, Hicks BD, Wang M, Hutchinson AA, Tucker M, Sampson J, Landi MT, Freedman ND, Gapstur S, Carter B, Hoover RN, Chanock SJ, Savage SA. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol. 2020;6:724–734. doi: 10.1001/jamaoncol.2020.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rogoża-Janiszewska E, Malińska K, Górski B, Scott RJ, Cybulski C, Kluźniak W, Lener M, Jakubowska A, Gronwald J, Huzarski T, Lubiński J, Dębniak T. Prevalence of germline TP53 variants among early-onset breast cancer patients from Polish population. Breast Cancer. 2021;28:226–235. doi: 10.1007/s12282-020-01151-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pilarski R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers (Basel) 2019;11 doi: 10.3390/cancers11060844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ngeow J, Eng C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb Perspect Med. 2020;10 doi: 10.1101/cshperspect.a036087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hendricks LAJ, Hoogerbrugge N, Schuurs-Hoeijmakers JHM, Vos JR. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet. 2021;99:219–225. doi: 10.1111/cge.13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Eissing M, Ripken L, Schreibelt G, Westdorp H, Ligtenberg M, Netea-Maier R, Netea MG, de Vries IJM, Hoogerbrugge N. PTEN Hamartoma Tumor Syndrome and Immune Dysregulation. Transl Oncol. 2019;12:361–367. doi: 10.1016/j.tranon.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee YR, Yehia L, Kishikawa T, Ni Y, Leach B, Zhang J, Panch N, Liu J, Wei W, Eng C, Pandolfi PP. WWP1 Gain-of-Function Inactivation of PTEN in Cancer Predisposition. N Engl J Med. 2020;382:2103–2116. doi: 10.1056/NEJMoa1914919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Klimkowski S, Ibrahim M, Ibarra Rovira JJ, Elshikh M, Javadi S, Klekers AR, Abusaif AA, Moawad AW, Ali K, Elsayes KM. Peutz-Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers (Basel) 2021;13 doi: 10.3390/cancers13205121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meserve EE, Nucci MR. Peutz-Jeghers Syndrome: Pathobiology, Pathologic Manifestations, and Suggestions for Recommending Genetic Testing in Pathology Reports. Surg Pathol Clin. 2016;9:243–268. doi: 10.1016/j.path.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 158.Daniell J, Plazzer JP, Perera A, Macrae F. An exploration of genotype-phenotype link between Peutz-Jeghers syndrome and STK11: a review. Fam Cancer. 2018;17:421–427. doi: 10.1007/s10689-017-0037-3. [DOI] [PubMed] [Google Scholar]
- 159.Onodera S, Nakamura Y, Azuma T. Gorlin Syndrome: Recent Advances in Genetic Testing and Molecular and Cellular Biological Research. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21207559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Olafsdottir T, Stacey SN, Sveinbjornsson G, Thorleifsson G, Norland K, Sigurgeirsson B, Thorisdottir K, Kristjansson AK, Tryggvadottir L, Sarin KY, Benediktsson R, Jonasson JG, Sigurdsson A, Jonasdottir A, Kristmundsdottir S, Jonsson H, Gylfason A, Oddsson A, Fridriksdottir R, Gudjonsson SA, Zink F, Lund SH, Rognvaldsson S, Melsted P, Steinthorsdottir V, Gudmundsson J, Mikaelsdottir E, Olason PI, Stefansdottir L, Eggertsson HP, Halldorsson BV, Thorsteinsdottir U, Agustsson TT, Olafsson K, Olafsson JH, Sulem P, Rafnar T, Gudbjartsson DF, Stefansson K. Loss-of-Function Variants in the Tumor-Suppressor Gene PTPN14 Confer Increased Cancer Risk. Cancer Res. 2021;81:1954–1964. doi: 10.1158/0008-5472.CAN-20-3065. [DOI] [PubMed] [Google Scholar]
- 161.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, McGee RB, Hines-Dowell S, Nuccio R, Quinn E, Shurtleff SA, Rusch M, Patel A, Becksfort JB, Wang S, Weaver MS, Ding L, Mardis ER, Wilson RK, Gajjar A, Ellison DW, Pappo AS, Pui CH, Nichols KE, Downing JR. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, Shurtleff S, Hedges DJ, Hamilton KV, Foy SG, Edmonson MN, Thrasher A, Bahrami A, Orr BA, Klco JM, Gu J, Harrison LW, Wang L, Clay MR, Ouma A, Silkov A, Liu Y, Zhang Z, Brady SW, Zhou X, Chang TC, Pande M, Davis E, Becksfort J, Patel A, Wilkinson MR, Rahbarinia D, Kubal M, Maciaszek JL, Pastor V, Knight J, Gout AM, Wang J, Gu Z, Mullighan CG, McGee RB, Quinn EA, Nuccio R, Mostafavi R, Gerhardt EL, Taylor LM, Valdez JM, Hines-Dowell SJ, Pappo AS, Robinson G, Johnson LM, Pui CH, Ellison DW, Downing JR, Zhang J, Nichols KE. Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discov. 2021;11:3008–3027. doi: 10.1158/2159-8290.CD-20-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kratz CP, Jongmans MC, Cavé H, Wimmer K, Behjati S, Guerrini-Rousseau L, Milde T, Pajtler KW, Golmard L, Gauthier-Villars M, Jewell R, Duncan C, Maher ER, Brugieres L, Pritchard-Jones K, Bourdeaut F. Predisposition to cancer in children and adolescents. Lancet Child Adolesc Health. 2021;5:142–154. doi: 10.1016/S2352-4642(20)30275-3. [DOI] [PubMed] [Google Scholar]
- 164.Fabius AWM, van Hoefen Wijsard M, van Leeuwen FE, Moll AC. Subsequent Malignant Neoplasms in Retinoblastoma Survivors. Cancers (Basel) 2021;13 doi: 10.3390/cancers13061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tu J, Huo Z, Yu Y, Zhu D, Xu A, Huang MF, Hu R, Wang R, Gingold JA, Chen YH, Tsai KL, Forcioli-Conti NR, Huang SXL, Webb TR, Su J, Bazer DA, Jia P, Yustein JT, Wang LL, Hung MC, Zhao Z, Huff CD, Shen J, Zhao R, Lee DF. Hereditary retinoblastoma iPSC model reveals aberrant spliceosome function driving bone malignancies. Proc Natl Acad Sci U S A. 2022;119:e2117857119. doi: 10.1073/pnas.2117857119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maciaszek JL, Oak N, Nichols KE. Recent advances in Wilms' tumor predisposition. Hum Mol Genet. 2020;29:R138–R149. doi: 10.1093/hmg/ddaa091. [DOI] [PubMed] [Google Scholar]
- 167.Sur ML, Armat I, Sur G, Pop DC, Samasca G, Lupan I, Timis TL, Florian IA, Sur D. Neurofibromatosis in Children: Actually and Perspectives. Children (Basel) 2022;9 doi: 10.3390/children9010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, Williams G, Bracamontes D, Messinger Y, Goodfellow PJ. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Guillerman RP, Foulkes WD, Priest JR. Imaging of DICER1 syndrome. Pediatr Radiol. 2019;49:1488–1505. doi: 10.1007/s00247-019-04429-x. [DOI] [PubMed] [Google Scholar]
- 170.Stewart DR, Best AF, Williams GM, Harney LA, Carr AG, Harris AK, Kratz CP, Dehner LP, Messinger YH, Rosenberg PS, Hill DA, Schultz KAP. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J Clin Oncol. 2019;37:668–676. doi: 10.1200/JCO.2018.78.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Plon SE, Lupo PJ. Genetic Predisposition to Childhood Cancer in the Genomic Era. Annu Rev Genomics Hum Genet. 2019;20:241–263. doi: 10.1146/annurev-genom-083118-015415. [DOI] [PubMed] [Google Scholar]
- 172.Sestini R, Bacci C, Provenzano A, Genuardi M, Papi L. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum Mutat. 2008;29:227–231. doi: 10.1002/humu.20679. [DOI] [PubMed] [Google Scholar]
- 173.Tabori U, Hansford JR, Achatz MI, Kratz CP, Plon SE, Frebourg T, Brugières L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin Cancer Res. 2017;23:e32–e37. doi: 10.1158/1078-0432.CCR-17-0574. [DOI] [PubMed] [Google Scholar]
- 174.Mangaonkar AA, Patnaik MM. Hereditary Predisposition to Hematopoietic Neoplasms: When Bloodline Matters for Blood Cancers. Mayo Clin Proc. 2020;95:1482–1498. doi: 10.1016/j.mayocp.2019.12.013. [DOI] [PubMed] [Google Scholar]
- 175.Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21:122–137. doi: 10.1038/s41568-020-00315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Szmyd B, Mlynarski W, Pastorczak A. Genetic predisposition to lymphomas: Overview of rare syndromes and inherited familial variants. Mutat Res Rev Mutat Res. 2021;788:108386. doi: 10.1016/j.mrrev.2021.108386. [DOI] [PubMed] [Google Scholar]
- 177.Samadder NJ, Giridhar KV, Baffy N, Riegert-Johnson D, Couch FJ. Hereditary Cancer Syndromes-A Primer on Diagnosis and Management: Part 1: Breast-Ovarian Cancer Syndromes. Mayo Clin Proc. 2019;94:1084–1098. doi: 10.1016/j.mayocp.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 178.Marmolejo DH, Wong MYZ, Bajalica-Lagercrantz S, Tischkowitz M, Balmaña J extended ERN-GENTURIS Thematic Group 3. Overview of hereditary breast and ovarian cancer (HBOC) guidelines across Europe. Eur J Med Genet. 2021;64:104350. doi: 10.1016/j.ejmg.2021.104350. [DOI] [PubMed] [Google Scholar]
- 179.Piombino C, Cortesi L, Lambertini M, Punie K, Grandi G, Toss A. Secondary Prevention in Hereditary Breast and/or Ovarian Cancer Syndromes Other Than BRCA. J Oncol. 2020;2020:6384190. doi: 10.1155/2020/6384190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pouptsis A, Swafe L, Patwardhan M, Stavraka C. Surgical and Systemic Treatment of Hereditary Breast Cancer: A Mini-Review With a Focus on BRCA1 and BRCA2 Mutations. Front Oncol. 2020;10:553080. doi: 10.3389/fonc.2020.553080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Al-Sarhani H, Gottumukkala RV, Grasparil ADS 2nd, Tung EL, Gee MS, Greer MC. Screening of cancer predisposition syndromes. Pediatr Radiol. 2022;52:401–417. doi: 10.1007/s00247-021-05023-w. [DOI] [PubMed] [Google Scholar]
- 182.Møller P, Stormorken A, Jonsrud C, Holmen MM, Hagen AI, Clark N, Vabø A, Sun P, Narod SA, Mæhle L. Survival of patients with BRCA1-associated breast cancer diagnosed in an MRI-based surveillance program. Breast Cancer Res Treat. 2013;139:155–161. doi: 10.1007/s10549-013-2540-z. [DOI] [PubMed] [Google Scholar]
- 183.Jacobson M, Coakley N, Bernardini M, Branco KA, Elit L, Ferguson S, Kim R. Risk reduction strategies for BRCA1/2 hereditary ovarian cancer syndromes: a clinical practice guideline. Hered Cancer Clin Pract. 2021;19:39. doi: 10.1186/s13053-021-00196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Machens A, Dralle H. Long-term outcome after DNA-based prophylactic neck surgery in children at risk of hereditary medullary thyroid cancer. Best Pract Res Clin Endocrinol Metab. 2019;33:101274. doi: 10.1016/j.beem.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 185.Mathiesen JS, Effraimidis G, Rossing M, Rasmussen ÅK, Hoejberg L, Bastholt L, Godballe C, Oturai P, Feldt-Rasmussen U. Multiple endocrine neoplasia type 2: A review. Semin Cancer Biol. 2022;79:163–179. doi: 10.1016/j.semcancer.2021.03.035. [DOI] [PubMed] [Google Scholar]
- 186.Eleje GU, Eke AC, Ezebialu IU, Ikechebelu JI, Ugwu EO, Okonkwo OO. Risk-reducing bilateral salpingo-oophorectomy in women with BRCA1 or BRCA2 mutations. Cochrane Database Syst Rev. 2018;8:CD012464. doi: 10.1002/14651858.CD012464.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Heemskerk-Gerritsen BAM, Jager A, Koppert LB, Obdeijn AI, Collée M, Meijers-Heijboer HEJ, Jenner DJ, Oldenburg HSA, van Engelen K, de Vries J, van Asperen CJ, Devilee P, Blok MJ, Kets CM, Ausems MGEM, Seynaeve C, Rookus MA, Hooning MJ. Survival after bilateral risk-reducing mastectomy in healthy BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat. 2019;177:723–733. doi: 10.1007/s10549-019-05345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Iyevleva AG, Imyanitov EN. Cytotoxic and targeted therapy for hereditary cancers. Hered Cancer Clin Pract. 2016;14:17. doi: 10.1186/s13053-016-0057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Thavaneswaran S, Rath E, Tucker K, Joshua AM, Hess D, Pinese M, Ballinger ML, Thomas DM. Therapeutic implications of germline genetic findings in cancer. Nat Rev Clin Oncol. 2019;16:386–396. doi: 10.1038/s41571-019-0179-3. [DOI] [PubMed] [Google Scholar]
- 190.Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wisniowski R, Siolek M, Dent R, Lubinski J, Narod S. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 191.Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, Owen J, Abraham J, Barrett S, Barrett-Lee P, Brown R, Chan S, Dowsett M, Flanagan JM, Fox L, Grigoriadis A, Gutin A, Harper-Wynne C, Hatton MQ, Hoadley KA, Parikh J, Parker P, Perou CM, Roylance R, Shah V, Shaw A, Smith IE, Timms KM, Wardley AM, Wilson G, Gillett C, Lanchbury JS, Ashworth A, Rahman N, Harries M, Ellis P, Pinder SE, Bliss JM. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med. 2018;24:628–637. doi: 10.1038/s41591-018-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Imyanitov EN. Cytotoxic and targeted therapy for BRCA1/2-driven cancers. Hered Cancer Clin Pract. 2021;19:36. doi: 10.1186/s13053-021-00193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gorodnova TV, Kotiv KB, Ivantsov AO, Mikheyeva ON, Mikhailiuk GI, Lisyanskaya AS, Mikaya NA, Guseynov KD, Bondarev NE, Matveyeva NS, Nekrasova EA, Sidoruk AA, Roman LD, Manikhas GM, Belyaev AM, Sokolenko AP, Berlev IV, Imyanitov EN. Efficacy of Neoadjuvant Therapy With Cisplatin Plus Mitomycin C in BRCA1-Mutated Ovarian Cancer. Int J Gynecol Cancer. 2018;28:1498–1506. doi: 10.1097/IGC.0000000000001352. [DOI] [PubMed] [Google Scholar]
- 194.Gorodnova TV, Sokolenko AP, Kondratiev SV, Kotiv KB, Belyaev AM, Berlev IV, Imyanitov EN. Mitomycin C plus cisplatin for systemic treatment of recurrent BRCA1-associated ovarian cancer. Invest New Drugs. 2020;38:1872–1878. doi: 10.1007/s10637-020-00965-8. [DOI] [PubMed] [Google Scholar]
- 195.Chan CY, Tan KV, Cornelissen B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin Cancer Res. 2021;27:1585–1594. doi: 10.1158/1078-0432.CCR-20-2766. [DOI] [PubMed] [Google Scholar]
- 196.Póti Á, Gyergyák H, Németh E, Rusz O, Tóth S, Kovácsházi C, Chen D, Szikriszt B, Spisák S, Takeda S, Szakács G, Szallasi Z, Richardson AL, Szüts D. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019;20:240. doi: 10.1186/s13059-019-1867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Stopsack KH. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur Urol. 2021;79:442–445. doi: 10.1016/j.eururo.2020.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Westphalen CB, Fine AD, André F, Ganesan S, Heinemann V, Rouleau E, Turnbull C, Garcia Palacios L, Lopez JA, Sokol ES, Mateo J. Pan-cancer Analysis of Homologous Recombination Repair-associated Gene Alterations and Genome-wide Loss-of-Heterozygosity Score. Clin Cancer Res. 2022;28:1412–1421. doi: 10.1158/1078-0432.CCR-21-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 200.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA Jr. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bellone S, Roque DM, Siegel ER, Buza N, Hui P, Bonazzoli E, Guglielmi A, Zammataro L, Nagarkatti N, Zaidi S, Lee J, Silasi DA, Huang GS, Andikyan V, Damast S, Clark M, Azodi M, Schwartz PE, Tymon-Rosario JR, Harold JA, Mauricio D, Zeybek B, Menderes G, Altwerger G, Ratner E, Alexandrov LB, Iwasaki A, Kong Y, Song E, Dong W, Elvin JA, Choi J, Santin AD. A phase 2 evaluation of pembrolizumab for recurrent Lynch-like versus sporadic endometrial cancers with microsatellite instability. Cancer. 2022;128:1206–1218. doi: 10.1002/cncr.34025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, García-Alfonso P, Neyns B, Luppi G, Cardin DB, Dragovich T, Shah U, Abdullaev S, Gricar J, Ledeine JM, Overman MJ, Lonardi S. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J Clin Oncol. 2022;40:161–170. doi: 10.1200/JCO.21.01015. [DOI] [PubMed] [Google Scholar]
- 203.Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, Uppaluri R, Ferguson C, Schmidt RE, Dahiya S, Ansstas G, Mardis ER, Dunn GP. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016;6:1230–1236. doi: 10.1158/2159-8290.CD-16-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Volkov NM, Yanus GA, Ivantsov AO, Moiseenko FV, Matorina OG, Bizin IV, Moiseyenko VM, Imyanitov EN. Efficacy of immune checkpoint blockade in MUTYH-associated hereditary colorectal cancer. Invest New Drugs. 2020;38:894–898. doi: 10.1007/s10637-019-00842-z. [DOI] [PubMed] [Google Scholar]
- 205.AlHarbi M, Ali Mobark N, AlMubarak L, Aljelaify R, AlSaeed M, Almutairi A, Alqubaishi F, Hussain ME, Balbaid AAO, Said Marie A, AlSubaie L, AlShieban S, alTassan N, Ramkissoon SH, Abedalthagafi M. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist. 2018;23:1401–1406. doi: 10.1634/theoncologist.2018-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pavelka Z, Zitterbart K, Nosková H, Bajčiová V, Slabý O, Štěrba J. Effective Immunotherapy of Glioblastoma in an Adolescent with Constitutional Mismatch Repair-Deficiency Syndrome. Klin Onkol. 2019;32:70–74. doi: 10.14735/amko201970. [DOI] [PubMed] [Google Scholar]
- 207.Fox E, Widemann BC, Chuk MK, Marcus L, Aikin A, Whitcomb PO, Merino MJ, Lodish M, Dombi E, Steinberg SM, Wells SA, Balis FM. Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res. 2013;19:4239–4248. doi: 10.1158/1078-0432.CCR-13-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, Worden F, Brose M, Patel J, Leboulleux S, Godbert Y, Barlesi F, Morris JC, Owonikoko TK, Tan DSW, Gautschi O, Weiss J, de la Fouchardière C, Burkard ME, Laskin J, Taylor MH, Kroiss M, Medioni J, Goldman JW, Bauer TM, Levy B, Zhu VW, Lakhani N, Moreno V, Ebata K, Nguyen M, Heirich D, Zhu EY, Huang X, Yang L, Kherani J, Rothenberg SM, Drilon A, Subbiah V, Shah MH, Cabanillas ME. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med. 2020;383:825–835. doi: 10.1056/NEJMoa2005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zheng X, Ji Q, Sun Y, Ge M, Zhang B, Cheng Y, Lei S, Shi F, Guo Y, Li L, Chen L, Shao J, Zhang W, Gao M. Efficacy and safety of selpercatinib in Chinese patients with advanced RET-altered thyroid cancers: results from the phase II LIBRETTO-321 study. Ther Adv Med Oncol. 2022;14:17588359221119318. doi: 10.1177/17588359221119318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20:1011–1022. doi: 10.1016/S1470-2045(19)30277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, Weiss B, Kim A, Bornhorst M, Shah AC, Martin S, Roderick MC, Pichard DC, Carbonell A, Paul SM, Therrien J, Kapustina O, Heisey K, Clapp DW, Zhang C, Peer CJ, Figg WD, Smith M, Glod J, Blakeley JO, Steinberg SM, Venzon DJ, Doyle LA, Widemann BC. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020;382:1430–1442. doi: 10.1056/NEJMoa1912735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Campbell BB, Galati MA, Stone SC, Riemenschneider AN, Edwards M, Sudhaman S, Siddaway R, Komosa M, Nunes NM, Nobre L, Morrissy AS, Zatzman M, Zapotocky M, Joksimovic L, Kalimuthu SN, Samuel D, Mason G, Bouffet E, Morgenstern DA, Aronson M, Durno C, Malkin D, Maris JM, Taylor MD, Shlien A, Pugh TJ, Ohashi PS, Hawkins CE, Tabori U. Mutations in the RAS/MAPK Pathway Drive Replication Repair-Deficient Hypermutated Tumors and Confer Sensitivity to MEK Inhibition. Cancer Discov. 2021;11:1454–1467. doi: 10.1158/2159-8290.CD-20-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tang JY, Ally MS, Chanana AM, Mackay-Wiggan JM, Aszterbaum M, Lindgren JA, Ulerio G, Rezaee MR, Gildengorin G, Marji J, Clark C, Bickers DR, Epstein EH Jr. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:1720–1731. doi: 10.1016/S1470-2045(16)30566-6. [DOI] [PubMed] [Google Scholar]
- 214.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Berkowitz N, Anak O, Niolat J, Jozwiak S. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014;15:1513–1520. doi: 10.1016/S1470-2045(14)70489-9. [DOI] [PubMed] [Google Scholar]
- 215.Geynisman DM, Kadow BT, Shuch BM, Boorjian SA, Matin SF, Rampersaud E, Milestone BN, Plimack ER, Zibelman MR, Kutikov A, Smaldone MC, Chen DY, Viterbo R, Joshi S, Greenberg RE, Malizzia L, McGowan T, Ross EA, Uzzo RG. Sporadic Angiomyolipomas Growth Kinetics While on Everolimus: Results of a Phase II Trial. J Urol. 2020;204:531–537. doi: 10.1097/JU.0000000000001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, Thamake S, Park EK, Perini RF, Linehan WM, Srinivasan R MK-6482-004 Investigators. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med. 2021;385:2036–2046. doi: 10.1056/NEJMoa2103425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Carril-Ajuria L, Colomba E, Cerbone L, Romero-Ferreiro C, Crouzet L, Laguerre B, Thibault C, Vicier C, de Velasco G, Fléchon A, Saldana C, Benusiglio PR, Bressac-de Paillerets B, Guillaud-Bataille M, Gaignard P, Scoazec JY, Richard S, Caron O, Escudier B, Albiges L. Response to systemic therapy in fumarate hydratase-deficient renal cell carcinoma. Eur J Cancer. 2021;151:106–114. doi: 10.1016/j.ejca.2021.04.009. [DOI] [PubMed] [Google Scholar]
- 218.Gleeson JP, Nikolovski I, Dinatale R, Zucker M, Knezevic A, Patil S, Ged Y, Kotecha RR, Shapnik N, Murray S, Russo P, Coleman J, Lee CH, Stadler ZK, Hakimi AA, Feldman DR, Motzer RJ, Reznik E, Voss MH, Chen YB, Carlo MI. Comprehensive Molecular Characterization and Response to Therapy in Fumarate Hydratase-Deficient Renal Cell Carcinoma. Clin Cancer Res. 2021;27:2910–2919. doi: 10.1158/1078-0432.CCR-20-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, Isakoff SJ, Ryan PD, Greene-Colozzi A, Gutin A, Sangale Z, Iliev D, Neff C, Abkevich V, Jones JT, Lanchbury JS, Hartman AR, Garber JE, Ford JM, Silver DP, Richardson AL. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sokolenko AP, Gorodnova TV, Bizin IV, Kuligina ES, Kotiv KB, Romanko AA, Ermachenkova TI, Ivantsov AO, Preobrazhenskaya EV, Sokolova TN, Broyde RV, Imyanitov EN. Molecular predictors of the outcome of paclitaxel plus carboplatin neoadjuvant therapy in high-grade serous ovarian cancer patients. Cancer Chemother Pharmacol. 2021;88:439–450. doi: 10.1007/s00280-021-04301-6. [DOI] [PubMed] [Google Scholar]
- 221.Mighton C, Lerner-Ellis JP. Principles of molecular testing for hereditary cancer. Genes Chromosomes Cancer. 2022;61:356–381. doi: 10.1002/gcc.23048. [DOI] [PubMed] [Google Scholar]
- 222.Kuligina ESh, Sokolenko AP, Mitiushkina NV, Abysheva SN, Preobrazhenskaya EV, Gorodnova TV, Yanus GA, Togo AV, Cherdyntseva NV, Bekhtereva SA, Dixon JM, Larionov AA, Kuznetsov SG, Imyanitov EN. Value of bilateral breast cancer for identification of rare recessive at-risk alleles: evidence for the role of homozygous GEN1 c.2515_2519delAAGTT mutation. Fam Cancer. 2013;12:129–132. doi: 10.1007/s10689-012-9575-x. [DOI] [PubMed] [Google Scholar]
- 223.Kurian AW. BRCA1 and BRCA2 mutations across race and ethnicity: distribution and clinical implications. Curr Opin Obstet Gynecol. 2010;22:72–78. doi: 10.1097/GCO.0b013e328332dca3. [DOI] [PubMed] [Google Scholar]
- 224.AlHarthi FS, Qari A, Edress A, Abedalthagafi M. Familial/inherited cancer syndrome: a focus on the highly consanguineous Arab population. NPJ Genom Med. 2020;5:3. doi: 10.1038/s41525-019-0110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Palmer JR, Polley EC, Hu C, John EM, Haiman C, Hart SN, Gaudet M, Pal T, Anton-Culver H, Trentham-Dietz A, Bernstein L, Ambrosone CB, Bandera EV, Bertrand KA, Bethea TN, Gao C, Gnanaolivu RD, Huang H, Lee KY, LeMarchand L, Na J, Sandler DP, Shah PD, Yadav S, Yang W, Weitzel JN, Domchek SM, Goldgar DE, Nathanson KL, Kraft P, Yao S, Couch FJ. Contribution of Germline Predisposition Gene Mutations to Breast Cancer Risk in African American Women. J Natl Cancer Inst. 2020;112:1213–1221. doi: 10.1093/jnci/djaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yadav S, LaDuca H, Polley EC, Hu C, Niguidula N, Shimelis H, Lilyquist J, Na J, Lee KY, Gutierrez S, Yussuf A, Hart SN, Davis BT, Chao EC, Pesaran T, Goldgar DE, Dolinsky JS, Couch FJ. Racial and Ethnic Differences in Multigene Hereditary Cancer Panel Test Results for Women With Breast Cancer. J Natl Cancer Inst. 2021;113:1429–1433. doi: 10.1093/jnci/djaa167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Monteiro AN, Bouwman P, Kousholt AN, Eccles DM, Millot GA, Masson JY, Schmidt MK, Sharan SK, Scully R, Wiesmüller L, Couch F, Vreeswijk MPG. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: best practices in functional analysis for clinical annotation. J Med Genet. 2020;57:509–518. doi: 10.1136/jmedgenet-2019-106368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Boonen RACM, Wiegant WW, Celosse N, Vroling B, Heijl S, Kote-Jarai Z, Mijuskovic M, Cristea S, Solleveld-Westerink N, van Wezel T, Beerenwinkel N, Eeles R, Devilee P, Vreeswijk MPG, Marra G, van Attikum H. Functional Analysis Identifies Damaging CHEK2 Missense Variants Associated with Increased Cancer Risk. Cancer Res. 2022;82:615–631. doi: 10.1158/0008-5472.CAN-21-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Evans DGR, van Veen EM, Byers HJ, Wallace AJ, Ellingford JM, Beaman G, Santoyo-Lopez J, Aitman TJ, Eccles DM, Lalloo FI, Smith MJ, Newman WG. A Dominantly Inherited 5' UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am J Hum Genet. 2018;103:213–220. doi: 10.1016/j.ajhg.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hansen TV, Ejlertsen B, Albrechtsen A, Bergsten E, Bjerregaard P, Hansen T, Myrhøj T, Nielsen PB, Timmermans-Wielenga V, Andersen MK, Jønson L, Nielsen FC. A common Greenlandic Inuit BRCA1 RING domain founder mutation. Breast Cancer Res Treat. 2009;115:69–76. doi: 10.1007/s10549-008-0060-z. [DOI] [PubMed] [Google Scholar]
- 231.Harboe TL, Eiberg H, Kern P, Ejlertsen B, Nedergaard L, Timmermans-Wielenga V, Nielsen IM, Bisgaard ML. A high frequent BRCA1 founder mutation identified in the Greenlandic population. Fam Cancer. 2009;8:413–419. doi: 10.1007/s10689-009-9257-5. [DOI] [PubMed] [Google Scholar]
- 232.Marchetti C, De Leo R, Musella A, D'Indinosante M, Capoluongo E, Minucci A, Benedetti Panici P, Scambia G, Fagotti A. BRCA Mutation Status to Personalize Management of Recurrent Ovarian Cancer: A Multicenter Study. Ann Surg Oncol. 2018;25:3701–3708. doi: 10.1245/s10434-018-6700-6. [DOI] [PubMed] [Google Scholar]
- 233.Morgan RD, Burghel GJ, Flaum N, Bulman M, Clamp AR, Hasan J, Mitchell CL, Schlecht H, Woodward ER, Lallo FI, Crosbie EJ, Edmondson RJ, Wallace AJ, Jayson GC, Evans DGR. Prevalence of germline pathogenic BRCA1/2 variants in sequential epithelial ovarian cancer cases. J Med Genet. 2019;56:301–307. doi: 10.1136/jmedgenet-2018-105792. [DOI] [PubMed] [Google Scholar]
- 234.AlDubayan SH, Pyle LC, Gamulin M, Kulis T, Moore ND, Taylor-Weiner A, Hamid AA, Reardon B, Wubbenhorst B, Godse R, Vaughn DJ, Jacobs LA, Meien S, Grgic M, Kastelan Z, Markt SC, Damrauer SM, Rader DJ, Kember RL, Loud JT, Kanetsky PA, Greene MH, Sweeney CJ, Kubisch C, Nathanson KL, Van Allen EM, Stewart DR, Lessel D Regeneron Genetics Center (RGC) Research Team. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) With Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019;5:514–522. doi: 10.1001/jamaoncol.2018.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ni VI, Ivantsov AO, Kotkova MA, Baskina SV, Ponomareva EV, Orlova RV, Topuzov EE, Kryukov KK, Shelekhova KV, Aleksakhina SN, Sokolenko AP, Imyanitov EN. Small fraction of testicular cancer cases may be causatively related to CHEK2 inactivating germ-line mutations: evidence for somatic loss of the remaining CHEK2 allele in the tumor tissue. Fam Cancer. 2021;20:49–53. doi: 10.1007/s10689-020-00190-5. [DOI] [PubMed] [Google Scholar]
- 236.Ring KL, Bruegl AS, Allen BA, Elkin EP, Singh N, Hartman AR, Daniels MS, Broaddus RR. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol. 2016;29:1381–1389. doi: 10.1038/modpathol.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cameron A, Chiarella-Redfern H, Chu P, Perrier R, Duggan MA. Universal Testing to Identify Lynch Syndrome Among Women With Newly Diagnosed Endometrial Carcinoma. J Obstet Gynaecol Can. 2020;42:137–143. doi: 10.1016/j.jogc.2019.06.018. [DOI] [PubMed] [Google Scholar]
- 238.Pearlman R, Frankel WL, Swanson BJ, Jones D, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, Goldberg RM, Paskett E, Shields PG, Freudenheim JL, Stanich PP, Lattimer I, Arnold M, Prior TW, Haut M, Kalady MF, Heald B, Paquette I, Draper DJ, Brell JM, Mahesh S, Weeman K, Bastola S, Zangmeister J, Gowda A, Kencana F, Malcolm A, Liu Y, Cole S, Bane C, Li C, Rehmus E, Pritchard CC, Shirts BH, Jacobson A, Cummings SA, de la Chapelle A, Hampel H. Prospective Statewide Study of Universal Screening for Hereditary Colorectal Cancer: The Ohio Colorectal Cancer Prevention Initiative. JCO Precis Oncol. 2021;5 doi: 10.1200/PO.20.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Bakhuizen JJ, Hogervorst FB, Velthuizen ME, Ruijs MW, van Engelen K, van Os TA, Gille JJ, Collée M, van den Ouweland AM, van Asperen CJ, Kets CM, Mensenkamp AR, Leter EM, Blok MJ, de Jong MM, Ausems MG. TP53 germline mutation testing in early-onset breast cancer: findings from a nationwide cohort. Fam Cancer. 2019;18:273–280. doi: 10.1007/s10689-018-00118-0. [DOI] [PubMed] [Google Scholar]
- 240.Evans DG, van Veen EM, Byers HJ, Evans SJ, Burghel GJ, Woodward ER, Harkness EF, Eccles DM, Greville-Haygate SL, Ellingford JM, Bowers NL, Pereira M, Wallace AJ, Howell SJ, Howell A, Lalloo F, Newman WG, Smith MJ. High likelihood of actionable pathogenic variant detection in breast cancer genes in women with very early onset breast cancer. J Med Genet. 2022;59:115–121. doi: 10.1136/jmedgenet-2020-107347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Beebe-Dimmer JL, Hathcock M, Yee C, Okoth LA, Ewing CM, Isaacs WB, Cooney KA, Thibodeau SN. The HOXB13 G84E Mutation Is Associated with an Increased Risk for Prostate Cancer and Other Malignancies. Cancer Epidemiol Biomarkers Prev. 2015;24:1366–1372. doi: 10.1158/1055-9965.EPI-15-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]