

Abstract
Rationale
The association between organ laterality abnormalities and ciliary ultrastructural defect or genotype in primary ciliary dyskinesia is poorly understood.
Objectives
To determine if there is an association between presence and/or type of laterality abnormality and ciliary ultrastructural defect or genotype.
Methods
Participants with primary ciliary dyskinesia in a multicenter, prospective study were grouped based on ciliary ultrastructural defect or genotype. In a retrospective analysis of these data, the association of ciliary ultrastructural defect or genotype and likelihood of a laterality abnormality was evaluated by logistic regression adjusted for presence of two loss-of-function versus one or more not–loss-of-function variants.
Results
Of 559 participants, 286 (51.2%), 215 (38.5%), and 58 (10.4%) were identified as having situs solitus, situs inversustotalis, and situs ambiguus, respectively; heterotaxy, defined as situs ambiguus with complex cardiovascular defects, was present in 14 (2.5%). Compared with the group with inner dynein arm defects with microtubular disorganization, laterality defects were more likely in the outer dynein arm defects group (odds ratio [OR], 2.07; 95% confidence interval [CI], 1.21–3.54; P < 0.01) and less likely in the normal/near normal ultrastructure group (OR, 0.04; 95% CI, 0.013–0.151; P < 0.01). Heterotaxy was present in 11 of 242 (4.5%) in the outer dynein arm defects group but 0 of 96 in the inner dynein arm defects with microtubular disorganization group (P = 0.038).
Conclusion
In primary ciliary dyskinesia, risk of a laterality abnormality differs by ciliary ultrastructural defect. Pathophysiologic mechanisms underlying these differences require further exploration.
Keywords: situs ambiguous, situs inversus totalis, laterality
Primary ciliary dyskinesia (PCD) (OMIM: 244400), a genetically heterogeneous condition caused by defects of motile cilia, is characterized by chronic sino-oto-pulmonary disease and organ laterality defects (1–3). Respiratory tract cilia consist of nine outer microtubule doublets surrounding a central pair of microtubules, resulting in the characteristic 9 + 2 configuration (1, 3). Establishment of the normal asymmetric left–right axis during embryonic gastrulation is a complex process regulated by motile nodal cilia, which lack a microtubule central pair (9 + 0) (4). During gastrulation, nodal cilia beat in a circular clockwise fashion, directing fluid leftward across the ventral node and resulting in a signaling gradient, which directs normal visceral organogenesis, termed situs solitus (SS) (4–8). Approximately 50% of patients with PCD develop abnormal organ arrangement if this gradient is not established (1, 2).
Individuals with PCD can have complete mirror image organ arrangement, known as situs inversus totalis (SIT). Others have organ laterality defects that are not mirror image, known as situs ambiguus (SA), occurring in at least 12% of individuals with PCD (9). The term SA has been used interchangeably with the term heterotaxy, though some define heterotaxy as strictly SA plus complex congenital heart defects (9).
Diagnosis of PCD relies on characteristic ciliary ultrastructural defects or pathogenic/likely pathogenic variants in one of more than 50 PCD-associated genes (biallelic variants for autosomal recessive genes) (1–3). Genotype–phenotype associations have recently emerged (10). Many genotypes linked with normal or near-normal ciliary ultrastructure, including pathogenic variants in genes encoding structural proteins in the central apparatus, radial spokes, nexin links, and basal bodies, as well as cytoplasmic proteins involved in ciliogenesis, are not essential for nodal ciliary function and thus do not affect organ laterality (1, 3, 11–14). Other PCD genes associated with specific ciliary ultrastructural defects, such as absent outer dynein arms (ODA), absent outer and inner dynein arms (ODA/IDA), or absent inner dynein arms with microtubular disorganization (IDA/MTD), are essential for nodal ciliary function and are associated with organ laterality defects. Little is known about the relationship between specific PCD genes and risk of laterality defects. We performed a retrospective analysis from our prospective multicenter cohort study of PCD (15, 16). Our first analysis evaluated the relationship between ultrastructural defect or genotype and laterality defects in individuals with PCD. Secondarily, we assessed whether variant functionality (loss of function [LOF] vs. not LOF) moderated this relationship. We hypothesized that risk of laterality defects differs by ciliary ultrastructural defect and associated genotypes.
Portions of these data were presented at the American Thoracic Society International Conference in 2021 (17).
Methods
Study Sites, Participants, and Procedures
Individuals of all ages with clinical features consistent with PCD were referred to one of nine sites in the Genetic Disorders of Mucociliary Clearance Consortium (GDMCC). The protocol was reviewed and accepted by central and site institutional review boards. Systematic evaluation included standardized clinical questionnaires, molecular genetic testing (2, 10), and ciliary biopsy for ultrastructural analysis using transmission electron microscopy (TEM), as previously described (10, 18, 19). Electron photomicrographs were reviewed centrally by three expert clinicians in a blinded fashion to define characteristic defects, including absent or truncated ODA, absent ODA/IDA, and IDA/MTD defects. PCD was diagnosed in participants with two pathogenic/likely pathogenic variants (henceforth referred to simply as “variants”) in a PCD-associated gene, hallmark TEM defects, or both. Each variant was classified as either LOF or not LOF according to American College of Medical Genetics guidelines (20).
Participants were grouped based on the hallmark ultrastructural defect associated with their genotype or, in those with no genetic diagnosis, grouped based on directly visualized ultrastructural defect. Categories included: 1) ODA defect; 2) ODA/IDA defect; 3) IDA/MTD defect; 4) normal ultrastructure associated with dynein axonemal heavy chain 11 (DNAH11) variants; and 5) normal/near-normal/other ultrastructure. Individuals with variants in DNAH11 appear to have normal ciliary ultrastructure when viewed under TEM; however, unlike variants in other genes that result in normal ultrastructure, DNAH11 encodes a component of the ODA (1). Therefore, these individuals were grouped separately from other variants that result in normal ultrastructure. Individuals with genes resulting in central complex defects, radial spoke defects, reduced number of multiple motile cilia, and nexin link defects were sorted into the normal/near-normal/other group.
Imaging reports (chest X-ray, chest computed tomography [CT], echocardiogram, abdominal CT, and/or abdominal ultrasound, as available) were reviewed by one investigator (A.T.B. or A.J.S.) to verify laterality defects (SIT and SA). Participants with SA were then subdivided into one of four groups using a congenital heart disease classification schema devised by Botto and colleagues (21) and later used by Shapiro and colleagues (9): 1) SA with complex cardiovascular malformation (heterotaxy); 2) SA with simple cardiac malformation; 3) SA without cardiac malformation; and 4) isolated laterality defect. Heterotaxy was defined as SA with a complex cardiovascular malformation. A portion of this study’s cohort includes participants from the Shapiro study (9). When no imaging was available, participants were assigned either SS or SIT based on physical examination findings (e.g., palpation of cardiac point of maximal impulse, palpation of abdomen).
Statistical Analysis
Descriptive statistics were used to report the participants’ demographic characteristics. Frequencies and percentages were used for categorical variables, and medians and ranges were used for continuous variables. The risk of laterality defects was evaluated among each ultrastructure group through multivariable analysis using either binomial (SIT + SA vs. SS) or multinomial (SIT vs. SA vs. SS) logistic regression adjusting for the presence of two LOF variants. Adjusted odds ratios (ORs) and 95% confidence intervals (CIs) were reported. The IDA/MTD group was chosen as the reference group, as it had the fewest individuals with laterality defects (aside from the normal/near-normal/other ultrastructure group, which could not be used because no individuals in this group had SA). Risk of laterality defects was then evaluated between specific genotypes compared with the coiled-coil domain containing 39 (CCDC39) + coiled-coil domain containing 40 (CCDC40) (IDA/MTD defects) group. Risk of heterotaxy for each ultrastructure group was also evaluated by multivariable regression; however, the ODA group was chosen as the reference group, because the IDA/MTD group had no individuals with heterotaxy. Fisher’s exact test was used to compare prevalence of heterotaxy in the ODA group to the IDA/MTD group.
Logistic regression was used to evaluate whether there was a difference in risk of laterality defects within each gene between those with two LOF variants and those with one or more not-LOF variants. Analyses were performed for only the three most prevalent genotypes because of small sample sizes in other genes. Significance was defined as P < 0.05. Analyses were conducted using SAS (version 9.4; SAS Institute Inc.).
Results
A total of 1,017 individuals were enrolled in the study; 559 (55%) had a diagnosis of PCD and were included in our analysis (Figure 1), which includes 305 participants from our prior publication (9). Among those 559 individuals, 247 (44%) were male, and median age at enrollment was 11.5 years (range, 0.1–82.3 yr) (Table 1). A total of 549 (98.2%) had either a chest X-ray or chest CT available for review (Figure 2). Only three individuals (0.5%) had no imaging available for review. SS was identified in 286 individuals (51.2%), SIT in 215 (38.4%), and SA in 58 (10.4%). A total of 242 (43.3%) had ODA defects, 96 (17.2%) had ODA/IDA defects, 96 (17.2%) had IDA/MTD defects, 82 (14.6%) had normal/near-normal/other ultrastructure, and another 43 (7.7%) had normal ultrastructure associated with DNAH11 variants. Situs distribution by ultrastructure group is shown in Table 2.
Figure 1.

Flowchart outlining the enrollment of participants. *Outer dynein arm (ODA) defect, ODA + inner dynein arm (IDA) defect, or IDA + microtubular disorganization defect. †Two pathogenic or likely pathogenic variants in a single primary ciliary dyskinesia (PCD)-causing gene. ‡Of the 46 genetically unresolved cases, 39 were tested by a panel of 28 or more genes plus whole-exome sequencing (WES) followed by targeted analysis of the known PCD-related genes (3). In addition, four had WES alone, two had panel testing alone, and one had no DNA available for testing. TEM = transmission electron microscopy.
Table 1.
Demographics by ciliary ultrastructural group
| ODA (n = 242) | ODA/IDA (n = 96) | IDA/MTD (n = 96) | Normal Ultrastructure Associated with DNAH11 (n = 43) | Normal/Near- Normal/Other (n = 82) | Total | |
|---|---|---|---|---|---|---|
| Male | 117 (48.3) | 46 (47.9) | 37 (38.5) | 13 (30.2) | 34 (41.4) | 247 (44.2) |
| Race | ||||||
| White | 206 (85.1) | 71 (74.0) | 79 (82.3) | 28 (65.1) | 67 (81.7) | 451 (80.7) |
| Black | 1 (0.4) | 4 (4.2) | 5 (5.2) | 2 (4.7) | 0 | 12 (2.1) |
| Asian | 22 (9.1) | 15 (15.6) | 4 (4.2) | 10 (23.3) | 5 (6.1) | 56 (10.0) |
| Mixed | 7 (2.9) | 5 (5.2) | 4 (4.2) | 1 (2.3) | 2 (2.4) | 19 (3.4) |
| Other | 6 (2.5) | 1 (1.0) | 2 (2.1) | 2 (4.7) | 4 (4.9) | 15 (2.7) |
| Not answered | 0 | 0 | 2 (2.1) | 0 | 4 (4.9) | 6 (1.1) |
| Ethnicity | ||||||
| Hispanic | 27 (11.2) | 8 (8.3) | 10 (10.4) | 5 (11.6) | 22 (26.8) | 72 (12.9) |
| Non-Hispanic | 213 (88.0) | 88 (91.7) | 85 (88.5) | 38 (88.4) | 60 (73.2) | 484 (86.6) |
| Not answered | 2 (0.8) | 0 | 1 (1.0) | 0 | 0 | 3 (0.5) |
| Age at enrollment, yr | 11.1 (0.1–82.3) | 12.9 (0.1–57.9) | 11.2 (0.1–61.8) | 12.3 (0.5–66.3) | 11.7 (0.8–64.5) | 11.5 (0.1–82.3) |
Definition of abbreviations: DNAH11 = dynein axonemal heavy chain 11; IDA = inner dynein arm; MTD = microtubular disorganization; ODA = outer dynein arm.
Data are presented as n (%) or median (range).
Figure 2.
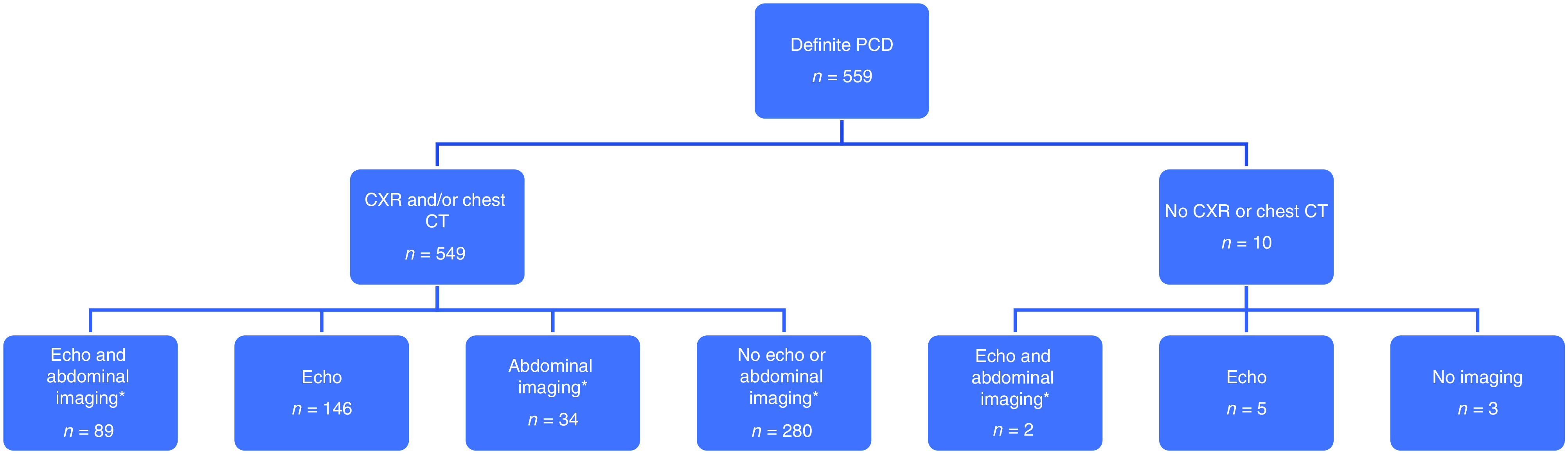
Flowchart outlining available imaging for participants. *Abdominal imaging includes abdominal ultrasound or abdominal CT. CT = computed tomography; CXR = chest X-ray; echo = echocardiogram; PCD = primary ciliary dyskinesia.
Table 2.
Situs distribution by ciliary ultrastructural group
| Ciliary Ultrastructure | SS (n = 286) | SIT (n = 215) | SA (n = 58) |
|---|---|---|---|
| ODA defect (n = 242) | 96 (39.7) | 113 (46.7) | 33 (13.6) |
| ODA/IDA defect (n = 96) | 40 (41.7) | 43 (44.8) | 13 (13.5) |
| IDA/MTD defect (n = 96) | 52 (54.2) | 36 (37.5) | 8 (8.3) |
| Normal ultrastructure associated with DNAH11 (n = 43) | 19 (44.2) | 20 (46.5) | 4 (9.3) |
| Normal/near-normal/other (n = 82) | 79 (96.3) | 3 (3.7) | 0 |
Definition of abbreviations: DNAH11 = dynein axonemal heavy chain 11; IDA = inner dynein arm; MTD = microtubular disorganization; ODA = outer dynein arm; SA = situs ambiguus; SIT = situs inversus totalis; SS = situs solitus.
Data are presented as n (%).
Variants in 35 PCD-associated genes were found within the cohort (Table 3). The five most prevalent genotypes were dynein axonemal heavy chain 5 (DNAH5) (n = 153, 27.4%), CCDC40 (n = 45, 8.1%), DNAH11 (n = 43, 7.7%), CCDC39 (n = 38, 6.8%), and dynein axonemal intermediate chain 1 (DNAI1) (n = 26, 4.7%). Forty-six individuals had a diagnosis based on abnormal ciliary ultrastructure alone.
Table 3.
Laterality defects and prevalence of two loss-of-function mutations by gene
| Ciliary Ultrastructure | Gene Mutation | SS | SIT | SA | Total (n) | 2 Loss-of-Function Mutations* |
|---|---|---|---|---|---|---|
| ODA defect | DNAH5 | 53 (34.6) | 75 (49) | 25 (16.3) | 153 | 86 (56.2) |
| DNAI1 | 14 (53.8) | 11 (42.3) | 1 (3.8) | 26 | 4 (15.4) | |
| CCDC103 † | 4 (28.6) | 7 (50) | 3 (21.4) | 14 | 0 | |
| DNAI2 | 7 (70) | 3 (30) | 0 | 10 | 8 (80) | |
| Other (CCDC114, ARMC4) | 6 (42.9) | 7 (50) | 1 (7.1) | 14 | 10 (71.4) | |
| No gene identified | 12 (48) | 10 (40) | 3 (12) | 25 | N/A | |
| ODA/IDA defect | SPAG1 | 6 (35.3) | 8 (47.1) | 3 (17.6) | 17 | 14 (82.4) |
| DNAAF4 | 11 (68.8) | 4 (25) | 1 (6.3) | 16 | 10 (62.5) | |
| DNAAF3 | 5 (45.5) | 6 (54.5) | 0 | 11 | 7 (63.6) | |
| DNAAF1 | 2 (22.2) | 6 (66.7) | 1 (11.1) | 9 | 5 (55.6) | |
| DNAAF5 | 4 (44.4) | 3 (33.3) | 2 (22.2) | 9 | 2 (22.2) | |
| LRRC6 | 5 (55.6) | 3 (33.3) | 1 (11.1) | 9 | 7 (77.8) | |
| Other (CFAP298, CFAP300, DNAAF2, PIH1D3, ZMYND10) | 5 (29.4) | 9 (52.9) | 3 (17.6) | 17 | 13 (76.5) | |
| No gene identified | 2 (25) | 4 (50) | 2 (25) | 8 | N/A | |
| IDA/MTD defect | CCDC40 | 26 (57.8) | 14 (31.1) | 5 (11.1) | 45 | 41 (91.1) |
| CCDC39 | 18 (47.4) | 18 (47.4) | 2 (5.3) | 38 | 31 (81.6) | |
| No gene identified | 8 (61.5) | 4 (30.8) | 1 (7.7) | 13 | N/A | |
| Normal ultrastructure associated with DNAH11 | DNAH11 | 19 (44.2) | 20 (46.5) | 4 (9.3) | 43 | 22 (51.2) |
| Normal/near-normal/other | HYDIN | 19 (100) | 0 | 0 | 19 | 19 (100) |
| RSPH4A | 16 (100) | 0 | 0 | 16 | 4 (25) | |
| RSPH1 | 15 (100) | 0 | 0 | 15 | 15 (100) | |
| CCNO | 9 (100) | 0 | 0 | 9 | 6 (66.7) | |
| Other (CCDC65, CCDC164, CFAP57, CFAP221, FOXJ1, GAS2L2, NEK10, OFD1, RPGR, RSPH3, RSPH9) | 20 (87) | 3 (13) | 0 | 23 | 19 (82.6) | |
| Total | 286 (51.2) | 215 (38.5) | 58 (10.4) | 559 | 323 (57.8) |
Definition of abbreviations: ARMC4 = armadillo repeat containing 4; CCDC39 = coiled-coil domain containing 39; CCDC40 = coiled-coil domain containing 40; CCDC65 = coiled-coil domain containing 65; CCDC103 = coiled-coil domain containing 103; CCDC114 = coiled-coil domain containing 114; CCDC164 = coiled-coil domain containing 164; CCNO = cyclin O; CFAP57 = cilia and flagella associated protein 57; CFAP221 = cilia and flagella associated protein 221; CFAP298 = cilia and flagella associated protein 298; CFAP300 = cilia and flagella associated protein 300; DNAAF1 = dynein axonemal assembly factor 1; DNAAF2 = dynein axonemal assembly factor 2; DNAAF3 = dynein axonemal assembly factor 3; DNAAF4 = dynein axonemal assembly factor 4; DNAAF5 = dynein axonemal assembly factor 5; DNAH5 = dynein axonemal heavy chain 5; DNAH11 = dynein axonemal heavy chain 11; DNAI1 = dynein axonemal intermediate chain 1; DNAI2 = dynein axonemal intermediate chain 2; FOXJ1 = forkhead box J1; GAS2L2 = growth arrest specific 2 like 2; HYDIN = hydrocephalus-inducing; IDA = inner dynein arm; LRRC6 = leucine-rich repeat containing protein 6; MTD = microtubular disorganization; N/A = not applicable; NEK10 = NIMA related kinase 10; ODA = outer dynein arm; OFD1 = orofaciodigital syndrome 1; PIH1D3 = PIH1 domain containing 3; RPGR = retinitis pigmentosa GTPase regulator; RSPH1 = radial spoke head component 1; RSPH3 = radial spoke head 3; RSPH4A = radial spoke head component 4A; RSPH9 = radial spoke head component 9; SA = situs ambiguus; SIT = situs inversus totalis; SPAG1 = sperm associated antigen 1; SS = situs solitus; ZMYND10 = zinc finger MYND-type containing 10.
Data are presented as n (%) unless otherwise noted.
Loss-of-function variants included nonsense, frameshift, start codon canonical splice site, large copy number changes, and full gene deletion variants. Not–loss-of-function mutations included missense, in-frame, and extended splice site (intronic and exonic) variants, which may be mild or have residual function.
In people with PCD who had normal/near-normal/other ultrastructure, nearly all individuals (79 of 82, 96.3%) were classified as having SS (Table 3). Three of the 82 (3.7%) individuals had SIT; two had variants in forkhead box J1 (FOXJ1) (autosomal dominant), and one had a variant in orofaciodigital syndrome type 1 (OFD1) (X-linked), both of which are previously reported to be associated with SIT (22, 23). No individuals in this group had SA.
Relative to the IDA/MTD group, the risk of any laterality defect (SIT + SA) was greater in the ODA group (OR, 2.07; 95% CI, 1.21–3.54; P < 0.01) but was not significantly different compared with the ODA/IDA group or the normal ultrastructure associated with DNAH11 group (OR, 1.61; 95% CI, 0.88–2.98; P = 0.13 and OR, 1.64; 95% CI, 0.77–3.5; P = 0.2, respectively) (Table 4 and Figure 3). The relative risk of SIT to SS was greater in the ODA group than in the IDA/MTD group (OR, 2.04; 95% CI, 1.16–3.60; P = 0.014). However, the relative risk of SA to SS was not significantly different for ODA versus the IDA/MTD group (OR, 2.19; 95% CI, 0.86–5.61; P = 0.1).
Table 4.
Odds ratio for laterality defect by ciliary ultrastructural group compared to reference inner dynein arm/microtubular disorganization group
| Ultrastructure Group | Laterality | OR | 95% CI |
|---|---|---|---|
| ODA | SIT + SA | 2.07 | 1.21–3.54 |
| SIT | 2.04 | 1.16–3.60 | |
| SA | 2.19 | 0.86–5.61 | |
| ODA/IDA | SIT + SA | 1.61 | 0.88–2.98 |
| SIT | 1.57 | 0.82–3.0 | |
| SA | 1.80 | 0.63–5.15 | |
| Normal ultrastructure associated with DNAH11 | SIT + SA | 1.64 | 0.77–3.50 |
| SIT | 1.74 | 0.78–3.86 | |
| SA | 1.30 | 0.33–5.07 | |
| Normal/near-normal/other | SIT + SA | 0.04 | 0.013–0.15 |
| SIT | 0.06 | 0.018–0.22 | |
| SA | 0 | 0 |
Definition of abbreviations: CI = confidence interval; DNAH11 = dynein axonemal heavy chain 11; IDA = inner dynein arm; ODA = outer dynein arm; OR = odds ratio; SA = situs ambiguus; SIT = situs inversus totalis.
Figure 3.

Risk of any laterality defect (situs inversus totalis + situs ambiguus) by ciliary ultrastructural group compared with reference inner dynein arm (IDA)/MTD group (n = 96). Horizontal bars represent 95% confidence interval (CI). *P < 0.01. †When CCDC103 is moved from the outer dynein arm (ODA) group to the ODA/IDA group, the odds ratio (OR) for a laterality defect in the ODA/IDA group becomes 1.97; 95% CI, 1.15–3.36; P = 0.014. The OR for laterality defect in the ODA group becomes 1.97; 95% CI, 1.15–3.62; P = 0.014. DNAH11 = dynein axonemal heavy chain 11; MTD = microtubular disorganization.
A comparison of the most prevalent genes from each ultrastructural defect group showed a similar relationship (Table 5). Relative to the CCDC39/CCDC40 group, the risk of any laterality defect (SIT + SA) was greater in the DNAH5 group (OR, 2.29; 95% CI, 1.3–4.0; P < 0.01) but was not significantly different compared with either the DNAI1 or coiled-coil domain containing 103 (CCDC103) groups (OR, 1.14; 95% CI, 0.45–2.88; P = 0.79 and OR, 3.43; 95% CI, 0.95–12.4; P = 0.06, respectively).
Table 5.
Risk of laterality defect by genotype compared to reference CCDC39/CCDC40 group
| Genotype | Laterality | OR | 95% CI |
|---|---|---|---|
| DNAH5 | SIT + SA | 2.33 | 1.32–4.08 |
| SIT | 1.77 | 1.01–3.12 | |
| SA | 1.84 | 0.74–4.58 | |
| DNAI1 | SIT + SA | 1.18 | 0.47–3.01 |
| SIT | 1.64 | 0.64–4.25 | |
| SA | 0.32 | 0.04–2.83 | |
| CCDC103 | SIT + SA | 3.61 | 0.99–13.08 |
| SIT | 2.42 | 0.73–8.0 | |
| SA | 2.03 | 0.42–9.89 | |
| DNAH11 | SIT + SA | 1.58 | 0.74–3.37 |
| SIT | 1.64 | 0.77–3.53 | |
| SA | 0.94 | 0.25–3.50 | |
| SPAG1 | SIT + SA | 2.10 | 0.71–6.22 |
| SIT | 1.45 | 0.51–4.16 | |
| SA | 2.28 | 0.52–9.94 |
Definition of abbreviations: CCDC39 = coiled-coil domain containing 39; CCDC40 = coiled-coil domain containing 40; CCDC103 = coiled-coil domain containing 103; CI = confidence interval; DNAH5 = dynein axonemal heavy chain 5; DNAI1 = dynein axonemal intermediate chain 1; DNAH11 = dynein axonemal heavy chain 11; OR = odds ratio; SA = situs ambiguus; SIT = situs inversus totalis; SPAG1 = sperm associated antigen 1.
The distribution of subcategories within SA is shown in Table 6. Of the 58 subjects with SA, 14 (23.7%) had SA with complex cardiovascular defects (heterotaxy), 11 (18.6%) had SA with simple cardiac defects, 22 (37.3%) had SA without cardiac defects, and 11 (18.6%) had isolated laterality defects. The distribution of heterotaxy by ultrastructural group was 11 (78.6%) in the ODA group, 2 (14.2%) in the normal ultrastructure associated with DNAH11 group, 1 (7.1%) in the ODA/IDA group, and none in the IDA/MTD group. Eight of the 14 (57.1%) with heterotaxy had mutations in DNAH5. There was no difference in risk of heterotaxy in the normal ultrastructure associated with DNAH11 group or the ODA/IDA group compared with the ODA group (OR, 1.06; 95% CI, 0.2–5.65; P = 0.95 and OR, 0.22; 95% CI, 0.03–1.84; P = 0.16, respectively). No individuals in the IDA/MTD group had heterotaxy, which was lower than the proportion with heterotaxy in the ODA group (4.5%) (Fisher exact test; P = 0.04).
Table 6.
Situs ambiguus distribution by gene
| Ciliary Ultrastructure | Gene Mutation | Heterotaxy | SA + Simple Cardiac Malformation | SA without Cardiac Malformation | Isolated Laterality Defect | Total SA |
|---|---|---|---|---|---|---|
| ODA defect | DNAH5 | 8 (32) | 5 (20) | 7 (28) | 5 (20) | 25 (43.1) |
| DNAI1 | 1 (100) | 0 | 0 | 0 | 1 (1.7) | |
| CCDC103 | 2 (66.7) | 0 | 1 (33.3) | 0 | 3 (5.2) | |
| DNAI2 | 0 | 0 | 0 | 0 | 0 | |
| No gene identified | 0 | 0 | 2 (66.7) | 1 (33.3) | 3 (5.2) | |
| Other (CCDC114, ARMC4) | 0 | 0 | 1 (100) | 0 | 1 (1.7) | |
| ODA/IDA defect | SPAG1 | 0 | 1 (33.3) | 2 (66.7) | 0 | 3 (5.2) |
| DNAAF4 | 0 | 1 (100) | 0 | 0 | 1 (1.7) | |
| DNAAF3 | 0 | 0 | 0 | 0 | 0 | |
| DNAAF1 | 0 | 0 | 1 (100) | 0 | 1 (1.7) | |
| DNAAF5 | 1 (50) | 0 | 0 | 1 (50) | 2 (3.4) | |
| LRRC6 | 0 | 0 | 1 (100) | 0 | 1 (1.7) | |
| Other (CFAP298, CFAP300, DNAAF2, PIH1D3, ZMYND10) | 0 | 2 (66.7) | 0 | 1 (33.3) | 3 (5.2) | |
| No gene identified | 0 | 0 | 2 (100) | 0 | 2 (3.4) | |
| IDA/MTD defect | CCDC40 | 0 | 2 (40) | 1 (20) | 2 (40) | 5 (8.6) |
| CCDC39 | 0 | 0 | 1 (50) | 1 (50) | 2 (3.4) | |
| No gene identified | 0 | 0 | 1 (100) | 0 | 1 (1.7) | |
| Normal ultrastructure associated with DNAH11 | DNAH11 | 2 (50) | 0 | 2 (50) | 0 | 4 (6.9) |
| Normal/near-normal/other | HYDIN | 0 | 0 | 0 | 0 | 0 |
| RSPH4A | 0 | 0 | 0 | 0 | 0 | |
| RSPH1 | 0 | 0 | 0 | 0 | 0 | |
| CCNO | 0 | 0 | 0 | 0 | 0 | |
| Other (CCDC65, CCDC164, CFPA57, CFAP221, FOXJ1, GAS2L2, NEK10, OFD1, RPGR, RSPH3, RSPH9) | 0 | 0 | 0 | 0 | 0 | |
| Total | 14 (24.1) | 11 (19.0) | 22 (37.9) | 11 (19.0) | 58 |
Definition of abbreviations: ARMC4 = armadillo repeat containing 4; CCDC39 = coiled-coil domain containing 39; CCDC40 = coiled-coil domain containing 40; CCDC65 = coiled-coil domain containing 65; CCDC103 = coiled-coil domain containing 103; CCDC114 = coiled-coil domain containing 114; CCDC164 = coiled-coil domain containing 164; CCNO = cyclin O; CFAP57 = cilia and flagella associated protein 57; CFAP221 = cilia and flagella associated protein 221; CFAP298 = cilia and flagella associated protein 298; CFAP300 = cilia and flagella associated protein 300; DNAAF1 = dynein axonemal assembly factor 1; DNAAF2 = dynein axonemal assembly factor 2; DNAAF3 = dynein axonemal assembly factor 3; DNAAF4 = dynein axonemal assembly factor 4; DNAAF5 = dynein axonemal assembly factor 5; DNAH5 = dynein axonemal heavy chain 5; DNAH11 = dynein axonemal heavy chain 11; DNAI1 = dynein axonemal intermediate chain 1; DNAI2 = dynein axonemal intermediate chain 2; FOXJ1 = forkhead box J1; GAS2L2 = growth arrest specific 2 like 2; HYDIN = hydrocephalus-inducing; IDA = inner dynein arm; LRRC6 = leucine-rich repeat containing protein 6; MTD = microtubular disorganization; NEK10 = NIMA related kinase 10; ODA = outer dynein arm; OFD1 = orofaciodigital syndrome 1; PIH1D3 = PIH1 domain containing 3; RPGR = retinitis pigmentosa GTPase regulator; RSPH1 = radial spoke head component 1; RSPH3 = radial spoke head 3; RSPH4A = radial spoke head component 4A; RSPH9 = radial spoke head component 9; SA = situs ambiguus; SPAG1 = sperm associated antigen 1; ZMYND10 = zinc finger MYND-type containing 10.
Data are presented as n (%).
Variant functionality (prevalence of two LOF variants) for each PCD-associated gene is shown in Table 3. Of the 513 individuals in the cohort with genetic variants identified, 323 (63%) had two LOF variants. There was a high prevalence of two LOF variants in CCDC39 (81.6%) and CCDC40 (91.1%) in the IDA/MTD group. The prevalence of two LOF variants in genes from the ODA group was 56.2% in DNAH5, 14.5% in DNAI1, 0% in CCDC103, and 80% in dynein axonemal intermediate chain 2 (listed in order of gene prevalence). Within DNAH5, there was no difference in risk of any laterality defect (SIT + SA) between those with two LOF variants and those with one or more not-LOF variants (OR, 1.23; 95% CI, 0.63–2.41; P = 0.54) (Table 7). Similarly, within CCDC39/40, there was no difference in risk of any laterality defect (SIT + SA) between those with two LOF variants and those with one or more not-LOF variants (OR, 1.07; 95% CI, 0.30–3.84; P = 0.91).
Table 7.
Risk of laterality defect within each genotype in those with two loss-of-function mutations compared to those with one or more not–loss-of-function mutations
| Genotype | Laterality | OR | 95% CI |
|---|---|---|---|
| DNAH5 | SIT + SA | 1.23 | 0.63–2.41 |
| SIT | 1.68 | 0.88–3.20 | |
| SA | 0.55 | 0.23–1.32 | |
| CCDC39/CCDC40 | SIT + SA | 1.07 | 0.30–3.84 |
| SIT | 1.11 | 0.30–4.16 | |
| SA | 0.91 | 0.10–8.36 | |
| DNAH11 | SIT + SA | 1.93 | 0.57–6.52 |
| SIT | 1.95 | 0.58–6.58 | |
| SA | 0.95 | 0.12–7.44 |
Definition of abbreviations: CCDC39 = coiled-coil domain containing 39; CCDC40 = coiled-coil domain containing 40; DNAH5 = dynein axonemal heavy chain 5; DNAH11 = dynein axonemal heavy chain 11; CI = confidence interval; OR = odds ratio; SA = situs ambiguus; SIT = situs inversus totalis.
Discussion
In this retrospective analysis of a multicenter prospective cohort study, participants with PCD secondary to an ODA defect and/or mutations in DNAH5 had an increased risk of laterality defects (SIT + SA), and specifically SA with complex cardiovascular disease (heterotaxy), compared with those with an IDA/MTD defect. Our study did not find a higher risk of laterality defects in those with ODA/IDA defects or normal ultrastructure associated with DNAH11 variants compared with those with IDA/MTD defects, although our power was limited by smaller sample sizes in those groups. The overall distribution of laterality defects within our study (51.2% SS, 38.4% SIT, 10.4% SA) was similar to that reported by Shapiro and colleagues (9) (47% SS, 41% SIT, 12% SA) as well as that reported by Best and colleagues (49% SS, 42% SIT, 9% SA) (24). Our study expands on these findings by analyzing the distribution and risk of laterality defects based on ciliary ultrastructure and/or genotype.
The high prevalence of laterality defects in the ODA group and specifically those with mutations in DNAH5 is also concordant with data presented by Nöthe-Menchen and colleagues (7), in which 65.9% of individuals with mutations in DNAH5 had a laterality defect. The true prevalence of SA, and specifically heterotaxy, in DNAH5 and other ODA defects may be even greater than we report in this cohort. One study showed that 40% of mouse fetuses with mutations in Dnahc5, the murine ortholog to DNAH5, have SA, many dying pre- or perinatally (25).
There are several possible explanations for the lower risk of laterality defects among those with IDA/MTD defects compared with other groups. Selection bias related to severity of lung disease could be present. Those with IDA/MTD defects are known to have more severe lung disease than others; thus, diagnosis in these individuals may be less contingent on presence of a laterality defect (10, 26). Other ultrastructural defects/genotypes with less severe lung disease may be missed or misdiagnosed in the absence of a laterality defect, which could cause an artificial increase in prevalence of laterality defects in people with ODA or ODA/IDA defects. Genes that encode components of the ODA (e.g., DNAH5) and those that act as “molecular rulers” (e.g., CCDC39 and CCDC40) have both been shown to affect nodal cilia (7, 27, 28). To date, however, no studies have directly compared function of nodal cilia in those with ODA defects to IDA/MTD defects, so this question is unanswered. In another theory, one could postulate that ODA defects, but not IDA/MTD defects, may preferentially affect nonmotile (primary) cardiac cilia, which are known to play a role in heart development and could therefore cause more laterality defects in the heart (i.e., heterotaxy). However, this possibility seems less likely, given that cardiac primary cilia do not normally contain either ODA or IDA elements (4, 29). Last, it seems plausible that variant functionality (two LOF variants vs. not-LOF variants) could influence embryonic nodal ciliary function and likelihood of a laterality defect. However, the IDA/MTD group had a greater prevalence of two LOF variants but lower prevalence of laterality defects than the ODA group, and within individual genes LOF variants were not associated with increased risk of laterality defects. This is in agreement with findings from Nöthe-Menchen and colleagues, who also reported no relationship between type of variant and occurrence of laterality defects (7).
Heterotaxy was present in 14 subjects in our cohort, but none had IDA/MTD defects. Notably, heterotaxy has not been described in the literature in individuals with IDA/MTD defects. Kennedy and colleagues reported two individuals (30) with IDA defects who had heterotaxy; however, this study used a different definition for heterotaxy. Of the two individuals labeled as having heterotaxy, neither had complex cardiac disease; one had isolated dextrocardia and the other had SA with simple cardiac defects (atrial septal defect and ventricular septal defect). One explanation for the lack of heterotaxy in those with IDA/MTD defects could again be selection bias, as these individuals may have heterotaxy but not survive long enough to receive a diagnosis of PCD, given their complex heart disease and greater lung involvement.
All subjects with heterotaxy in our study were <18 years of age. The most likely explanation for this phenomenon is that with technologic advances, more individuals with complex congenital heart disease are surviving past infancy but still have a markedly shortened lifespan. The prevalence of SA in PCD, including heterotaxy, is therefore likely higher than current estimates. When analyzing only individuals <18 years of age, the prevalence of SA increases by 16% and the prevalence of heterotaxy increases by 48%.
Those with mutations in CCDC103 were noted to have a particularly high prevalence of laterality defects. Of the 14 individuals with pathogenic variants in CCDC103, seven had SIT and three had SA, including two with heterotaxy. None of the individuals with PCD caused by CCDC103 mutations had two LOF variants. It is possible that this phenomenon contributes in some way to the high prevalence of laterality defects within this group; however, the small sample size of individuals with CCDC103 variants prevents us from forming any conclusions currently. CCDC103 has been categorized as a dynein arm attachment factor that results in ODA defects; however, there is some evidence that certain variants result in both ODA and IDA defects, and others result in normal ciliary ultrastructure (31–33). In our analysis, CCDC103 was included in the ODA group; however, when moved to the ODA/IDA group, the likelihood of any laterality defect (SIT + SA) remained significant for ODA versus IDA/MTD and became significant for ODA/IDA versus IDA/MTD (see footnotes in Table 3 and Figure 3).
Most individuals with variants that result in normal/near-normal/other ciliary ultrastructure had no laterality defects, as has previously been reported (1, 9, 24). These variants typically encode ciliary components that are not required for proper function of the nodal monocilia; thus, normal situs occurs in most of these genotypes. However, we found that variants in two particular genes, OFD1 and FOXJ1, result in normal-appearing ciliary ultrastructure but can lead to laterality defects, as has been previously reported (3, 22, 23).
Although DNAH11 variants appear to be associated with normal ultrastructure under TEM, these findings differ from most PCD disease-causing genes with normal/near-normal/other ultrastructure in that it frequently results in laterality defects, similar to genes with ODA defects noted on TEM. This finding is consistent with the role of DNAH11 of encoding a protein of the ODA complex, which appears to be integral to the function of nodal cilia.
There are limitations to our study. Some patients did not have cardiac or abdominal imaging, and it is possible that laterality defects (i.e., intestinal malrotation, polysplenia, asplenia, or minor congenital heart defects) could have been missed, which would have resulted in an artificially reduced prevalence of SA. In addition, 46 individuals lacked a genetic diagnosis, which limited our genotype–phenotype analysis.
In summary, we report a greater likelihood of laterality defects in individuals with PCD who have ODA defects than those who have IDA/MTD defects. Almost all heterotaxy was found in individuals with ODA defects. Additional studies are needed to elucidate the underlying mechanisms explaining the unequal distribution of laterality defects between PCD ultrastructure and genotype groups.
Acknowledgments
Acknowledgment
The authors thank Dr. Jaclyn Stonebraker and Elizabeth Schecterman for technical assistance and Dr. Hong Dang for bioinformatics assistance. They also thank Dr. Shrikant Mane and Dr. Francesc Lopez-Giraldez from the Yale Center for Mendelian Genomics (UM1 HG006504) for providing whole-exome sequencing and bioinformatics support, and the GDMCC coordinators as well as the patients and families who participated.
Footnotes
Supported by the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network (RDCRN) grant U54HL096458. RDCRN is an initiative of the Office of Rare Diseases Research (ORDR) funded through a collaboration between NCATS and National Heart, Lung, and Blood Institute (NHLBI) grant R01HL071798. Other support includes National Institutes of Health (NIH)/NHLBI and the UNC Children’s Research Institute Carolina for the Kids Grant Program. Supported in part by the intramural research program of the NHLBI, NIH (K.N.O.).
Author Contributions: S. D. Davis, M.R.K., T.W.F., J.J.A., S.D.S., S. D. Dell, K.N.O., C.E.M., M.R., A.J.S., K.M.S., N.A.C., M.W.L., and M.A.Z.: recruited and evaluated patients at GDMCC sites. A.T.B., S. D. Davis, and M.W.L.: designed study. A.T.B., S. D. Davis, M.A.Z., M.R.K., and M.W.L.: prepared manuscript. L.L. and F.-C.L.: performed statistical analysis.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia: recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med . 2013;188:913–922. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leigh MW, Ferkol TW, Davis SD, Lee HS, Rosenfeld M, Dell SD, et al. Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolescents. Ann Am Thorac Soc . 2016;13:1305–1313. doi: 10.1513/AnnalsATS.201511-748OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wallmeier J, Nielsen KG, Kuehni CE, Lucas JS, Leigh MW, Zariwala MA, et al. Motile ciliopathies. Nat Rev Dis Primers . 2020;6:77. doi: 10.1038/s41572-020-0209-6. [DOI] [PubMed] [Google Scholar]
- 4. Koefoed K, Veland IR, Pedersen LB, Larsen LA, Christensen ST. Cilia and coordination of signaling networks during heart development. Organogenesis . 2014;10:108–125. doi: 10.4161/org.27483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hirokawa N, Tanaka Y, Okada Y, Takeda S. Nodal flow and the generation of left-right asymmetry. Cell . 2006;125:33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 6. Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell . 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 7. Nöthe-Menchen T, Wallmeier J, Pennekamp P, Höben IM, Olbrich H, Loges NT, et al. Randomization of left-right asymmetry and congenital heart defects: the role of DNAH5 in humans and mice. Circ Genom Precis Med . 2019 doi: 10.1161/CIRCGEN.119.002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A-/- mice analysis. J Cell Biol . 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, Olivier KN, et al. Genetic Disorders of Mucociliary Clearance Consortium Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest . 2014;146:1176–1186. doi: 10.1378/chest.13-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis SD, Ferkol TW, Rosenfeld M, Lee HS, Dell SD, Sagel SD, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med . 2015;191:316–324. doi: 10.1164/rccm.201409-1672OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bustamante-Marin XM, Horani A, Stoyanova M, Charng WL, Bottier M, Sears PR, et al. Mutation of CFAP57, a protein required for the asymmetric targeting of a subset of inner dynein arms in Chlamydomonas, causes primary ciliary dyskinesia. PLoS Genet . 2020;16:e1008691. doi: 10.1371/journal.pgen.1008691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bustamante-Marin XM, Yin WN, Sears PR, Werner ME, Brotslaw EJ, Mitchell BJ, et al. Lack of GAS2L2 causes PCD by impairing cilia orientation and mucociliary clearance. Am J Hum Genet . 2019;104:229–245. doi: 10.1016/j.ajhg.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chivukula RR, Montoro DT, Leung HM, Yang J, Shamseldin HE, Taylor MS, et al. A human ciliopathy reveals essential functions for NEK10 in airway mucociliary clearance. Nat Med . 2020;26:244–251. doi: 10.1038/s41591-019-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lucas JS, Davis SD, Omran H, Shoemark A. Primary ciliary dyskinesia in the genomics age. Lancet Respir Med . 2020;8:202–216. doi: 10.1016/S2213-2600(19)30374-1. [DOI] [PubMed] [Google Scholar]
- 15.Genetics of primary ciliary dyskinesia; [accessed 2022 Jan 5]. Available from: https://ClinicalTrials.gov/show/NCT02389049.
- 16.Rare genetic disorders of the breathing airways; [accessed 2022 Jan 5]. Available from: https://ClinicalTrials.gov/show/NCT00323167
- 17.Barber AT, Shapiro AJ, Davis SD, Ferkol TW, Atkinson JJ, Sagel SD, et al. Relationship between genotype and laterality defects in primary ciliary dyskinesia. In D2. D002 Cystic Fibrosis and Primary Ciliary Dyskinesia. Oral presentation at: American Thoracic Society 2021 International Conference. May 14–19, 2021; San Diego, CA (presented virtually)
- 18. Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med . 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 19. Olin JT, Burns K, Carson JL, Metjian H, Atkinson JJ, Davis SD, et al. Genetic Disorders of Mucociliary Clearance Consortium Diagnostic yield of nasal scrape biopsies in primary ciliary dyskinesia: a multicenter experience. Pediatr Pulmonol . 2011;46:483–488. doi: 10.1002/ppul.21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med . 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A, National Birth Defects Prevention Study Seeking causes: classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol . 2007;79:714–727. doi: 10.1002/bdra.20403. [DOI] [PubMed] [Google Scholar]
- 22. Hannah WB, DeBrosse S, Kinghorn B, Strausbaugh S, Aitken ML, Rosenfeld M, et al. The expanding phenotype of OFD1-related disorders: hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol Genet Genomic Med . 2019;7:e911. doi: 10.1002/mgg3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wallmeier J, Frank D, Shoemark A, Nöthe-Menchen T, Cindric S, Olbrich H, et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am J Hum Genet . 2019;105:1030–1039. doi: 10.1016/j.ajhg.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Best S, Shoemark A, Rubbo B, Patel MP, Fassad MR, Dixon M, et al. Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax . 2019;74:203–205. doi: 10.1136/thoraxjnl-2018-212104. [DOI] [PubMed] [Google Scholar]
- 25. Tan SY, Rosenthal J, Zhao XQ, Francis RJ, Chatterjee B, Sabol SL, et al. Heterotaxy and complex structural heart defects in a mutant mouse model of primary ciliary dyskinesia. J Clin Invest . 2007;117:3742–3752. doi: 10.1172/JCI33284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis SD, Rosenfeld M, Lee HS, Ferkol TW, Sagel SD, Dell SD, et al. Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am J Respir Crit Care Med . 2019;199:190–198. doi: 10.1164/rccm.201803-0548OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J, et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet . 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugrue KF, Zohn IE. Mechanism for generation of left isomerism in Ccdc40 mutant embryos. PLoS One . 2017;12:e0171180. doi: 10.1371/journal.pone.0171180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol . 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 30. Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation . 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 31. Fassad MR, Patel MP, Shoemark A, Cullup T, Hayward J, Dixon M, et al. Clinical utility of NGS diagnosis and disease stratification in a multiethnic primary ciliary dyskinesia cohort. J Med Genet . 2020;57:322–330. doi: 10.1136/jmedgenet-2019-106501. [DOI] [PubMed] [Google Scholar]
- 32. Panizzi JR, Becker-Heck A, Castleman VH, Al-Mutairi DA, Liu Y, Loges NT, et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat Genet . 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shoemark A, Moya E, Hirst RA, Patel MP, Robson EA, Hayward J, et al. High prevalence of CCDC103 p.His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax . 2018;73:157–166. doi: 10.1136/thoraxjnl-2017-209999. [DOI] [PMC free article] [PubMed] [Google Scholar]