

SUMMARY
Tumor suppression by TP53 involves cell autonomous and non-cell autonomous mechanisms. TP53 can suppress tumor growth by modulating immune system functions, however, the mechanistic basis for this activity is not well understood. We report that p53 promotes the degradation of the DNA exonuclease TREX1, resulting in cytosolic dsDNA accumulation. We demonstrate that p53 requires the ubiquitin ligase TRIM24 to induce TREX1 degradation. The cytosolic DNA accumulation resulting from TREX1 degradation activates the cytosolic DNA sensing cGAS/STING pathway, resulting in induction of Type I interferons. TREX1 overexpression sufficed to block p53 activation of the cGAS/STING pathway. p53 mediated induction of type I interferon (IFNB1) is suppressed by cGAS/STING knockout and p53’s tumor suppressor activities are compromised by loss of signaling through the cGAS/STING pathway. Thus, our study reveals that p53 utilizes the cGAS/STING innate immune system pathway for both cell intrinsic and cell extrinsic tumor suppressor activities.
Graphical Abstract
eTOC blurb
Ghosh et. al. report that the tumor suppressor, TP53, activates the innate immune response to suppress tumor growth. TP53 promotes the degradation of the cytosolic DNA exonuclease TREX1, resulting in cytoplasmic DNA accumulation and activation of the cGAS/STING pathway. The absence of cGAS or STING compromises p53’s tumor suppressor activity.
INTRODUCTION
The product of the tumor suppressor gene, TP53, was initially identified as a cellular protein that interacts with viral proteins.1-4 Viral proteins often target cellular pathways that govern immune responses as a means of evading detection.5-7 WTp53 has been postulated to play a role in the control of the immune system, although the mechanism(s) and consequences of WTp53 induced crosstalk between tumor and immune cells is insufficiently understood.8-14 Given its multifaceted ability to suppress tumor growth and progression, the high frequency of TP53 mutations in human cancer contributes to the acquisition of several hallmarks of oncogenesis, among which is immune evasion.15 The cGAS/STING cytosolic DNA sensing pathway has emerged as a key mediator of the innate immune response.16 DNA is normally present in the nucleus and mitochondria, and the presence of cytosolic DNA is a danger-associated molecular pattern (DAMP) that is recognized by the pattern recognition receptor (PRR) cGAS.17 In homeostatic conditions, DNA exonucleases degrade cytoplasmic DNA to prevent the inappropriate activation of the cGAS/STING pathway.18,19 Cytoplasmic DNA is detected by cGAS, which becomes enzymatically active and produces a second messenger, cGAMP, which in turn triggers activation of downstream effectors of the pathway, STING/TBK1/IRF3.20-22 Importantly, epigenetic silencing of cGAS in cancer cells disables the innate immune response to the accumulation of cytosolic DNA.23-26 Loss of function in the cGAS pathway contributes to tumor development through cell-intrinsic and –extrinsic mechanisms.26-30 Mutant p53 has been shown to suppress downstream signaling from cGAS/STING pathway, thereby facilitating immune evasion.31 In contrast, wildtype p53 has been implicated in activating or contributing to downstream signaling from cGAS/STING although the mechanism remains unknown.31,32 Herein we report that WTp53 promotes the degradation of the DNA exonuclease TREX1, thereby resulting in the accumulation of cytoplasmic DNA which triggers activation of the cGAS/STING pathway. Notably, our data indicate that WTp53 employs the pathogen recognition receptor cGAS for tumor suppression.
RESULTS
WTp53 activates the cGAS/STING innate immune pathway
Cytosolic double stranded DNA is sensed by cGAS which then produces the secondary messenger molecule cGAMP, which then activates STING and promotes TBK1 autophosphorylation and phosphorylation of STING and IRF3 resulting in the induction of the type I interferon response (Fig. 1A). We previously reported that mutant p53 can suppress downstream signaling from the cGAS/STING pathway.31 In contrast, we found that wildtype p53 activates the pathway.31 We extended this observation by examining whether wildtype p53 regulated downstream signaling in several different cell models in which we modulated p53 expression through different approaches. In three different mouse cell lines, we observed that wildtype p53 correlated with increased signaling of the cGAS/STING pathway as indicated by the increased phosphorylation of TBK1 substrates (phospho-TBK1, phospho-IRF3 and phospho-STING). Comparison of MEFs derived from p53 wildtype (p53+/+) or null (p53−/−) mice showed a clear positive correlation between WTp53 and cGAS/STING pathway. (Fig. 1B) We engineered p53 null 4T1 breast cancer cell line to inducibly express WTp53 and we observed that WTp53 expression increased TBK1 substrate phosphorylation. (Fig. 1C) Moreover, p53 knockdown with two different shRNAs reduced TBK1 substrate phosphorylation in CT26 murine colorectal cancer cells. (Fig. 1D) Our data indicates that WTp53 stimulates the cGAS/STING pathway.
Figure 1: WTp53 activates the cGAS/STING innate immune pathway.

(A) Schematic diagram shows cytosolic DNA sensing pathway and the phosphorylation events of TBK1, STING and IRF3. Western blot analysis of (B) p53+/+ and p53 −/− mouse embryonic fibroblast (MEFs). (C) P53 null mouse 4T1 cells engineered to inducibly express WTp53. (D) P53 was knocked down using two different shRNAs in CT26 cells and cell lysates were analyzed by western blot. (E-F) Heat maps show the differential gene expression as accessed by Nanostring (n=3) in (E) A549 shControl and shp53 and (F) H1299 Uninduced or induced WTp53. (G) p53+/+ and p53−/− MEFs were treated with 2 μg/ml of HT-DNA for 3 h and harvested for western blot analysis. (H) P53 null H1299 cells were engineered to inducibly express WT53. Cells were treated for 24 h with Doxycycline to induce p53 and then treated with 2 μg/ml of HT-DNA for 3 h and subjected to western blot analysis. (I and J) P53 null H1299 and 4T1 cells were induced to express WTp53, and A549 shControl or shp53 cells were treated with 2 μg/ml HT-DNA for 18 h, and cells were harvested for RT-PCR analysis of IFNB1 mRNA (I) or the conditioned medium was harvested for ELISA detection of secreted IFNB1 (J). (K) Representative CLSM images of IRF3 in H1299 cells induced to express WTp53 and treated with HT-DNA for 3 h. Scale bar=10 μm.
Quantification graphs: In all panels, error bars represent mean with standard deviation. p values are based on Student’s t test. ***p < 0.001, **p < 0.01, *p < 0.05, ns=non-significant. See also Figure S1.
Since WTp53 activated the cGAS/STING pathway, we investigated if this resulted in differences in expression of interferon stimulated genes (ISG) via Nanostring analysis. We observed that WTp53 (in A549 and H1299 cells) had a high correlation with increased expression of ISGs. (Fig. 1E and 1F) We further validated the upregulation of key ISGs using real-time RT-PCR analysis of IFNB1, IFIT1, ISG15 and CXCL10 in H1299 induced WTp53, p53+/+ or p53−/− MEFs and 4T1 induced WTp53 cells. (Fig. S1A-S1C)
The basal activity of the cGAS/STING pathway is due to the presence of endogenous cytoplasmic DNA, and transfection of sheared Herring Testes DNA (HT-DNA) can augment pathway activation. To determine if WTp53 also contributed to the response to HT-DNA, we transfected MEFs (p53+/+ vs. p53−/−), H1299 (with inducible WTp53), 4T1 (with inducible WTp53) and A549 (shControl vs. shp53) with HT-DNA and assessed the impact on the cGAS/STING pathway. In MEFs, we observed that HT-DNA induced more pronounced phosphorylation of TBK1 substrates in p53+/+ MEFs as compared to p53−/− MEFs. (Fig. 1G) Similarly, in WTp53 inducible H1299 cells, we observed a modest induction of TBK1 substrate phosphorylation in HT-DNA transfected uninduced cells, and this was markedly increased in cells induced to express WTp53. (Fig. 1H) Increased phosphorylation of TBK1 substrates was also observed in 4T1 cells in which we induced WTp53 and treated with HT-DNA. (Fig. S1D) Conversely, shRNA knockdown of p53 in A549 cells strongly reduced HT-DNA activation of the cGAS/STING pathway. (Fig. S1E) Assessment of 5 different siRNAs targeting p53 confirmed that p53 knockdown in A549 cells resulted in reduced TBK1 substrate phosphorylation, thus ruling out an off-target effects of the shRNA. (Fig. S1F) Our results indicate that WTp53 can stimulate basal and augment agonist activation of the cGAS/STING pathway.
To determine if increased TBK1 activity in response to WTp53 expression resulted in activation of IRF3’s transcriptional activity, we performed RT-PCR analysis of its canonical transcriptional target, interferon beta 1 (IFNB1). WTp53 induction in H1299 and 4T1 cells increased basal levels of IFNB1 mRNA and also caused a higher induction in response to HT-DNA treatment. (Fig. 1I) Conversely, shRNA knockdown of p53 in A549 cells reduced both the basal expression and HT-DNA induction of IFNB1. (Fig. 1I) Of note we previously showed from the TCGA breast cancer data that WTp53 correlates more with the interferon responsive genes (ISGs) than mutant p53.31 In agreement with the transcriptional induction of IFNB1, we observed higher amounts of secreted IFNB1 in cells induced to express p53 (H1299 and 4T1), and reduced IFNB1 secretion in response to p53 knockdown (A549). (Fig. 1J) These results indicate that WTp53 mediated stimulation of the cGAS/STING pathway results in IRF3’s functional activation.
Inactive IRF3 resides in the cytoplasm and upon its phosphorylation by TBK1, translocate to the nucleus.33,34 To corroborate our findings that WTp53 activates IRF3, we assessed its subcellular localization. In unstimulated cells, we observed that GFP-tagged IRF3 was primarily located in the cytoplasm. (Fig. 1K) Induction of WTp53 alone was sufficient to promote approximately 20% of cells to exhibit nuclear accumulation of GFP-IRF3. HT-DNA treatment induced GFP-IRF3 nuclear translocation in 45% of the uninduced cells and this was further increased to 90% upon WTp53 induction (Fig. 1K and S1G). As a parallel approach to examine IRF3’s localization, we performed subcellular fractionation and detected IRF3 by western blot. In both H1299 and 4T1, WTp53 induction resulted in increased phospho- and total IRF3 accumulation in the nuclear fraction (Fig. S1H). Collectively, our data strongly suggests that WTp53 induces basal and agonist mediated innate immune response.
WTp53 promotes cGAS/STING/IRF3 mediated apoptosis
Previously, it was reported that potent activation of STING results in apoptosis.32 Since we observed that WTp53 could not only stimulate basal but also augment HT-DNA mediated cGAS/STING signaling, we performed FACS analysis to determine if it also dictated the cellular response to activation of the pathway using annexin V-FITC/PI staining. (Fig. S2A) In uninduced H1299 cells, we observed that HT-DNA transfection induced apoptosis in a dose dependent manner, which further increased in cells induced to express WTp53. (Fig. 2A) To determine if the sensitization to HT-DNA affected the kinetics of cell death, we performed a time-course experiment. We found that HT-DNA treatment induced apoptosis in a temporal manner and WTp53 induction accelerated this process. (Fig. 2B) Of note, we utilized a WTp53 cDNA that carries a proline at codon 72, a polymorphism that has been shown to minimally induce apoptosis.35 In agreement, we observed that WTp53 expression alone did not induce apoptosis. (Fig. 2A) Furthermore, we also treated A549 shControl or shp53 cells with HT-DNA and found that p53 knockdown blunted the apoptotic response to HT-DNA treatment. (Fig. 2C) To further confirm that HT-DNA mediate cell death is apoptosis, we treated cells with the caspase inhibitor (ZVED) after HT-DNA treatment and found ZVED treatment blunted the pro-apoptotic effect of HT-DNA treatment. (Fig. S2B) We also found that the combination of HT-DNA treatment with p53 expression induced PARP cleavage. (Fig. S2C) Thus, our data suggests that WTp53’s potent stimulation of cGAS/STING signaling surpasses the threshold required for apoptosis to occur.
Figure 2: WTp53 promotes CGAS/STING/IRF3 mediated apoptosis.

Graphical representation of apoptosis quantification by flow cytometry of H1299 induced WTp53 (A) treated with increasing amount of HT-DNA for 24 h and (B) HT-DNA was treated for different time as indicated. (C) shControl or shp53 A549 cells were treated with HT-DNA for 24 h and subjected to FACS analysis for apoptosis. H1299 doxycycline inducible WTp53 cells were stably knockout for cGAS, STING and IRF3. Representative graphs indicate quantification analysis of apoptotic death analyzed by flow cytometry of (D) non-target (NT), cGASKO and STINGKO (E) non-target (NT) and IRF3KO H1299 cells induced WTp53 treated with 2 μg/ml or 4 μg/ml of HT-DNA for 24 h. Cells were harvested, stained with Annexin V-FITC and PI and subjected to flow cytometry analysis. (E-F) Non-target (NT), cGASKO and STINGKO H1299 cells induced WTp53 and subjected to (E) Western blot (F) RT-PCR analysis for IFNB1 mRNA. (G) Non-target (NT) and IRF3KO H1299 cells induced WTp53 cells were subjected to RT-PCR analysis for IFNB1 mRNA.
Quantification graphs: In all panels, error bars represent mean with standard deviation. p values are based on Student’s t test. ***p < 0.001, **p < 0.01, ns=non-significant. See also Figure S2
Thus, we sought to determine if the enhanced apoptotic response in WTp53 induced cells treated with HT-DNA was indeed due to activation of the cGAS/STING pathway. We used CRISPR-Cas9 to knockout cGAS, STING and IRF3 and tested how loss of these genes affected WTp53’s sensitization to HT-DNA. As expected, cGAS knockout reduced the population of cells undergoing apoptosis in response to HT-DNA. Importantly, cGAS knockout completely abrogated the ability of WTp53 to sensitize to HT-DNA treatment. (Fig. 2D) Additionally, both STING and IRF3 knockout cells also rescued the cells from WTp53 sensitization to activation of the pathway. (Fig. 2D and 2E) The p53 transcriptional target IFI16 has been postulated to be a cytosolic DNA sensor and thus we assessed if it was involved in this context. We found that unlike cGAS, IFI16 is dispensable for WTp53 activation of the cGAS/STING pathway and sensitization to HT-DNA. (Fig. S2D) To further assess whether other nucleic acids (like RNA) have a similar effect as DNA, we compared the response to HT-DNA and Poly (I:C) a synthetic analog of double-stranded RNA treatment. Our data shows that WTp53 only augments the response to HT-DNA and not Poly (I:C) treatment. (Fig. S2E) Taken together our data indicates that WTp53 specifically sensitizes to HT-DNA via stimulation of the cGAS/STING pathway.
Since we observed that WTp53 stimulated IFNB1 mRNA expression, and WTp53 has previously been shown to indirectly regulate IFNB1 expression through IRF7 and IRF9, we checked if this was dependent on the cGAS/STING pathway.12 Loss of either cGAS or STING resulted in reduced IFNB1 mRNA levels and abrogated WTp53 induction of IFNB1 mRNA. (Fig. 2E and 2F) The involvement of this pathway was further supported by the observation that IRF3 knockout cells also failed to induce IFNB1 mRNA in response to WTp53. (Fig. 2G and S2F) In contrast, IFI16 is not required for WTp53 to stimulate the cGAS/STING pathway since IFI16 knockout had no effect on the stimulation TBK1 substrate phosphorylation or the induction of IFNB1 mRNA. (Fig. S2G and S2H) Taken together our data suggests that WTp53 mediated induction of Type I interferon response is cGAS, STING and IRF3 dependent.
WTp53 promotes TREX1 degradation resulting in cytosolic DNA accumulation
Our western blot analysis did not indicate that p53 was stimulating the pathway by inducing expression of the components of the cGAS/STING pathway and thus we assessed if it was regulating the activity of cGAS. In both H1299 and 4T1, WTp53 induction resulted in increased cGAMP, the second messenger produced upon agonist activation of cGAS.30 (Fig. 3A and 3B) We speculated that the activation of cGAS enzymatic activity by WTp53 is likely due to an increased presence of an endogenous cGAS agonist and thus we assessed the levels of cytoplasmic DNA using either an anti-dsDNA antibody or PicoGreen staining. In A549 cells, we detected cytoplasmic DNA, which decreased after p53 knockdown. (Fig. 3C and 3D) Similarly, detection of cytoplasmic DNA by PicoGreen staining exhibited a similar trend of reduced cytoplasmic DNA after p53 knockdown. (Fig. 3E and 3F) Conversely, WTp53 expression in H1299 cells resulted in increased detection of cytoplasmic DNA by anti-dsDNA antibody and Picogreen staining. (Fig. 3G-3J) We also observed that WTp53 induction in 4T1 cells increased the level of cytoplasmic DNA detected with PicoGreen staining. (Fig. S3A and S3B) To confirm that we were detecting dsDNA in the cytoplasm, we treated cells with dsDNAse or RNase. Our results show that treatment with dsDNAase, but not RNase, eliminated the detection of cytoplasmic DNA. (Fig. S3C and S3D) Since cGAS can also be activated by micronuclei, we also attempted to detect the presence of micronuclei but did not find any in either uninduced or induced p53 conditions. (Fig. S3E) These results suggest that WTp53 stimulates cGAS activity by increasing the abundance of cytoplasmic dsDNA.
Figure 3: WTp53 promotes TREX1 degradation resulting in cytosolic DNA accumulation.

Representative graphs indicate cellular cGAMP levels in (A) H1299 and (B) 4T1 cells with inducible WTp53. (C) Representative confocal microscopy images of cytosolic dsDNA in A549 shControl or shp53 cells as detected with a dsDNA specific antibody. (D) Graphical representation shows dsDNA quantitation in A549 shControl or shp53 cells. (E) Confocal live-cell imaging of A549 shControl or shp53 cells stained with 3 μl/ml PicoGreen (green) for 1 hr and counter stained with Hoechst33342 for 15 min. (F) Graphical representation shows PicoGreen quantitation in the indicated cohorts. (G) Representative confocal images of H1299 cells expressing inducible WTp53 stained with dsDNA specific antibody. (H) Graphical representation shows dsDNA quantitation in H1299 induced WTp53 cells. (I) Representative confocal live-cell imaging of H1299 induced WTp53 cells stained with PicoGreen and counter stained with Hoechst33342. (J) Graphical representation shows PicoGreen quantitation in the indicated cohorts. (K-M) Representative graphs indicates % exonuclease activity in (K) A549 (shControl or shp53) (L) H1299 inducible WTp53 cells and (M) 4T1 inducible WTp53 cells. (N) A549 shControl or shp53 cells were subjected to western blot. (O-P) Representative Immunoblots of (O) H1299 cells induce WTp53 and (P) p53+/+ and p53 −/− mouse embryonic fibroblast (MEFs). (Q) In H1299 cells, Myc-TREX1 was co-transfected with increasing amount of HA-WTp53 and subjected to western blot analysis. (R) H1299 cells were induced with doxycycline to express WTp53 for 24 hrs and treated with MG132 (20 μM) for 6 hrs after which cells were harvested for western blot analysis.
Quantification graphs: In all panels, error bars represent mean with standard deviation. p values are based on Student’s t test. ***p < 0.001, **p < 0.01, ns=non-significant. Scale Bar 10 μm. See also Figure S3.
Since cytoplasmic dsDNA is degraded by exonucleases, we assessed if WTp53 regulated cellular exonuclease activity. WTp53 knockdown in A549 cells resulted in an approximately 2.5 fold increase in exonuclease activity, whereas WTp53 induction in H1299 cells resulted in an almost 4 fold decrease. (Fig. 3K and 3L) Similarly, WTp53 induction in 4T1 cells resulted a reduction in exonuclease activity by 50%. (Fig. 3M) The 3′→5′ cytosolic exonuclease, TREX1, is the predominant exonuclease, accounting 60 to 70% of total cellular exonuclease activity and thus we assessed its expression.18,36-38 TREX1 protein levels were induced after p53 knockdown in A549 and CT26 cells, and reduced in H1299 and 4T1 cells upon p53 induction. (Fig. 3N, S3F and 3O, S3G) Comparison of p53+/+ vs. p53−/− MEFs showed increased TREX1 protein in the latter. (Fig. 3P) Of note, we did not see any change in TREX1 protein after induction of p53R248W in H1299 or mutant p53 knockdown in MDA-MB-231 cells. (Fig. S3H) Analysis of TREX1 mRNA levels in A549 and H1299 cells indicated that WTp53 did not alter its expression, suggesting that it instead controls TREX1 protein turnover. (Fig. S3I) Since WTp53 can induce senescence and TREX1 downregulation occurs in senescent cells, we assessed by β-galactosidase (β-Gal) staining if WTp53 induction in 4T1 cells resulted in senescence.39,40 We did not detect β-Gal staining in 4T1 cells induced to express WTp53 for four days. In contrast, we observed a modest induction of β-Gal staining when we concurrently induced WTp53 and treated with the DNA damaging agent doxorubicin. These data suggest that WTp53 is able to downregulate TREX1 protein independently of its ability to induce senescence (Fig. S3J). We performed cycloheximide chase experiments to determine if this was the case. The half-life of the GFP-TREX1 and endogenous TREX1 protein in uninduced H1299 cells was approximately 10.8 hours and 6.9 hours respectively, whereas in WTp53 induced cells it was reduced to 5.5 hours for GFP-TREX1 and 4.7 hours for TREX1, thus indicating a faster turnover. (Fig. S3K and S3L) We co-transfected Myc-TREX1 with increasing amounts of HA-WTp53 and observed a dose dependent reduction in Myc-TREX1. (Fig. 3Q) The reduction in endogenous TREX1 levels was rescued by MG132 treatment, indicating that WTp53 promotes TREX1 degradation through the proteasomal pathway. (Fig. 3R) We reasoned that if MG132 treatment rescued TREX1 in the presence of WTp53, it should alter cytosolic DNA accumulation. As anticipated we found WTp53 mediate induction of cytosolic DNA was reduced down by MG132 treatment. (Fig. S3M and S3N) To confirm that reduction in TREX1 protein levels in response to p53 was sufficient to trigger the cGAS/STING pathway, we transfected cells with two different doses of siRNA to knockdown TREX1. We observed that even a modest reduction in TREX1 protein was sufficient to trigger this pathway. (Fig. S3O) These results suggests that WTp53 induces the accumulation of cytoplasmic dsDNA by promoting TREX1 degradation.
TRIM24 is an ubiquitin ligase for TREX1
To date, an E3 ubiquitin ligase that promotes TREX1 degradation has not been identified. We speculated that a known p53 transcriptional target with E3 ubiquitin ligase activity might be involved in TREX1 degradation. There are 6 well studied E3 ubiquitin ligases that are p53 transcriptional targets: MDM2, COP1, PIRH2, TRIM24, FBXW7 and SIAH. We transfected H1299 cells with siRNAs targeting these ubiquitin ligases and found that TRIM24 knockdown potently induced TREX1 protein. (Fig. S4A) We also observed a modest induction in TREX1 after FBXW7 knockdown but it also significantly induced TREX1 mRNA. In contrast, TRIM24 knockdown only induced TREX1 protein and had no effect on its mRNA and thus we further assessed if TRIM24 controlled TREX1 turnover. (Fig. S4B) We confirmed in A549 cells that siRNA mediated TRIM24 knockdown induces TREX1. (Fig. 4A) Co-transfection of Myc-TREX1 with Flag-TRIM24 resulted in reduced Myc-TREX1 protein that could be rescued by MG132, indicating that TRIM24 promotes TREX1 degradation through the proteasome. (Fig. 4B) TRIM24’s ability to promote TREX1 degradation required the ring domain since its deletion prevented TREX1 degradation. (Fig. S4C) Additionally, co-immunoprecipitation studies showed that Flag-TRIM24 co-precipitated with GFP-TREX1, suggesting that TRIM24 directly interacts with TREX1 to promote its degradation. (Fig. 4C) Next we performed ubiquitination assays to determine if TRIM24 can promote TREX1 ubiquitination. Transfection of GFP-TREX1 alone resulted in a modest amount of ubiquitination and this was greatly enhanced by co-transfection of Flag-TRIM24. (Fig. 4D) To gain insight into the TREX1 lysine residues required for TRIM24 mediated degradation, we mutated six different TREX1 lysines (K66R, K75R, K160R, K175R, K271R, K277R) and we also deleted the c-terminal region (dCTR). Our degradation experiment showed that lysine mutants K66, K160, K175, K271, K277 and dCTR are protected from TRIM24. (Fig. S4D) Since TREX1 can also translocate to the nucleus, which we predict would result in cytoplasmic DNA accumulation, we examined if WTp53 altered its subcellular localization.41 We detected TREX1 in the cytoplasm and its subcellular localization did not change in response to p53. (Fig. S4E) TRIM24 has previously been shown to be a p53 transcriptional target.42,43 We confirmed that WTp53, and not mtp53 (p53R248W) bound to the TRIM24 promoter and induced TRIM24 mRNA. (Fig. S4F) We observed that despite the induction of TRIM24 mRNA by WTp53, the TRIM24 protein levels did not change. (Fig. S4G and S4H) TRIM24 has been reported to promote its own auto-ubiquitination and degradation.42,43 To test this in our H1299 inducible WTp53 system, we treat H1299 induced WTp53 cells with MG132. We observed that MG132 could prevent the downregulation of TRIM24, supporting the previous studies suggesting that it promotes its auto-degradation. (Fig. S4H) To determine if WTp53 requires TRIM24 to induce TREX1 degradation, we used siRNA to knockdown TRIM24 in H1299 cells transfected with empty vector or a HA-p53 expression vector. We observed that TREX1 downregulation by p53 could be rescued by TRIM24 knockdown, and thus we concluded that TRIM24 mediates TREX1 degradation in response to WTp53. (Fig. 4E)
Figure 4: TRIM24 is an ubiquitin ligase for TREX1.

(A) A549 cells were transfected with two different siRNA for TRIM24 and subjected to western blot analysis. (B) H1299 cells were co-transfected with Myc-TREX1 and Flag-TRIM24 for 24 h. and prior to harvesting cells were treated with MG-132 for 6 h. (C) p53 null H1299 cells were co-transfected with GFP-TREX1 and Flag-TRIM24. Prior to harvesting, cells were treated with MG-132 for 6 h. Cells were lysed and GFP-TREX1 was immunoprecipitated from the whole cell lysate. Lysates and immunoprecipitates (IP) were analyzed by western blotting. (D) H1299 cells were co-transfected with GFP-TREX1, poly ubiquitin and Flag-TRIM24 for 24 h. Prior to harvesting, cells were treated with MG-132 for 6 h. Cells were harvested under denaturing conditions by using boiling 1% SDS buffer and lysates were then immunoprecipitated and were processed for Western blotting. (E) TRIM24 knockdown (SiTRIM24) H1299 cells were transfected with HA-WTp53 and subjected to western blot. (F) H1299 induced WTp53 cells were stably overexpress GFP-TREX1 or GFP-TREX1 D18N. Cells were induced to express WTp53 and subjected to western blot. (G) Representative confocal microscopic immunofluorescence (IF) images are showing cytosolic dsDNA in H1299 induced WTp53 overexpress GFP-TREX1 or GFP-TREX1 D18N. (H) Graphical representation shows dsDNA quantitation in the indicated cells. (I) Graphical representation of apoptosis quantification by flow cytometry of H1299 induced WTp53 cells were stably overexpress GFP-TREX1 or GFP-TREX1 D18N cells that were treated with 2ug/ml of HT-DNA for 24 hrs. (J) Representative CLSM images are showing cytosolic dsDNA in parental or Rho° H1299 induced WTp53. (K) Graphical representation shows dsDNA quantitation in the indicated cells. (L) Parental or Rho° H1299 induced WTp53 and subjected to western blot analysis.
Quantification graphs: In all panels, error bars represent mean with standard deviation. p values are based on Student’s t test. ***p < 0.001, **p < 0.01, ns=non-significant. Scale Bar 10 μm. See also Figure S4.
Our data thus far implicate downregulation of TREX1 protein and exonuclease activity as the mechanism by which WTp53 activates the cGAS/STING pathway and sensitizes cells to agonist induced apoptosis. To test this directly, we co-expressed WTp53 with wildtype TREX1 or its catalytically dead mutant TREX1 D18N, and assessed TBK1 substrate phosphorylation and the accumulation of cytoplasmic DNA. We observed that wildtype TREX1, but not the inactive TREX1 D18N mutant, was able to block the phosphorylation of TBK1 substrates in response to WTp53 expression. (Fig. 4F) Furthermore, wildtype TREX1 overexpression reduced the appearance of cytoplasmic DNA in response to WTp53 and this was dependent on its catalytic activity. (Fig. 4G and 4H) Furthermore, wildtype TREX1 (and not the D18N mutant) suppressed the induction of IFNB1 mRNA by WTp53. (Fig. S4I) The observation that WTp53 could augment the activation of cGAS/STING resulting in apoptosis, led us to determine if TREX1 could prevent this from occurring. Expression of WTp53 and treatment with HT-DNA resulted in approximately 55% of the cells undergoing apoptosis. Consistent with our observations that TREX1 could block the activation of the cGAS/STING pathway by WTp53, we observed that TREX1 overexpression suppressed WTp53’s ability to sensitize to HT-DNA. (Fig. 4I) The protection afforded by TREX1 overexpression required its catalytic activity since the D18N mutant was unable to prevent apoptosis. Our data indicate that downregulation of TREX1 by WTp53 augments cGAS/STING pathway activation and sensitizes to innate immune signaling induced apoptosis.
Endogenous cytoplasmic DNA has been shown to originate from either the nucleus or mitochondria. To test if cytoplasmic DNA originated from the mitochondria, we cultured H1299 cells with media supplemented with a low-concentration of ethidium bromide for 2 weeks in order to generate Rho° cells depleted of mitochondrial DNA.44 (Fig.S4J) We found that WTp53 induction in Rho° cells had reduced accumulation of cytoplasmic DNA. (Fig. 4J, 4K and S4K) Moreover, WTp53 failed to activate the cGAS/STING pathway as TBK1 and its substrate phosphorylation was not induced after p53 induction in the Rho° cells. (Fig. 4L) To further substantiate that the mitochondrial permeability transition pore was required for WTp53 to activate the cGAS/STING pathway, we treated cells with cyclosporin A (Cys A), which prevents pore formation by binding to cyclophilin.40-42 Treatment with cyclosporin A blocked WTp53 activation of the cGAS/STING pathway in a dose dependent manner. (Fig. S4L) Taken together, our data suggests that WTp53 requires the mitochondria to activate the cGAS/STING pathway.
Pharmacological activation of p53 triggers the de-repression of endogenous retroviruses and this has been proposed to trigger interferon expression through a putative mechanism involving PAMP RNA sensing machinery.11 We observed that nutlin treatment stimulated phosphorylation of TBK1 and the downregulation of TREX1 in a dose dependent manner indicating pharmacological activation of endogenous p53 induces the cGAS/STING pathway. (Fig. S4M) The WTp53 transcriptional target MAVS is a mitochondrial protein that is a downstream effector for various PAMP RNA sensing machinery, and can activate TBK1 and interferon expression.45-47 To determine if WTp53 stimulated TBK1 through a PAMP RNA sensing mechanism in a MAVS-dependent manner, we generated MAVS knockout cells with CRISPR/Cas9. Deletion of MAVS had a minimal impact on the activation of TBK1 by WTp53. (Fig. S4N) Our data reinforce the notion that accumulation of cytoplasmic DNA leaked from the mitochondria underlies the activation of the cGAS/STING pathway by WTp53.
WTp53 promotes cGAS/STING dependent antitumor immune response.
Our data led us to posit that WTp53 works through cGAS to regulate tumor growth by recruitment of immune cells. To test this we generated 4T1 WTp53 inducible cells with a non-targeting or cGAS-targeting CRISPR vector and determined the effect of cGAS loss on WTp53’s control of tumor growth. Consistent with our results showing that cGAS is required for p53 to activate the pathway and induce IFNB1, we observed that WTp53 induction did not induce TBK1 substrate phosphorylation or IFNB1 mRNA in cGAS KO 4T1 cells. (Fig. 5A and 5B) We injected 4T1 inducible WTp53/cGAS knockout cells into the mammary fat pad of female BALB/c mice. We observed that cGASKO tumors grew faster than the control, uninduced tumors. In contrast, doxycycline induction of WTp53 expression resulted in a sustained lack of tumor growth. Surprisingly, WTp53 induced growth suppression was dependent on cGAS since after an approximately 3-day delay in growth, the WTp53-induced/cGASKO tumors resumed growing. (Fig. 5C, 5D, S5A and S5B) Immunophenotyping analysis revealed that the blunted growth of the WTp53 induced tumors correlated with a dramatic increase in tumor infiltration by CD45+, CD4+, CD8+ and NK cells. (Fig. 5E-5H) In WTp53-induced/cGASKO tumors, infiltration by these types of immune cells was similar to control tumors, suggesting that cGAS is required for WTp53 to recruit these cells.
Figure 5: WTp53 promotes cGAS/STING dependent antitumor immune response.

(A) Immunoblots of 4T1 doxycycline inducible WTp53 non-target (NT) or cGAS knockout cells. (B) RT-PCR analysis for IFNB1 in 4T1 doxycycline inducible WTp53 non-target (NT) or cGAS knockout cells. (C) 4T1 inducible WTp53 non-target or cGASKO cells were injected into the mammary gland of immunocompetent female BALB/c mice (n=5). Mice were given doxycycline orally to induce WTp53 on the indicated days. Tumor growth was monitored and measured using slide calipers. All mice were sacrificed on day 23 and graphical quantification represents the tumor growth rate. (D) Tumor volume of the indicated 4T1 tumor cohorts on Day 23. (E-H) Representative graphs showed FACS quantification of (E) CD45+ T-lymphocytes (F) CD3+CD4+ T-helper (G) CD3+CD8+ cytotoxic T-lymphocyte and (H) NK cells per milligram (mg) of 4T1 induced WTp53 non-target or cGASKO tumor tissues (n=5). 4T1 doxycycline inducible WTp53 non-target (NT) or STING knockout cells were subjected to (I) western blot or (J) RT-PCR for IFNB1. (K) 5 x 104 4T1 inducible WTp53 non-target (NT) or STINGKO cells were injected into the mammary gland of immunocompetent female BALB/c mice (n=4). Mice were given doxycycline orally to induce WTp53 on the indicated days (upward arrows). Tumor growth was monitored and measured using slide calipers. All mice were sacrificed on day 23 and graphical quantification represents the tumor growth rate in BALB/c mice. (L) Tumor volume of the indicated 4T1 tumor cohorts on Day 23. (M-P) Representative graphs showed FACS quantification of (M) CD45+ T-lymphocytes (N) CD3+CD4+ T-helper (O) CD3+CD8+ cytotoxic T-lymphocyte and (P) NK cells per milligram (mg) of 4T1 induced WTp53 non-target or STINGKO tumor tissues (n=4).
Quantification graphs: In all panels, error bars represent mean with standard error mean. In scatter dot plots, each dot represent one mouse, p values are based on Student’s t test. ***p < 0.001, **p < 0.01, *p < 0.05, ns=non-significant. See also Figure S5.
To further assess the contribution of the immune system to WTp53 growth suppression in vivo, we repeated the experimental approach using NOD/SCID mice. In these immunocompromised mice, the growth advantage observed with the cGASKO cells was largely eliminated, indicating that the immune response was required for the observed differences in tumor growth in the syngeneic model. (Fig. S5C-S5E) WTp53 induction slowed tumor growth as compared to the other groups, however, unlike in the BALB/c mice, the tumors continued to grow and did not plateau. The lack of sustained growth arrest suggests that the immune system has a key role in p53-mediated growth suppression. Importantly, cGASKO again rescued the reduced tumor growth of the WTp53 induced tumors. Of note we also assessed if p53 induction promotes apoptosis resulting in restricted tumor growth. Our TUNEL staining in 4T1 induced WTp53 tumors from both BALB/c and NOD/SCID mice showed that there was no significant changes in the TUNEL staining indicating tumor growth suppression by p53 induction is not due to apoptosis. (S5F and S5G) These data suggest that cGAS is a downstream effector of WTp53’s growth inhibitory function through both cell-autonomous and non-cell autonomous mechanisms.
cGAS has been shown to regulate cellular phenotypes through STING-dependent and independent mechanisms.48 The pronounced requirement for cGAS in WTp53 tumor suppression led us to investigate if STING was similarly involved. We used CRISPR/Cas9 to generate WTp53 inducible 4T1 cells that lack STING (STINGKO). Similar to the cGASKO data above, STINGKO cells failed to induce TBK1 substrate phosphorylation or IFNB1 mRNA in response to WTp53 expression. (Fig. 5I, 5J) As expected, we observed that WTp53 reduced proliferation in vitro and that STINGKO cells had a slightly higher proliferation rate but it was not statistically significant different from control cells. (Fig. S5H) When injected into syngeneic mice, the STINGKO tumors grew slightly faster than the control tumors but the difference was not statistically significant. (Fig. 5K, 5L) As before , we observed that WTp53 expression induced a stable suppression of tumor growth. Strikingly, STING knockout rescued WTp53 induced tumor growth suppression. (Fig. 5K, 5L and S5I-S4J) In keeping with a role for the cGAS/STING pathway in the recruitment of immune cells by WTp53, we observed that STING KO prevented the spike in TILs in the WTp53-induced/STINGKO tumors. (Fig. 5M-5P)
To further assess the immune system involvement in WTp53 mediated tumor growth suppression in vivo, we injected 4T1 STINGKO inducible WTp53 cells into immunodeficient NOD/SCID mice. Whereas WTp53 induced tumors grew slower, STING KO tumors grew faster but not in a statistically significant manner compared to the non-targeted control tumors. (Fig. S5K-S5M) STINGKO partially rescued WTp53 mediated tumor growth suppression suggesting that like cGAS, STING also is a downstream effector of WTp53’s growth inhibitory function. Taken together, our study reveals that WTp53 engagement of the cGAS/STING pathway is critical for its tumor suppressor activities through tumor cell autonomous and non-cell-autonomous mechanisms.
TREX1 deficiency induces a WTp53 dependent antitumor immune response
Genetic perturbation of TREX1 function results in a severe autoimmune response due to the cellular inability to clear cytosolic DNA.19,49-52 This autoimmune response due to TREX1 deficiency can be rescued by loss of either cGAS, STING or IRF3, indicating that these genes are epistatic.19,51,53-56 Since WTp53 positively regulates the cGAS/STING pathway, we considered the possibility that WTp53 knockdown may also negate the immunostimulatory effect of TREX1 loss. We used CT26 cells to test this possibility in vivo in a syngeneic BALB/c mouse tumor model. We generated CT26 cells with shRNA mediated knockdown of either p53, TREX1 alone or both. (Fig. S6A) Western blot analysis of these different shRNA knockdown cells showed a decrease in cGAS/STING pathway upon p53 knockdown. As expected, cells with TREX1 knockdown had elevated activity of the cGAS/STING pathway as indicated by the increased phosphorylation of TBK1 substrates and higher IFNB1 mRNA expression. The combined knockdown of p53 and TREX1 reverted the cGAS/STING pathway activity and IFNB1 mRNA back to baseline control levels. (Fig. S6B) Next, we injected CT26 shControl, shp53, shTREX1 and shp53/shTREX1 cells subcutaneously in BALB/c mice and monitored tumor growth. The hyperactivation of the cGAS/STING pathway resulted in a severe growth delay in the TREX1 knockdown tumors. (Fig. 6A) In contrast, p53 knockdown cells produced tumors that grew faster than the control. Strikingly, the combined knockdown of TREX1 and p53 resulted in tumors that grew at the same rate as the control tumors. (Fig. 6A and S6C, S6D) The observed differences in tumor growth in vivo did not reflect differences in cell proliferation as we did not observe any significant differences in vitro. (Fig. S6E) Additionally, when these same cells were injected into immunocompromised NOD/SCID mice, there was no difference in tumor growth, suggesting that an intact immune system was required to differentially control tumor growth. (Fig. 6B and S6F, S6G) To gain insight into this observation we performed immune-phenotyping to detect the presence of tumor infiltrating immune cells (Fig. S6H). WTp53 knockdown reduced the intratumoral presence of CD45+, CD4+, CD8+ and NK cells. In contrast, TREX1 knockdown tumors had a completely opposite phenotype and showed massive infiltration by those immune cells types. Importantly, the combined knockdown of TREX1 and p53 resulted in an immune infiltration phenotype similar to control tumors. (Fig. 6C-6F) Thus, p53 loss is similar to cGAS/STING/IRF3 loss and is sufficient to negate the effect of TREX1 deficiency.
Figure 6: TREX1 deficiency induces a WTp53 dependent antitumor immune response.

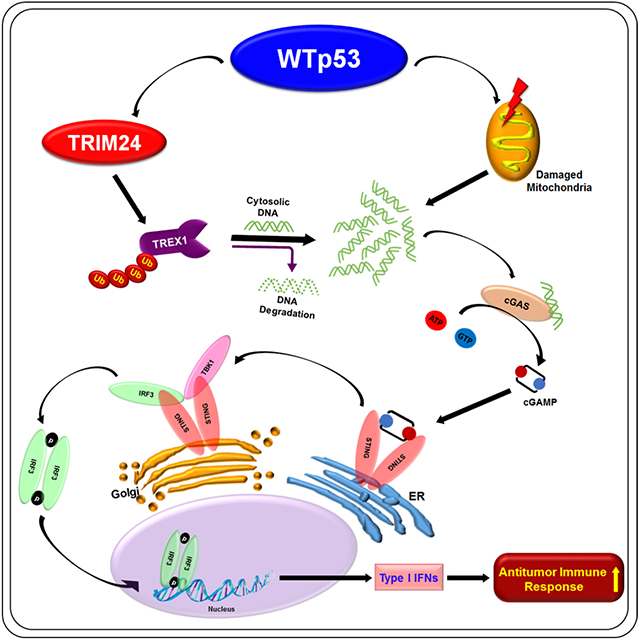
(A) CT26 shControl, shp53, shTREX1 and shp53/shTREX1 cells were injected with matrigel subcutaneously into immunocompetent female BALB/c mice (n=4). Tumor growth was monitored and measured using slide calipers. All mice were sacrificed on day 21 and graphical quantification represents the tumor growth rate in mice. (B) CT26 shp53, shTREX1 or shp53/shTREX1 cells were subcutaneously injected into the of immunodeficient NOD/SCID mice (n=5). All mice were sacked on day 14 and graphical quantification represents the tumor growth rate. (C-F) Representative graphs showed FACS quantification of (C) CD45+ T-lymphocytes (D) CD3+CD4+ T-helper (E) CD3+CD8+ cytotoxic T-lymphocyte and (F) NK cells per milligram (mg) of indicated tumor tissues (n=4) from BALB/c mice. (G) TREX1 expression in different tumor tissues in the Tumor Immune Estimation Resource (TIMER) database (http://timer.cistrome.org/). (*p < 0.05, **p < 0.01, ***p < 0.001). (H) Schematic representation of WTp53 promotes TRIM24 mediated TREX1 degradation that cause cytosolic DNA accumulation and activation of innate immune pathway to induce antitumor immune response.
Quantification graphs: In all panels, error bars represent mean with standard deviation. p values are based on Student’s t test. ***p < 0.001, **p < 0.01, ns=non-significant. See also Figure S6.
Since we observed that TREX1 could prevent the activation of the cGAS/STING pathway by p53, we surmised that TREX1 might function as an oncogene in human cancers and thus we performed a pan-cancer analysis of its expression using TCGA and GEPIA data sets. In comparison to normal tissue, TREX1 was overexpressed in a variety of cancers. (Figure 6G and S6I). Of note we found that LUAD and LUSC have elevated TREX1 expression relative to tumor tissue, for which we can only speculate that TREX1 has an important role in the physiology of the lung, perhaps to control pathogen invasion. We also analyzed the Human Protein Atlas (HPA) database and found TREX1 was highly overexpressed in the tumor tissue when compared with the normal tissue. (Fig. S6J) Furthermore, when we analyzed TCGA human database to compare survival between low and high TREX1 expressing colorectal and breast cancer patients, we observed that high TREX1 expression portended poor long-term survival in WTp53 tumors only as we did not find any significant correlation in mutant p53 tumors. (Figure S6K, S6L and S6M) We also stratified colorectal tumors based on their p53 status and found that WTp53 positively correlates with TREX1 mRNA whereas mutant p53 tumors do not have a significant correlation with TREX1 mRNA. (Fig. S6N and S6O) We also found TRIM24 negatively correlated with TREX1 in both WTp53 and mutant p53 tumors. (Fig. S6P and S6Q) These data suggest that elevated TREX1 expression contributes to tumor aggressiveness.
DISCUSSION
The tumor suppressor p53 has been shown to regulate a variety of cellular processes that contribute to its tumor suppressor activity. Although studies have shown that p53 induced crosstalk between tumor cells and the immune system can lead to cancer immunoediting, the underlying mechanism(s) are not well understood. Recently, it was shown that pharmacological activation of p53 with nutlin induced expression of endogenous retroviruses (ERVs) and the accumulation of double stranded RNAs (dsRNA) in the cytoplasm.11 The dsRNA was associated with the stimulation of Type I interferons, which was purportedly a result of activation of innate immune response RNA sensors.11 However, our data show that MAVS knockout minimally impacted activation of TBK1 by WTp53 suggesting that MAVS-dependent RNA sensing machinery is only partially involved. Notably, loss of TREX1 results in the accumulation of single-stranded DNA derived from retroelements.19 Thus, TREX1 deficiency can activate the cGAS DNA sensing pathway and the production of type I interferons.19 Importantly, nutlin treatment promoted TREX1 degradation and TBK1 activation, indicating that pharmacological activation of p53 recapitulates the signaling to the cGAS/STING pathway that we observed using different approaches. Moreover, TREX1 overexpression is sufficient to override the stimulation of the cGAS/STING pathway by WTp53, which lends credence to our interpretation that the key activating event is the accumulation of cytoplasmic DNA resulting from TREX1 degradation (Fig. 6H). This is further supported by the observation that WTp53 can lower the threshold for HT-DNA activation of the pro-apoptotic activity of the cGAS/STING pathway, and this can be reversed by TREX1 overexpression. Importantly, we found that the p53 transcriptional target, TRIM24, promotes the ubiquitin mediated degradation of TREX1, thus bolstering our conclusion that p53 is a negative regulator of TREX1. An additional line of evidence is the observation that cells deficient for mitochondrial DNA (Rho° cells) have reduced cytoplasmic DNA accumulation and fail to induce the cGAS/STING pathway in response to WTp53 induction. Since we did not observe an increased incidence of micronuclei in WTp53 induced cells, these data also reinforce the notion that WTp53 activation of the cGAS/STING pathway primarily requires the release of mitochondrial and not nuclear DNA. Our in vivo studies support the notion that WTp53’s regulation of TREX1, cGAS and STING mechanistically drives its ability to modulate immune cell activity. Strikingly, our data also revealed that cGAS and STING play an integral role in WTp53 tumor growth suppression in vivo, independently of their effects on the immune system, suggesting that loss of cGAS or STING compromises WTp53 function(s).
We observed that p53 lowered the threshold for apoptosis induction by HT-DNA, a cGAS agonist. Therefore, it is possible that combining pharmacological activators of p53 with cGAS or STING agonists could enhance the immunogenicity of tumor cells and their subsequent elimination. Furthermore, we found that TREX1 overexpression suppressed the activation of the cGAS/STING pathway. Based on the observation that TREX1 is overexpressed in a number of human cancers (excluding LUAD and LUSC), our data suggest that the development of TREX1 inhibitors has the potential to restore antitumor immune responses in different types of cancers.57 Moreover, targeting TREX1 in tumors that have lost wildtype p53 may render them immunologically “hot” allowing for immune cell dependent clearance. Taken together, our study establishes danger-associated molecular pattern (DAMP) sensing as a component of WTp53’s tumor suppressor activity.
Limitations of the Study
One limitation of our study is that WTp53 activation of the cGAS/STING pathway may be a dynamic, multi-step process, which we do capture using our experimental approach. Thus, further studies are required to assess if kinetic changes occur beyond what we have described. Our data indicates that in cGAS or STING knockout cells p53 induction does not restrict tumor growth to the same extent as in the control (NT) WTp53 induced tumors. Our current study does not answer how p53 tumor suppressor activity is compromised in the cGAS or STING null cells.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Luis A. Martinez (luis.martinez@stonybrookmedicine.edu).
Material Availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact. This includes plasmids and proteins. All reagents will be made available on request after completion of a Material Transfer Agreement.
Data and Code Availability
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request. All the original data for western blots is available at Mendeley (DOI: 10.17632/y84khfx263.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
4-6 weeks old male or female BALB/c, NOD/SCID mice were purchased from Envigo. All the experiments with mice were conducted in Stony Brook University animal care facility and in accordance with the Institutional Animal Care and Use Committee.
Cell Lines
All the cell lines were purchased from ATCC and cultured according to the manufacturer’s instructions. H1299, A549 (Human), 4T1, CT26 (Mouse) cells were cultured in complete Roswell Park Memorial Institute (RPMI) medium supplemented with 10% Fetal Bovine Serum (FBS). HEK293T was cultured in complete Dulbecco's Modified Eagle Medium (DMEM) medium. MEFs (p53−/−, p53+/+) were isolated in Dr. Tomoo Iwakuma Lab, University of Kansas Medical Center, according to the protocol described earlier and were cultured in complete DMEM medium.58
METHOD DETAILS
Cloning and Plasmids
Human shp53 pLKO.1 was a kind gift from Robert Weinberg (Addgene #19119). Non-targeting control and mouse p53 shRNA and TREX1 shRNA were cloned into pLKO.1 (Addgene #24150 and #8453) vector. Doxycycline inducible WTp53 was cloned in PCW57-MCS1-2A-MCS2 (Addgene # 71782). cGAS, STING, IRF3, IFI16 gRNAs were cloned into lentiCRISPRv2-puro (Addgene # 98290) as previously described.59 GFP-TREX1 (Addgene #27219) and GFP-TREX1 D18N (Addgene #27220) cDNAs were cloned into the lentivirus vector PLVX-puro.
pCMVHA-WTp53 was generated by PCR amplification of p53 from IMR90 lung fibroblast cDNA and cloned into pCMV-HA (Clontech). PCDNA3-GFP-IRF3 was a kind gift from Nancy Reich (Stony Brook University). All the constructs used in the study were confirmed by DNA sequencing. The sequences for sgRNAs, shRNAs, siRNAs and primers for PCR mutagenesis used in this study are listed in Supplementary Table S1.
Lentiviral particles production in 293FT cells.
For shRNA knockdown lentiviral particle was generated by transfecting 293T cells with 1.5 μg of ps-PAX2 (Addgene #12260), 0.5 μg of pCMV-VSV-G (Addgene # 8454) and 2 μg of plasmid of gene of interest using Lipofectamine 2000 following the manufacturer’s protocol. Viral supernatant was collected post 48 hrs and 72 hrs of transfection. Target cells were infected with the viral particle using polybrene (5 μg/mL) overnight and selection was performed with puromycin or hygromycin.
Generation of knock out cells using CRISPR-Cas9
To generate lentiviruses for transduction, HEK293T cells were transfected with plasmid(s) encoding guide RNAs targeting selected genes and packaging vectors (pCMV-VSV-G and psPAX2) using standard Lipofectamine 2000 transfection method. Culture supernatants were collected at 48 and 72 h post-transfection and used for infection of the targeted cells with polybrene (5 μg/ml). Cells were selected with puromycin for 10 days.
Cytoplasmic and nuclear fractionation
Doxycycline inducible WTp53 H1299 and 4T1 cells were culture in 60mm dishes and doxycycline was added for 24 hrs to induce p53. Cells were washed with PBS, harvested with trypsin-EDTA, and washed twice with PBS to remove traces of trypsin and growth medium. Cytoplasmic and nuclear fractionation was performed using the subcellular protein fractionation kit (Thermo Scientific, 78840) according to the manufacturer’s instructions.
Cell proliferation assay.
CT26 shControl or shp53 cells (3,000) and 4T1 induced WTp53 STINGKO cells (1000) were seeded on a 96-well plate and cell proliferation was detected for the next five days. Viable cells were measured by CellTiter-Blue® Cell Viability Assay kit (Promega) according to the manufacturer’s protocol. Briefly, 20 μl of cell titer blue reagent was directly added to the culture medium and incubated at 37°C for 4 h and plates were shaken for 10 sec and the fluorescence reading were obtained by reading the plate at 570/590 nm by Molecular Device Spectra Max M5 instrument.
Senescence assay
H1299 inducible WTp53 cells were seeded on to the 60mm cell culture dish and after 24 hrs cells were treated with Doxycycline to induce WTp53 alone or with Doxorubicin for four days. After the treatment, β-galactosidase activity was assessed according to the manufacturer’s protocol with a β-Galactosidase Staining Kit (Cell Signaling #9860). All the images were captured in the Nikon Ti microscope and β-Galactosidase-positive cells were counted in at least ten different fields.
Real-time quantitative PCR (RT-PCR).
Cells were cultured according to the experiment and total RNA were collected in RLT buffer (Qiagen) and isolated using the Qiagen mini RNA isolation kit. RNA quantity and quality were confirmed with a NanoDrop ND-1000 spectrophotometer, cDNA was synthesized using 500 ng of total RNA using oligo (dT) primers and Reverse Transcriptase (Quanta). Real-time qRT-PCR was performed in Bio-Rad CFX96 Touch real-time PCR detection system using Universal SYBr Green Supermix (Bio-Rad). Gene-specific primers sequences are listed in Supplementary Table S1.
IFN Beta measurement by ELISA.
H1299 and 4T1 doxycycline inducible WTp53 cells (106) and A549 shp53 cells (106) were seeded on a 60 mm dish and after 24 hrs cells were transfected with HT-DNA (4 μg) for 18 hrs and cellular conditioned media were collected analyzed using VeriKine Human or mouse IFN Beta ELISA Kit. Quantification of IFNB1 concentration was performed in triplicates according to the manufacturer protocol and the reading was taken at 450 nm by Molecular Device Spectra Max M5 instrument and calculated using an IFNB1 standard curve.
Nanostring gene Analysis
H1299 inducible WTp53 cells were treated with doxycycline to induce WTp53 for 24 hrs and A549 shControl and shp53 cells were harvested and lysed in RLT (Qiagen) buffer. RNA was extracted using the manufacturer’s protocol. Samples were prepared according to the manufacturer’s protocols for the Nanostring nCounter Autoimmune Profiling Panel (NanoString, Seattle, WA, USA). A list of genes and target probe sequences can be found at www.nanostring.com. Cartridges were run on the nCounter Sprint Profiler. Transcripts were analyzed using the nSolver software and R studio V3.6.
cGAMP ELISA.
Cytosolic cGAMP was measured using Cayman Chemical 2′ 3′-cGAMP ELISA Kit according to manufacturer’s protocol. For cells quantification, 10 million cells were harvested, washed with PBS, Cells were lysed using lysis buffer from Thermo (as recommended by the Manufacturer). Each cell lysate was examined according to the manufacturer’s instructions.
siRNA mediated transient knockdown
Cells (2X105) were plated onto a 6-well plate and after 24 hrs siRNAs of selected genes were transfected using Lipofectamine RNAiMAX (Invitrogen) using the manufacturer’s protocol. Cells were harvested after 48 hrs and processed for western blot or RT-PCR analysis.
Immunofluorescence.
H1299 cells with inducible WTp53 cells stably transfected with GFP-IRF3 were grown onto 1% gelatin pre-coated glass coverslips and after all the treatment cells were washed twice with DPBS and counter stained with Hoechst 33342 and mounted with the ProLong Gold Antifade Reagent. Images were captured with a Nikon Ti epifluorescence microscope and processed using Nikon AR software. For immunofluorescence, cells were grown on glass cover slides and after all the treatment cells were washed twice with DPBS and fixed with 4% paraformaldehyde for 15 mins at room temperature. Cells were then permeabilized with 0.1% triton X-100 and block in 2% BSA for 45 mins. Cells were then incubated with corresponding primary antibodies overnight at 4°C. Cells were washed twice and incubated with secondary antibody for 2 hrs at room temperature. Secondary antibody washed counterstained by DAPI and mount on slides with Fluoromount G. Cells were stained with 3 μl/ml PicoGreen for 1 hr at 37°C and counter stained by Hoechst 33342. Staining was examined under a Leica confocal scanning microscope equipped with a 100× oil-immersion objective.
Mitochondrial DNA depleted Rho° cell preparation
H1299 inducible WTp53 cells were cultured in tetracycline free RPMI supplemented with 500 ng/ml ethidium bromide (Sigma-Aldrich), 50 μg/ml uridine and 1 mM sodium pyruvate for 12 Days. Mitochondrial depletion was checked under CLSM using Mitotracker Red dye.
Chromatin Immunoprecipitation (ChIP)
H1299 inducible WTp53 or p53R248W cells were plated in 15-cm plates and grown to 80% confluence. Cells were induced with Doxycycline to express p53 and the ChIP assay was performed using ChIP-IT Express Enzymatic kit (Active Motif #53009 CA, USA) according to the manufacturer’s protocols. Briefly, the chromatin was extracted from 1 × 107 formalin-fixed cells and chromatin was immunoprecipitated using 5 μg anti-p53 antibody (DO1). The amount of precipitated DNA was measured using NanoDrop 1000 (Thermo Fisher Scientific). The immunoprecipitated chromatin was subjected to RT-PCR analysis using SYBR Green PCR Kit.
Flow cytometry.
Cells were seeded in a 6-well plate and after all the treatment cells were washed twice with PBS, trypsinized and re-suspended in 100 μl of binding buffer and further incubated with Annexin-V FITC or Annexin-V Pacific blue and Propidium iodide for 15 min in the dark at room temperature. Prior to flow cytometric analysis, 400 μl of binding buffer was added and cells were immediately subjected for the FACS analysis for the number of apoptotic cells. Data was generated using Cole Parmer Cytoflex.
Preparation of single cells from tumors for Tumor Immune Profiling
For analyzing tumor associated immune cell populations, tumors were excised, finely minced and incubated in RPMI 1640 containing 0.5 mg/ml collagenase D (Worthington), 0.01 mg/ml DNase I (Roche), and 0.5 mg/ml Dispase (Worthington) for 30 min at 37°C on a shaking platform. Post digestion, the cells were filtered using 70 μm cell strainer. Samples were pelleted at 1000 rcf for 5 mins at 4C and washed with PBS. The red blood cells (RBCs) of the samples were lysed using ACK lysis buffer (Invitrogen). Samples were washed with FACS buffer (PBS+2% FBS) and subjected to processing for flow cytometric analysis. Samples were incubated with Fc blocking antibody (BioLegend) for 15 mins at room temperature and subjected to live dead staining as well as cell surface marker staining using fluorochrome-conjugated monoclonal antibodies. Antibodies are listed below and were purchased from BioLegend, eBioscience and R&D: CD45 (clone 30-F11), CD3ε (145-2C11), CD4 (RM4.5), CD8 (53-6.7), B220 (RA3-6B2), MHC class II (M5/114.15.2) CD11b (M1/70), CD11c (N418), Ly6G (1A8), F4/80 (BM8), CD206 (MMR). Stained cells were washed with cold PBS +1% FBS and kept on ice until analysis. Gating was based on live cells and live cell populations were discriminated initially via CD45/SSC scatterplots, and the different cell populations were defined based on our gating strategy. Fluorescence minus One (FMO) controls were performed as well. Stained cells were acquired using BD LSRII flow cytometry (BD) and analyzed using the Kalluza software.
Immunoblotting, Ubiquitination and Immunoprecipitation.
To prepare cell lysates for western blotting, the cells were lysed on the dish using RIPA (0.5% SDS, 0.1% Sodium Deoxycholate, 0.5% NP40, 1 mM EDTA, in PBS pH 7.4 and filter-sterilize) buffer supplemented with protease and phosphatase inhibitors, scraped and placed into microcentrifuge tubes, sonicated and centrifuged at 13,000g for 10 mins at 4 °C to remove insoluble material. Protein concentration was determined using the Micro BCA Protein Assay kit (Pearce) and equal amounts of protein were resolved on 8 or 10% Bis-Tris polyacrylamide gels, transferred to a PVDF membrane blocked with 5% milk and incubated with primary antibody over night at 4C. For co-immunoprecipitation of proteins, cells were washed with PBS, harvested and lysed in immunoprecipitation buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05 mM EDTA, 1% NP40 and 10% glycerol). Lysate was clarified by centrifugation at 20,000g (4°C) for 20 min, pre-cleared with protein-G agarose (KPL) for 2 h at 4 °C and then immunoprecipitated overnight with the corresponding antibodies.
In vivo animal experiments.
Mice were anesthetized using Isoflurane and CT26 shControl or shp53 cells (50,000) in 0.1 ml PBS were injected with matrigel into mice. Mice were monitored and tumor volume was measured manually using slide calipers every other day till day 21 when all the mice were sacked and the tumors were harvested.
4T1 inducible WTp53 cGAS or STING KO cells were trypsinized washed twice with PBS and 50000 cells in PBS were injected at the mammary gland after anaesthetizing the mice. When the tumors reaches 100 mm3, mice were given doxycycline (20 mg/kg) orally every other day to induce WTp53. Tumors were monitored and volume was measured manually using slide calipers till day 21 when all the mice were sacked and the tumors were processed for further experiment.
Quantification and Statistical Analysis
All the experiments were repeated at least three times unless otherwise mentioned in the figure legends. The statistical differences in all assays including Fold difference in mRNA, cell proliferation and growth, flow cytometry and tumor growth between different samples and/or treatments were analyzed by two-tailed Student’s t-tests using Microsoft Excel 2013 and all the graphs were made on GraphPad Prism 8 (GraphPad Software) and presented as Mean ±SD or Mean ±SEM. The gene expression analysis was carried out by NanoString using their nSolver software. Statistical significance was set at P < 0.05, unless otherwise stated in the text. All experiments were carried out with at least three biological replicates otherwise mentioned in the figure legend. The numbers of animals used are described in the corresponding figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| STING (D2P2F) | Cell Signaling | Cat# 13647, RRID:AB_2732796 |
| TBK1 / NAK | Cell Signaling | Cat# 3504, RRID:AB_2255663 |
| IRF3 | Cell Signaling | Cat# 4302, RRID:AB_1904036 |
| Phospho-STING (Ser366) | Cell Signaling | Cat# 19781, RRID:AB_2737062 |
| Phospho-TBK1 (Ser172) (D52C2) | Cell Signaling | Cat# 5483, RRID:AB_10693472 |
| Phospho-IRF-3 (Ser396) (D6O1M) | Cell Signaling | Cat# 29047, RRID:AB_2773013 |
| TP53 | Santa Cruz Biotechnology | Cat# sc-126, RRID:AB_628082 |
| p53 (CM5) | Leica | Cat# sc-6243, RRID:AB_653753 |
| GAPDH | GeneTex | Cat# GTX100118, RRID:AB_1080976 |
| Anti-beta-Actin-Peroxidase antibody | Sigma-Aldrich | Cat# A3854, RRID:AB_262011 |
| c-Myc | Santa Cruz Biotechnology | Cat# sc-40, RRID:AB_627268 |
| HA-probe (F-7) | Santa Cruz Biotechnology | Cat# sc-7392, RRID:AB_627809 |
| GFP (B-2) | Santa Cruz Biotechnology | Cat# sc-9996, RRID:AB_627695 |
| Flag-HRP | Sigma-Aldrich | Cat # A8592, RRID:AB_439702 |
| anti-mouse CD335 (NKp46) antibody | BioLegend | Cat# 137611, RRID:AB_10915472 |
| anti-mouse CD45 | Biolegend | Cat# 103131, RRID:AB_893344 |
| CD3e Monoclonal Antibody (145-2C11) | Thermo Fisher Scientific | Cat# 25-0031-81, RRID:AB_469571 |
| CD4 Monoclonal Antibody (RM4-5) | Thermo Fisher Scientific | Cat# 17-0042-81, RRID:AB_469322 |
| anti-mouse CD8a | BioLegend | Cat# 100707, RRID:AB _312746 |
| TREX1 | Abcam | Cat# ab185228, RRID:AB_2885196 |
| TRIM24 | Proteintech | Cat# 14208-1-AP, RRID:AB_2256646 |
| V5-probe (E10) | Santa Cruz Biotechnology | Cat# sc-81594, RRID:AB_1131162 |
| cGAS | Cell Signaling | Cat# 15102, RRID:AB_2732795 |
| Anti-Lamine B1 | Abcam | Cat# ab16048, RRID:AB_443298 |
| dsDNA | Santa Cruz Biotechnology | Cat# sc-58749, RRID:AB_783088 |
| IFI-16 (1G7) | Santa Cruz Biotechnology | Cat# sc-8023, RRID:AB_627775 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 | Invitrogen | Cat# 2082816 |
| Lipofectamine RNA iMAX | Invitrogen | Cat# 13778-150 |
| PicoGreen | Invitrogen | Cat# P7589 |
| Cyclosporine A | ApexBio | Cat# B1922 |
| Ethidium bromide | Sigma-Aldrich | Cat# 15585-04 |
| HT-DNA | Sigma-Aldrich | Cat# D6898 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| Sodium pyruvate | Gibco | Cat# 11360-070 |
| Hoechst 33342 | ThermoFisher Scientific | Cat# H3570 |
| DAPI | ThermoFisher Scientific | Cat# 62248 |
| FITC Annexin V/ PI | BioLegend | Cat# 640914 |
| Doxycycline hydrochloride | Fisher Scientific | Cat# BP2653-5 |
| Halt™ Protease Inhibitor Cocktail | ThermoFisher Scientific | Cat# 78429 |
| Fluoromount G | SuthernBiotech | Cat# 0100-01 |
| Pro Long™ Gold Antifade | ThermoFisher Scientific | Cat# P10144 |
| BSA | GoldBiotechnology | Cat# A-420 |
| Doxorubicin hydrochloride | Sigma | Cat# D1515 |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Pierce | Cat# 23235 |
| Mouse IFN Beta ELISA Kit | pbl | Cat# 42400-1 |
| Human IFN Beta ELISA Kit | pbl | Cat# 41410-1 |
| SsoAdvanced Universal SYBR® Green | Bio-Rad | Cat #1725274 |
| QIAprep spin Miniprep Kit | Qiagen | Cat# 27106 |
| Reasy Mini Kit | Qiagen | Cat# 74104 |
| CellTiter-Blue® Cell Viability Assay | Promega | Cat# G8081 |
| AnnexinV-FITC/PI apoptosis detection kit | BiLegend | Cat# 640914 |
| Clarity Max Western ECL Substrate | Bio-Rad | Cat# 1705061 |
| Exonuclease activity assay kit | VioVision | Cat# K175-100 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | ThermoFisher Scientific | Cat# 78835 |
| cGAMP ELISA Kit | Cayman chemical | Cat# 501700 |
| ChIP-IT Express Enzymatic | Active Motif | Cat# 53009 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat# 9860 |
| Experimental Models: Cell Lines | ||
| A549 | ATCC | N/A |
| 4T1 | ATCC | N/A |
| CT26 | ATCC | N/A |
| H1299 | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| BALB/c | Envigo | Cat # 4702F |
| NOD/SCID | Envigo | Cat # 1700M |
| Deposited data | ||
| Uncropped Western Blot Data | Mendeley | DOI: 10.17632/y84khfx263.1 |
| Software and Algorithms | ||
| GraphPad Prism 8.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| Excel 2016 | Microsoft | https://www.office.com/ |
| ImageJ 1.52a | Wayne Rasband, NIH | https://imagej.net/ |
| BD FACS DIVA 6.2 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-sorters/facsaria-iii |
| Kaluza | Beckman Coulter Life Sciences | https://www.beckman.com/flow-cytometry/software/kaluza |
| NIS-elements AR 5.02.01 | Nikon | https://www.microscope.healthcare.nikon.com/ |
| Leica TCS SP8 X confocal | Leica | https://www.leica-microsvstems.com |
Highlights.
Wild type p53 (WTp53) activates the cytosolic DNA sensing cGAS/STING pathway
WTp53 requires TRIM24 to promote TREX1 degradation
TREX1 degradation causes cytosolic DNA accumulation and activation of cGAS/STING
Loss of cGAS or STING compromises WTp53’s tumor suppressor activity
ACKNOWLEDGMENTS
The authors wish to acknowledge the Stony Brook Cancer Center for expert assistance with the Nanostring Data analysis. We would like to acknowledge the technical support provided by the Research Flow cytometry core facility, Department of Pathology, Stony Brook Renaissance School of Medicine.
FUNDING
This work was supported by NCI CA166974-07A1, the New York State Empire Investment Program, the Stony Brook Renaissance School of Medicine and Cancer Center, the TRO Carol M. Baldwin Award, the Stony Brook Cancer Center Bahl IDEA Award, and the Lynn November Pilot Funds for Therapeutic Development in Aggressive Breast Cancer and the John C. Dunphy Private Foundation Cancer Innovation Fund from the Stony Brook Cancer Center. D.C.M. is supported by NIH/NCI K22CA226033, a grant from the American Pulse Association, and startup funds from the Stony Brook Cancer Center (Stony Brook, NY) and Bahl Center for Metabolomics and Imaging (Stony Brook, NY). We thank Dr. Nancy C. Reich (Stony Brook University, NY, USA) for providing us with the pcDNA3 GFP-IRF3 clone.
Footnotes
DECLARATION OF INTERESTS
Authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kress M, May E, Cassingena R & May P (1979) Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31, 472–483, doi: 10.1128/JVI.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lane DP & Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263, doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 3.Linzer DI & Levine AJ (1979) Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52, doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 4.DeLeo AB et al. (1979) Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A 76, 2420–2424, doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Z et al. (2015) Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112, E4306–4315, doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau L, Gray EE, Brunette RL & Stetson DB (2015) DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571, doi: 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 7.Roetman JJ, Apostolova MKI & Philip M (2022) Viral and cellular oncogenes promote immune evasion. Oncogene 41, 921–929, doi: 10.1038/s41388-021-02145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue W et al. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660, doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blagih J et al. (2020) Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep 30, 481–496 e486, doi: 10.1016/j.celrep.2019.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo G, Yu M, Xiao W, Celis E & Cui Y (2017) Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res 77, 2292–2305, doi: 10.1158/0008-5472.CAN-16-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X et al. (2021) Pharmacological activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting anti-tumor immunity. Cancer Discov, doi: 10.1158/2159-8290.CD-20-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz-Fontela C et al. (2008) Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med 205, 1929–1938, doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine AJ (2020) P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int J Mol Sci 21, doi: 10.3390/ijms21020541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miciak J & Bunz F (2016) Long story short: p53 mediates innate immunity. Biochim Biophys Acta 1865, 220–227, doi: 10.1016/j.bbcan.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D & Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Sun L, Wu J, Du F, Chen X & Chen ZJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791, doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T & Chen ZJ (2018) The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299, doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson SR, Hemphill WO, Hudson T & Perrino FW (2020) TREX1 - Apex predator of cytosolic DNA metabolism. DNA Repair (Amst) 94, 102894, doi: 10.1016/j.dnarep.2020.102894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stetson DB, Ko JS, Heidmann T & Medzhitov R (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598, doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J et al. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830, doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S et al. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630, doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka Y & Chen ZJ (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5, ra20, doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia T, Konno H & Barber GN (2016) Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 76, 6747–6759, doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 24.Xia T, Konno H, Ahn J & Barber GN (2016) Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297, doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konno H et al. (2018) Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051, doi: 10.1038/s41388-017-0120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Queiroz N, Xia T, Konno H & Barber GN (2019) Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol Cancer Res 17, 974–986, doi: 10.1158/1541-7786.MCR-18-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dou Z et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406, doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo SR et al. (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842, doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng L et al. (2014) STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852, doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ablasser A & Chen ZJ (2019) cGAS in action: Expanding roles in immunity and inflammation. Science 363, doi: 10.1126/science.aat8657. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh M et al. (2021) Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell, doi: 10.1016/j.ccell.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gulen MF et al. (2017) Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427, doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald KA et al. (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4, 491–496, doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 34.McWhirter SM et al. (2004) IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A 101, 233–238, doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dumont P, Leu JI, Della Pietra AC 3rd, George DL & Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33, 357–365, doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 36.Brucet M et al. (2007)Structure of the dimeric exonuclease TREX1 in complex with DNA displays a proline-rich binding site for WW Domains. J Biol Chem 282, 14547–14557, doi: 10.1074/jbc.M700236200. [DOI] [PubMed] [Google Scholar]
- 37.Mazur DJ & Perrino FW (2001) Structure and expression of the TREX1 and TREX2 3' --> 5' exonuclease genes. J Biol Chem 276, 14718–14727, doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 38.Mazur DJ & Perrino FW (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3'-->5' exonucleases. J Biol Chem 274, 19655–19660, doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 39.Itahana K, Dimri G & Campisi J (2001) Regulation of cellular senescence by p53. Eur J Biochem 268, 2784–2791, doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi A et al. (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9, 1249, doi: 10.1038/s41467-018-03555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orebaugh CD et al. (2013) The TREX1 C-terminal region controls cellular localization through ubiquitination. J Biol Chem 288, 28881–28892, doi: 10.1074/jbc.M113.503391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain AK, Allton K, Duncan AD & Barton MC (2014) TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol Cell Biol 34, 2695–2709, doi: 10.1128/MCB.01705-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allton K et al. (2009) Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A 106, 11612–11616, doi: 10.1073/pnas.0813177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King MP & Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503, doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T et al. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6, 981–988, doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 46.Meylan E et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172, doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 47.Seth RB, Sun L, Ea CK & Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682, doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 48.Yang H, Wang H, Ren J, Chen Q & Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114, E4612–E4620, doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang YG, Lindahl T & Barnes DE (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886, doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 50.Crow YJ et al. (2006) Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38, 917–920, doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 51.Gall A et al. (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131, doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morita M et al. (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3'-->5' DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24, 6719–6727, doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gray EE, Treuting PM, Woodward JJ & Stetson DB (2015) Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol 195, 1939–1943, doi: 10.4049/jimmunol.1500969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao N et al. (2019) cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J Autoimmun 100, 84–94, doi: 10.1016/j.jaut.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Ablasser A et al. (2014) TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192, 5993–5997, doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 56.Hasan M et al. (2017) Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U S A 114, 746–751, doi: 10.1073/pnas.1611113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hemphill WO et al. (2021) TREX1 as a Novel Immunotherapeutic Target. Front Immunol 12, 660184, doi: 10.3389/fimmu.2021.660184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parrales A et al. (2016) DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol 18, 1233–1243, doi: 10.1038/ncb3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shalem O et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request. All the original data for western blots is available at Mendeley (DOI: 10.17632/y84khfx263.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.