

Abstract
Tyrosine phosphorylation of CAS (Crk-associated substrate, p130Cas) has been implicated as a key signaling step in integrin control of normal cellular behaviors, including motility, proliferation, and survival. Aberrant CAS tyrosine phosphorylation may contribute to cell transformation by certain oncoproteins, including v-Crk and v-Src, and to tumor growth and metastasis. The CAS substrate domain (SD) contains 15 Tyr-X-X-Pro motifs, which are thought to represent the major tyrosine phosphorylation sites and to function by recruiting downstream signaling effectors, including c-Crk and Nck. CAS makes multiple interactions, direct and indirect, with the tyrosine kinases Src and focal adhesion kinase (FAK), and as a result of this complexity, several plausible models have been proposed for the mechanism of CAS-SD phosphorylation. The objective of this study was to provide experimental tests of these models in order to determine the most likely mechanism(s) of CAS-SD tyrosine phosphorylation by FAK and Src. In vitro kinase assays indicated that FAK has a very poor capacity to phosphorylate CAS-SD, relative to Src. However, FAK expression along with Src was found to be important for achieving high levels of CAS tyrosine phosphorylation in COS-7 cells, as well as recovery of CAS-associated Src activity toward the SD. Structure-functional studies for both FAK and CAS further indicated that FAK plays a major role in regulating CAS-SD phosphorylation by acting as a docking or scaffolding protein to recruit Src to phosphorylate CAS, while a secondary FAK-independent mechanism involves Src directly bound to the CAS Src-binding domain (SBD). Our results do not support models in which FAK either phosphorylates CAS-SD directly or phosphorylates CAS-SBD to promote Src binding to this site.
CAS (Crk-associated substate, p130Cas) was first recognized as a tyrosine-phosphorylated protein in cells transformed by v-Crk or v-Src (27, 38, 52) and later characterized as a docking protein containing multiple protein-protein interaction domains, including a Src-homology 3 (SH3) domain at the N terminus, a Src-binding domain (SBD) near the C terminus, and a large interior substrate domain (SD) (40, 47, 52). The CAS SH3 domain may function as a molecular switch regulating CAS tyrosine phosphorylation since it interacts with tyrosine kinases focal adhesion kinase (FAK) (22, 47, 48) and the FAK-related kinase PYK2 (also known as CAKβ, RAFTK, and CADTK) (3, 42) and also with tyrosine phosphatases PTP-1B (35) and PTP-PEST (18). The SBD represents a second site of CAS interaction with tyrosine kinases and consists of a proline-rich motif, RPLPSPP (amino acid residues 639 to 645 in mouse CAS), that can interact with the SH3 domains of Src-family kinases (SFKs) and a nearby tyrosine phosphorylation site (Tyr-668 and/or Tyr-670) that can promote an interaction with the Src-homology 2 (SH2) domain of SFKs (37, 40). CAS-SD, the major region of tyrosine phosphorylation, is characterized by 15 tyrosines present in Tyr-X-X-Pro (YXXP) motifs. When phosphorylated, most YXXP motifs conform to the binding consensus for the Crk SH2 domain (pYDxP) (13), and thus one or more of these sites likely mediates CAS interactions with v-Crk (10, 40) and its normal counterpart the SH2/SH3 adaptor c-Crk (29, 64). The SH2-mediated binding of Crk to CAS promotes subsequent signaling events through proteins associated with the Crk SH3 domain(s), including C3G and DOCK180, which stimulate guanine nucleotide exchange on Rap1 and Rac1, respectively (19, 23, 28, 31, 60). CAS tyrosine phosphorylation also promotes SH2-mediated interactions with the adaptor Nck (57) and the SH2-containing inositol 5′-phosphatase 2 (SHIP2) (49), which may also act as downstream effectors in CAS signaling.
Functionally, CAS has been linked to the organization of the actin cytoskeleton and the regulation of cell motility, growth, and survival. Mice lacking CAS die at about embryonic day 12 with numerous developmental defects, including a heart abnormality associated with disorganized cardiocyte myofibrils and Z disks, while fibroblasts derived from CAS null embryos exhibit disorganized actin stress fibers (24). In cultured fibroblasts, CAS localizes to focal adhesions and undergoes tyrosine phosphorylation in response to integrin-mediated cell adhesion (6, 41, 46). The recruitment of c-Crk to tyrosine-phosphorylated CAS appears to be a key step in integrin control of cell migration. In COS cell expression studies, formation of a CAS/Crk complex enhances haptotactic cell migration and promotes cell invasion through collagen (15, 16, 29) while rates of cell spreading and migration are enhanced upon CAS reexpression in CAS null fibroblasts (25). The ability of FAK to promote cell migration has been linked to its ability to bind CAS and promote CAS tyrosine phosphorylation (14, 45). CAS/Crk coupling has also been linked to activation of the Rac-JNK (c-Jun N-terminal kinase) pathway (17), which may contribute to the anchorage requirement for cell cycle progression (43) and cell survival (2, 16). Increased CAS tyrosine phosphorylation has also been observed during integrin-mediated cellular uptake of the enteropathogenic bacterium Yersinia pseudotuberculosis (65) and type 2 and type 5 adenoviruses (33) and following stimulation of certain receptor tyrosine kinases, G-protein-coupled receptors, and B- and T-cell antigen receptors (reviewed in reference 44).
The original descriptions of CAS as a putative v-Src substrate and a binding target for the SH2 domain of v-Crk suggest a possible involvement of deregulated CAS tyrosine phosphorylation in oncogenic transformation. Indeed, CAS null cells are resistant to transformation by oncogenic Src but can be transformed by Src when CAS is reexpressed (24). Although Src transformation does not require maximal levels of CAS tyrosine phosphorylation (9), the CAS phosphatase PTP-1B can inhibit transformation by v-Src (as well as v-Crk and v-Ras) (36), suggesting that CAS-SD tyrosine phosphorylation may be an important event in transformation by certain oncoproteins. CAS exhibits abnormally high phosphotyrosine levels in fibroblasts transformed by ornithine decarboxylase or Ras (5) and in carcinoma cells selected for metastatic potential (29). Interestingly, increased expression of the human CAS ortholog (BCAR1) has been linked to breast cancer progression and tamoxifen resistance (8, 62).
The fact that CAS interacts with members of both the Src and FAK kinase families creates uncertainty regarding the mechanism by which integrin-regulated CAS-SD tyrosine phosphorylation is achieved. Previous studies utilizing knockout fibroblast cell lines have implicated SFKs as playing a critical role. Thus, CAS tyrosine phosphorylation is greatly reduced in cells lacking either Src or Fyn or in a Src/Fyn/Yes triple knockout and is restored when Src is reexpressed (21, 30, 53, 57, 64), while cells lacking the SFK negative regulator C-terminal Src kinase (CSK) exhibit elevated CAS phosphotyrosine (53, 64). In contrast, the level of CAS phosphotyrosine was reported to be near-normal in FAK null cells (53, 64), suggesting that FAK may not be a critical CAS kinase. However, a later study showed that inducible FAK reexpression in these cells caused a several-fold increase in adhesion-dependent CAS tyrosine phosphorylation (45). PYK2 is upregulated in FAK null cells (45, 58) and may compensate partially for the absence of FAK in promoting CAS tyrosine phosphorylation (4). Further complicating the issue of the relative roles played by FAK versus SFKs in promoting CAS-SD tyrosine phosphorylation is the observation that SFKs not only bind to the CAS-SBD but also interact through their SH2 domains with FAK at its Tyr-397 autophosphorylation site (47, 56, 66). This interaction can aid in activating SFKs by releasing autoinhibitory intramolecular interactions (55), and FAK could therefore be viewed as a docking or scaffolding protein that functions to recruit and activate SFKs to phosphorylate CAS and other substrates. Yet another complication stems from observations that Src bound to the FAK Tyr-397 site phosphorylates the FAK kinase domain activation loop which enhances FAK catalytic activity (12, 45, 51). Given these intricacies, it is not surprising that several models for the mechanism of CAS-SD tyrosine phosphorylation by FAK and/or Src have been proposed (11, 40, 57, 59, 64; see Fig. 1), and uncertainty remains regarding this signaling event.
FIG. 1.

Models for CAS substrate domain phosphorylation by FAK or Src (see text for details). SD, substrate domain; SBD, Src-binding domain; 2, SH2 domain; 3, SH3 domain; Y, tyrosine; encircled P, phosphorylated tyrosine; visage, kinase domain.
The objective of this study was to clarify the mechanism of CAS-SD tyrosine phosphorylation by FAK and/or Src by testing existing models. By analyzing CAS phosphorylation in vitro by FAK or Src and by evaluating FAK and CAS functional requirements for achieving CAS tyrosine phosphorylation in a CAS-FAK-Src complex reconstituted in COS-7 cells, we determined that FAK plays a major role in regulating CAS-SD phosphorylation through its ability to recruit Src to phosphorylate CAS while a second FAK-independent mechanism involves Src recruited directly to the CAS-SBD.
MATERIALS AND METHODS
Antibodies, plasmids, and cells.
The anti-CAS rabbit polyclonal antiserum Cas-B (22) was generously provided by Amy Bouton, University of Virginia. The monoclonal antibody against CAS (here designated CAS-TL) and horseradish peroxidase (HRP)-conjugated anti-mouse immunoglobulin G (IgG) were obtained from BD Transduction Laboratories (San Diego, Calif.). Anti-FAK polyclonal antibody C20 was from Santa Cruz Biotechnology (Santa Cruz, Calif.). Anti-FAK pTyr-397 (pFAK397) was from Biosource International (Hopkinton, Mass.). Anti-Src monoclonal antibody 327 was from Oncogene Research Products (Cambridge, Mass.). Anti-phosphotyrosine monoclonal antibody 4G10 was from Upstate Biotechnology (Lake Placid, N.Y.). Monoclonal antibody 12CA5 that recognizes the hemagglutinin (HA) epitope was from Boehringer Mannheim (Indianapolis, Ind.). Monoclonal antibody 9E10 against the c-Myc epitope was generously provided by Kathy Gould, Vanderbilt University. Rabbit anti-mouse IgG was obtained from Jackson ImmunoResearch Laboratories (West Grove, Pa.).
pRc/CMV-based plasmids for expression of mouse FAK variants tagged either at their C termini with an HA epitope tag (FAKHA) or at their N termini with a c-Myc tag (mycFAK) have been described (12, 48, 67). pRc/CMV plasmids for the expression of mouse n-Src variants, either unmutated wild-type (WT) or kinase-dead (KD; arginine substitution for the conserved kinase domain Lys-303), were described previously (48). A pRc/CMV plasmid for expressing mouse c-Src, lacking the n-Src SH3 domain insert, was constructed for this study. Plasmid pRc/CMV-CASmyc(WT) for the expression of wild-type mouse CAS with a C-terminal c-Myc epitope tag was described previously (14). Standard methods were used to construct additional plasmids for expression of CAS mutants with a C-terminal Myc tag. pRc/CMV-CASmyc(ΔSH3) was made by introducing ClaI and HpaI sites in codons 5 to 7 and 64 and 65, respectively, and then digesting with these enzymes and religating to remove the intervening sequence which encodes the SH3 domain. pRc/CMV-CASmyc(mPR) was made by introducing a SacII site in codons 642 to 644 such that the RPLPSPP SH3 binding motif was changed to the sequence RAAASPP. pRc/CMV-CASmyc(F668/F670) was made by changing codons for SBD tyrosines 668 and 670 to encode phenylalanine. Plasmid pGEX-CAS(SD) was constructed by subcloning an XhoI-HindIII fragment of the mouse CAS cDNA, encoding amino acid residues 33 to 545, including the entire SD, into pGEX-KG (20). The GST-CasSD fusion protein encoded by pGEX-CasSD was expressed in Escherichia coli cells and purified by affinity chromatography using glutathione-agarose beads (20).
COS-7 cells and mouse embryo fibroblasts were obtained and cultured as described previously (12, 45).
Assays for kinase activity toward CAS in FAK immunoprecipitates.
Expression plasmids for mycFAK variants (WT, F397, or R454) and n-Src variants (WT or KD) were transfected into COS-7 cells (1.5 μg of each plasmid per 60-mm-diameter dish of subconfluent cells) using Lipofectamine (Life Technologies, Rockville, Md.). Cell lysates were prepared 48 h after transfection in NP-40 buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1% aprotinin, 50 mM NaF, and 0.1 mM Na3VO4). The bicinchoninic acid protein assay (Pierce, Rockford, Ill.) was used to determine protein concentration, and equal amounts of total protein (∼300 μg) from the lysates were brought to 1 ml in NP-40 buffer and used for immunoprecipitation with 2 μg of monoclonal antibody 9E10, followed by 10 μg of rabbit anti-mouse IgG. The immune complexes formed were precipitated using 25 μl of a 50% slurry of protein A-Sepharose beads (Zymed Laboratories, San Francisco, Calif.) which had been preadsorbed with mouse CAS protein (CAS beads). (To prepare CAS beads, mouse embryo fibroblasts were lysed in radioimmunoprecipitation assay [RIPA] buffer [NP-40 buffer supplemented with 1% sodium deoxycholate and 0.1% sodium dodecyl sulfate {SDS}] and then were adjusted to a total protein concentration of 5 mg/ml and subjected to immunoprecipitation using 5 μg of CAS-TL monoclonal antibody, followed by 50 μg of rabbit anti-mouse IgG and then 500 μl of a 50% slurry of protein A-Sepharose. The CAS beads were then washed extensively in RIPA buffer before being resuspended in NP-40 buffer.) CAS beads containing mycFAK immune complexes were washed extensively in NP-40 buffer and divided for further analysis. One-third of the beads were utilized for immunoblot assessments of FAK recovery or coprecipitating Src, while the rest were used for in vitro kinase reactions.
For the kinase reactions, beads were washed in kinase assay buffer (50 mM PIPES [pH 7.0], 10 mM MnCl2, 1 mM dithiothreitol [DTT]) and then equally divided and incubated for 30 min at room temperature with intermittent agitation in 30 μl of kinase assay buffer containing 0.25 μCi of [γ-32P]ATP/ml (4,500 Ci/mmol; ICN, Irvine, Calif.) and in the presence or absence of 44 μM PD161430 (45, 67). Reactions were stopped by boiling samples for 5 min in 100 μl of SDS boiling buffer (0.2% SDS, 50 mM Tris-Cl [pH 7.5], 5 mM EDTA, 10 mM DTT). After boiling, the beads were pelleted, the supernatant was collected and adjusted to the composition of RIPA buffer, and then CAS was again immunoprecipitated, essentially as described above. Reimmunoprecipitation was necessary to remove FAK, which autophosphorylates and has an electrophoretic mobility similar to CAS. The beads were resuspended in 2× SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer, CAS was resolved by SDS–7% acrylamide PAGE, and CAS phosphorylation was visualized by autoradiography of the dried gel and quantitated by phosphorimage analysis. Portions of the reimmunoprecipitated CAS samples were also subjected to immunoblot analysis to evaluate CAS recovery.
Assays for CAS tyrosine phosphorylation and CAS-associated kinase activity following FAK-Src-CAS complex formation in COS-7 cells.
Plasmids (1 to 1.5 μg each) expressing Src (n-Src or c-Src), FAK (HA tagged or untagged), and in some cases, Myc-tagged CAS, were transfected into COS-7 cells (alone or in combinations) as described above. For each transfection, 3 μg of total plasmid DNA was used, with empty vector making up the additional transfected DNA when needed. Cell lysates were prepared in NP-40 buffer and adjusted to equal protein concentrations as described above. A portion of each lysate (25 μl containing ∼25 μg of protein) was used to assess expression of the exogenous FAK or Src proteins or Tyr-397-phosphorylated FAK by immunoblot analysis while the remaining lysate was used for CAS immunoprecipitation. When Myc-tagged CAS variants were expressed, immunoprecipitation was accomplished using the 9E10 antibody essentially as described above for Myc-tagged FAK. In experiments where Myc-tagged FAK variants were expressed, CAS was not coexpressed and the endogenous COS-7 cell CAS was immunoprecipitated using the CAS-TL antibody essentially as described above for the preparation of CAS beads. CAS immune complexes were recovered from the NP-40 lysates on protein A-Sepharose beads, and the beads were divided for immunoblot assessment of CAS recovery and phosphotyrosine content or for kinase reactions as described above.
Immunoblot analyses.
Proteins from whole-cell lysates or isolated by immunoprecipitation were resolved by SDS–7% acrylamide PAGE, transferred to Immobilon-P (Millipore Corp., Bedford, Mass.), and subjected to immunoblot analysis with antibodies FAK C-20, 12CA5, 9E10, Src-327, CAS-TL, CAS-B, pFAK397, or 4G10. Blots were blocked from 2 h to overnight in Tris-buffered saline containing 0.2% Tween 20 (TBST) and either 3% bovine serum albumin–1% ovalbumin (for 4G10) or a 5% nonfat dry milk solution (for all others). After blocking, blots were incubated for 2 h in primary antibody solutions prepared in blocking buffer prior to washing extensively in TBST. Detection was performed with enhanced chemiluminescence using either HRP-conjugated goat anti-mouse IgG or HRP-conjugated protein A (BD Transduction Laboratories) and detection reagents from Amersham Pharmacia Biotech (Piscataway, N.J.). Concentrations of primary antibodies were as follows: C-20, 0.4 μg/ml; 12CA5, 1.0 μg/ml; 9E10, 3.8 μg/ml; Src-327, 1.0 μg/ml; CAS-TL, 0.5 μg/ml; Cas-B, 1:500 dilution of whole antiserum; pFAK397, 0.25 μg/ml; 4G10, 1.0 μg/ml.
Tryptic phosphopeptide mapping and phosphoamino acid analysis.
CAS proteins were labeled in vitro with 32P either as described above or by using baculovirus-produced c-Src or FAK as described previously (12, 45). GST-CAS(SD) was phosphorylated using baculoviral Src. The 32P-labeled proteins were separated by SDS-PAGE, eluted from the gel, and subjected to two-dimensional tryptic phosphopeptide mapping analysis and phosphoamino acid analysis on thin-layer cellulose plates (7). For phosphopeptide mapping, electrophoretic separation was carried out for 30 min at 1,000 V in pH 1.9 buffer. Chromatographic separation was carried out for ∼20 h in phosphochromo buffer.
RESULTS
Models for CAS substrate domain phosphorylation.
Depicted in Fig. 1 are five alternative models proposed for CAS-SD tyrosine phosphorylation by Src or FAK. According to model A (11, 40), Src phosphorylates CAS-SD after its SH3 domain binds to the RPLPSPP motif in CAS-SBD. Once bound, Src may phosphorylate the nearby SBD tyrosine(s) 668 and/or 670 and undergo an additional SH2-mediated interaction with these sites to stabilize the interaction. The other four models emphasize FAK and Src cooperativity in promoting CAS-SD phosphorylation. In model B (59), FAK phosphorylates CAS-SBD tyrosines 668 and/or 670, driving an SH2-mediated recruitment of Src which then phosphorylates CAS-SD. The Src SH3 domain interaction with the SBD could further stabilize the interaction. Models C to E each invoke phosphorylation of FAK Tyr-397 as a key step. In model C (57), Src plays a noncatalytic bridging role whereby the interaction of the Src SH2 domain with FAK pTyr-397 and the simultaneous interaction of the Src SH3 domain with its binding site in the CAS-SBD recruits FAK to phosphorylate CAS-SD. In model D (12, 45), FAK directly phosphorylates CAS-SD, but only after its activation loop Tyr-576/Tyr-577 site has been phosphorylated by Src bound to the pTyr-397 site. Finally, in model E (64), FAK is viewed as playing a docking role whereby CAS bound to FAK becomes phosphorylated by Src bound to the pTyr-397 site. The goal of our study was to determine which of the alternative mechanism(s) depicted in Fig. 1 are most likely to play substantial roles in regulating CAS-SD tyrosine phosphorylation.
CAS is poorly phosphorylated by FAK, relative to Src bound to FAK.
Since direct phosphorylation of CAS by FAK is central to models B to D (Fig. 1), initial studies were undertaken to determine the capacity of FAK to directly phosphorylate CAS by using an in vitro assay of FAK immunoprecipitates. Since FAK activation loop phosphorylation may be important for maximizing FAK kinase activity toward CAS, it was important to conduct these assays under conditions where the FAK activation loop tyrosines are highly phosphorylated. Therefore, wild-type mouse FAK with an N-terminal Myc epitope tag (WT-mycFAK) was expressed in COS-7 cells together with an unmutated mouse n-Src (neuronal isoform) which is sufficient to promote phosphorylation of the FAK activation loop tyrosines 576 and 577 (51). The coexpressed Src is catalytically active under these conditions, probably because the negative regulation brought about by the intramolecular interaction of its SH2 domain with the C-terminal phosphotyrosine (Tyr-535 for n-Src) is released by its interaction with the FAK Tyr-397 autophosphorylation site. WT-mycFAK was then immunoprecipitated on protein A-Sepharose beads by using an antibody against the Myc tag and assayed for kinase activity toward CAS (which had been preadsorbed to the beads) by incorporation of 32P from [γ-32P]ATP, followed by autoradiography. As shown in Fig. 2C, lane 5, substantial CAS phosphorylation by the WT-mycFAK (coexpressed with Src) immunoprecipitate was observed, which was measured by phosphorimage analysis to be ∼75 times above the background level obtained from a control assay of CAS beads incubated with lysates from cells transfected with the vector only (compare to Fig. 2C, lane 1). Phosphoamino acid analysis showed that only tyrosine residues were phosphorylated in the reaction (data not shown). Interestingly, when a kinase-dead mutant n-Src (KD-Src) was coexpressed instead of the unmutated n-Src, the activity associated with the WT-mycFAK immunoprecipitate was only slightly above the background (Fig. 2C, compare lane 5 with lane 3). The requirement for Src kinase activity could indicate that the observed FAK-associated activity toward CAS was dependent upon FAK activation loop phosphorylation by Src, as suggested by model D. However, other results from our analysis shown in Fig. 2 clearly indicated that the vast majority of the kinase activity toward CAS in these assays was not due to FAK itself but rather to coprecipitating Src bound to the FAK pTyr-397 site. Thus, CAS phosphorylation was substantially reduced (by greater than 10-fold) when the WT-mycFAK (+Src) assays were performed in the presence a Src-selective inhibitor (PD161430) which has no effect on FAK activity (45, 67) (Fig. 2C, compare lanes 5 and 6). Similar experiments using F397-mycFAK, which does not bind and coprecipitate Src (Fig. 2B, lane 4), resulted in little CAS phosphorylation (Fig. 2C, compare lanes 7 and 5). Finally, immunoprecipitates of the KD R454-mycFAK, when coexpressed with n-Src, showed similar PD161430-sensitive CAS phosphorylation as observed for WT-mycFAK (Fig. 2C, compare lanes 9 and 10 with lanes 5 and 6). Although the R454 mutant is defective in Tyr-397 autophosphorylation, coexpressed Src can phosphorylate this site (12) to promote its own SH2-mediated interaction, which is evident from the coprecipitation (Fig. 2B, lane 5).
FIG. 2.

CAS phosphorylation by FAK immunoprecipitates is largely due to coprecipitating Src. Myc-tagged FAK variants (either WT, F397, or R454) were expressed in COS-7 cells along with either WT n-Src or a kinase-dead (KD) n-Src mutant, as indicated. Control cells were transfected with empty expression plasmid (−). Cell lysates were incubated with anti-Myc antibody 9E10, and immunoprecipitates (IP) were formed with protein A-Sepharose beads that had been preadsorbed with mouse CAS protein which served as substrate for kinase reactions. After incubation with the lysates, the beads were split three ways and utilized for immunoblot (IB) assessment of FAK recovery (A) or coprecipitating Src recovery (B) or for kinase assays (C). The kinase reactions, utilizing [γ-32P]ATP, were carried out in the presence or absence of 44 μM PD161430, a Src-selective inhibitor. Reactions were stopped by boiling in SDS buffer, and CAS was reimmunoprecipitated to eliminate FAK, which comigrates with CAS on gels. After SDS-PAGE, CAS phosphorylation was visualized by autoradiography and quantitated by phosphorimage analysis. (D) The reimmunoprecipitated CAS was also subjected to immunoblot analysis to show near-equal recovery.
The data shown in Fig. 2 indicate that coprecipitating Src accounts for >90% of the kinase acitivity toward CAS measured in WT-FAK immunoprecipitates from lysates where wild-type n-Src was expressed. That the remaining activity is due to FAK is supported by the observation that PD161430 blocked all of the activity in the sample containing R454-mycFAK (+Src) (Fig. 2C, compare lanes 6 and 10). Also, activity measured in the presence of PD161430 is enhanced by coexpression with WT n-Src but not by the KD mutant (Fig. 2C, compare lanes 6 and 8 with lane 4), suggesting some enhancement of FAK activity as a result of Src-mediated phosphorylation of the FAK activation loop (model D). Nevertheless, these findings as a whole emphasize that FAK is a very inefficient CAS kinase relative to Src. This conclusion is reinforced when one considers that fewer Src molecules, relative to FAK, are likely to be recovered in the FAK immunoprecipitates.
CAS phosphorylation by Src and FAK is qualitatively similar, with all major sites residing in CAS-SD.
According to model B, FAK selectively phosphorylates Tyr-668 and/or Tyr-670 in the CAS-SBD while Src phosphorylates the CAS-SD. To assess possible qualitative differences in CAS phosphorylation by Src versus FAK, we generated two-dimensional tryptic phosphopeptide maps from CAS phosphorylated in vitro by the immunoprecipitate of WT-mycFAK coexpressed with n-Src in the absence or presence of PD161430 (i.e., CAS labeled as in Fig. 2C, lanes 5 and 6). The map in Fig. 3A represents CAS phosphorylation in the absence of PD161430 (largely due to Src), and the map in Fig. 3B represents CAS phosphorylation in the presence of PD161430 (largely due to FAK). The maps are very similar, with each showing a cluster of three apparent phosphopeptide spots (labeled 1 to 3) having similar electrophoretic and chromatographic mobilities, two other major spots (labeled 4 and 5), and a number of less intense spots (labeled 5 to 10). These spots comigrated when the samples were mixed (Fig. 3C), attesting to their identity. Although the maps shown in Fig. 3A and B are notable for their overall similarity, differences in minor spots are evident, with spots 6 to 8 and 10 appearing relatively less intense in Fig. 3B. Other minor spots are evident in one or the other of the maps shown in Fig. 3A and B, which have not been given a numerical label as they were not consistently observed. Not surprisingly, a map obtained for CAS phosphorylated in vitro using baculoviral-expressed c-Src (B-Src) closely resembles the map obtained from the in vitro labeling reaction using the WT-FAK (+Src) immunoprecipitate (Fig. 3, compare D to A and the mix in E). However, we have been unable to phosphorylate CAS to sufficient levels to permit mapping using a baculoviral-expressed FAK.
FIG. 3.

Tryptic phosphopeptide maps of mouse CAS phosphorylated in vitro. 32P-labeled CAS proteins phosphorylated in vitro by various kinase preparations were separated by SDS-PAGE, recovered from gels, and subjected to two-dimensional tryptic phosphopeptide mapping analysis. Electrophoretic separation (horizontal arrows) was in pH 1.9 buffer and chromatographic separation (vertical arrows) was in phosphochromo buffer. (A and B) CAS phosphorylated by WT-FAK immunoprecipitates from COS-7 cell lysates where n-Src was coexpressed in the absence (A) or presence (B) of the PD161439 Src inhibitor (i.e., lanes 5 and 6 in Fig. 2C). (C) Mix of samples A and B. (D) CAS phosphorylated by recombinant baculovirus-expressed c-Src (B-Src). (E) Mix of samples A and D. (F) GST-CAS(SD) phosphorylated by B-Src. For each map, ∼500 Cerenkov cpm were loaded on the thin-layer cellulose plates, and autoradiographic exposure was for 5 to 7 days.
To determine which of the phosphopeptides evident in the CAS maps shown in Fig. 3A to E represent the SD region, we also obtained a map from a bacterially expressed glutathione S-transferase (GST) fusion protein containing the entire SD region of mouse CAS (residues 33 to 545) but lacking the C-terminal region including the SBD. The GST-CAS(SD) protein was labeled by in vitro phosphorylation using B-Src. The map of GST-CAS(SD) is essentially identical to the maps generated for full-length CAS (Fig. 3, compare panel F to panels A and D), indicating that all major sites of full-length CAS phosphorylation under these conditions fall within the CAS-SD region. Apparently, none of the major phosphopeptide spots resolved from full-length CAS represent the SBD Tyr-668/670 sites, since no such spots were missing in the GST-CAS(SD) map. Thus, the data shown in Fig. 2 and 3 indicate that FAK and Src may essentially phosphorylate the same CAS-SD tyrosine sites, although Src activity toward these sites is at least 10-fold greater than FAK activity.
FAK and Src cooperate to promote CAS-SD tyrosine phosphorylation in cells.
To conduct the assays shown in Fig. 2, FAK and CAS were bound to beads via separate antibody linkages, and it is possible that this artificial arrangement restricted the ability of FAK to directly phosphorylate the CAS-SBD or CAS-SD. Therefore, we wanted to next assess the ability of FAK to phosphorylate CAS under conditions where FAK was directly bound to CAS via normal cellular interactions. To this end, COS-7 cells were transfected with three separate plasmids, in various combinations, expressing either Myc-tagged WT-CAS (CASmyc), HA-tagged WT-FAK (FAKHA), or untagged n-Src. After 2 days, CASmyc was immunoprecipitated with the anti-Myc antibody and subjected to phosphorylation analysis by immunoblotting for phosphotyrosine and by measuring phosphorylation by coprecipitating kinases. When CASmyc was expressed alone, it did not become measurably tyrosine phosphorylated in the cells and coprecipitated very little kinase activity (Fig. 4C to E, lanes 1). Coexpression with FAKHA alone had a very minor effect on CASmyc cellular phosphotyrosine levels and associated kinase activity (Fig. 4A to D, lanes 2; 4E, lanes 3 and 4), while expression with n-Src alone resulted in a more substantial increase in these properties (Fig. 4B to D, lanes 3; 4E, lanes 5 and 6). However, expression of FAK and n-Src together led to a dramatic increase in both CASmyc cellular phosphotyrosine content and associated kinase activity (Fig. 4D, lane 4; 4E, lanes 7 and 8). The total CAS-associated activity was measured by phosphorimage analysis to be ∼8-fold greater when FAKHA and n-Src were coexpressed relative to when n-Src was expressed alone (Fig. 4E, compare lane 5 to lane 7). The CAS-associated activity was substantially blocked by the Src inhibitor PD161430 (Fig. 4E) while phosphoamino acid analysis indicated that phosphorylation under these conditions occurred essentially on tyrosine (not shown). To further characterize this activity, a tryptic phosphopeptide map was obtained for CASmyc labeled under conditions identical to the sample shown in Fig. 4E, lane 7, where CASmyc was immunoprecipitated from COS-7 lysates following coexpression with both FAKHA and n-Src. As a control, a map was also obtained for immunoprecipitated CASmyc, prepared from lysates where neither FAK nor n-Src were coexpressed, that was phosphorylated by B-Src. These maps are very similar to one another (Fig. 5, compare panels A and B) and closely resemble the maps for nontagged mouse CAS or GST-CAS(SD) phosphorylated by exogenously added Src or FAK/Src complex shown in Fig. 3. Since CAS phosphorylation by the associated coprecipitating kinases is a good indication of phosphorylation occurring in the cells, the map of the CAS-associated activity indicates that CAS tyrosine phosphorylation taking place in the COS-7 cells occurs substantially within the SD, while the SBD may not represent a major site of cellular phosphorylation.
FIG. 4.

FAK and n-Src cooperate to promote cellular CAS tyrosine phosphorylation and enhance CAS-associated kinase activity. Myc-tagged CAS (CASmyc) was expressed in COS-7 cells either alone, with HA-tagged FAK (FAKHA), with n-Src, or with both FAKHA and n-Src, and mycCAS phosphotyrosine content and associated kinase activity were determined. Equal total protein amounts of the cell lysates were used for immunoblot (IB) assessment of either FAKHA (A) or Src (B) expression using anti-HA antibody 12CA5 or anti-Src antibody 327, respectively. The remaining lysates (equal protein amounts) were used to immunoprecipitate (IP) CASmyc by using anti-Myc antibody 9E10 for the CAS phosphorylation analyses. (C) Near-equal CASmyc recovery was indicated by immunoblotting the immunoprecipitates with CAS-TL antibody. (D) Cellular phosphotyrosine levels of CASmyc were determined by immunoblotting equal portions of the CASmyc immunoprecipitates with antiphosphotyrosine antibody 4G10. (E) Kinase reactions were carried out in the presence of [γ-32P]ATP and either the presence (+) or absence (−) of 44 μM PD161430 on another equal portion of the mycCAS immunoprecipitates. Reactions were stopped by boiling in SDS buffer and CASmyc was reimmunoprecipitated to eliminate comigrating FAK. After SDS-PAGE, CAS phosphorylation was visualized by autoradiography.
FIG. 5.
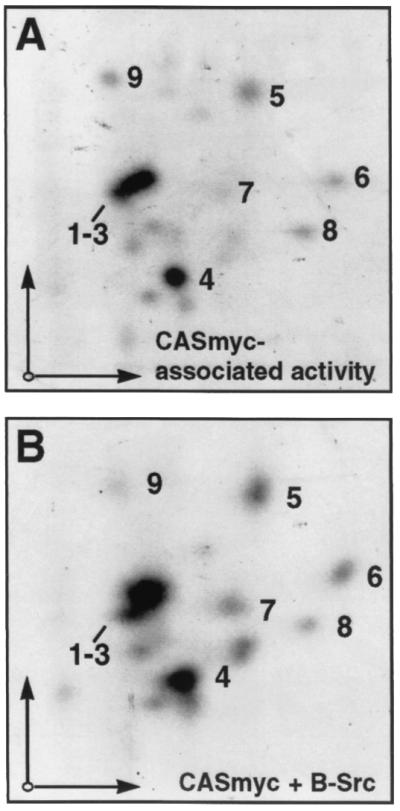
Tryptic phosphopeptide maps of CASmyc phosphorylated by coprecipitating kinases. (A) WT-CASmyc coexpressed in COS-7 cells with FAK and n-Src and phosphorylated by coprecipitating kinases (i.e., lane 7 in Fig. 4E). (B) CASmyc expressed alone in COS-7 cells and phosphorylated by baculovirus-expressed Src (B-Src). For each map, ∼250 Cerenkov cpm were loaded on the thin-layer cellulose plates and autoradiographic exposure was for 7 days. Electrophoretic and chromatographic separation conditions were the same as for the maps shown in Fig. 3.
The n-Src neuronal isoform used in our experiments contains a six-amino-acid insert in the nSrc loop of the SH3 domain, and it is possible that this insertion results in a reduced binding affinity for CAS compared to the nonneuronal c-Src SH3 domain, such that c-Src could have been much more effective in promoting CAS tyrosine phosphorylation in the absence of FAK expression. To test this possibility, we compared the ability of n-Src versus c-Src to promote CASmyc tyrosine phosphorylation following coexpression in COS-7 cells in the presence or absence of FAKHA. As shown in Fig. 6, c-Src behaved similarly to n-Src in this assay and promoted only a low level of CAS tyrosine phosphorylation in the absence of FAKHA but a much higher level when FAKHA was also expressed. Taken together, the data shown in Fig. 4 to 6 strongly support a FAK/Src cooperative mechanism for CAS-SD tyrosine phosphorylation wherein FAK acts by recruiting Src (either n-Src or c-Src) to CAS (model E). CAS tyrosine phosphorylation induced by Src alone could reflect the FAK-independent mechanism where Src is recruited directly to the CAS-SBD (model A).
FIG. 6.

c-Src behaves similarly to n-Src in cooperating with FAK to promote CAS tyrosine phosphorylation. c-Src, which lacks the six-amino-acid nSrc insert in the SH3 domain, was compared to n-Src in its ability to promote CASmyc tyrosine phosphorylation in transfected COS-7 cells both in the presence or absence of FAKHA. See Fig. 4A to D legend for details. IB, immunoblot; IP, immunoprecipitate.
FAK-promoted CAS phosphorylation is independent of FAK kinase activity, but requires Tyr-397 and site(s) for CAS-SH3 binding.
We next tested the structural requirements for FAK in its ability to cooperate with Src to promote CAS tyrosine phosphorylation. Myc-tagged FAK variants (either WT, F397, R454, F576/F577 [phenylalanine substitutions for activation loop tyrosines], or A712/A715/A873/A876 [mPR; alanine substitutions for prolines in both CAS-SH3 binding sites]) were individually expressed in COS-7 cells along with n-Src, and immunoprecipitates of the endogenous CAS protein were analyzed for phosphotyrosine content and associated kinase activity. Consistent with our earlier results, WT-mycFAK expression increased the cellular phosphotyrosine content of endogenous CAS to a level well above that observed when n-Src was expressed alone (Fig. 7A to E, compare lanes 2 and 1). Again, the FAK-promoted cellular CAS tyrosine phosphorylation was also reflected as increased PD161430-sensitive CAS-associated kinase activity (Fig. 7F, compare lanes 1 and 2 to 3 and 4). Expression of the kinase-dead R454-mycFAK variant had the same effect as expressing WT-FAK (Fig. 7E, lane 4, and F, lane 7), indicating that FAK's own kinase activity is not required for the response. The activation loop mutant F576/F577-mycFAK, which exhibits reduced kinase activity relative to WT-FAK (45), also efficiently promoted cellular CAS tyrosine phosphorylation (Fig. 7E, lane 5) and increased the CAS-associated kinase activity (Fig. 7F, lane 9), although the latter was somewhat reduced relative to the activity promoted by WT-and R454-mycFAK. We previously reported that the F576/F577 FAK mutation impairs the ability of Src to enhance FAK and CAS coimmunoprecipitation (48), suggesting that Src-mediated phosphorylation of the FAK activation loop may act to stabilize the SH3-mediated FAK/CAS interaction. The reduced recovery of CAS-associated kinase activity observed when F576/F577-mycFAK is expressed could be a reflection of this instability.
FIG. 7.

FAK structure/function requirements for FAK/Src-promoted CAS tyrosine phosphorylation. n-Src was expressed in COS-7 cells either alone or with a Myc-tagged FAK variant (either WT, F397, R454, F576/F577, or mPR [A712/A715/A873/A876]) and endogenous CAS phosphotyrosine content and associated kinase activity were assessed. Equal total protein amounts of the cell lysates were used for immunoblot (IB) assessment of either mycFAK expression using anti-Myc antibody 9E10 (A), FAK Tyr-397 phosphorylation using phosphospecific antibody pFAK397 (B), or Src expression using anti-Src antibody 327 (C). The remaining lysates (equal protein amounts) were used to immunoprecipitate (IP) endogenous CAS using CAS-TL antibody for the CAS phosphorylation analyses. (D) Near-equal CAS recovery was indicated by immunoblotting the immunoprecipitates with CAS-TL antibody. (E) Cellular phosphotyrosine levels of CAS were determined by immunoblotting equal portions of the CAS immunoprecipitates with anti-phosphotyrosine antibody 4G10. (F) Kinase reactions were carried out in the presence of [γ-32P]ATP and either the presence (+) or absence (−) of 44 μM PD161430 on another equal portion of the CAS immunoprecipitates. Reactions were stopped by boiling in SDS buffer, and CAS was reimmunoprecipitated to eliminate comigrating FAK. After SDS-PAGE, CAS phosphorylation was visualized by autoradiography.
The substantial inhibition of CAS-associated activity by PD161430 indicates that FAK is acting to recruit Src to CAS. Indeed, neither cellular CAS phosphotyrosine content nor CAS-associated kinase activity were obviously enhanced by expressing either F397-mycFAK, which cannot undergo phosphorylation to promote SH2-mediated binding to Src (Fig. 7E, lane 3, and F, lane 5), or mPR-mycFAK, which cannot undergo SH3-mediated binding to CAS (Fig. 7E, lane 6, and F, lane 11). The mPR mutation actually acted to reduce CAS cellular phosphotyrosine and associated kinase activity relative to what was observed when n-Src was expressed alone. This dominant-negative activity indicates that stable interactions may form between mPR-FAK and n-Src (and endogenous SFKs), effectively sequestering the SFKs away from CAS.
As controls for these experiments, equal portions of the total cellular lysates were used for immunoblot analysis to establish that the FAK variants (Fig. 7A) and n-Src (Fig. 7C) were expressed to similar levels and that CAS recovery was equivalent (Fig. 7D) for each experimental condition. Analysis of the cell lysates with an antibody that specifically recognizes the phosphorylated FAK Tyr-397 site showed that the R454 and F576/F577 mutants become highly phosphorylated on Tyr-397 (Fig. 7B), presumably by n-Src, indicating that FAK autophosphorylation activity is dispensable for Tyr-397 phosphorylation under these experimental conditions. Thus, our analysis of FAK mutants indicates that FAK must interact with both the CAS SH3 domain and with the Src SH2 domain at the phosphorylated Tyr-397 site in order to promote Src-mediated CAS tyrosine phosphorylation, consistent with model E.
CAS-SH3 domain is required for FAK-promoted CAS tyrosine phosphorylation, but not PXXP motif or Tyr-668/Tyr-670 sites in CAS-SBD.
We also tested the requirements for CAS protein interaction domains in the ability of CAS to become tyrosine phosphorylated when coexpressed with FAK and/or n-Src. Either WT-CASmyc or one of three CASmyc variants (ΔSH3, SH3 domain deletion; mPR, RPLPSPP motif in SBD changed to RAAASPP; and FF668/670, phenylanine substitutions for SBD Tyr-668/670 residues) were expressed in COS-7 cells either alone or with WT-FAK and/or n-Src. Immunoprecipitates of the CASmyc variants were then prepared and analyzed for cellular phosphotyrosine levels by antiphosphotyrosine immunoblotting. As before (Fig. 4), WT-CASmyc did not become detectably tyrosine phosphorylated when expressed alone or when expressed only with FAK and was only modestly phosphorylated when expressed with n-Src alone, but became much more highly phosphorylated when expressed together with both FAK and n-Src (Fig. 8D, lanes 1 to 4). The ΔSH3 CAS mutant also became tyrosine phosphorylated when coexpressed with n-Src to an extent similar to what was observed for WT-CAS (Fig. 8D, compare lanes 7 and 3). However, unlike WT-CAS, ΔSH3-CAS failed to become more extensively phosphorylated when FAK was also expressed along with n-Src (Fig. 8D, compare lanes 8 and 4). The ability of FAK to promote CAS tyrosine phosphorylation was not compromised by either the mPR or the FF668/670 mutations (Fig. 8D, lanes 12 and 16). However, both mPR- and FF668/670-CASmyc were resistant to tyrosine phosphorylation when only n-Src was expressed (Fig. 8D, compare lanes 11, 12, 15, and 16 to lanes 3 and 4). Thus, the analysis of CAS mutants indicates a requirement for both Src SH3 and SH2 binding sites in the CAS-SBD in the limited tyrosine phosphorylation promoted by Src alone (indicative of model A), while the more extensive phosphorylation cooperatively promoted by FAK and Src is largely independent of the CAS-SBD but requires the CAS-SH3 domain (indicative of model E).
FIG. 8.

CAS structure/function requirements for FAK/Src-promoted CAS tyrosine phosphorylation. Myc-tagged CAS (CASmyc) variants (either WT, ΔSH3 [deletion of SH3 domain], mPR [RAAASPP mutation of RPLPSPP motif], or F668/F670) were expressed in COS-7 cells either alone, with FAK (untagged), with n-Src, or with both FAK and n-Src, and the resulting phosphotyrosine content of the CASmyc variants was assessed. Equal total protein amounts of the cell lysates were used for immunoblot (IB) assessment of either FAK (A) or Src (B) expression using anti-FAK C20 antibody or anti-Src antibody 327, respectively. The remaining lysates (equal protein amounts) were used to immunoprecipitate (IP) CASmyc using anti-Myc antibody 9E10 for the CAS phosphotyrosine analyses. (C) Near-equal CASmyc recovery was indicated by immunoblotting the immunoprecipitates with CAS-B antibody. (D) Cellular phosphotyrosine levels of CASmyc were determined by immunoblotting equal portions of the immunoprecipitates with anti-phosphotyrosine antibody 4G10.
DISCUSSION
CAS-SD tyrosine phosphorylation is an important event in integrin control of cell behavior, but understanding the mechanism by which this is achieved is complicated by the dual interactions CAS makes with the tyrosine kinases FAK and Src and by the direct binding and regulatory interactions made between FAK and Src. As a consequence of this complexity, five plausible models have been proposed for CAS-SD phosphorylation by FAK or Src (Fig. 1). In this study, we used biochemical and structure/function approaches to test these models. Our results indicate that CAS-SD tyrosine phosphorylation is most efficiently achieved by a FAK/Src cooperative mechanism whereby CAS bound via its SH3 domain to FAK becomes phosphorylated by Src bound to the FAK pTyr-397 site (model E). FAK therefore plays a major role as a docking protein that acts to bring Src to its substrate CAS. Our results also support a FAK-independent mechanism of CAS-SD phosphorylation by Src resulting from direct recruitment of Src to the CAS-SBD (model A), although this mechanism appears to account for substantially less CAS phosphorylation than occurs through Src bound to FAK. Our findings that FAK exhibits very weak catalytic activity toward CAS, relative to Src, and does not appear to uniquely phosphorylate CAS tyrosines, including those in the SBD, downplay other FAK/Src cooperative models involving direct phosphorylation of CAS by FAK.
Model E, along with models B to D, predict that FAK and Src act cooperatively to promote CAS-SD phosphorylation. Accordingly, we found that expression of both kinases in COS-7 cells results in substantially higher levels of cellular CAS phosphorylation and CAS-associated kinase activity recovered in immunoprecipitates than is achieved when either kinase is expressed alone (Fig. 4 and 6). Models E and D, but not B and C, also predict that FAK's role in promoting CAS phosphorylation is dependent on its ability to directly interact with both Src and CAS, and this prediction is supported by our findings that FAK mutants unable to bind either Src (F397) or CAS (A712/A715/A873/A876 [mPR]) impaired FAK enhancement of CAS tyrosine phosphorylation (Fig. 7) as well as did the CAS ΔSH3 mutant which cannot bind FAK (Fig. 8). In support of model D, suggested by past findings that the FAK F576/F577 mutation results in diminished FAK activity (12, 45), we found that Src expression enhances CAS phosphorylation by FAK immunoprecipitates when assayed in the presence of a Src inhibitor (Fig. 2). However, other results strongly support model E over model D. Thus, the vast majority of kinase activity toward CAS recovered in FAK immunoprecipitates is due to coprecipitating Src rather than FAK itself (Fig. 2), and FAK's own kinase activity is not critical for FAK-promoted CAS phosphorylation in COS-7 cells (Fig. 7). Initial support for model E came from the observation that expression of a CD2-FAK chimeric protein caused elevated CAS phosphotyrosine levels that were aberrantly maintained when cells were held in suspension, while the F397 and R454 mutants of CD2-FAK were unable to achieve this response (64). Also consistent with model E are findings of enhanced adhesion-dependent CAS phosphotyrosine induced by expression of WT-FAK, but not by F397- or mPR-FAK mutants (14, 45). A mechanism similar to model E may also regulate tyrosine phosphorylation of paxillin (54, 61).
In addition to the FAK/Src cooperative mechanism of model E, our results also support the FAK-independent mechanism depicted in model A, whereby CAS-SD phosphorylation is due to Src recruited directly to CAS via its SH3 and SH2 domain interactions with the SBD. Thus, even in the absence of FAK, we found that expression of Src in COS-7 cells promoted some CAS tyrosine phosphorylation—measured to be ∼10 to 20% of that induced when FAK is coexpressed with Src (Fig. 4). Consistent with model A, this activity is retained for the CAS ΔSH3 mutant but is reduced in the CAS-SBD mutants, mPR and F668/F670 (Fig. 8). The loss of activity in the CAS-F668/F670 mutant is consistent with the notion that Src, once initially bound by its SH3 domain, phosphorylates the Tyr668/670 site to further stabilize its interaction by SH2 binding. Other studies supporting model A have shown that CAS-SBD mutations disrupt Src/CAS coimmunoprecipitation and reduce CAS tyrosine phosphorylation (40) and that the Src-SH3/CAS-SBD interaction is sufficient to recruit and activate Src to phosphorylate CAS-SD (11). Also, recovery of CAS-associated activity from Crk-transformed cells was found to be greatly reduced by mutation of the SH3-binding site in the CAS-SBD (40). Thus, model A appears to be an alternative mechanism for CAS-SD phosphorylation and could be the major mechanism under conditions where FAK is not associated with CAS or is not highly phosphorylated at the Tyr-397 site.
In our studies we primarily used the n-Src neuronal isoform, which contains a short insert in the SH3 domain. It could be argued that this insert may impair the binding affinity for the CAS-SBD relative to the nonneuronal c-Src SH3 domain, as indeed appears to be the case for certain other SH3 ligands (50, 63). If the affinity of the n-Src SH3 domain for the CAS-SBD site is considerably less than that of nonneuronal c-Src, then the relative contribution made by c-Src bound directly to CAS could be substantially greater than is observed for n-Src. However, when we directly compared the ability of n-Src versus c-Src to promote CAS tyrosine phosphorylation we found that c-Src behaves essentially the same as n-Src in this regard. Thus, both Src isoforms were able to promote CAS tyrosine phosphorylation to only a low level in the absence of FAK and, for both, the coexpression of FAK led to a dramatic enhancement of CAS phosphotyrosine levels (Fig. 6). Thus the argument that FAK plays an important role by recruiting Src to phosphorylate CAS need not be restricted to neuronal cell types where n-Src is expressed.
Several results from the study indicated that model B, in which FAK phosphorylates the CAS-SBD tyrosines 668 and 670 to promote Src binding, is unlikely to make a significant contribution to CAS-SD phosphorylation. Most notable was the finding that the CAS 668 and 670 tyrosines are not required for FAK/Src-enhanced CAS phosphorylation (Fig. 8). Also, phosphopeptide mapping did not provide evidence for this site being among the major sites phosphorylated by FAK (Fig. 3), and FAK kinase activity was found to be dispensible for FAK-enhanced CAS phosphorylation while the Src-binding Tyr-397 site was required (Fig. 7). Model B stems from observations showing that FAK stimulates CAS-SD phosphorylation following coexpression of the two proteins in COS-1 cells, while FAK appeared capable only of directly phosphorylating the Tyr-668 and 670 site (59). The same study found that the ability of FAK to promote CAS-SD phosphorylation required CAS Tyr-668 and 670 and FAK kinase activity but was independent of FAK Tyr-397, which is in contrast to our own observations. We cannot explain this discrepency, but we note that the earlier study (59) analyzed the effects on CAS phosphorylation of expressing only FAK with CAS. Given the intricate nature of the interactions between CAS, FAK, and Src and the capacity for mutual catalytic activation by FAK and Src, a full assessment of the mechanism of CAS phosphorylation should consider the combined action of both kinases. Indeed, we found FAK expression alone resulted in very little CAS tyrosine phosphorylation compared to when both FAK and Src were expressed. While it is conceivable that the model B mechanism could account for the minimal CAS phosphorylation induced when FAK alone is expressed, the much greater capacity for CAS-SD phosphorylation by Src bound to FAK indicate that model B is unlikely to play a significant role in CAS signaling.
Our results are also inconsistent with model C, whereby Src acts as a bridge to recruit FAK to phosphorylate CAS. According to model C, the ability of the Src SH3 domain to bind the CAS-SBD is critical for FAK/Src-promoted CAS-SD phosphorylation. Yet it is clear from our analysis of CAS mutants (Fig. 8) that this is not the case. The ability of FAK and Src to cooperatively promote CAS tyrosine phosphorylation is little affected by the mPR mutation in the CAS-SBD. Rather, it is the CAS ΔSH3 mutation that abolishes this phosphorylation response, indicating the importance of a direct CAS-FAK interaction, as does the finding that the FAK-mPR mutation, which disrupts the FAK-CAS interaction (22, 48), eliminates FAK-promoted CAS tyrosine phosphorylation in the presence of Src (Fig. 7). Model C also predicts a major role for the kinase activity of FAK, rather than Src activity, but FAK kinase activity is not required for FAK to promote CAS phosphorylation (Fig. 7). Also, we have observed that FAK does not efficiently promote cellular CAS tyrosine phosphorylation when coexpressed with KD-Src (data not shown). Model C stemmed from the observation that expression in Src null cells of a truncated Src protein that lacks its kinase domain but includes both SH3 and SH2 domains is able to enhance cellular CAS phosphotyrosine levels, coupled with data showing that immunoprecipitates of FAK from Src null cells still are able to phosphorylate CAS with high efficiency (57). While the mechanism by which the truncated Src promotes CAS tyrosine phosphorylation remains uncertain, coprecipitating SFKs, i.e., Fyn and Yes, could have accounted for the bulk of the CAS kinase activity observed in the FAK immunoprecipitates from Src null cells. Even if full-length Src could act as a bridge to bring FAK to CAS, it seems unlikely that the meager FAK catalytic activity toward CAS, relative to that of Src, would critically impact CAS-SD phosphorylation and downstream signaling events.
The complexity of the CAS tryptic phosphopeptide maps (four or five major spots and several additional minor spots) indicate that CAS is phosphorylated at multiple tyrosine sites by the FAK/Src complex. These sites appear to all lie in or very near the SD region, as indicated by the essentially identical appearance of the maps for full-length mouse CAS and the GST-CAS-SD fusion protein (Fig. 3). It will be of future interest to determine which YXXP tyrosines account for the observed phosphopeptides and if other tyrosines in the region are phosphoacceptor sites. It was somewhat surprising that our maps did not provide evidence for a major phosphopeptide representing the Tyr668/Tyr670 site. Previous studies have shown that mutation of the Tyr-668 equivalent in rat CAS can reduce the interaction between CAS and Src following their coexpression in COS-1 cells (40) and that the C-terminal region of CAS containing Y668/670 is efficiently phosphorylated by a baculoviral Src preparation, enhancing its ability to bind Src in a blot overlay assay (10). In light of these studies, we do not discount the possibility that the Tyr-668/670 site was phosphorylated in our in vitro kinase reactions but was insufficiently labeled to give rise to a major phosphopeptide spot.
While our studies indicated that CAS-SD tyrosine phosphorylation by FAK and Src is primarily achieved by the mechanism represented by model E, with model A having a secondary role when FAK is present, the relative importance of these and other mechanisms could be different for other complexes containing members of the FAK, Src, and CAS families. Since FAK and the FAK-related kinase PYK2 appear to have differences in substrate specificity (34), it is possible that PYK2 could be a more efficient CAS kinase than FAK such that models B to D could be more important. In addition, there are substantial differences in the SBDs of CAS and the two CAS-related proteins HEF1/CAS-L (32, 39) and Efs/Sin (1, 26), which could impact on the SD phosphorylation mechanism. For example, the binding site for the Src SH3 domain is not conserved in HEF1/CAS-L. Thus, the relative importance of the various possible mechanisms for SD phosphorylation of CAS family members by SFKs and FAK/PYK2 kinases will need to be examined on an individual basis.
ACKNOWLEDGMENTS
We thank Amy Bouton (University of Virginia) for a generous supply of the Cas-B antibody and David Fry (Parke-Davis) for providing the PD161430 Src inhibitor.
This study was supported by Public Health Service grant R01-GM49882 from the National Institute of General Medical Sciences (S.K.H.) and by training grant T32-CA78136 from the National Cancer Institute Training Program in Breast Cancer Research (P.J.R.).
REFERENCES
- 1.Alexandropoulos K, Baltimore D. Coordinate activation of c-Src by SH3- and SH2-binding sites on a novel p130Cas-related protein, Sin. Genes Dev. 1996;10:1341–1355. doi: 10.1101/gad.10.11.1341. [DOI] [PubMed] [Google Scholar]
- 2.Almeida E A, Ilic D, Han Q, Hauck C R, Jin F, Kawakatsu H, Schlaepfer D D, Damsky C H. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH2-terminal kinase. J Cell Biol. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Astier A, Avraham H, Manie S N, Groopman J, Canty T, Avraham S, Freedman A S. The related adhesion focal tyrosine kinase is tyrosine-phosphorylated after β1 integrin stimulation in B cells and binds to p130cas. J Biol Chem. 1997;272:228–232. doi: 10.1074/jbc.272.1.228. [DOI] [PubMed] [Google Scholar]
- 4.Astier A, Manie S N, Avraham H, Hirai H, Law S F, Zhang Y, Golemis E A, Fu Y, Druker B J, Haghayeghi N, Freedman A S, Avraham S. The related adhesion focal tyrosine kinase differentially phosphorylates p130Cas and the Cas-like protein, p105HEF1. J Biol Chem. 1997;272:19719–19724. doi: 10.1074/jbc.272.32.19719. [DOI] [PubMed] [Google Scholar]
- 5.Auvinen M, Paasinen-Sohns A, Hirai H, Andersson L C, Hölttä E. Ornithine decarboxylase- and ras-induced cell transformations: reversal by protein tyrosine kinase inhibitors and role of pp130Cas. Mol Cell Biol. 1995;15:6513–6525. doi: 10.1128/mcb.15.12.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouton A H, Burnham M R. Detection of distinct pools of the adapter protein p130CAS using a panel of monoclonal antibodies. Hybridoma. 1997;16:403–411. doi: 10.1089/hyb.1997.16.403. [DOI] [PubMed] [Google Scholar]
- 7.Boyle W J, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 8.Brinkman A, van der Flier S, Kok E M, Dorssers L C J. BCAR1, a human homologue of the adapter protein p130Cas, and antiestrogen resistance in breast cancer cells. J Natl Cancer Inst. 2000;92:112–120. doi: 10.1093/jnci/92.2.112. [DOI] [PubMed] [Google Scholar]
- 9.Burnham M R, Harte M T, Bouton A H. The role of SRC-CAS interactions in cellular transformation: ectopic expression of the carboxy terminus of CAS inhibits SRC-CAS interactions but has no effect on cellular transformation. Mol Carcinog. 1999;26:20–31. doi: 10.1002/(sici)1098-2744(199909)26:1<20::aid-mc3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 10.Burnham M R, Harte M T, Richardson A, Parsons J T, Bouton A H. The identification of p130cas-binding proteins and their role in cellular transformation. Oncogene. 1996;12:2467–2472. [PubMed] [Google Scholar]
- 11.Burnham M R, Bruce-Staskal P J, Harte M T, Weidow C L, Ma A, Weed S A, Bouton A H. Regulation of c-Src activity and function by the adaptor CAS. Mol Cell Biol. 2000;20:5865–5878. doi: 10.1128/mcb.20.16.5865-5878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calalb M B, Polte T R, Hanks S K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src-family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantley L C, Songyang Z. Specificity in recognition of phosphopeptides by src-homology 2 domains. J Cell Sci Suppl. 1994;18:121–126. doi: 10.1242/jcs.1994.supplement_18.18. [DOI] [PubMed] [Google Scholar]
- 14.Cary L A, Han D C, Polte T R, Hanks S K, Guan J L. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheresh D A, Leng J, Klemke R L. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J Cell Biol. 1999;146:1107–1116. doi: 10.1083/jcb.146.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho S Y, Klemke R L. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol. 2000;149:223–236. doi: 10.1083/jcb.149.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolfi F, Garcia-Guzman M, Ojaniemi M, Nakamura H, Matsuda M, Vuori K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc Natl Acad Sci USA. 1998;95:15394–15399. doi: 10.1073/pnas.95.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garton A J, Burnham M R, Bouton A H, Tonks N K. Association of PTP-PEST with the SH3 domain of p130cas; a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene. 1997;15:877–885. doi: 10.1038/sj.onc.1201279. [DOI] [PubMed] [Google Scholar]
- 19.Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, Kaibuchi K, Matsui H, Hatase O, Takahashi H, Kurata T, Matsuda M. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15:6746–6753. doi: 10.1128/mcb.15.12.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan K L, Dixon J E. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 21.Hamasaki K, Mimura T, Morino N, Furuya H, Nakamoto T, Aizawa S I, Morimoto C, Yazaki Y, Hirai H, Nojima Y. Src kinase plays an essential role in integrin-mediated tyrosine phosphorylation of Crk-associated substrate p130Cas. Biochem Biophys Res Commun. 1996;222:338–343. doi: 10.1006/bbrc.1996.0745. [DOI] [PubMed] [Google Scholar]
- 22.Harte M T, Hildebrand J D, Burnham M R, Bouton A H, Parsons J T. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honda H, Oda H, Nakamoto T, Honda Z, Sakai R, Suzuki T, Saito T, Nakamura K, Nakao K, Isikawa T, Katsuki M, Yazaki Y, Hirai H. Ćardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nat Genet. 1998;19:361–365. doi: 10.1038/1246. [DOI] [PubMed] [Google Scholar]
- 25.Honda H, Nakamoto T, Sakai R, Hirai H. P130Cas, an assembling molecule of actin filaments, promotes cell movement, cell migration, and cell spreading in fibroblasts. Biochem Biophys Res Commun. 1999;262:25–30. doi: 10.1006/bbrc.1999.1162. [DOI] [PubMed] [Google Scholar]
- 26.Ishino M, Ohba T, Sasaki H, Sasaki T. Molecular cloning of a cDNA encoding a phosphoprotein, Efs, which contains a Src homology 3 domain and associates with Fyn. Oncogene. 1995;11:2331–2338. [PubMed] [Google Scholar]
- 27.Kanner S B, Reynolds A B, Vines R R, Parsons J T. Monoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinases. Proc Natl Acad Sci USA. 1990;87:3328–3332. doi: 10.1073/pnas.87.9.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klemke R L, Leng J, Molander R, Brooks P C, Vuori K, Cheresh D A. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klinghoffer R A, Sachsenmaier C, Cooper J A, Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 1999;18:2459–2471. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knudsen B S, Feller S M, Hanafusa H. Four proline-rich sequences in the guanine nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J Biol Chem. 1994;269:32781–32787. [PubMed] [Google Scholar]
- 32.Law S F, Estojak J, Wang B, Mysliwiec T, Kruh G, Golemis E A. Human enhancer of filamentation 1, a novel p130cas-like docking protein, associates with focal adhesion kinase and induces pseudohyphal growth in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:3327–3337. doi: 10.1128/mcb.16.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li E G, Stupack D G, Brown S L, Klemke R, Schlaepfer D D, Nemerow G R. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275:14729–14735. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Dy R C, Cance W G, Graves L M, Earp H S. Interactions between two cytoskeleton-associated tyrosine kinases: calcium-dependent tyrosine kinase and focal adhesion tyrosine kinase. J Biol Chem. 1999;274:8917–8924. doi: 10.1074/jbc.274.13.8917. [DOI] [PubMed] [Google Scholar]
- 35.Liu F, Hill D E, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the src homology 3 domain of p130Cas. J Biol Chem. 1996;271:31290–31295. doi: 10.1074/jbc.271.49.31290. [DOI] [PubMed] [Google Scholar]
- 36.Liu F, Sells M A, Chernoff J. Transformation suppression by protein tyrosine phosphatase 1B requires a functional SH3 ligand. Mol Cell Biol. 1998;18:250–259. doi: 10.1128/mcb.18.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manie S N, Astier A, Haghayeghi N, Canty T, Druker B J, Hirai H, Freedman A S. Regulation of integrin-mediated p130Cas tyrosine phosphorylation in human B cells. A role for p59Fyn and SHP2. J Biol Chem. 1997;272:15636–15641. doi: 10.1074/jbc.272.25.15636. [DOI] [PubMed] [Google Scholar]
- 38.Mayer B J, Hamaguchi M, Hanafusa H. A novel oncogene with structural similarity to phospholipase C. Nature. 1998;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 39.Minegishi M, Tachibana K, Sato T, Iwata S, Nojima Y, Morimoto C. Structure and function of Cas-L, a 105-kD Crk-associated substrate-related protein that is involved in β1 integrin-mediated signaling in lymphocytes. J Exp Med. 1996;184:1365–1375. doi: 10.1084/jem.184.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamoto T, Sakai R, Ozawa K, Yazaki Y, Hirai H. Direct binding of C-terminal region of p130Cas to SH2 and SH3 domains of Src kinase. J Biol Chem. 1996;271:8959–8965. doi: 10.1074/jbc.271.15.8959. [DOI] [PubMed] [Google Scholar]
- 41.Nojima Y, Morino N, Mimura T, Hamasaki K, Furuya H, Sakai R, Sato T, Tachibana K, Morimoto C, Yazaki Y, Hirai H. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. J Biol Chem. 1995;270:15398–15402. doi: 10.1074/jbc.270.25.15398. [DOI] [PubMed] [Google Scholar]
- 42.Ohba T, Ishino M, Aoto H, Sasaki T. Interaction of two proline-rich sequences of cell adhesion kinase β with SH3 domains of p130Cas-related proteins and a GTPase activating protein, Graf. Biochem J. 1998;330:1249–1254. doi: 10.1042/bj3301249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oktay M, Wary K K, Dans M, Birge R B, Giancotti F G. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J Cell Biol. 1999;145:1461–1469. doi: 10.1083/jcb.145.7.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Neill G M, Fashena S J, Golemis E A. Integrin signalling: a new Cas(t) of characters enters the stage. Trends Cell Biol. 2000;10:111–119. doi: 10.1016/s0962-8924(99)01714-6. [DOI] [PubMed] [Google Scholar]
- 45.Owen J D, Ruest P J, Fry D W, Hanks S K. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petch L A, Bockholt S M, Bouton A, Parsons J T, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 SRC substrate. J Cell Sci. 1995;108:1371–1379. doi: 10.1242/jcs.108.4.1371. [DOI] [PubMed] [Google Scholar]
- 47.Polte T R, Hanks S K. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polte T R, Hanks S K. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130Cas) are elevated in cytoskeletal-associated fractions following adhesion and Src transformation: requirements for Src kinase activity and FAK proline-rich motifs. J Biol Chem. 1997;272:5501–5509. doi: 10.1074/jbc.272.9.5501. [DOI] [PubMed] [Google Scholar]
- 49.Prasad N, Topping R S, Decker S J. SH2-containing inositol 5′-phosphatase SHIP2 associates with the p130cas adapter protein and regulates cellular adhesion and spreading. Mol Cell Biol. 2001;21:1416–1428. doi: 10.1128/MCB.21.4.1416-1428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren R, Mayer B J, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 51.Ruest P J, Roy S, Shi E, Mernaugh R L, Hanks S K. Phosphospecific antibodies reveal focal adhesion kinase activation loop phosphorylation in nascent and mature focal adhesions and requirement for the autophosphorylation site. Cell Growth Differ. 2000;11:41–48. [PubMed] [Google Scholar]
- 52.Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and c-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakai R, Nakamoto T, Ozawa K, Aizawa S, Hirai H. Characterization of the kinase activity essential for tyrosine phosphorylation of p130Cas in fibroblasts. Oncogene. 1997;14:1419–1426. doi: 10.1038/sj.onc.1200954. [DOI] [PubMed] [Google Scholar]
- 54.Schaller M D, Parsons J T. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaller M D, Hildebrand J D, Parsons J T. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol Biol Cell. 1999;10:3489–3505. doi: 10.1091/mbc.10.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaller M D, Hildebrand J D, Shannon J D, Fox J W, Vines R R, Parsons J T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlaepfer D D, Broome M A, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sieg D J, Ilic D, Jones K C, Damsky C H, Hunter T, Schlaepfer D D. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK-cell migration. EMBO J. 1998;17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tachibana K, Urano T, Fujita H, Ohashi Y, Kamiguchi K, Iwata S, Hirai H, Morimoto C. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase—a putative mechanism for the integrin-mediated tyrosine phosphorylation of Crk-associated substrates. J Biol Chem. 1997;272:29083–29090. doi: 10.1074/jbc.272.46.29083. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, Matuoka K, Takenawa T, Kurata T, Nagashima K, Matsuda M. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci USA. 1994;91:3443–3447. doi: 10.1073/pnas.91.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas J W, Cooley M A, Broome J M, Salgia R, Griffin J D, Lombardo C R, Schaller M D. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–36692. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- 62.van der Flier S, Brinkman A, Look M P, Kok E M, Meijer-van Gelder M E, Klijn J G M, Dorssers L C J, Foekens J A. Bcar1/p130Cas protein and primary breast cancer: prognosis and response to tamoxifen treatment. J Natl Cancer Inst. 2000;92:120–127. doi: 10.1093/jnci/92.2.120. [DOI] [PubMed] [Google Scholar]
- 63.Viguera A R, Arrondo J L R, Musacchio A, Saraste M, Serrano L. Characterizatiion of the interaction of natural proline-rich peptides with five different SH3 domains. Biochemistry. 1994;33:10925–10933. doi: 10.1021/bi00202a011. [DOI] [PubMed] [Google Scholar]
- 64.Vuori K, Hirai H, Aizawa S, Ruoslahti E. Induction of p130cas signaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weidow C L, Black D S, Bliska J B, Bouton A H. CAS/Crk signalling mediates uptake of Yersinia into human epithelial cells. Cell Microbiol. 2000;2:549–560. doi: 10.1046/j.1462-5822.2000.00079.x. [DOI] [PubMed] [Google Scholar]
- 66.Xing Z, Chen H C, Nowlen J K, Taylor S J, Shalloway D, Guan J L. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X, Chattopadhyay A, Ji Q S, Owen J D, Ruest P J, Carpenter G, Hanks S K. Focal adhesion kinase promotes phospholipase C-γ1 activity. Proc Natl Acad Sci USA. 1999;96:9021–9026. doi: 10.1073/pnas.96.16.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]