

SUMMARY
Modes of somatodendritic transmission range from rapid synaptic signaling to protracted regulation over distance. Somatodendritic dopamine secretion in the midbrain leads to D2 receptor-induced modulation of dopamine neurons on the timescale of seconds. Temporally imprecise release mechanisms are often presumed to be at play, and previous work indeed suggested roles for slow Ca2+ sensors. We here use mouse genetics and whole-cell electrophysiology to establish that the fast Ca2+ sensor synaptotagmin-1 (Syt-1) is important for somatodendritic dopamine release. Syt-1 ablation from dopamine neurons strongly reduces stimulus-evoked D2 receptor-mediated inhibitory postsynaptic currents (D2-IPSCs) in the midbrain. D2-IPSCs evoked by paired stimuli exhibit less depression, and high-frequency trains restore dopamine release. Spontaneous somatodendritic dopamine secretion is independent of Syt-1, supporting that its exocytotic mechanisms differ from evoked release. We conclude that somatodendritic dopamine transmission relies on the fast Ca2+ sensor Syt-1, leading to synchronous release in response to the initial stimulus.
In brief
Lebowitz et al. show that synaptotagmin-1 is important for somatodendritic dopamine release triggered by single action potentials but not for spontaneous release or release evoked by high-frequency trains. Hence, multiple Ca2+ sensors mediate actionpotential-evoked somatodendritic release in the midbrain with crucial roles for the fast, synchronous sensor synaptotagmin-1.
Graphical Abstract
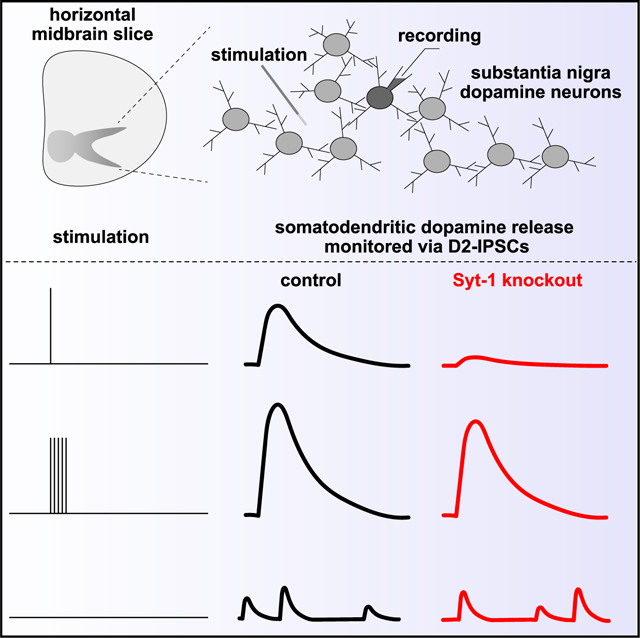
INTRODUCTION
Neuronal somata and dendrites are equipped with machinery to detect, transduce, and process neurotransmitter signals. However, many neurons also release neurotransmitters and neuromodulators through vesicular exocytosis from these compartments.1,2 Somatodendritic exocytosis is involved in processes ranging from synaptic-like local signaling to paracrine regulation over long distances.3–5 Distinct release profiles may be established by varying individual components of the exocytotic pathway, for example the transmitter-containing vesicles, the protein machinery that triggers exocytosis, or the structural organization of secretory sites at the somatodendritic membrane. The molecular organization and function of the various somatodendritic release pathways may be shared or specific depending on the neurotransmitter, necessitating detailed investigation of each transmitter.
An important form of somatodendritic exocytosis is that of dopamine in the midbrain, which underlies a transmission mode that controls the excitability of dopamine neurons.6–10 We here focused on dissecting the Ca2+-triggering mechanisms of somatodendritic dopamine release. At conventional synapses, release has been classified by its temporal relationship to action potential firing. Synchronous release is completed within milliseconds of presynaptic depolarization and is mediated by the fast Ca2+ sensors synaptotagmin-1 (Syt-1), Syt-2, and Syt-9.11–15 Asynchronous release relies on the buildup of residual Ca2+, persists for more than tens of milliseconds, and is triggered by additional Ca2+ sensors that include Syt-7.16–20 Spontaneous release is independent of action potential firing, and its reliance on Ca2+ entry has remained uncertain, yet Ca2+ sensors including Syt-1 are important regulators of this form of release.15,20–24
In somatodendritic dopamine transmission, activation of D2 G protein-coupled receptors on dopamine neurons elicits an inhibitory postsynaptic current (D2-IPSC) that is carried by K+ ions through the activation of G protein-gated inwardly rectifying K+ (GIRK) channels.7,9,10 A prevailing model has been that somatodendritic dopamine exocytosis uses distinct protein machinery compared with axonal dopamine release.25,26 Specifically, while synchronous dopamine release from axons relies on Syt-1 and is rapid and precise,27–29 somatodendritic release may have a different Ca2+ dependence, may rely on slower Ca2+ sensors, and may depend on Syt-7 and Syt-4.30–33 This is consistent with some, but not all, aspects of somatodendritic dopamine signaling. For example, D2 receptor activation necessitates ~100 μM dopamine,34 suggesting the need for synchronous exocytosis such that the extracellular dopamine concentration increases rapidly. In agreement, knockout of RIM, a protein that co-organizes release-ready vesicles with Ca2+ entry to accelerate release,35–41 strongly impairs evoked somatodendritic dopamine transmission.42 These data suggest rapid somatodendritic dopamine release mechanisms, but a suitable fast Ca2+ sensor has not been identified with confidence.
Here, we find that the fast Ca2+ sensor Syt-1 is important for evoked somatodendritic dopamine release. Conditional knockout of Syt-1 from dopamine neurons strongly reduced D2-IPSCs elicited by single stimuli, likely due to a decrease in vesicular release probability. This deficit was restored by viral re-expression of Syt-1. High-frequency stimulus trains induced robust D2-IPSCs after Syt-1 ablation, indicating that an additional release pathway is activated. Lastly, Syt-1 knockout did not impair spontaneous D2-IPSCs and neither did lowering the extracellular Ca2+ concentration. We conclude that Syt-1 is a Ca2+ sensor for synchronous somatodendritic dopamine release. Additional sensors must be present in these mice to mediate transmission during repetitive activity after Syt-1 deletion, likely via asynchronous release. Spontaneous somatodendritic dopamine release is independent of Ca2+ and of the fast Ca2+ sensor Syt-1.
RESULTS
Syt-1 knockout strongly impairs somatodendritic dopamine release evoked by single stimuli
Of the known Ca2+ sensors, Syt-1 and Syt-7 are expressed in dopamine neurons.43,44 We recently established that Syt-1 is a fast Ca2+ sensor for triggering axonal dopamine release.29 Based on these data and on the properties of somatodendritic dopamine transmission, we hypothesized that Syt-1 triggers somatodendritic dopamine secretion. We performed electrophysiological recordings in brain slices (Figure 1A)10,42 of mice with genetic ablation of Syt-1 selectively in dopamine neurons. We generated these mice though intercrossing of conditional, floxed Syt-1 mice45 with DATIRES—Cre mice46 (Figure 1B) as described before.29,47 In the following experiments, conditional knockout (cKO) mice for Syt-1 (Syt-1 cKODA) are mice with two floxed alleles for Syt-1 and a heterozygote DATIRES—Cre allele, and Syt-1 control mice are littermate or age-matched mice from the same crossings with two wild type alleles for Syt-1 and a heterozygote DATIRES—Cre allele. The basal electrical properties of dopamine neurons, including the tonic firing rate, series resistance, and capacitance, were not altered in brain slices of Syt-1 cKODA mice (Figure S1).
Figure 1. Somatodendritic dopamine release is strongly decreased in Syt-1 cKODA mice.
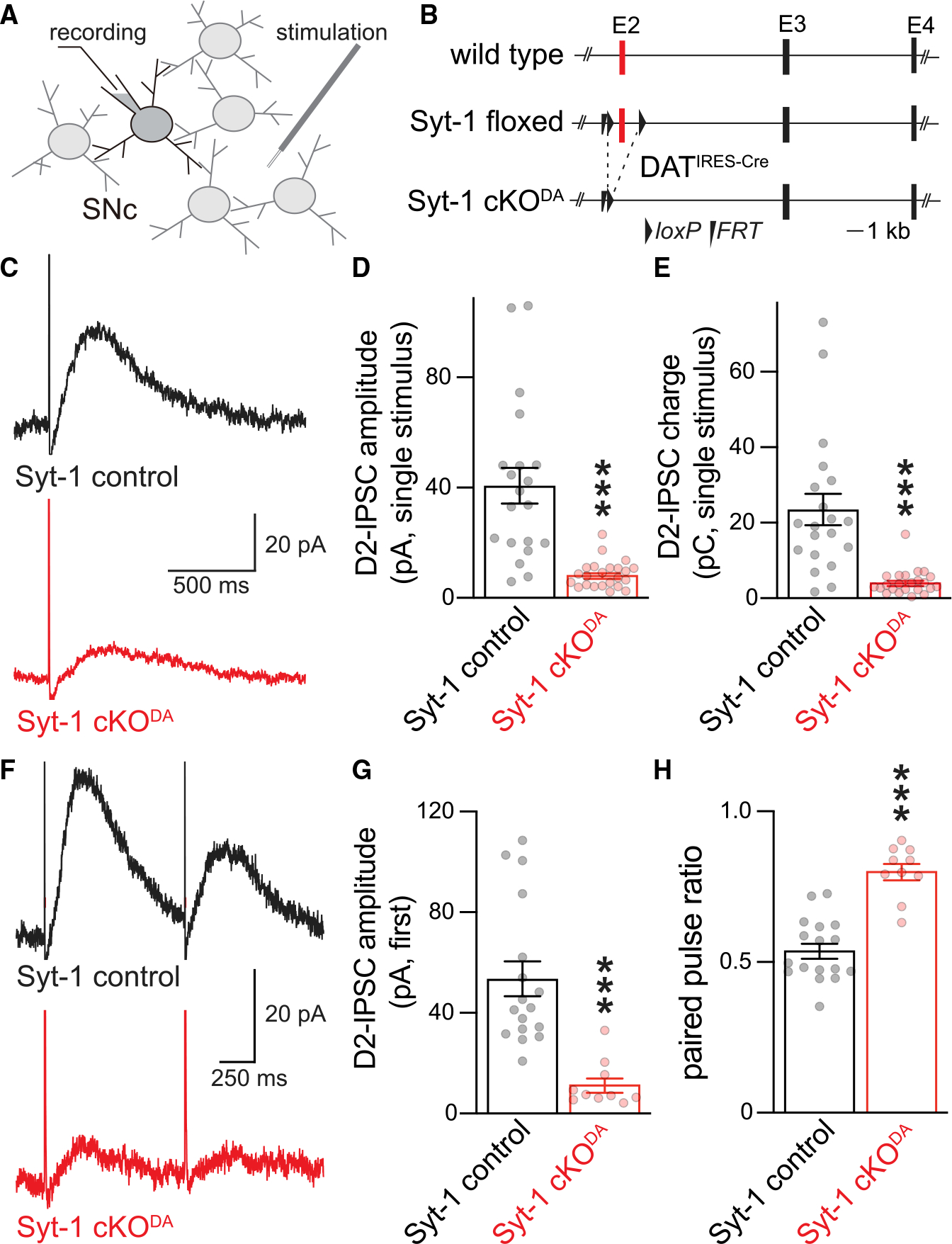
(A and B) Schematic of recording (A) in the substantia nigra pars compacta (SNc) and of Syt-1 cKODA mice (B) generated as described before.29 Exon numbers refer to coding exons.
(C–E) Example traces (C) and quantification of amplitude (D) and charge (E, area under the curve) of D2-IPSCs evoked by a single stimulus. Syt-1 control 20 cells/6 mice, and Syt-1 cKODA 23 cells/6 mice.
(F–H) Example traces of D2-IPSCs evoked by two electrical stimuli with a 1 s interval (F), and quantification of the first peak amplitude (G) and ratio of the amplitude of the second to the first D2-IPSC (H, paired pulse ratio). Syt-1 control 17 cells/7 mice, and Syt-1 cKODA 10 cells/4 mice. Data are mean ± SEM; ***p < 0.005 as determined by Mann Whitney rank-sum tests (D, E, and G) or an unpaired Student’s t test (H). For basal electrical properties, see Figure S1; for D2-IPSC rise times, see Figure S2.
We determined if evoked somatodendritic dopamine release relies on Syt-1 (Figures 1 and S2). Release was monitored via D2-IPSCs, and we focused on the substantia nigra pars compacta (SNc) because axon collaterals are likely absent in the SNc, different from the ventral tegmental area (VTA).48–50 Single electrical stimuli were applied through a stimulation electrode that was positioned ~50–100 μm away from a dopamine neuron recorded in whole-cell voltage clamp and held at −55 mV (Figure 1A). The average D2-IPSC amplitude and charge transfer evoked with a single stimulus were decreased by ~80% in Syt-1 cKODA mice, establishing that evoked somatodendritic dopamine release relies on Syt-1 (Figures 1C–1E). We next quantified the ratio of D2-IPSCs in response to two consecutive stimuli (paired pulse ratios) at 1 Hz (Figures 1F–1H). Previous work has shown that with paired stimuli, the second D2-IPSC is strongly depressed, similar to axonal dopamine release measured with different approaches.27,51,52 In slices of Syt-1 control mice, the ratio was 0.5 and was increased to 0.8 in Syt-1 cKODA mice (Figures 1G and 1H). In synaptic physiology, paired pulse ratios are often used to estimate vesicular release probability, and they are inversely correlated with it.53 Following this model, somatodendritic dopamine release has a high vesicular release probability that is mediated by the presence of Syt-1. Altogether, these experiments establish that evoked somatodendritic dopamine release is strongly decreased after Syt-1 ablation and suggest that Syt-1 is an important Ca2+ sensor for it.
Syt-1 re-expression restores somatodendritic dopamine release in Syt-1 cKODA mice
It is possible that Syt-1 is needed early in dopamine neuron development for maturation of the somatodendritic release machinery but that it does not directly act as a Ca2+ sensor for secretion. We sought to address this possibility by restoring somatodendritic release through re-expression of Syt-1 in adult Syt-1 cKODA mice. We constructed an adeno-associated virus (AAV) for Cre-dependent co-expression of Syt-1 and the fast channelrhodopsin variant oChIEF-citrine54 using a 2A-based strategy (Figure 2A). The virus was produced as serotype 9 (AAV9) and was injected into the midbrain of Syt-1 cKODA mice to generate Syt-1 cKODA + Syt-1 mice. Syt-1 control mice were injected with a virus for Cre-dependent oChIEF-citrine expression (Figure 2A).
Figure 2. Syt-1 re-expression restores somatodendritic dopamine release in Syt-1 cKODA mice.
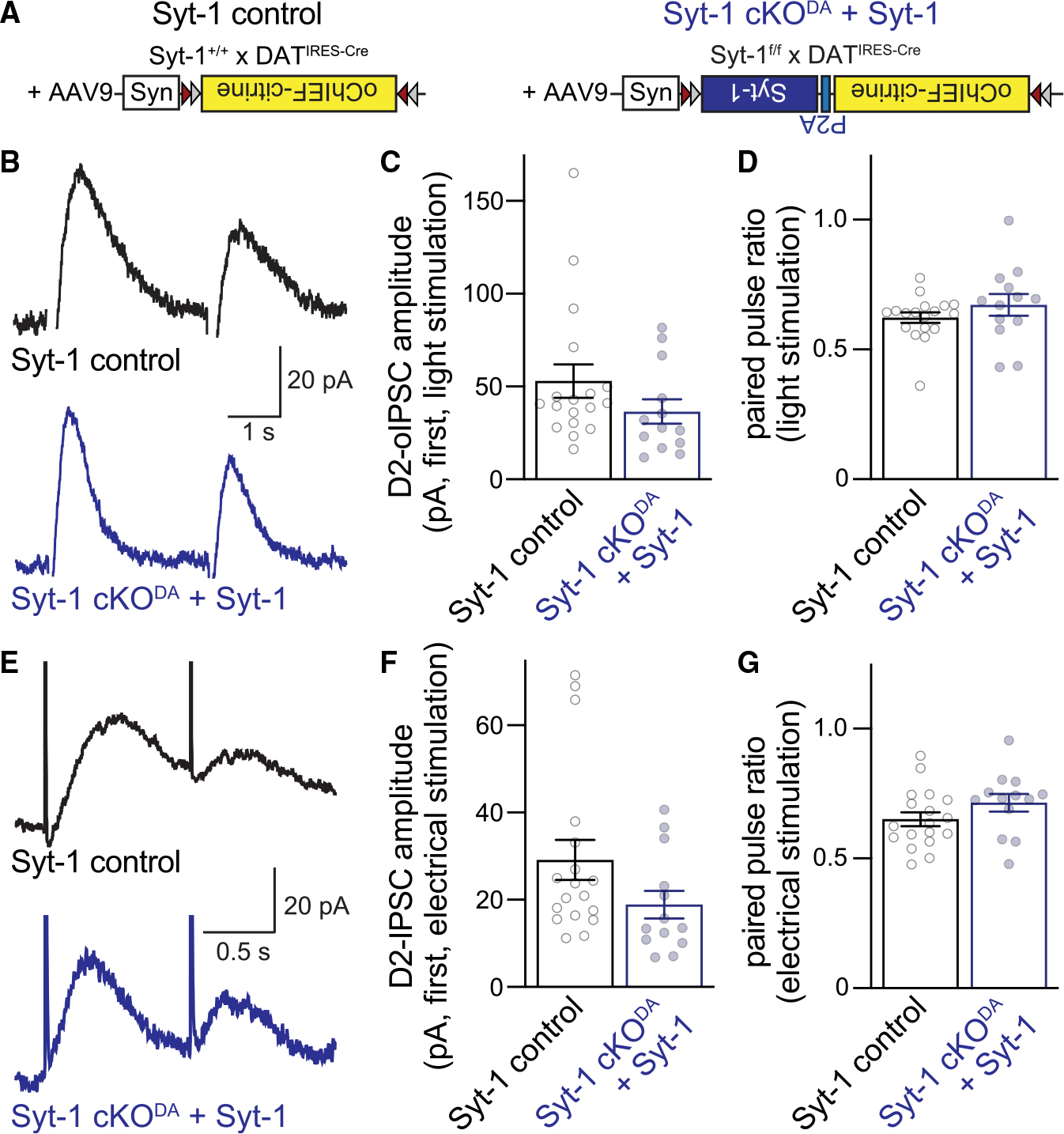
(A) Strategy of rescue experiments.
(B–D) Example traces (B) and quantification of amplitude (C) and paired pulse ratio (D) of optogenetically induced D2-oIPSCs (two stimuli at 0.4 Hz) in control mice expressing oChIEF-citrine (Syt-1 control) and Syt-1 cKODA mice co-expressing oChIEF-citrine and Syt-1 (Syt-1 cKODA + Syt-1). Syt-1 control 18 cells/6 mice, and Syt-1 cKODA + Syt-1 13 cells/5 mice.
(E–G) As in (B)–(D) but for pairs of electrical stimuli (at 1 Hz) in the same cells; n as in (B)–(D).
Data are mean ± SEM; no significant differences were detected with Mann Whitney rank-sum tests (C, D, and F) or an unpaired Student’s t test (G).
After 20 to 30 days of expression, we performed slice recordings in SNc areas with strong oChIEF-citrine fluorescence. To verify that the recorded neuron received inputs from dopamine neurons expressing the virus, two pulses of blue light were applied 2.5 s apart, and the optically induced D2 receptor-mediated IPSCs (D2-oIPSCs) were quantified (Figures 2B–2D). D2-oIPSCs were readily detected in both conditions, and the paired pulse ratio was indistinguishable. Hence, Syt-1 re-expression might restore somatodendritic dopamine release in Syt-1 cKODA mice. To test this, we measured electrically evoked D2-IPSCs (Figures 2E–2G) from the same cells used to record D2-oIPSCs (Figures 2B–2D). Robust D2-IPSCs were present in both conditions, and the paired pulse ratio was indistinguishable between Syt-1 control and Syt-1 cKODA + Syt-1 mice. In these analyses, we only included neurons with a D2-oIPSC equal to or greater than the D2-IPSC generated by a single electrical stimulus to select for cells with robust viral expression in the inputs. Altogether, the data indicate that Syt-1 re-expression restores most release in Syt-1 cKODA mice and support that Syt-1 operates as a Ca2+ sensor for somatodendritic dopamine secretion.
High-frequency trains induce robust somatodendritic release in Syt-1 cKODA mice
At conventional synapses, stimulus trains induce asynchronous glutamate or GABA release after Syt-1 knockout, and this asynchronous release is often sufficient to maintain synaptic charge transfer.55,56 A release component that is Syt-1 independent was also detected for axonal dopamine release after Syt-1 ablation, but its detection with amperometry in striatal slices required blocking dopamine reuptake or strong depolarization via KCl.29 In vivo microdialysis, a method that samples over long time periods and large striatal space, revealed robust in vivo dopamine levels in Syt-1 cKODA mice.29 These observations might suggest that asynchronous release is sufficient to maintain dopamine secretion in response to repetitive stimulation after removing the fast Ca2+ sensor.
We probed whether repetitive stimulation induced somatodendritic dopamine release in the absence of Syt-1. First, we evoked D2-IPSCs with 5 pulses at 40 Hz in Syt-1 cKODA and control mice (Figures 3A–3D). These stimuli induced robust D2-IPSCs in Syt-1 cKODA slices, revealing that short trains restore a substantial amount of dopamine release. When we compared the relative increase of the 5-pulse D2-IPSC (Figure 3B) with the single-pulse D2-IPSC (Figure 1D, recorded from the same cells), we found a significantly greater increase in Syt-1 cKODA mice (Figure 3D). In Syt-1 control neurons, the 5-pulse D2-IPSC was ~3-fold greater than the single-pulse D2-IPSC, consistent with the depression observed in response to paired stimuli (Figure 1G). In Syt-1 cKODA neurons, the 5-pulse D2-IPSC was increased ~9-fold over the single-pulse D2-IPSC, establishing a strong enhancement. These data indicate that repetitive stimulation, likely via buildup of residual Ca2+, triggers release through engaging additional Ca2+ sensors, at least in the Syt-1 cKODA mice.
Figure 3. Somatodendritic dopamine release in response to stimulus trains.
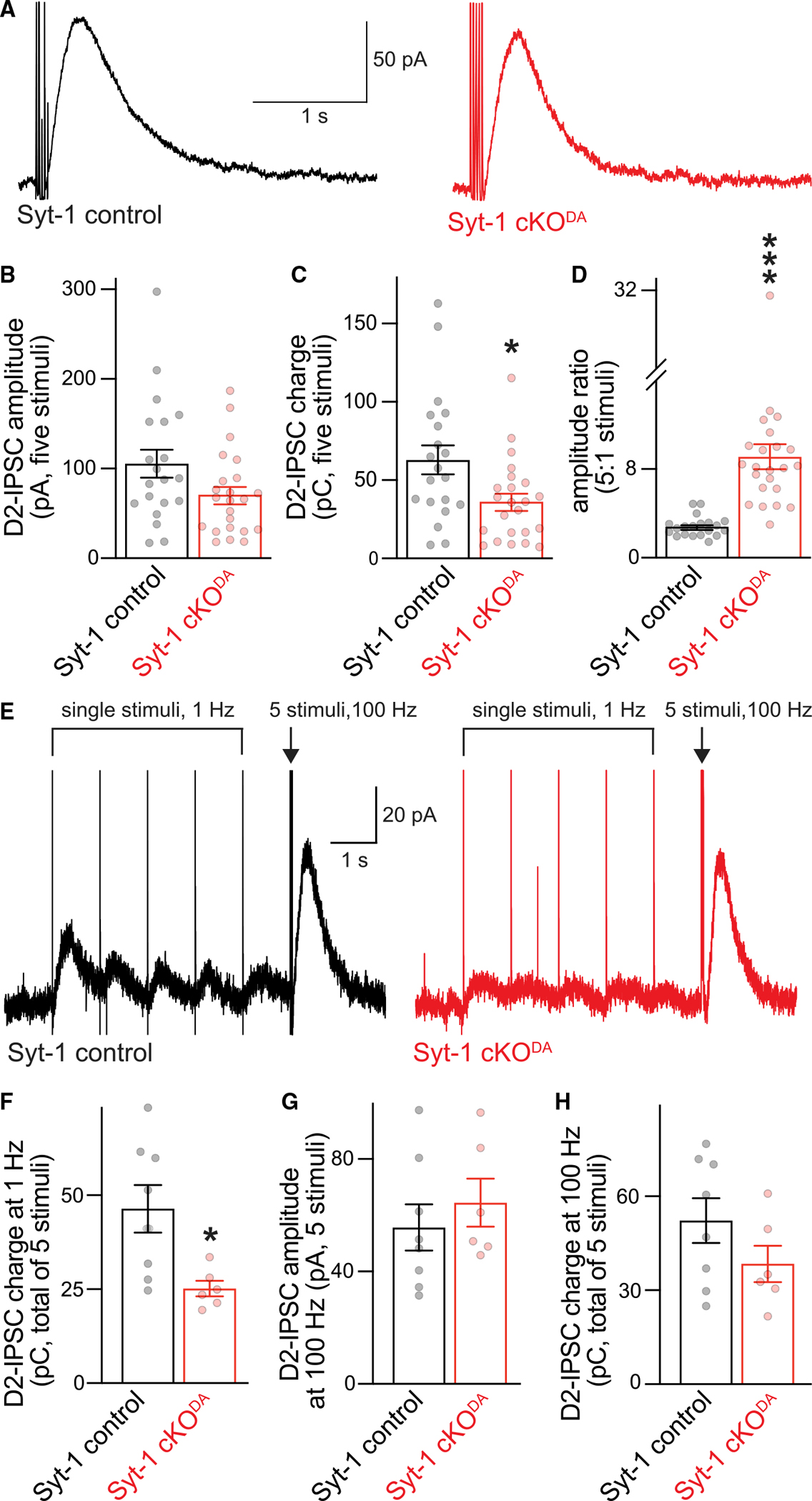
(A–D) Example traces (A) and quantification of amplitude (B) and charge (C, area under the curve) of D2-IPSCs evoked with a train of five electrical stimuli at 40 Hz, and ratio of the 5- to 1-stimulus D2-IPSC (D, single-stimulus amplitudes are from Figure 1C and were recorded in the same cells). Syt-1 control 20 cells/6 mice, and Syt-1 cKODA 23 cells/6 mice.
(E–H) Example traces of D2-IPSCs evoked by 5 stimuli at 1 Hz followed by a 5-stimulus train at 100 Hz (E), and quantification of the summed 5-stimulus D2-IPSC charge at 1 Hz (F), and the 5-stimulus amplitude (G) and charge (H) at 100 Hz. Syt-1 control 8 cells/3 mice, and Syt-1 cKODA 6 cells/2 mice.
Data are mean ± SEM; *p < 0.05, ***p < 0.005 as determined by Mann Whitney rank-sum tests (B–D) or unpaired Student’s t tests (F–H).
The D2-IPSC can be depressed with 1-Hz stimulation, and it builds up rapidly when the stimulation frequency is switched to 100 Hz following this depression.51 We investigated this by comparing Syt-1 cKODA with Syt-1 control mice (Figures 3E–3H). The overall charge during a 5-stimulus 1-Hz train was moderately decreased in Syt-1 cKODA slices compared with controls (Figure 3F). The D2-IPSCs evoked by 100-Hz 5-stimulus trains that followed the fifth stimulus at 1 Hz were comparable between Syt-1 cKODA and control slices (Figures 3G and 3H), similar to the 5-stimulus D2-IPSC at 40 Hz (Figures 3A–3D). These data support that a release pathway that is less dependent on Syt-1 is activated at high stimulus frequencies. Another fast, synchronous Ca2+ sensor is unlikely to mediate these D2-IPSCs because this sensor would be activated by single stimuli, which is largely excluded by the strong impairment of single-pulse D2-IPSCs (Figure 1). Instead, a slower, higher-affinity sensor is likely at play, possibly Syt-7 via mediating asynchronous release.31
Spontaneous somatodendritic dopamine release is Syt-1 independent
Midbrain dopamine neurons have spontaneous somatodendritic release that persists in the absence of action potential firing.42,57 This is similar to conventional synapses, where miniature release events occur, are suppressed by the fast Ca2+ sensor Syt-1, and may be regulated by alternate Ca2+ sensors.15,20–24 The exocytotic pathway for spontaneous somatodendritic dopamine release has not been well characterized, but it is vesicular and occurs at fusion sites that do not require the release site scaffold RIM.42,57
With the goal to improve detection of spontaneous D2-IPSCs, we overexpressed D2 receptors in the midbrain with AAVs (Figure 4A). We first tested whether evoked release is altered by D2 receptor overexpression (Figure S3). In both control and Syt-1 cKODA neurons, evoked D2-IPSCs and their paired pulse ratios were similar to the ones without overexpression (Figures 1, S3). D2 receptor overexpression greatly facilitated the detection of spontaneous events compared with previously published studies (Figure 4B).42,57 We measured spontaneous D2-IPSCs at baseline, in low extracellular Ca2+, and after blockade of action potential firing. In Syt-1 cKODA neurons, the frequencies and amplitudes of spontaneous D2-IPSCs were similar to controls (Figures 4B–4D). In both genotypes, reducing extracellular Ca2+ from 2.4 to 0.5 mM did not decrease event frequency or amplitude, nor did blockade of sodium channels with 1 μM TTX. We conclude that spontaneous somatodendritic dopamine release is independent of Syt-1. This is surprising because Syt-1 removal typically increases spontaneous event frequency at regular synapses.15,22,24,58 These results underscore that evoked and spontaneous somatodendritic fusion events may not use the same release machinery42 and bolster the model that distinct secretory pathways mediate evoked and spontaneous somatodendritic dopamine release.
Figure 4. Spontaneous somatodendritic dopamine release is not detectably impaired by Syt-1 cKODA.
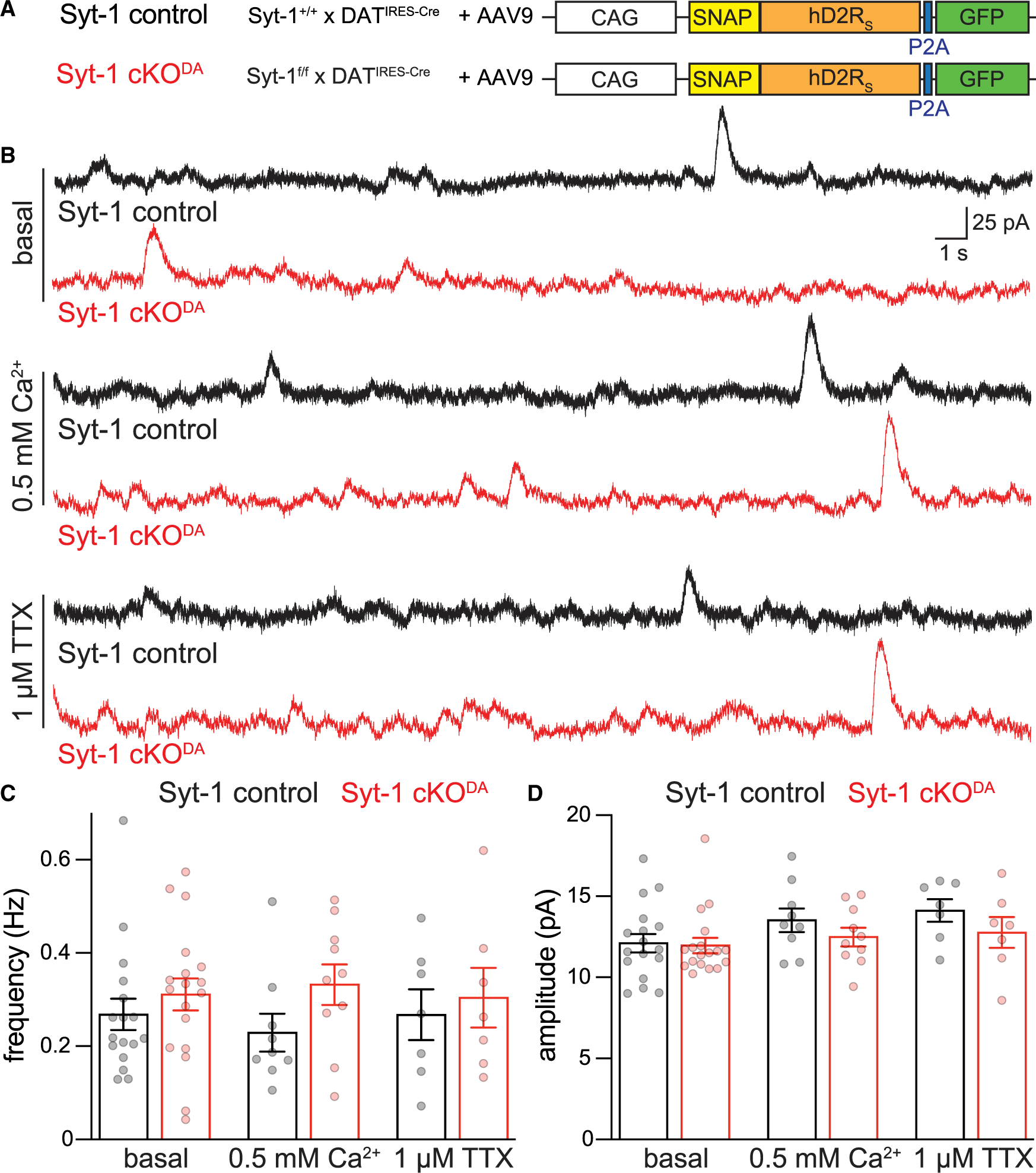
(A) Strategy for AAV-mediated overexpression of D2 receptors (human, short version [hD2RS]) in the midbrain.
(B–D) Example traces (B) and quantification of frequency (C) and amplitude (D) of spontaneous D2-IPSCs recorded in brain slices after viral expression of D2 receptors in basal recording conditions, after reducing extracellular Ca2+ from 2.4 to 0.5 mM, or following blockade of sodium channels with 1 μM TTX. Basal: Syt-1 control 17 cells/7 mice, and Syt-1 cKODA 18 cells/5 mice; 0.5 mM Ca2+: Syt-1 control 9 cells/4 mice, and Syt-1 cKODA 10 cells/5 mice; 1 μM TTX: Syt-1 control 7 cells/6 mice, and Syt-1 cKODA 7 cells/4 mice.
Data are mean ± SEM; no significant differences were detected with two-way ANOVA. For evoked D2-IPSCs after D2 receptor overexpression, see Figure S3.
DISCUSSION
We assessed Ca2+-sensing mechanisms for the triggering of somatodendritic dopamine release and found that the fast Ca2+ sensor Syt-1 is important. Genetic ablation of Syt-1 in dopamine neurons strongly decreased D2-IPSCs evoked by a single stimulus, and viral re-expression of Syt-1 restored D2-IPSCs. D2-mediated transmission depresses during repetitive stimulation, supporting the model of a high initial release probability. After knockout of Syt-1, release during high-frequency stimulus trains was nearly normal, suggesting that Syt-1-independent release mechanisms respond to Ca2+ buildup. Spontaneous D2-IPSCs were unaffected by ablation of Syt-1, adding to a growing body of evidence indicating that spontaneous and evoked dopamine release are mediated by separate exocytotic pathways.
Comparison with previous work on somatodendritic and axonal release
Our work relates to earlier and recent studies on somatodendritic dopamine release. Based on the distinct Ca2+ dependence of somatodendritic and axonal dopamine release, and relying on knockdown experiments in cultured dopamine neurons, it was proposed that Syt-4 and Syt-7 are involved.32,59 Indeed, constitutive double knockout of Syt-4 and Syt-7, but not knockout of Syt-7 alone, impaired somatodendritic dopamine release measured by voltammetry in brain slices.30 It is noteworthy that vertebrate Syt-4 is unlikely a Ca2+ sensor as it lacks key Ca2+-binding sites.60,61 Two recent studies have confirmed roles for Syt-7 in somatodendritic release. One study found that the strong depression of D2-IPSCs during stimulus trains was alleviated by constitutive knockout of Syt-7.62 The other31 used Syt-7 antibodies or constitutive Syt-7 knockout to assess D2-IPSCs that may arise from autocrine signaling in which a dopamine neuron senses its own released dopamine.63 Both manipulations impaired this form of transmission during short stimulus trains. Furthermore, Syt-1 antibodies applied through the recording pipette impaired this autocrine transmission in response to a single stimulus, but it remained uncertain whether the antibody blocked Syt-1 function and whether Syt-1 controls the more commonly detected non-autocrine D2-IPSCs that we assessed here. Finally, Syt-1 knockout mice were also studied with voltammetry using either optogenetic or electrical stimulation, and mixed effects were observed with uncertain contributions of Syt-1 to somatodendritic release depending on the stimulation method.64 Altogether, these recent studies support roles for Syt-7 in somatodendritic release. However, they did not allow for a decisive identification of the necessity and identity of the synchronous Ca2+ sensor for somatodendritic release.
The effect of Syt-1 deletion on evoked somatodendritic dopamine release (Figures 1, 2, and 3) resembles release deficits seen at classical synapses. At hippocampal synapses, Syt-1 ablation strongly impairs synchronous release, but the charge transfer during stimulus trains is maintained, and the resulting transmission is sufficient to support at least some aspects of behavior, for example memory acquisition.55,56 The reliance on a fast Ca2+ sensor for midbrain dopamine transmission is counterintuitive since the speed of information transfer of this transmission mode is limited by G protein signaling, making it much slower than fast synaptic transmission.9,65,66 A possible explanation is that synchronous dopamine release is needed to establish the high concentration of dopamine, ~100 μM, that is necessary for D2 receptor activation.34,67
Distinct release pathways for multiple modes of somatodendritic dopamine transmission
Our work defines the Ca2+-triggering mechanism for synchronous somatodendritic dopamine release in response to the initial stimulus. The data also show that during stimulus trains, dopamine release recovers in the absence of Syt-1. Based on these findings, and building on previous work, we propose that somatodendritic dopamine release occurs through three exocytotic pathways. The first pathway is fast and synchronous and relies on the Ca2+ sensor Syt-1 (Figures 1 and 2) and the release site organizer RIM.42 The second pathway is important during repetitive activity, for example burst firing, and it does not depend on Syt-1 (Figure 3). Secretion of this second pathway occurs at release sites organized by RIM, as RIM ablation abolishes it.42 Some studies suggest that Syt-7 is involved.30–32,62 This transmission mode is likely mediated by asynchronous release in response to build up of Ca2+ during repetitive activity, consistent with roles of Syt-7, a Ca2+ sensor for asynchronous release at synapses.16,17,20 Finally, previous work42,57 and our data (Figure 4) uncover the presence of a spontaneous dopamine release pathway that is independent of action potential firing and is not sensitive to lowering extracellular Ca2+ or to removing Syt-1 or RIM.
Together, these studies reveal the distinct involvement of specific proteins, Syt-1, Syt-7, and RIM, in synchronous, asynchronous, and spontaneous somatodendritic dopamine release. It is interesting to assess how these distinct molecular requirements could reflect distinct cell biological pathways for secretion. While Syt-1 is a key component of synaptic vesicles,68,69 Syt-7 is not localized on synaptic vesicles but instead at the target membrane or on different intracellular membrane compartments.70,71 The identity of the intracellular compartment for somatodendritic dopamine secretion remains uncertain. Early studies established that somatodendritic dopamine release depends on SNARE proteins25,72 and on Ca2+,10,32,33,59,73,74 indicating that exocytosis underlies this form of release. Classical work further suggested that tubulo-vesicular structures, rather than small synaptic vesicles, may be involved.75–77 Together, these studies and our work raise the possibility that multiple membrane compartments mediate somatodendritic dopamine release: small synaptic vesicles with Syt-1 may account for the initial synchronous component, while release in response to high-frequency trains, and possibly spontaneous release, may be supported by tubulo-vesicles or other compartments. This model may also explain why the asynchronous component is difficult to detect in experiments assessing axonal release.29 It is possible that axonal release is dominated by small clear vesicles, which densely occupy the dopamine axon.78–80
Limitations of the study
Our work reveals important roles for Syt-1 in somatodendritic dopamine release. One limitation to the interpretation of our work is that morphological characterization of the underlying signaling structure remains difficult. Release site architecture and release-receptor assemblies have not been visualized, the intracellular membrane compartments that undergo exocytosis remain uncertain, and it was not possible to determine whether exogenously expressed proteins, for example D2 receptors or Syt-1, localize like the endogenous counterparts. In the VTA, but less likely in the SNc, axon collaterals might be present as well. Hence, models on the molecular and cellular organization of this release mode can currently only be inferred from functional experiments like the one presented here, characterizing transmission deficits after gene knockout. Localizing Syt-1 and other proteins for somatodendritic transmission remains a future goal. In addition, the D2-IPSC measurements used here differ from axonal release measurements with electrochemical detection and from measurements of synaptic transmission monitored via ionotropic receptors. It is difficult to directly compare effect magnitudes across methods, and it remains uncertain whether classical concepts of synaptic transmission, for example on the inverse relationship of paired pulse ratios and vesicular release probability, fully apply. Nevertheless, our experiments establish that Syt-1 is critical for somatodendritic dopamine release evoked by single action potentials.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Pascal S. Kaeser (kaeser@hms.harvard.edu).
Materials availability
Plasmids generated for this study will be shared within limits of respective material transfer agreements. AAVs are exhaustible and will be shared as long as they are available and within limits of respective material transfer agreements. Mouse mutant alleles are publicly available as outlined in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| (+)-MK 801 maleate | Hello Bio | Cat# HB0004 |
| NBQX | Hello Bio | Cat# HB0442 |
| CGP55845 hydrochloride | Hello Bio | Cat# HB0960 |
| Picrotoxin | Hello Bio | Cat# HB0506 |
| Tetrodotoxin citrate | Tocris | Ca# 1069 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T cells | ATCC | Cat#: CRL-3216 RRID: CVCL_0063 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Syt-1 floxed; generated from C57BL/6Ntac-Syt1tm1a(EUCOMM)Wtsi/WtsiCnrm | Zhou et al.45 | EMMA ID EM:06829, RRID:IMSR_EM:06829; the identifier refers to the line before flp recombination |
| Mouse: B6.SJL-Slc6a3tm1.1(cre)Bkmm/J; DATIRES–Cre | Backman et al.46 | JAX 006660, RRID: IMSR_JAX:006660 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pAAV-hSyn-FLEX-oChIEF-citrine-P2A-Syt-1 | This paper; Kaeser lab, plasmid was used to generate AAVs. | lab plasmid code (LPC): p869 |
| pAAV-hSyn-FLEX-oChIEF-citrine | Addgene, Lin et al.,54 plasmid was used to generate AAVs. | Plasmid# 50973, RRID:Addgene 50973; LPC: p901 |
| pFB-CAG-SNAP-hD2Rshort-P2A-GFP | This paper; ViroVek, plasmid was used to generate AAVs. | LPC: p1038 |
|
| ||
| Software and algorithms | ||
|
| ||
| AxoGraph | AxoGraph | RRID:SCR_014284; https://www.axograph.com |
| LabChart | AD Instruments | https://www.adinstruments.com/products/labchart |
| Prism 9 | GraphPad | RRID: SCR_002798; https://www.graphpad.com |
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Conditional knockout mice for Syt-1 in dopamine neurons were generated through mouse breeding as described before,29 crossing Syt-1floxed mice45 with DATIRES—Cre mice,46 and striatal phenotypes were described.29,47 Syt-1floxed mice were generated from a gene targeting experiment (C57BL/6Ntac-Syt1tm1a(EUCOMM)Wtsi/WtsiCnrm) for coding exon 2 (if non-coding exons are included in the numbering, the corresponding exon number is exon 6),45,81 and further characterized in.82–84 In all experiments, Syt-1 cKODA mice are mice homozygote for Syt-1 floxed (Syt-1f/f) alleles and with a heterozygote allele for DATIRES—Cre; Syt-1 control mice are littermate mice or age-matched mice from the same breedings with homozygote Syt-1 wild type alleles (Syt-1+/+) and a heterozygote allele for DATIRES—Cre. Male and female adult mice (25–45 weeks old) were used for all experiments. Genotype comparisons were performed by an experimentalist blind to genotype, and unblinding of genotypes was done after experiments and analyses were completed. All animal experiments were performed according to protocols approved by the respective Animal Care and Use Committee at Harvard University and at Oregon Health and Science University.
Cell lines
HEK293T cells were purchased from ATCC (CRL-3216, RRID: CVCL_0063, an immortalized, authenticated cell line of female origin). The cells were expanded and stored as stock vials in liquid nitrogen. They were thawed for use and were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (Atlas Biologicals F-0500-D) and 1% Penicillin-Streptomycin. The cells were grown in a tissue culture incubator set to 37°C and 5% CO2, and they were passaged every 2 to 3 days at a ratio of 1:5 to 1:7. Each batch of cells was replaced after ~20 passages with cells thawed from a fresh stock vial.
METHOD DETAILS
Molecular biology and production of AAV viruses
Adeno-associated viruses (AAVs) were either generated in the lab or by ViroVek (Hayward, CA). All AAVs were of the AAV9 serotype and genomic titers ranged from 1.43 × 1012 to 2.15 × 1013 viral genome copies/mL. Cre-dependent AAVs were injected in the substantia nigra for expression of oChIEF-citrine with or without Syt-1. For expression oChIEF-citrine alone, pAAV-hSyn-FLEX-oChIEF-citrine54 (RRID:Addgene_50973, LPC: p901) was used; the following protein is encoded (numbered in subscript according to amino acid sequences of GenBank KF437396.1 for oChIEF and GenBank MK301208.1 for citrine): oChIEF,1MVSR … YESS359-LE-citrine,1MVSKG … ELYK239. For co-expression of oChIEF and synaptotagmin-1, we constructed an AAV vector, pAAV-hSyn-FLEX-oChIEF-citrine-P2A-Syt-1 (LPC: p869) producing citrine-tagged oChIEF and rat synaptotagmin-1. The following two proteins are encoded (numbered in subscript according to amino acid sequences of GenBank KF437396.1 for oChIEF, GenBank MK301208.1 for citrine, and NCBI Ref Seq NM_001033680.2 for Syt-1): oChIEF,1MVSR … YESS359-LEG-citrine,2VSKG … ELYK239-GAPGSGATNFSLLKQAGDVEENPG and PAPL-Syt-1,2VSAS … AVKK421. AAVs were generated in HEK293T cells in the lab using calcium phosphate transfection. Cells were collected and lysed 72 h after transfection. AAV viral particles were extracted and purified from the 40% layer after iodixanol gradient ultracentrifugation. An AAV encoding the short version of the human D2 receptor (hD2RS) was generated by ViroVek (Hayward, CA). A plasmid encoding an SNAP-tagged (at the N-terminus) hD2RS and a cytosolic GFP (via a P2A sequence) was generated and provided to ViroVek. This plasmid was subcloned to generate pFB-CAG-SNAP-hD2Rshort-P2A-GFP (LPC: p1038). The following two proteins are encoded (numbered in subscript according to amino acid sequences of GenBank KM043782.1 for the SNAPtag, NCBI Ref Seq NM_016574.4 for hD2Rshort, and GenBank MF405194.1 for GFP): MKTIIALSYIFCLVFADYKDDDDA-SNAPtag,172MDKD … PGLG353-hD2Rshort,1MDPL … ILHC414-SGSGATNFSLLKQAGDVEENPG and P-GFP,1MVSK … ELYK239. After verification, the plasmid was used by ViroVek for AAV production.
Stereotaxic viral delivery
Mice (postnatal days 56–115) were anesthetized and placed in a stereotaxic frame for midbrain injections of AAV viruses. Anesthesia was maintained throughout surgery with isoflurane. 120 nL of AAV was injected bilaterally into the ventral midbrain at a rate of 0.1 μL/min using an UltraMicroPump3 microinjection syringe pump (World Precision Instruments, Sarasota FL) controlled by a Wizard 211 digital readout system (ANILAM Inc., Jamestown NY). Injection coordinates were (in mm): 2.3 posterior from bregma; ± 1.3 lateral from midline; 4.5 below pia. Post-operative analgesia was provided following approved protocols. Experiments with AAV-mediated D2 receptor expression were performed 10 to 20 days after surgery. Experiments with oChIEF-citrine expression (and for the rescue condition Syt-1 co-expression) were performed 20 to 30 days after surgery.
Brain slice preparation
Mice (25–45 weeks old) were anesthetized with isoflurane and decapitated, and the brain was collected in 32–35°C warm Krebs buffer containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 25 NaHCO3, and 11 Dextrose. The Krebs buffer used for extraction, cutting, and recovery contained 10 μM MK-801 to prevent NMDA-mediated excitotoxic damage. 222 μm thick horizontal slices containing the midbrain were cut using a vibrating microtome in warm Krebs buffer bubbled with 95% O2/5% CO2 and slices were allowed to recover in the same buffer at 30°C for ≥30 min before recordings. Prior to recordings, slices were hemisected along the midline to record from each side separately. For recording, the hemisected slice was transferred to the recording chamber, which was continuously perfused at 2 to 3 mL/min with bubbled Krebs buffer at 34°C. All experiments were completed within 7 h of slicing.
Electrophysiological recordings in brain slices
SNc dopamine neurons were identified by their morphology and by their localization lateral to the medial terminal nucleus of the accessory optic system. D2-IPSCs were recorded following previously established protocols.10,42,57 Specifically, experiments were conducted in Krebs buffer with the addition of NBQX (600 nM), CGP55845 (300 nM), and picrotoxin (100 μM). Recordings were made using glass pipettes (1.0–2.5 MΩ) filled with an internal solution containing (in mM): 100 K-methanesulphonate, 20 NaCl, 1.5 MgCl2, 10 HEPES (K), 2 ATP, 0.2 GTP, 10 phosphocreatine, and 10 BAPTA (4K). The recorded cells exhibited ~1–5 Hz spontaneous firing as assessed in cell-attached configuration, and firing rates were recorded and analyzed in a subset of experiments in cell attached mode with an amplitude threshold. After break-in, cells were voltage-clamped at −55 mV and resistance and capacitance were monitored; cells with a series resistance ≥12 MΩ were discarded. A focal monopolar or bipolar stimulation electrode was used to stimulate the cells with a current of ~50–250 μA once per min. The typical distance of the stimulation electrode to the recorded cell was 50–100 μm. For assessment of evoked release, the stimulation intensity was set such that a D2-IPSC was detected in response to five stimuli at 40 Hz, and only cells with a detectable 5-stimulus D2-IPSC were included. The number and frequency of electrical stimuli varied, and the stimulation protocol is described in each figure; if multiple stimulus protocols were used for a given cell, the order was randomized but stimulation intensity was kept identical across protocols. For optogenetic stimulation, a 470 nm LED (Thorlabs) was used to deliver paired flashes of blue light (2 mW, 25 ms, 0.4 Hz) through a 40× water-immersion objective directly over the recorded cell. As with electrical stimulation, paired light flashes were delivered once per minute. All data reported for stimulated D2-IPSCs reflect the average of three consecutive sweeps. Analyses of spontaneous D2-IPSCs were constrained to the intervals between stimuli or after stimulus protocols were completed. These recordings were performed in the same solutions as described above for most cells; for a subset of cells, 1 μM TTX was added or the extracellular solution contained 0.5 mM CaCl2 and 5 mM MgCl2 instead of 2.4 CaCl2 and 1.2 MgCl2. Data acquisition was performed with AxoGraph (Berkley, CA) and recordings were monitored with LabChart (AD instruments, Colorado Springs, CO). D2-IPSC amplitudes were measured as the average of a 20 ms window centered around the peak outward current determined in AxoGraph. The charge transfer was quantified as the area under the curve starting at stimulation for 1.5 s, at which time the D2-IPSC returned to baseline. Paired pulse ratios were calculated as the ratio of the second D2-IPSC peak to the first. In rescue experiments, we used optogenetic activation to assess the D2-oIPSC for each recorded cell. We then used electrical stimulation to assess rescue and only included cells for which the D2-oIPSC amplitude was equal to or greater than that of the D2-IPSC generated by a single electrical stimulus to select for cells with robust viral expression in the inputs. Spontaneous D2-IPSCs were identified using a sliding template in AxoGraph as described previously.42,57 The template was based on the shape of the D2-IPSC and used the following parameters: a 50 ms baseline, a rise time (10–90%) of 50 ms, and a decay t of 200 ms. The threshold for event detection was 3.7 times the standard deviation of baseline noise for events that fit the waveform of the sliding template, and events had to be separated by a minimum of 300 ms. For all experiments examining spontaneous D2-IPSCs and several experiments examining evoked D2-IPSCs, sulpiride was added at the end of the recording period to inhibit D2 receptors, which led to a block of spontaneous and evoked D2-IPSCs, respectively, as established before.10,57 Data acquisition and analyses were conducted by experimenters blind to genotype.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are displayed as mean ± SEM with individual values as circles, and statistical significance is denoted as *p < 0.05, **p < 0.01, and ***p < 0.005 in the figures. Representative traces except for those of spontaneous D2-IPSCs are the average of three consecutive sweeps. Statistical testing was performed in GraphPad Prism 9 (GraphPad, San Diego, CA). For statistical analyses, each cell was considered an observation because the technical nature of these recordings precluded getting sufficient numbers of cells per mouse for averaging per mouse. Datasets were tested for normality with a Shapiro-Wilk test. For comparison of two groups with a single variable, normally distributed data were assessed with unpaired Student’s t-tests, and non-normally distributed data with Mann-Whitney rank sum tests. For spontaneous D2-IPSC analyses, a two-way ANOVA was used. Sample sizes, statistical tests used, and significance are reported for all data in the corresponding figures and figure legends.
Supplementary Material
Highlights.
Somatodendritic dopamine release is assessed in Syt-1 knockout mice via D2-IPSCs
Single action potential-evoked D2-IPSCs strongly depend on the fast Ca2+ sensor Syt-1
High-frequency trains induce similar D2-IPSCs in Syt-1 knockout and control mice
Spontaneous somatodendritic dopamine release is independent of Syt-1
ACKNOWLEDGMENTS
We thank all members of the Kaeser and Williams laboratories for insightful discussions and feedback. This work was supported in part by the NIH (R01NS103484 and R01NS083898, P.S.K., and R01DA004523 and T32DA 007262, J.T.W.), an Alice and Joseph Brooks fellowship (A.B.), and Harvard Medical School (P.S.K.).
Footnotes
DECLARATION OF INTERESTS
Claire Qiao is currently a graduate student at Peking University.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111915.
REFERENCES
- 1.Kennedy MJ, and Ehlers MD (2011). Mechanisms and function of dendritic exocytosis. Neuron 69, 856–875. 10.1016/j.neuron2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pang ZP, and Südhof TC (2010). Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 22, 496–505. 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacson JS, and Strowbridge BW (1998). Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron 20, 749–761. 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 4.Cheramy A, Leviel V, and Glowinski J (1981). Dendritic release of dopamine in the substantia nigra. Nature 289, 537–542. [DOI] [PubMed] [Google Scholar]
- 5.Son SJ, Filosa JA, Potapenko ES, Biancardi VC, Zheng H, Patel KP, Tobin VA, Ludwig M, and Stern JE (2013). Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron 78, 1036–1049. 10.1016/j.neuron.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ludwig M, Apps D, Menzies J, Patel JC, and Rice ME (2016). Dendritic release of neurotransmitters. In Comprehensive Physiology (John Wiley & Sons, Inc.), pp. 235–252. 10.1002/cphy.c160007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ford CP (2014). The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282, 13–22. 10.1016/j.neuroscience.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crocker AD (1997). The regulation of motor control: an evaluation of the role of dopamine receptors in the substantia nigra. Rev. Neurosci. 8, 55–76. 10.1515/REVNEURO.1997.8.1.55. [DOI] [PubMed] [Google Scholar]
- 9.Gantz SC, Ford CP, Morikawa H, and Williams JT (2018). The evolving understanding of dopamine neurons in the substantia nigra and ventral tegmental area. Annu. Rev. Physiol. 80, 219–241. 10.1146/annurev-physiol-021317-121615. [DOI] [PubMed] [Google Scholar]
- 10.Beckstead MJ, Grandy DK, Wickman K, and Williams JT (2004). Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42, 939–946. 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Mashimo T, and Südhof TC (2007). Synaptotagmin-1, −2, and −9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54, 567–581. 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC, et al. (2001). Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49. 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 13.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, and Südhof TC (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. [DOI] [PubMed] [Google Scholar]
- 14.Chapman ER, Hanson PI, An S, and Jahn R (1995). Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 270, 23667–23671. [DOI] [PubMed] [Google Scholar]
- 15.Broadie K, Bellen HJ, DiAntonio A, Littleton JT, and Schwarz TL (1994). Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc. Natl. Acad. Sci. USA 91, 10727–10731. 10.1073/pnas.91.22.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen H, Linhoff MW, McGinley MJ, Li G-L, Corson GM, Mandel G, and Brehm P (2010). Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc. Natl. Acad. Sci. USA 107, 13906–13911. 10.1073/pnas.1008598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, and Südhof TC (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC, and Sudhof TC (2007). A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682. 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao J, Gaffaney JD, Kwon SE, and Chapman ER (2011). Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147, 666–677. 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaeser PS, and Regehr WG (2014). Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 76, 333–363. 10.1146/annurev-physiol-021113-170338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong APH, Goriounova NA, Habets RLP, Takai Y, Borst JG, et al. (2010). Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327, 1614–1618. 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Pang ZP, Shin O-H, and Südhof TC (2009). Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat. Neurosci. 12, 759–766. 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, and Südhof TC (2011). Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron 70, 244–251. 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Littleton JT, Stern M, Schulze K, Perin M, and Bellen HJ (1993). Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 74, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 25.Bergquist F, Niazi HS, and Nissbrandt H (2002). Evidence for different exocytosis pathways in dendritic and terminal dopamine release in vivo. Brain Res. 950, 245–253. 10.1016/S0006-8993(02)03047-0. [DOI] [PubMed] [Google Scholar]
- 26.Rice ME, and Patel JC (2015). Somatodendritic dopamine release: recent mechanistic insights. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, Kershberg L, Wang J, Schneeberger S, and Kaeser PS (2018). Dopamine secretion is mediated by sparse active zone-like release sites. Cell 172, 706–718.e15. 10.1016/j.cell.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee A, Imig C, Balakrishnan K, Kershberg L, Lipstein N, Uronen R-L, Wang J, Cai X, Benseler F, Rhee JS, et al. (2022). Molecular and functional architecture of striatal dopamine release sites. Neuron 110, 248–265.e9. 10.1016/j.neuron.2021.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee A, Lee J, Nemcova P, Liu C, and Kaeser PS (2020). Synaptotagmin-1 is the Ca2+ sensor for fast striatal dopamine release. Elife 9, e58359. 10.7554/eLife.58359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delignat-Lavaud B, Ducrot C, Kouwenhoven W, Feller N, and Trudeau LÉ (2022). Implication of synaptotagmins 4 and 7 in activity-dependent somatodendritic dopamine release in the ventral midbrain. Open Biol. 12, 210339.. 10.1098/rsob.210339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hikima T, Witkovsky P, Khatri L, Chao MV, and Rice ME (2022). Synaptotagmins 1 and 7 play complementary roles in somatodendritic dopamine release. J. Neurosci. 42, 3919–3930. 10.1523/JNEUROSCI.2416-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendez JA, Bourque M-J, Fasano C, Kortleven C, and Trudeau L-E (2011). Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J. Biol. Chem. 286, 23928–23937. 10.1074/jbc.M111.218032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen BT, Patel JC, Moran KA, and Rice ME (2011). Differential calcium dependence of axonal versus somatodendritic dopamine release, with characteristics of both in the ventral tegmental area. Front. Syst. Neurosci. 5, 39. 10.3389/fnsys.2011.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ford CP, Phillips PEM, and Williams JT (2009). The time course of dopamine transmission in the ventral tegmental area. J. Neurosci. 29, 13344–13352. 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, and Südhof TC (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng L, Kaeser PS, Xu W, and Südhof TC (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller M, Liu KSY, Sigrist SJ, and Davis GW (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 32, 16574–16585. 10.1523/JNEURO-SCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Y, Babai N, Kaeser P, Südhof TC, and Schneggenburger R (2015). RIM1 and RIM2 redundantly determine Ca 2+ channel density and readily releasable pool size at a large hindbrain synapse. J. Neurophysiol. 113, 255–263. 10.1152/jn.00488.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gracheva EO, Hadwiger G, Nonet ML, and Richmond JE (2008). Direct interactions between C. elegans RAB-3 and Rim provide a mechanism to target vesicles to the presynaptic density. Neurosci. Lett. 444, 137–142. 10.1016/j.neulet.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han Y, Kaeser PS, Südhof TC, and Schneggenburger R (2011). RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69, 304–316. 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaeser PS, Deng L, Fan M, and Südhof TC (2012). RIM genes differentially contribute to organizing presynaptic release sites. Proc. Natl. Acad. Sci. USA 109, 11830–11835. 10.1073/pnas.1209318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson BG, Cai X, Wang J, Bunzow JR, Williams JT, and Kaeser PS (2019). RIM is essential for stimulated but not spontaneous somatodendritic dopamine release in the midbrain. Elife 8, e47972. 10.7554/eLife.47972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, et al. (2015). Architecture of the synaptotagmin–SNARE machinery for neuronal exocytosis. Nature 525, 62–67. 10.1038/nature14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- 47.Kershberg L, Banerjee A, Kaeser PS Protein composition of axonal dopamine release sites in the striatum. 2022. ELife. 10.1101/2022.08.31.505994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wassef M, Berod A, and Sotelo C (1981). Dopaminergic dendrites in the pars reticulata of the rat substantia nigra and their striatal input. Combined immunocytochemical localization of tyrosine hydroxylase and anterograde degeneration. Neuroscience 6, 2125–2139. 10.1016/0306-4522(81)90003-8. [DOI] [PubMed] [Google Scholar]
- 49.Bayer VE, and Pickel VM (1990). Ultrastructural localization of tyrosine hydroxylase in the rat ventral tegmental area: relationship between immunolabeling density and neuronal associations. J. Neurosci. 10, 2996–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deutch AY, Goldstein M, Baldino F, and Roth RH (1988). Telencephalic projections of the A8 dopamine cell group. Ann. N. Y. Acad. Sci. 537, 27–50. 10.1111/j.1749-6632.1988.tb42095.x. [DOI] [PubMed] [Google Scholar]
- 51.Beckstead MJ, Ford CP, Phillips PEM, and Williams JT (2007). Presynaptic regulation of dendrodendritic dopamine transmission. Eur. J. Neurosci. 26, 1479–1488. 10.1111/j.1460-9568.2007.05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong W-H, Folk RW, Broussard GJ, Liang R, Jang MJ, et al. (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360, eaat4422. 10.1126/science.aat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. 10.1146/annurev.physiol.64.092501.11454764/1/355. [DOI] [PubMed] [Google Scholar]
- 54.Lin JY, Lin MZ, Steinbach P, and Tsien RY (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J. 96, 1803–1814. 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maximov A, and Südhof TC (2005). Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48, 547–554. 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 56.Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, and Südhof TC (2012). Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 73, 990–1001. 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gantz SC, Bunzow JR, and Williams JT (2013). Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron 78, 807–812. 10.1016/j.neuron.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouazza-Arostegui B, Camacho M, Brockmann MM, Zobel S, and Rosenmund C (2022). Deconstructing synaptotagmin-1’s distinct roles in synaptic vesicle priming and neurotransmitter release. J. Neurosci. 42, 2856–2871. 10.1523/JNEUROSCI.1945-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen BT, and Rice ME (2001). Novel Ca2+ dependence and time course of somatodendritic dopamine release: substantia nigra versus striatum. J. Neurosci. 21, 7841–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai H, Shin O-H, Machius M, Tomchick DR, Südhof TC, and Rizo J (2004). Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat. Struct. Mol. Biol. 11, 844–849. 10.1038/nsmb817. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, and Chapman ER (2010). Rat and Drosophila synaptotagmin 4 have opposite effects during SNARE-catalyzed membrane fusion. J. Biol. Chem. 285, 30759–30766. 10.1074/jbc.M110.137745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kissiwaa SA, Lebowitz JJ, Engeln KA, Bowman AM, Williams JT, and Jackman SL (2021). Synaptotagmin-7 enhances phasic dopamine release. Preprint at bioRxiv. 10.1101/2021.10.17.464710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hikima T, Lee CR, Witkovsky P, Chesler J, Ichtchenko K, and Rice ME (2021). Activity-dependent somatodendritic dopamine release in the substantia nigra autoinhibits the releasing neuron. Cell Rep. 35, 108951. 10.1016/j.celrep.2021.108951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delignat-Lavaud B, Kano J, Ducrot C, Massé I, Mukherjee S, Giguère N, Moquin L, Lévesque C, Nanni SB, Bourque M-J, et al. (2021). The calcium sensor synaptotagmin-1 is critical for phasic axonal dopamine release in the striatum and mesencephalon, but is dispensable for basic motor behaviors in mice. Preprint at bioRxiv. 10.1101/2021.09.15.460511. [DOI] [Google Scholar]
- 65.Liu C, Goel P, and Kaeser PS (2021). Spatial and temporal scales of dopamine transmission. Nat. Rev. Neurosci. 22, 345–358. 10.1038/s41583-021-00455-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biederer T, Kaeser PS, and Blanpied TA (2017). Transcellular nanoalignment of synaptic function. Neuron 96, 680–696. 10.1016/j.neuron.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Condon AF, Robinson BG, Asad N, Dore TM, Tian L, and Williams JT (2021). The residence of synaptically released dopamine on D2 autoreceptors. Cell Rep. 36, 109465. 10.1016/j.celrep.2021.109465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bixby JL, and Reichardt LF (1985). The expression and localization of synaptic vesicle antigens at neuromuscular junctions in vitro. J. Neurosci. 5, 3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. (2006). Molecular anatomy of a trafficking organelle. Cell 127, 831–846. 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 70.Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, Lao Y, and Südhof TC (2001). Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron 30, 459–473. 10.1016/s0896-6273(01)00290-2. [DOI] [PubMed] [Google Scholar]
- 71.Chakrabarti S, Kobayashi KS, Flavell R.a., Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, and Andrews NW (2003). Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J. Cell Biol. 162, 543–549. 10.1083/jcb200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fortin GD, Desrosiers CC, Yamaguchi N, and Trudeau LE (2006). Basal somatodendritic dopamine release requires snare proteins. J. Neurochem. 96, 1740–1749. 10.1111/j.1471-4159.2006.03699.x. [DOI] [PubMed] [Google Scholar]
- 73.Chen BT, Moran KA, Avshalumov MV, and Rice ME (2006). Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca channels contrasted with strong regulation of axonal dopamine release. J. Neurochem. 96, 645–655. 10.1111/j.1471-4159.2005.03519.x. [DOI] [PubMed] [Google Scholar]
- 74.Ford CP, Gantz SC, Phillips PEM, and Williams JT (2010). Control of extracellular dopamine at dendrite and axon terminals. J. Neurosci. 30, 6975–6983. 10.1523/JNEUROSCI.1020-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nirenberg MJ, Chan J, Liu Y, Edwards RH, and Pickel VM (1996). Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 16, 4135–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pickel VM, Chan J, and Nirenberg MJ (2002). Region-specific targeting of dopamine D2-receptors and somatodendritic vesicular monoamine transporter 2 (VMAT2) within ventral tegmental area subdivisions. Synapse 45, 113–124. 10.1002/syn.10092. [DOI] [PubMed] [Google Scholar]
- 77.Mercer L, del Fiacco M, and Cuello AC (1979). The smooth endoplasmic reticulum as a possible storage site for dendritic dopamine in substantia nigra neurones. Experientia 35, 101–103. 10.1007/BF01917903. [DOI] [PubMed] [Google Scholar]
- 78.Liu C, and Kaeser PS (2019). Mechanisms and regulation of dopamine release. Curr. Opin. Neurobiol. 57, 46–53. 10.1016/j.conb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uchigashima M, Ohtsuka T, Kobayashi K, and Watanabe M (2016). Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc. Natl. Acad. Sci. USA 113, 4206–4211. 10.1073/pnas.1514074113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wildenberg G, Sorokina A, Koranda J, Monical A, Heer C, Sheffield M, Zhuang X, McGehee D, and Kasthuri B (2021). Partial connectomes of labeled dopaminergic circuits reveal non-synaptic communication and axonal remodeling after exposure to cocaine. Elife 10, e71981. 10.7554/eLife.71981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Südhof TC, and Brunger AT (2017). The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548, 420–425. 10.1038/nature23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kochubey O, Babai N, and Schneggenburger R (2016). A synaptotagmin isoform switch during the development of an identified CNS synapse. Neuron 90, 984–999. 10.1016/j.neuron.2016.04.038. [DOI] [PubMed] [Google Scholar]
- 84.Bouhours B, Gjoni E, Kochubey O, and Schneggenburger R (2017). Synaptotagmin2 (Syt2) drives fast release redundantly with Syt1 at the output synapses of parvalbumin-expressing inhibitory neurons. J. Neurosci. 37, 4604–4617. 10.1523/JNEUROSCI.3736-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.