

Abstract
Interleukin-4 (IL-4) is a signature cytokine pivotal in Type 2 helper T cell (Th2) immune response, particularly in allergy and hypersensitivity. Interestingly, IL-4 increases endogenous levels of prostaglandin D2 (PGD2) and its metabolites, Δ12-prostaglandin J2 (Δ12-PGJ2) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), collectively called cyclopentenone PGs (CyPGs). However, the therapeutic role of IL-4 in hematologic malignancies remains unclear. Here, we employed a murine model of acute myeloid leukemia (AML), where human MLL-AF9 fusion oncoprotein was expressed in hematopoietic progenitor cells, to test the effect of IL-4 treatment in vivo. Daily intraperitoneal treatment with IL-4 at 60 μg/kg/d significantly alleviated the severity of AML, as seen by decreased leukemia-initiating cells (LICs). The effect of IL-4 was mediated, in part, by the enhanced expression of hematopoietic- PGD2 synthase (H-PGDS) to effect endogenous production of CyPGs, through autocrine and paracrine signaling mechanisms. Similar results were seen with patient-derived AML cells cultured ex vivo with IL-4. Use of GW9662, a peroxisome proliferator-activated receptor gamma (PPARγ) antagonist, suggested endogenous CyPGs-PPARγ axis mediated p53-dependent apoptosis of LICs by IL-4. Taken together, our results reveal a beneficial role of IL-4 treatment in AML suggesting a potential therapeutic regimen worthy of clinical trials in AML patients.
Keywords: AML, IL-4, H-PGDS, prostaglandin, PPARγ
Introduction
AML is characterized by abnormal and immature hematopoietic cells in the bone marrow, peripheral blood, and other tissues.1 Recent developments in cytogenetics have provided a better understanding of the molecular heterogeneity of AML. These findings have led to the development of new targeted treatments for AML. Unfortunately, these treatments have fallen short of effectively bringing about long-term remission and survival benefits. Due to the persistence of leukemia-initiating cells (LICs) that originate from normal hematopoietic stem or progenitor cells (HSPCs) via mutational events,2 patients tend to relapse from the minimal residual disease (MRD). Therefore, novel therapies that preferentially target LICs to terminate the source of AML serve as a promising approach.
Studies have shown that the tumor microenvironment (TME) plays an important role in the prognosis of AML.3 By manipulating the signaling pathways in the TME, novel therapies have been proposed for the treatment of AML. For example, inhibition of IL-1β signaling by targeting IRAK1 impedes leukemic cell proliferation in MLL-AF9-driven AML both in vitro and in vivo.4 Similarly, blockage of CCL3-CCR1/5 or CXCL12-CXCR4 in vivo reduced leukemia burden and prolonged survival in MLL-AF9-induced AML mice.5
IL-4 is a cytokine produced by T-cells and granulocytes during Th2 immunity, which is generally considered as tumor-promoting. Blocking the interaction of IL-4 with its receptor (IL-4R) has been a target for developing novel therapies for various cancers.6–8 However, multiple in vitro studies have also found that IL-4 may exert an anti-leukemic effect in AML.9–11 Thus, given the intriguing contradictory nature of in vitro and in vivo effects of IL-4, it is difficult to predict its therapeutic potential for AML.12 Direct clinical data on the efficacy of IL-4 treatment in AML are lacking. But a phase I/II study in patients with chronic lymphocytic leukemia (CLL) found that administration of IL-4 might induce an anti-apoptotic effect on leukemic cells.13 Another study using an MLL-AF9-driven murine model treated with IL-4 reported a slight reduction of tumor burden and a limited survival benefit of IL-4 in AML.14 We extended these initial reports with a more in-depth mechanistic study and prolonged treatment duration with IL-4 to explore its long-term effect on AML.
Previous studies in our laboratory have demonstrated that cyclooxygenase (COX)-derived endogenous Δ12-PGJ2 and 15d-PGJ2, collectively called CyPGs, were able to target LICs by apoptosis in a murine model of chronic myeloid leukemia (CML).15 Interestingly, these bioactive lipid mediators were also up-regulated in healthy bone marrow-derived macrophages (BMDMs) by IL-4 stimulation.16 Based on these observations, we examined if IL-4 treatment had any beneficial effects in the remission of AML through the endogenous production of CyPGs leading to significant improvement in prognosis. Using a murine model of MLL-AF9-driven AML17 and AML patient-derived cells, we demonstrate the anti-leukemic effect of IL-4 occurs via endogenous CyPGs and activation of the nuclear receptor PPARγ to induce p53-dependent apoptosis of LICs. This study provides a compelling rationale for future clinical trials with IL-4 in AML patients.
Materials and Methods
Generation of experimental AML murine model
Retroviral stocks were generated by transfecting phoenix-eco cells (ATCC CRL −3214) with MSCV-MLL-AF9 plasmid (gift from Dr. Ravi Bhatia, University of Alabama, Birmingham) using TransIT 293 reagent (Mirus Bio, Madison, WI, USA). Briefly, bone marrow cells were flushed out of femurs and tibias isolated from B6.SJL-Ptprca CD45.1 donor mice. Lineage negative (Lin−) cells were isolated using EasySep™ mouse hematopoietic progenitor cell isolation kit (Stem cell technologies, Vancouver, BC, Canada) following red blood cell lysis with ACK lysis buffer (155 mM NH4Cl, 12 mM KHCO3, 0.1mM EDTA-2Na). Isolated Lin− cells were stained with fluorescent antibodies PE-Cy7-conjugated anti-mouse Ly-6A/E (Sca-1) (BD Pharmingen™, Franklin Lakes, NJ, USA) and APC-conjugated anti-mouse CD117 (c-Kit) (Biolegend, San Diego, CA, USA). Hematopoietic stem cells (HSCs) were fluorescence-activated cell sorted (FACS) as Lin−Sca-1+c-Kit+ population. Sorted HSCs were transduced with MSCV-MLL-AF9 retrovirus in conditioned media for 6 hours. Primary transplantation was performed by retro-orbital injection of retrovirus-transduced CD45.1+ HSCs into sub-lethally irradiated (475 Rads) CD45.2+ recipient mice. Upon onset of the disease, mice were euthanized, and isolated splenocytes were transplanted into non-irradiated secondary CD45.2 recipients (7–8 weeks old) (Fig. S1A). CD45.1 mice were from our in-house breeding colony, while C57BL/6 CD45.2 mice were purchased from Taconic Biosciences, Hudson, NY. All mice were fed normal chow and provided with Milli-Q water ad libitum. All experiments were preapproved by the Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee (IBC).
IL-4 treatment regimen
One week after secondary transplantation, recipient mice were treated daily with recombinant mouse (rm)IL-4 at 60 μg/kg/d (Gemini Bio, West Sacramento, CA, USA) intraperitoneally for three weeks. At the end of the treatment, all mice were euthanized. The dose of rmIL-4 was derived from the United States Food and Drug Administration (FDA) guidelines from a human phase II trial where IL-4 was given to patients with B-cell lymphoma or leukemia.9, 18, 19 Inhibition of PPARγ was achieved via injecting GW9662 at 1 mg/kg (Cayman Chemical, Ann Arbor, MI, USA) intraperitoneally into mice every other day following secondary transplantation with or without concurrent rmIL-4 treatment.
In vitro treatment
Splenocytes were isolated from either normal or AML mice and treated with rmIL-4 (10 ng/mL, 24hr) or CyPGs (Δ12-PGJ2, 15d-PGJ2, and Δ12-PGJ3 at 100 nM in sterile PBS for 24hr) and incubated at 37 °C and 5% CO2. Δ12-PGJ2 and 15d-PGJ2 were purchased from Cayman Chemical, Ann Arbor, MI, USA. Δ12-PGJ3 was prepared in our laboratory as described previously.20 All three compounds were prepared fresh before use in sterile PBS and concentrations were calculated by liquid chromatography-mass spectrometry (not shown). For mRNA expression analysis, primary AML splenocytes were treated with rmIL-4 (10 ng/mL, 24hr); for protein expression analysis, primary AML splenocytes were treated with HQL79 (50 μM) and GW9662 (10 μM) in the presence of rmIL-4 (50 ng/mL) and incubated at 37°C and 5% CO2 for 72 h. For the analyses of apoptosis and viability, primary AML splenocytes were treated with rmIL-4 (0, 50, 100 ng/mL) for 24, 48, and 72 h. At the time point of 48 h, GW9662 (10 μM) was added to the treatment of 50 ng/mL rmIL-4. HQL79 and GW9662 were purchased from Cayman Chemical, Ann Arbor, MI, USA, and reconstituted in cell-culture grade dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA).
Survival analysis
Primary AML splenocytes (> 95 % CD45.1+ cells) were treated with rmIL-4 (100 ng/mL, 0 ng/mL as control) for 24 h and then transplanted into recipient mice. Survival was followed up to 60 days post transplantation. Mice exhibiting hypothermia, leukocytosis, spontaneous bleeding, or cachexia, were euthanized per the IACUC at Penn State.
Complete blood counts
At the end point of rmIL-4 treatment, approximately 25 μL of peripheral blood was collected retro-orbitally to enumerate complete blood cell (CBC) counts on a Hemavet 950FS (Drew Scientific, Miami Lakes, FL, USA). Leukocytosis was also monitored by evaluating the peripheral blood in primary transplant recipient mice.
ELISA
Whole blood samples were collected by performing cardiac puncture at endpoint. Sera were isolated by centrifugation for 15 min at 8 000 g, 4 ˚C and used to measure 15d-PGJ2 with a specific ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer’s instructions and previously validated in our laboratory for specificity. Absorbance values were measured in a Biotek plate reader at 405 nm. Concentrations of 15d-PGJ2 in sera were calculated based on a standard curve generated by a four-parameter logistic curve fitting program as recommended by the provider.
AML patient cells
Deidentified AML cells isolated from peripheral blood of AML patients with written informed consent were from the tissue bank at the Penn State Cancer Institute of Penn State University College of Medicine, Hershey, PA. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood from patients pre-diagnosed as AML with WHO classification system by density gradient centrifugation and cultured in SFEM media (Stem cell technologies, Vancouver, BC, Canada) supplemented with 100 ng/mL human recombinant (Hr)-SCF, 100 ng/mL Hr-FLT3L, 20 ng/mL Hr-IL-3, 20 ng/mL Hr-G-CSF, 20 ng/mL Hr-GM-CSF (Shenandoah Biotechnology, Warminster, PA, USA), and 1% penicillin/streptomycin in the absence or presence of 100 ng/mL Hr-IL-4 (R&D Systems, Minneapolis, MN, USA) and incubated at 37°C and 5% CO2 for 60 hours. These experiments were preapproved by the Institutional Review Board and Institutional Biosafety Committee at Penn State University.
Flow cytometry analysis
At endpoint, freshly dissected spleens were weighed, and nucleated cells were collected from bone marrows and spleens from mice at endpoint. Bone marrow cells were flushed out from femurs and tibias using PBS containing 1% penicillin/streptomycin and 2% FBS. Both bone marrow and splenic single cell suspensions were subjected to red blood cell lysis with sterile ACK lysis buffer. Single cell suspensions were subjected to magnetic bead sorting to isolate hematopoietic progenitor cells as per the manufacturer’s instructions followed by staining with PE-Cy7-conjugated anti-mouse Ly-6A/E (Sca-1) and APC-conjugated anti-mouse CD117 (c-Kit) fluorescent antibodies. Raw data of Lin− cell numbers were collected followed by validation of LICs as Lin−CD45.1+Sca-1−c-Kit+ population on a BD Accuri C6 flow cytometer and analyzed using FlowJo v10 (BD FlowJo, Ashland, OR, USA).
Patient AML cells were harvested at the end of treatment, washed with cell staining buffer, resuspended in Annexin V binding buffer, and stained with FITC-conjugated anti-human CD34, APC-conjugated anti-human CD38 (Biolegend, San Diego, CA, USA), PE-Cy7-conjugated anti-human CD123, and PE-conjugated Annexin V (BD Biosciences, Franklin Lakes, NJ, USA). Apoptosis was analyzed in patient AML blasts as CD34+CD38−CD123+Annexin V+ populations on a BD Accuri C6.
Primary AML splenocytes treated by rmIL-4 (in combination of GW9662) were harvested and stained with APC-conjugated Annexin V (BD Pharmingen™, Franklin Lakes, NJ, USA) and propidium iodide (PI) (Biolegend, San Diego, CA, USA). Cell viability, early and late apoptosis, and necrosis were evaluated by analyzing the Annexin V−PI−, Annexin V+PI−, Annexin V+PI+, and Annexin V−PI+ populations, respectively.
Live/dead cell analysis
Primary AML splenocytes treated by rmIL-4 were harvested and stained with Acridine orange (AO) and PI (Nexcelom Bioscience, Lawrence, MA, USA). Live/dead cells were analyzed and imaged using a Cellometer K2 (Nexcelom Bioscience, Lawrence, MA, USA).
Western immunoblotting
Total cellular protein was extracted from bone marrow, spleen, lungs, and other in vitro experiments using Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Rockford, IL, USA) containing protease inhibitor cocktail and 5 mM sodium orthovanadate (Sigma-Aldrich, St. Louis, MO, USA). Cell lysates were incubated on ice for 30 min and then centrifuged for 10 min at 12 000 g, 4 ˚C. Supernatants were quantified using a BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA) for protein concentration. Equal amount of protein was loaded onto SDS-PAGE gels and then transferred onto nitrocellulose membranes. Blots were incubated in the following primary antibodies: P53 (1:1 000, Cell Signaling Technology, 2524S), CASPASE-3 (1:2 000, Cell Signaling Technology, 9662S), H-PGDS (1:1 000, Cayman Chemical, 160013), COX-1 (1:1 000, Cayman Chemical, 160109), COX-2 (1:1 000, Cayman Chemical, 160106), and normalized to β-actin (Fitzgerald, 10R-2927) expression. Appropriate secondary antibodies either goat anti-rabbit and goat anti-mouse (Thermo Fisher Scientific, Rockford, IL, USA) were used. Blots developed with West Pico reagent (Thermo Fisher Scientific, Rockford, IL, USA) were scanned in an imager (G:Box Chemi) with GeneSys software program (Version 1.5.7.0). Experiments were performed in at least biological triplicate. Densitometry analysis was carried out using Image J software program (National Institutes of Health, Bethesda, MD).
Real-time PCR
Total RNA was extracted from bone marrow, spleen, and lung tissue of mice and patient-derived AML cells using the TRI reagent (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions. RNA concentration was measured using Nanodrop One and used to synthesize cDNA using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Rockford, IL, USA). cDNA was used to perform quantitative PCR (qPCR) using PerfeCTa qPCR SuperMix Master Mix and PerfeCTa SYBR® Green SuperMix Reagent (Quanta Biosciences, Beverly, MA, USA) in a StepOne Plus Real-time PCR system (Applied Biosystems) using prevalidated TaqMan probes: 18S (Hs99999901_s1), Ccl11 (Mm00441238_m1), Cd36 (Mm00432403_m1), CDKN1A (Hs00355782_m1), Cox-1 (Mm00477214_m1), Cox-2 (Mm004478374_m1), Hpgds (Mm00479846_m1), Il4 (Mm00445259_m1), Il13 (Mm00434204_m1), Mrc1 (Mm01329362_m1), PGDS (Hs00183950_m1), TP53 (Hs01034249_m1), and Trp53 (Mm01731290_g1) and primers: mouse Vegfa (Forward 5’-CTGCTCTCTTGGGTGCACTG-3’; Reverse 5’-GCAGCCTGGGACCACTTG-3’), mouse Vegfb (Forward 5’-GAAGCCAGACAGGGTTGCC-3’; Reverse 5’-GATGGATGATGTCAGCTGGGG-3’), mouse Vegfc (Forward 5’-GGTCCATCCACCATGCACTT-3’; Reverse 5’-TTTGCCTTCAAAAGCCTTGACC-3’), mouse Vegfd (Forward 5’-CGAACATGGACCAGTGAAGGATT-3’; Reverse 5’-CCACAGCTTCCAGTCCTCAG-3’), mouse Egf (Forward 5’-GAGGTCCGCTAGAGAAATGTCAA-3’; Reverse 5’-TGGGGCATGTGCAGTGATAG-3’), mouse Pparg (Forward 5’-AGGAGCCTGTGAGACCAACA-3’; Reverse 5’-TCACCGCTTCTTTCAAATCTTGTC-3’), mouse Ppard (Forward 5’-AAGTTTTGGCAGGAGCTGGG-3’; Reverse 5’-GCGCAGATGGACTGCCTTTA-3’), and mouse Gapdh (5’-AGGTCGGTGTGAACGGATTTG-3’; Reverse 5’-TGTAGACCATGTAGTTGAGGTCA-3’). Data were analyzed using the method of Livak and Schmittgen21 and normalized to 18S ribosomal RNA (rRNA).
Histology
Mouse lungs were isolated at the endpoint and fixed in 10% (v/v) buffered formalin followed by embedding in paraffin for sectioning. Sections were stained with hematoxylin and eosin (H&E) and Periodic acid–Schiff (PAS) reagent. Slides were blinded for pathological evaluation. Images were obtained on a microscope installed with AxioVision software for histological analysis.
Statistical analysis
Results were analyzed using GraphPad Prism version 6 (GraphPad) and presented as the mean ± SEM. For comparison between two groups, unpaired two-tailed Student t test was utilized. For the comparison of patient AML cells treated with/without Hr-IL-4, paired two-tailed Student t test was applied. To compare the survival of AML mice, log-rank (Mantel-Cox) test was adopted. Variation from these analyses and replicate numbers are described in figure legends.
Results
IL-4 administration reduces leukemogenesis in AML mice
To establish a murine model of AML, bone marrow cells from CD45.1 mice were isolated and sorted for Lin−Sca-1+c-Kit+ cells in the bone marrow that served as donor cells. These cells were transduced to express human MLL-AF9 fusion oncogene and subsequently transplanted into sub-lethally irradiated CD45.2 recipients (Fig. S1A). Recipient mice were euthanized when peripheral white blood cells (WBCs) of recipients increased to 50 K/μL and splenocytes were assessed for the frequency of CD45.1+ cells. To test the effect of IL-4 treatment, we performed a secondary transplantation by retro-orbital injection of 0.45 × 106 primary CD45.1+ leukemic cells per mouse into CD45.2 recipients. rmIL-4 in vivo treatment was administered to secondary recipients one week post transplantation, allowing the engraftment of leukemic cells (Fig.1A).
Figure 1. IL-4 administration reduces leukemogenesis in AML mice.
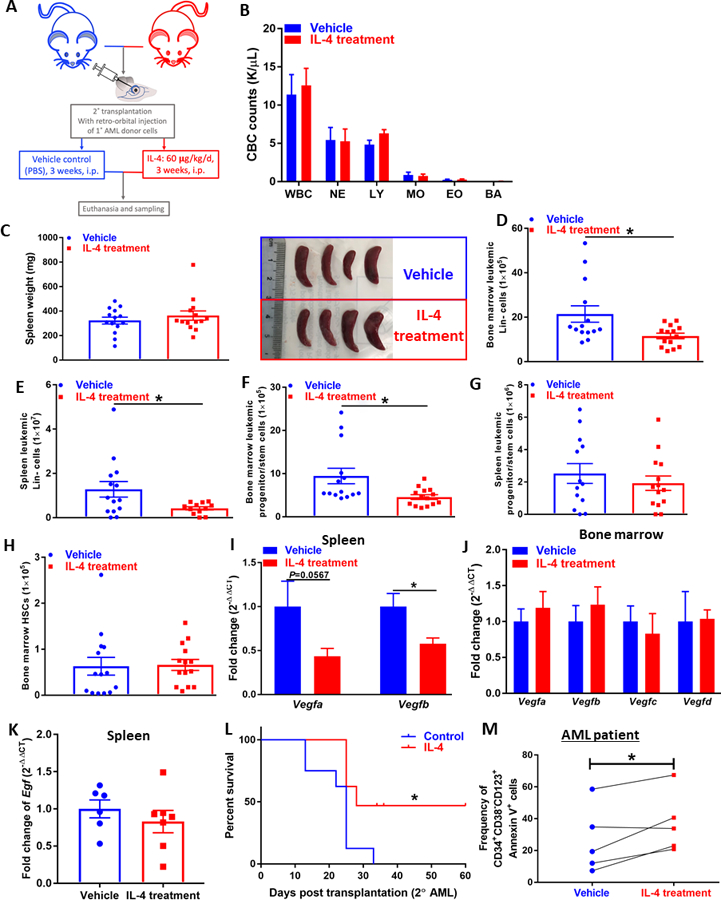
(A) Recipients of secondary transplants of primary AML cells were dosed intra-peritoneally daily with rmIL-4 (60 μg/kg) or PBS (vehicle control) for three weeks following euthanasia. (B) CBC counts (K/𝛍L peripheral blood) at endpoint, total WBC, eosinophil (EO), monocyte (MO), neutrophil (NE), lymphocyte (LY), basophil (BA) profile of AML mice treated with vehicle or IL-4. (n=14). (C) Representative image and spleen weights (mg) of AML mice. (D)(E) Flow cytometric analysis of total leukemic cells identified by CD45.1+ in the Lin− population of bone marrow (D) and spleen (E) of AML mice. (n=14). (F)(G) Flow cytometric analysis of LICs identified as CD45.1+sca-1−c-kit+ in the Lin− population of bone marrow (F) and spleen (G) of AML mice. (n=14). (H) Flow cytometric analysis of normal HSCs identified as CD45.1−sca-1+c-kit+ in the Lin− population of bone marrow of AML mice. (n=14). (I)(J) Expression of VEGF genes (Vegfa, Vegfb, Vegfc, Vegfc) assessed by qPCR analysis in the spleen (I) and bone marrow (J) of AML mice with/without IL-4 treatment. Data were normalized to Gapdh expression. (n=6~8). (K) Expression of Egf assessed by qPCR analysis in the spleen of AML mice with/without IL-4 treatment. Data were normalized to Gapdh expression. (n=6~7). (L) Survival analysis of mice transplanted with IL-4-treated primary AML cells. Primary AML cells were cultured ex vivo for 24 h with rmIL4 (100 ng/mL) or without rmIL-4 (control), and then transplanted into mice (n=8 per group). (M) Flow cytometric analysis of apoptotic leukemic blasts identified as Annexin V+CD34+CD38−CD123+ population in patient AML cells treated with vehicle or IL-4 (n=5). Data represent mean± SEM in B-H, unpaired two-tailed Student t test was utilized; Log-rank (Mantel-Cox) test was used in L and paired two-tailed Student t test was applied to panel M; *, P < 0.05; **, P < 0.01.
At the endpoint, mice were euthanized and analyzed for CBC counts in the peripheral blood. Both PBS- and IL-4-treated groups exhibited signs of leukemia including leukocytosis (Fig. 1B) and splenomegaly (Fig. 1C), whereas no apparent differences induced by IL-4 treatment were observed. We sought to investigate the infiltration of leukemic cells in the bone marrow and spleen as a measure of leukemogenesis. A striking decrease of total leukemic cells was observed in the bone marrow (Fig. 1D) and spleen (Fig. 1E) upon IL-4 treatment clearly indicating disease remission. This effect was demonstrated to be derived from the targeted elimination of LICs,22 identified as Lin−CD45.1+Sca-1−c-Kit+ population (Fig. S1B) in the bone marrow (Fig. 1F) and a decreased trend in spleen (Fig. 1G). To determine any possible defects in normal stem cell functions because of long-term IL-4 administration, recipient HSCs in the bone marrow were also enumerated (Fig. 1H). No changes in normal HSC numbers were observed because of long-term use of IL-4, indicating that IL-4 treatment does not impair normal HSC function in AML mice. Surprisingly, we also found decreased expression of vascular endothelial growth factor (VEGF) family of genes (i.e. Vegfa and Vegfb) in the spleen (Fig. 1I) but not bone marrow (Fig. 1J) of IL-4-treated AML mice, implying that IL-4 treatment may have a beneficial impact in AML by restricting splenic angiogenesis.23 In addition, lack of expression of Vegfc and Vegfd as well as unchanged epidermal growth factor (Egf) expression (Fig. 1K) in the spleen of AML mice treated with IL-4 suggested that lymphangiogenesis24–26 was not impacted by IL-4 stimulation. More importantly, we also tested the survival benefits of IL-4 in AML mice and found that IL-4-treatment exhibited significantly improved survival comparing to their counterparts (Fig. 1L). Furthermore, PBMCs isolated from AML patients treated with Hr-IL-4 exhibited significant apoptosis, which reinforces the potential clinical application of IL-4 in AML (Fig. 1M).
IL-4 treatment activates prostaglandin metabolism in AML mice
Previous studies indicated IL-4 treatment of BMDMs enhanced Cox-1 expression and increased production of 15d-PGJ2, via Fes-Akt-mTORC axis.16 Furthermore, we also demonstrated that CyPGs, including series 2 (PGJ2) induced by selenium supplementation15 or series 3 (PGJ3) induced by eicosapentaenoic acid (EPA) supplementation,17 alleviated leukemia by targeting leukemic cells in murine models of CML and AML, where BCR-ABL and MLL-AF9 fusion oncoproteins were expressed in HSCs, respectively. Here, we examined the effect of IL-4 on prostaglandin metabolism in bone marrow and spleen from AML mice. Expression of three key enzymes, COX-1, COX-2, and H-PGDS, at the RNA and protein levels, were found to be increased by IL-4 treatment in the bone marrow of AML mice. While the transcript levels of Cox1 and Hpgds were significantly increased by IL-4 by two- and five-folds, respectively, compared to the PBS-treated AML mice, expression of Cox2 only showed an increased trend in the bone marrow upon IL-4 treatment (Fig. 2A). We failed to see a similar effect in the spleen from AML mice treated with IL-4 (Fig. 2B). At the protein level, we observed significantly increased expression of H-PGDS, but not COX-1 or COX-2 in the bone marrow (Fig. 2C). A similar phenomenon was also observed in spleen samples (Fig. 2D) from AML mice treated with IL-4. The unchanged COX-1 protein expression in IL-4 treated AML cells indicate that AML cells differ in their response to IL-4 stimulation from that of BMDMs that we reported earlier.14 Four out of the five AML patient cells treated with IL-4 also showed enhanced expression of PGDS (Fig. 2E). H-PGDS catalyzes the conversion of PGH2 to PGD2.27, 28 The increased expression of H-PGDS at both transcript and protein levels correlated well its downstream CyPG metabolite, 15d-PGJ2 in the serum of AML mice upon IL-4 in vivo treatment (Fig. 2F). This result suggests that IL-4 in vivo treatment may negatively impact the outcome of AML by prompting a systemic enhancement in endogenous CyPG production via upregulation of H-PGDS.
Figure 2. IL-4 supplement activates prostaglandin metabolism.
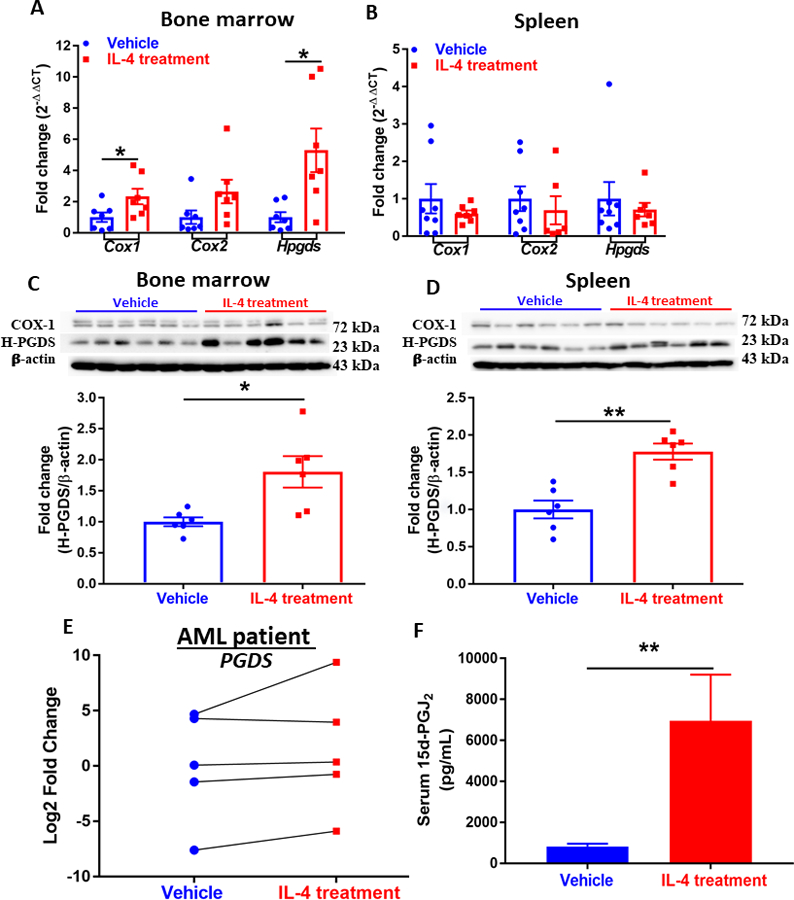
(A)(B) Expression of genes (Cox1, Cox2, and Hpgds) in the prostanoid biosynthesis pathway assessed by qPCR analysis in the bone marrow (A) and spleen (B) of AML mice. Data were normalized to 18S rRNA expression. (n=6~8). Western blot showing expression of COX-1 and H-PGDS in cells isolated from bone marrow (C) and spleen (D) of AML mice. Densitometry was done by normalizing to vehicle control and relative to β-actin for H-PGDS. Data shown are mean ± SEM per group, each group has at least three replicates. (n=6). (E) Expression of PGDS in patient-derived AML cells treated with vehicle or IL-4 (n=5). (F) Serum level of 15d-PGJ2 of AML mice treated with vehicle (n=10) or IL-4 (n=8) measured by ELISA. Data represent mean± SEM in A-D and F, unpaired two-tailed Student t test was utilized; and paired two-tailed Student t test was applied to E; *, P < 0.05; **, P < 0.01.
In addition, IL-4 stimulation of splenic cells from leukemic mice ex vivo showed increased expression of Hpgds at both transcriptional and translational levels (Fig. S2A, B), while Cox2 expression was decreased. Corroborating our in vivo findings, these data reiterated the importance of intrinsic COX/H-PGDS axis in AML as an intracellular mechanism mediating IL-4’s effects. To further address the potential contribution of the TME, we focused on splenocytes isolated from normal healthy mice to study the impact of IL-4. Here, in vitro treatment of normal primary splenocytes with IL-4 also up-regulated COX-1 and H-PGDS but down-regulated COX-2 (Fig. S3A-D), which corroborated with the in vivo observations suggesting a potential mechanism of paracrine CyPGs derived from the TME targeting leukemic cells. Furthermore, these IL-4-enhanced CyPGs may form a positive forward loop with IL-4 given that in-vitro treatment of primary splenocytes with CyPGs induced the expression of Il4 and Il13 (Fig. S4A and S4B). Intriguingly, in-vitro treatment of normal primary splenocytes with IL-4 also induced endogenous expression of Il4 and Il13 (Fig. S3E).
IL-4 administration induces apoptosis via p53 and caspase activation in AML mice
To further dissect the molecular mechanism of IL-4-induced effect on AML, we examined the modulation of classical pro-apoptotic genes. The administration of IL-4 significantly up-regulated the expression of p53 (Tpr53) mRNA and protein in the bone marrow and spleen from AML mice (Fig. 3A-C). In the patient AML blasts, IL-4 treatment increased the expression of TP53 as well as CDKN1A (Fig. 3D and 3E), which encodes p21 that mediates p53-dependent apoptosis.29 In the p53-dependent apoptotic cascade, apoptosis is executed by caspases to coordinate the cell death program.30 Thus, we examined the expression of Caspase-3, which showed a general increase in the bone marrow (Fig. 3F) and spleen (Fig. 3G) upon IL-4 treatment. Furthermore, since caspase-9 activates caspase-3 by proteolytic cleavage, the cleaved form of caspase-3 indicated a significant increase in response to IL-4 treatment (Fig. 3H and 3I). IL-4 treatment resulted in decreased viability of leukemic cells (Fig. S5A and S5B), which led to significantly reduced cell numbers (Fig. S5C and S5D) at various time points. Further analysis suggested that IL-4 treatment enhanced early and late apoptosis (Fig. S5E and S5F) in addition to necrosis (Fig. S5G). Taken together, these results imply that administration of IL-4 leads to apoptosis of leukemic cells by activating caspases in a p53-dependent manner. Interestingly, treatment of cells with HQL-79, a selective H-PGDS inhibitor, abrogated the pro-apoptotic effect of IL-4 as seen by the down-regulated expression of P53 and CASPASE-3 (Fig. S2B), further demonstrating the critical role of endogenous CyPGs in IL-4-induced apoptosis in AML.
Figure 3. IL-4 administration induces apoptosis via P53 and caspase activation.
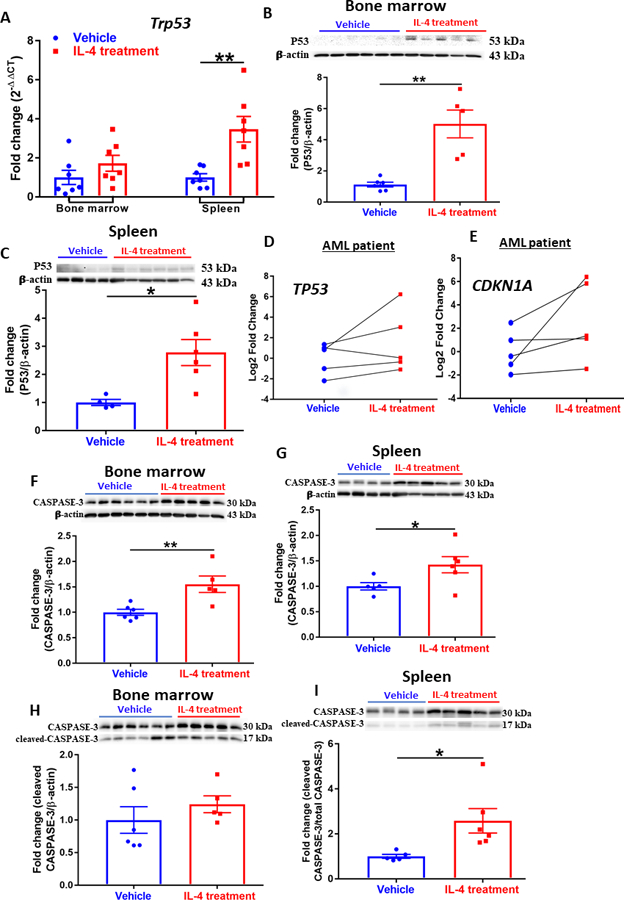
(A) Expression of Trp53 assessed by qPCR analysis in the bone marrow and spleen of AML mice. Data were normalized to 18S rRNA expression. (n=6~7). (B)(C) Western blot showing expression of P53 in cells isolated from bone marrow (B) and spleen (C) of AML mice. Densitometry was done by normalizing to vehicle control and relative to β-actin. (n=4~6). (D)(E) Expression of TP53 (D) and CDKN1A (E) assessed by qPCR analysis in patient AML cells treated with vehicle or IL-4 (n=5); each line represents a single patient sample treated with vehicle or IL-4. Data were normalized to 18S rRNA expression. (F)(G) Western blot showing expression of CASPASE-3 in cells isolated from bone marrow (F) and spleen (G) of AML mice. Densitometry was done by normalizing to vehicle control and relative to β-actin; n=4~6. (H)(I) Western blot showing expression of cleaved CASPASE-3 in cells isolated from bone marrow (H) and spleen (I) of AML mice. Densitometry was done by normalizing to vehicle control and relative to total Caspase-3; n=4~6. (A-C, F-I) Data shown are mean ± SEM per group; paired two-tailed Student t test was applied to D and E; and unpaired two-tailed Student t test was utilized for other panels; *, P < 0.05; **, P < 0.01.
Antagonism of PPAR𝛄 abrogates the anti-leukemic effect of IL-4 in AML mice
To address the link between IL-4 and CyPGs, we examined two receptors that share the same ligands, i.e., membrane bound chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2; Gpr44) and the intracellular receptor PPARγ. First, normal splenic cells treated with 13,14-dihydro-15-keto-PGD2 (DK-PGD2), a specific ligand for CRTH2, enhanced the expression of IL-4 and IL-13, which implies that signaling mediated by CRTH2 may also play a role in the feed forward loop increasing IL-4 and CyPG expression (data not shown). Second, expression of PPARγ (but not PPARδ) (Fig. S6A-D) and its downstream genes, Cd36 and Mrc1 (Fig. S6E and S6F), in bone marrow and spleen in AML mice were increased with IL-4 treatment, indicating a possible involvement of PPARγ activation via CyPGs during IL-4-treatment.
To further examine the role of PPARγ activation in IL-4-mediated apoptosis of LICs, we utilized GW9662, a potent antagonist of PPARγ, to treat primary AML cells in vitro in the presence of IL-4. Interestingly, GW9662 abrogated the up-regulation of P53 and CASPASE-3 induced by IL-4 (Fig. S2B), which led us to investigate this phenomenon in vivo. In vivo treatment of GW9662 in AML mice along with IL-4 (Fig. 4A) inhibited the activity of PPARγ, as seen by decreased expression of Cd36 (Fig. S6G) and Mrc1 (Fig. S6H), in the bone marrow and spleen. Strikingly, antagonism of PPARγ by GW9662 exacerbated leukocytosis (Fig. 4B) and splenomegaly (Fig. 4C). Consistently, the leukemic cells were observed to be increased by GW9662 in bone marrow (Fig. 4D) and spleen (Fig. 4E), offsetting the effect of IL-4. In addition, primitive leukemic cells within the Lin− population were also significantly elevated by GW9662, compromising the anti-leukemic effect of IL-4 treatment in AML mice (Fig. 4F and 4G). Interestingly, GW9662 treatment alone seemed to worsen the disease, indicating the central role of PPARγ activation by IL-4.
Figure 4. Antagonism of PPAR𝛄 offsets the anti-leukemic effect of IL-4 in AML mice.
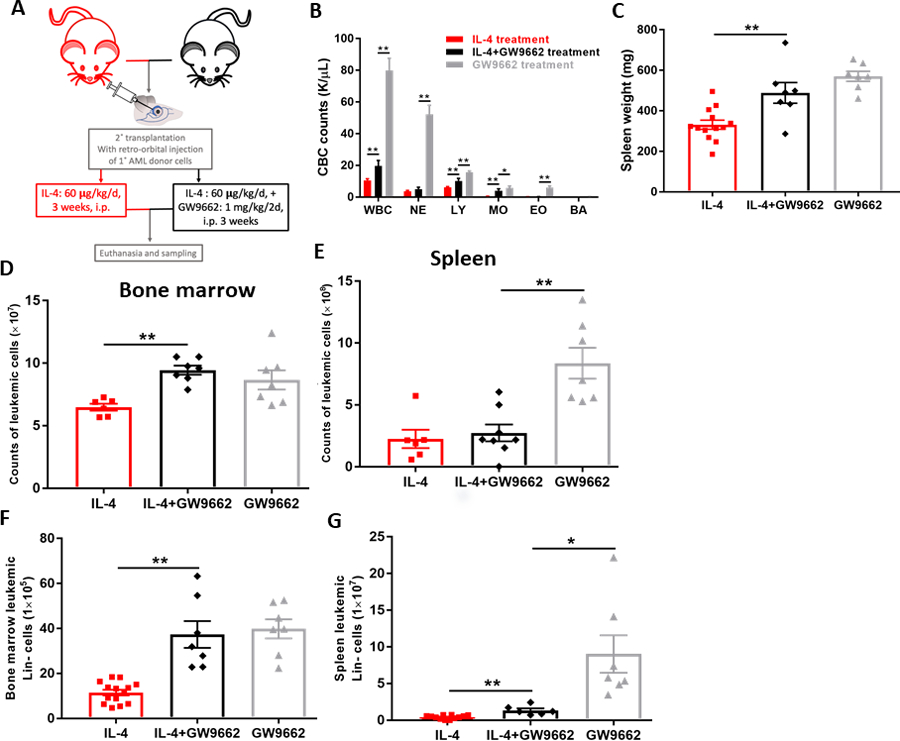
(A) Recipients of primary AML cells were dosed intra-peritoneally daily with rmIL-4 (60 μg/kg) in PBS suspension for three weeks with/without GW9662 (1mg/kg/2d, 14 doses). Euthanasia was done at the endpoint. (B) CBC counts (K/𝛍L peripheral blood) at endpoint, total WBC, EO, MO, NE, LY, BA profile of AML mice treated with IL-4 or IL-4+GW9662. (C) Spleen weights (mg) of AML mice. (D)(E) Cell counts of leukemic cells (CD45.1+) in bone marrow (D) and spleen (E) of AML mice. (F)(G) Flow cytometric analysis of total leukemic cells identified by CD45.1+ in the Lin− population of bone marrow (F) and spleen (G) of AML mice. Data shown are mean ± SEM per group, each group has at least seven biological replicates; unpaired two-tailed Student t test and outlier test were utilized; *, P < 0.05; **, P < 0.01.
Inhibition of PPARγ dampens apoptosis triggered by IL-4 in AML mice
We previously demonstrated the role of PPARγ in the ligand-dependent regulation of H-PGDS expression in macrophages.31 Based on these studies, we speculated the existence of a positive feedback loop involving PPARγ and its endogenous ligands, CyPGs, as key mediators of IL-4-dependent effects. Along these lines, we found that antagonism of PPARγ by GW9662 decreased the expression of H-PGDS enhanced by in-vitro (Fig. S2B) and in vivo (Fig. 5A and 5B) IL-4 in AML. Given that IL-4-stimulated CyPGs activated PPARγ as presented above (Fig. 2F, S5A, and S5B), our studies clearly illustrate that a positively reinforcing loop involving PPARγ and CyPGs was triggered by IL-4 in AML. Consistent with the in vitro result in AML cells (Fig. S2B), IL-4-induced expression of P53 and Caspase-3 activation (Fig. 5E and 5F) was considerably attenuated by GW9662 (Fig. 5C-F). Correspondingly, IL-4-induced apoptosis and necrosis in AML cells was blocked by the inhibition of PPARγ by GW9662 (Fig. 5G-I), leading to increased leukemic cell viability (Fig. 5J).
Figure 5. Antagonism of PPAR𝛄 interferes with the prostaglandin metabolism and apoptosis activated by IL-4 in AML mice.
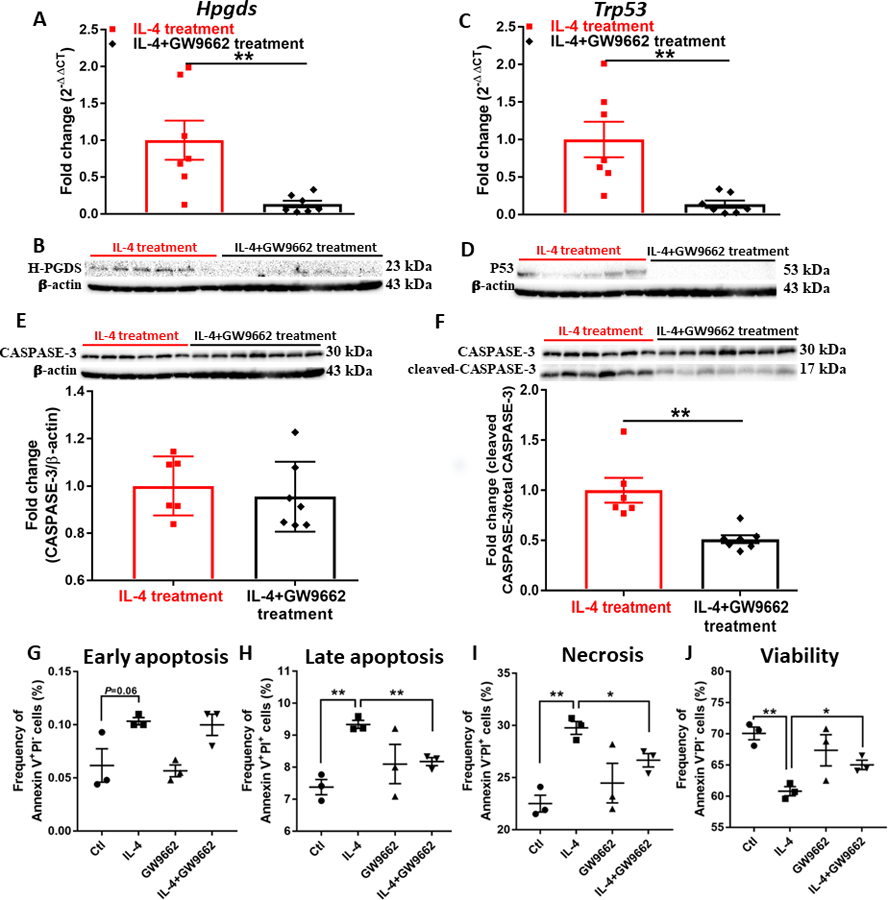
(A)(C) Expression of Hpgds (A) and Trp53 (C) assessed by qPCR analysis in the bone marrow of AML mice. Data were normalized to 18S rRNA expression. (B)(D) Western blot showing expression of H-PGDS (B) and P53 (D) in cells isolated from bone marrow of AML mice. (E) Western blot showing expression of CASPASE-3 in cells isolated from bone marrow of AML mice. Densitometry was done by normalizing to IL-4-treated group and relative to β-actin. (F) Western blot showing expression of cleaved CASPASE-3 in cells isolated from bone marrow of AML mice. Bands were normalized to IL-4-treated group and relative to total Caspase-3. (G-J) Flow cytometric analysis of AML cells treated by 50 ng/mL IL-4 and 10 μM GW9662 for 48 h; (G) early apoptosis, (H) late apoptosis, (I) necrosis, and (J) viability were identified as Annexin V+PI−, Annexin V+PI+, Annexin V−PI+, and Annexin V−PI− fractions, respectively. Data shown are mean ± SEM per group, n=6~7 for A-F, n=3 for G-J; unpaired two-tailed Student t test was utilized; *, P < 0.05; **, P < 0.01.
IL-4 supplement induces no apparent pulmonary toxicity
To evaluate the toxicological effect of long-term IL-4 administration on lungs, we examined lung inflammation by histology with H&E and PAS staining. The H&E sections were scored for airway damage, inflammation (leukocytic interstitial infiltrates and intravascular segmented neutrophilia with margination), and the presence of leukemia (intravascular blasts, peribronchiolar/perivascular infiltration, and alveolar septal involvement) (Table 1 and Fig. 6A). Airway damage was not observed. Although AML mice treated with IL-4 showed increased leukocyte infiltration and neutrophilia in lung associated with increased expression of Ccl11 (Fig. 6B),32 which could be indicative of inflammation, they nevertheless exhibited reduced metastasis of leukemia into lung compared to their PBS counterparts. More importantly, PAS-stained sections were examined for thickness of epithelium, hyperplasia of goblet cells, and formation of mucus (Fig. 6C). Hyperplasia of mucus-producing cells lining the epithelium was not observed in either group upon PAS staining. In addition, mucous secretion was generally not evident in bronchioles even after prolonged IL-4 administration. Mice in either group failed to show any clinical signs of asthma or airway hyperresponsiveness symptoms throughout the experiment. We also failed to detect any expression of IL-4 or IL-13 in the lungs of IL-4-treated mice (not shown). Overall, although inflammatory cell infiltration was observed in IL-4 treatment, these data clearly support the absence of overt pulmonary toxicity with long-term administration of IL-4 in AML mice.
Table 1.
Histological analysis of pulmonary pathology with IL-4 treatment
| Treatment | Airway damage | Leukocytic interstitial infiltrates | Marginating intravascular segmented neutrophilia (>3 in medium vessels) | Intravascular blasts | Peribronchiolar/peri vascular leukemia infiltration | Leukemia alveolar septal involvement | Leukemia (0 or 1, E=equivocal) |
|---|---|---|---|---|---|---|---|
| Vehicle #1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vehicle #2 | 0 | 1 | 0 | 1 | 0 | 0 | 1 |
| Vehicle #3 | 0 | 1 | 1 | 1 | 0 | 0 | 1 |
| Vehicle #4 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| IL-4 #1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| IL-4 #2 | 0 | 1 | 1 | E | 0 | 0 | E |
| IL-4 #3 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| IL-4 #4 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
0=absent, 1=present
Lung sections from IL-4 treated mice (n=4) and their counterparts (n=4) were stained with H&E dye. A blinded histological analysis was performed on the sections to score the pulmonary inflammation and leukemia infiltration.
Figure 6. IL-4 supplement induces no apparent pulmonary toxicity.
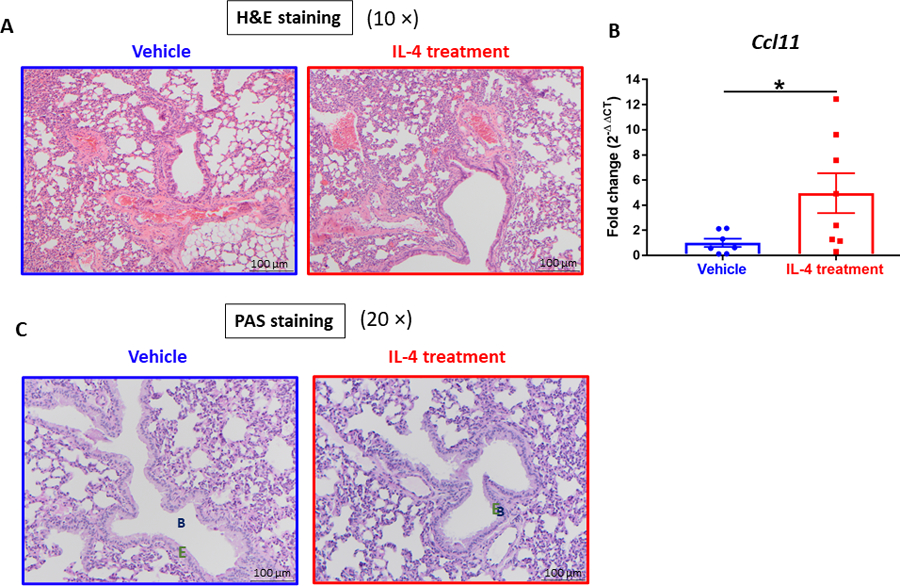
(A) Representative images of lungs from AML mice by H&E stain. (B) Expression of Ccl11 assessed by qPCR analysis in the lungs of AML mice. (n=7~8). (C) Representative images of lungs from AML mice by PAS stain. B: Bronchiole. E: epithelium. Data were normalized to 18S rRNA expression. Data shown are mean ± SEM per group, each group has at three replicates; unpaired two-tailed Student t test was utilized; *, P < 0.05; **, P < 0.01.
Discussion
Literature is replete with studies investigating the possible therapeutic role of various cytokines in hematopoietic malignancies.33–35 Available data with AML have implicated several candidates, such as IL-1, IL-4, and IL-6.14, 36, 37 Here, we demonstrate that IL-4 treatment alleviates the severity and benefits the survival of AML by inducing apoptosis of LICs in an MLL-AF9-induced murine model. Specifically, our studies indicate that this effect is mediated by coordinated autocrine and paracrine actions of CyPGs and activation of PPARγ upon IL-4 stimulation.
Our understanding of the conventional cancer immunity suggests the importance of anti-tumor activities of CD8+ cytotoxic T cells, CD4+ Th1 cells, as well as NK cells.38 In contrast, Th2 cells are generally considered as tumor-promoting cells. However, in hematologic cancers, the role of Th2 immune response may paradoxically assist with anti-tumor properties, which may be mediated by IL-4.39 Our finding that IL-4 induces AML cell apoptosis via p53-caspase activation provides support for this notion. A previous in vivo study tested the anti-leukemic role of IL-4 on MLL-AF9-induced AML mice and described a slightly improved survival in mice with a ten-day treatment of IL-4 injection (60 μg/kg per day).14 Moreover, IL-4 was reported to negatively regulate cancer cell growth in other in vitro studies of AML and CML, and acute lymphocytic leukemia (ALL) and CLL.40–42 Although these studies reveal an overall inhibitory effect of IL-4 in hematopoietic malignancies, there is clearly a deficit in our understanding of the mechanism of action of IL-4 in AML.
We believe that the underpinnings of the pro-apoptotic effect of IL-4 on AML cells demonstrated here are associated with complex mechanisms involving multiple signaling pathways. Our previous studies described a post-transcriptional and translational control of COX-1 expression by IL-4 led us to connect the activation of the intracellular nuclear receptor, PPARγ, by endogenous CyPGs.15, 43 Such an activation of PPARγ by CyPGs was seen in the form of increased transcriptional activity of PPARγ, as evidenced by the augmented expression of PPARγ downstream target genes following IL-4 stimulation. On the other hand, antagonism of PPARγ by GW9662 abrogated this effect and blocked the up-regulation of P53 expression and caspase activation induced by IL-4 stimulation, leading to diminished apoptosis of AML cells. Apart from the pro-apoptotic effect exerted by PPARγ activation, previous studies from our laboratory have shown that the Hpgds proximal promoter contains two confirmed PPAR-response elements (PPREs), suggesting the existence of a positive feed forward loop between PPARγ activation and HPGDS expression leading to endogenous CyPGs production.31 Intriguingly, this loop may assist in the maintenance of IL-4-induced pro-apoptotic effect in AML through an autocrine signaling via constitutive production of CyPGs, thereby further activating PPARγ. Furthermore, our data also revealed that CyPG metabolism was elevated by IL-4 treatment, suggesting a plausible paracrine signaling in which neighboring cells in TME provide CyPGs to induce apoptosis in AML cells. Perhaps the most interesting observation was that CyPGs could further enhance IL-4 expression in the normal splenic cells. It is plausible that IL-4 generated in the TME can in turn act on leukemic cells, further reinforcing the effect of IL-4 on AML cells. Besides, the distinct expression of COX-1 upon IL-4 stimulation between normal cells and leukemic cells suggests that cancer cells may respond differently to IL-4 which results in a different fate from normal cells, reflecting in the LICs and HSCs in the bone marrow of IL-4-treated mice. Taken together, these findings imply that IL-4 treatment can activate both intrinsic and extrinsic signaling cascades to create a “reservoir” of CyPGs in leukemia-infiltrated organs, where CyPGs also activate the expression of IL-4 in the TME to further complement the effect of exogenous IL-4.
Although we have identified PPARγ as a key effector, it is very likely that there are other signaling pathways that could be activated by CyPGs, including CRTH2, Akt,44, 45 tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL),46, 47 and microRNA-15548–50 that are all currently being investigated and could provide a more complete understanding of the role of CyPGs in AML. Importantly, four out of five patient AML cells showed significantly enhanced expressions of PGDS, TP53, and CDKN1A, highlighting the translation potential of our findings. However, further analysis into the background of these patients suggested that the only patient sample, which showed resistance to IL-4 treatment, presented more cytogenetic mutations than the other four, including chromosomal deletion (−5) and P53 mutation that could have a bearing on these results. As shown in previous studies,51, 52 deletion of long arm of chromosome 5, del(5q), leads to an unfavorable prognosis with poor survival and complete remission in AML. And del(5q) was associated with P53 mutation in AML mice and AML patients.53, 54 Taken together, it is very likely that potential unresponsiveness to IL-4 treatment may occur in AML patients with mutations commonly considered as being unfavorable; but recognizing the specific IL-4-unresponsive or -resistant mutations is imperative for the translational application of IL-4 in AML patients.
Although IL-4 is well studied in allergy and asthma, there is a general lack of data available regarding the susceptibility of IL-4-treated patients to allergies or asthma. Even though histological analysis of lung sections showed neutrophilia and leukocyte infiltration, it is clear that CyPGs at supraphysiological concentrations do not cause airway hyperresponsiveness, 55 and increased leukocytes could also signal timely resolution via efferocytosis of neutrophils by macrophages.56 That said, although IL-4 is a responder cytokine necessary for allergic inflammation, IL-4 itself cannot initiate allergy or asthma in the absence of any antigen. Furthermore, long-term IL-4 treatment did not affect the number of HSCs, which clearly demonstrated no apparent effects on normal HSCs supporting a convalescent hematopoiesis. In addition, mice treated with IL-4 did not exhibit any cachectic signs throughout the entire duration of the experiment compared to their counterparts. Though these findings support the therapeutic use of IL-4 in AML, further investigation is needed to fully understand the safety as well as its pharmacokinetics and pharmacodynamics. In summary, our results highlight a new potentially attractive therapeutic use of IL-4 in AML that involves endogenous CyPGs and PPARγ.
Supplementary Material
Figure 7. Proposed mechanism of IL-4-induced apoptosis in AML.
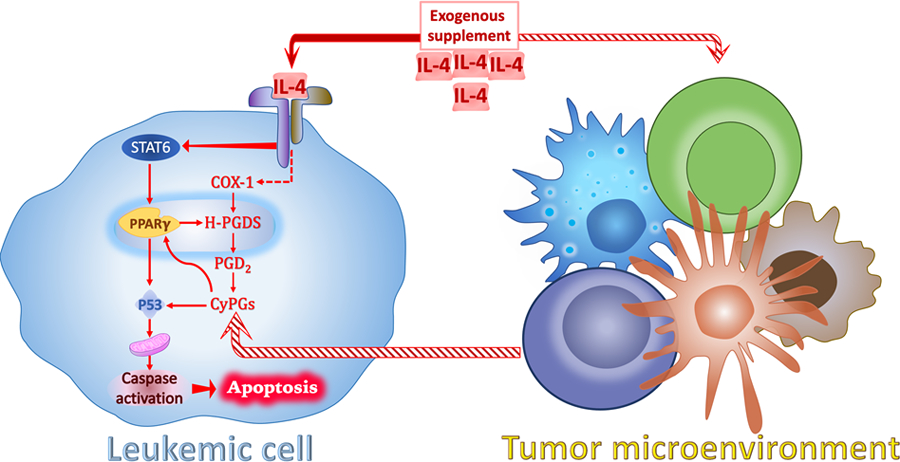
Upon stimulation by IL-4, leukemic cells and neighboring cells produce increased CyPGs via COX-1/H-PGDS/PGD2 axis as autocrine and paracrine secretions. CyPGs activate apoptotic signaling pathway (P53 and Caspase-3) by targeting PPARγ and/or P53 which leads to the remission of AML. IL-4 receptor-mediated signaling activates STAT6, which coordinates with PPARγ to up-regulate the expression of H-PGDS. Thus, PPARγ acts as a receptor for CyPGs to enhance endogenous production of CyPGs to partake in the anti-leukemic effects.
Acknowledgements
We thank Brian Dawson and Sarah Neering at the Flow Cytometry Core Facility of Penn State Huck Institutes of the Life Sciences for cell sorting, and current and former members in Prabhu and Paulson laboratories for timely help. This work was supported, in part, by grants from the American Institute for Cancer Research, NIH R01 DK077152, UDSA-NIFA Hatch project (# 4771; accession #0000005) to KSP and RFP.
This study is original research and has not been previously published or submitted for publication elsewhere while under consideration.
Abbreviations:
- AML
acute myeloid leukemia
- ALL
acute lymphocytic leukemia
- AO
acridine orange
- BA
basophil
- BMDM
bone marrow-derived macrophages
- CBC
complete blood cell
- CLL
chronic lymphocytic leukemia
- CML
chronic myeloid leukemia
- COX
cyclooxygenase
- CRTH2
chemoattractant receptor-homologous molecule expressed on Th2 cells
- CyPG
cyclopentenone prostaglandin
- DK-PGD2
13,14-dihydro-15-keto-prostaglandin D2
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- DMSO
dimethyl sulfoxide
- EGF
epidermal growth factor
- EO
eosinophil
- EPA
eicosapentaenoic acid
- FACS
fluorescence-activated cell sorting
- FDA
Food and Drug Administration
- H-PGDS
hematopoietic-PGD2 synthase
- H&E
hematoxylin and eosin
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem or progenitor cell
- IL-4
Interleukin-4
- LIC
leukemia-initiating cell
- Lin
lineage negative
- LY
lymphocyte
- MO
monocyte
- MRD
minimal residual disease
- NE
neutrophil
- PAS
Periodic acid–Schiff
- PBMC
peripheral blood mononuclear cell
- PGD2
prostaglandin D2
- Δ12-PGJ2
Δ12-prostaglandin J2
- PI
propidium iodide
- PPARγ
peroxisome proliferator-activated receptor gamma
- PPRE
PPAR-response element
- qPCR
quantitative PCR
- rRNA
ribosomal RNA
- Th2
Type 2 helper T cell
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- VEGF
vascular endothelial growth factor
- WBC
white blood cell
Footnotes
Conflict of Interest
The authors declare no competing financial interests.
This investigation focuses on acute myeloid leukemia, where we demonstrate: 1) IL-4 induces apoptosis of LICs in AML mice and patient AML blasts; and 2) IL-4-dependent PPARγ activation by endogenous CyPGs leads to LIC apoptosis via autocrine and paracrine mechanisms. These findings provide evidence for IL-4 to be a novel therapy for leukemia patients.
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
References
- 1.Norris D, Stone J. WHO classification of tumours of haematopoietic and lymphoid tissues Geneva: WHO. 2008; [Google Scholar]
- 2.Jordan CT. The leukemic stem cell. Best practice & research Clinical haematology 2007;20(1):13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett AJ. Acute myeloid leukaemia and the immune system: implications for immunotherapy. Br J Haematol Jan 2020;188(1):147–158. doi: 10.1111/bjh.16310 [DOI] [PubMed] [Google Scholar]
- 4.Liang K, Volk AG, Haug JS, et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell Jan 12 2017;168(1–2):59–72 e13. doi: 10.1016/j.cell.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang R, Feng W, Wang H, et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett Jan 28 2020;469:151–161. doi: 10.1016/j.canlet.2019.10.032 [DOI] [PubMed] [Google Scholar]
- 6.Bankaitis KV, Fingleton B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin Exp Metastasis Dec 2015;32(8):847–56. doi: 10.1007/s10585-015-9747-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puri RK, Leland P, Kreitman RJ, Pastan I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. Int J Cancer Aug 15 1994;58(4):574–81. doi: 10.1002/ijc.2910580421 [DOI] [PubMed] [Google Scholar]
- 8.Venmar KT, Carter KJ, Hwang DG, Dozier EA, Fingleton B. IL4 receptor ILR4alpha regulates metastatic colonization by mammary tumors through multiple signaling pathways. Cancer Res Aug 15 2014;74(16):4329–40. doi: 10.1158/0008-5472.CAN-14-0093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pena-Martinez P, Eriksson M, Ramakrishnan R, et al. Interleukin 4 induces apoptosis of acute myeloid leukemia cells in a Stat6-dependent manner. Leukemia Mar 2018;32(3):588–596. doi: 10.1038/leu.2017.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jansen JH, Fibbe WE, Wientjens GJ, Willemze R, Kluin-Nelemans JC. Inhibitory effect of interleukin-4 on the proliferation of acute myeloid leukemia cells with myelo-monocytic differentiation (AML-M4/M5); the role of interleukin-6. Leukemia Apr 1993;7(4):643–5. [PubMed] [Google Scholar]
- 11.McKenna HJ, Smith FO, Brasel K, et al. Effects of flt3 ligand on acute myeloid and lymphocytic leukemic blast cells from children. Exp Hematol Feb 1996;24(2):378–85. [PubMed] [Google Scholar]
- 12.Tao M, Li B, Nayini J, et al. In vivo effects of IL-4, IL-10, and amifostine on cytokine production in patients with acute myelogenous leukemia. Leuk Lymphoma Mar 2001;41(1–2):161–8. doi: 10.3109/10428190109057966 [DOI] [PubMed] [Google Scholar]
- 13.Lundin J, Kimby E, Bergmann L, Karakas T, Mellstedt H, Osterborg A. Interleukin 4 therapy for patients with chronic lymphocytic leukaemia: a phase I/II study. Br J Haematol Jan 2001;112(1):155–60. doi: 10.1046/j.1365-2141.2001.02525.x [DOI] [PubMed] [Google Scholar]
- 14.Martinez PEP, Chapellier M, Eriksson M, Ramakrishnan R, Hogberg C, Jaras M. Interleukin-4 Is a Negative Regulator of Acute Myeloid Leukemia Cells. Blood Dec 2014;124(21) [Google Scholar]
- 15.Finch ER, Tukaramrao DB, Goodfield LL, Quickel MD, Paulson RF, Prabhu KS. Activation of PPARγ by endogenous prostaglandin J2 mediates the antileukemic effect of selenium in murine leukemia. Blood 2017;129(13):1802–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shay AE, Diwakar BT, Guan BJ, Narayan V, Urban JF Jr., Prabhu KS. IL-4 up-regulates cyclooxygenase-1 expression in macrophages. J Biol Chem Sep 1 2017;292(35):14544–14555. doi: 10.1074/jbc.M117.785014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finch ER, Kudva AK, Quickel MD, et al. Chemopreventive Effects of Dietary Eicosapentaenoic Acid Supplementation in Experimental Myeloid Leukemia. Cancer Prev Res (Phila) Oct 2015;8(10):989–99. doi: 10.1158/1940-6207.CAPR-15-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiernik PH, Dutcher JP, Yao X, et al. Phase II study of interleukin-4 in indolent B-cell non-Hodgkin lymphoma and B-cell chronic lymphocytic leukemia: a study of the Eastern Cooperative Oncology Group (E5Y92). J Immunother Nov-Dec 2010;33(9):1006–9. doi: 10.1097/CJI.0b013e3181f5dfc5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Food, Administration D. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Center for Drug Evaluation and Research (CDER) 2005;7(0.001) [Google Scholar]
- 20.Hegde S, Kaushal N, Ravindra KC, et al. Delta12-prostaglandin J3, an omega-3 fatty acid-derived metabolite, selectively ablates leukemia stem cells in mice. Blood Dec 22 2011;118(26):6909–19. doi: 10.1182/blood-2010-11-317750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods Dec 2001;25(4):402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 22.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature Aug 17 2006;442(7104):818–22. doi: 10.1038/nature04980 [DOI] [PubMed] [Google Scholar]
- 23.Carmeliet P VEGF as a key mediator of angiogenesis in cancer. Oncology 2005;69 Suppl 3:4–10. doi: 10.1159/000088478 [DOI] [PubMed] [Google Scholar]
- 24.Achen MG, Jeltsch M, Kukk E, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci U S A Jan 20 1998;95(2):548–53. doi: 10.1073/pnas.95.2.548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joukov V, Pajusola K, Kaipainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J Apr 1 1996;15(7):1751. [PMC free article] [PubMed] [Google Scholar]
- 26.Scavelli C, Vacca A, Di Pietro G, Dammacco F, Ribatti D. Crosstalk between angiogenesis and lymphangiogenesis in tumor progression. Leukemia. Jun 2004;18(6):1054–8. doi: 10.1038/sj.leu.2403355 [DOI] [PubMed] [Google Scholar]
- 27.Urade Y, Fujimoto N, Ujihara M, Hayaishi O. Biochemical and immunological characterization of rat spleen prostaglandin D synthetase. J Biol Chem Mar 15 1987;262(8):3820–5. [PubMed] [Google Scholar]
- 28.Christ-Hazelhof E, Nugteren DH. Purification and characterisation of prostaglandin endoperoxide D-isomerase, a cytoplasmic, glutathione-requiring enzyme. Biochim Biophys Acta Jan 29 1979;572(1):43–51. doi: 10.1016/0005-2760(79)90198-x [DOI] [PubMed] [Google Scholar]
- 29.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature Dec 16 1993;366(6456):701–4. doi: 10.1038/366701a0 [DOI] [PubMed] [Google Scholar]
- 30.Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans Nov 2001;29(Pt 6):684–8. doi: 10.1042/0300-5127:0290684 [DOI] [PubMed] [Google Scholar]
- 31.Gandhi UH, Kaushal N, Ravindra KC, et al. Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) γ. Journal of Biological Chemistry 2011;286(31):27471–27482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huaux F, Gharaee-Kermani M, Liu T, et al. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am J Pathol Dec 2005;167(6):1485–96. doi: 10.1016/S0002-9440(10)61235-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrmann F, Andreeff M, Gruss HJ, Brach MA, Lubbert M, Mertelsmann R. Interleukin-4 inhibits growth of multiple myelomas by suppressing interleukin-6 expression. Blood Oct 15 1991;78(8):2070–4. [PubMed] [Google Scholar]
- 34.Kawakami M, Kawakami K, Kioi M, Leland P, Puri RK. Hodgkin lymphoma therapy with interleukin-4 receptor-directed cytotoxin in an infiltrating animal model. Blood May 1 2005;105(9):3707–13. doi: 10.1182/blood-2004-08-3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Defrance T, Fluckiger AC, Rossi JF, Magaud JP, Sotto JJ, Banchereau J. Antiproliferative effects of interleukin-4 on freshly isolated non-Hodgkin malignant B-lymphoma cells. Blood Feb 15 1992;79(4):990–6. [PubMed] [Google Scholar]
- 36.Givon T, Slavin S, Haran-Ghera N, Michalevicz R, Revel M. Antitumor effects of human recombinant interleukin-6 on acute myeloid leukemia in mice and in cell cultures. Blood May 1 1992;79(9):2392–8. [PubMed] [Google Scholar]
- 37.Mitchell K, Barreyro L, Todorova TI, et al. IL1RAP potentiates multiple oncogenic signaling pathways in AML. J Exp Med Jun 4 2018;215(6):1709–1727. doi: 10.1084/jem.20180147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian F, Misra S, Prabhu KS. Selenium and selenoproteins in prostanoid metabolism and immunity. Crit Rev Biochem Mol Biol Dec 2019;54(6):484–516. doi: 10.1080/10409238.2020.1717430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akashi K The role of interleukin-4 in the negative regulation of leukemia cell growth. Leuk Lymphoma Feb 1993;9(3):205–9. doi: 10.3109/10428199309147371 [DOI] [PubMed] [Google Scholar]
- 40.Luo HY, Rubio M, Biron G, Delespesse G, Sarfati M. Antiproliferative effect of interleukin-4 in B chronic lymphocytic leukemia. J Immunother (1991) Dec 1991;10(6):418–25. doi: 10.1097/00002371-199112000-00005 [DOI] [PubMed] [Google Scholar]
- 41.Manabe A, Coustan-Smith E, Kumagai M, et al. Interleukin-4 induces programmed cell death (apoptosis) in cases of high-risk acute lymphoblastic leukemia. Blood Apr 1 1994;83(7):1731–7. [PubMed] [Google Scholar]
- 42.Akashi K, Shibuya T, Harada M, et al. Interleukin 4 suppresses the spontaneous growth of chronic myelomonocytic leukemia cells. J Clin Invest Jul 1991;88(1):223–30. doi: 10.1172/JCI115281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci Jan 1 2008;13:1813–26. doi: 10.2741/2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim EH, Na HK, Kim DH, et al. 15-Deoxy-Delta(12,14)-prostaglandin J(2) induces COX-2 expression through Akt-driven AP-1 activation in human breast cancer cells: a potential role of ROS. Carcinogenesis Apr 2008;29(4):688–695. doi: 10.1093/carcin/bgm299 [DOI] [PubMed] [Google Scholar]
- 45.Shin SW, Seo CY, Han H, et al. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin Cancer Res Sep 1 2009;15(17):5414–25. doi: 10.1158/1078-0432.CCR-08-3101 [DOI] [PubMed] [Google Scholar]
- 46.Han H, Shin SW, Seo CY, et al. 15-Deoxy-delta 12,14-prostaglandin J2 (15d-PGJ 2) sensitizes human leukemic HL-60 cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through Akt downregulation. Apoptosis Nov 2007;12(11):2101–14. doi: 10.1007/s10495-007-0124-2 [DOI] [PubMed] [Google Scholar]
- 47.Hasegawa H, Yamada Y, Komiyama K, et al. A novel natural compound, a cycloanthranilylproline derivative (Fuligocandin B), sensitizes leukemia cells to apoptosis induced by tumor necrosis factor related apoptosis-inducing ligand (TRAIL) through 15-deoxy-Delta 12, 14 prostaglandin J2 production. Blood Sep 1 2007;110(5):1664–74. doi: 10.1182/blood-2007-01-068981 [DOI] [PubMed] [Google Scholar]
- 48.Kim S, Lee ES, Lee EJ, et al. Targeted eicosanoids profiling reveals a prostaglandin reprogramming in breast Cancer by microRNA-155. J Exp Clin Cancer Res Jan 25 2021;40(1):43. doi: 10.1186/s13046-021-01839-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narayan N, Bracken C, Ekert P. MicroRNA-155 expression and function in AML: An evolving paradigm. Journal article. Experimental Hematology. 2018;62:1–6. Elsevier. doi: 10.1016/j.exphem.2018.03.007 [DOI] [PubMed] [Google Scholar]
- 50.Narayan N, Morenos L, Phipson B, et al. Functionally distinct roles for different miR-155 expression levels through contrasting effects on gene expression, in acute myeloid leukaemia. Leukemia Apr 2017;31(4):808–820. doi: 10.1038/leu.2016.279 [DOI] [PubMed] [Google Scholar]
- 51.Grimwade D, Walker H, Harrison G, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood Sep 1 2001;98(5):1312–20. doi: 10.1182/blood.v98.5.1312 [DOI] [PubMed] [Google Scholar]
- 52.Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood Dec 15 2000;96(13):4075–83. [PubMed] [Google Scholar]
- 53.Stoddart A, Fernald AA, Wang J, et al. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood Feb 13 2014;123(7):1069–78. doi: 10.1182/blood-2013-07-517953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volkert S, Kohlmann A, Schnittger S, Kern W, Haferlach T, Haferlach C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer May 2014;53(5):402–10. doi: 10.1002/gcc.22151 [DOI] [PubMed] [Google Scholar]
- 55.Kudva AK, Kaushal N, Mohinta S, et al. Evaluation of the stability, bioavailability, and hypersensitivity of the omega-3 derived anti-leukemic prostaglandin: Delta(12)-prostaglandin J3. PLoS One 2013;8(12):e80622. doi: 10.1371/journal.pone.0080622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol Oct 30 2014;7(2):a016311. doi: 10.1101/cshperspect.a016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.