

SUMMARY
The gut microbiota influences acetylation on host histones by fermenting dietary fiber into butyrate. Although butyrate could promote histone acetylation by inhibiting histone deacetylases, it may also undergo oxidation to acetyl-coenzyme A (CoA), a necessary cofactor for histone acetyltransferases. Here, we find that epithelial cells from germ-free mice harbor a loss of histone H4 acetylation across the genome except at promoter regions. Using stable isotope tracing in vivo with 13C-labeled fiber, we demonstrate that the microbiota supplies carbon for histone acetylation. Subsequent metabolomic profiling revealed hundreds of labeled molecules and supported a microbial contribution to host fatty acid metabolism, which declined in response to colitis and correlated with reduced expression of genes involved in fatty acid oxidation. These results illuminate the flow of carbon from the diet to the host via the microbiota, disruptions to which may affect energy homeostasis in the distal gut and contribute to the development of colitis.
In brief
Stable isotope tracing by Lund et al. demonstrates that the gut microbiota provisions fiber-derived carbon to host epithelial cells in support of histone acetylation and fatty acid metabolism. Intestinal inflammation interferes with this metabolic connection and the expression of genes related to fatty acid oxidation.
Graphical Abstract
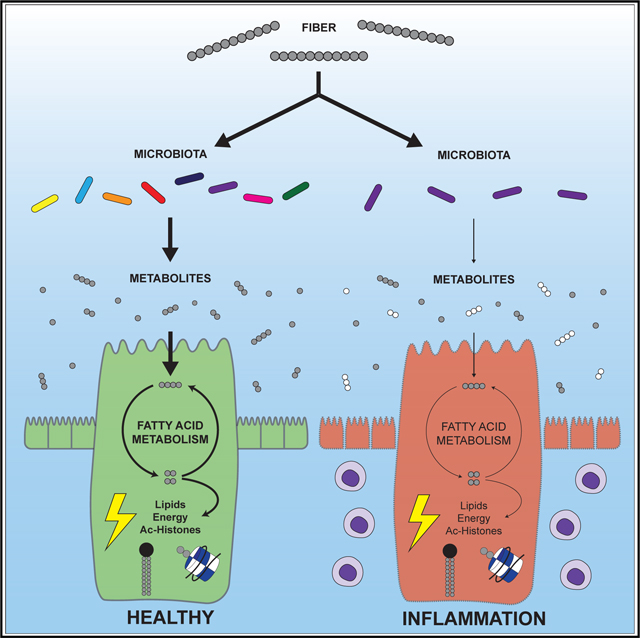
INTRODUCTION
The distal gut is inhabited by the microbiota, an enormous community of microbes that forms a dynamic ecosystem with its host organism. Interactions within and across kingdoms have a significant impact on host physiology. For instance, the microbiota has roles in preventing enteric infections,1–4 regulating the immune system5–9 and intestinal epithelium,10–14 and assisting with nutrient acquisition.15,16 Undesirable alterations in the microbiota lead to a state known as dysbiosis.2,16–22 Characterized by a loss of diversity and an outgrowth of the facultatively anaerobic Proteobacteria over the obligate anaerobes that typically predominate, dysbiosis is a hallmark of several human diseases, especially ulcerative colitis and Crohn disease.23–26 This correlation motivates the study of host-microbiota interactions in pursuit of understanding the processes that maintain gut homeostasis and how these processes become corrupted in disease.
Host-microbiota interactions depend in part on small-molecule metabolites that can function as receptor ligands, enzyme inhibitors, or metabolic precursors.27–32 This microbial metabolome is related to the composition of the microbiota and host diet.33–37 Fermentation of dietary fiber by the microbiota generates high concentrations of the short-chain fatty acids (SCFAs) acetate, propionate, and butyrate.38–40 Butyrate has received much attention because of its ability to inhibit histone deacetylases (HDACs).41 HDACs represent one of the many classes of chromatin-modifying enzymes that modulate the post-translational modification status of histone proteins. Together with DNA methylation and non-coding RNA, histone modifications dictate the usage of the information encoded in the underlying DNA sequence, thereby contributing to gene regulation in an epigenetic (i.e., above genetics) fashion.42 Histones, which form octameric scaffolds that associate with 147 bp of DNA to organize eukaryotic chromatin into nucleosomes, are subject to a myriad of post-translational modifications, among which lysine acetylation and methylation are the most appreciated and well studied.43–47 These modifications, added and removed by “writer” and “eraser” enzymes, regulate chromatin activity in part by recruiting “reader” proteins to promote a biological outcome, such as transcriptional activation or repression. Thus, inhibition of HDACs by butyrate has the potential to affect gene expression. Previous work has shown that butyrate, produced primarily by obligate anaerobes, enforces immunological tolerance in the gut through acetylation of histone H3 near the Foxp3 gene, thereby supporting the differentiation of regulatory T cells.48
Beyond acting as an HDAC inhibitor, butyrate also functions as a receptor ligand and an energy source. Butyrate and other SFCAs bind to numerous GPCRs.49 The ameliorative effect of a high-fiber diet on experimentally induced colitis has been linked to GPR43 and GPR109A.50 Butyrate may also activate intracellular receptors, such as PPARγ.19,51 Microbiota-dependent PPARγ signaling in epithelial cells promotes mitochondrial β-oxidation, which is essential for maintaining anaerobic conditions and homeostasis in the gut lumen. This metabolism of gut epithelial cells depends heavily on microbial SCFAs, which serve as readily available carbon sources that can undergo β-oxidation to generate acetyl- coenzyme A (CoA), thereby fueling the tricarboxylic acid (TCA) cycle and ATP production.52,53 Since acetyl-CoA is the donor substrate for histone acetyltransferases (HATs), butyrate also has the potential to affect histone acetylation patterns through its metabolism.52 As proposed by Donohoe et al., whether butyrate acts as an indirect activator of HATs or a direct inhibitor of HDACs likely depends on the propensity of cells for fatty acid metabolism, which is expected to be high in the case of gut epithelial cells.
Stable isotope labeling presents an attractive approach to study the contribution of different substrates to downstream metabolic pathways and protein modifications. Using this method, carbon from the medium-chain fatty acid octanoate was recently traced to the acetyl groups of histones.54 Although early studies with radioactive isotopes were instrumental in establishing the importance of butyrate metabolism in the distal gut,55–58 their measurement of 14CO2 only accounts for the contribution of butyrate oxidation to the TCA cycle without consideration of any alternative fates of butyrate-derived acetyl-CoA, such as its use for histone acetylation. Since acetyl-CoA occupies a central hub in cell metabolism, the microbiota has the potential to support many additional metabolic pathways in gut epithelial cells. Accordingly, subsequent studies provided evidence of radioisotope incorporation into lipids and histones, although the identities of these labeled molecules were not further resolved.59,60 A more recent investigation with stable isotopes, in which Escherichia coli was labeled externally with 13C-glucose and then administered to uncolonized mice, demonstrated the remarkable extent to which microbially derived compounds enter host tissues.61 However, there is a need for alternative strategies to more fully model the metabolic transformations and ecological interactions that occur in an intact microbiota concentrated in the distal gut with nourishment from the host diet.
In the present study, we aimed to broadly investigate the influence of the microbiota on histone acetylation patterns in the distal gut and provide direct evidence of a metabolic connection between the microbiota and host epithelial cells that feeds into histone acetylation. Through deep profiling of histone modifications with mass spectrometry (MS) and subsequent chromatin immunoprecipitation sequencing (ChIP-seq) analysis, we find that germ-free (GF) mice have a genome-wide loss of histone H4 acetylation except near transcription start sites. Supporting a metabolic contribution of the microbiota to histone acetylation, we show through isotope tracing experiments that histone acetyl groups contain carbon derived from butyrate and dietary fiber. Finally, we combine our in vivo isotope tracing approach with untargeted metabolomics and complementary high-throughput analyses in a mouse model of intestinal inflammation, revealing how inflammation disrupts integration between the microbiota and host fatty acid metabolism. Overall, these findings highlight how microbial metabolism of dietary fiber contributes to host cell metabolism and histone acetylation in the distal gut.
RESULTS
Acetylation of histone H4 across gene bodies is reduced in the absence of the microbiota
To assess the impact of the microbiota on histone modifications in the distal gut, we prepared histone extracts from cecal and colonic epithelial cells isolated from either conventional (Conv) or GF female mice and analyzed them by bottom-up MS, an approach that is capable of detecting hundreds of uniquely modified histone peptides (Figure 1A).
Figure 1. Acetylation of histone H4 across gene bodies is reduced in the absence of the microbiota.

(A) Schematic of experimental design. Epithelial cells were isolated from the colons and ceca of conventional (Conv) or germ-free (GF) mice. Histone post-translational modifications were profiled by MS. Acetylated H4 was profiled in colonic epithelial cells by ChIP-seq in parallel with gene expression profiling by RNA-seq.
(B) The relative abundances of the unmodified and acetylated forms of the histone H4 tail peptide (4GKGGKGLGKGGAKR17) are plotted for epithelial cells from ceca or colons of GF versus Conv mice (mean ± SEM, n = 5). The abundances of the mono-, di-, and tri-acetylated forms (ac1, ac2, ac3), were obtained by summing the relative abundances of all peptides carrying a given number of acetyl groups regardless of the specific positions of the acetyl groups. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed Welch’s t test comparing GF and conventional for a given tissue. See also Figure S1.
(C) Heatmaps and averaged profile plots of the reference-normalized H4ac4 ChIP signal across all genes for two biological replicates from conventional or GF mice. Profile plots span from 3 kb upstream of the transcription start site (TSS) to 3 kb downstream of the transcription end site (TES). The distance between the TSS and TES is scaled to a uniform length except for 3 kb downstream of the TSS and upstream of the TES, denoted by the internal tick marks. See also Figure S2A.
(D) Volcano plot of fold change versus statistical significance for the expression of 12,094 genes in colonic epithelial cells from conventional and germ-free mice. Significant differences (q < 0.05, abs(log2 FC) > mean +2 × SD) are highlighted in green and blue. Adjusted p values exceeding 1 × 10−20 are plotted at 1 × 10−20. n = 5. ns, not significant. See also Figure S2B.
Overall, we identified 47 histone peptides with statistically significant differences (Figures S1A and S1B). Compared with their colonized counterparts, mice lacking a microbiota had notable changes in the relative abundances of the modified forms of a peptide derived from the N-terminal tail of histone H4 (Figures 1B,S1A, and S1B). This peptide (4GKGGKGLGKGGAKR17) contains four acetylation sites at H4K4,H4K8, H4K12,and H4K16. Overall levels of mono-, di-, and tri-acetylation on this peptide were significantly reduced in the ceca and colons of GF mice with a corresponding increase in the levels of the unmodified peptide from roughly 50% to over 60% (Figure 1B). Overall, our results demonstrate that the microbiota supports histone H4 acetylation in epithelial cells from the cecum and colon, consistent with a previous study that examined whole colon tissue.62
We next performed ChIP-seq to localize the reduction in H4 acetylation to specific genomic regions. Given that global alterations in histone marks may obscure the detection of quantitative changes by standard ChIP-seq approaches, we included a spike-in of exogenous chromatin from Drosophila S2 cells to serve as an external reference.63 Consistent with the reduction in H4 acetylation observed by MS, the proportion of sequencing reads originating from the exogenous chromatin was higher in the GF samples, indicating less enrichment of mouse chromatin (Figure S2A). We observed a modest but consistent reduction in H4 acetylation at gene bodies across the genome rather than a more significant loss at specific loci in GF mice (Figure 1C). Transcription start sites (TSSs) were insulated from this loss and appeared to become even more prominently marked by H4 acetylation. Despite the overall decrease in gene body acetylation, GF mice had relatively small and balanced numbers of 125 upregulated and 161 downregulated genes (Figures 1D; Table S1), similar to previous work.64 Pathway analysis showed an enrichment for small-molecule and ion-transporter activity, oxidoreductase activity, and brush-border and extracellular-matrix components among these differentially expressed genes (Figure S2B).
Acetylated histones contain carbon derived from butyrate and dietary fiber
A lack of microbiota-dependent butyrate, and therefore a release on the inhibition of HDACs, could explain the reduced levels of H4 acetylation in GF mice. However, in addition to its activity as an HDAC inhibitor, butyrate constitutes a major energy source for gut epithelial cells via its oxidation to acetyl-CoA, the donor substrate for HATs.52,53 Thus, a lack of butyrate may also decrease acetyl-CoA levels and HAT activity, leading to a reduction in histone H4 acetylation in GF mice. To determine whether butyrate contributes to histone acetylation through metabolism, we incubated Caco2 cells with 1 mM 13C-labeled butyrate and then assayed isotope incorporation into acetylated histone peptides by MS (Figure 2A). As depicted in Figure 2B, the isotope label appeared on acetylated histones within 30 min, reaching a level that then plateaued over 24 h. Mass spectra of precursor ions showed a shifted isotopomer distribution with increased isotopomer intensities beginning with the m + 2 peak, consistent with the presence of acetyl groups composed of two 13C atoms (Figure S3A). Isotope incorporation ranged from less than 1% to almost 40% and was dictated mainly by the total number of acetyl groups on the peptide (Figure 2B). The multiply acetylated forms (ac2, ac3, ac4) of the histone H4 tail peptide attained higher levels of labeling than its mono-acetylated forms (ac1), possibly because the former is more likely to originate from more accessible regions of chromatin with greater HAT activity. Starving cells of alternative carbon sources, such as glucose, potentiated the isotopic incorporation from butyrate (Figure S3B).
Figure 2. Acetylated histones contain carbon derived from butyrate and dietary fiber.

(A) Outline of isotope tracing approach with U-13C-butyrate in cell culture.
(B) Caco2 cells were incubated with 1 mM U-13C-butyrate over time. Isotope incorporation into histone acetyl groups was determined by MS (mean ± SD, n = 2–4 replicate MS runs). The mono-, di-, tri-, and tetra-acetylated forms (ac1, ac2, ac3, ac4) of the histone H4 tail peptide are plotted in separate windows. Within each window, the different combinations of labeled and unlabeled acetyl groups are plotted in different colors. The number of labeled acetyl groups is denoted by asterisks (e.g., ac4** indicates four total acetyl groups, of which two are 13C-labeled). The 0-h time point represents control cells incubated with 1 mM unlabeled butyrate for 24 h. Similar results were obtained with HEK293 cells. See also Figures S3A and S3B.
(C) Outline of isotope tracing approach with U-13C-inulin in mice.
(D) Mice were orally gavaged with 13C-inulin or PBS once per day for up to 3 days. Histones were extracted from colonic epithelial cells harvested 24 h after the last gavage. Isotope incorporation is plotted over time for various forms of the acetylated histone H4 tail peptide (mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed Welch’s t test. See also Figures S3C and S3E.
(E) Mice received two daily gavages of labeled (13C) or unlabeled (Un) inulin with or without antibiotic (Abx) treatment. Isotope incorporation for the acetylated histone H4 tail peptide is plotted across the different treatment groups (mean ± SD, n = 2). The dosage of inulin (milligrams) is indicated by numbers. *p < 0.05 by unpaired, two-tailed Welch’s t test. See also Figures S3D, S3F, and S4C.
To test whether microbially derived products, such as butyrate, serve as carbon sources for histone acetylation in a more physiological model, we treated mice with isotope-labeled, fermentable fiber (U-13C-inulin). Inulin is known to be metabolized into SCFAs, including butyrate.65,66 Thus, we predicted that the microbiota would ferment inulin into butyrate and other products, which would then be oxidized by gut epithelial cells into acetyl-CoA for use by HATs (Figure 2C). In line with this hypothesis, we detected isotope incorporation into acetylated histone H4 as well as histone H3 after gavaging mice with 13C-inulin (Figures 2D, S3C, and S3E). Incorporation ranged from 5% to 10% and occurred in a dose- and time-dependent manner (Figures 2D, 2E, and S3C–S3F). Demonstrating a dependence on the microbiota, antibiotics suppressed isotope incorporation (Figures 2E, S3D, and S3F). These results provide evidence that microbiota-dependent products generated from dietary fiber can act as carbon sources for histone acetylation.
Inflammation disrupts the composition, activity, and compartmentalization of the gut microbiota and its associated molecules
Next, we endeavored to trace carbon flow through the intermediary molecules connecting fiber fermentation by the microbiota to acetyl-CoA metabolism and histone acetylation in host cells. Additionally, since inflammatory bowel diseases are commonly associated with alterations in cellular metabolism and the microbiota,23–25,55–58,67 we also aimed to determine whether inflammation has a negative impact on carbon flux from the microbiota to gut epithelial cells, which could more broadly affect the metabolic pathways upstream of histone acetylation. To this end, we performed our in vivo isotope tracing approach in conjunction with colitis induced by dextran sodium sulfate (DSS) and turned our attention from histones to small-molecule metabolites (Figure 3A). We also carried out additional high-throughput analyses in parallel to gain more insight into the factors, such as host gene expression, that could influence the gut metabolome and carbon transfer.
Figure 3. Inflammation disrupts the composition, activity, and compartmentalization of the gut microbiota and its associated molecules.

(A) Outline of isotope tracing approach in colitis model. Mice were treated with or without DSS to induce colitis. On days 6 and 7, 13C-labeled or unlabeled inulin was delivered by gavage. Tissues were harvested on day 8 for analysis.
(B) Change in relative weight over time. Mean ± SEM, n = 6. *p < 0.05, **p < 0.01 by unpaired, two-tailed Welch’s t test.
(C) Overall metabolite levels were analyzed in mice receiving Un inulin. principal-component analysis (PCA) was performed using 12,505 features from untargeted metabolomics. Features with missing area values were assigned an arbitrary value of 1. Dotted ellipses indicate 95% confidence intervals.
(D) Heatmap of metabolite features showing differential abundance across tissues or disease (3,345 features, q < 0.05 by two-way ANOVA). See also Figures S4A, S4B, and S4D.
Female mice treated with DSS developed colitis, as evident from weight loss (Figure 3B). We profiled tissue extracts of control and DSS mice with a liquid chromatography (LC)-MS-based platform for untargeted metabolomics, allowing measurement of overall metabolite levels as well as the incorporation of 13C atoms. Focusing first on general metabolite levels, we found that each tissue had a distinct metabolome in control mice, although the colon and cecum were closely related (Figures 3C; Table S2). The cecal contents were most disparate from the other tissues, which is not surprising given the capacity of the microbiota to perform metabolic transformations beyond that encoded in the host genome and that are kept compartmentalized by the epithelial barrier. The effect of this barrier is especially apparent by comparing metabolite features in the cecal contents versus other tissues in control mice, which display trends of almost complete restriction to, or exclusion from, the lumen (Figures 3D and S4A). DSS had the greatest impact on the metabolome of the cecal contents, causing a shift in the direction of the colon and cecum (Figure 3C). This change is likely driven by a compromised epithelial barrier, allowing leakage of host molecules into the lumen, or impairment in the normal output of the microbiota due to disease-associated alterations in its activity or composition. Epitomizing the latter case, deoxycholic acid, hydroxyindoleacetic acid, and urobilinogen, all of which are known to be dependent on the microbiota,68–70 declined significantly in the cecal contents of DSS mice (Figure S4A). Conversely, lactic acid and palmitoylcarnitine, which are more associated with host metabolism, became significantly elevated in the cecal contents of DSS mice (Figure S4A). We observed a small number of features showing upregulation in DSS cecal contents beyond the levels seen in control tissues, possibly representing colitis-induced molecules from the microbiota or molecules modified by the inflammatory response. These features included caprolactam, dipropylnitrosamine, nitrotyrosine, and ornithine (Figure S4A).
The gut metabolome is determined in part by the composition of the microbiota, which is known to become altered in colitis. Thus, we also performed 16S rRNA sequencing on a subset of samples to assess community composition in control and DSS mice and correlate this information with the metabolomics data. In control mice treated with 13C-inulin, a single operational taxonomic unit (OTU), matching perfectly to Dubosiella newyorkensis (strain NYU-BL-A4) from the Erysipelotrichaceae family of the Firmicutes phylum,71 accounted for roughly 90% of the entire community (Figures S4B and Table S3). This unexpected result contrasts with the high levels of diversity associated with a typical microbiota. Although inulin has been associated with the growth of Erysipelotrichaceae,72,73 a similar expansion did not occur in the previous inulin experiment, which showed the expected degree of heterogeneity in the community (Figure S4C). Thus, we attributed this skewed community structure to idiosyncrasies in this group of mice or housing conditions at the time. Despite the continued dominance of Erysipelotrichaceae, increases in organisms from the Enterobacteriaceae, Bacteroidaceae, and Verrucomicrobiaceae families (Figure S4B) were observed in DSS mice. The outgrowth of Proteobacteria, which includes Enterobacteriaceae, is a common occurrence in patients with inflammatory bowel disease.23–25
To complement the 16S data, we characterized the metaproteome of the cecal contents by MS, which led to the identification of over 1,000 host proteins and almost 400 proteins from the highly abundant D. newyorkensis (Figures S4D; Table S4). Comparing control and DSS mice, host proteins generally became more abundant in the cecal contents of the latter, while microbial proteins followed the opposite trend (Figure S4C). Again, this is consistent with a loss of epithelial barrier integrity in the DSS mice and a decline in the relative levels or activities of microbes associated with control mice.
Dietary fiber is a carbon source for hundreds of metabolites
Next, we compared female mice treated with 13C-labeled versus unlabeled inulin to identify metabolite features containing carbon derived from dietary fiber. Across all tissues, we detected isotopic labeling on nearly 300 features (Figure 4; Table S2). Molecules in the cecal contents were generally labeled to a greater extent than those in the cecal or colon tissue, consistent with a requirement for microbial processing before the labeled carbon becomes available to host cells. Only minor amounts of labeling surfaced in liver tissue. As expected, 13C atoms from inulin appeared on intermediates from glycolysis and the TCA cycle, including pyruvate, alpha-ketoglutarate, succinate, and malate (Figures S5A and S5C), in agreement with a contemporary study.74 Also consistent with previous literature, inulin underwent fermentation into SCFAs65,66,74 (Figure S5B). For instance, roughly 12% of propionate demonstrated an isotopic shift to the m + 2 isotopomer. Aside from its catabolic breakdown, inulin contributed to the biosynthesis of amino acids, nucleotide derivatives, and NAD (Figures S5D–S5F). In general, closely related molecules had similar isotopomer distributions, as highlighted by alpha-ketoglutarate, glutamate, and glutamine.
Figure 4. Dietary fiber is a carbon source for hundreds of metabolites.

Heatmap of 13C isotope incorporation for 288 metabolite features classified as labeled. The color scale represents the log2 of the relative exchange rate (RER), except in cases where RER < 1, which were assigned a value of log2(1) = 0. NA values were replaced with the row minimum. See also Figures S5 and S6.
Inflammation interferes with the contribution of fiber to host fatty acid metabolism
One of the more intriguing findings from the in vivo isotope tracing analysis involved molecules bearing long-chain acyl groups, including free fatty acids, membrane lipids, and acylcarnitines. We noted the presence of inulin-derived carbon in several long-chain fatty acids (LCFAs), such as hydroxypentadecanoic acid, from the cecal contents of control mice (Figure 5A). The isotopomer distributions for these LCFAs showed a pattern characteristic of the sequential transfer of two 13C atoms from fully labeled acetyl-CoA to an elongating acyl chain. LCFAs can be used anabolically for membrane biosynthesis or catabolically for fatty acid oxidation. Providing evidence for the former, we detected isotopic labeling of several lipids in both the cecal contents as well as the gut tissues (Figure 5B). However, a portion of these acyl groups is likely transported into mitochondria for fatty acid oxidation since long-chain acylcarnitines from the cecum and colon also acquired the isotopic label (Figure 5C). Interestingly, DSS-induced colitis curtailed the labeling of these acylated molecules (Figures 5A–5C). Treatment with non-inflammatory high-molecular-weight DSS,75 which also led to the establishment of a distinct community composition, did not interfere with the labeling of these molecules (Figure S6). Overall, these results indicate that the microbiota utilizes dietary fiber to construct long-chain acyl groups and that inflammation interferes with either the transfer of these pre-assembled molecules into the cecal tissue or their de novo synthesis in gut epithelial cells using carbon flowing from the microbiota.
Figure 5. Inflammation interferes with the contribution of fiber to host fatty acid metabolism.

(A–C) Isotopomer distributions for metabolites with long-chain acyl groups, including LCFAs (A), membrane lipids (B), and acylcarnitines (C). Metabolites were isolated from the cecal contents, cecum, or colon. The monoisotopic peak (m + 0) and isotopomers past the m + 6 peak are omitted for clarity. The distributions are corrected for the relative abundances of naturally occurring isotopomers, meaning that unlabeled samples are expected to have no signal beyond the m + 0 peak. Mean ± SEM, n = 6. See also Figure S6.
Inflammation suppresses cecal expression of genes supporting fatty acid metabolism
To correlate changes in our metabolomics data with changes in host gene expression, we performed RNA sequencing (RNA-seq) in parallel. Compared with the colon, colitis had a greater impact on gene expression in the cecum, which displayed a transcriptional signature of inflammation based on upregulated expression of genes related to NOD-like receptor (NLR) and interleukin (IL)-23 signaling, such as Il1b, Il6, Il17a, Il17f, Tnf, and Ccl2 (Figure 6A and S7A; Tables S5 and S6). S100a8 and S100a9, which together form calprotectin, a classical marker of colitis, were also upregulated. Concomitant with upregulated expression of inflammatory genes, DSS mice had downregulated expression of genes related to fatty acid metabolism (Figure 6B), including members of the carnitine shuttle system (Cpt1a, Cpt2, Crat), acyl-CoA dehydrogenases (Acads, Acadm, Acadvl), and other enzymes directly responsible for catabolizing fatty acids into acetyl-CoA (Ehhadh, Hadh, Hadha, Hadhb). Colitis also reduced the expression of peroxisomal genes (Figure S7B). Like mitochondria, peroxisomes are capable of fatty acid oxidation and have particular importance in the catabolism of certain fatty acids, such as very-long-chain fatty acids (VLCFAs), branched chain fatty acids, and dicarboxylic fatty acids. These changes in gene expression may reflect changes in the relative proportions of epithelial cells and inflammatory cells in the cecum. Overall, we find that colitis disrupted the expression of genes supporting cecal fatty acid metabolism, correlating with the reduced flux of inulin-derived carbon to acylated molecules.
Figure 6. Inflammation suppresses cecal expression of genes supporting fatty acid metabolism.

(A) PCA of whole cecum and colon tissue based on 15,825 transcriptomic features (n = 6, three each from Un and 13C inulin groups).
(B) Heatmap of core enrichment genes related to fatty acid oxidation based on gene set enrichment analysis (GSEA) of the cecal transcriptome. The relative weight change for each mouse is also indicated at the top. See also Figure S7.
DISCUSSION
Host-microbiota interactions are known to influence many physiological processes in the gut. Here, we initially focused on the impact of the microbiota on host epigenetics in the cecum and colon, finding that the lack of this important community leads to a reduction in histone H4 acetylation across the genome except at TSSs. Through isotope tracing experiments with butyrate in cell culture and dietary fiber in female mice, we demonstrate that butyrate and other products of microbial metabolism act as carbon sources for histone acetylation. Using a metabolomics approach, we also show a broader contribution of microbial metabolism to host physiology, as evident by our tracking of the isotope label to acylated molecules. We also document direct evidence that this microbiota-dependent metabolic link to histone acetylation operates in vivo. Finally, we observe that inflammation interferes with carbon flux from the microbiota to host fatty acid metabolism.
Many studies have demonstrated that the microbiota promotes histone acetylation.48,62 Our results showing higher levels of acetylation on histone H4 in conventional versus GF mice agree with those of a previous publication.62 The positive effect of the microbiota on histone acetylation has been linked to fiber fermentation, which produces SCFAs like butyrate.48,62 However, butyrate may not accumulate to significant levels in gut epithelial cells since they are adapted to derive their energy from SCFAs through fatty acid oxidation. Thus, rather than supporting histone acetylation via inhibition of a negative regulator (HDACs), the microbiota may activate a positive regulator (HATs) via provision of substrates that feed the production of acetyl-CoA, the donor substrate for HATs. These two mechanisms of butyrate-mediated promotion of histone acetylation were demonstrated by experiments showing that silencing of ATP citrate lyase prevents increases in histone H3 acetylation at low but not high doses of butyrate.52 Earlier studies in cell culture suggested that butyrate may act as a carbon donor for histone acetylation based on incorporation of 14C into histones and the detection of 13C-labeled acetate hydrolyzed from histones.52,59 Our isotope tracing analysis with 13C-labeled butyrate in cell culture, combined with the high-resolution analysis by MS, extends these results and shows unequivocally that the acetyl groups of histones contain carbon derived from butyrate. Importantly, using 13C-labeled fiber in a more physiological setting, we obtained direct evidence that this microbiota-dependent metabolic link to histone acetylation operates in vivo as well. The extent to which butyrate functions as an inhibitor of HDAC activity versus a supplier of acetyl-CoA in support of HAT activity remains unclear but likely relates to its concentration and the rate at which cells metabolize it.52 Given the recent and unexpected observation that HDAC activity is higher in intestinal epithelial cells from conventional, butyrate-sufficient mice versus GF, butyrate-deficient mice, HDAC inhibition may not be the primary activity of butyrate in the gut.76
Although a global decrease in H4 acetylation in GF mice has been noted previously,62 we have expanded upon these earlier findings by performing ChIP-seq and RNA-seq to ascertain where this reduction occurs in the genome and the consequences for gene expression. These results showed a loss of H4 acetylation across the genome, particularly at gene bodies. This finding mirrors what has been recently described for H4 acetylation in cells treated with an HDAC inhibitor.77 Despite the global loss of H4 acetylation in GF mice, we find that this histone modification becomes more pronounced at TSSs. However, these epigenetic changes appear to have only minor effects on transcription since we find relatively small and balanced numbers of upregulated and downregulated genes in GF mice. This is consistent with a prior analysis comparing chromatin accessibility in conventional and GF mice, which found that the anatomical location along the intestinal tract but not colonization status influenced the epigenetic landscape.64 Instead, variations in gene expression due to colonization status were proposed to arise from microbiota-dependent transcription factors acting on a pre-configured epigenetic landscape established during early development. While some differences in the genomic patterns of H3K4me1 and H3K27ac were noted in a subsequent study of GF mice, a more remarkable observation was the increased binding of the nuclear receptors HNF4A and HNF4G to areas of chromatin in GF mice that had similar patterns of accessibility and histone modifications, again emphasizing an important contribution of stimulus-dependent transcription factors in controlling gene expression.78 In this regard, it is especially noteworthy that microbial metabolites can act as ligands for nuclear receptors.31,79,80
The contribution of the microbiota to histone acetylation through metabolism prompted us to examine isotope incorporation more broadly using metabolomics to determine how microbial metabolism of dietary fiber supplies precursors to host cells. The idea that the microbiota supports host metabolism in the gut has been suggested by the transfer of isotope-labeled carbon from butyrate to CO2 in cultures of primary colonocytes as well as established cell lines.53,55–60 Some studies additionally tracked the isotope label to acetyl-CoA, histones, and lipids.14,59,60 Consistent with the microbiota supporting host metabolism, GF mice display signs of energy deprivation in the colon.53 Notably, this energy deficit could be rescued by introducing a normal microbiota or a single butyrate-producing organism.
Bringing clinical significance to this metabolic connection, similar observations of impaired butyrate oxidation and mitochondrial function have been reported in the case of ulcerative colitis in humans and DSS-induced colitis in mice.56–58,67,81,82 Building on these in vitro and ex vivo analyses, we conducted isotope tracing in vivo with 13C-labeled fiber and obtained definitive evidence of carbon transfer from the microbiota to the host under physiological conditions. A recent study similarly employed 13C-labeled fiber to interrogate the metabolic pathways of the microbiota using an in vitro system,83 which affords more control over labeling but cannot address potential interactions with host tissues like in vivo approaches employed by us and others.74,84 Among our most significant findings, we detected incorporation of the isotope label into the acyl chains of palmitoylcarnitine and membrane lipids. These results support the notion that the microbiota acts as a carbon source for acetyl-CoA, which can then be used by gut epithelial cells to drive the TCA cycle, acetylate proteins, or construct long-chain acyl-CoA for lipid synthesis (Figure 7). Although we did not address precisely which members of the microbiota are responsible for provisioning carbon to the host in our studies, we suspect that the dominant Erysipelotrichaceae (Figure S4B) might be one possibility. Given that the microbiota is typically heterogeneous, the unexpected outgrowth of this single taxon represents a potential caveat in our metabolomics data. However, the similar isotopic distributions for the metabolites presented in Figure S6B from the 13C-H2O and 13C-DSShi groups, despite two distinct community compositions (Figure S6A), suggests that additional taxa beyond Erysipelotrichaceae are also capable of mediating carbon transfer to the host. Furthermore, we observed isotopic labeling of histone acetylation in a diverse community without Erysipelotrichaceae expansion (Figures 2E and S4C). Thus, this metabolic link between the microbiota and host may be functionally conserved and resilient to at least some variations in community composition, which is in line with a landmark study showing conservation of functional gene categories in the microbiota despite variations in taxonomy.85
Figure 7. Proposed model linking dietary fiber and the microbiota to fatty acid metabolism in gut epithelial cells.

Dietary fiber is processed by the microbiota to generate precursors, such as SCFAs, for fatty acid metabolism in host epithelial cells. The resulting acetyl- and acyl-CoA can be utilized to acetylate histones, synthesize lipids, or fuel the TCA cycle for ATP production. Long-chain fatty acids from the microbiota may also contribute to these processes. In the absence of a microbiota, GF mice must rely on other sources of metabolic precursors. Similarly, inflammation interferes with carbon flow from fiber to the host via the microbiota.
Our observation that DSS-induced colitis interfered with the isotopic labeling of acylated molecules aligns with earlier work correlating colitis with deficient fatty acid metabolism. A major question is whether this deficiency is a primary factor in the development of ulcerative colitis. Insufficient energy production could compromise the function of epithelial cells, causing a breakdown of the epithelium and a consequent immune response to microbial products that leak into host tissues. Although it is unclear what might trigger an initial decline in fatty acid metabolism, antibiotic usage, low-fiber diets, or infections could all reduce the ability of the microbiota to produce the SCFAs that fuel epithelial cell metabolism.86 Excessive regeneration of the epithelium may also favor the non-oxidative metabolism of undifferentiated cells.86,87 On the other hand, deficient metabolism could represent a promoting factor or even a simple consequence rather than a primary cause of ulcerative colitis since the microbial communities associated with the disease and an inflamed epithelium may have respectively lower capacities for SCFA production and fatty acid oxidation.
The labeling of long-chain acyl groups also raises the question of their origin and whether LCFAs might complement SCFAs as energy sources in the gut. One possibility accounting for these labeled acyl groups involves the oxidation of labeled SCFAs to acetyl-CoA, which could then be used by host cells to synthesize LCFAs. Alternatively, the labeled acyl groups may come directly from microbial lipids or LCFAs, which could be made available to host cells through membrane vesicles or the action of secreted phospholipases. In support of LCFAs originating directly from the microbiota, a recent study imaged the transfer of lipids from bacteria to mammalian cells.88 Besides their assimilation into host membranes, these microbial lipids could also be oxidized for energy, as indicated by our detection of labeled palmitoylcarnitine. Indeed, the same energy deficiency observed in GF mice53 has not been described, to our knowledge, in mice lacking Scad activity, which is necessary for the utilization of SCFAs. This suggests that SCFAs may not constitute the only pool of microbiota-dependent carbon sources in the gut.
Sequencing approaches have facilitated our understanding of the community structure and coding potential of the microbiota, which is estimated to exceed the gene content of the human genome by a factor of 150×.89 Further insight into how the microbiota influences host physiology necessitates the complementary study of microbial proteins and small molecules, representing the functional output of the microbiota. Overall, the current study contributes to our knowledge of host-microbiota interactions by using isotopic labeling to demonstrate a connection between microbial fermentation of dietary fiber and host fatty acid metabolism, which may be important for gut homeostasis.
Limitations of the study
Since the colonic epithelium is a heterogeneous collection of cells that includes enteroendocrine cells, goblet cells, Panethlike cells, intestinal stem cells, transit-amplifying (TA) cells, and absorptive colonocytes, our measurements of histone acetylation and metabolites reflect contributions from all of these subpopulations. Colonocytes and TA cells likely represent the major subpopulations with reported frequencies ranging from 30% to 60% and 10% to 40%, respectively.90,91 These frequencies may vary between GF and Conv mice. Additionally, while the untargeted metabolomics platform provided us with an unbiased profile of the isotopic labeling of small-molecule metabolites, the use of synthetic standards and targeted methods could provide additional confidence in compound identification as well as information about the positions of the labeled carbon atoms.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagent should be directed to and will be fulfilled by the lead contact, Benjamin A. Garcia (bagarcia@wustl.edu).
Materials availability
This study did not generate new, unique reagents.
Data and code availability
The ChIP-seq and RNA-seq data have been deposited at NCBI GEO: GSE179233. The mass spectrometry proteomics data for the colitis study have been deposited to the ProteomeXchange Consortium via the PRIDE92 partner repository with the dataset identifier PXD026959 and 10.6019/PXD026959. (PRIDE: PXD026959) The metabolomics data are available from MassIVE: MSV000087752. All datasets are publicly available as of the date of publication. All data reported in this paper will be shared by the lead contact by request.
Raw data and analysis scripts have been deposited online (Mendeley Data: https://doi.org/10.17632/sdj5j3ffy7.1) and are publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All experiments were performed with approval from the Institutional Animal Care and Use Committee. Unless otherwise noted, mice were C57BL/6J females from Jackson Labs, age 7–8 weeks. Germ-free mice were provided by the Penn Gnotobiotic Mouse Facility. Animals were euthanized by CO2 asphyxiation followed by cervical dislocation. Colons and ceca were dissected, flushed with sterile phosphate-buffered saline (PBS), splayed open longitudinally, and washed again with PBS for use in epithelial cell isolation. For the inulin labeling time course, mice were acclimated to an irradiated AIN76a diet (Research Diets, D10001) for 3 days and then orally gavaged with 100 mg of U-13C-labeled inulin (Isolife), dissolved in 100 μL PBS, once per day for up to 3 days. Control mice received PBS alone or unlabeled inulin. One day after the last gavage, colon tissue was harvested for epithelial cell isolation. For the antibiotic experiment, mice were acclimated to Splenda water (10 mg/mL) over 3 days and then treated with or without antibiotics (1 mg/mL ampicillin, 1 mg/mL neomycin, 0.5 mg/mL metronidazole, 0.5 mg/mL vancomycin) in the Splenda drinking water and switched to the AIN76a diet. On days 5 and 6, mice received an oral gavage of 10, 50, or 100 mg of U-13C-inulin or 100 mg unlabeled inulin. On day 7, colon tissue was harvested for epithelial cell isolation. For the colitis experiments, mice (8–10 wks) were acclimated to the AIN76a diet over the preceding 3 days and then had colitis induced with 2% dextran sodium sulfate (MP Bio, 36–50 kDa) in the drinking water on day 0. Non-inflammatory high molecular weight DSS (Sigma, 500 kDa) was also used in some cases. On days 6 and 7, mice received an oral gavage of 100 mg of U-13C-labeled or unlabeled inulin (IsoLife) dissolved in 200 μL PBS. On day 8, mice were sacrificed by cervical dislocation after a 4 hour fast. Livers, colons, ceca, and cecal contents were harvested and flash frozen in liquid nitrogen.
Cell culture
Standard conditions for the culture of HEK293 (female human embryonic kidney) and Caco2 (male human colon adenocarcinoma) cells consisted of DMEM supplemented with 10% FBS or FetalPlex and 1X penicillin/streptomycin in a humidified incubator at 37°C. Starved conditions consisted of DMEM without glucose or pyruvate (but with glutamine) supplemented with 1% dialyzed FBS and antibiotics. For isotope labeling experiments, cells were seeded in 5 or 10 cm dishes, allowed to grow for 24–48 hours, and then treated with 1 mM unlabeled or U-13C-labeled sodium butyrate (Sigma). Cell monolayers were rinsed with PBS, trypsinized, washed again in PBS, and flash frozen in liquid nitrogen. Cell lines were not authenticated.
METHOD DETAILS
Epithelial cell isolation
To prepare epithelial cell pellets, colons and ceca were incubated in PBS containing 30 mM EDTA and 1.5 mM DTT for 20 minutes at 4°C with occasional inversion. Tissue was subsequently transferred to PBS containing 30 mM EDTA, incubated for 10 mins at 37°C, and shaken vigorously for 1–2 minutes until the mesenchyme separated from the epithelium. The mesenchyme was removed and epithelial cells were pelleted at 1500 rpm for 5 minutes at 4°C and washed in PBS. In some cases, cell pellets were further incubated with dispase (Corning, 0.3 U/ml) in PBS (100 μL dispase per 50 mL PBS) at 37°C for 10 min and then quenched with 1/10 volume of FBS containing DNase (10 ul DNase per ml FBS) followed by washing in PBS. Cell pellets were flash frozen in liquid nitrogen and stored at −80°C until use. For ChIP-seq analysis, cells were cross-linked prior to freezing as indicated below.
Histone extraction and analysis by LC-MS/MS
Histones were isolated from the nuclei of frozen cell pellets by acid extraction and derivatized with propionic anhydride similarly as described previously.93 Briefly, cell pellets were resuspended in nuclear isolation buffer (NIB) containing 15 mM Tris pH 7.5, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, and 250 mM sucrose supplemented with 1 mM DTT, 10 mM sodium butyrate, 500 μM AEBSF, 5 nM microcystin, and 0.2% NP-40 Alternative. After allowing lysis to proceed on ice for 10 mins, nuclei were pelleted at 500 × g for 5 mins at 4°C, washed twice in NIB without detergent, and then extracted in 0.4 N H2SO4 for 2–4 hrs at 4°C with rotation. Insoluble debris was pelleted by centrifugation at 3400 × g for 5 mins at 4°C and proteins were precipitated overnight on ice by adding 1 volume of trichloroacetic acid to 3 volumes of supernatant. Precipitated proteins were pelleted as above, washed in 0.1% HCl in acetone and then acetone, and allowed to air dry before resuspending in 100 mM NH4HCO3. For derivatization, 1 volume of 25% propionic anhydride in 2-propanol was added to 2 volumes of sample containing 5–20 μg of protein. Ammonium bicarbonate salt was also added for buffering purposes. After incubation for 15 mins at 37°C, samples were dried in a speed vac and derivatized a second time as above. Derivatized samples were then resuspended in 100 mM NH4HCO3 and digested overnight at room temperature with 1 μg trypsin per 20 μg of protein. After two additional rounds of derivatization, digested peptides were desalted with C18 stage tips for analysis by mass spectrometry.
Histone peptides were resolved with EasyLC 1000 or Dionex UltiMate 3000 LC systems fitted with 75 μm i.d. x 15–20 cm fused silica columns (Polymicro Tech) packed with ReproSil-Pur 120 C18-AQ (3 μm, Dr. Maisch GmbH) and connected in line with a mass spectrometer (Thermo Elite, Velos, Fusion, QE, or QE-HF). The chromatography conditions generally consisted of a linear gradient from 5 to 33% solvent B (0.1% formic acid in 80% acetonitrile) in solvent A (0.1% formic acid in water) over 45 mins and then 33 to 98% solvent B over 5 mins at a flow rate of 300 nL/min. The mass spectrometer was programmed for data-independent acquisition (DIA). One acquisition cycle consisted of a full MS scan, 8 DIA MS/MS scans of 50 m/z isolation width starting from 325 m/z, a second full MS scan, and 8 more DIA MS/MS scans to reach 1125 m/z. Typically, full MS scans were acquired in the Orbitrap mass analyzer across 300–1100 m/z at a resolution of 60,000 in positive profile mode with a maximum injection time of 100 ms and an AGC target of 2e5. MS/MS data from CID or HCD fragmentation was collected in the ion trap (when available) or the Orbitrap. These scans typically used an NCE of 30, an AGC target of 1e4, and a maximum injection time of 50 ms. Histone MS data, including isotope incorporation, were analyzed with EpiProfile94 and further processed in R to filter for peaks with consistent retention times.
Metabolomics
Frozen tissues were lyophilized and then homogenized in 80% methanol with bead beating (Precellys homogenizer, Bertin Technologies) at a target concentration of 5–10 mg tissue per 500 μL of solvent. For tissues with limited quantities, a minimum of 250 μL solvent was used. For lyophilized tissues exceeding 10 mg, bead beating was used to ground them into a dry powder, of which roughly 10 mg was used for homogenization. Aliquots of the homogenates were treated with additional methanol and then centrifuged to precipitate proteins. The supernatants were dried under nitrogen and then resuspended in HPLC mobile phases for resolution on C18 or HILIC columns in line with a Thermo Orbitrap ID-X mass spectrometer. For C18 chromatography, extracted samples were injected onto a Waters Acquity BEH C18 column (2.1 × 150 mm, 1.7 μm) at 65°C using a 13 min linear gradient from 99% solvent A (0.1% formic acid in water) to 100% solvent B (acetonitrile with 0.1% formic acid) at a flow rate of 0.6 mL/min on a Thermo Vanquish UHPLC. The mass spectrometer was run in ESI+/− modes from 60–1000 Da at a resolution of 120,000. For HILIC, extracted samples were injected onto an Agilent Poroshell HILIC-Z column (2.1 × 100 mm, 1.9 μm) at 30°C using a 10 min linear gradient from 95% solvent B (acetonitrile) to 70% solvent A (water with 10 mM ammonium acetate pH 9 and 0.1% medronic acid) at a flow rate of 0.4 mL/min to separate highly polar metabolites. Each sample was assigned a randomly generated number to determine injection order.
Pooled samples were injected periodically throughout the runs for QC. Data-dependent MS/MS was collected on separate injections of pooled samples, including tissue-specific pools, both with and without the use of AcquireX. For these MS/MS runs, a full MS scan was collected in the Orbitrap every 0.6 seconds in the scan range of 550–1000 m/z with quadrupole isolation with AGC set to 1e5, resolution set to 60,000, and maximum injection time set to 50 ms. Between the full MS scans, MS/MS scans were collected in the Orbitrap at a resolution of 15,000. Quadrupole isolation was used with a window of 1.5 m/z. The AGC was set to 2e4 and the maximum injection time was set to 22 ms. Precursor ions were fragmented by HCD activation with stepped collision energies of 20, 35, and 50%. Dynamic exclusion was set to 2.5 seconds and the intensity threshold for MS/MS was set to 2e4.
CompoundDiscoverer (CD) was used for data processing and isotope incorporation analysis. Additional analyses were performed in R. For the analysis of overall metabolite levels, peak areas from CD were first multiplied by a correction factor to standardize extraction conditions to 20 mg/mL and then normalized by log2 transformation. The relative exchange rate (RER) calculated by CD was used to identify features with 13C incorporation based on consistently higher RER in labeled versus unlabeled conditions and a low background RER in the unlabeled conditions (log2[labeled/unlabeled] > 1; p < 0.01 from unpaired, two-tailed t test; maximum mean RER for unlabeled <1; maximum median RER for labeled >2). In cases where the log2 FC or p value was undefined, a feature was considered as labeled if the mean RER for unlabeled conditions within a given tissue was zero and the mean RER for the labeled condition was greater than 2. In the experiment with high MW DSS, the criteria were set as: log2[labeled/unlabeled] > 1; p < 0.05 from unpaired, two-tailed t test; maximum mean RER for unlabeled <0.5; maximum median RER for labeled >0.5). For features with undefined log2 FC or p values, the mean RER for unlabeled conditions within a given tissue was required to be <0.1 and the mean RER for the labeled condition was required to be greater than 1. In some cases, features previously identified as labeled were manually retrieved.
ChIP-seq and RNA-seq
Cross-linking was performed by treatment with 1% formaldehyde in PBS for 5 mins at room temperature, followed by quenching with 125 mM glycine for an additional 5 mins at room temperature. Cross-linked pellets were then washed in PBS and flash frozen until use. Nuclear lysates were prepared by resuspending cells in lysis buffer 1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1× protease inhibitors) and rotating for 10 mins at 4°C. After centrifugation at 3000 rpm for 5 mins at 4°C, the cell pellet was resuspended in lysis buffer 2 (10 mM Tris-HCl pH 8, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 13 protease inhibitors) and rotated for 10 mins at room temperature. Cells were pelleted and then resuspended in lysis buffer 3 (10 mM Tris-HCl pH 8, 100 mM NaCl, 1 mm EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.1% N-lauroylsarcosine, 1× protease inhibitors) for chromatin shearing with a Covaris S220 focused ultrasonicator (8.4W, 200 peak power, 200 cycles/burst, 900 seconds) in a 4°C water bath. After sonication, Triton X-100 was added to 1% final concentration and the lysate was cleared by centrifugation at top speed in a microfuge for 10 mins at 4°C. Magnetic protein G beads were washed with blocking solution (0.5% BSA in PBS) and then incubated with rabbit anti-serum against acetylated histone H4 (Millipore 06–866) or normal rabbit anti-serum in blocking solution for several hours at 4°C with rotation. Beads were then washed in blocking solution and then added to nuclear lysates containing a spike-in of Drosophila S2 chromatin (cells generously provided by P. Pascual-Garcia), prepared as above. After overnight incubation at 4°C, beads with captured chromatin were washed with 3 × 1 mL RIPA wash buffer (50 mM HEPES-KOH pH 7.5, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% sodium deoxycholate) and 1 × 1 mL of final wash buffer (10 mM Tris-HCl pH 8, 50 mM NaCl, 1 mM EDTA). Beads were eluted with 200 μL elution buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS) for 30 min at 65°C. The supernatant was then incubated at 65°C overnight to reverse cross-links. After dilution with one volume of TE, RNase was added to 0.2 mg/mL and incubated at 37°C for 2 hrs. Proteinase K was then added to 0.2 mg/mL and incubated at 55°C for 2 hrs. DNA was purified by phenol-chloroform extraction with subsequent ethanol precipitation. Purified DNA was resuspended in 10 mM Tris pH 8 and prepared for sequencing using the NEBNext Ultra II DNA library kit for Illumina (New England Biolabs). Paired-end sequencing with 75 cycles and a 6-cycle index read was performed on the Illumina NextSeq500 system.
Total RNA was extracted from epithelial cell pellets and frozen tissues using the RNeasy kit (Qiagen). The QIAshredder column was used for epithelial cell pellets. For frozen tissues from the colitis study, homogenization was carried out in the provided lysis buffer using bead beating (Precellys homogenizer, Bertin Technologies). The NEBNext Ultra or Ultra II Directional RNA Library Prep Kit for Illumina with the mRNA magnetic isolation module (New England Biolabs) was used to prepare sequencing libraries from 1 μg of total RNA.
Sequencing reads were aligned with STAR (v2.5.2a)95 (ChIP-seq parameters: –outFilterMultimapNmax 20 –outFilterMismatchNmax 999 –alignMatesGapMax 1000000; RNA-seq parameters: –outFilterType BySJout –outFilterMultimapNmax 20 –alignSJoverhangMin 8 –alignSJDBoverhangMin 1 –outFilterMismatchNmax 999 –alignIntronMax 1000000) to the mm10 genome assembly or the mm10 genome concatenated with the Drosophila dm6 genome in the case of the ChIP with the exogenous spike-in. Alignments were filtered for uniquely mapping reads with samtools.96 Transcriptome data was analyzed quantitatively with HTSeq97 and DESEQ2.98 Pathway analysis was performed using GSEA99 or the ClusterProfiler and ReactomePA packages in R.100,101 For ChIP, normalization to the exogenous reference chromatin in lieu of sequencing depth was performed as described previously.102 Briefly, downsampling factors, corrected for any variations in reference chromatin abundance in the input samples, were calculated and applied to each sample such that equal numbers of Drosophila reads were obtained across all samples with the largest factor being equal to 1. BigWig files were generated using an in-house script. DeepTools was used for additional analysis and visualization.103
Metaproteomics
Protein precipitates from the metabolite extractions of cecal contents were resolubilized in 8 M urea, 0.1 M NaCl, and 50 mM Tris pH 8 supplemented with protease inhibitors. Insoluble debris was pelleted by centrifugation. Roughly 20 μg of soluble protein was reduced with 5 mM DTT for 30 mins at room temperature and then alkylated with 20 mM iodoacetamide for 30 mins at room temperature in the dark. After adding four volumes of 0.1 M ammonium bicarbonate, proteins were digested with 1 μg trypsin overnight at 37°C and then desalted with C18 stage tips for mass spectrometry analysis. As for the histone analysis, peptides were separated by reverse-phase chromatography with an online EasyLC1000 nano-LC system. The gradient consisted of 2 to 30% solvent B (80% acetonitrile with 0.1% formic acid) over 52 mins, 30 to 60% solvent B over 24 mins, and 60 to 90% solvent B over 2 mins at a flow rate of 300 nL/min. Water with 0.1% formic acid served as solvent A. The mass spectrometer (Thermo QE) was operated in data-dependent acquisition (DDA) mode. A full scan in positive profile mode was acquired over the range of 300–1400 m/z at a resolution of 70,000, AGC target of 1e6, and maximum IT of 100 ms. The top 15 precursor ions were selected for HCD fragmentation at NCE 30 and MS/MS scans were collected at a resolution of 17,500 with an AGC target of 1e5, a maximum IT of 50 ms, and an isolation width of 2.0 m/z in centroid mode. Only ions with charge states between +2 and +6 were considered. Dynamic exclusion was set to 40 seconds. The minimum AGC target was set to 1e3.
Metaproteomics data was analyzed with ProteomeDiscoverer using a custom metaproteome constructed by concatenating the mouse proteome with the proteomes of bacteria from the most abundant OTUs based on 16S sequencing. For database searches, trypsin was set as the protease with up to 2 missed cleavages. The mass tolerances were 10 ppm for precursors and 0.02 Da for fragments. Carbamidomethylation (+57.021 Da to Cys) was specified as a fixed modification while oxidation (+15.995 Da to Met), carbamylation (+43.006 Da to Lys and peptide N-termini), acetylation (+42.011 Da to protein N-termini), methionine loss (−131.040 Da to protein N-termini), and methionine loss with acetylation (−89.030 Da to protein N-termini) were set as variable modifications. The Percolator node was used to control for false-positive PSMs. The data was further analyzed in R with the MSStats package.104
16S sequencing
Aliquots of cecal contents were submitted to the Microbiome Sequencing Core at the Weill Cornell School of Medicine for analysis. DNA was extracted using the Promega Maxwell RSC 48 system and amplified with the 515F and 926R primers, which target the V4 and V5 regions of the 16S rRNA gene, following the protocol from the Earth Microbiome Project (https://earthmicrobiome.org/protocols-and-standards/16s/, https://doi.org/10.17504/protocols.io.nuudeww).105,106 Sequencing (250 bp paired-end reads) was performed on the Illumina MiSeq platform. After de-multiplexing, paired reads were merged using USEARCH software (v 11.0.667)107 with the usearch -fastq_mergepairs command with options -fastq_maxdiffs 5 -fastq_pctid 90 -fastq_minovlen 17-fastq_minmergelen 300. PhiX reads were removed with using usearch filter_phix. Reads were then filtered for quality with usearch -fastq_filter -fastq_maxee 1.0 and clustered by usearch -cluster_otus using default settings. Merged, pre-filtered reads were mapped to OTUs by usearch -otutab with default settings. Taxonomic prediction was performed with usearch -sintax -strand both -sintax_cutoff 0.8 using the RDP 16S training set (v16) as a reference database.108,109 Later analyses utilized a workflow involving Cutadapt,110 Dada2,111 and QIIME2.112 The relative abundances of OTUs and other taxonomies were further analyzed in R. Unpaired t-tests without the assumption of equal variances were used to detect statistically significant differences between two groups of interest (H2O and DSS). One-way ANOVA or Kruskal-Wallis tests were used to detect differences between multiple groups of interest.
QUANTIFICATION AND STATISTICAL ANALYSIS
Group sizes, measurements of center and spread, statistical tests, and thresholds for significance are indicated in the figure legends. The R scripts used for analysis are available online at Mendeley Data (Lund, Peder (2022), “Stable Isotope Tracing from the Microbiota to Acetylated Histones and Metabolites”, Mendeley Data, V1, https://doi.org/10.17632/sdj5j3ffy7.1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-acetyl-histone H4, rabbit polyclonal | Millipore | Cat# 06–866; RRID: AB_310270 |
|
| ||
| Biological samples | ||
|
| ||
| Drosophila S2 cells | Maya Capelson lab | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| AIN76a diet | Research Diets | Cat# D10001 |
| Inulin from chicory, unlabeled | Isolife | Cat# N-10302 |
| Inulin from chicory, U-13C-labeled | Isolife | Cat# U-10302 |
| Inulin from chicory, unlabeled | Sigma | Cat# I2255 |
| Dextran sodium sulfate, 36–50 kDa | MP Bio | Cat# 160110 |
| Dextran sodium sulfate, 500 kDa | Sigma | Cat# 31395 |
| Sodium butyrate, 13C4-labeled | Sigma | Cat# 488380 |
| Propionic anhydride | Sigma | Cat# 240311 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NEBNext Ultra II DNA library kit for Illumina | New England Biolabs | Cat# E7645 |
| NEBNext Ultra Directional RNA library kit for Illumina | New England Biolabs | Cat# E7420 |
| NEBNext Ultra II Directional RNA library kit for Illumina | New England Biolabs | Cat# E7765 |
|
| ||
| Deposited data | ||
|
| ||
| Acetylated histone H4 ChIP-seq data | This paper | GEO: GSE179233 |
| Colonic epithelial cell RNA-seq data | This paper | GEO: GSE179233 |
| DSS colitis RNA-seq data | This paper | GEO: GSE179233 |
| DSS colitis metaproteomics data | This paper | PRIDE: PXD026959 |
| DSS colitis metabolomics data | This paper | MassIVE: MSV000087752 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293 cells | Benjamin Garcia lab | N/A |
| Caco2 cells | Gary Wu lab | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Labs | N/A |
| Mouse: C57BL/6J, germ-free | Penn Gnotobiotic Mouse Facility | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| EpiProfile | Yuan et al.94 | N/A |
| RStudio, v1.2.5033 | RStudio | https://www.rstudio.com/ |
| STAR, v2.5.2a | Dobin et al.95 | N/A |
| HTSeq | Anders et al.97 | https://htseq.readthedocs.io/en/master/ |
| DESEQ2 | Anders and Huber98 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA | Subramanian et al.99 | https://www.gsea-msigdb.org/gsea/index.jsp |
| R package, ClusterProfiler | Yu et al.100 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R package, ReactomePA | Yu and He101 | https://bioconductor.org/packages/release/bioc/html/ReactomePA.html |
| DeepTools | Ramírez et al.103 | https://deeptools.readthedocs.io/en/develop/ |
| ProteomeDiscoverer | Thermo | N/A |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| R package, MSStats | Choi et al.104 | https://bioconductor.org/packages/release/bioc/html/MSstats.html |
| Compound Discoverer | Thermo | N/A |
| USEARCH | Edgar and Bateman107 | https://www.drive5.com/usearch/ |
| Cutadapt v3.4 | Martin110 | https://cutadapt.readthedocs.io/en/stable/# |
| Dada2 v1.22.0 | Callahan et al.111 | https://benjjneb.github.io/dada2/index.html |
| QIIME2 v2021.8.0 | Bolyen et al.112 | https://qiime2.org/ |
| samtools | Li et al.96 | http://www.htslib.org/ |
Highlights.
Acetylation of histone H4 is reduced in epithelial cells of germ-free mice
The microbiota metabolizes fiber to generate carbon for histone acetylation
Inflammation disrupts carbon flow from the microbiota to host fatty acid metabolism
ACKNOWLEDGMENTS
Funding is generously acknowledged from the Crohn’s and Colitis Foundation (RFA 598467 to P.J.L.), the Crohn’s and Colitis Foundation Microbiome Initiative, the PennCHOP Microbiome Program, the Penn Center for Nutritional Science and Medicine (PenNSAM), The Rockefeller University (C.D.A.), the Shapiro-Silverberg Fund for the Advancement of Translational Research at The Rockefeller University (L.A.G.), the St. Jude Children’s Research Hospital Chromatin Consortium, the Host-Microbial Analytic and Repository Core of the Center for Molecular Studies in Digestive and Liver Diseases (P30 DK050306), and the National Institutes of Health (T32CA009140 to P.J.L.; F32GM134560 and K99GM143550 to L.A.G.; P01CA196539, R01AI118891, and R01HD106051 to B.A.G.; R01GM40922 to C.D.A.). The authors wish to thank Johayra Simithy, Sophie Trefely, Kevin Janssen, Simone Sidoli, Greg Donahue, Pau Pascual-Garcia, Nathaniel W. Snyder, and Meenakshi Bewtra for assistance. P.J.L. acknowledges Warren Pear for additional support and mentoring.
Footnotes
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111809.
REFERENCES
- 1.Ghosh S, Dai C, Brown K, Rajendiran E, Makarenko S, Baker J, Ma C, Halder S, Montero M, Ionescu VA, et al. (2011). Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am. J. Physiol. Gastrointest. Liver Physiol 301, G39–G49. 10.1152/ajpgi.00509.2010. [DOI] [PubMed] [Google Scholar]
- 2.Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, and Sonnenburg JL (2014). Gut microbiota-produced succinate promotes C. Difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16, 770–777. 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, and Sonnenburg JL (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99. 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bäumler AJ, and Sperandio V.(2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93. 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic T reg cell homeostasis. Science 341, 569–573. 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda K, and Littman DR (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. 10.1038/nature18848. [DOI] [PubMed] [Google Scholar]
- 7.Thaiss CA, Zmora N, Levy M, and Elinav E.(2016). The microbiome and innate immunity. Nature 535, 65–74. 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 8.Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites andhost immunity. Nat. Rev. Immunol 16, 341–352. 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gensollen T, Iyer SS, Kasper DL, and Blumberg RS (2016). How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, and Medzhitov R.(2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241. 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, and Diefenbach A.(2009). RORgt and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol 10, 83–91. 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443. 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. (2015). Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671. 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, and Stappenbeck TS (2016). The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165, 1708–1720. 10.1016/j.cell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O, Subramanian S, Manary MJ, Trehan I, Jorgensen JM, et al. (2016). Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 351, aad3311. 10.1126/science.aad3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonnenburg JL, and Bäckhed F.(2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiffany CR, and Bäumler AJ (2019). Dysbiosis: from fiction to function. Am. J. Physiol. Gastrointest. Liver Physiol 317, G602–G608. 10.1152/ajpgi.00230.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillis CC, Hughes ER, Spiga L, Winter MG, Zhu W, Furtado de Carvalho T, Chanin RB, Behrendt CL, Hooper LV, Santos RL, and Winter SE (2018). Dysbiosis-associated change in host metabolism generates lactate to support Salmonella growth. Cell Host Microbe 23, 54–64.e6. 10.1016/j.chom.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, et al. (2017). Microbiota-activated PPAR-g signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, and Finlay BB (2007). Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–129. 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonnenburg ED, and Sonnenburg JL (2014). Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metabol. 20, 779–786. 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, Timmis KN, and Schreiber S.(2004). Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53, 685–693. 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, and Pace NR (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785. 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni J, Wu GD, Albenberg L, and Tomov VT (2017). Gut microbiota and IBD: causation or correlation? Nat. Rev. Gastroenterol. Hepatol 14, 573–584. 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy M, Thaiss CA, and Elinav E.(2016). Metabolites: messengers between the microbiota and the immune system. Genes Dev. 30, 1589–1597. 10.1101/gad.284091.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, and Rudensky AY (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, et al. (2017). Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549, 48–53. 10.1038/nature23874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyamoto J, Mizukure T, Park SB, Kishino S, Kimura I, Hirano K, Bergamo P, Rossi M, Suzuki T, Arita M, et al. (2015). A gut microbial metabolite of linoleic acid, 10-hydroxy-cis-12-octadecenoic acid, ameliorates intestinal epithelial barrier impairment partially via GPR40-MEK-ERK pathway. J. Biol. Chem 290, 2902–2918. 10.1074/jbc.M114.610733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klepsch V, Moschen AR, Tilg H, Baier G, and Hermann-Kleiter N(2019). Nuclear receptors regulate intestinal inflammation in the context of IBD. Front. Immunol 10, 1070. 10.3389/fimmu.2019.01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melhem H, Kaya B, Ayata CK, Hruz P, and Niess JH (2019). Metabolite-Sensing G protein-coupled receptors connect the diet-microbiota-metabolites Axis to inflammatory bowel disease. Cells 8, 450. 10.3390/cells8050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcobal A, Kashyap PC, Nelson TA, Aronov PA, Donia MS,Spormann A, Fischbach MA, and Sonnenburg JL (2013). A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 7, 1933–1943. 10.1038/ismej.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. (2008). Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 105, 2117–2122. 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schroeder BO, and Bäckhed F.(2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med 22, 1079–1089. 10.1038/nm.4185. [DOI] [PubMed] [Google Scholar]
- 36.Martin FPJ, Dumas ME, Wang Y, Legido-Quigley C, Yap IKS, Tang H, Zirah S, Murphy GM, Cloarec O, Lindon JC, et al. (2007). A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol. Syst. Biol 3, 112. 10.1038/msb4100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker A, Pfitzner B, Neschen S, Kahle M, Harir M, Lucio M, Moritz F, Tziotis D, Witting M, Rothballer M, et al. (2014). Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J. 8, 2380–2396. 10.1038/ismej.2014.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilán CG, and Salazar N.(2016). Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol 7, 185. 10.3389/fmicb.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koh A, De Vadder F, Kovatcheva-Datchary P, and Bäckhed F.(2016). From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345. 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 40.Morrison DJ, and Preston T.(2016). Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microb. 7, 189–200. 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Candido E, Reeves R, and Davie JR (1978). Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14, 105–113. 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 42.Allis CD, and Jenuwein T.(2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet 17, 487–500. 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 43.Fan J, Krautkramer KA, Feldman JL, and Denu JM (2015). Metabolic regulation of histone post-translational modifications. ACS Chem. Biol 10, 95–108. 10.1021/cb500846u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouzarides T.(2007). Chromatin modifications and their function. Cell 128, 693–705. 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Berger SL (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 46.McGinty RK, and Tan S.(2015). Nucleosome structure and function. Chem. Rev 115, 2255–2273. 10.1021/cr500373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, and Garcia BA (2015). Comprehensive catalog of currently documented histone modifications. Cold Spring Harbor Perspect. Biol 7, a025064. 10.1101/cshperspect.a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 49.Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, et al. (2002). The orphan g protein-coupled receptors gpr41 and gpr43 are activated by propionate and other short chain carboxylic acids*. J. Biol. Chem 278, 11312–11319. 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 50.Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, Ian McKenzie C, Hijikata A, Wong C, et al. (2015). Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun 6, 6734–6815. 10.1038/ncomms7734. [DOI] [PubMed] [Google Scholar]
- 51.Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, Koppen A, Mudde K, Haenen D, Al-Lahham S, et al. (2013). Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor. Mol. Cell Biol 33, 1303–1316. 10.1128/mcb.00858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, and Bultman SJ (2012). The warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626. 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, and Bultman SJ (2011). The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metabol. 13, 517–526. 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, and Hirschey MD (2016). Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 17, 1463–1472. 10.1016/j.celrep.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roediger WE (1980). Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 21, 793–798. 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roediger WEW (1980). The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet 316, 712–715. 10.1016/S0140-6736(80)91934-0. [DOI] [PubMed] [Google Scholar]
- 57.Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, and Murthy SN (2000). Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut 46, 493–499. 10.1136/gut.46.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, and Williams NS (1994). Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis. Gut 35, 73–76. 10.1136/gut.35.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andriamihaja M, Chaumontet C, Tome D, and Blachier F.(2009). Butyrate metabolism in human colon carcinoma cells: implications concerning its growth-inhibitory effect. J. Cell. Physiol 218, 58–65. 10.1002/jcp.21556. [DOI] [PubMed] [Google Scholar]
- 60.Leschelle X, Delpal S, Goubern M, Blottière HM, and Blachier F.(2000). Butyrate metabolism upstream and downstream acetyl-CoA synthesis and growth control of human colon carcinoma cells. Eur. J. Biochem 267, 6435–6442. 10.1046/j.1432-1327.2000.01731.x. [DOI] [PubMed] [Google Scholar]
- 61.Uchimura Y, Fuhrer T, Li H, Lawson MA, Zimmermann M, Yilmaz B, Zindel J, Ronchi F, Sorribas M, Hapfelmeier S, et al. (2018). Antibodies set boundaries limiting microbial metabolite penetration and the resultant mammalian host response. Immunity 49, 545–559.e5. 10.1016/j.immuni.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, and Denu JM (2016). Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 64, 982–992. 10.1016/j.molcel.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 64.Camp JG, Frank CL, Lickwar CR, Guturu H, Rube T, Wenger AM, Chen J, Bejerano G, Crawford GE, and Rawls JF (2014). Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 24, 1504–1516. 10.1101/gr.165845.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deroover L, Verspreet J, Luypaerts A, Vandermeulen G, Courtin CM, and Verbeke K.(2017). Wheat bran does not affect postprandial plasma short-chain fatty acids from13C-inulin fermentation in healthy subjects. Nutrients 9, E83. 10.3390/nu9010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butts CA, Paturi G, Tavendale MH, Hedderley D, Stoklosinski HM, Herath TD, Rosendale D, Roy NC, Monro JA, and Ansell J.(2016). The fate of 13C-labelled and non-labelled inulin predisposed to large bowel fermentation in rats. Food Funct. 7, 1825–1832. 10.1039/c5fo01056j. [DOI] [PubMed] [Google Scholar]
- 67.Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, Jurickova I, Braun T, Novak E, Bauman L, Collins MH, et al. (2019). Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun 10, 38–13. 10.1038/s41467-018-07841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamoud AR, Weaver L, Stec DE, and Hinds TD (2018). Bilirubin in the liver–gut signaling Axis. Trends Endocrinol. Metab 29, 140–150. 10.1016/j.tem.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million M, et al. (2013). Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144, 967–977. 10.1053/j.gastro.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grüner N, and Mattner J.(2021). Bile acids and microbiota: multifaceted and versatile regulators of the liver–gut axis. Int. J. Mol. Sci 22, 1397–1417. 10.3390/ijms22031397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cox LM, Sohn J, Tyrrell KL, Citron DM, Lawson PA, Patel NB, Iizumi T, Perez-Perez GI, Goldstein EJC, and Blaser MJ (2017). Description of two novel members of the family erysipelotrichaceae: ileibacterium valens gen. nov., sp. nov. and Dubosiella newyorkensis, gen. nov., sp. nov., from the murine intestine, and emendation to the description of Faecalibacterium rodentium. Int. J. Syst. Evol. Microbiol 67, 1247–1254. 10.1099/ijsem.0.001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Catry E, Bindels LB, Tailleux A, Lestavel S, Neyrinck AM, Goossens JF, Lobysheva I, Plovier H, Essaghir A, Demoulin JB, et al. (2018). Targeting the gut microbiota with inulin-type fructans: preclinical demonstration of a novel approach in the management of endothelial dysfunction. Gut 67, 271–283. 10.1136/gutjnl-2016-313316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li LL, Wang YT, Zhu LM, Liu ZY, Ye CQ, and Qin S.(2020). Inulin with different degrees of polymerization protects against diet-induced endotoxemia and inflammation in association with gut microbiota regulation in mice. Sci. Rep 10, 978. 10.1038/s41598-020-58048-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeng X, Xing X, Gupta M, Keber FC, Lopez JG, Lee YCJ, Roichman A, Wang L, Neinast MD, Donia MS, et al. (2022). Gut bacterial nutrient preferences quantified in vivo. Cell 185, 3441–3456.e19. 10.1016/J.CELL.2022.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kitajima S, Takuma S, and Morimoto M.(2000). Histological analysis of murine colitis induced by dextran sulfate sodium of different molecular weights. Exp. Anim 49, 9–15. 10.1538/EXPANIM.49.9. [DOI] [PubMed] [Google Scholar]
- 76.Wu SE, Hashimoto-Hill S, Woo V, Eshleman EM, Whitt J, Engleman L, Karns R, Denson LA, Haslam DB, and Alenghat T.(2020). Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature 586, 108–112. 10.1038/s41586-020-2604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slaughter MJ, Shanle EK, Khan A, Chua KF, Hong T, Boxer LD, Allis CD, Josefowicz SZ, Garcia BA, Rothbart SB, et al. (2021). HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell Rep. 34, 108638. 10.1016/j.celrep.2020.108638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davison JM, Lickwar CR, Song L, Breton G, Crawford GE, and Rawls JF (2017). Microbiota regulate intestinal epithelial gene expression by suppressing the transcription factor Hepatocyte nuclear factor 4 alpha. Genome Res. 27, 1195–1206. 10.1101/gr.220111.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ranhotra HS, Flannigan KL, Brave M, Mukherjee S, Lukin DJ, Hirota SA, and Mani S.(2016). Xenobiotic receptor-mediated regulation of intestinal barrier function and innate immunity. Nucl. Receptor Res 3, 101199. 10.11131/2016/101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duszka K, and Wahli W.(2018). Enteric microbiota–gut–brain axis from the perspective of nuclear receptors. Int. J. Mol. Sci 19, E2210. 10.3390/ijms19082210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith SA, Ogawa SA, Chau L, Whelan KA, Hamilton KE, Chen J, Tan L, Chen EZ, Keilbaugh S, Fogt F, et al. (2021). Mitochondrial dysfunction in inflammatory bowel disease alters intestinal epithelial metabolism of hepatic acylcarnitines. J. Clin. Invest 131, e133371. 10.1172/JCI133371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Preter V, Arijs I, Windey K, Vanhove W, Vermeire S, Schuit F, Rutgeerts P, and Verbeke K.(2012). Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm. Bowel Dis 18, 1127–1136. 10.1002/ibd.21894. [DOI] [PubMed] [Google Scholar]
- 83.Deng P, Valentino T, Flythe MD, Moseley HNB, Leachman JR, Morris AJ, and Hennig B.(2021). Untargeted stable isotope probing of the gut microbiota metabolome using 13C-labeled dietary fibers. J. Proteome Res 20, 2904–2913. 10.1021/acs.jproteome.1c00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, Trefely S, Fernandez S, Carrer A, et al. (2020). Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591. 10.1038/s41586-020-2101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2008). A core gut microbiome in obese and lean twins. Nature 457, 480–484. 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Litvak Y, Byndloss MX, and Bäumler AJ (2018). Colonocyte metabolism shapes the gut microbiota. Science 362, eaat9076. 10.1126/science.aat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, Chávez-Arroyo A, Lokken KL, Tsolis RM, Winter SE, and Bä umler AJ (2016). Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 353, 1249–1253. 10.1126/science.aag3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL, Gewirtz AT, Worgall TS, and Ley RE (2020). Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat. Commun 11, 2471–2511. 10.1038/s41467-020-16274-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brügger MD, Valenta T, Fazilaty H, Hausmann G, and Basler K.(2020). Distinct populations of crypt-associated fibroblasts act as signaling hubs to control colon homeostasis. PLoS Biol. 18, e3001032. 10.1371/JOURNAL.PBIO.3001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, Kleshchevnikov V, Dabrowska M, Pritchard S, Bolt L, et al. (2021). Cells of the human intestinal tract mapped across space and time. Nature 597, 250–255. 10.1038/S41586-021-03852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lund PJ, Kori Y, Zhao X, Sidoli S, Yuan ZF, and Garcia BA(2019). Isotopic labeling and quantitative proteomics of acetylation on histones and beyond. Methods Mol. Biol 1977, 43–70. 10.1007/978-1-4939-9232-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yuan Z-F, Lin S, Molden RC, Cao X-J, Bhanu NV, Wang X, Sidoli S, Liu S, and Garcia BA (2015). EpiProfile quantifies histone peptides with modifications by extracting retention time and intensity in high-resolution mass spectra. Mol. Cell. Proteomics 14, 1696–1707. 10.1074/mcp.M114.046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/BIOINFORMATICS/BTP352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anders S, Pyl PT, and Huber W.(2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anders S, and Huber W.(2010). Differential expression analysis forsequence count data. Genome Biol. 11, R106. 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu G, and He Q-Y (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst 12, 477–479. 10.1039/C5MB00663E. [DOI] [PubMed] [Google Scholar]
- 102.Fursova NA, Blackledge NP, Nakayama M, Ito S, Koseki Y, Farcas AM, King HW, Koseki H, and Klose RJ (2019). Synergy between variant PRC1 complexes defines polycomb-mediated gene repression. Mol. Cell 74, 1020–1036.e8. 10.1016/j.molcel.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T.(2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choi M, Chang C-Y, Clough T, Broudy D, Killeen T, MacLean B, and Vitek O.(2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- 105.Ul-Hasan S, Bowers RM, Figueroa-Montiel A, Licea-Navarro AF, Beman JM, Woyke T, and Nobile CJ (2019). Community ecology across bacteria, archaea and microbial eukaryotes in the sediment and seawater of coastal Puerto Nuevo, Baja California. PLoS One 14, e0212355. 10.1371/JOURNAL.PONE.0212355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Caporaso JG, Ackermann G, Apprill A, Bauer M, Berg-Lyons D, Betley J, Fierer N, Fraser L, Fuhrman JA, Gilbert JA, et al. (2018). EMP 16S Illumina Amplicon Protocol. protocols.io. 10.17504/protocols.io.nuudeww. [DOI] [Google Scholar]
- 107.Edgar RC, and Bateman A.(2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. 10.1093/BIOINFORMATICS/BTQ461. [DOI] [PubMed] [Google Scholar]
- 108.Edgar RC (2016). SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. Preprint at bioRxiv. 10.1101/074161. [DOI] [Google Scholar]
- 109.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. 10.1093/NAR/GKT1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin M.(2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 111.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/NMETH.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. 10.1038/S41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-seq and RNA-seq data have been deposited at NCBI GEO: GSE179233. The mass spectrometry proteomics data for the colitis study have been deposited to the ProteomeXchange Consortium via the PRIDE92 partner repository with the dataset identifier PXD026959 and 10.6019/PXD026959. (PRIDE: PXD026959) The metabolomics data are available from MassIVE: MSV000087752. All datasets are publicly available as of the date of publication. All data reported in this paper will be shared by the lead contact by request.
Raw data and analysis scripts have been deposited online (Mendeley Data: https://doi.org/10.17632/sdj5j3ffy7.1) and are publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.