

SUMMARY
Kinesins drive the transport of cellular cargoes as they walk along microtubule tracks; however, recent work has suggested that the physical act of kinesins walking along microtubules can stress the microtubule lattice. Here, we describe a kinesin-1 KIF5C mutant with an increased ability to generate damage sites in the microtubule lattice as compared with the wild-type motor. The expression of the mutant motor in cultured cells resulted in microtubule breakage and fragmentation, suggesting that kinesin-1 variants with increased damage activity would have been selected against during evolution. The increased ability to damage microtubules is not due to the enhanced motility properties of the mutant motor, as the expression of the kinesin-3 motor KIF1A, which has similar single-motor motility properties, also caused increased microtubule pausing, bending, and buckling but not breakage. In cells, motor-induced microtubule breakage could not be prevented by increased α-tubulin K40 acetylation, a post-translational modification known to increase microtubule flexibility. In vitro, lattice damage induced by wild-type KIF5C was repaired by soluble tubulin and resulted in increased rescues and overall microtubule growth, whereas lattice damage induced by the KIF5C mutant resulted in larger repair sites that made the microtubule vulnerable to breakage and fragmentation when under mechanical stress. These results demonstrate that kinesin-1 motility causes defects in and damage to the microtubule lattice in cells. While cells have the capacity to repair lattice damage, conditions that exceed this capacity result in microtubule breakage and fragmentation and may contribute to human disease.
Graphical Abstract
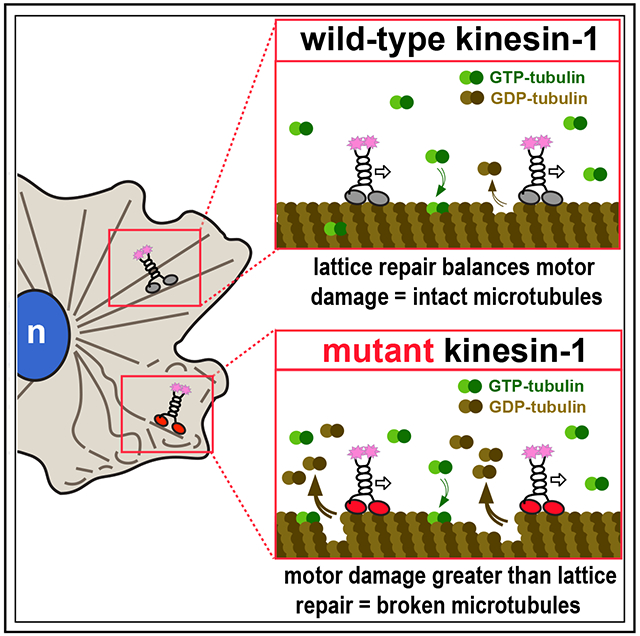
In brief
Budaitis et al. demonstrate that kinesin-1 damages the microtubule while stepping along tubulin subunits. Motor-induced damage makes microtubules sensitive to mechanical stress. Although motor-induced damage can be repaired, excessive motor-induced damage results in microtubule breakage and fragmentation in cells and may contribute to disease.
INTRODUCTION
Kinesins are a superfamily of proteins that carry out diverse microtubule-dependent processes such as cell division, cell motility, axonal transport, and cilium assembly and function .[1] Kinesin proteins are defined by the presence of a kinesin motor domain that contains sequences for nucleotide and microtubule binding. The conventional kinesin function of cargo transport involves processive motility, wherein the kinesin motor domain converts the energy of ATP hydrolysis into force and directed motion along the microtubule surface. Much work has focused on understanding the molecular mechanisms by which kinesins generate processive motility and has revealed critical contributions for motor protein dimerization, ATP binding and hydrolysis, and conformational changes of the neck linker (NL).2–4
Recent work has raised the possibility that the physical act of motors walking on microtubules creates stress in the microtubule lattice. In microtubule gliding assays, high densities of kinesin-1 motors can cause breakage of Taxol-stabilized microtubules and their splitting into protofilaments.5–8 Cryoelectron and fluorescence microscopy analyses revealed that the kinesin-1 motor domain, when in its strong microtubule-binding state (ATP-bound or apo), induces a change in the conformation of tubulin subunits within the GDP lattice.9–12 This conformational change can manifest to adjacent tubulin subunits and positively influence subsequent kinesin-binding events in the same region of the microtubule.12,13
Microtubule gliding assays involve motors working in teams while anchored to a solid substrate, and it has been unclear whether the processive motility of single kinesin motors walking on the microtubule could also create stress on the lattice. Recent work using in vitro motility assays demonstrated that motility of the mammalian kinesin-1 motor KIF5B, the yeast kinesin-8 motor Klp3, and yeast cytoplasmic dynein results in microtubule destruction via breakage and depolymerization.14 Although it was widely known that members of the kinesin-8 and kinesin-13 families can catalyze conformational changes to tubulin subunits that result in filament disassembly at the ends of microtubules,15 this was the first demonstration of motor stepping-induced destruction occurring along the shaft of the microtubule.
Although these findings have implications for the stability and function of microtubule networks in cells,16 whether processive motility of kinesin and/or dynein motors creates stress and/or defects in the microtubule lattice in cells has not been determined. Here, we describe a kinesin-1 mutant that causes microtubule destruction when expressed in cells. The identification of this destructive kinesin-1 mutant arose from our recent work investigating the role of the NL in force generation of kinesin-1 and kinesin-3 motors.17–20 The NL is a short and flexible segment that links the kinesin motor domain to the first coiled coil (the neck coil) that drives dimerization of kinesin proteins.3 We generated a series of truncations in the rat kinesin-1 motor KIF5C and then focused on a variant that lacks half of the coverstrand (deletion of amino acids 2–6, hereafter referred to as Δ6). Using in vitro single-molecule assays, we found that Δ6 motors display reduced force generation (stall force ~1 pN) but enhanced motility properties (velocity, processivity, and landing rate). These results support the hypothesis that the strength of NL docking involves a trade-off between speed and force generation.18,20,21
Surprisingly, we found that expression of Δ6 motors resulted in destruction of the microtubule network in cells. Using in vitro assays, we show that Δ6 motors generate increased damage along the microtubule lattice as compared with the wild-type (WT) kinesin-1. Although soluble tubulin can repair lattice damage induced by both WT and mutant motors, the increased damage caused by Δ6 results in microtubule that are sensitive to mechanical stress. These findings suggest that the Δ6 mutant is an unnatural or rogue motor whose activity would have been selected against during evolution. These results also indicate that cells must repair lattice defects to prevent microtubule breakage and fragmentation as motor-induced damage results in microtubules that are unable to bear compressive loads.
RESULTS
Truncation of the kinesin-1 coverstrand results in reduced force output but enhanced motility
During force generation, the NL responds to ATP binding and hydrolysis to dock along the side of the motor domain in two steps (Figure S1). The first step involves zippering of the first half of the NL (β9) to the coverstrand (β0) to form a two-stranded β-sheet called the cover-neck bundle (CNB). The second step involves binding of the second half of the NL (β10) within a docking pocket to latch the NL in place. The length of the coverstrand varies across kinesin families17 and deletion of the coverstrand of Drosophila kinesin-1 severely impairs motility and force generation.18 To examine how the length of the coverstrand influences CNB formation and force generation for mammalian kinesin-1, we created a series of truncations that successively shorten the coverstrand (Figure S1D) in the context of a constitutively active version of rat KIF5C containing aa 1–560 (RnKIF5C(1–560)). In initial experiments, all truncations displayed increases in velocity and processivity in single-molecule motility assays (Figure S1D), and thus, for the purposes of brevity and clarity, only the results of truncation KIF5C(1–560)-Δ6, which removes half of the coverstrand (Figure 1A), will be reported here.
Figure 1. Truncation of the coverstrand results in reduced kinesin-1 force output but enhanced motility.
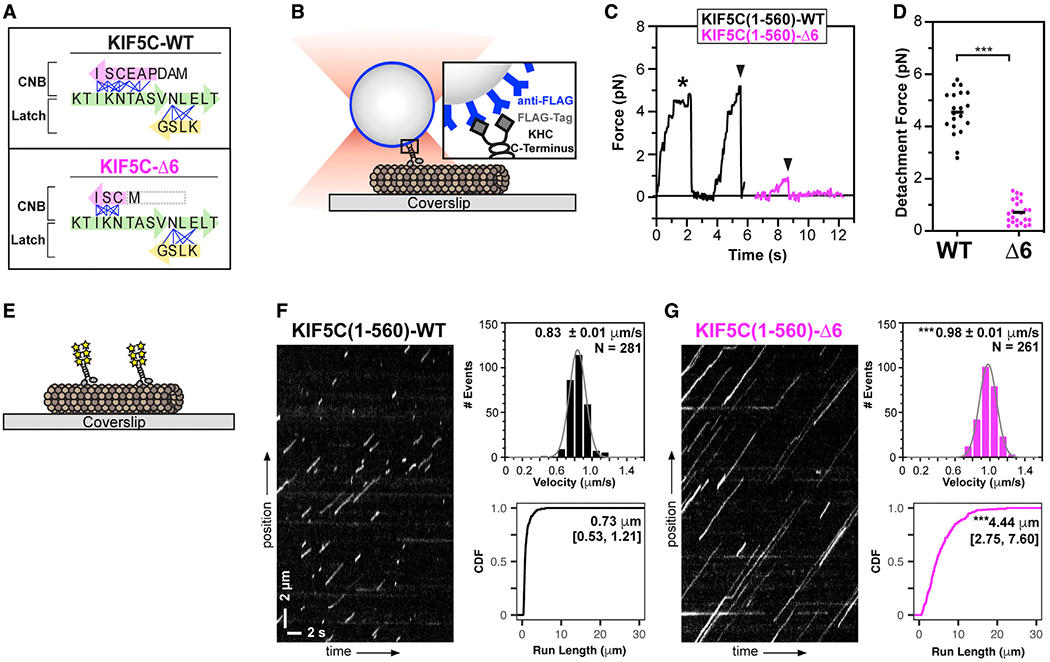
(A) Cartoon schematic of key structural elements involved in kinesin-1 NL docking. The first half of the NL(β9, green) interacts with the coverstrand (β0, magenta) to form the cover-neck bundle (CNB). The second half of the NL (β10, green) interacts with β7 (yellow) of the core motor domain for NL latching. Residue-residue contacts for NL docking are depicted as blue lines. The coverstrand residues missing in the Δ6 truncation are indicated by a gray box. See also Figures S1A–S1D.
(B–D) Motility under load.
(B) Schematic of assay. FLAG-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors were attached to beads functionalized with anti-FLAG antibodies and subjected to optical trap assays.
(C) Representative force versus time records of beads driven by single KIF5C(1–560)-WT (black) or KIF5C(1–560)-Δ6 (magenta) motors. The asterisk indicates a stall event; black arrowheads indicate abrupt detachment events.
(D) Detachment forces. Each dot indicates the detachment force of a motility event and the black line indicates the mean value for the population. n > 20 events for each construct; ***p < 0.001 (Student’s t test).
(E–G) Motility under unloaded conditions.
(E) Schematic of assay. The motility of 3xmCit-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors along Taxol-stabilized microtubules was determined in single-molecule motility assays.
(F and G) Representative kymographs with time displayed on the x axis (scale bars, 2 s) and distance displayed on the y axis (scale bars, 2 μm). Motor velocities are plotted as histograms with the mean ± SEM indicated at the top right. ***p < 0.001 compared with the WT protein (two-tailed Welch’s t test). Run lengths are plotted as cumulative distribution functions (CDFs) with median [quartiles] indicated at the top right. ***p < 0.001 compared with the WT protein (Kolmogorov-Smirnov test). N = 281 or 261 motility events across three independent experiments.
We used a custom-built optical trap apparatus with nanometer-level spatial resolution to assess the effect of the Δ6 coverstrand truncation on kinesin-1’s force output. FLAG-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 kinesin-1 motors were bound to anti-FLAG-coated beads and subjected to standard single-molecule trapping assays (Figure 1B). Individual KIF5C(1–560)-WT motors were motile in the absence of load, stalled on the microtubule when approaching the detachment force, and detached from the microtubule at an average force of 4.6 ± 0.8 pN (Figures 1C and 1D), consistent with previous studies.18,20,21 In contrast, KIF5C(1–560)-Δ6 mutant motors detached from the microtubule before stalling and at much lower loads than WT motors, with a mean detachment force of 0.7 ± 0.4 pN (Figures 1C and 1D). The reduced force output of the KIF5C(1–560)-Δ6 protein is similar to that of kinesin-1 motors with point mutations in the coverstrand that impair CNB formation.18,20
We then examined the motility properties of KIF5C(1–560)-Δ6 mutant motors under unloaded conditions. KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors tagged with three tandem monomeric citrine fluorescent proteins (3xmCit) were added to flow chambers containing Taxol-stabilized microtubules, and their single-molecule motility was examined using total internal reflection fluorescence (TIRF) microscopy (Figure 1E). Individual KIF5C(1–560)-WT motors underwent directed motility with speed (0.83 ± 0.01 μm/s) and processivity (median 0.73 μm [quartiles 0.53, 1.21 μm]) (Figure 1F), comparable with those of previous work.18,20 In contrast, individual KIF5C(1–560)-Δ6 motors were faster (0.98 ± 0.01 μm/s) and more processive (4.4 μm [2.75, 7.60 μm]) (Figure 1G). The enhanced motility properties of the KIF5C(1–560)-Δ6 protein are similar to those of kinesin-1 motors with point mutations in the coverstrand,20 suggesting that mutations which shorten and/or impair CNB formation are tolerated by the motor when stepping under no load.
Collectively, these results support the model that the coverstrand plays a critical mechanical role for single kinesin motors to step under load. These results also highlight how subtle changes in the coverstrand can act as a molecular gear shift, where motor speed and processivity come at the cost of robust force production.3,17–22
Expression of Δ6 mutant motors results in the destruction of the microtubule network in cells
We next set out to test whether the coverstrand truncation Δ6 impacts the ability of kinesin motors to work in teams to drive cargo transport in cells. We intended to use an organelle dispersion assay (Figure S2A); however, we were surprised to find that COS-7 cells expressing KIF5C(1–560)-Δ6 had mislocalized Golgi elements (Figure S2B). We also noticed that the KIF5C(1–560)-Δ6 motors decorated highly curved and knotted microtubules in cells (Figure S2B). We thus considered the possibility that expression of KIF5C(1–560)-Δ6 may cause unexpected changes to the microtubule network in cells.
We compared the organization of the microtubule network in cells expressing KIF5C(1–560)-Δ6 with that of the cells expressing KIF5C(1–560)-WT (Figures 2A and 2B). We quantified the organization of the microtubule network using three different parameters. First, to quantify microtubule destruction, the number of microtubule fragments per cell was determined and plotted against the motor expression level (Figure S3A). Second, to quantify loss of the microtubule network, the total length of the microtubule network within a 100 × 100 pixel (10.6 × 10.6 μm) box at the cell periphery was determined and plotted against motor expression level (Figures S3B and S3C). Third, to quantify the density of the microtubule network, we determined the number and size (area) of microtubule-enclosed spaces in the 100 × 100 pixel box at the cell periphery (Figures S3B and S3C).
Figure 2. Expression of KIF5C(1–560)-Δ6 results in microtubule destruction in cells.
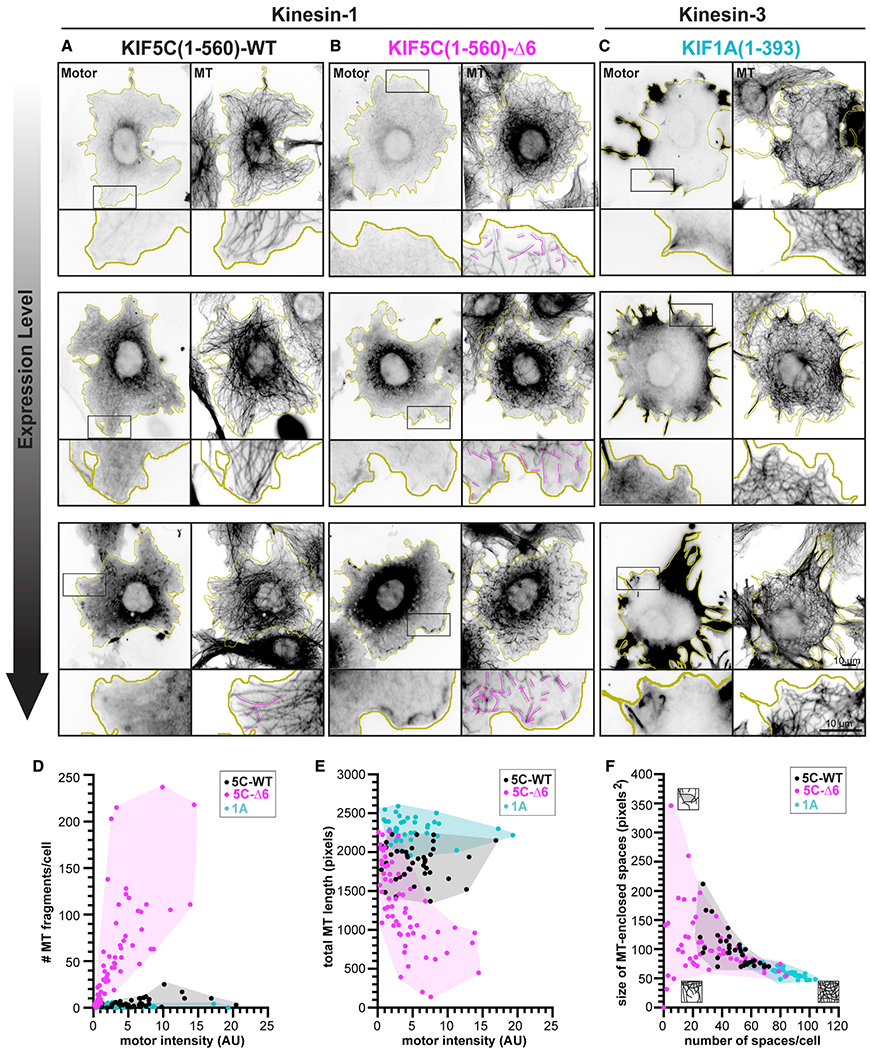
(A–C) Representative images of the microtubule (MT) network in cells expressing low, medium, or high levels of the indicated kinesin motor. COS-7 cells were transfected with plasmids encoding for the expression of 3xmCit-tagged (A) KIF5C(1–560)-WT, (B) KIF5C(1–560)-Δ6, or (C) KIF1A(1–393) motors and then fixed and stained with an antibody against β-tubulin. Yellow lines indicate the periphery of each cell; black boxes indicate regions shown below each image at higher magnification; and magenta traces indicate MT fragments. Scale bars, 10 μm. See also Figures S2, S4, and S5 and Video S3.
(D–F) Quantification of changes in the MT network. See also Figure S3. Each dot represents one cell. n > 39 cells each across 2 independent trials.
(D) Number of MT fragments/cell versus motor expression level.
(E) Total length of MTs in a 100 × 100 pixel box (10.6 × 10.6 μm, 10,000 total pixels) at the cell periphery versus motor expression level.
(F) Density of MTs in a 100 × 100 pixel box (10.6 × 10.6 μm, 10,000 total pixels) at the cell periphery as described by the number and size of MT-enclosed spaces. Cartoons depict the density of the MT network at each extreme (MTs are black, spaces enclosed by MTs are gray). AU, arbitrary units.
The microtubule network in cells expressing kinesin-1 KIF5C(1–560)-WT displayed a characteristic radial array that extends to the cell periphery, with a small number of microtubule fragments observed in cells expressing high levels of the WT motor (Figure 2A, magenta lines). In contrast, the microtubule network in cells expressing kinesin-1 KIF5C(1–560)-Δ6 mutant motors was dramatically disrupted. Microtubule fragments were observed at the periphery even in cells expressing low levels of KIF5C(1–560)-Δ6 motors (Figure 2B magenta lines; Figure 2D). Cells expressing the KIF5C(1–560)-Δ6 mutant motors also displayed a dramatic loss of microtubules at the cell periphery (Figures 2B and 2E) and a corresponding decrease in the number of microtubule-enclosed spaces in the cell periphery (Figures 2B and 2F).
To test whether it is the altered motility properties of KIF5C(1–560)-Δ6 mutant motors (Figure 1) that results in microtubule destruction, we tested whether expression of other kinesin motors with similar motility properties results in alteration of the microtubule network in COS-7 cells. To do this, we expressed the constitutively active kinesin-3 motor KIF1A(1–393) that has comparable single-motor motility properties (fast and superprocessive motility and detaches from microtubule at low force).19,23,24 However, expression of KIF1A(1–393) did not lead to the destruction of the microtubule network (Figure 2C). In these cells, the microtubule network was not fragmented (Figure 2D) but rather, expression of KIF1A(1–393) appeared to facilitate microtubule polymerization as the total microtubule length increased and the size of microtubule-enclosed spaces decreased (Figures 2E and 2F). Collectively, these results suggest that destruction of the microtubule network in cells is not solely a result of the enhanced motility properties of Δ6 mutant motors, as the microtubule network is not destroyed in cells expressing superprocessive KIF1A motors.
To rule out the possibility that the effects of the KIF5C(1–560)-Δ6 mutant motor are restricted to COS-7 cells, we expressed KIF5C(1–560)-Δ6 in human hTERT-RPE, MRC-5, and U-2-OS cells and in mouse NIH-3T3 cells. In each case, expression of Δ6 motors led to the fragmentation and/or loss of the microtubule network (Figure S4). Furthermore, microtubule destruction requires processive motility of KIF5C(1–560)-Δ6 as expression of a monomeric version (KIF5C(1–339)-Δ6) did not alter the organization or morphology of the microtubule network (Figure S5).
KIF5C(1–560)-Δ6 activity leads to buckling, knotting, and breakage of microtubules
We used live-cell imaging to uncover the events leading to destruction of the microtubule network in cells expressing KIF5C(1–560)-Δ6 motors. We imaged microtubule dynamics at the periphery of COS-7 cells expressing EGFP-α-tubulin and quantified the pausing, bending, and breaking of microtubules before they underwent catastrophe and shrinkage. Microtubules in control cells and in cells expressing KIF5C(1–560)-WT motors underwent periods of growth and then paused at the cell periphery before undergoing catastrophe and shrinkage (Figure 3A). In these cells, the majority of pauses lasted <30 s (Figure 3F), and although the paused microtubules often became bent in response to compressive forces (25.4% of microtubules in untransfected cells and 44.5% in KIF5C(1–560)-WT cells; Figure 3G), the pausing and bending rarely led to microtubule breakage (Figure 3G). In contrast, expression of KIF5C(1–560)-Δ6 mutant motors or KIF1A(1–393) motors resulted in a dramatic increase in the time of pausing at the cell periphery such that the majority of microtubule pauses lasted >150 s (Figures 3C and 3F). Furthermore, the paused microtubules showed an increased tendency to bend (56.7% of microtubules in KIF5C(1–560)-Δ6 cells and 59.1% in KIF1A(1–393) cells; Figure 3G). Together, the increased pause time and bending resulted in highly curled microtubules (Figure 3H) that tend to loop back toward the center of the cell and become knotted with one another (Videos S1 and S2).
Figure 3. KIF5C(1–560)-Δ6 leads to increased bending, buckling, and breakage of microtubules in cells.
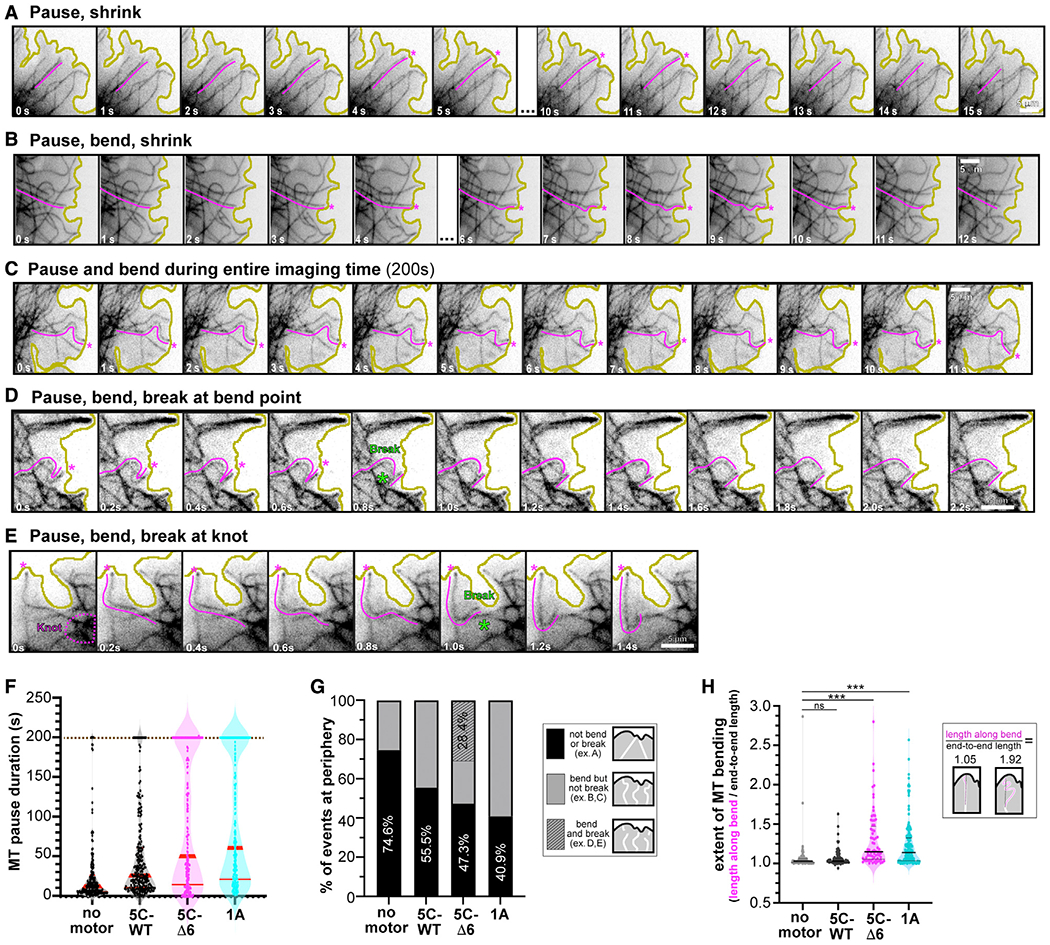
(A–E) Representative images of the types of microtubule growth events observed at the cell periphery. Magenta lines highlight the microtubule whose behavior was quantified, and magenta asterisks indicate time points of pausing at the plasma membrane. Green asterisks indicate the time frame when breakage occurred. Yellow lines indicate the periphery of the cell. Scale bars, 5 μm. See also Videos S1, S2, and S3.
(F and G) Quantification of microtubule fates from videos of cells expressing no motor, KIF5C(1–560)-WT, KIF5C(1–560)-Δ6, or KIF1A(1–393) motors together with EGFP-αtubulin. No motor, n = 181 events; KIF5C(1–560)-WT, n = 238 events; KIF5C(1–560)-Δ6, n = 167 events; KIF1A(1–393), n = 242 events across >7 cells per condition.
(F) The amount of time that a microtubule paused at the cell periphery (regardless of bending) before catastrophe. Each dot represents an individual microtubule event. Brown dotted line, imaging time limit. Red lines, median [quartiles]: no motor, 11.8s [5.6, 24.5]; KIF5C(1–560)-WT, 24.4 s [10.6, 55.6]; KIF5C(1–560)-Δ6, 46.2 s [14.2,180.1]; KIF1A(1–393), 55.5 s [20.2,180.0].
(G) The fate of the microtubule was scored as not bending or breaking (black, example in A), bending but not breaking (gray, examples in B and C), or bending and breaking (diagonal stripe, examples in D and E) before catastrophe.
(H) The extent of microtubule bending was quantified as the ratio of the length along the bent microtubule over the end-to-end length of that microtubule. Each dot represents an individual microtubule. No motor, n = 90 events, median 1.03 [quartiles 1.01, 1.06]; KIF5C(1–560)-WT, n = 115 events, 1.03 [1.01,1.07]; KIF5C(1–560)-Δ6, n = 72 events, 1.15 [1.05,1.48]; KIF1A(1–393), n = 163 events, 1.14 [1.04,1.33]. In total, 7 or more cells across 2–3 experiments per condition. ns, not significant and ***p < 0.001 (Kolmogorov-Smirnov test) as compared with no motor.
Expression of both KIF5C(1–560)-Δ6 and KIF1A(1–393) motors resulted in increased microtubule bending, looping, and buckling; however, only microtubules in cells expressing the KIF5C(1–560)-Δ6 motors were observed to break into fragments (Figure 3G). Microtubule breakage was observed to occur after persistent bending and buckling of a microtubule at the cell periphery (Figure 3D) as well as at locations of “knotted” microtubules (Figure 3E). The fragments released after microtubule breakage often underwent depolymerization, and the cumulative effect of microtubule fragmentation and depolymerization was a loss of microtubules at the periphery of the cell (Figure 2; Video S3). Overall, these results suggest that the activity of KIF5C(1–560)-Δ6 motors leads to bending, buckling, knotting, and breakage of microtubules, a phenomenon most noticeable at the cell periphery, and the eventual destruction of the microtubule network in cells.
Microtubule acetylation is not sufficient to protect microtubules from KIF5C(1–560)-Δ6 destruction
Recent work has demonstrated that acetylation of α-tubulin at lysine 40 (αTub-K40ac) protects microtubules from mechanical stress.25,26 We thus tested whether increasing αTub-K40ac can protect microtubules from destruction caused by the activity of KIF5C(1–560)-Δ6 motors. We cotransfected COS-7 cells with plasmids for expression of α-tubulin acetyltransferase (αTAT1) and either KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors. Expression of αTAT1 resulted in a dramatic increase in the levels of αTub-K40ac (Figures S6A–S6C), as expected,27,28 which provided a partial but statistically significant resistance to motor-induced microtubule breakage, although the net change in microtubule length was not significantly different in the absence and presence of αTAT1 (Figures 4B and 4C).
Figure 4. Expression of αTAT1 drives increased microtubule acetylation but does not protect the microtubule network from KIF5C(1–560)-–6 destruction.
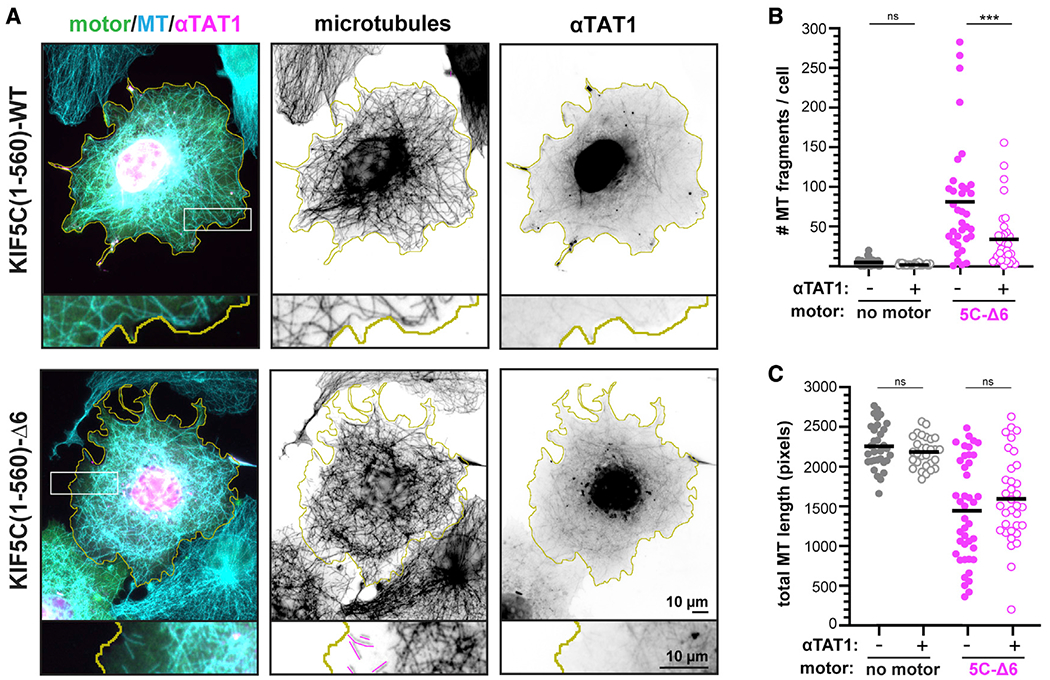
(A) Representative images of the microtubule (MT) network in cells expressing KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 together with αTAT1. The cells were fixed and stained with antibodies against total β-tubulin (microtubules) or αTub-K40ac (see also Figures S6A–S6C). Yellow lines indicate the periphery of each cell, white boxes indicate regions shown below each image at higher magnification, and magenta traces indicate microtubule fragments. Scale bars, 10 μm.
(B and C) Quantification of the microtubule network. Each dot represents one cell.
(B) Number of microtubule fragments per cell.
(C) Total length of microtubules in a 100 × 100 pixel box (10.6 × 10.6 μm, 10,000 total pixels) at the cell periphery. ***p < 0.001 (Student’s t test); ns, not significant.
As an alternative strategy to assess the impact of αTub-K40ac on motor-driven microtubule destruction, we treated cells with the deacetylase inhibitor trichostatin A (TSA) to block the activity of the α-tubulin deacetylase HDAC6.29 Cells treated with TSA had a substantial increase in αTub-K40ac levels (Figures S6D–S6F); however, expression of KIF5C(1–560)-Δ6 mutant motors still caused destruction of the microtubule network (Figure S6F). We thus conclude that “softening” of microtubules by α-tubulin K40 acetylation is not sufficient to protect microtubules from damage and breakage caused by the motility of KIF5C(1–560)-Δ6 motors.
KIF5C(1–560)-Δ6 motors fail to promote rescue events in microtubule dynamics assays
To gain an understanding of how KIF5C(1–560)-Δ6 impacts microtubules and leads to their destabilization and/or destruction, we turned to in vitro assays. We started with a microtubule dynamics assay to explore how the activity of KIF5C(1–560)-WT and KIF5C(1–560)-Δ6 motors leads to the destruction of growing microtubules. Microtubules were grown from GMPCPP-tubulin seeds in presence of soluble GTP-tubulin and either 3xmCit-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors (Figure 5A). In the absence of motor, classic microtubule dynamics of growth, catastrophe, and shrinkage were observed (Figure 5B). After catastrophe, the shrinking microtubules rarely underwent rescue events (resumption of microtubule growth) but rather depolymerized all the way back to the GMPCPP seed (Figure 5C).
Figure 5. KIF5C(1–560)-WT but not KIF5C(1–560)-Δ6 promotes microtubule rescue and overall microtubule growth.
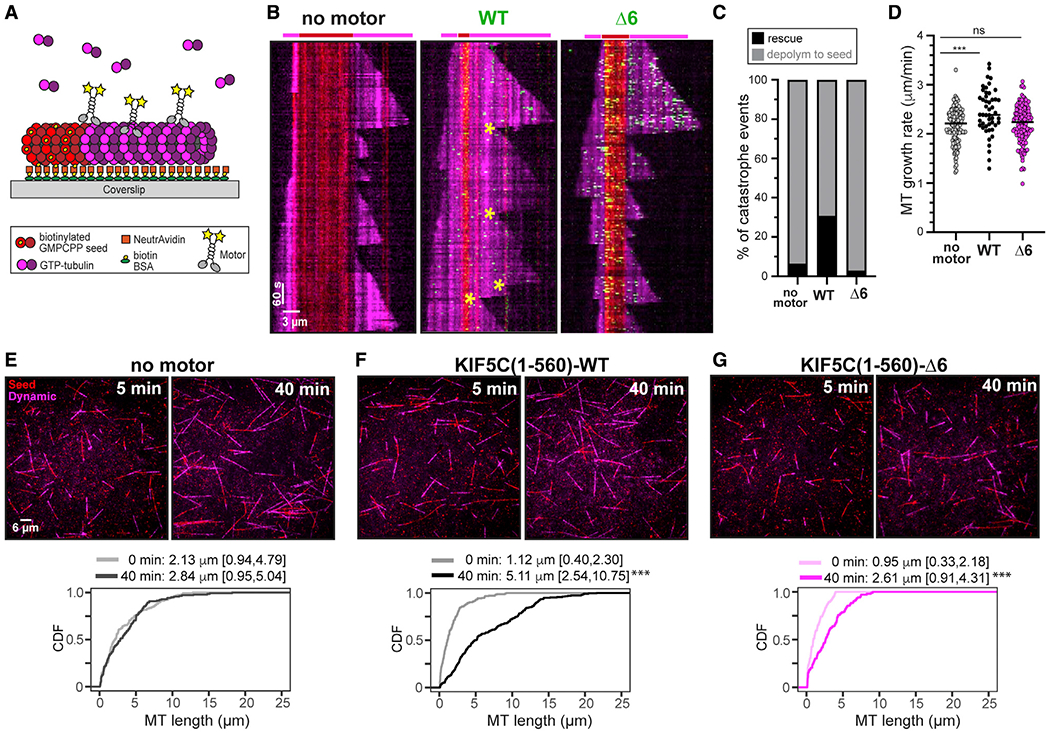
(A) Schematic of microtubule dynamics assay. Microtubules were polymerized from biotinylated GMPCPP-tubulin seeds in the presence of 10.7 μM tubulin and in the absence of motor or presence of 36 nM 3xmCit-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors.
(B) Representative kymographs. Red, biotinylated GMPCPP microtubule seeds; magenta, dynamic microtubules; green, KIF5C motors. Time is displayed on the y axis (scale bars, 60 s) and distance on the x axis (scale bars, 3 μm). Yellow asterisks mark rescue events.
(C and D) Quantification of (C) frequency of microtubule rescue and (D) microtubule growth rates.
(C) From videos of dynamic microtubules, events were scored as a catastrophe followed by depolymerization to the GMPCPP seed (gray) or catastrophe followed by a rescue event and new microtubule growth (black). N > 170 microtubules for each condition across two independent trials.
(D) Each spot indicates a microtubule growth event. No motor, 2.16 ± 0.36 μm/min (mean ± SD); WT, 2.45 ± 0.46 μm/min; Δ6, 2.20 ± 0.39 μm/min. ***p < 0.0001 (Tukey’s multiple comparisons test). See also Figures S1E–S1G.
(E–G) Representative images at 5 and 40 min time points. Red, GMPCPP seeds; magenta, dynamic microtubules. Scale bars, 6 μm. Graphs display cumulative distribution functions (CDFs) of microtubule lengths at the beginning (0 min) and end (40 min) of the assay. n > 102 each microtubules across two independent trials. Data reported as median [quartiles]. ***p < 0.001 as compared with the 0 min time point (Kolmogorov-Smirnov test).
KIF5C(1–560)-WT motility resulted in a dramatic increase in the number of rescue events (Figures 5B and 5C) and a slight but significant increase in microtubule growth rate (Figure 5D). Together, the increased rescue and growth led to a dramatic increase in the overall length of microtubules (Figure 5F). In contrast, the activity of KIF5C(1–560)-Δ6 motors did not lead to an increase in the number of rescue events (Figure 5C) or microtubule growth rates (Figure 5D), although there was an increase in the overall length of microtubules (Figure 5G). Given the Δ6 motor’s higher affinity for and processivity along microtubules (Figures 1 and S1E), we repeated the microtubule dynamics assays with varying concentrations of WT and Δ6 motors. An increase in the amount of KIF5C(1–560)-WT motors (to stoichiometrically match the activity of 36 nM Δ6 motor) caused a slight decrease in the rescue and growth rates (Figures S1F and S1G), suggesting that too much WT motor may be detrimental to microtubule growth. A decrease in the amount of KIF5C(1–560)-Δ6 motors (to stoichiometrically match the activity of 36 nM WT motor) resulted in a slight increase in microtubule rescues but no change in the microtubule growth rate (Figures S1F and S1G). These results indicate that although the activity of the WT motor alters the microtubule lattice in a way that promotes microtubule rescue and growth, the activity of the Δ6 motor becomes incompatible with rescue events.
KIF5C(1–560)-Δ6 causes more damage to the microtubule lattice than the WT protein
We considered several possible explanations for why KIF5C(1–560)-Δ6 motors are less able to promote rescue events for dynamic microtubules. One possibility is that the weak force output of KIF5C(1–560)-Δ6 motors (Figure 1D) results in little damage to the microtubule and thus only rare tubulin repair events that can trigger rescue events. An alternative possibility is that WT and Δ6 motors induce equivalent amounts of damage to the lattice, but the defects induced by KIF5C(1–560)-Δ6 motors cannot be repaired simply by the addition of soluble tubulin. A third possibility is that KIF5C(1–560)-Δ6 motors generate more damage than the WT motor such that the repair events are insufficient to maintain microtubule stability.
To test these possibilities, we performed microtubule repair assays.14,30 We defined a repair event as the incorporation of new tubulin subunits into a pre-existing lattice at sites of motor-induced damage. We generated GDP-lattice microtubules (GMPCPP-tubulin seeds and caps) and added purified KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors (Figure 6A). Given the Δ6 motor’s higher affinity for and processivity along microtubules (Figures 1 and S1E), we added more KIF5C(1–560)-WT motors to ensure similar activity of WT and mutant motors in the assay. In the presence of both KIF5C(1–560)-WT and KIF5C(1–560)-Δ6 motors, regions of tubulin incorporation into the GDP-microtubule lattice could be detected (Figure 6B). For both WT and Δ6, an increase in motor concentration resulted in more and larger damage sites (Figures 6B and 6C); however, the damage sites induced by KIF5C(1–560)-Δ6 activity were dramatically larger at each stoichiometric concentration (6 nM WT motor versus 2 nM Δ6 motor and 18 nM WT motor versus 6 nM Δ6 motor) (Figure 6C). These results indicate that both KIF5C(1–560)-WT and KIF5C(1–560)-Δ6 motors generate defects in the microtubule lattice that can be repaired by the incorporation of soluble tubulin, but the KIF5C(1–560)-Δ6 motor induces larger damage sites that are insufficient to maintain microtubule integrity.
Figure 6. KIF5C(1–560)-Δ6 induces more microtubule damage than KIF5C(1–560)-WT.
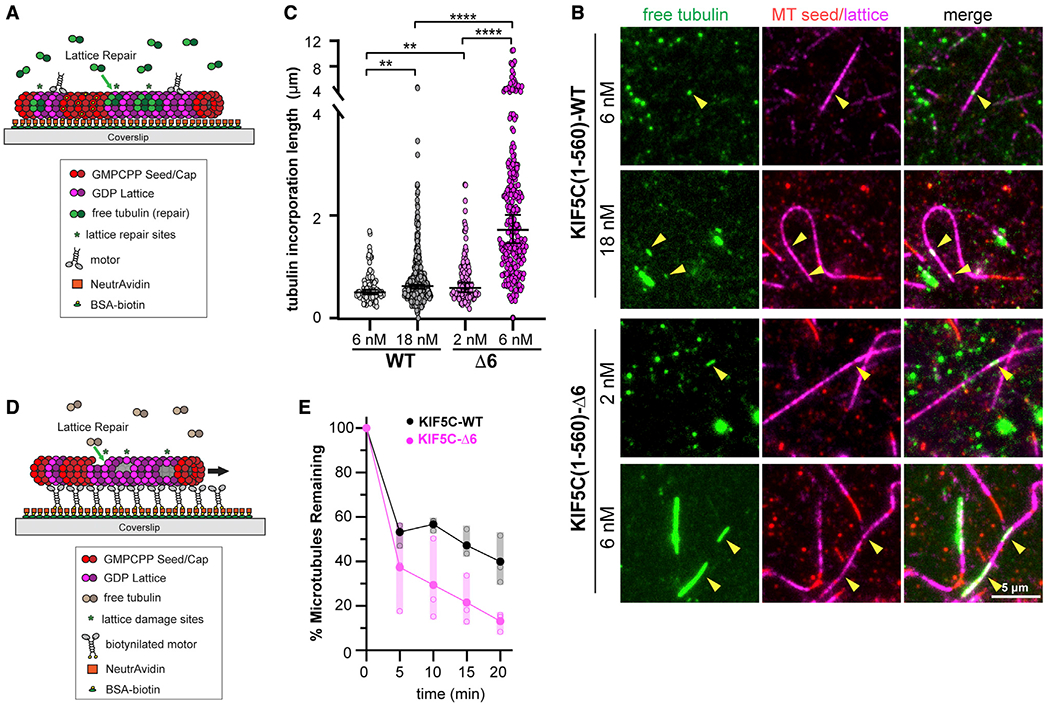
(A–C) Microtubule repair assay.
(A) Schematic of assay. GDP-tubulin microtubules (GMPCPP seeds and GMPCPP caps) were attached to a coverslip. Purified Halo-FLAG-tagged KIF5C(1–560)-WT (6 or 18 nM) or KIF5C(1–560)-Δ6 (2 or 6 nM) motors were added to the flow chamber in the presence of 10 μM 488 nm labeled soluble tubulin. Static images were obtained after 7 min of free tubulin incorporation into motor-driven damage sites.
(B) Representative images. Green, microtubule repair sites; red, GMPCPP seeds; magenta, GDP-tubulin lattice. Arrowheads indicate microtubule repair sites. Scale bars, 5 μm.
(C) Quantification of the length of microtubule repair sites. WT, n = 375; Δ6, n = 246 repair sites across three independent experiments. **p < 0.01 and ****p < 0.0001 (two-tailed t test).
(D and E) Microtubule destruction assay.
(D) Schematic of assay. Biotinylated KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors attached to a coverslip drive the gliding of GDP-tubulin microtubules (GMPCPP seeds and GMPCPP caps) in the presence of 7 μM soluble unlabeled tubulin.
(E) Quantification of motor-driven microtubule destruction over time. The total length of microtubules per field of view was measured, and the percentage of microtubules remaining in the chamber at the indicated time points was calculated and plotted as a dot plot. Black, KIF5C(1–560)-WT; magenta, KIF5C(1–560)-Δ6. Solid dots indicate the average loss of microtubule length across three independent trials, and open dots indicate loss of microtubule length for an individual trial. See also Figure S7A.
To extend these results, we carried out microtubule destruction assays,5–8,14 in which anchored motors cause stress on the lattice and microtubule destruction in the presence of limiting soluble tubulin. KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors were adhered to the surface of coverslips, and then, GDP-microtubules (GMPCPP-tubulin seeds and caps) were added in the presence of soluble tubulin (Figure 6D). Although the activity of both KIF5C(1–560)-WT and KIF5C(1–560)-Δ6 motors resulted in the gradual destruction of microtubules over time (Figures 6E and S7A), the destruction caused by KIF5C(1–560)-Δ6 motors occurred at a faster rate and to a much greater extent than the damage caused by the activity of WT motors (Figures 6E and S7A). These results support the hypothesis that the KIF5C(1–560)-Δ6 motors generate more damage to the microtubule lattice than KIF5C(1–560)-WT motors and that the resulting microtubules are more sensitive to mechanical stress.
Finally, to verify that KIF5C(1–560)-Δ6 causes more damage to the microtubule lattice than KIF5C(1–560)-WT motors, we incubated motors and microtubules on grids for 5 min in the absence of soluble tubulin. After blotting and washing, the microtubules were visualized by negative stain electron microscopy. In the absence of motor, long straight microtubules were observed, regardless of the polymerization condition (Taxol, GMPCPP-tubulin, or GTP-tubulin) (Figures 7 and S7B). In the presence of KIF5C(1–560)-WT motors, defects and breaks in the lattice were occasionally observed, whereas in the presence of KIF5C(1–560)-Δ6 motors, nearly every microtubule showed some type of defect or break in the lattice (Figures 7 and S7B). Together, these results support the hypothesis that KIF5C(1–560)-Δ6 motors generate more damage to the microtubule lattice and that the damage makes microtubules susceptible to mechanical stress and breakage.
Figure 7. KIF5C(1–560)-Δ6 induces more microtubule damage than KIF5C(1–560)-WT.
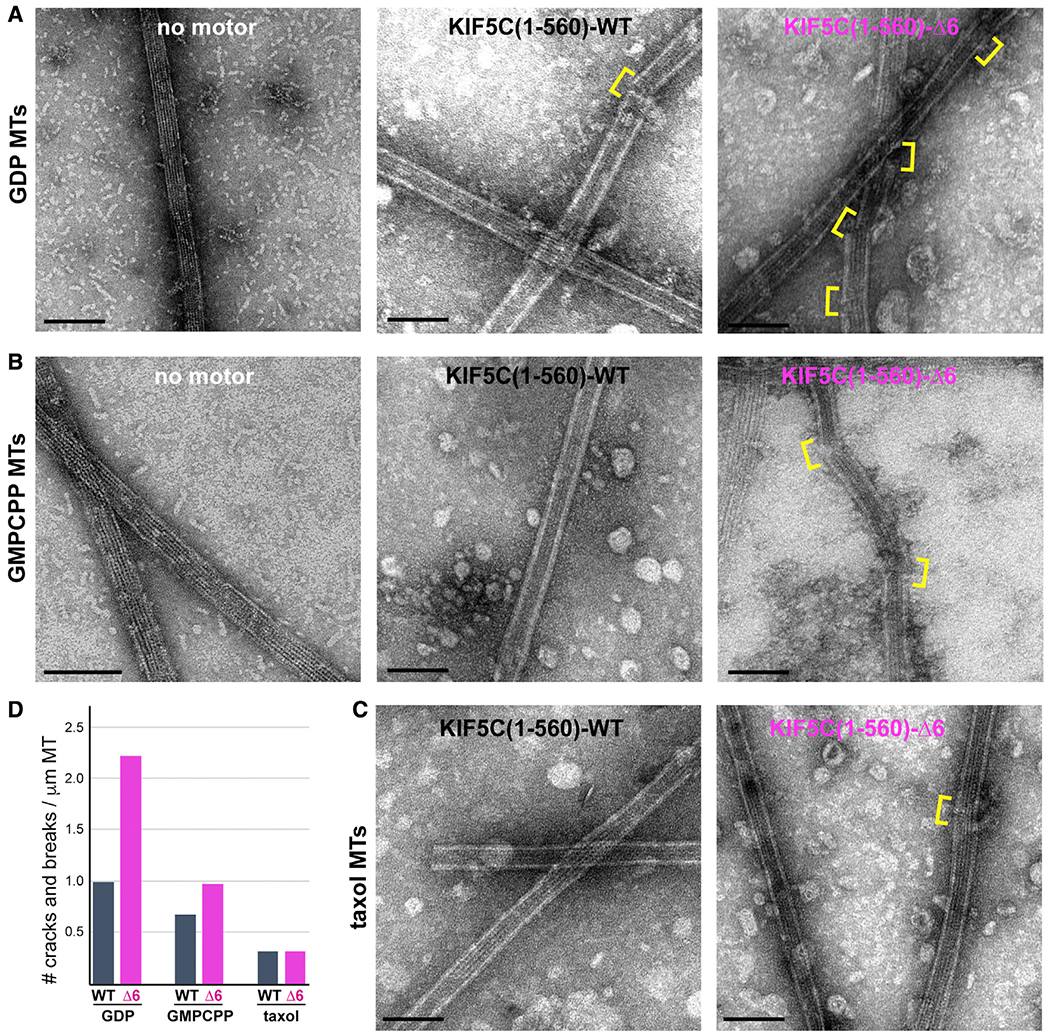
(A–C) GDP-tubulin microtubules (A, GDP MTs), GMPCPP-tubulin microtubules (B, GMPCPP MTs), or Taxol-stabilized microtubules (C, Taxol MTs) were incubated on grids with no motor or with purified Halo-FLAG-tagged KIF5C(1–560)-WT or KIF5C(1–560)-Δ6 motors for 5 min before blotting and staining with uranyl formate. Yellow brackets indicate breaks or cracks in the lattice. See also Figure S7B.
(D) Quantification of the number of breaks or cracks per μm of microtubule. GDP MTs, n = 26 breaks across 45 MTs for WT and 39 breaks across 30 MTs for Δ6; GMPCPP MTs, n = 21 breaks across 55 MTs for WT and 70 breaks across 128 MTs for Δ6; Taxol MTs, n = 5 breaks across 29 MTs for WT and 14 breaks across 77 MTs for Δ6. Scale bars, 100 nm.
DISCUSSION
The length of the coverstrand is optimized for kinesin-1 motility and force generation
The mechanistic basis of ATP-dependent force generation by motor proteins continues to be of great interest.3 For kinesin-1 proteins, zippering of the coverstrand (β0) with the NL (β9-β10) to form a two-stranded CNB is a critical first step in generation of a power stroke.17,22 CNB formation has also been observed structurally for members of the kinesin-2, kinesin-3, kinesin-5, and kinesin-6 families21,31–37 and is critical for kinesin-3 force generation.19
For CNB formation, variations in both the sequence and length of the coverstrand are thought to dictate kinesin-specific force and motility properties.21 Previous investigations of how the length of the coverstrand impacts force generation showed that deletion of the entire coverstrand segment of kinesin-1 abrogated force output.18 Here, we show that even a partial truncation of the coverstrand results in a kinesin-1 motor that is severely compromised in force generation as KIF5C(1–560)-Δ6 motors detached from the microtubule at low opposing forces (<1 pN; Figure 1).
Shortening of the coverstrand also resulted in kinesin-1 motors with enhanced motility properties (velocity, processivity, and landing rate) under unloaded conditions (Figure 1), likely due to allosteric effects of NL docking on the microtubule- and nucleotide-binding regions of the motor domain. Thus, a weakening of CNB formation via the point mutations in Khalil et al.18 and Budaitis et al.20 or partial deletion of the coverstrand (Figure 1) enables the motor to move with greater speed and processivity under unloaded conditions, whereas a complete loss of CNB formation impairs the motor’s ability to efficiently undergo processive motility.18 These findings support the hypothesis that the sequence and length of the coverstrand have been optimized in order to balance the motility and force generation properties of processive kinesins.18,20
Kinesin-1 motility causes damage to the microtubule lattice that can be repaired by soluble tubulin
Using in vitro reconstitution assays with GDP-tubulin microtubules, we demonstrate that while stepping, kinesin-1 motor proteins induce defects in the microtubule lattice that can be repaired by the incorporation of soluble tubulin, consistent with recent reports.14,30 Although both KIF5C(1–560)-WT and KIF5C(1–560)-Δ6 motors cause lattice damage that can be repaired by soluble tubulin (Figure 6), the activity of KIF5C(1–560)-Δ6 motors results in an increase in the size of the lattice repair sites compared with the KIF5C(1–560)-WT motor (Figure 6). In the microtubule dynamics assay, lattice repair in response to KIF5C(1–560)-WT activity enables rescue events that lead to an overall increase in microtubule mass (Figure 5), as previously reported by Andreu-Carbóet al.30 However, the excessive damage caused by KIF5C(1–560)-Δ6 motors and insufficient repair of those defects result in an inability of depolymerizing microtubules to undergo rescue events (Figure 5).
How does kinesin-1 cause damage to the microtubule lattice? Expression of monomeric KIF5C(1–339)-Δ6 does not result in microtubule breakage in cells (Figure S5), and no damage occurs in the presence of ADP in the microtubule destruction assay, indicating that lattice damage requires the motility of dimeric kinesin-1 motors. Our finding that kinesin-3 KIF1A motility does not cause microtubule damage indicates that there is something about the mechanochemical cycle of kinesin-1 that induces damage in the microtubule lattice. KIF1A spends the majority of its mechanochemical cycle in a one head-bound state, whereas kinesin-1 spends a majority of its mechanochemical cycle with both of its motor domains (heads) strongly bound to the microtubule (two head-bound state).24,38 A critical clue comes from recent reports that in the strong binding state (apo or AMPPNP), kinesin-1 motor domains push on and rotate tubulin subunits, thereby creating stress within the microtubule lattice.11,12 The expansion and contraction of tubulin subunits as a dimeric kinesin-1 steps on and releases from the microtubule is thus thought to drive a loss of subunits from the microtubule lattice.
How is the damage behavior enhanced in constructs with weakened NL docking? The shortened CNB makes the motor less able to withstand external load, as measured in optical trap experiments; however, the shortened CNB still exerts allosteric effects on microtubule- and nucleotide-binding regions of the motor domain, as observed in the single-molecule motility assay (Figure 1). These allosteric effects may alter the motor-microtubule interface to cause increased pushing on and rotation of tubulin subunits by the Δ6 motor domains. Alternatively, the shortened CNB may enable the KIF5C(1–560)-Δ6 motor protein to generate more interhead tension and/or more quickly cycle past the vulnerable one head-bound state as compared with the WT motor. Further structural and kinetic work is required to understand how weakening of CNB, by point mutation or truncation, alters the chemomechanical cycle of kinesin-1 and leads to an enhanced ability of kinesin-1 to generate stress within the microtubule lattice.
Motor-induced damage makes microtubules sensitive to mechanical stress
Microtubules can bear significant compressive loads and are often bent and buckled in cells; however, bending and buckling are not sufficient to cause microtubule breakage.39–45 Indeed, we observed that microtubule bending in cells can occur upon polymerization against the plasma membrane yet rarely leads to microtubule breakage. Increasing the extent of microtubule looping, bending, and buckling is also not sufficient to cause microtubule breakage. We show that the kinesin-3 KIF1A(1–393) motor accumulates at the plus ends of the microtubules in the cell periphery and increases the time of microtubule pausing, and this, in turn, results in increased microtubule bending and buckling (Figures 2 and 3). The fact that a dramatic increase in microtubule bending does not lead to microtubule breakage in cells has also been observed upon increased microtubule sliding, driven by overactivation of kinesin-1 or kinesin-1446–49 or overexpression of microtubule bundling proteins of the MAP65/PRC1/Ase1 family.50
Although KIF5C(1–560)-Δ6 also increases microtubule pausing, the resulting increase in microtubule bending and buckling is followed by breakage and fragmentation. Expression of other KIF5C variants with truncation (Δ3, Δ7, Δ8, and Δ9) or mutation20 of the coverstrand also causes microtubule destruction in cells (Figure S1D). A kinesin-induced loss of tubulin subunits from the microtubule lattice reduces the microtubule’s flexural rigidity and “softens” the microtubule, as noted by Kabir et al.6 For microtubules that are not experiencing mechanical stress, e.g., microtubule dynamics assays, a little bit of damage can promote microtubule rescue but too much damage prevents rescues and is detrimental to microtubule polymerization. For microtubules that experience mechanical stress, e.g., in microtubule gliding assays (Figure 6), on EM grids (Figure 7), and in cells (Figures 2 and 3), the excessive defects induced by KIF5C(1–560)-Δ6 result in microtubules that are “too soft” and undergo breakage and fragmentation.
One mechanism that protects microtubules from breakage in cells is acetylation of α-tubulin at K40. However, we find that increased αTub-K40ac is unable to protect microtubules from breakage resulting from KIF5C(1–560)-Δ6 damage. αTub-K40ac weakens interactions between tubulin subunits in neighboring protofilaments and enhances microtubule flexibility, making the microtubule resistant to repeated mechanical stress.25,26 Thus, αTub-K40ac is most effective in the context of a fully intact microtubule where intertubulin contacts are maximized. KIF5C(1–560) motility removes tubulin subunits from the microtubule lattice, rendering αTub-K40ac ineffective. In contrast to αTub-K40ac, Taxol protects microtubules from motor-induced damage, as demonstrated by Triclin et al.14 and recapitulated here (Figure 7). In this respect, a recent report demonstrating that UNC-45A can bind to the lattice of Taxol-stabilized microtubules, causing bends and kinks that lead to breakages along the microtubule length, is quite interesting.51 Further work is required to understand how Taxol-induced strengthening of longitudinal tubulin contacts makes microtubules resistant to kinesin-induced but not UNC45A-induced damage.
Repair of motor-induced damage in cells
Our findings extend previous reports of motor-induced lattice damage in reconstituted systems14,30 by demonstrating that motor-induced damage occurs in cells. One important corollary of our work is that repair of lattice defects is essential to prevent microtubule breakage and fragmentation in cells. Indeed, the lattice damage and subsequent repair induced by the WT protein could be part of a positive feedback loop that results in strong and stable microtubule tracks for directed transport, as suggested by Théry et al.16 and Andreu-Carbó et al.30 Defects along cellular microtubules can be observed by cryoelectron microscopy,52–54 and their repair is likely to be particularly important in cardiomyocytes where microtubules bear significant compressive loads55 and in neurons where the microtubules span the length of the axon. Indeed, microtubule curling and disorganization have been observed in aging, after injury, and in certain animal models of axonopathies.49
A second important corollary is that cells have a limited capacity to repair or resolve lattice defects, and conditions that exceed this capacity, such as expression of highly destructive KIF5C(1–560)-Δ6 motors, result in microtubule breakage and fragmentation. Our identification of Δ6 as a kinesin motor that enhances microtubule damage in cells enables future work exploring whether lattice repair mechanisms become limiting for kinesin and/or tubulin mutations that lead to neurodegenerative diseases. Our findings also open a path to exploring whether and which factors contribute to recognition and repair of microtubule damage in cells. These factors include the end binding (EB) proteins that accumulate at damage sites in vitro55,56 and can accumulate as islands on the microtubule lattice in cells,57 CLIP-170 (cytoplasmic linker protein of 170 kDa) and related proteins that promote microtubule rescues58,59 and CLASPs (cytoplasmic linker associated proteins) that can bind tubulin heterodimers and oligomers as well as microtubules and have been proposed to contribute to microtubule repair.60,61
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kristen Verhey (kjverhey@umich.edu).
Materials availability
Plasmids generated in this study will be deposited to Addgene.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
COS-7 [male Ceropithecus aethiops (African green monkey) kidney fibroblast, RRID: CVCL_0224] and NIH 3T3 [male Mus musculus embryo fibroblast, RRID: CVCL_0594] cells were grown in in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% (vol/vol) Fetal Clone III (HyClone) and 2 mM GlutaMAX (L-alanyl-L-glutamine dipeptide in 0.85% NaCl, Gibco). MRC-5 (male Homo sapiens fetal lung fibroblast, RRID: CVCL_0440) and U2OS (female Homo sapiens osteosarcoma, RRID: CVCL_0042) cells were grown in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% (vol/vol) FBS (HyClone) and 2 mM GlutaMAX (Gibco). hTERT-RPE1 cells (female Homo sapiens retinal pigment epithelium, RRID: CVCL_4388) were grown in DMEM/F12 with 10% (vol/vol) FBS (HyClone), 0.5 mg/ml hygromycin B, and 2 mM GlutaMAX (Gibco). All cell lines were purchased from American Type Culture Collection and grown at 37°C with 5% (vol/vol) CO2. All cells lines are checked annually for mycoplasma contamination and COS-7 cells were authenticated through mass spectrometry (the protein sequences exactly match those in the Ceropithecus aethiops genome).
METHOD DETAILS
Plasmids and Adenoviral vectors
A truncated, constitutively active kinesin-1 [rat KIF5C(1-560)] was used. Coverstrand truncation mutants were generated by PCR and all plasmids were verified by DNA sequencing. The truncated, constitutively active version of the kinesin-3 motor KIF1A contains the first 393 amino acids of rat KIF1A, which includes the neck coil sequence for dimerization, followed by a Leucine zipper (LZ) sequence to maintain the dimer state.23 Motors were tagged with three tandem monomeric Citrine fluorescent proteins (3xmCit) or HALO-FLAG tags for single-molecule imaging assays, a FLAG tag for optical trapping assays, monomeric NeonGreen (mNG)-FRB for Golgi dispersion assays, or were biotinylated via an AviTag (aa sequence GLNDIFEAQKIEWHE) and co-expression with HA-BirA for microtubule gliding assays. The mouse αTATI coding sequence (NP_001136216) was cloned into the vector pmCit-C1. Constructs coding for FRB (DmrA) and FKBP (DmrC) sequences were obtained from ARIAD Pharmaceuticals and are now available from Takara Bio. Plasmids encoding monomeric NeonGreen were obtained from Allele Biotechnology. The adenovirus plasmid encoding EGFP-tubulin (pShuttle-EGFP-tubulin) was a gift from Torsten Wittmann (Addgene plasmid #24327, RRID: Addgene_24327) and adenovirus was produced by the University of Michigan Vector Core.
Transfection and lysate preparation
24 hr after seeding, COS-7 cells were transfected with plasmids using TransIT-LT1 transfection reagent (Mirus) and Opti-MEM Reduced Serum Medium (Gibco). 24 hr after transfection, the cells were trypsinized and harvested by centrifugation at 3000 x g at 4°C for 3 min. The pellet was resuspended in cold 1X PBS, centrifuged at 3000 x g at 4°C for 3 min, and the pellet was resuspended in 50 μL of cold lysis buffer [25 mM HEPES/KOH, 115 mM potassium acetate, 5 mM sodium acetate, 5 mM MgCl2, 0.5 mM EGTA, and 1% (vol/vol) Triton X-100, pH 7.4] with 1 mM ATP, 1 mM phenylmethylsulfonyl fluoride, and 1% (vol/vol) protease inhibitor cocktail (P8340, Sigma-Aldrich). Lysates were clarified by centrifugation at 20,000 x g at 4°C for 10 min and lysates were snap frozen in 5 μL aliquots in liquid nitrogen and stored at −80°C.
Imaging of fixed and live cells
For fixed cells, 24 hr post-transfection, the cells were rinsed with PBS and fixed in 3.7% (vol/vol) paraformaldehyde (ThermoFisher Scientific) in PBS for 10 min at room temperature. Fixed cells were permeabilized in 0.2% Triton X-100 in PBS for 5 min and blocked with 0.2% fish skin gelatin in PBS for 5 min. Primary and secondary antibodies were applied in 0.2% fish skin gelatin in PBS for 1 hr at room temperature in the dark. Primary antibodies: Ms anti-β-tubulin (clone E7, Developmental Studies Hybridoma Bank, 1:2000), Ms anti-αTubK40ac (clone 6-11B-1, Sigma T7451; 1:10,000), Rb anti-giantin (Biolegend #924302, 1:200). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories and used at 1:500 dilution. Nuclei were stained with 10.9 mM 40,6-diamidino-2-phenylindole (DAPI) and the coverslips were mounted using Prolong Gold (Invitrogen). Images were acquired on an inverted epifluorescence microscope (Nikon TE2000E) with a 40× 0.75 NA, a 60xv1.40 NA oil-immersion, or a 100× 1.40 NA objective and a CoolSnap HQ camera (Photometrics).
Image analysis was carried out using Fiji/ImageJ (National Institutes of Health). Three parameters were calculated. First, the number of microtubule fragments/cell was scored manually where a fragment was defined as a short section of microtubule with both ends visible (Figure S5A). The motor expression level in the same cell was determined as the whole-cell fluorescence intensity (arbitrary fluorescent units). Second, a 100x100 pixel (10.6x10.6 mm, 10,000 total pixels) ROI box was placed in a flat region of the cell periphery and positioned to be touching the edge of the cell. The microtubule image in this ROI was thresholded, despeckled, and skeletonized (Figure S5B). After verifying that the final imaged matched the original image, the total microtubule length in the box was calculated as the number of pixels with fluorescence signal (Figure S5C). Third, using the same 100x100 pixel box, the number and size of the spaces completely enclosed by microtubules was calculated as a description of the density of the microtubules in that region (Figure S5C).
For live-cell imaging of dynamic microtubules, cells in glass-bottom dishes (Matek) were infected with adenovirus for expression of EGFP-α-tubulin. 24 hr later the cells were transfected and Janelia Fluor 552 (JF552, 50 nM, Janelia Materials) ligand was added to label Halo-tagged motors. 16 h post-transfection, the cells were washed and then incubated in Leibovitz’s L-15 medium (Gibco) and imaged at 37°C in a temperature-controlled and humidified stage-top chamber (Tokai Hit). Live-cell imaging was performed on an inverted TIRF microscope Ti-E/B (Nikon) equipped with the perfect focus system, a 100× 1.49 NA oil immersion TIRF objective (Nikon), three 20-mW diode lasers (488 nm, 561 nm, and 640 nm), and an electron-multiplying charge-coupled device detector (iXon X3DU897; Andor). The angle of illumination was adjusted for maximum penetration of the evanescent field into the cell. Image acquisition was controlled with Elements software (Nikon).
Microtubule growth events at the cell periphery were scored from videos of cells expressing no motor, KIF5C(1-560)-WT, KIF5C(1-560)-Δ6, or KIF1A(1-393) motors. Two types of scoring were applied to each event. First, the pause time of a microtubule at the cell periphery before catastrophe and depolymerzation was calculated. Pausing was defined as a microtubule immobilized with respect to the plasma membrane regardless of continued growth and bending of the microtubule. Second, the behavior of the microtubule was categorized as no bending or breaking, bending but not breaking, or bending and breaking before catastrophe and depolymerization. Bending was defined as a deviation from straight line and the amount of bending (regardless of breaking) was quantified as the length of the bent microtubule divided by the end-to-end length of that microtubule.
Optical trapping assays
Bovine brain tubulin (Cytoskeleton TL238) was reconstituted in 25 mL BRB80 buffer [80 mM PIPES (Sigma P-1851), 1 mM EGTA (Sigma E-4378), 1 mM MgCl2 (Mallinckrodt H590), pH adjusted to 6.9 with KOH] supplemented with 1 mM GTP (Cytoskeleton BST06) and kept on ice. 13 mL PEM104 buffer (104 mM PIPES, 1.3 mM EGTA, 6.3 mM MgCl2, pH adjusted to 6.9 with KOH) was mixed with 2.2 mL 10 mM GTP, 2.2 mL DMSO, and 4.8 mL of 10 mg/mL tubulin and microtubules were polymerized by incubation for 40 min at 37°C. Subsequently, 2 mL of stabilization solution [STAB: 38.6 mL PEM80, 0.5 mL 100 mM GTP, 4.7 mL 65 g/L NaN3 (Sigma S-8032), 1.2 mL 10 mM Taxol (Cytoskeleton TXD01), 5 mL DMSO (Cytoskeleton)] was added to the stock microtubule solution at room temperature.
Optical trap assays were performed as described previously.20,70,71 0.44 μm anti-FLAG-coated beads were prepared by crosslinking anti-FLAG antibodies to carboxy polystyrene beads (Spherotech) via EDC chemistry. Lysates containing FLAG-tagged motors were diluted in assay buffer [AB: P12 buffer (12 mM PIPES (Sigma P-1851), 1 mM EGTA (Sigma E-4378), 1 mM MgCl2 (Mallinckrodt H590), pH adjusted to 6.9 with KOH), 1 mM DTT (Sigma Aldrich), 20 mM Taxol (Cytoskeleton), 1 mg/mL casein (Blotting-Grade Blocker, Biorad), 1 mM ATP (Sigma Aldrich)] and then incubated with gently sonicated anti-FLAG beads to allow binding for 1 hr at 4°C on a rotator in the presence of oxygen scavenging reagents (5 mg/mL b-D-glucose (Sigma Aldrich), 0.25 mg/mL glucose oxidase (Sigma Aldrich), and 0.03 mg/mL catalase (Sigma Aldrich).
A flow cell that holds a volume of ~15 μL was assembled using a microscope slide, etched coverslips, and double-sided sticky tape. Before assembly, etched coverslips were incubated in a solution of 100 μL poly-l-lysine (PLL, Sigma P8920) in 30 mL ethanol for 15 min. The coverslip was then dried with a filtered air line. After flow cell assembly, microtubules were diluted 150 times from the stock in a solution of PemTax (1 μL 10 mM Taxol in 500 μL P12). The diluted microtubules were added to the flow cell and allowed to adhere to the PLL surface for 10 min. Unbound microtubules were then washed out with 20 μL PemTax. A solution of casein (Blotting-Grade Blocker, Biorad 1706404) diluted in PemTax (1:8 mixture) was then added to the flow cell and allowed to incubate for 10 min to block the remainder of the surface to prevent non-specific binding. After the incubation, the flow cell was washed with 50 μL PemTax and 80 μL assay buffer (AB). 20 μL of the bead/motor incubation was then added to the flow cell.
Optical trapping measurements were obtained using a custom-built instrument with separate trapping and detection systems.18 Briefly, beads were trapped with a 1,064 nm laser that was coupled to an inverted microscope with a 100x/1.3 NA oil-immersion objective. Bead displacements from the trap center were recorded at 3 kHz and further antialias filtered at 1.5 kHz. To ensure that we were at the single molecule limit for the motility assay, the protein-bead ratio was adjusted such that only 5-10% of trapped beads showed microtubule binding. A motor-coated bead was trapped in solution and subjected to position calibration and trap stiffness using Labview routines. Afterward, the bead was brought close to a surface-bound microtubule to allow for binding. Bead position displacement and force generation were measured for single motor-bound beads. Detachment force measurements include motility events where single motors reached a plateau stall before detachment and events where the motor abruptly detached from the microtubule. Detachment forces are plotted as a dot plot where each dot indicates the maximum detachment force of an event and the mean for each construct is indicated by a black horizontal line. Statistical differences between the maximum detachment force of wild type and mutant motors were calculated by using a two-tailed unpaired Student’s t test.
Unloaded, single-molecule motility assays
Microtubules were polymerized (unlabeled and HiLyte-647-labeled porcine brain tubulin, Cytoskeleton #T240 and #TL670M) in BRB80 buffer (80 mM Pipes/KOH pH 6.8, 1 mM MgCl2, 1 mM EGTA) supplemented with 2 mM GTP and 2 mM MgCl2 and incubated for 60 min at 37°C. Taxol (Cytoskeleton) in prewarmed BRB80 was added to 2 μM and incubated for 60 min. Microtubules were stored in the dark at room temperature for up to 2 weeks. Flow cells were prepared by attaching a #1.5 coverslip (ThermoFisher Scientific) to a glass slide (ThermoFisher Scientific) using double-sided tape. Microtubules were diluted in fresh BRB80 buffer supplemented with 10 μM taxol, infused into flow cells, and incubated for four minutes to allow for nonspecific absorption to the glass. Flow cells were incubated with (i) blocking buffer [30 mg/mL casein in P12 buffer (12 mM Pipes/KOH pH 6.8, 1 mM MgCl2, 1 mM EGTA) supplemented with 10 μM taxol] for four minutes and then (ii) motility mixture (0.5–1.0 μL of COS-7 cell lysate, 25 μL P12 buffer, 15 μL blocking buffer, 1 mM ATP, 0.5 μL 100 mM DTT, 0.5 μL of 20 mg/mL glucose oxidase, 0.5 μL of 8 mg/mL catalase, and 0.5 μL 1 M glucose). Flow chambers were sealed with molten paraffin wax and imaged on an inverted Nikon Ti-E/B TIRF microscope with a perfect focus system, a 100 × 1.49 NA oil immersion TIRF objective, three 20 mW diode lasers (488 nm, 561 nm, and 640 nm) and EMCCD camera (iXon+ DU879; Andor). Images were acquired every 50 ms for 30 s at room temperature. Acquisition was controlled using Nikon Elements software.
Motility data were analyzed by first generating maximum intensity projections to identify microtubule tracks (width = 3 pixels) and then generating kymographs in Fiji/ImageJ (National Institutes of Health). Only motility events that lasted for at least three frames were analyzed. Furthermore, events that ended as a result of a motor reaching the end of a microtubule were included; therefore, the reported run lengths for highly processive motors are likely to be an underestimation. The velocities were binned, plotted as a histogram, fit to a Gaussian, and a two-tailed Welch’s t test was used to assess whether velocity distributions were significantly different between motors. The cumulative distributions of motor run lengths were fit to an exponential (WT motor) or gamma (D6 motor) distribution and a Kolmogorow-Smirnov test was used to assess whether run length distributions were significantly different between motors.
Protein expression and purification
COS-7 cells were transfected with plasmids for expression of KIF5C(1-560)-WT-Halo-Flag or KIF5C(1–560)-Δ6-Halo-Flag and the protein was fluorescently labelled by the inclusion of 50 nM JF552 Halo ligand (Janeila Materials) in the growth medium. Cells from two 10cm dishes were harvested 24 h after transfection and lysed in 1 ml lysis buffer [25 mM HEPES, 115 mM KOAc, 5 mM NaOAc, 5 mM MgCl2, 0.5 mM EGTA, 1% Triton X-100, pH to 7.4 with KOH] supplemented with protease inhibitor cocktail, 1 mM PMSF, 1 mM ATP, and 1 mM DTT. After centrifugation at 16,000xg for 10 min at 4°C, the supernatant was incubated with 50 μl anti-Flag M2 agarose beads (Sigma-Aldrich) with rotation for 1.5 h at 4°C. The beads were washed with wash buffer (150 mM KCl, 20 mM Imidazole pH 7.5, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA) supplemented with protease inhibitor cocktail, 1 mM PMSF, 1 mM DTT, and 3 mM ATP, and washed again with wash buffer supplemented with protease inhibitor cocktail, 1 mM PMSF and 1 mM DTT. The protein was eluted with 80 μl BRB80 buffer (80 mM PIPES/KOH pH6.8, 1 mM MgCl2, 1 mM EGTA) supplemented with protease inhibitor cocktail, 1 mM PMSF, 0.5 mM DTT, 0.1 mM ATP, 0.5 mg/ml 3xFlag peptide (Sigma-Aldrich) for 1h. The protein was collected as the supernatant after centrifugation at 1,500xg for 5 min at 4°C. Aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
Microtubule dynamics assay
Microtubule seeds were prepared by polymerizing 25 μM tubulin (Cytoskeleton) consisting of 6% biotinylated-tubulin (Cytoskeleton) and 6% fluorescent (X-Rhodamine or HiLyte647) tubulin (Cytoskeleton) in the presence of the nonhydrolyzable GTP analogue GMPCPP (Jena Bioscience) in BRB80 buffer and 2.5 mM MgCl2 for 35 min at 37 °C. The seeds were sedimented by centrifugation at 90,000 rpm for 5 min at 25 °C (Beckman Coulter). The microtubule pellet was resuspended in warm BRB80 buffer and microtubule seeds were stored in the dark at room temperature.
A flow chamber (~10 μl volume) was assembled by attaching a clean #1.5 coverslip (Thermo Fisher Scientific) to a glass slide (Thermo Fisher Scientific) with two stripes of double-sided tape. Microtubule seeds were immobilized by sequential incubation with: (i) 1 mg/ml BSA-biotin (Sigma-Aldrich), (ii) blocking buffer (1 mg/ml BSA in BRB80), (iii) 0.5 mg/ml NeutrAvidin (Thermo Fisher), (iv) blocking buffer, (v) short GMPCPP-stabilized microtubule seeds, and (vi) blocking buffer. Microtubule growth was initiated by flowing in 10.7 μM tubulin containing 7% Hilyte647–labeled tubulin (Cytoskeleton) together with 36 nM motor proteins in the reaction buffer (BRB80 with 1 mM GTP, 2.5 mM ATP, 0.1 mg/ml BSA, 1 mg/ml casein, 1 mM MgCl2, 0.1% methylcellulose, and oxygen scavenging mix [1 mM DTT, 10 mM glucose, 0.1 mg/ml glucose oxidase, and 0.08 mg/ml catalase]). The flow cells were sealed with molten paraffin wax and imaged by TIRF microscopy. The temperature was set at 37°C in a temperature-controlled chamber (Tokai Hit) and time-lapse images were acquired in 488-nm, 561-nm, and 640-nm channels at a rate of every 5 s for 15 min. Maximum-intensity projections were generated and kymographs (width = 3 pixels) were generated using Fiji/ImageJ and displayed with time on the x-axis and distance on the y-axis. From kymographs, the total number of growth events resulting in catastrophe were determined and scored as either an event resulting in complete depolymerization to the GMPCPP seed or a rescue event followed by new microtubule growth before reaching the GMPCPP seed. The fraction of catastrophe events resulting in rescue are plotted as a stacked bar plot for N > 170 microtubules across two independent experiments.
To quantify overall microtubule growth over the course of imaging, still images were acquired in 488-nm, 561-nm, and 640-nm channels at 0 and 40 minutes. The total length of microtubules in each field of view was measured using Fiji/ImageJ (National Institutes of Health) and the measurements were summed across 4 fields of view for each time point from two independent experiments.
Microtubule repair assay
Microtubule seeds were attached to the surface of a flow chamber by sequential incubation with: (i) 1 mg/ml BSA-biotin for 3 min, (ii) blocking buffer, (iii) 0.5 mg/ml NeutrAvidin for 3 min, (iv) blocking buffer, (v) GMPCPP-stabilized microtubule seeds (18% x-rhodamine tubulin) for 3 min, and (vi) blocking buffer. Microtubules were polymerized from the seeds by incubating 26 μM tubulin [gift of R. Ohi (University of Michigan) with 12.5% HiLyte647-tubuiln (Cytoskeleton)] and 1 mM GTP in imaging buffer [BRB80 buffer supplemented with 0.1% methylcellulose, 1 mg/ml casein, 3 mM MgCl2, 6 mM DTT and oxygen scavenger mix (16 mM glucose, 0.7 mg/ml catalase and 0.3 mg/ml glucose oxidase)] for 15 min. at 37°C. Microtubules were capped by incubating with 13 μM unlabeled tubulin and 1 mM GMPCPP in imaging buffer for 5 min at 37°C. Wash buffer was flowed in to depolymerize the dynamic tubulin structures grown on the stabilizing GMPCPP cap. Subsequently, a mix containing10 μM HiLyte488-tubulin, 1 mM GTP, 5 mM ATP, and purified motors in imaging buffer was flowed in. To achieve equivalent densities of KIF5C(1-560)-WT and KIF5C(1-560)-Δ6 on the microtubules in this assay, we calculated their relative affinities for GDP microtubules in the microtubule dynamics assay (# motors/mm GDP-microtubule/time frame). As KIF5C(1-560)-Δ6 displayed a 3-fold higher density than KIF5C(1-560)-WT on GDP microtubules, a higher concentration of KIF5C(1-560)-WT-Halo-Flag (18 nM) than KIF5C(1-560)-Δ6-Halo-Flag (6 nM) was incubated with the microtubules in this assay. After incubation of motors with microtubules in the presence of soluble tubulin for 7 min at 37°C, the flow chamber was washed with blocking buffer to remove unincorporated HiLyte488-tubulin and unbound motors and then 13 μM unlabeled tubulin was added to prevent microtubule depolymerization. The chamber was sealed with molten paraffin wax, and images were collected on a Nikon Ti-E/B TIRF microscope equipped with a 100X 1.49 N.A. oil immersion TIRF objective (Nikon), three 20 mW diode lasers (488 nm, 561 nm and 640 nm), and EMCCD detector (iXon X3DU897, Andor). Microtubule repair sites were defined as sites of Hi-Lyte488-tubulin incorporation site flanked by HiLyte647-tubulin containing microtubule lattice on both sides. The length of incorporation sites was quantified using Fiji/imageJ2.
Microtubule destruction assay
Biotinylated GMPCPP-seeds and biotinylated KIF5C(1-560)-Avitag motors were attached to the surface of a flow chamber by sequential incubation (5 min each) with (i) 1 mg/ml BSA-biotin, (ii) wash buffer (1 mg/ml BSA in BRB80), (iii) 0.5 mg/ml NeutrAvidin, (iv) wash buffer, (v) GMPCPP-seeds (6% HiLyte647-tubulin, 6% biotin-tubulin) in BRB80, (vi) wash buffer, (vii) cell lysates containing 75 nM biotinylated motors in BRB80 with 3.5 mM ADP, 35 mM glucose, 0.1 U Hexokinase to prevent motors from binding to soluble tubulin or GMPCPP-seeds, and (viii) wash buffer with 3.5 mM ADP. Next the chamber was placed at 37°C in a temperature-controlled and humidified stage-top incubator (Tokai Hit). GDP-lattice microtubules were assembled in the chamber by polymerization of GTP-tubulin for 30 min [7 uM bovine brain tubulin (gift of R. Ohi, University of Michigan) with 6% HiLyte488-tubulin (Cytoskeleton)] in BRB80 with GTP, methylcellulose, BSA, DTT, and ADP (to prevent motor-tubulin binding). Next, GMPCPP-caps were assembled by polymerization for 1.5 min [7 uM bovine brain tubulin (gift of R. Ohi, University of Michigan) with 11% HiLyte647-tubuiln (Cytoskeleton)] in BRB80 with 2.6 mM GMPCPP, methylcellulose, BSA, and ADP (to prevent motor-tubulin binding). The flow chamber was then washed twice with wash buffer [P12 buffer (12 mM Pipes/KOH pH 6.8, 1 mM MgCl2, 1 mM EGTA) containing ADP] and images of microtubules in 8 fields of view were obtained (0 min time point). Since microtubule destruction does not occur in the presence of ADP, the microtubules are considered to be full length at this 0 min time point. To initiate microtubule gliding by the surface-attached kinesin motors, Motility Mix was added to the chamber [25 μl P12 buffer, 15 ml of 15 mg/mL BSA in P12, 2 μl of 10 mg/mL casein/P12, 0.5 μl of the following: 1 M DTT, 100 mM MgCl2, 1 M glucose, 8 mg/mL catalase, 20 mg/mL glucose oxidase, and supplemented with 2 mM ATP and 7 μM unlabeled tubulin (gift of R. Ohi, University of Michigan)]. Images of microtubules in 8 fields of view were obtained every 5 min (5,10,15,20 min time points) using a Ti-E/B inverted TIRF microscope (Nikon) equipped with a 100x 1.49 N.A. oil immersion TIRF objective (Nikon), three 20 mW diode lasers (488 nm, 561 nm and 640 nm), and EMCCD detector (iXon X3DU897, Andor). The total length of microtubules in each field of view was measured using Fiji/ImageJ (National Institutes of Health) and the measurements were summed across 8 fields of view for each time point. Data are from three independent experiments.
Negative staining and electron microscopy
Taxol-stabilized GDP microtubules, GMPCPP microtubules, and GDP-microtubules were prepared as described.56 For taxol-stabilized GDP microtubules, 10 μl of glycerol-free tubulin (Cytoskeleton) at 100 μM was incubated for 1 hr at 37°C in BRB80 supplemented with 1mM GTP and 10% DMSO. 20 μl of BRB80 with taxol (warmed up to 37°C) was added to bring the final taxol concentration in the mixture to 20μM. The microtubules were incubated at room temperature overnight and then pelleted through 100μl cushion (BRB80, 60% glycerol, and 20 μM Taxol) at 50,000 × g for 45 min at 30°C in a TLA100 rotor. The microtubule pallet was washed twice with BRB80, 20μM taxol, and resuspended in 50μl BRB80, 20 μM Taxol. For GMPCPP microtubules, 20 μl of 100 μM glycerol-free tubulin in BRB80 supplemented with 1mM GMPCPP and 1mM DTT was incubated on ice for 5-10 min and then transferred to 37°C for 1 hr. The microtubules were pelleted by centrifugation at 50,000 × g for 10 min at 30°C, resuspended in 50 μl ice-cold BRB80 + 1mM DTT and incubated on ice for 30 min with pipetting up and down every five min to depolymerize microtubules. GMPCPP at final concentration of 1 mM was added to the depolymerized tubilin mixture and incubated on ice for another 10 min, then incubated overnight at 37 °C. GMPCPP-microtubules were pelleted through 100μl cushion buffer (BRB80,60% glycerol) at 50,000 × g for 45 min at 30°C, washed twice with BRB80, and resuspended in 50 μl BRB80. For GDP-microtubules, 20 μl of 100 μM glycerol-free tubulin was incubated at 37 °C for 1 hr in BRB80 supplemented with 1mM GTP and 10% DMSO. The microtubules were pelleted by centrifugation through a cushion (BRB80,60% glycerol, and 1mM GTP) at 50,000 × g for 45 min at 30°C, washed twice in BRB80, 1mM GTP and 10% DMSO, and resuspended in the same buffer.
Copper grids with Formvar carbon film (FCF400-CU, EMS) were cleaned using PELCO easiGlow at 5 mAmp for 30 seconds. 3μl of each microtubule at 1μM (based on original tubulin dimer) in its resuspension buffer was incubated on the glow-discharged grid for 1 minute. Then 5 μl of 25nM KIF5C(1-560)-WT or KIF5C(1-560)-Δ6 in BRB80 buffer supplemented with 4 mM ATP was added to the microtubules on the grid and incubated at 25 °C, 100% humidity for another 5 min. Temperature and humidity were set using a water bath. The extra solution was blotted from the grids using calcium-free Whatman filter papers and followed by negative staining with 0.75% uranyl formate. Imagining was performed at room temperature with Morgagni transmission electron microscope (FEI) equipped with a CCD camera and an acceleration voltage of 100 kV (22,000x magnification, 2.1 Å/pixel) and a T12 equipped with a (30,000x magnification, 1.2 Å/pixel). For each condition, images were collected blind and the number of breaks and cracks in the microtubules was quantified using Appion software and the total length of microtubules was quantified using Fiji/ImageJ. The data are reported as the number of breaks and cracks per μM of microtubule.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of each assay is described with the methodology of the assay in Method details section. Statistical analyses were performed and Graphs were generated using Prism software (GraphPad 8.0.0). The mean and standard error are described in the main text and/or the figures. The number of values examined, the experimental replicates, and the statistical tests applied are described in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-beta-tubulin, clone E7 | DSHB | catalog #E7; RRID: AB_528499 |
| Anti-Alpha-Tubulin-K40ac, Clone 6-11B-1 | Sigma-Aldrich | catalog #T7451; RRID: AB_609894 |
| anti-giantin | BioLegend | catalog #924302; RRID: AB_291560 |
| anti-FLAG tag agarose beads, clone M2 | Sigma-Aldrich | cat# A2220 |
| anti-FLAG tag | ThermoFisher | cat# MA1-91878-BTIN; RRID: AB_2537620 |
| Biological samples | ||
| porcine brain tubulin | Cytoskeleton | cat# T240 |
| bovine brain tubulin | R. Ohi lab | lab purified |
| biotinylated tubulin | Cytoskeleton | cat# T333P |
| fluorescent tubulins (HiLyte488, X-Rhodamine, HiLyte647) | Cytoskeleton | cat# TL488M, TL620M, TL670M |
| 3xFLAG peptide | Sigma-Aldrich | cat# F4799 |
| Chemicals, peptides, and recombinant proteins | ||
| Taxol | Cytoskeleton | cat# TXD01 |
| Neutravidin | ThermoFisher | cat# 3100 |
| GMP-CPP | Jena Sciences | cat# JBS-NU-405S |
| biotin-BSA | Sigma-Aldrich | cat# A8549 |
| Janelia Fluor 552 (JF552) | Janelia Materials | cat# JF552 |
| trichostatin A | Sigma-Aldrich | cat# T1952 |
| Experimental models: Cell lines | ||
| COS-7 | ATCC | RRID: CVCL_0224 |
| hTERT-RPE1 | ATCC | RRID: CVCL_4388 |
| MRC-5 | ATCC | RRID: CVCL_0440 |
| NIH-3T3 | ATCC | RRID: CVCL_0594 |
| U-2OS | ATCC | RRID: CVCL_0042 |
| Recombinant DNA | ||
| plasmid pN1-RnKIF5C(1-560)-3xmCit | Cai et al.62 | lab plasmid |
| plasmid pN1-RnKIF5C(1-560)-Flag | Budaitis et al.20 | lab plasmid |
| plasmid pN1-RnKIF5C(1-560)-Halo-Flag | this study | lab plasmid |
| plasmid pN1-RnKIF5C(1-560)-mNeonGreen | Norris et al.63 | lab plasmid |
| plasmid pN1-RnKIF5C(1-560,Δ6)-3xmCit | this study | lab plasmid |
| plasmid pN1-RnKIF5C(1–560,Δ6)-Flag | this study | lab plasmid |
| plasmid pN1-RnKIF1A(1-560,Δ6)-Halo-Flag | this study | lab plasmid |
| plasmid pN1-RnKIF5C(1-560,Δ6)-mNeonGreen | this study | lab plasmid |
| plasmid pN1-RnKIF1A(1-393)-3xmCit | Hammond et al.64 | lab plasmid |
| plasmid pN1-RnKIF1A(1-393)-Flag | Budaitis et al.19 | lab plasmid |
| plasmid pN1-RnKIF1A(1-393)-Halo-Flag | Budaitis et al.19 | lab plasmid |
| plasmid pN1-RnKIF1A(1-393)-mNeonGreen | Budaitis et al.19 | lab plasmid |
| plasmid pC1-mCitrine-MmalphaTAT | Soppina et al.65 | lab plasmid |
| plasmid pN1-RnKIF5C(1-560)-mNG-FRB | Schimert et al.66 | lab plasmid |
| plasmid pN1-GMAP210-mRFP-FKBP | Engelke et al.67 | lab plasmid |
| adenovirus pShuttle-EGFP-tubulin | Kumar et al.68 | Addgene#24327 |
| Software and algorithms | ||
| Fiji/ImageJ | Schneider et al.69 | https://fiji.sc/ |
| Prism 8.0 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| carboxy polystyrene beads | Spherotech | cat# CP-10-10 |
| copper grids | EMS | cat# FCF400-CU |
Highlights.
Kinesin-1 motors create damage sites in the microtubule lattice while stepping
Mutation of kinesin-1’s coverstrand enhances its ability to damage microtubules
Motor-induced damage causes microtubules to break in response to mechanical stress
Cells must repair motor-induced lattice damage to prevent microtubule breakage
ACKNOWLEDGMENTS
We are grateful to David Sept, Ryoma Ohi, Morgan DeSantis, and members of their laboratories for helpful discussions and reagents. This work was supported by grants from the National Institutes of Health to K.J.V. (R01GM070862 and R35GM131744) and a grant from National Science Foundation to M.J.L. (1330792). B.G.B. was supported by the Cellular and Molecular Biology Training grant T32-GM007315 from the National Institutes of Health, a Graduate Research Fellowship (DGE 1256260) from the National Science Foundation, an EDGE Fellowship from the Endowment of Basic Sciences at the University of Michigan Medical School, and a Rackham Predoctoral Fellowship from the Horace H. Rackham School of Graduate Studies at the University of Michigan. D.N.R. was supported by the National Science Foundation Graduate Research Fellowship Program under grant no. 1445197.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cub.2022.04.020.
DECLARATION OF INTERESTS
K.J.V. is a member of the Current Biology Advisory Board.
REFERENCES
- 1.Hirokawa N, and Tanaka Y (2015). Kinesin superfamily proteins (KIFs): various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res 334, 16–25. [DOI] [PubMed] [Google Scholar]
- 2.Hunter B, and Allingham JS (2020). These motors were made for walking. Protein Sci. 29, 1707–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang W, and Karplus M (2019). Structural basis for power stroke vs. Brownian ratchet mechanisms of motor proteins. Proc. Natl. Acad. Sci. USA 116, 19777–19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Zhang H, Geng Y, and Ji Q (2020). How kinesin-1 utilize the energy of nucleotide: the conformational changes and mechanochemical coupling in the unidirectional motion of kinesin-1. Int. J. Mol. Sci 21, 6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dumont EL, Do C, and Hess H (2015). Molecular wear of microtubules propelled by surface-adhered kinesins. Nat. Nanotechnol 10, 166–169. [DOI] [PubMed] [Google Scholar]
- 6.Kabir AMR, Sada K, and Kakugo A (2020). Breaking of buckled microtubules is mediated by kinesins. Biochem. Biophys. Res. Commun 524, 249–254. [DOI] [PubMed] [Google Scholar]
- 7.Reuther C, Diego AL, and Diez S (2016). Kinesin-1 motors can increase the lifetime of Taxol-stabilized microtubules. Nat. Nanotechnol 11, 914–915. [DOI] [PubMed] [Google Scholar]
- 8.VanDelinder V, Adams PG, and Bachand GD (2016). Mechanical splitting of microtubules into protofilament bundles by surface-bound kinesin-1. Sci. Rep 6, 39408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krebs A, Goldie KN, and Hoenger A (2004). Complex formation with kinesin motor domains affects the structure of microtubules. J. Mol. Biol 335, 139–153. [DOI] [PubMed] [Google Scholar]
- 10.Morikawa M, Yajima H, Nitta R, Inoue S, Ogura T, Sato C, and Hirokawa N (2015). X-ray and cryo-EM structures reveal mutual conformational changes of kinesin and GTP-state microtubules upon binding. EMBO J. 34, 1270–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peet DR, Burroughs NJ, and Cross RA (2018). Kinesin expands and stabilizes the GDP-microtubule lattice. Nat. Nanotechnol 13, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shima T, Morikawa M, Kaneshiro J, Kambara T, Kamimura S, Yagi T, Iwamoto H, Uemura S, Shigematsu H, Shirouzu M, et al. (2018). Kinesin-binding-triggered conformation switching of microtubules contributes to polarized transport. J. Cell Biol 217, 4164–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muto E, Sakai H, and Kaseda K (2005). Long-range cooperative binding of kinesin to a microtubule in the presence of ATP. J. Cell Biol 168, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Triclin S, Inoue D, Gaillard J, Htet ZM, DeSantis ME, Portran D, Derivery E, Aumeier C, Schaedel L, John K, et al. (2021). Self-repair protects microtubules from destruction by molecular motors. Nat. Mater 20, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friel CT, and Welburn JP (2018). Parts list for a microtubule depolymerising kinesin. Biochem. Soc. Trans 46, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Théry M, and Blanchoin L (2021). Microtubule self-repair. Curr. Opin. Cell Biol 68, 144–154. [DOI] [PubMed] [Google Scholar]
- 17.Hwang W, Lang MJ, and Karplus M (2008). Force generation in kinesin hinges on cover-neck bundle formation. Structure 16, 62–71. [DOI] [PubMed] [Google Scholar]
- 18.Khalil AS, Appleyard DC, Labno AK, Georges A, Karplus M, Belcher AM, Hwang W, and Lang MJ (2008). Kinesin’s cover-neck bundle folds forward to generate force. Proc. Natl. Acad. Sci. USA 105, 19247–19252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budaitis BG, Jariwala S, Rao L, Yue Y, Sept D, Verhey KJ, and Gennerich A (2021). Pathogenic mutations in the kinesin-3 motor KIF1A diminish force generation and movement through allosteric mechanisms. J. Cell Biol 220. e202004227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budaitis BG, Jariwala S, Reinemann DN, Schimert KI, Scarabelli G, Grant BJ, Sept D, Lang MJ, and Verhey KJ (2019). Neck linker docking is critical for kinesin-1 force generation in cells but at a cost to motor speed and processivity. eLife 8, e44146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesse WR, Steiner M, Wohlever ML, Kamm RD, Hwang W, and Lang MJ (2013). Modular aspects of kinesin force generation machinery. Biophys. J 104, 1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang W, Lang MJ, and Karplus M (2017). Kinesin motility is driven by subdomain dynamics. eLife 6, e28948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soppina V, Norris SR, Dizaji AS, Kortus M, Veatch S, Peckham M, and Verhey KJ (2014). Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc. Natl. Acad. Sci. USA 111, 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaniewski TM, Gicking AM, Fricks J, and Hancock WO (2020). A kinetic dissection of the fast and superprocessive kinesin-3 KIF1A reveals a predominant one-head-bound state during its chemomechanical cycle. J. Biol. Chem 295, 17889–17903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portran D, Schaedel L, Xu Z, Théry M, and Nachury MV (2017). Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol 19, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Théry M, and Nachury MV (2017). Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 356, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, and Gaertig J (2010). MEC-17 is an alpha-tubulin acetyltransferase. Nature 467, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shida T, Cueva JG, Xu Z, Goodman MB, and Nachury MV (2010). The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 107, 21517–21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, and Yao TP (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. [DOI] [PubMed] [Google Scholar]
- 30.Andreu-Carbó M, Fernandes S, Velluz MC, Kruse K, and Aumeier C (2022). Motor usage imprints microtubule stability along the shaft. Dev. Cell 57, 5–18.e8. [DOI] [PubMed] [Google Scholar]
- 31.Atherton J, Farabella I, Yu IM, Rosenfeld SS, Houdusse A, Topf M, and Moores CA (2014). Conserved mechanisms of microtubule-stimulated ADP release, ATP binding, and force generation in transport kinesins. eLife 3, e03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atherton J, Yu I-M, Cook A, Muretta JM, Joseph A, Major J, Sourigues Y, Clause J, Topf M, Rosenfeld SS, et al. (2017). The divergent mitotic kinesin MKLP2 exhibits atypical structure and mechano-chemistry. eLife 6, e27793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulet A, Behnke-Parks WM, Sindelar CV, Major J, Rosenfeld SS, and Moores CA (2012). The structural basis of force generation by the mitotic motor kinesin-5. J. Biol. Chem 287, 44654–44666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goulet A, Major J, Jun Y, Gross SP, Rosenfeld SS, and Moores CA (2014). Comprehensive structural model of the mechanochemical cycle of a mitotic motor highlights molecular adaptations in the kinesin family. Proc. Natl. Acad. Sci. USA 111, 1837–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J, Zhang Y, Wang S, Huo L, Lou J, and Feng W (2018). Structural delineation of the neck linker of Kinesin-3 for processive movement. J. Mol. Biol 430, 2030–2041. [DOI] [PubMed] [Google Scholar]
- 36.von Loeffelholz O, Peña A, Drummond DR, Cross R, and Moores CA (2019). Cryo-EM structure (4.5-A) of yeast kinesin-5-microtubule complex reveals a distinct binding footprint and mechanism of drug resistance. J. Mol. Biol 431, 864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varela PF, Chenon M, Velours C, Verhey KJ, Ménétrey J, and Gigant B (2021). Structural snapshots of the kinesin-2 OSM-3 along its nucleotide cycle: implications for the ATP hydrolysis mechanism. FEBS Open Bio 11, 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mickolajczyk KJ, and Hancock WO (2017). Kinesin processivity is determined by a kinetic race from a vulnerable one-head-bound state. Biophys. J 112,2615–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bicek AD, Tüzel E, Demtchouk A, Uppalapati M, Hancock WO, Kroll DM, and Odde DJ (2009). Anterograde microtubule transport drives microtubule bending in LLC-PK1 epithelial cells. Mol. Biol. Cell 20, 2943–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kent IA, Rane PS, Dickinson RB, Ladd AJ, and Lele TP (2016). Transient pinning and pulling: a mechanism for bending microtubules. PLoS One 11, e0151322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brangwynne CP, MacKintosh FC, Kumar S, Geisse NA, Talbot J, Mahadevan L, Parker KK, Ingber DE, and Weitz DA (2006). Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol 173, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odde DJ, Ma L, Briggs AH, DeMarco A, and Kirschner MW (1999). Microtubule bending and breaking in living fibroblast cells. J. Cell Sci 112, 3283–3288. [DOI] [PubMed] [Google Scholar]
- 43.Pallavicini C, Monastra A, Bardeci NG, Wetzler D, Levi V, and Bruno L (2017). Characterization of microtubule buckling in living cells. Eur. Biophys. J 46, 581–594. [DOI] [PubMed] [Google Scholar]
- 44.Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, and Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang N, Naruse K, Stamenović D, Fredberg JJ, Mijailovich SM, Tolić-Nørrelykke IM, Polte T, Mannix R, and Ingber DE (2001). Mechanical behavior in living cells consistent with the tensegrity model. Proc. Natl. Acad. Sci. USA 98, 7765–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jolly AL, Kim H, Srinivasan D, Lakonishok M, Larson AG, and Gelfand VI (2010). Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc. Natl. Acad. Sci. USA 107, 12151–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oladipo A, Cowan A, and Rodionov V (2007). Microtubule motor Ncd induces sliding of microtubules in vivo. Mol. Biol. Cell 18, 3601–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Randall TS, Yip YY, Wallock-Richards DJ, Pfisterer K, Sanger A, Ficek W, Steiner RA, Beavil AJ, Parsons M, and Dodding MP (2017). A small-molecule activator of kinesin-1 drives remodeling of the microtubule network. Proc. Natl. Acad. Sci. USA 114, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hahn I, Voelzmann A, Liew YT, Costa-Gomes B, and Prokop A (2019). The model of local axon homeostasis - explaining the role and regulation of microtubule bundles in axon maintenance and pathology. Neural Dev. 14, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Portran D, Zoccoler M, Gaillard J, Stoppin-Mellet V, Neumann E, Arnal I, Martiel JL, and Vantard M (2013). MAP65/Ase1 promote microtubule flexibility. Mol. Biol. Cell 24, 1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habicht J, Mooneyham A, Hoshino A, Shetty M, Zhang X, Emmings E, Yang Q, Coombes C, Gardner MK, and Bazzaro M (2021). UNC-45A breaks the microtubule lattice independently of its effects on non-muscle myosin II. J. Cell Sci 134, jcs248815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atherton J, Stouffer M, Francis F, and Moores CA (2018). Microtubule architecture in vitro and in cells revealed by cryo-electron tomography. Acta Crystallogr. D Struct. Biol 74, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atherton J, Stouffer M, Francis F, and Moores CA (2022). Visualising the cytoskeletal machinery in neuronal growth cones using cryo-electron tomography. J. Cell. Sci 135, jcs259234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaedel L, Triclin S, Chrétien D, Abrieu A, Aumeier C, Gaillard J, Blanchoin L,Théry M, and John K (2019). Lattice defects induce microtubule self-renewal. Nat. Phys 15, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldblum RR, McClellan M, White K, Gonzalez SJ, Thompson BR, Vang HX, Cohen H, Higgins L, Markowski TW, Yang TY, et al. (2021). Oxidative stress pathogenically remodels the cardiac myocyte cytoskeleton via structural alterations to the microtubule lattice. Dev. Cell 56, 2252–2266.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vemu A, Szczesna E, Zehr EA, Spector JO, Grigorieff N, Deaconescu AM, and Roll-Mecak A (2018). Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 361, eaau1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mustyatsa VV, Kostarev AV, Tvorogova AV, Ataullakhanov FI, Gudimchuk NB, and Vorobjev IA (2019). Fine structure and dynamics of EB3 binding zones on microtubules in fibroblast cells. Mol. Biol. Cell 30, 2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henrie H, Bakhos-Douaihy D, Cantaloube I, Pilon A, Talantikite M, Stoppin-Mellet V, Baillet A, Poüs C, and Benoit B (2020). Stress-induced phosphorylation of CLIP-170 by JNK promotes microtubule rescue. J. Cell Biol 219. e201909093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Forges H, Pilon A, Cantaloube I, Pallandre A, Haghiri-Gosnet AM, Perez F, and Poüs C (2016). Localized mechanical stress promotes microtubule rescue. Curr. Biol 26, 3399–3406. [DOI] [PubMed] [Google Scholar]
- 60.Lawrence EJ, Arpag G, Norris SR, and Zanic M (2018). Human CLASP2 specifically regulates microtubule catastrophe and rescue. Mol. Biol. Cell 29, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aher A, Rai D, Schaedel L, Gaillard J, John K, Liu Q, Altelaar M, Blanchoin L, Thery M, and Akhmanova A (2020). CLASP mediates microtubule repair by restricting lattice damage and regulating tubulin incorporation. Curr. Biol 30, 2175–2183.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai D, McEwen DP, Martens JR, Meyhofer E, and Verhey KJ (2009). Single molecule imaging reveals differences in microtubule track selection between kinesin motors. PLoS Biol. 7, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Norris SR, Núñez MF, and Verhey KJ (2015). Influence of fluorescent tag on the motility properties of kinesin-1 in single-molecule assays. Biophys. J 108, 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hammond JW, Cai D, Blasius TL, Li Z, Jiang Y, Jih GT, Meyhofer E, and Verhey KJ (2009). Mammalian kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol. 7, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soppina V, Herbstman JF, Skiniotis G, and Verhey KJ (2012). Luminal localization of alpha-tubulin K40 acetylation by cryo-EM analysis of fab-labeled microtubules. PLoS One 7, e48204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schimert KI, Budaitis BG, Reinemann DN, Lang MJ, and Verhey KJ (2019). Intracellular cargo transport by single-headed kinesin motors. Proc. Natl. Acad. Sci. USA 116, 6152–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Engelke MF, Winding M, Yue Y, Shastry S, Teloni F, Reddy S, Blasius TL, Soppina P, Hancock WO, Gelfand VI, and Verhey KJ (2016). Engineered kinesin motor proteins amenable to small-molecule inhibition. Nat. Commun 7, 11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar P, Lyle KS, Gierke S, Matov A, Danuser G, and Wittmann T (2009). GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol 184, 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reinemann DN, Norris SR, Ohi R, and Lang MJ (2018). Processive kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Curr. Biol 28, 2356–2362.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reinemann DN, Sturgill EG, Das DK, Degen MS, Vörös Z, Hwang W, Ohi R, and Lang MJ (2017). Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Curr. Biol 27, 2810–2820.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.