

Abstract
A puzzle of autophagy in neurons is that, unlike in other cells, it is not robustly induced by inhibition of mammalian target of rapamycin (mTOR). A new study now solves this conundrum and establishes that myotubularin-related phosphatase 5 limits the induction of neuronal autophagy by mTOR inhibitors.
Macroautophagy (hereafter referred to as autophagy) captures cytoplasmic cargo into autophagosomes for clearance in lysosomes1. This pathway is critically important for neuronal function and survival. In fact, knockout of key autophagy genes causes neurodegeneration in mice, and mutations in autophagy genes are linked to neurodegenerative disorders in humans2,3. Moreover, proteins that are prone to aggregation in neurodegenerative diseases are substrates for autophagy, sparking strong interest in autophagy as a therapeutic target2. Thus, a key question in the field is: how can autophagy in neurons be manipulated to enhance clearance of protein aggregates and promote neuronal viability in neurodegenerative disease? Conventional methods of inducing autophagy have limited effects in neurons compared with non-neuronal cells such as astrocytes4–7. However, the factors that confer resistance to these autophagy inducers in neurons have remained unknown. In a new study published in this issue of Current Biology, Chua et al.8 solve this conundrum and identify myotubularin-related phosphatase 5 (MTMR5) as an autophagy suppressor that is enriched in neurons (Figure 1A,B).
Figure 1. Autophagy regulation by MTMR5 and MTMR2 in iNeurons and other cell types.
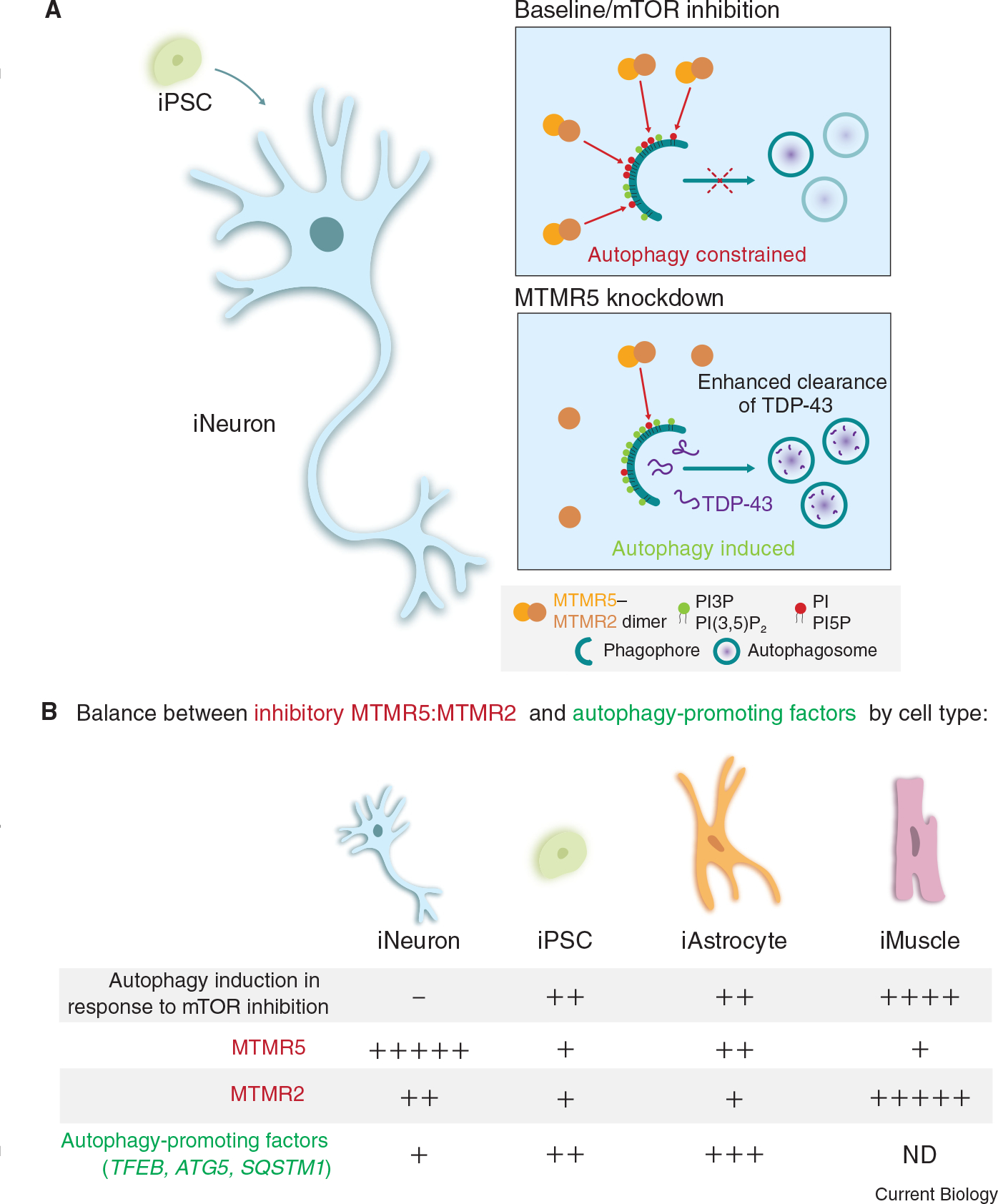
(A) In iNeurons differentiated from iPSCs, MTMR5 and MTMR2 antagonize the formation of autophagosomes, thereby desensitizing iNeurons to mTOR inhibition. This suppression is a result of the formation of MTMR5–MTMR2 heterodimers that reduce PI3P levels, thereby preventing the assembly of autophagy-initiating factors (top panel). Reduction of MTMR5 expression allows for autophagy induction and enhanced clearance of cargoes such as TDP-43 (bottom panel). (B) Cell-type-specific stoichiometries between factors that inhibit and promote autophagy may account for the differential regulation of autophagy in response to mTOR inhibition in iNeurons compared with iPSCs, iAstrocytes and iMuscle cells. iNeurons do not upregulate autophagy in response to mTOR inhibition and have higher levels of MTMR5–MTMR2 and lower levels of autophagy-promoting factors compared with the other cell types examined. By contrast, iPSCs, iAstrocytes, and iMuscle cells induce autophagy in response to mTOR inhibition and this effect may be due to varying proportions of lower levels of autophagy-inhibitory factors relative to higher levels of autophagy-promoting factors. ND, not determined.
To study how autophagy is regulated in neurons compared with non-neuronal cells, Chua et al.8 used an induced pluripotent stem cell (iPSC) line edited with CRISPR–Cas9 to tag endogenous LC3, a marker for autophagic organelles, with EGFP at the amino terminus. These iPSCs were then differentiated into glutamatergic forebrain-like neurons (iNeurons), astrocytes (iAstrocytes), or skeletal muscle cells (iMuscle cells), and autophagy levels were compared across these cell types in response to pharmacological inhibition of mammalian target of rapamycin (mTOR) with Torin1. mTOR is a serine/threonine kinase of the phosphatidylinositol 3-kinase (PI3K)-related kinase family that negatively regulates autophagy by phosphorylating and suppressing the function of various targets, including ULK1 and ATG13, components of the ULK1 complex, which initiate autophagosome formation, and TFEB, a transcription factor that activates the expression of autophagy and lysosomal genes9. Inhibition of mTOR prevents these inhibitory phosphorylation events, which induces autophagy in various cell types, but has surprisingly only modest effects in neurons5,6,10. Indeed, Chua et al.8 found that Torin1 robustly increased the number of autophagosomes in iAstrocytes, iMuscle cells, and the parental iPSC line, but not in iNeurons (Figure 1B). Torin1 effectively reduced mTOR activity in iNeurons, as evidenced by reduced phosphorylation of downstream targets (such as S6 and 4E-BP1, factors that promote protein synthesis), but was not sufficient to stimulate autophagosome formation. Thus, unlike iPSCs, iAstrocytes, and iMuscle cells, iNeurons are resistant to autophagy induction by mTOR inhibition, indicating the existence of neuron-specific mechanisms for regulating autophagy.
To elucidate the molecular determinants regulating autophagy in neurons, Chua et al.8 compared the rates of synthesis and turnover of mRNA transcripts in iNeurons, parental iPSCs and fibroblasts. They found that the transcript SBF1, encoding the protein MTMR5, was synthesized at similar rates but had greater stability in iNeurons than in fibroblasts and undifferentiated iPSCs. Measurements of SBF1 mRNA and MTMR5 protein confirmed fivefold higher expression levels in iNeurons compared with the parental iPSC line. MTMR5 is a member of the MTMR family of phosphatases, which remove the 3-phosphate from membrane phosphoinositides, thereby converting phosphatidylinositol 3-phosphate (PI3P) and PI(3,5)P2 into PI and PI5P, respectively11. Since PI3P serves as a platform for the assembly of autophagy initiation complexes1, MTMRs can function as autophagy suppressors12,13. MTMR5 is catalytically inactive as a phosphatase, but dimerizes with and enhances the phosphatase activity of MTMR214. Accordingly, Chua et al.8 find that MTMR2 transcript expression is also enriched in iNeurons compared with the parent iPSCs. Thus, MTRMR5–MTMR2 heterodimers emerge as a candidate that might restrict the induction of autophagy by mTOR inhibition in neurons.
Strikingly, modulation of MTMR5 expression can tune autophagy levels. Overexpression of MTMR5 in iPSCs attenuated the induction of autophagy by Torin1. Thus, MTMR5 is sufficient to suppress autophagy and increase resistance to Torin1-mediated induction of autophagy in iPSCs, similar to iNeurons. Conversely, knockdown of MTMR5 (or its binding partner MTMR2) in iNeurons unlocked a sensitivity to Torin1, enabling iNeurons to increase autophagy in response to Torin1 (Figure 1A). This effect required the class III PI3K VPS34, which phosphorylates PI to generate PI3P, a phospholipid important for autophagosome biogenesis. PI3P levels would be reduced by high levels of MTMR5–MTMR2 heterodimers15.
To further define the effects of MTMR5 and MTMR2 knockdown on autophagy induction in iNeurons by Torin1, Chua et al.8 measured the levels of p62/SQSTM1, a receptor for selective autophagy that recruits ubiquitinated substrates to the autophagosome16. In this process, p62 is incorporated into autophagosomes and is degraded. The authors found that knockdown of MTMR5 or MTMR2 reduced steady-state levels of p62. Moreover, blocking lysosome function captured more p62 in the absence of MTMR5 or MTMR2 compared with the control. Thus, more p62 is routed to the lysosome for degradation in the absence of MTMR5 or MTMR2, indicating increased autophagic flux. These findings are consistent with mTOR inhibition more effectively inducing autophagy in neurons in the absence of MTMR5 or MTMR2.
Chua et al.8 also investigated the effect of MTMR5 or MTMR2 knockdown on the turnover of TDP-43, an RNA-binding protein involved in the pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. For this experiment, the authors fused TDP-43 to Dendra2, a photoconvertible tag used to optically label the TDP-43 population at the start of the experiment and track its lifetime. They found that knockdown of MTMR5 enhanced the turnover of TDP-43, an effect that could be blocked largely by inhibiting lysosome function. Thus, reducing MTMR5 stimulates proteolytic clearance of TDP-43. Similar effects were observed with knockdown of MTMR2. Interestingly, knockdown of MTMR5 was sufficient to achieve these effects under baseline conditions, and treatment with Torin1 did not further enhance the proteolytic degradation of TDP-43. Thus, the optical pulse labeling of autophagy substrates enabled higher sensitivity for discerning changes in autophagic flux.
Importantly, this study provides key insights into the mechanistic complexity underlying cell-type-specific regulation of autophagy in neurons compared with non-neuronal cells. The authors suggest that the regulation of autophagy is influenced by a cell-type-specific stoichiometry of inhibitory factors, including the MTMR5:MTMR2 ratio, antagonized by autophagy-promoting factors (Figure 1B). For example, the parental iPSC line has lower levels of SBF1 and MTMR2 transcripts, and their corresponding proteins, than iNeurons. The parental iPSC line also has higher levels of transcripts for autophagy-promoting factors (e.g. TFEB, ATG5, and SQSTM1). In combination, these features may enable iPSCs to be more sensitive to autophagy stimulation by Torin1. iAstrocytes, however, express moderate levels of MTMR5, yet are able to robustly activate autophagy in response to Torin1. One possible reason for this effect is that iAstrocytes express lower levels of MTMR2 mRNA and protein compared with iNeurons, and higher levels of autophagy-promoting factors compared with iNeurons. Interestingly, iMuscle cells express the most MTMR2 protein of the cell types examined, yet display the largest induction of autophagy with Torin1. This effect may be explained by the significantly lower expression levels of SBF1 and MTMR5 in iMuscle cells compared with iNeurons, which would result in reduced activity of MTMR2. Thus, levels of both MTMR5 and MTMR2 appear to play a role in regulating autophagy in different cell types. Further complexity is revealed upon examination of iMotor Neurons. Unlike iNeurons, iMotor Neurons show induction of autophagy with Torin1, indicating distinct mechanisms for autophagy regulation that are specific to neuronal subtypes. Interestingly, iMotor Neurons express similar levels of SBF1 as iNeurons. However, the expression of autophagy-promoting factors in iMotor Neurons is strikingly higher than in iNeurons. Thus, the relative stoichiometry of autophagy-promoting and -inhibitory factors may underlie the cell-type-specific sensitivities of autophagy induction to various stimuli. Moreover, the precise combination of factors contributing to this ratio is likely cell-type specific.
Accumulating evidence indicates that autophagy serves a variety of functions in neurons depending on neuronal subtype and developmental stage. How do these findings from Chua et al.8 relate to the diversity of functions for autophagy in neurons? The unique complement of autophagy-promoting versus -inhibitory factors across neuronal subtypes confers a differential sensitivity of autophagy to diverse stimuli. Moreover, these subtype-specific stoichiometries may render a differential susceptibility of neurons to the progression of neurodegenerative disease. For example, Chua et al.8 suggest that autophagy-promoting factors such as TFEB may have a more central role in regulating autophagy in iMotor Neurons. In support of this model, Cunningham et al.17 found that dysfunction of the autophagy–lysosomal pathway underlies motor neuron degeneration in models of ALS, and these phenotypes were partially caused by a defect in the nuclear import of TFEB. Autophagy can also be regulated by mTOR-independent pathways, such as calcineurin-mediated dephosphorylation of TFEB to induce nuclear translocation of TFEB18. But the contributions of these pathways to autophagic activity in neurons and different neuronal subtypes remain to be explored. Another outstanding question that emerges from this study is why is the regulation of autophagy in neurons so complex? It may be that the unique properties of neurons (such as their post-mitotic state and exceptionally long lifetime) require them to be more selective in the volume and identity of material to be degraded19. Furthermore, evidence suggests that neuronal proteostasis is regulated by crosstalk with neighboring astrocytes20. Future studies will need to elucidate the impact of astrocytes on the regulation of autophagy in neurons to control proteostasis in the brain. In sum, the study by Chua et al.8 illuminates the complexity of regulating autophagy across cell types. This knowledge will help define more targeted therapeutic approaches to mitigate neurodegeneration by manipulating autophagy in a manner specific to neuron subtype. In this way, autophagy could be upregulated specifically in the selectively vulnerable neuronal populations characteristic of each neurodegenerative disease.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Nakatogawa H (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 21, 439–458. [DOI] [PubMed] [Google Scholar]
- 2.Chua JP, De Calbiac H, Kabashi E, and Barmada SJ (2022). Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 18, 254–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulkarni VV, and Maday S (2018). Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 78, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kulkarni A, Dong A, Kulkarni VV, Chen J, Laxton O, Anand A, and Maday S (2020). Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy 16, 1651–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, and Finkbeiner S (2010). A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 107, 16982–16987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox JH, Connor T, Chopra V, Dorsey K, Kama JA, Bleckmann D, Betschart C, Hoyer D, Frentzel S, Difiglia M, et al. (2010). The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington’s disease. Mol. Neurodegener. 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moruno-Manchon JF, Uzor NE, Ambati CR, Shetty V, Putluri N, Jagannath C, McCullough LD, and Tsvetkov AS (2018). Sphingosine kinase 1-associated autophagy differs between neurons and astrocytes. Cell Death Dis. 9, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chua JP, Bedi K, Paulsen MT, Ljungman M, Tank EMH, Kim ES, McBride JP, Colón-Mercado JM, Ward ME, Weisman LS, and Barmada SJ (2022). Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons. Curr. Biol. 32,, 2581–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maday S, and Holzbaur EL (2016). Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 36, 5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raess MA, Friant S, Cowling BS, and Laporte J (2017). WANTED — Dead or alive: Myotubularins, a large disease-associated protein family. Adv. Biol. Regul. 63, 49–58. [DOI] [PubMed] [Google Scholar]
- 12.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, and Noda T (2010). Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 11, 468–478. [DOI] [PubMed] [Google Scholar]
- 13.Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, Laporte J, and Deretic V (2009). Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 28, 2244–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SA, Vacratsis PO, Firestein R, Cleary ML, and Dixon JE (2003). Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc. Natl. Acad. Sci. USA 100, 4492–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palamiuc L, Ravi A, and Emerling BM (2020). Phosphoinositides in autophagy: current roles and future insights. FEBS J. 287, 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham KM, Maulding K, Ruan K, Senturk M, Grima JC, Sung H, Zuo Z, Song H, Gao J, Dubey S, et al. (2020). TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. eLife 9, e59419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, and Berman SB (2011). Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 20, 927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorrbaum AR, Kochen L, Langer JD, and Schuman EM (2018). Local and global influences on protein turnover in neurons and glia. eLife 7, e34202. [DOI] [PMC free article] [PubMed] [Google Scholar]