

Conspectus
Retrosynthetic analysis emerged in the 1960s as a teaching tool with profound implications. Its educational value can be appreciated by a glance at total synthesis manuscripts over 50 years later, most of which contain a retrosynthesis on page one. Its vision extended to computer language—a pioneering idea in the 20th century that continues to expand the frontiers today. The same principles that guide a student to evaluate, expand and refine a series of bond dissections can be programmed, so that computer assistance can perform the same tasks but at faster speeds.
The slow step in the synthesis of complex structures, however, is seldom route design. Compression of molecular information into close proximity (Cm/Å3) requires exploration and empiricism, a close connection between theory and experiment. Here, retrosynthetic analysis guides the choice of experiment, so that the most simplifying—but often least assured—disconnection is prioritized: a high-risk, high reward strategy. Acceleration of total synthesis by retrosynthetic software may be put to better use, counterintuitively, by target design.
Compared to the 1960s, retrosynthetic analysis in the 21st century finds itself among computers of unimaginable power and a biology that is increasingly molecular. Put together, the logic of retrosynthesis, the insight of structural biology and the predictions of computation have inspired us to imagine an integration of the three. The synthetic target is treated as dynamic—a constellation of related structures—in order to find the nearest congener with the closest affinity but the shortest synthetic route. Such an approach merges synthetic design with structural design toward the goal of improved access for improved function.
In this Account, we detail the evolution of our program from its inception in traditional natural product (NP) total synthesis to its current expression through the lens of chemical informatics: a view of NPs as aggregates of molecular parameters that define single points in a chemical space. Early work on synthesis and biological annotation of apparent metal pool binders and non-selective covalent electrophiles (asmarine alkaloids, isocyanoterpenes, Nuphar dimers) gave way to NPs with well-defined protein targets. The plant metabolite salvinorin A (SalA) potently and selectively agonizes the kappa-opioid receptor (KOR), rapidly penetrates the brain and represents an important lead for next-generation analgesics and antipruritics. To synthesize and diversify this lead, we adopted what we now call a dynamic approach. Deletion of a central methyl group stabilized the SalA scaffold, opened quick synthetic access and retained high potency and selectivity. The generality of this idea was then tested against another neuroactive class. As an alternative hypothesis to TrkB channels, we proposed that the so-called “neurotrophic” Illicium terpenes may bind to gamma-aminobutyric acid (GABA)-gated ion channels to cause weak, chronic excitation. Syntheses of (−)-jiadifenolide, 3,6-dideoxy-10-hydroxypseudoanisatin, (−)-11-O-debenzoyltashironin, (−)-bilobalide and (−)-picrotoxinin (PXN) allowed this hypothesis to be probed more broadly. Feedback from protein structure and synthetic reconnaissance led to a dynamic retrosynthesis of PXN and the identification of 5MePXN, a moderate GABAAR antagonist with greater aqueous stability available in 8 steps from dimethylcarvone. We expect this dynamic approach to synthetic target analysis to become more feasible in the coming years and hope the next generation of scientists finds this approach helpful to address problems at the frontier of chemistry and biology.
Graphical Abstract
Introduction
Molecular features like high stereochemical content, complex bond networks and diverse heteroatom content remain challenging to synthesize from simple feedstocks. Yet biosynthetic pathways produce these motifs with ease. The evolutionary selection for molecular complexity4 may relate to by higher specificity of protein binding5 and greater aqueous solubility.6,7 Complexity may lower clinical trial attrition, potentially due to reduced off-target effects5,8 like Cyp450 inhibition. Parametrizations using substructural features9 have supported a correlation between evolutionary optimization and therapeutic design: “drug” chemical space (MDDR and World Drug Index) is optimized away from commercial building blocks (aka “synthetic”9 space) and towards natural products (NP space) (Figure 1a).
Figure 1.
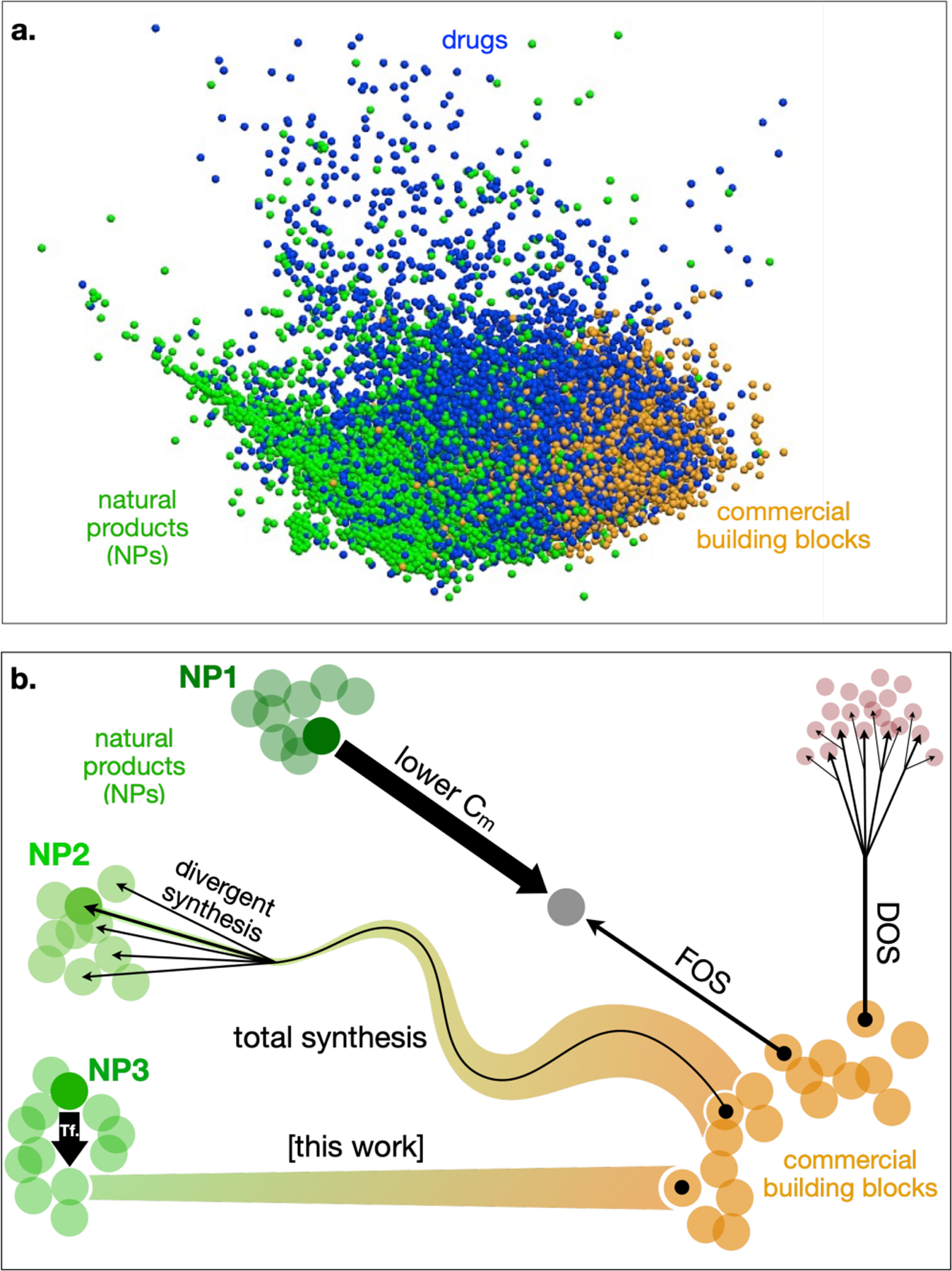
Abstract visualizations of chemical space. a. NPs (•), drugs (•) and commercial building blocks (•) differentiated by ChemGPS using a unique parameterization of chemical space. Adapted with permission from ref. 9. Copyright 2007 Swiss Chemical Society. b. Different methods to navigate from commercial building blocks to complex molecule space using chemical synthesis.
Different strategies have emerged to navigate from commercial building blocks to complex chemical space (Figure 1b). NP total synthesis identifies effective routes to NPs using transforms matched to existing chemical reactions or imagines theoretical methods that are then invented.10 Divergent functionalization from a simpler intermediate allows population of multiple nodes proximal to the NP.11 Function-oriented synthesis (FOS) identifies the key pharmacophoric groups or molecular interactions necessary for biomolecular target binding, and reduces the NP complexity for efficient synthesis.12 Diversity-oriented synthesis (DOS) recognizes the value of molecular complexity, but emphasizes the greater potential for complex non-NP space to identify novel human therapeutic targets for which there is no natural selection pressure; synthesis of large, diverse libraries increases the likelihood of novel target and lead identification.13 Like DOS and FOS, our alternative model2 of navigating to NP space recognizes that a given NP structure is unlikely to be ideal for human use. Rapid access to a close congener, however, may accelerate scaffold optimization and render the space useful. This approach merges traditional retrosynthetic analysis with modeled NP•receptor binding. Here, a small, judicious movement in chemical space from a target NP is made. This movement is chosen to maintain complexity, chemical space location and NP•receptor interaction, but unravel an efficient route to building blocks and improve upon NP properties. We refer to this strategy as dynamic retrosynthetic analysis, since the NP target is no longer treated as static. Below, we detail how this perspective evolved over time and provided us a valuable problem-solving tool for complex molecule synthesis.
Non-specific targets
The value of NP space to chemistry and biology was the founding principle of our lab about 10 years ago. We sought to develop general methods to access stereochemically-complex carbocycles and heterocycles that represented jumping-off points for biological investigation. These interests converged on two general problems (Figure 2). First, we identified many methodological gaps in the covalent “folding” of linear motifs and sought to expand the existing repertoire of reactions.14 Second, we were interested in pharmacophores that had been poorly characterized but held the potential to react covalently with a protein target.15 Like many total synthesis endeavors, these objectives led to new methods and biological discoveries, but also caused us to reconsider how total synthesis could best interface with biological research. The asmarines, Nuphar dimers and isocyanoterpenes contained pharmacophores demonstrated to react covalently in non-biological contexts.16 We aimed to access and assess the mechanism of action (MOA) of each class. Each chemotype embodied a synthetic challenge that had not been solved efficiently or at all. And each was considered a candidate for a covalent folding approach. This fundamental problem in chemistry—selective guidance of one cyclization reaction path of a linear chain among multiple options—has remained generally unsolved.17 The asmarine and isocyanoterpene scaffolds might form from audacious cation-olefin cyclizations, and the Nuphar dimers might from the polyhydroamination of an unsaturated amine. We demonstrated proof-of-principle for each strategy, effecting short syntheses of the dart frog nAChR antagonists18 and the strained, acid-sensitive terpene funebrene.19 However, the idea of advancing this basic methodology through more challenging total syntheses to biological interrogation and medicinal chemistry seemed unrealistic. The tail-to-head terpene cyclization literature alone was a graveyard of broken research threads. It has been gratifying to see other groups17 carry the torch far beyond what we imagined and advance this fascinating, fundamental chemistry.
Figure 2.
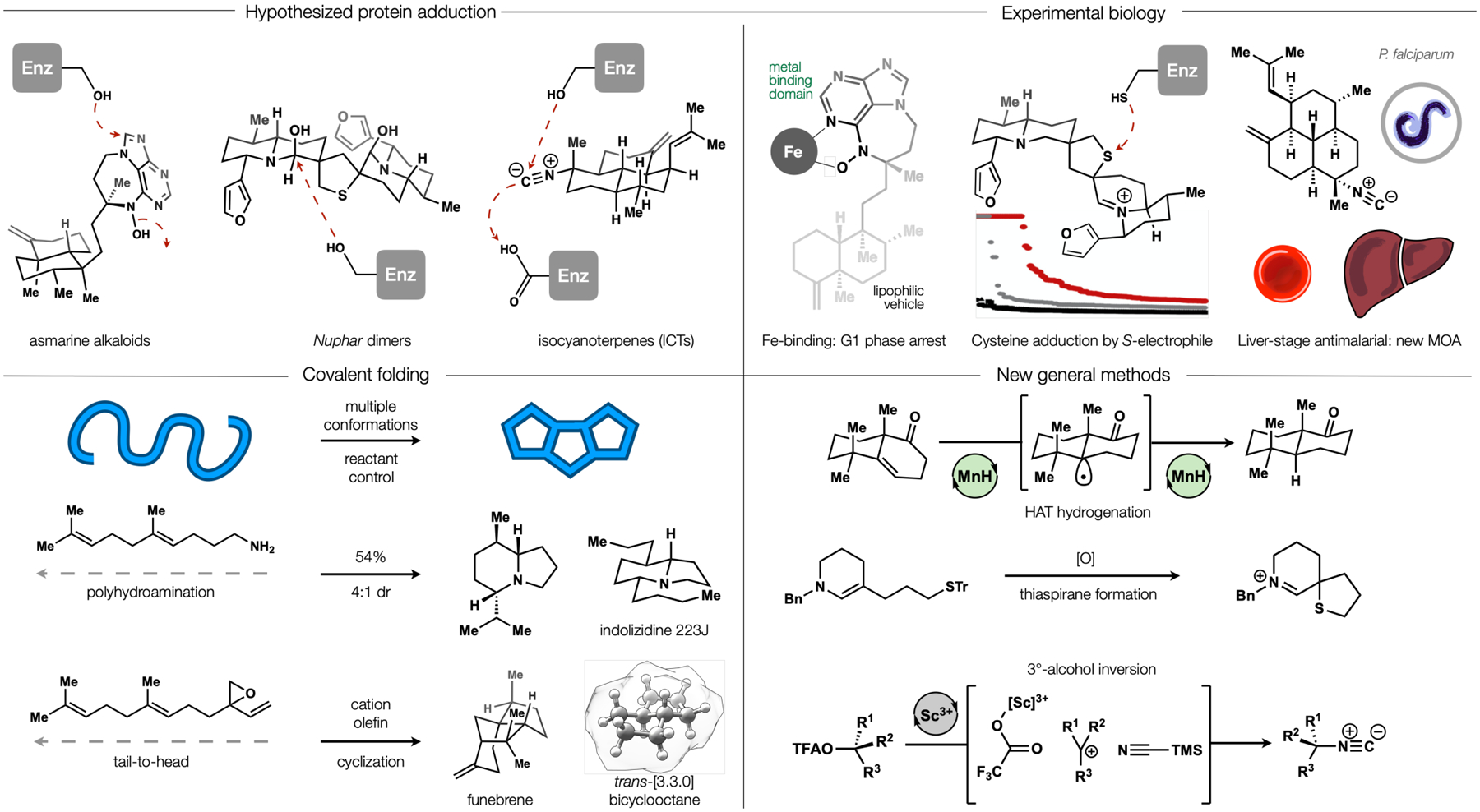
Synthetic methods enabled total syntheses, which illuminated biological MOA associated with the asmarines, Nuphar thiaspiranes and isocyanoterpenes.
Although covalent folding approaches proved impractical, alternative routes produced valuable methods and allowed biological annotation. For example, a stereochemical problem in the asmarine alkaloids provided the inspiration for HAT hydrogenation.20 We reasoned according to the Hammond postulate that a trans-ring product would be favored via an early transition state that resembled the lowest ground state of a pyramidalized radical intermediate (Figure 2, lower right). To our surprise and gratification, HAT hydrogenation has become a helpful reaction for many groups.21b The proposal of an outer-sphere metal hydride hydrogen atom transfer (MHAT) elementary step to form the initial radical led to a broader program in catalysis, reviewed elsewhere.21
New methods were also developed to access the Nuphar dimers, whose unique thiaspirane pharmacophore inspired a simple method for its synthesis via the oxidative cyclization of enamine thioethers.22 Similarly, isocyanoterpenes that exhibit potent antimalarial activity contain a common stereogenic tertiary isonitrile that we found could be best installed by a stereospecific SN1 reaction.23 This unusual reaction allows high-yielding inversion of aliphatic tertiary alcohols with minimal elimination or racemization,24 a singularity within the unstabilized carbocation literature.
Chemical syntheses of each of these chemotypes enabled annotation of their biological activity. The asmarines were reported to exhibit potent cytotoxic activity against HT29 cells but the MOA was unknown. The N-hydroxyaminopurine pharmacophore had been observed to accept nucleophiles on the aromatic ring with concomitant N–O bond cleavage,16a and we wondered if this reactivity underlay its phenotypic effects. Interrogation of its MOA,25 however, revealed a correlation between arrest at G1 phase, direct effects on iron-responsive proteins and 1:1 binding of ferric iron, which, taken together, were best explained by depletion of ribonucleotide reductase. We determined that the neoclerodane portion served as a lipophilic vehicle and the seven-membered ring enforced an iron-binding tautomer, which allowed us to “reverse engineer” the complex metabolite and create a simple equivalent.
The Nuphar dimers had been proposed by LaLonde to accept nucleophiles at a latent, sulfur-stabilized iminium carbon.16b We observed that, in contrast, the sulfur could act as an electrophilic site and initiate a retrodimerization cascade to release a potent warhead, resulting in extensive adduction of proteinaceous cysteines according to Cravatt’s isoTOP proteomics assay.22
Finally, we showed that the isocyanoterpenes are unlikely to exhibit toxicity against Plasmodia sp. by inhibition of heme detoxification alone.26 This mechanism had been proposed by analogy to quinine-based antimalarials and the affinity of isonitriles for transition metals, such as the iron in heme. In blood-stage malaria, the parasites catabolize hemoglobin, causing the release of heme, which must be neutralized by crystallization; disruption of this process causes parasite death. In collaboration with the Winzeler group, we found that the isocyanoterpenes are active in liver-stage parasites, which lack heme catabolism, suggesting an alternative mechanism of parasite death.26
Each of these studies illustrated the confluence of NP synthesis, reaction methodology and biological discovery, yet each also identified an apparently non-selective MOA. Since one benefit of NP-level complexity is specificity, this sloppiness proved unsatisfying. Instead we sought targets that either possessed known binding sites or were likely to. This choice to consider the binding of small molecule and macromolecular receptor changed the way our group thought about total synthesis.
Specific target 1: KOR
Conversations with Professor Laura Bohn at the Scripps graduate school retreat in 2013 and a follow-up at Scripps Florida in 2014 highlighted the significance of the S. divinorum metabolite salvinorin A (SalA).27 As the active constituent in the recreational hallucinogen salvia, SalA had been annotated as a kappa-opioid receptor (KOR) agonist in 2001.28 This came as a real surprise. No binding occurred at the 5-HT2A serotonin receptor, a typical target of hallucinogens. And high affinity binding to the KOR was observed in the absence of a basic nitrogen, which had been thought necessary for high affinity ligands of opioid receptors. Its absence was hypothesized to explain the high selectivity over mu-, delta- and nociception-opioid receptors. In contrast to these other family members, the KOR has emerged as a target for next generation pain therapeutics; SalA co-emerged as an important starting point for medicinal chemistry.27 Since this topic has been reviewed in several journals, we provide a personal perspective and conceptual overview below.
The therapeutic potential of SalA required a synthetic route that was competent for medicinal chemistry, which we hoped to advance for many years. “Medchem-ready” routes are precedented but uncommon in total synthesis; claims of practicality can lack substantiation by SAR discovery or optimization towards functional endpoints. A few retrosynthetic sketches of SalA indicated that elaboration of an intact A-ring (Figure 3), followed by iterative cyclization of B and C could form the bond network quickly. However, forays into this route alerted us to the difficulty of the C20 methyl group: it prevented simple aldol reaction at C10, jeopardized the stability of intermediates and steered the stereochemistry away from SalA. Looking into the basis of intermediate stereocontrol led us to a similar problem in SalA itself: isolation work noted the acid- and base-instability of SalA, leading Munro to characterize the degradation product as 8-epi-SalA.29 The driving force for this counterintuitive trans-anti-trans to trans-anti-cis epimerization was not explained in the literature. One reference calculated a reasonable 2.2 kcal/mol preference for 8-epi-SalA (Keq ~ 40),30 whereas another claimed no energy difference at all (Keq = 1), which implied a kinetic isomerization.31 One reference invoked a simple enolization/protonation,32 whereas another suggested a C8/C9 cleavage via retro-Michael and re-addition.33 We favored a thermodynamic enolization, and suggested the driving force involved 1,3-diaxial strain between the angular methyl (C20) and the benzylic C12 hydrogen. Deletion of C20 might remove our synthetic obstacles and also improve SalA itself by removing the structural liability of C8 epimerization, which reduces potency over 250-fold.34
Figure 3.
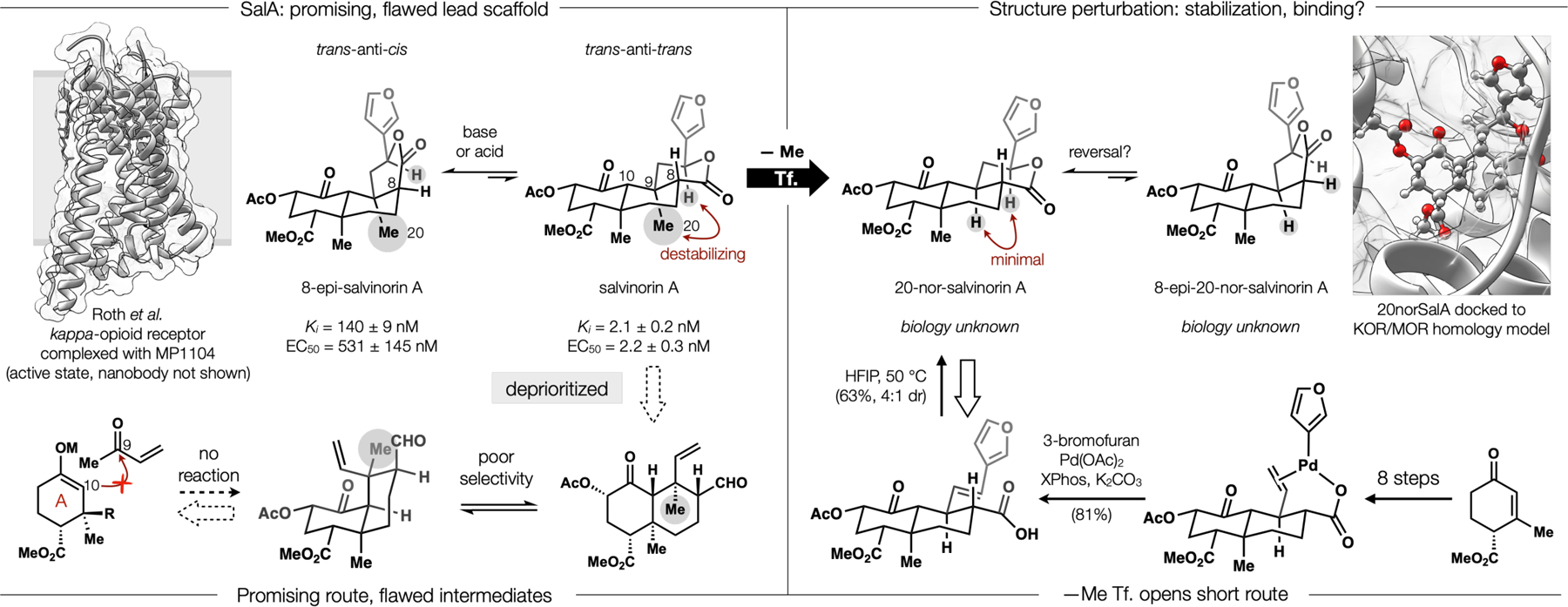
Retrosynthetic analysis of SalA identified a common feature (C20 methyl) that destabilized the target, destabilized intermediates and obstructed synthesis; its deletion improved the target and maintained binding.
Jeremy Roach, the graduate student working towards SalA, eagerly agreed to pursue this unorthodox strategy, despite its inherent risk: deletion of C20 might disrupt binding. Although the change seemed minor, unrelated medicinal campaigns have noted surprising gains in potency upon methyl addition, and correspondinglosses upon deletion (called the “magic methyl” effect).35 Besides a direct interaction with the binding site, C19/C20 diaxial interactions bend the entire scaffold out of plane, so that C20 deletion causes a flattening—effects that might affect binding. We reached out to Seva Katritch (USC) and former Scripps colleague Ray Stevens (USC), who calculated binding using a hybrid MOR/KOR homology model (this was early 2015 and the active conformation of KOR shown in Figure 336 had not yet been published) and suggested 20norSalA should bind like SalA. Further, we expected methyl deletion to facilitate synthesis: enolate addition to an aldehyde exceeds the rate of ketone addition, and the absence of Me-Me repulsion would favor a trans-ring fusion in synthetic intermediates. All of these assumptions proved correct.1
Upon reflection, the SalA project changed how we thought about the field of NP total synthesis, whose value is justified mainly in three ways: complexity, methodology, biology (Figure 4). First, NP complexity stimulates innovative problem solving. 20norSalA, however, is not an NP, but contains similar complexity and information density as SalA (466 vs. 479 mcbits, and 1.32 mcbits/Å3 vs. 1.29 mcbits/Å3, respectively). Yet, its synthesis is not analogous to any known route to any similar structure, whereas many syntheses of NPs are close iterations of a prior route. So, complexity-derived novelty is achieved in a non-NP target. Second, many total syntheses stimulate new methods to solve key problems. 20norSalA, while not a NP, required the invention of a carboxylate-directed Heck reaction that proved general37 to engage hindered alkenes at rates far exceeding standard Heck conditions. The final lactonization appeared to proceed via hydrogen-bond mediated internal protonation/ ion pair collapse. Other routes to the NP, SalA, lack new methods. Third: biology, that ever-present, seldom-prosecuted rationale for total synthesis. Here we found that the function of 20norSalA is almost identical to SalA: its potency, its selectivity index over the other GPCR opioid receptors, its brain residence time, its effect on itch. A small collection of aryl analogs demonstrated that the 3-furyl substituent could be replaced with a phenyl and maintain high affinity, overturning the idea that a hydrogen-bonding heterocycle was necessary.38 20norSalA was, however, more stable than SalA. The major change between SalA and 20norSalA was not in form or function, but in disconnection. This change illustrated in a plain way the difference between structural and synthetic complexity (see 5MePXN below for a second example).
Figure 4.
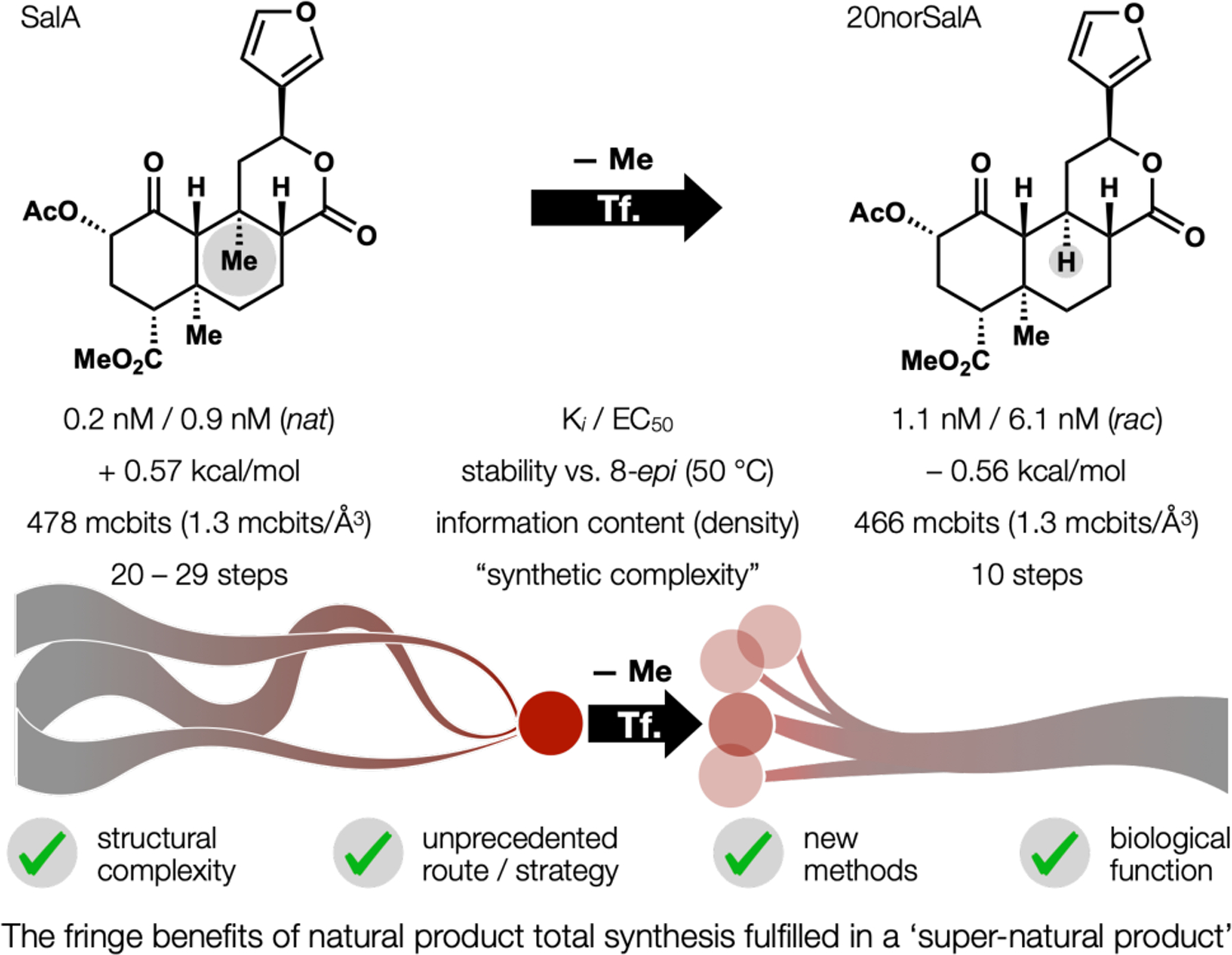
Evaluation of 20norSalA, which fulfills the direct goals and fringe benefits of total synthesis.
Dynamics: a general model
We anticipate this type of thinking is generalizable because the same heuristics that guide retrosynthetic analysis would guide this dynamic analysis (Figure 5). Here the practice of total synthesis is visualized as the connection of remote, NP space to proximal synthetic or commercial ($) space. The NP loci are visualized as distant because certain molecular properties (Fsp3, stereocenters, oxygen content, rings, information density, etc.) are far from commercial materials, both in synthetic steps but also in quantified molecular parameters. NP congeners and possible analogs then form a cloud of structures (tgt) around the NP (TGT) locus. Each tgt can be thought of as connected by a theoretical transform (Tf.) that alters the TGT structure, but maintains the molecular parameters that distinguish the NP. One Tf. and therefore one tgt may provide a shorter retrosynthetic/synthetic path than another tgt or the TGT itself. If this sounds familiar, that’s because these ideas describe the fundamental process of retrosynthetic analysis.
Figure 5.
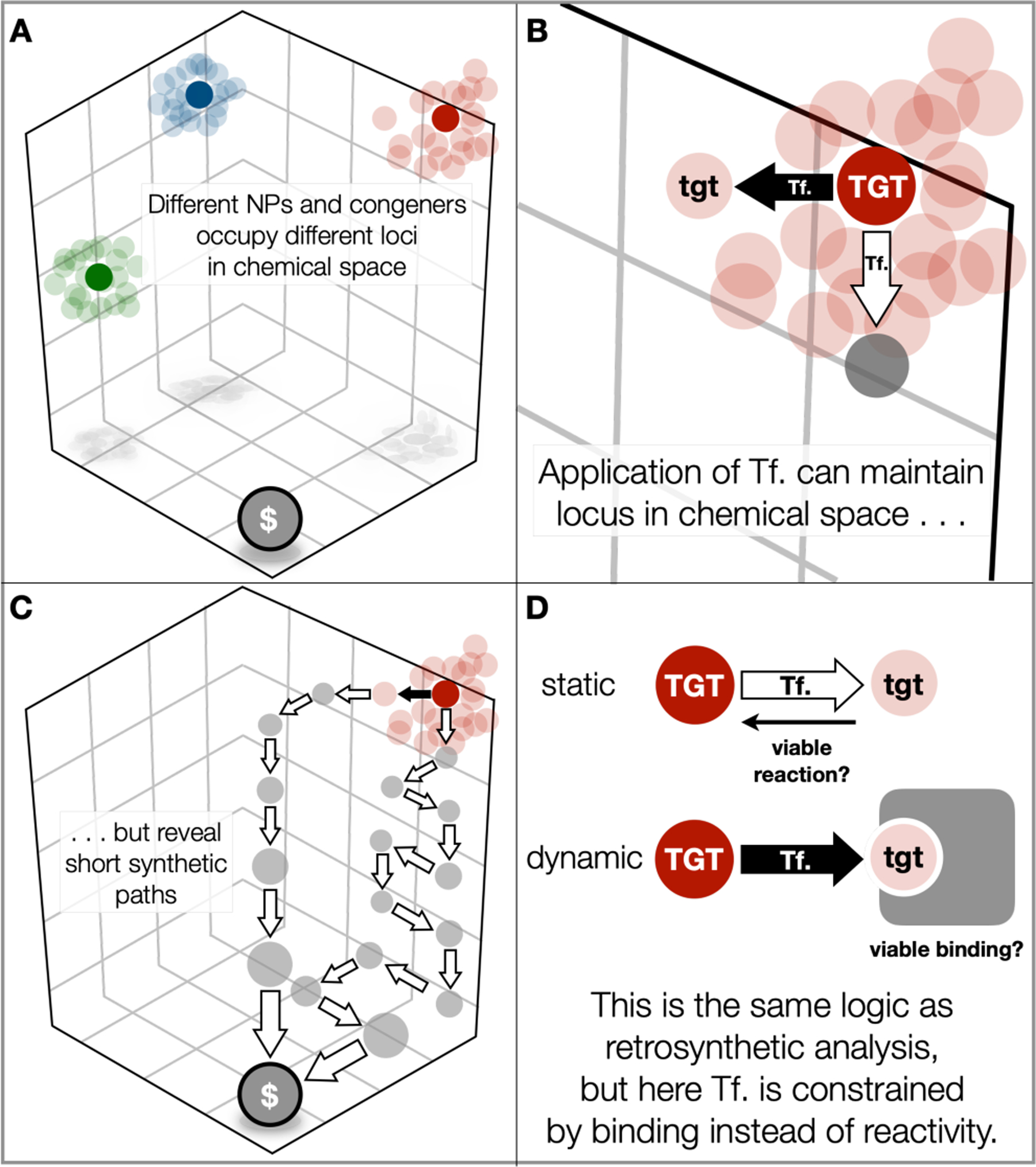
Conceptualization of both static and dynamic retrosynthetic analysis as movement through chemical space. A. Viewing NPs as clusters of related structures in chemical space; B. Viewing the relationships between nodes as transforms (Tf.); C. As in traditional retrosynthetic analysis, a Tf. can alter retrosynthetic paths. D. Dynamic analysis constrains the transform by function rather than reactivity.
The difference between traditional retrosynthetic analysis and this thought experiment is placement of a constraint on the tgt instead of the Tf. Normally, the TGT is static and the Tf. corresponds to a chemical reaction from an intermediate tgt. If the TGT is dynamic, then the Tf. no longer is constrained by reactivity and instead may be considered a point mutation. The constraint, however, is one of function: will the modified tgt maintain the binding of the TGT? The ultimate goal of this approach is to discover short, straightforward and diversifiable routes to valuable NPs and their analogs, where practicality is proven by application. While the slow march of methodology may eventually surpass a specific route, this conceptualization always stays one step ahead of methodology by incorporating new reactions into the algorithm and finding still shorter routes. This abstract argument for the benefit of a dynamic analysis is hard to prove or disprove, and comparison of synthetic routes from different groups across different decades is fraught with ambiguities. Below we describe a more straightforward comparison from within our own group that does not require abstraction or unfair comparison.
Specific target 2: GABAAR
A small series of sesquiterpenes from the Illicium genus (Figure 6) has stimulated an unusual amount of activity from the chemical synthesis community (>50 synthesis papers as of October 2020). Interest in this class may have arisen from three characteristics: aesthetics of the complex polyoxygenated architectures; a neurite outgrowth phenotype assigned by the isolation chemists (perhaps better described as neuroprotective due to higher cell number versus control39); and practical benefits of chemical synthesis over isolation. These trace metabolites occur at parts-per-million levels, translating to single-digit milligram quantities of pure material from multiple kilograms of plant mass (0.00008–0.00016% yield).40–43 Along with many other groups,44 we entered the fray to solve this material access problem. The results were a short, gram-scale synthesis45 using a new method for butenolide heterodimerization46 that enabled the first biological interrogations of the class,47–49 the first in vivo studies of any member49 and a mechanistic model to understand how neurotropic effects might occur.48
Figure 6.
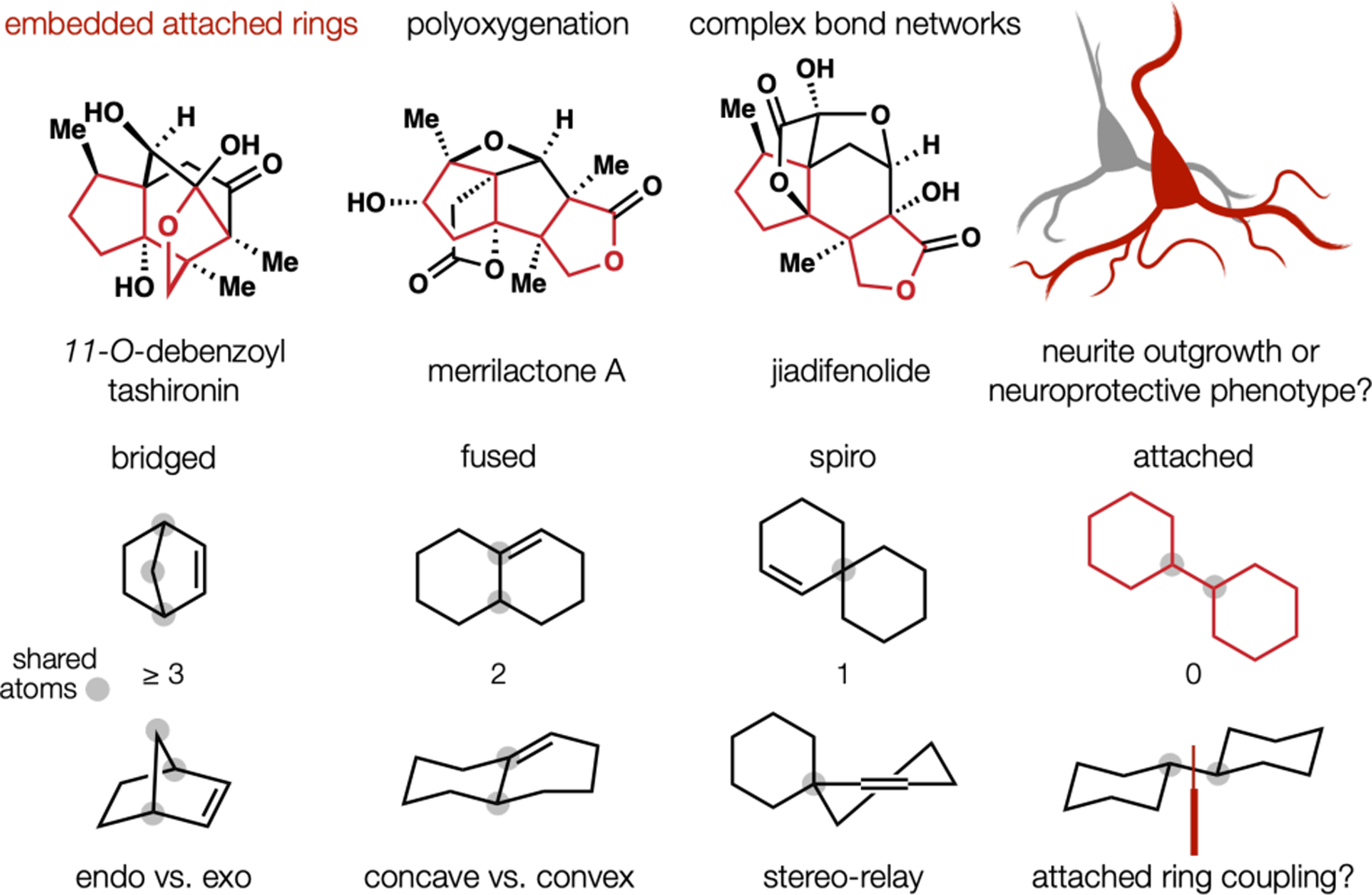
Dissection of the Illicium terpenes via an unorthodox attached ring coupling.
Each of our five total syntheses within this program incorporated oxidation states within the starting materials, a strategy that emerged from the first synthesis, (−)-jiadifenolide. Coincidently, this design suited the long-term biological goals of the program: to match a combinatorial array of receptors, described below. This design also contrasted other work, which increased the scaffold oxidation state iteratively;44 the creative extreme is embodied by Maimone’s oxidative remodeling of cedrene to Illicium congeners.50 Access to pre-oxidized building blocks might become possible by retrosynthetic interception of an attached-ring intermediate, which we hoped to form directly from two similarly-sized building blocks. We held this design casually due to the unlikelihood of its success.
Attached-rings are not typical sub-targets because stereocenters at the bridgeheads are challenging to set, being subject to neither cyclic nor acyclic stereocontrol. Direct sp3-sp3 coupling of two different polysubstituted rings is nearly unprecedented.51 Our high oxidation-state subtarget (tgt, Figure 7, top) was extremely hindered and, we thought, unlikely to form.
Figure 7.
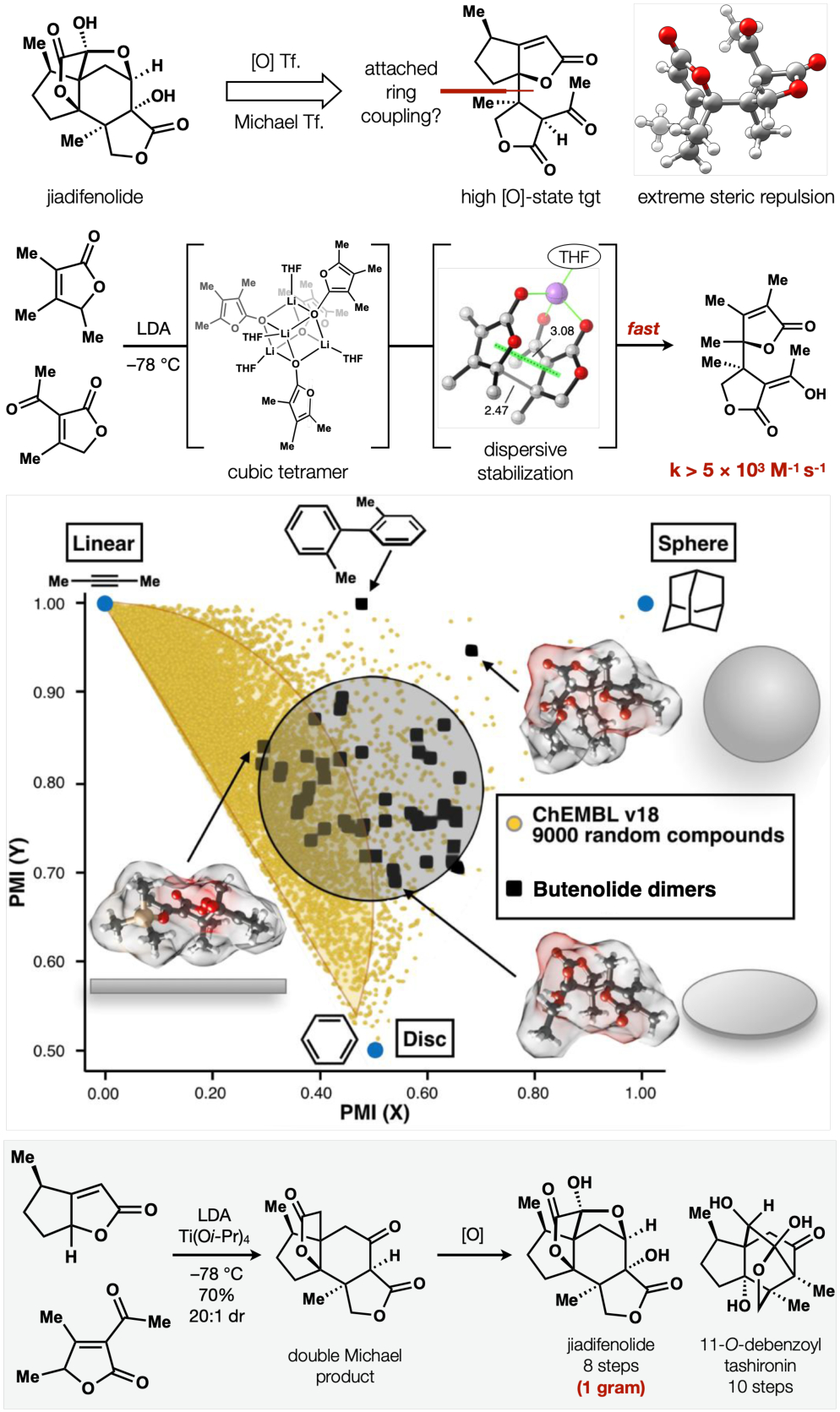
Butenolide heterocoupling: driving forces and applications to cGAS/STING inhibitors and terpenes.
In spite of our expectations, we discovered that polysubstituted butenolides could stereo-, regio- and heteroselectively dimerize to form hindered attached-rings. The reaction exhibited remarkable kinetics, with half-lives at −78 °C of <2 seconds at 100 μM, corresponding to a bimolecular rate constant near 6 × 103 M−1 s−1 and an activation barrier lower than 8 kcal/mol (in contrast, lithium enolates add to benzaldehydes52 with rate constants near 5 M−1 s−1). Rates, competition experiments, radical traps and Hammett analysis arrived at two alternatives: single-electron transfer (SET) or electrophile-induced deaggregation of the enolate—a cubic tetramer according to Collum’s 6Li Job plot analysis.53 Calculations from the Houk group excluded SET as prohibitively high-energy but indicated that reactions of the monomeric enolate might possess a barrier of only 4.4 kcal/mol, a result of π-π interactions that strengthen in the transition state. This productive π-stacking may explain why all other fully-substituted attached ring heterocouplings involve indole substrates. Further, we can extrapolate general design principles to access other attached-ring motifs, which are the tetrahedral equivalents of biaryl rings. According to analysis by Principal Moment of Inertia (PMI), these complex attached rings embody diverse shapes, in contrast to typical synthetic compounds found in collections like ChEMBL (overlay from ref. 54). The value of this collection was demonstrated by the Lairson lab, who determined several members to selectively antagonize the cGAS/STING signaling pathway, a cellular sensor of cytosolic DNA with roles in inflammation and cancer.46
Importantly, this butenolide heterodimerization enabled a one-step, high-yielding, stereoselective assembly of the entire skeleton of the Illicium terpenes with nearly all the oxidation states embedded—a strategy we aimed to imitate in later work. The upshot was easy access to these trace metabolites and the ability to interrogate the mechanism of neurotrophic behavior. (−)-Jiadifenolide became our work-horse compound since we could procure 1 gram of material in a single pass. A clue to the neurotrophic phenotype of jiadifenolide came from its origin in the Illicium genus.47
The evolution of plants has responded to insect predation with terpenoid metabolites that target the nervous system. Phylogenetic relationships between insect and mammalian neuron receptors can cause cross-activity of terpenoids: this is one reason plants can be poisonous to humans.55 For example, the shikimi plant of the Illicium genus produces anisatin, a toxin whose skeleton resembles jiadifenolide. We wondered if neurotrophic activity might relate to an overlap in function. This hypothesis required three non-obvious connections: structural homology, a signaling model to explain neurotrophic effects, and actual target engagement. Jiadifenolide and anisatin do not conspicuously overlap in space, although they are similar in oxygenation and volume. As it happens, anisatin shares the picrotoxinin (PXN) binding site in the channel of the gamma-aminobutyric acid A receptor (GABAAR),56 a mammalian ligand-gated ion channel homologous to the insect RDL receptor. Picrotoxinin and jiadifenolide, despite their unrelated skeletons, overlap very well and indicate the potential for jiadifenolide and the other neurotrophic terpenes to antagonize the GABAAR.47 Is there a potential connection between this target and the neurite outgrowth phenotype?
We think there is. Now-classic studies on neuron function uncovered a link between synaptic activity and enhanced growth, as a result of sustained membrane depolarization.57,58 Agonism of GABAARs inhibits depolarization whereas their antagonism promotes it, a result of unbalanced excitation from cation-selective channels. The consequence can be hyperexcitation of the cell (Figure 8), as the threshold potential is reached more frequently (refs 47 and 48 contain an overview with primary references). Depolarization induces the opening of voltage-gated calcium channels; chronic depolarization then sustains intracellular calcium at elevated levels. If intracellular calcium is too low, cells depend on trophic factors, without which death occurs. Calcium that is too high can be toxic to neurons. Therefore, there is a sweet-spot or “calcium set-point” at which growth is optimal and sustained.59 And this growth is correlated to voltage-gated calcium channels specifically, as opposed to ligand gated calcium channels like NMDA receptors.60
Figure 8.
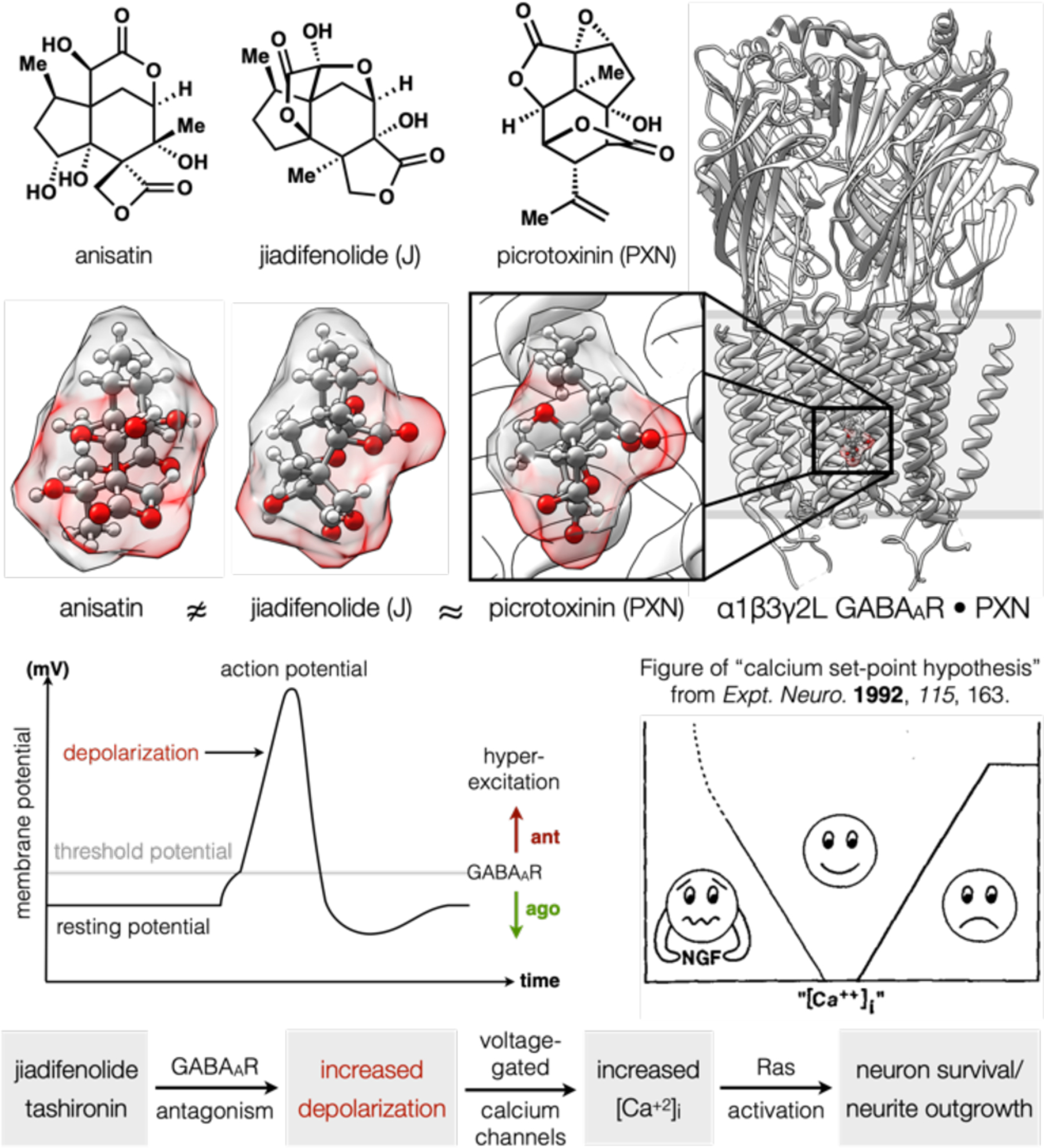
An alternative hypothesis to explain neurotrophic effects of the Illicium terpenes. Figure of “calcium set-point hypothesis” reproduced with permission from ref 60. Copyright 1992 Elsevier.
The Illicium terpenes may elicit a neurotrophic phenotype through the mechanism outlined in Figure 8,61 which we have begun to interrogate by experiment. This hypothesis depends on the ability of the “neurotrophic” terpenes to antagonize GABAARs. A collaboration with Rok Cerne, Jeff Schkeryantz, Jeff Witkin (Eli Lilly) and Marisa Roberto (Scripps) showed this to be the case. Both jiadifenolide and 11-O-debenzoyltashironin caused hyperexcitation of cultured cortical neurons and antagonized GABA-evoked currents in both primary cultures and recombinant receptors.48 Whereas in vitro assays revealed weak current block from the neurotrophic terpenes, acute slices of brain tissue showed block of miniature inhibitory postsynaptic currents (mIPSCs) at 1 μM and lower.62 Remarkably, jiadifenolide does not cause convulsion—common with GABAAR antagonists—and instead induces an antipsychotic phenotype.49 The reason for this discrepancy in apparent potency is not clear, but could be due to distribution, current kinetics, or selective binding to a small population of receptor subtypes.
Potential correlation of Illicium terpene GABAAR antagonism to [Ca2+]i and in vitro neurotrophic effects suggests several exciting research directions, including the potential to target individual GABAAR subtypes. These ion channels are a combinatorial assembly of 5 protein subunits whose compositions reflect the developmental state of neurons63 and their location in the brain. We hope to match the combinatorial assembly of these receptors with a combinatorial assembly of antagonists, whereby two low information-density, high oxidation-state building blocks are merged to high information-density, high oxidation-state products. NP channel binders tend to occupy small volumes yet contain high information content64—multiple rings, oxygen atoms, stereocenters—leading to very high information density (Cm/Å3).62
We successfully applied this strategy to the synthesis of two other antagonists, the non-convulsive Ginkgo metabolite bilobalide (BB)62,65 and the convulsive Anamirta metabolite PXN (Figure 9).3 Both syntheses begin from high oxidation state building blocks (Gasteiger partial charges of oxygen-bearing carbons in red),66 which can be stereoselectively merged to provide rapid entry to high oxidation state targets. BB emerges from a novel, enantioselective, diastereoselective Reformatsky reaction, and PXN from an α-selective, stereoselective aldol reaction (tert-alcohol chemistry inconsequential).
Figure 9.
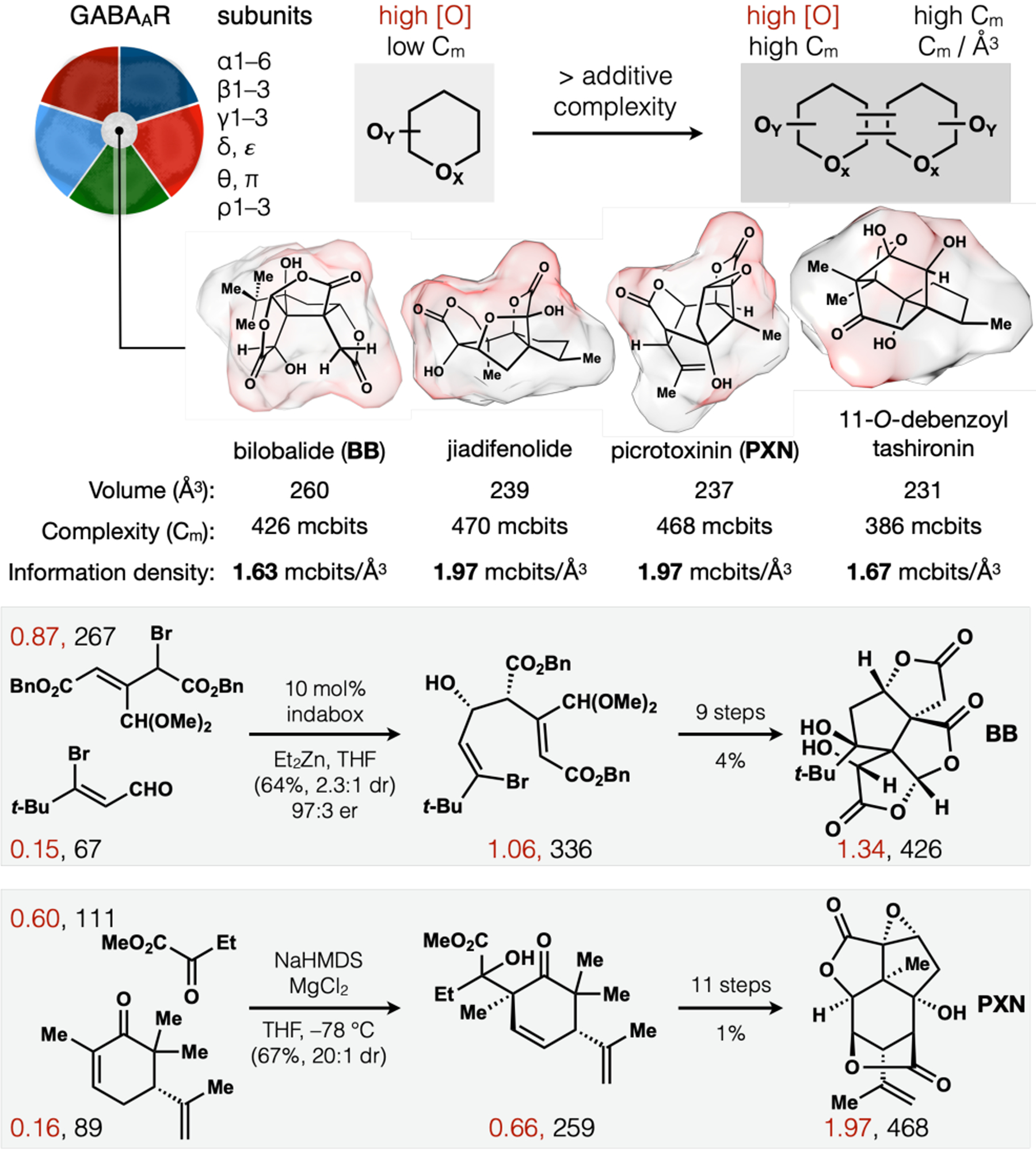
High oxidation-state building blocks (Gasteiger partial charges in red) merged to access complex antagonists (Cm in black) of GABAAR, a combinatorial receptor.
The high information content of these intermediates (Cm score64 in black) reflects existing oxygenation patterns and encoded patterns, so that elaboration to the target is straightforward. The overall yield to reach PXN, however, suffered due to competitive reactions of unstable intermediates, reflecting the instability of PXN itself. Treatment of the target as dynamic revealed a shorter, higher-yielding route to a more stable congener.
Correct diastereoselectivity in the aldol fragment coupling required a symmetrizing gem-dimethyl motif, which nevertheless required a lengthy, low-yielding excision. We realized, however, that retaining a methyl in the target would compress the synthesis length (Figure 10), even though the complexity of target increased (468 to 480 mcbits). Its value, however, depended on retained affinity for the GABAAR, a possibility supported by calculations from Tim Carpenter (LLNL). 5MePXN is accessed via Suarez oxidation of a 1,3-diaxial alcohol-methyl pair and cleavage of a bromoether protecting group common across PXN syntheses. The precursor can be obtained in multigram quantities during a single scale-up via tandem dihydroxylation/lactonization of the corresponding alkene, which is only 4 straightforward steps away from the aldol product of Figure 9. The revised synthesis decreases step burden from 12 to 8, and amplifies overall yield from 0.6% to 8%! While we reached PXN in the shortest route yet reported, this robust, scalable and concise route to 5MePXN, a non-NP, may hold more value. First, 5MePXN resists hydrolysis at pH 8, whereas PXN degrades almost completely.3 Second, 5MePXN engages the GABAAR according to radioligand (3H-TBOB) displacement, albeit with a reduced potency (IC50 = 9 μM) compared to PXN (IC50 = 0.2 μM). Third, and most importantly, this route provides a robust synthetic branch from dimethylcarvone to a PXN analog almost identical to PXN and related only by a methylation (+Me) transform (Tf.). A quick route to the chemical space of PXN can enable rapid diversification to picrotoxane congeners, whose semisyntheses have required as many steps as 5MePXN itself.67 This powerful synthetic platform allows interrogation, for the first time, of ion-channel selectivity among PXN analogs.
Figure 10.
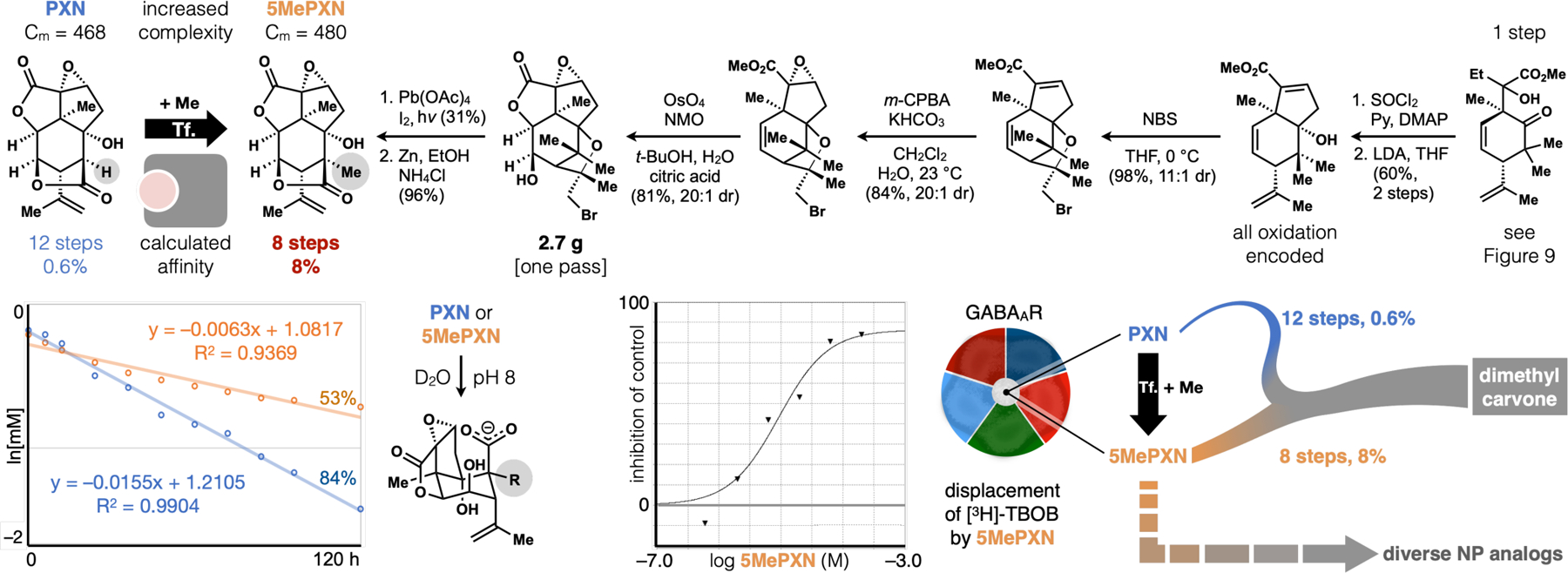
Me-addition to PXN increases complexity (ΔCm = +12) but shortens the synthetic route, providing a robust supply of a complex GABAAR antagonist (5MePXN) with improved stability towards hydrolysis.
Conclusion
Chemical synthesis enables the study and modulation of biological function using atomic-resolution changes in structure.68 Its application to complex targets like NPs holds value for interrogation of biological MOA, methods development and populating remote, valuable areas of chemical space for high-throughput screens.69 Traditional NP synthesis holds the target as static, even if the ultimate goal is divergency and diversity;11 yet target diversification (navigation of local chemical space) does not require access to the NP itself. A dynamic approach may better suit these goals by providing faster access to a particular locus. Its ease derives from the larger search space: each different target congener—with its own degree of synthetic complexity—contains a similar number of viable transforms compared to the static target.
Dynamic approaches may benefit trainees as much as static approaches. Anecdotally, the decision of a trainee to undertake a total synthesis project honors an unwritten Faustian pact, where incredible labor and self-sacrifice are exchanged for commensurate rewards in training, career prospects, self-fulfillment, and discovery. Yet while many of the downsides of this “deal with the devil” relate to a cultural emphasis on reaching a singular, immutable target, its benefits—both to the individual, and to the broader scientific community—typically result from working in complex, NP space, not necessarily on the NP itself. Traditional total synthesis yields exceptional training, but risks irrelevance.70
Dynamic analysis also carries risk. However, this risk is not open-ended, but a yes-or-no question answerable by synthesis: is function maintained? Loss of binding still delivers knowledge as a structure-activity relationship. And successful binding gives way to longer-term goals of functional endpoints, like restriction to the peripheral nervous system (SalA) or receptor selectivity (PXN). It provides practitioners with the opportunity to problem solve, discover and develop methods, but also to practice target design.
We have successfully applied dynamic retrosynthetic analysis twice, first with the KOR agonist salvinorin A and second with GABAAR antagonist picrotoxinin. Deletion of a methyl from the former, and addition of a methyl to the latter, significantly decreased synthetic burden, retained target engagement and stabilized each scaffold. In both cases, structural complexity remained nearly identical to the NP (−3% Cm SalA; +3% Cm PXN), and both modifications increased chemical stability. Currently, we are making inroads into diversification of this privileged chemical space towards functional endpoints, enabled by robust supplies of these complex scaffolds.
Because the basic precepts of dynamic retrosynthetic analysis are identical to static retrosynthetic analysis, there is reason to believe that automated searches can deliver viable targets and routes. Automation requires two modules: retrosynthetic software and ligand docking. Retrosynthetic software has significantly advanced in recent years,71 but the addition of diverse point mutations, followed by a search of these numerous alternative targets, would demand substantial computational power. The second module already exists: in silico docking is routinely employed to estimate binding affinity of large libraries. A merger of these two modules would enable the constellation of related compounds to be docked in silico and analyzed for short synthetic routes.
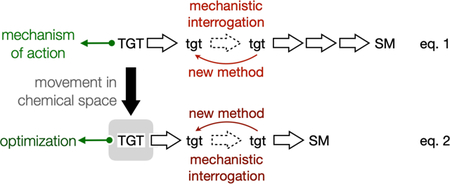 |
This Account described how our research model changed over time to incorporate a new tool—dynamic retrosynthetic analysis—into our repertoire of problem-solving techniques. This tool merely extends our initial framework by viewing NP synthesis through the lens of chemical informatics. When the lab began, we adopted a traditional model (eq. 1), employing the heuristics of retrosynthetic analysis10 to uncover possible synthetic routes and identify methodological gaps, which led to reaction invention, mechanistic interrogation and biological discovery. Through the lens of chemical informatics, there is nothing especially remarkable about a single NP, even if its immediate chemical space holds great value. Equation 1 can become equation 2, where a nearly identical target reveals a better path and a more effective means of structure optimization. This dynamic approach leverages advances in structural biology and gains in computation to bring greater relevance to natural product synthesis in the modern era. Dynamic analysis has propelled our research in rewarding directions; we hope its ideas guide the next generation even further.
ACKNOWLEDGMENT
We gratefully acknowledge the students, postdocs and faculty, past and present, who have left indelible marks on our research program.
Funding Sources
This work was supported by grants from the NIH (GM122606), NSF (1856747, 1955922) and a generous Skaggs Fellowship to S.W.
Biographies
Stone Woo received his undergraduate education at the University of Sydney. Currently, he is a graduate student in the Shenvi lab exploring complexity-rich chemical space in the context of natural product total synthesis.
Ryan Shenvi was born and raised in Wilmington, Delaware. He earned his PhD with Phil Baran at the Scripps Research Institute, where he is now Professor in the Department of Chemistry. Postdoctoral research with E. J. Corey inspired the approach to natural product synthesis described in this Account.
REFERENCES
- (1).Roach JJ; Sasano Y; Schmid CL; Zaidi S; Katrich V; Stevens RC; Bohn LM; Shenvi RA “Dynamic Strategic Bond Analysis Yields a 10-step Synthesis of 20-nor-Salvinorin A, a Potent κ-OR Agonist” ACS Cent. Sci 2017, 3, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]; The workflow of this project—retrosynthetic analysis, structure perturbation, calculated receptor affinity, experimentation through synthesis—changed the way our lab thought about natural product synthesis.
- (2).Huffman BJ; Shenvi RA Natural Products in the “Marketplace”: Interfacing Synthesis and Biology J. Am. Chem. Soc 2019, 141, 3332–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]; Here we outline the basic ideas of dynamic retrosynthetic analysis.
- (3).Crossley SWM; Tong G; Lambrecht MJ; Burdge HE; Shenvi RA “Synthesis of (−)-Picrotoxinin by Late-Stage Strong Bond Activation” J. Am. Chem. Soc 2020, 142, 11376–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work afforded the second example of a dynamic retrosynthetic approach and gave rapid entry to PXN chemical space.
- (4).Van Hattum H; Waldmann H “Biology-Oriented Synthesis: Harnessing the Power of Evolution” J. Am. Chem. Soc 2014, 136, 11853. [DOI] [PubMed] [Google Scholar]
- (5).Clemons PA; Bodycombe NE; Carrinski HA; Wilson JA; Shamji AF; Wagner BK; Koehler AN; Schreiber SL Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ishikawa M; Hashimoto Y Improvement in Aqueous Solubility in Small Molecule Drug Discovery Programs by Disruption of Molecular Planarity and Symmetry. J. Med. Chem 2011, 54, 1539. [DOI] [PubMed] [Google Scholar]
- (7).For a concise summary and case studies, see; Caille S; Cui S; Faul MM; Mennen SM; Tedrow JS; Walker SD “Molecular Complexity as a Driver for Chemical Process Innovation in the Pharmaceutical Industry” J. Org. Chem 2019, 84, 4583 and references therein. [DOI] [PubMed] [Google Scholar]
- (8).Kombo DC; Tallapragada K; Jain R; Chewning J; Mazurov AA; Speake JD; Hauser TA; Toler S “3D Molecular Descriptors Important for Clinical Success” J. Chem. Inf. Mod 2013, 53, 327. [DOI] [PubMed] [Google Scholar]
- (9).Wetzel S; Schuffenhauer A; Roggo S; Ertl P; Waldmann H “Cheminformatic Analysis of Natural Products and their Chemical Space” Chimia 2007, 61, 355. [Google Scholar]
- (10).Corey EJ and Cheng XM, The Logic of Chemical Synthesis, J. Wiley, New York, 1989. [Google Scholar]
- (11).Boger DL; Brotherton CE “Total Syntheses of Azafluoranthene Alkaloids: Rufescine and Imeluteine” J. Org. Chem 1984, 49, 4050. [Google Scholar]
- (12).Wender PA; Verma VA; Paxton TJ; Pillow TH “Function-Oriented Synthesis, Step-Economy, and Drug Design” Acc. Chem. Res 2008, 41, 40. [DOI] [PubMed] [Google Scholar]
- (13).Gerry CJ; Schreiber SL “Recent achievements and current trajectories of diversity-oriented synthesis” Curr. Op. Chem. Bio 2020, 56, 1. [DOI] [PubMed] [Google Scholar]
- (14).For an outstanding overview, see:; Lehmann JW; Blair DJ; Burke MD “Towards the generalized iterative synthesis of small molecules” Nat. Rev. Chem 2018, 2, 0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For an excellent review of covalent inhibitors, see:; Bauer RA “Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies” Drug Disc. Today 2015, 20, 1061. [DOI] [PubMed] [Google Scholar]
- (16).For demonstration of covalent reactions of the asmarine N-hydroxyaminopurine pharmacophore, see:; Yosief T; Rudi A; Kashman Y “Asmarines A–F, Novel Cytotoxic Compounds from the Marine Sponge Raspailia Species” J. Nat. Prod 2000, 63, 299; [DOI] [PubMed] [Google Scholar]; For suggestions that the Nuphar dimers react covalently at carbon, see:; LaLonde RT “Intramolecular iminium ion-sulfide charge-transfer association: a recurring theme in the study of thiaspirane alkaloids” Acc. Chem. Res 1980, 13, 39; [Google Scholar]; Martin Schnermann proposed that isocyanoterpenes may react covalently with carboxylates to exhibit anti-malarial activity, as summarized in; (d) Schnermann MJ; Shenvi RA Nat. Prod. Rep 2015, 32, 543; and by analogy to [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Restorp P; Rebek J Jr. “Reaction of Isonitriles with Carboxylic Acids in a Cavitand: Observation of Elusive Isoimide Intermediates” J. Am. Chem. Soc 2008, 130, 11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tiefenbacher has advanced the most powerful, general strategies for covalent folding of farnesyl and cyclofarnesyl substrates:; Zhang Q; Tiefenbacher K “Sesquiterpene Cyclizations inside the Hexameric Resorcinarene Capsule: Total Synthesis of δ Selinene and Mechanistic Studies” Angew. Chem. Int. Ed 2019, 58, 12688. [DOI] [PubMed] [Google Scholar]
- (18).Pronin SV; Tabor MG; Jansen DJ; Shenvi RA “A Stereoselective Hydroamination Transform to Access Polysubstituted Indolizidines” J. Am. Chem. Soc 2012, 134, 2012. [DOI] [PubMed] [Google Scholar]
- (19).Pronin SV; Shenvi RA “Synthesis of highly strained terpenes by non-stop tail-to-head polycyclization” Nat. Chem 2012, 4, 915. [DOI] [PubMed] [Google Scholar]
- (20).Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA “Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol” J. Am. Chem. Soc 2014, 136, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Crossley SWM; Obradors C; Martinez RM; Shenvi RA “Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins” Chem. Rev 2016, 116, 8912; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green SA; Crossley SWM; Matos JLM; Vásquez-Céspedes S; Shevick SL; Shenvi RA “The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer” Acc. Chem. Res 2018, 51, 2628; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matos JLM; Green SA; Shenvi RA Markovnikov Functionalization by Hydrogen Atom Transfer, Organic Reactions, 2019, 100, 383; [Google Scholar]; (d) Shevick SL; Wilson CV; Kotesova S; Kim D; Holland PL; Shenvi RA “Catalytic hydrogen atom transfer to alkenes: a roadmap for metal hydrides and radicals” Chem. Sci 2020, DOI: 10.1039/D0SC04112B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tada N; Jansen DJ; Mower MP; Blewett MM; Umotoy JC; Cravatt BF; Wolan DW; Shenvi RA “Synthesis and Sulfur Electrophilicity of the Nuphar Thiaspirane Pharmacophore” ACS Cent. Sci 2016, 2, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pronin SV; Shenvi RA “Synthesis of a Potent Antimalarial Amphilectene” J. Am. Chem. Soc 2012, 134, 19604. [DOI] [PubMed] [Google Scholar]
- (24).Pronin SV; Reiher CA; Shenvi RA “Stereoinversion of tertiary alcohols to tertiary-alkyl isonitriles and amines” Nature 2013, 501, 195. [DOI] [PubMed] [Google Scholar]
- (25).Lambrecht MJ; Kelly JW; Shenvi RA “Mechanism of Action of the Cytotoxic Asmarine Alkaloids” ACS Chem. Biol 2018, 13, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lu H-H; Pronin SV; Antonova-Koch Y; Meister S; Winzeler EA; Shenvi RA “Synthesis of (+)-7,20-Diisocyanoadociane and Liver-Stage Antiplasmodial Activity of the ICT Class” J. Am. Chem. Soc 2016, 138, 7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).For recent reviews, see:; (a) Roach JJ; Shenvi RA “A review of salvinorin analogs and their kappa-opioid receptor activity” Bioorg. Med. Chem. Lett 2018, 28, 1436; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hill SJ; Brion AUCM; Shenvi RA “Chemical syntheses of the salvinorin chemotype of KOR agonist” Nat. Prod. Rep 2020, 37, 1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Roth BL; et al. “Salvinorin A: A potent naturally occurring nonnitrogenous κ opioid selective agonist” Proc. Natl. Acad. Sci. U. S. A 2002, 99, 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Munro TA; Duncan KK; Staples RJ; Xu W; Liu-Chen L-Y; Béguin C; Carlezon WA Jr.; Cohen BM “8-epi-salvinorin B: crystal structure and affinity at the κ opioid receptor” Beilstein J. Org. Chem 2007, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Harding WW; Schmidt M; Tidgewell K; Kannan P; Holden KG; Dersch CM; Rothman RB; Prisinzano TE “Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Selective modification of the furan ring” Bioorg. Med. Chem. Lett 2006, 16, 3170. [DOI] [PubMed] [Google Scholar]
- (31).Nozawa M; Suka Y; Hoshi T; Suzuki T; Hagiwara H “Total Synthesis of the Hallucinogenic Neoclerodane Diterpenoid Salvinorin A” Org. Lett 2008, 10, 1365. [DOI] [PubMed] [Google Scholar]
- (32).Munro TA; Rizzacasa MA; Roth BL; Toth BA; Yan F Studies toward the Pharmacophore of Salvinorin A, a Potent κ Opioid Receptor Agonist J. Med. Chem 2005, 48, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Valdes III LJ; Butler WM; Hatfield GM; Paul AG; Koreeda M “Divinorin A, a Psychotropic Terpenoid, and Divinorin B from the Hallucinogenic Mexican Mint Salvia divinorum” J. Org. Chem 1984, 49, 4716. [Google Scholar]
- (34).Beǵuin C; et al. “Synthesis and in vitro evaluation of salvinorin A analogues: effect of configuration at C(2) and substitution at C(18).” Bioorg. Med. Chem. Lett 2006, 16, 4679. [DOI] [PubMed] [Google Scholar]
- (35).Methyl addition might have increased potency since the effect of ligand modification can be unpredictable. All the same, the risk of pursuing a target of NP-like complexity with no prior experimental data about its activity was at the forefront of our minds from day one, perhaps driven by a cultural distrust of docking … one might say pier pressure.
- (36).Che T; Majumdar S; Zaidi SA; Ondachi P; McCorvy JD; Wang S; Mosier PD; Uprety R; Vardy E; Krumm BE; Han GW; Lee MY; Pardon E; Steyaert J; Huang X-P; Strachan RT; Tribo AR; Pasternak GW; Carroll FI; Stevens RC; Cherezov V; Katritch V; Wacker D; Roth BL “Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor” Cell 2018, 172, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huffman TR; Wu Y; Emmerich A; Shenvi RA “Intermolecular Heck Coupling with Hindered Alkenes Directed by Potassium Carboxylates” Angew. Chem. Int. Ed 2019, 58, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cunningham CW; Rothman RB; Prisinzano TE “Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A” Pharmacol. Rev 2011, 63, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shoji M; Nishioka M; Minato H; Harada K; Kubo M; Fukuyama Y; Kuzuhara T “Neurotrophic activity of jiadifenolide on neuronal precursor cells derived from human induced pluripotent stem cells” Biochem. Biophys. Res. Comm 2016, 470, 798. [DOI] [PubMed] [Google Scholar]
- (40).Kubo M; Okada C; Huang J-M; Harada K; Hideaki H; Fukuyama Y “Novel Pentacyclic seco-Prezizaane-Type Sesquiterpenoids with Neurotrophic Properties from Illicium jiadifengpi” Org. Lett 2009, 22, 5190. [DOI] [PubMed] [Google Scholar]
- (41).Huang J-M; Yokoyama R; Yang C-S; Fukuyama Y “Structure and Neurotrophic Activity of seco-Prezizaane-Type Sesquiterpenes from Illicium merrillianum” J. Nat. Prod 2001, 64, 428. [DOI] [PubMed] [Google Scholar]
- (42).Huang J-M; Yokoyama R; Yang C-S; Fukuyama Y “Merrilactone A, a novel neurotrophic sesquiterpene dilactone from Illicium merrillianum” Tetrahedron Lett. 2000, 41, 6111. [Google Scholar]
- (43).Yokoyama R; Huang J-M; Yang C-S; Fukuyama Y “New seco-Prezizaane-Type Sesquiterpenes, Jiadifenin with Neurotrophic Activity and 1,2-Dehydroneomajucin from Illicium jiadifengpi” J. Nat. Prod 2002, 65, 527. [DOI] [PubMed] [Google Scholar]
- (44).Condakes ML; Novaes LFT; Maimone TJ “Contemporary Synthetic Strategies toward seco-Prezizaane Sesquiterpenes from Illicium Species” J. Org. Chem 2018, 83, 14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lu H-H; Martinez MD; Shenvi RA “An eight-step gram-scale synthesis of (−)-jiadifenolide” Nat. Chem 2015, 7, 604. [DOI] [PubMed] [Google Scholar]
- (46).Huffman BJ; Chen S; Schwarz JL; Plata RE; Chin EN; Lairson LL; Houk KN; Shenvi RA “Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists” Nat. Chem 2020, 12, 310. [DOI] [PubMed] [Google Scholar]
- (47).Shenvi RA “Neurite outgrowth enhancement by jiadifenolide: possible targets” Nat. Prod. Rep 2016, 33, 535. [DOI] [PubMed] [Google Scholar]
- (48).Ohtawa M; Krambis MJ; Cerne R; Schkeryantz JM; Witkin JM; Shenvi RA “Synthesis of (−)-11-O-Debenzoyltashironin: Neurotrophic Sesquiterpenes Cause Hyperexcitation” J. Am. Chem. Soc 2017, 139, 9637. [DOI] [PubMed] [Google Scholar]
- (49).Witkin JM; Shenvi RA; Li X; Gleason SD; Weiss J; Morrow D; Catow JT; Wakulchik M; Ohtawa M; Lu H-H; Martinez MD; Schkeryantz JM; Carpenter TS; Lightstone FC; Cerne R “Pharmacological characterization of the neurotrophic sesquiterpene jiadifenolide reveals a non-convulsant signature and potential for progression in neurodegenerative disease studies” Biochem. Pharmacol 2018, 155, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hung K; Condakes ML; Novaes LFT; Harwood SJ; Morikawa T; Yang Z; Maimone TJ “Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes” J. Am. Chem. Soc 2019, 141, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).For remarkable exceptions to the rule, see:; (a) Movassaghi M; Ahmad OK; Lathrop SP “Directed Heterodimerization: Stereocontrolled Assembly via Solvent-Caged Unsymmetrical Diazene Fragmentation” J. Am. Chem. Soc 2011, 133, 13002; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fuchs JR; Funk RL “Total Synthesis of (±)-Perophoramidine” J. Am. Chem. Soc 2004, 126, 5068. [DOI] [PubMed] [Google Scholar]
- (52).Kolonko KJ; Wherritt DJ; Reich HJ “Mechanistic Studies of the Lithium Enolate of 4-Fluoroacetophenone: Rapid-Injection NMR Study of Enolate Formation, Dynamics, and Aldol Reactivity” J. Am. Chem. Soc 2011, 133, 16774. [DOI] [PubMed] [Google Scholar]
- (53).Renny JS; Tomasevich LL; Tallmadge EH; Collum DB “Method of Continuous Variations: Applications of Job Plots to the Study of Molecular Associations in Organometallic Chemistry” Angew. Chem. Int. Ed 2013, 52, 11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Brown DG; Boström J “Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?” J. Med. Chem 2016, 59, 4443. [DOI] [PubMed] [Google Scholar]
- (55).Buckingham SD; Ihara M; Sattelle DB; Matsuda K “Mechanisms of Action, Resistance and Toxicity of Insecticides Targeting GABA Receptors” Curr. Med. Chem 2017, 24, 2935. [DOI] [PubMed] [Google Scholar]
- (56).Ikeda T; Ozoe Y; Okuyama E; Nagata K; Honda H; Shono T; Narahashi T “Anisatin modulation of the γ-aminobutyric acid receptor-channel in rat dorsal root ganglion neurons” Br. J. Pharmacol 1999, 7, 1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Harris WA “Neural activity and development” Ann. Rev. Physiol 1981, 43, 689. [DOI] [PubMed] [Google Scholar]
- (58).Gallo V; Kingsbury A; Balázs R; Jørgensen OS “The Role of Depolarization in the Survival and Differentiation of Cerebellar Granule Cells in Culture” J. Neurosci 1987, 7, 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Johnson EM; Koike T; Franklin J “A “Calcium Set-Point Hypothesis” of Neuronal Dependence on Neurotrophic Factor” Expt. Neurol 1992, 115, 163. [DOI] [PubMed] [Google Scholar]
- (60).Ghosh A; Carnahan J; Greenberg ME “Requirement for BDNF in Activity-Dependent Survival of Cortical Neurons” Science 1994, 263, 1618. [DOI] [PubMed] [Google Scholar]
- (61).Vaillant AR; Mazzoni I; Tudan C; Boudreau M; Kaplan DR; Miller FD “Depolarization and Neurotrophins Converge on the Phosphatidylinositol 3-Kinase-Akt Pathway to Synergistically Regulate Neuronal Survival” J. Cell Biol 1999, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Demoret RM; Baker MA; Ohtawa M; Chen S; Lam C-C; Khom S; Roberto M; Forli S; Houk KN; Shenvi RA “Synthesis and Mechanistic Interrogation of Ginkgo biloba Chemical Space En Route to (−)-Bilobalide” J. Am. Chem. Soc 2020, 142, 18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Harada K; Matsuoka H; Fujihara H; Ueta Y; Yanagawa Y; Inoue M “GABA Signaling and Neuroactive Steroids in Adrenal Medullary Chromaffin Cells” Front. Cell. Neurosci 2016, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Böttcher T “An Additive Definition of Molecular Complexity” J. Chem. Inf. Model 2016, 56, 462. [DOI] [PubMed] [Google Scholar]
- (65).Baker MA; Demoret RM; Ohtawa M; Shenvi RA “Concise asymmetric synthesis of (−)-bilobalide” Nature 2019, 575, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
- (67).Krische MJ; Trost BM “Transformations of the Picrotaxanes: The Synthesis of Corianin and Structural Analogues from Picrotoxinin” Tetrahedron 1998, 54, 7109. [Google Scholar]
- (68).Boger DL “The Difference a Single Atom Can Make: Synthesis and Design at the Chemistry–Biology Interface” J. Org. Chem 2017, 82, 11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Dandapani S; Marcaurelle LA “Grand challenge commentary: Accessing new chemical space for ‘undruggable’ targets” Nat. Chem. Bio 2010, 6, 861. [DOI] [PubMed] [Google Scholar]
- (70).As a reviewer commented: “failure to reach a certain target structure but design and synthesis of an even better or equally good molecule in fewer steps does not make students ‘chicken’ … it elevates them.”
- (71).A shrewd approach to NP space exploration might leverage computational workflows as valuable tools with limitations, “not … used for their own sake but because they enable discoveries that are otherwise inaccessible to human researchers” [Coley C, Trends in Chemistry 2020. DOI: 10.1016/j.trechm.2020.11.004]. [DOI] [Google Scholar]