

Abstract
Most tumor cells depend upon activation of the ribonucleoprotein enzyme telomerase for telomere maintenance and continual proliferation. The catalytic activity of this enzyme can be reconstituted in vitro with the RNA (hTR) and catalytic (hTERT) subunits. However, catalytic activity alone is insufficient for the full in vivo function of the enzyme. In addition, the enzyme must localize to the nucleus, recognize chromosome ends, and orchestrate telomere elongation in a highly regulated fashion. To identify domains of hTERT involved in these biological functions, we introduced a panel of 90 N-terminal hTERT substitution mutants into telomerase-negative cells and assayed the resulting cells for catalytic activity and, as a marker of in vivo function, for cellular proliferation. We found four domains to be essential for in vitro and in vivo enzyme activity, two of which were required for hTR binding. These domains map to regions defined by sequence alignments and mutational analysis in yeast, indicating that the N terminus has also been functionally conserved throughout evolution. Additionally, we discovered a novel domain, DAT, that “dissociates activities of telomerase,” where mutations left the enzyme catalytically active, but was unable to function in vivo. Since mutations in this domain had no measurable effect on hTERT homomultimerization, hTR binding, or nuclear targeting, we propose that this domain is involved in other aspects of in vivo telomere elongation. The discovery of these domains provides the first step in dissecting the biological functions of human telomerase, with the ultimate goal of targeting this enzyme for the treatment of human cancers.
A fundamental difference between normal somatic cells and malignant cells is the ability of the latter to proliferate beyond the normally defined set of cell divisions, through a process known as cellular immortalization. The ability of cancer cells to become immortal is linked to the replication of chromosome termini or telomeres. Telomeres are DNA-protein structures that protect chromosome ends from degradation and inappropriate recombination (8). The DNA portion of this structure in most eukaryotes is comprised of tandem repeats of a short G-rich sequence that extends past the complementary C strand, forming a 3′G-rich overhang that can adopt higher-ordered structures (8, 23). During DNA replication in normal human somatic cells, there is a loss of telomeric DNA, which eventually elicits a growth arrest signal in cultured cells termed senescence (26, 28, 55). If such a signal is disrupted, as it is in transformed cells, further telomere shortening eventually denudes chromosome ends of its protective DNA, leading to a period of crisis characterized by massive genomic instability and cell death (12, 55). Telomere loss may therefore serve as a protective mechanism to prevent sustained proliferation of abnormal cells that have a neoplastic predisposition.
Most cancer cells overcome the proliferative blockade of telomere shortening through activation of the normally dormant telomerase enzyme (3, 58). Human telomerase is a reverse transcriptase containing a ∼127-kDa catalytic protein (hTERT) (27, 32, 41, 47) that reverse transcribes the template region of the associated RNA subunit (hTR) (18) onto the 3′ end of telomeric DNA, thereby elongating telomeres. Normally, somatic cells express only the hTR subunit (2, 18), but during tumorigenesis the hTERT gene is illegitimately activated, restoring telomerase activity, preventing further telomere shortening and thereby immortalizing cells (14, 33, 35, 41, 47, 48). hTERT is both required for the tumorigenic transformation of normal cells (16, 24, 54) and the continual proliferation of cancer cells (20, 25, 64). Since telomerase is activated in as many as ∼85% of tumors but is absent in most normal tissues (3, 58), inhibition of hTERT could represent a specific means of targeting a broad range of cancers. Understanding how hTERT functions in human cells could be important for developing antitelomerase therapies.
Enzyme catalysis can be reconstituted in vitro with hTERT and hTR, suggesting that these subunits form the core of a more complex holoenzyme (4–7, 40, 43, 60, 61); however, the exact stochiometry of this core complex is uncertain. Biochemical purification of telomerase activity from the ciliate Euplotes suggests that the enzyme is composed of a single RNA, catalytic protein subunit, and associated protein (38). However, accumulating evidence suggests that telomerase may be a multimeric complex. For example, certain template mutations of the RNA were found to be copied in yeast and human cells only when a wild-type telomerase complex was present (51, 52, 60), and telomerase activity was immunoprecipitated with catalytically inactive hTERT fragments produced in telomerase-positive cells (7).
TERT proteins from a variety of organisms are defined by a large central catalytic domain, encompassing approximately one third to one half of the protein, which contains reverse transcriptase motifs essential for catalysis (46). C-terminal to this domain is a short highly divergent region, where the comparison of yeast and human proteins reveals little to no obvious sequence conservation or functional similarity (5, 7, 19; S. S. R. Banik et al., unpublished data). On the other hand, the N terminus of yeast telomerase contains four domains termed I, II, III, and the T-motif that are essential for yeast viability, with the latter two domains being necessary for RNA binding (9, 19, 63). In Tetrahymena spp., the N terminus is also essential for telomerase activity and a 321-amino-acid region encompassing the T-motif and domains II and III (as defined in yeast) can bind the telomerase RNA (36). Similarly, in vitro, deletion of the first 350 amino acids of human TERT abolishes telomerase activity, and a large 287-amino-acid N-terminal fragment of hTERT that maps to RNA-binding regions in the yeast and Tetrahymena protein has been shown to bind hTR (5, 7). More recently, an alignment of ∼500 amino acids of the N terminus from an array of phylogenic TERT proteins identified five amino acids that are identical, clustering in three regions termed GQ, CP, and QFP, which overlap with yeast domains I, II, and III, respectively (63). Thus, the N terminus may contain evolutionarily conserved regions essential for RNA binding and telomerase activity.
Recent studies suggest that telomere elongation by hTERT involves more than the association with hTR or catalytic activity. Addition of a double hemagglutinin (HA) epitope tag to the C terminus of hTERT (hTERT-HA) results in a catalytically active enzyme that cannot maintain telomere length or immortalize cells in vivo (13, 49, 66). More recently, three different alanine substitution mutations in the N terminus of yeast catalytic subunit of telomerase, Est2p, have been found to dissociate catalytic from biological activity (19, 63). The biological function disrupted by these mutations is uncertain, since telomerase activity appears to be regulated at multiple levels in vivo. For example, the enzyme must localize to the nucleus to be functional, a process recently shown to be regulated during T-cell activation (39), possibly by phosphorylation or association of the protein 14-3-3 (39, 56). Telomerase is also targeted specifically to telomeres, and in yeast this process is mediated through a number of proteins (17, 22, 31, 50, 53, 65). Lastly, telomere elongation is known to be cell cycle regulated and tightly coupled to the synthesis of the complementary C strand (1, 15, 53). Biochemical analysis of in vitro reconstituted enzyme activity would not be expected to identify domains of TERT responsible for most of these other cellular functions. To elucidate the domains of hTERT required for such functions in human cells, we studied the consequence of mutations to hTERT in vivo. In addition to identifying domains essential for catalytic activity, we discovered a domain essential for another cellular function of telomerase. This DAT domain is dispensable for catalytic activity, but is required for in vivo telomerase function. This represents the first domain of hTERT linked to the biological regulation of telomerase.
MATERIALS AND METHODS
Plasmids.
By using pairs of complementary oligonucleotides bearing the sequence AATGCTGCTATACGATCG (encoding for the sequence NAAIRS for the sense oligonucleotides) in place of the sequence encoding the six amino acids to be mutated (flanked on either side by 15 nucleotides complementary to native hTERT sequence), each NAAIRS substitution was introduced into either the EcoRI-MluI or the MluI-NcoI fragment of a N-terminal FLAG-tagged hTERT (FLAG-hTERT) by QuikChange site-directed mutagenesis (Stratagene). Accordingly, 90 separate oligonucleotide pairs were used to systematically substitute every six amino acids from the +2 position up to +547, with the exception of position +260. Mutated regions were sequenced to confirm correct substitution. To create retroviral constructs, all 90 of the mutated fragments were removed and cloned back into EcoRI-MluI or MluI-NcoI sites of full-length FLAG-hTERT cloned in the EcoRI-SalI sites of the plasmid pBluescript SK(−) (Stratagene), after which the mutated open reading frame was excised and cloned into the EcoRI-SalI sites in the retroviral vector pBabehygro (45). To create in vitro expression constructs, selected mutants were similarly extracted and cloned into the EcoRI-MluI or MluI-NcoI sites of an N- and a C-terminal FLAG-tagged hTERT cDNA (FLAG-hTERT-FLAG) that was inserted in the plasmid pCIneo (Promega). GST-pCIneo was made by digesting pGEX-4T1 (Amersham Pharmacia Biotech) with SspI and SalI and then cloning this fragment into pCIneo digested with EcoRI (blunted with Mung bean nuclease) and SalI. GST-hTERT-pCIneo (wild type; positions +50, +92, and +152) was produced by introducing FLAG-hTERT into GST-pCIneo with EcoRI and SalI and then removing the FLAG sequence by PCR cloning with primers 5′-CGAATTCCAAACCGCCCCCTCCTTCCGCCAG and 5′-GTCCACGCGTCCTGCCCG. GST-hTERT-pCIneo (+386 and +512) were made by inserting MluI and SalI fragments, digested from corresponding FLAG-hTERT-pBabehygro plasmids, into wild-type GST-hTERT-pCIneo cut with MluI and SalI.
The hTR-expressing plasmid pBluescriptSK-hTR was created by inserting the EcoRI-digested hTR PCR product, generated by amplifying plasmid phTRA (10) with primers 5′-CGGAATTCGGGTTGCGGAGGG and 5′-CGGAATTCGCATGTGTGAGCCGAGTCCTGG into the same site downstream of the T7 promoter in pBluescript SK(−) (Stratagene).
Cell culture and apoptosis assays.
The simian immunodeficiency virus (SV40) T/t-Ag transformed human embryonic kidney cell line HA5 (59) was infected at population doubling (pd) ∼51 to 56 with the amphotropic retroviruses derived from the above-described pBabehygro constructs encoding each of the 90 NAAIRS mutant FLAG-hTERT cDNAs or, as controls, wild-type FLAG-hTERT or no insert, after which stable polyclonal populations were selected in media supplemented with 100 μg of hygromycin B (Sigma)/ml as previously described (13). A population doubling of 0 was arbitrarily assigned to the first confluent plate under selection. Cells were continually passaged at 1:4 or 1:8 under selection until either crisis or until the culture divided more than 2.5 times longer than vector control cell lines. Crisis was defined as the period when cultures failed to become confluent within 25 days and exhibited massive cell death.
For apoptosis studies, infected HA5 cell lines were split 1:4 or 1:8, and 3 days later the adherent cells were trypsinized and pooled with nonadherent cells from the media. These cells were washed twice in cold 1× phosphate-buffered saline (PBS) and stained with annexin V and propidium iodide according to manufacturer's instructions using the Annexin V-FITC Apoptosis Detection Kit II (Pharmingen). Flow analysis was performed at the Duke Comprehensive Cancer Center Flow Cytometry Shared Resource facility by using a FACSCaliber (Becton Dickinson).
hTERT mRNA detection, telomerase activity, and telomere length assays.
For quantitative reverse transcription-PCR (RT-PCR), total RNA from each of the described infected HA5 cells was isolated with the RNAzol reagent according the manufacturer's instructions (Teltest), and 250 ng of RNA was RT-PCR amplified to detect either total hTERT or PBGD mRNA by using the LightCycler TeloTAGGG hTERT Quantification Kit and LightCycler (Roche). hTERT signals were normalized to PBGD mRNA levels, and the number of hTERT transcript was determined by using a standard curve generated from RT-PCR of known concentrations of in vitro-transcribed hTERT mRNA, in accord with the manufacturer's instructions (Roche). Conversion to transcript per cell was determined based on the number of cell equivalents of RNA assayed.
To specifically detect endogenous or ectopic hTERT mRNA or the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA, the RNA described above was amplified by using semiquantitative RT-PCR as previously described (24) with primers specific for the following: endogenous hTERT, 5′-ACTCGACACCGTGTCACCTA and 5′-GTGACAGGGCTGCTGGTGTC; ectopic hTERT, 5′-GACACACATTCCACAGGTCG and 5′-GACTCGACACCGTGTCACCTAC; or GAPDH, 5′-GAGAGACCCTCACTGCTG and 5′-GATGGTACATGACAAGGTGC. Reaction products were resolved on 10% polyacrylamide gels, dried, and exposed to a phosphorimager screen.
To detect telomerase activity, lysates were isolated from infected HA5 cells at two different passages, protein concentration was measured by Bradford assay (Bio-Rad), lysates were diluted in the lysis buffer to a concentration of 0.1 μg/μl, and 0.2 μg was assayed for telomerase activity by using the telomeric repeat amplification protocol as previously described (34). As a negative control, duplicate reactions were heat treated at 85°C for 2 min to inactivate telomerase. Reaction products were resolved on 10% polyacrylamide gels, dried, and exposed to a phosphorimager screen to quantitate enzyme activity as previously described (34).
Telomeres were visualized by Southern hybridizing 10 μg of HinfI and RsaI restriction enzyme-digested genomic DNA with the 32P-labeled telomeric (C3TA2)3 oligonucleotide exactly as previously described (12), with the exception that washes were performed with 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate).
Western blot and indirect immunofluorescence.
To analyze mutant hTERT protein expression, 293T cells were transiently transfected with pCIneo or pCIneo-FLAG-hTERT-FLAG constructs by calcium phosphate transfection method (21). Cells were collected at ∼48 h posttransfection and lysed in 1× PBS, 5 mM EDTA, 0.2% NP-40, 10% glycerol, 1 mM benzamidine, 1 μg of pepstatin A/ml, 1 μg of leupeptin/ml, 1.5 μg of aprotinin/ml, 0.1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, and 1 mM Na3VO4. The protein concentration was measured by Lowry assay (Bio-Rad), and 30 μg of soluble lysate was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membrane (Millipore), and blocked with TBST (1× TBS [50 mM Tris-HCl, pH 7.4; 150 mM NaCl]–0.02% Tween 20)–5% milk. Blots were incubated with either anti-FLAG M2 mouse monoclonal antibody (Sigma) or anti-actin (C-2) mouse monoclonal antibody (Santa Cruz Biotechnology Inc.) and the goat anti-mouse immunoglobulin G-horseradish peroxidase (Santa Cruz Biotechnology, Inc.) diluted in TBST–5% milk. Blots were washed three times for 6 min each time in 1× TBS or TBST, and protein was detected with ECL Reagent according to the manufacturer's protocol (Amersham Pharmacia Biotech).
Localization of hTERT proteins was visualized in the human osteosarcoma cell line, U2OS, by indirect immunofluorescence. A total of 2 μg of pCIneo, pCIneo-FLAG-hTERT-FLAG wild-type, or NAAIRS mutant +92 and +122 constructs were transiently transfected into U2OS cells by calcium phosphate and examined ∼36 h posttransfection. Cells were fixed with 3% paraformaldehyde–2% sucrose, permeablized with 1× PBS–0.2% Triton X-100, and blocked with PBTN (1× PBS, 0.1% Triton X-100, 5% goat serum). Ectopic hTERT was detected by anti-FLAG M2 mouse monoclonal antibody recognized by a goat anti-mouse antibody conjugated with fluorescein isothiocyanate (Jackson ImmunoResearch) diluted in PBTN. Nuclei were stained with 2 μg of Hoechst 33258 (Sigma)/ml. Cells were examined at ×400 magnification on a Nikon Eclipse TE300 light microscope.
hTR-hTERT and hTERT-hTERT coimmunoprecipitations.
hTR was expressed and 32P labeled with the T7-coupled Maxiscript Kit (Ambion) by using 1 μg of linearized pBluescriptSK-hTR. Unincorporated nucleotides were removed by using a G-25 Minispin Column (Amersham Pharmacia Biotech). 35S-labeled proteins were produced by using the T7 quick coupled TNT System (Promega) from plasmids pCIneo-FLAG-hTERT-FLAG; pCIneo-FLAG-hTERT-FLAG-NAAIRS +50, +152, +386, or +512; and pCMV-HDAC1-FLAG in the presence of 3 μl of hTR RNA.
For coimmunoprecipitations, 4.4 μg of the M2 anti-FLAG monoclonal antibody was prebound to 25 μl of GammaBind G-Sepharose (Amersham Pharmacia Biotech) in S-100 buffer (9 mM Tris, pH 7.5; 0.9 mM MgCl2; 0.9 mM EGTA, pH 8; 1.5 mM dithiothreitol; 0.5% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}; 10% glycerol; 1 mM benzamidine; 0.1 mM phenylmethylsulfonyl fluoride) in the presence of blocking agents (100 ng of bovine serum albumin /ml, 100 ng of casein/ml, 100 ng of tRNA/ml, 250 ng of yeast total RNA/ml, 100 ng of glycogen/ml) as previously described with minor modifications (44). Coated beads were added to completed TNT reactions, diluted with S-100 buffer in a final volume of 750 μl, and incubated for 1 h at room temperature in the presence of 200 U of RNasin (Promega) and nonspecific blocking agents described above. The beads were washed three times with prechilled S-100 buffer, heated in SDS buffer, and resolved by SDS-PAGE.
For the immunoprecipitation of hTERT-hTERT complexes, wild-type and NAAIRS-substituted (+50, +152, +386, or +512) FLAG-hTERT-FLAG and N-terminal glutathione S-transferase (GST)-tagged hTERT were separately transcribed and translated as described above in the presence of 0.5 μl of [35S]methionine and 1 μl of cold methionine, or 4 μl of [35S]methionine, respectively, supplemented with 20 pmol of Ts oligonucleotide (34) and 1 μl of trace-labeled hTR RNA expressed in vitro with the Maxiscript Kit (Ambion) by using 0.17 μl of [32P]UTP and 6 μM cold UTP. Reactions were incubated 30°C for 40 min, mixed with the appropriate reaction, and then incubated for an additional 60 min at 30°C. Reactions were immunoprecipitated with the anti-Flag M2 monoclonal antibody as described above. The reciprocal complex made with 35S-labeled GST-hTERT and trace labeled FLAG-hTERT-FLAG was also immunoprecipitated with 1 μg of the Z-5 anti-GST antibody (Santa Cruz Biotechnology, Inc.). As controls, HDAC1-FLAG and GST were immunoprecipitated in the presence of GST-hTERT.
RESULTS
Identification of functional domains in the N terminus of hTERT by mutational analysis.
To define domains essential for telomerase function, we generated a panel of 90 individual tandem NAAIRS substitution mutations within the N terminus of hTERT, beginning immediately after the initiating methionine and terminating at the conserved T-motif (46). NAAIRS substitution mutagenesis presumably has only minor effects on protein structure, since substitutions do not alter protein length and the NAAIRS sequence has the unique ability to adopt multiple structural conformations (62). Moreover, this mutagenesis approach has been successfully employed to map the pocket region of pRB (57), as well as locate C-terminal domains within hTERT (Banik et al., unpublished). The panel of NAAIRS substitution mutants was introduced into telomerase-negative HA5 cells by retroviral infection. HA5 cells are human embryonic kidney cells transformed with the SV40 T-Ag gene, which lack hTERT expression and lose telomeric DNA every cell division until they reach crisis and die (12, 59). The proliferative potential of these cells can therefore serve as a reliable indicator of the biological consequence of hTERT mutations, since stable expression of biologically active versions of hTERT restores telomerase activity, stabilizes telomere length, and immortalizes HA5 cells (see reference 13 and also below). The resulting HA5 infected cell lines were assayed for telomerase function in vitro by assessing telomerase enzyme activity and telomerase function in vivo by determining if the infected cells bypass crisis induced by telomere shortening.
To verify expression of hTERT mutants, we used quantitative RT-PCR to detect hTERT mRNA. This method was chosen because overexpression of hTERT by the retroviral promoter in HA5 cells produces undetectable levels of protein, as assessed by Western blotting with an anti-FLAG antibody (not shown). RNA was isolated from all 90 stably infected cell lines and RT-PCR amplified with primers specific for hTERT transcripts (Fig. 1A). Vector control-infected HA5 cells were found to have extremely low levels of hTERT mRNA, corresponding to ∼1 transcript per 100 cells, which is >150-fold lower than that observed in tumor cell lines by quantitative SAGE analysis (37). Consistent with low hTERT expression, HA5 cells do not have readily detectable levels of telomerase activity, lose telomeric DNA, and fail to immortalize (12). Cell lines stably infected with FLAG-hTERT mutant constructs expressed hTERT mRNA at variable levels. However, in every case the expression was several orders of magnitude higher than vector cell lines when normalized with the housekeeping gene PBGD and was comparable to that detected in wild-type FLAG-hTERT-infected cells (Fig. 1A and Table 1).
FIG. 1.

Expression and telomerase activity of N-terminal hTERT mutants. (A) Total RNA was isolated from HA5 cell lines stably infected with vector (▵), FLAG-hTERT (▪, or FLAG-hTERT NAAIRS substitution mutants representative of nonessential (+212: ○), essential (+158, □), slow-growth (+110, ⧫), and biologically essential (+128, ●) and RT-PCR amplified with primers specific for hTERT by quantitative, real-time RT-PCR. The amount of transcript detected by fluorescence with FRET probes is plotted in arbitrary units against each PCR cycle (top panel). The housekeeping PBGD transcript was similarly measured to verify equivalent RNA addition per reaction (bottom panel), while H2O (◊) was assayed in both reactions as a negative control. (B) A total of 0.2 μg of lysate prepared from the described HA5 cell lines was assayed for telomerase activity by TRAP assay. As a control, a portion of the lysate was heat treated (HT) to inactivate telomerase prior to assaying. The internal standard (IS) served as a positive control for PCR amplification. Catalytic activity for each sample was normalized with the internal standard and is expressed as a percentage of wild-type FLAG-hTERT activity, indicated as follows: ++ (>60%), + (60 to 15%), +/− (<15%), and − (extremely low or no detectable activity). Domain refers to the location of the mutant, as described in the text. Life span (M, mortal; I, immortal; S, slow growth) as defined in the text. (C) Biologic activity of hTERT mutants was measured by serially passaging HA5 cell lines to determine whther cells entered crisis like vector or immortalized like wild-type hTERT. Representative clones are shown: vector (▴), FLAG-hTERT (▪), +212 (○), +50 (□), +14 (⋄), and +128 (▵). (D) Telomere length of representative HA5 cells infected with NAAIRS mutants that result in an immortal, slow-growth, or finite life span was determined by releasing the terminal restriction fragments of genomic DNA isolated from the described cell lines at early passage (pd 2 to 3) with the restriction enzymes HinfI and RsaI. These fragments were resolved and detected by Southern hybridization with a telomeric probe. ✽, Sample +212 was underloaded. Domain refers to the location of the mutant, as described in the text.
TABLE 1.
hTERT expression and catalytic and biologic telomerase activity for the panel of N-terminal hTERT mutants
| Domain and mutanta | hTERT expression (105)b | Telomerase in vitro activityc | Life spand |
|---|---|---|---|
| Vector | 0.00001 | − | M |
| Wild type | 5 | ++ | I |
| I-A | |||
| +2 | 6 | + | S |
| +8 | 4 | − | M |
| +14 | 15 | + | S |
| +20 | 2 | − | M |
| +26 | 1 | − | M |
| +32 | 3 | ++ | S |
| +38 | 9 | +/− | M |
| +44 | 2 | +/− | M |
| +50f | 7 | − | M |
| +56 | 4 | − | M |
| +62 | 2 | ++ | I |
| DAT | |||
| +68 | 3 | ++ | M |
| +74 | 1 | + | M |
| +80 | 8 | + | M |
| +86 | 3 | ++ | S/M |
| +92 | 2 | ++ | M |
| +98 | 5 | ++ | M |
| +104 | 7 | ++ | S |
| +110 | 11 | ++ | S |
| +116 | 1 | ++ | S |
| +122 | 1 | ++ | M |
| +128 | 6 | ++ | M |
| I-B | |||
| +134 | 1 | +/− | M |
| +140 | 1 | +/− | M |
| +146 | 1 | +/− | M |
| +152f | 10 | − | M |
| +158 | 6 | − | M |
| +164 | 3 | − | M |
| +170 | 2 | − | M |
| L1 | |||
| +176 | 1 | ++ | I |
| +182 | 3 | ++ | I |
| +188 | 0.4 | ++ | I |
| +194 | 1 | ++ | I |
| +200 | 1 | ++ | I |
| +206 | 3 | ++ | I |
| +212 | 0.4 | ++ | I |
| +218 | 0.3 | ++ | I |
| +224 | 1 | ++ | I |
| +230 | 0.4 | ++ | I |
| +236 | 3 | ++ | I |
| +242 | 1 | ++ | I |
| +248 | 1 | ++ | I |
| +254 | 1 | ++ | I |
| +266 | 1 | ++ | I |
| +272 | 2 | ++ | I |
| +278 | 2 | ++ | I |
| +284 | 5 | ++ | I |
| +290 | 3 | ++ | I |
| +296 | 5 | ++ | I |
| +302 | 5 | ++ | I |
| +308 | 4 | ++ | I |
| +314 | 5 | ++ | I |
| +320 | 4 | ++ | I |
| +326 | 6 | + | I |
| +332 | 5 | + | I |
| +338 | 4 | ++ | I |
| +344 | 5 | ++ | I |
| II | |||
| +350 | 8 | +/− | S |
| +356 | 3 | − | M |
| +362 | 8 | +/− | S |
| +368 | 3 | ++ | I |
| +374 | 4 | ++ | I |
| +380 | 6 | +/− | M |
| +386e | 9 | − | M |
| +392 | 6 | − | M |
| +398 | 20 | + | S |
| +404 | 6 | − | M |
| L2 | |||
| +410 | 7 | ++ | I |
| +416 | 4 | ++ | I |
| +422 | 5 | ++ | I |
| +428 | 2 | ++ | I |
| +434 | 6 | ++ | I |
| +440 | 3 | ++ | I |
| III | |||
| +446 | 5 | +/− | M |
| +452 | 3 | − | M |
| +458 | 3 | +/− | M |
| +464 | 6 | − | M |
| +470 | 1 | − | M |
| +476 | 1 | − | M |
| +482 | 1 | − | M |
| +488 | 2 | +/− | M |
| +494 | 2 | − | M |
| +500 | 3 | ++ | I |
| +506 | 4 | +/− | M |
| +512e | 2 | − | M |
| +518 | 5 | +/− | M |
| +524 | 4 | ++ | I |
| +530 | 4 | − | M |
| +536 | 4 | − | M |
| +542 | 4 | − | M |
Each mutant is named after the starting position of the substitution in the native hTERT peptide sequence (i.e., +2 is 2PRAPRC to 2NAAIRS). Essential domains (I-A, I-B, II, and III), biologically essential domain (DAT), and nonessential linker regions (L1 and L2) are shown left of columns for mutant positions.
Fold overexpression of hTERT transcripts compared to vector control after samples were normalized for RNA content with the transcript level of the housekeeping gene PBGD.
In vitro telomerase activity for each mutant was determined by normalizing the activity to the internal standard and then expressed as a percentage of wild-type FLAG-hTERT activity as follows: ++ (>60%), + (60 to 15%), +/− (<15%), and − (extremely low or no detectable activity). At least two separate lysates were tested for each mutant.
Polyclonal HA5 cells overexpressing FLAG-hTERT mutants were serially passaged to determine the life span. Lifespan was defined by similarity to growth of vector (negative control) and wild-type FLAG-hTERT (positive control) cell lines. Mutants that grew like vector eventually underwent crisis and are termed M (mortal), mutants that continually divide at a rate similar to that of the wild-type FLAG-hTERT are termed I (immortal), and mutants that continually divide at a slower rate compared to that of the wild type are termed S (slow growth).
Mutants shown to have reduced hTR binding.
Mutants shown to have wild-type hTR binding.
Having confirmed that hTERT mutants were equivalently overexpressed, we next characterized each mutant cell line for in vitro telomerase activity and extended life span as a measure of in vivo telomerase function. As described in detail below, mutant hTERT proteins gave rise to four distinct phenotypes: nonessential (catalytically and biologically active), essential (catalytically and biologically inactive), slow growth (catalytically active, biologically impaired), and biologically essential (catalytically active, biologically dead). Compilation of the different phenotypes with the respective mutation position revealed clustering along the primary amino acid sequence (Table 1; see also Fig. 3), implying distinct domains within the N terminus. Specifically, we defined four domains (I-A, I-B, II, and III) that are essential for catalytic activity, two nonessential or linker regions (L1 and L2), and one biologically essential domain (Fig. 3). As discussed below, based on the ability of mutations within the biologically essential domain to separate in vivo and in vitro telomerase function, we have named this last domain the “dissociates activities of telomerase” (DAT) domain.
FIG. 3.

Domain structure of the N terminus of hTERT. Secondary structure of the N terminus of hTERT as predicted by the Jprep2 program (http://jura.ebi.ac.uk:8888/) is shown, cylinders represent α-helices or β-sheets. Essential domains I-A, I-B, II, and III, as well as the T-motif, are denoted above structure prediction. Shaded regions denote the DAT domain; L1 and L2 define the nonessential linker regions. Two structured regions, outside of defined domains, are indicated by dashed lines. Essential domains I, II, and III found in the N terminus of Est2p (19) or conserved regions GQ, CP, and QFP identified in TERT proteins by alignment (63) are shown below structure prediction.
Linker regions are dispensable for telomerase activity.
A total of 39 separate hTERT mutants were found to be phenotypically similar to wild-type hTERT, when expressed in HA5 cells. Lysates from the HA5 cells expressing these mutants contained high to moderate levels of catalytic activity, as measured by the ability of these extracts to elongate a single-stranded oligonucleotide with telomeric repeats (Fig. 1B and Table 1). It is formally possible that this activity was due to spurious activation of the endogenous hTERT gene, which would not be distinguished with the hTERT primers used to confirm FLAG-hTERT expression (Fig. 1A). To rule out this possibility, mRNA was isolated from representative cell lines containing NAAIRS substitutions in each of the nonessential regions at early passage, as well as at very late passage (a point after crisis of vector control cells), when the endogenous gene would be expected to be activated. This RNA was then RT-PCR amplified with primers specific for either endogenous or ectopic hTERT mRNA. Endogenous hTERT was found at neither early nor late (postcrisis) passage, despite clear expression of the ectopic TERT and a control housekeeping gene (Fig. 2), indicating that the observed telomerase activity in these mutants was a direct result of ectopic expression of the hTERT mutants.
FIG. 2.

Absence of endogenous hTERT expression in telomerase-positive HA5 cell lines. Total RNA collected from HA5 cells at either early passage (pd 2 to 3) (A) or at late passage (pd >39) (B) was analyzed by RT-PCR with primers specific for either endogenous or ectopic hTERT or GAPDH (control for RNA content). Results with nonessential (+212, +326, +422, and +524), slow-growth (+14, +110, and +398), and biologically essential (+128) mutants are shown. CWR and LNCaP are prostate cancer cell lines expressing endogenous hTERT and serve as positive controls for endogenous and a negative control for ectopic hTERT expression. A water sample is used to control for contaminating DNA in the reaction mix.
Consistent with the high levels of activity, HA5 cell lines stably expressing each of these 39 mutants were, like wild-type hTERT-infected cells, able to bypass crisis and continued to proliferate in culture (Fig. 1C and Table 1). Additionally, cell lines expressing representative mutants had larger telomeres (∼6 kbp) compared to vector control HA5 cells (∼4.5 kbp) at early passage (Fig. 1D). The two regions mapped by these mutants may serve as linkers, since these regions are, by all known biological criteria, nonessential for telomerase function and have little predicted secondary structure (Fig. 3).
N-terminal essential domains.
Cell lysates from 34 independent hTERT mutants had extremely low or no detectable catalytic activity (Fig. 1B and Table 1). Like vector-infected cells, HA5 cells stably expressing these hTERT mutants lost telomeric DNA (Fig. 1D) and succumbed to crisis after undergoing a limited number of cell divisions (Fig. 1C and Table 1). Thus, loss of enzyme activity rendered these mutants biologically inactive, and hence these mutants define regions of hTERT that are essential. Mapping these essential mutants to hTERT sequence revealed a clustering in four regions (Fig. 3), which align with essential domains I, II, and III defined by mutational analysis in yeast (19), or homology blocks GQ, CP, and QFP (63). We note that, in humans, domain I is actually separated into two halves, which we term I-A and I-B, by a novel domain dispensable for telomerase in vitro enzyme activity (see below).
Since hTERT and hTR reconstitute a fully active enzyme in vitro, we reasoned that the absence of activity in essential domain mutants could be due to protein instability or loss in hTR interaction. To address the first possibility, we transiently overexpressed NAAIRS mutants from the four different essential domains in 293T cells to determine whether these mutants were produced at levels comparable to the wild type. Western blots indicated that there were no substantial differences in protein levels between the wild type and essential hTERT mutants (Fig. 4A) or noticeable degradation products (data not shown). Based on these findings, we propose that poor protein expression is not a major factor for reduction in catalytic activity of essential domain hTERT mutants. However, since hTERT protein is ectopically expressed at far higher levels transiently in 293T cells compared to stably in HA5 cells, we cannot rule out the possibility that a slight reduction in protein levels, not detected in 293T cells, could have an impact on telomerase activity when expressed in HA5 cells. The absence of telomerase activity in the described cell lines could also be argued to be due to low hTR levels. We discount this possibility since both telomerase-positive and -negative cell lines were derived from the same cells and because we assayed for telomerase activity in polyclonal populations, which are unlikely to have uniformly lower hTR expression in telomerase-negative cells compared to the similarly derived telomerase-positive cells.
FIG. 4.

Protein stability and hTR binding of mutants within essential domains of hTERT. (A) Lysates from 293T cells transiently transfected with FLAG-hTERT-FLAG, wild type, or the indicated NAAIRS mutants were resolved by SDS-PAGE and examined by anti-FLAG Western blotting. An anti-actin Western blot was used to ensure equal protein loading. (B) Binding of hTR with hTERT was examined in vitro by coimmunoprecipitating 35S-labeled FLAG-hTERT-FLAG (F-hTERT-F) with purified 32P-labeled hTR by using anti-FLAG antibodies. Immunoprecipitates were separated by SDS-PAGE and exposed to autoradiograph. Input hTR was diluted 1/1,000 and hTERT 1/10 for visualization. (C) Binding of hTR with FLAG-hTERT-FLAG protein containing NAAIRS substitutions (+50, +152, +386, and +512) in essential domains I-A, I-B, II, and III, respectively, was similarly examined. As a control for nonspecific interactions, HDAC1-FLAG (HDAC1-F) was immunoprecipitated in the presence of labeled hTR. The positions of F-hTERT-F, HDAC1-F, and hTR are indicated left of gel.
Since a large deletion of the first 350 amino acids of hTERT abolishes both hTR binding and telomerase activity in vitro (7), we next addressed whether N-terminal essential domains are involved in hTR-binding. 32P-radiolabeled hTR was incubated with 35S-labeled double FLAG epitope-tagged hTERT generated in vitro and immunoprecipitated with an anti-FLAG antibody to assess the hTR association (Fig. 4B). In this in vitro system, hTERT specifically interacted with hTR. The FLAG-tagged hTERT protein, but not the irrelevant FLAG-tagged protein HDAC1, coimmunoprecipitated hTR, despite the fact that HDAC1 was readily immunoprecipitated with the same antibody. Similarly, hTERT containing representative NAAIRS substitution in essential domains I-A and I-B (+50 and +152, respectively) also interacted with hTR, although these mutants are telomerase negative. However, immunoprecipitates of hTERT containing representative NAAIRS mutants within essential domains II and III (+386 and +512, respectively), which are essential for telomerase activity, showed a clearly visible two- to fourfold reduction in hTR binding. Since these mutants were expressed at levels equivalent to that of wild-type hTERT, we propose that domains II and III are critical for stable interaction between hTERT and hTR and that disruption of this interaction resulted in loss of enzyme activity.
hTERT mutants that only partially restore telomerase function.
HA5 cells expressing nine different hTERT mutants were found to have impaired growth dynamics but nevertheless a greatly extended life span (Fig. 1C and Table 1). All but two (mutants +350 and +362) of these slow-growth mutant cell lines contained comparable levels of in vitro catalytic activity to cells infected with wild type or with nonessential mutants that had a similarly extended, if not immortal, life span (Fig. 1B and Table 1). Representative HA5 cell lines expressing these mutants did not have detectable levels of endogenous hTERT at early or late passage, indicating that enzyme activity was not due to activation of the endogenous hTERT gene (Fig. 2). Each of these mutant proteins could also be transiently expressed in 293T cells at levels equivalent to wild-type hTERT with no apparent proteolysis (Fig. 5A and data not shown). Although we cannot rule out that the proteins would behave identically when expressed in HA5 cells, the fact that most of these mutant are also highly telomerase positive argues against a loss of protein expression underlying the phenotypes of these mutants. Lastly, this slow-growth phenotype was reproducible. We infected HA5 cells again with three randomly chosen hTERT mutants that gave rise to the slow-growth phenotype (+32, +86, and +116); all had slow-growth, of which two continued to proliferate beyond crisis of vector control cells (not shown). Taken together, these data indicate that the slow-growth phenotype is directly related to the mutations in hTERT.
FIG. 5.

Expression and cell viability of slow-growth hTERT mutants. (A) Anti-FLAG Western blot of slow-growth (+14 and +110) and nonessential (+212 and +422) hTERT mutants transiently expressed in 293T cells. Equal loading is shown by the anti-actin blot. (B) Viability of slow-growth and immortal HA5 cells was determined by flow cytometry of annexin V and propidium iodide double-stained cells. The percentages shown are averages for three independent experiments.
Slow-growth mutants lost substantial amounts of telomeric DNA at early passage; closely resembling telomere sizes found in telomerase-negative cells rather than in wild-type- or nonessential hTERT mutant-infected cell lines (Fig. 1D). The presence of shortened telomeres at early passage suggests the possibility that these cell populations teeter on the edge of extinction, with only a small fraction of cells having functional telomeres at any one time. One prediction of such a model is that the slow growth would result from increased cell death in the population. To test this prediction, we double stained late-passage HA5 cell cultures stably expressing three independent slow-growth mutants or, as controls, wild-type or nonessential mutants of hTERT, with annexin V, a marker of early apoptosis, and propidium iodide, an indicator of late-stage apoptosis (Fig. 5B). Two slow-growth mutants (+14 and +110) showed a >3-fold increase in the amount of double-stained, late-stage apoptotic cells, while the remaining mutant cell line (+398) exhibited higher levels of annexin V staining compared to normal immortalized HA5 cells. In every case, HA5 cells infected with slow-growth mutants had a lower amount of unstained, nonapoptotic cells than those infected with wild type or with nonessential hTERT mutants. Therefore, the apparent slow growth found in HA5 cells infected with the described hTERT mutants was a result of reduced viability of cells within the population.
The novel DAT domain is essential for in vivo telomerase function.
A region of hTERT comprised of a series of eight NAAIRS substitution mutants were discovered to be dispensable for in vitro enzyme catalysis but essential for biological activity. Lysates from HA5 cells containing these mutants had high to moderate levels of in vitro telomerase activity (Fig. 1B and Table 1) and yet lost significant amounts of telomeric DNA (Fig. 1D) and were mortal (Fig. 1C and Table 1). Molecular characterization revealed that all of these mutants were overexpressed in HA5 cells (Fig. 1A) and representative mutants generated stable protein, at least when transiently expressed in 293T cells (Fig. 6A). Although it is formally possible that the stability of the same mutants may be much lower in HA5 cells, the fact that DAT domain mutations bestow high levels of telomerase activity in HA5 cells argues against this possibility. The biologically essential phenotype was also reproducible, since HA5 cells reinfected with three independent DAT domain mutants (+68, +92, and +128) demonstrated the same phenotype (not shown). Thus, mutations in the DAT domain disrupt functions distinct from those we have so far characterized by NAAIRS substitution analysis.
FIG. 6.
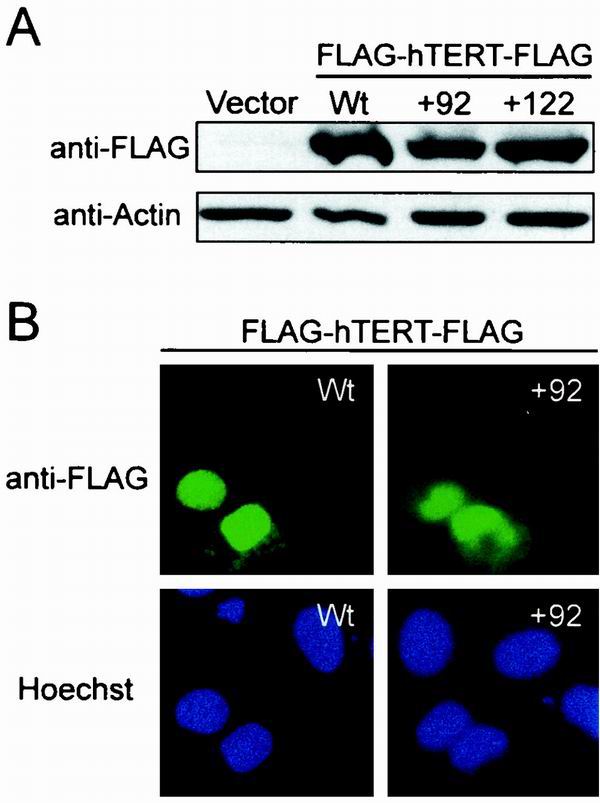
Protein stability and nuclear localization of hTERT with mutations in the DAT domain. (A) Anti-FLAG Western blot of lysates from 293T cells transiently transfected with biologically essential hTERT mutants +92, +122, wild-type hTERT, or control vector. The anti-actin blot shows equal protein loading. (B) Subcellular localization of DAT domain mutants transiently expressed in U2OS cells by indirect immunofluorescence. Localization of FLAG-hTERT-FLAG was visualized with an anti-FLAG antibody recognized by a fluorescein isothiocyanate-conjugated secondary antibody (green). Hoechst was used to stain nuclei (blue).
In vitro telomerase activity can be detected in lysates derived from human and yeast cells regardless of cell cycle progression (15, 30). However, telomeres are elongated in a cell cycle-dependent fashion in Saccharomyces cerevisiae (15), implying a regulation of the biological activity of telomerase. Indeed, binding of 14-3-3 proteins to hTERT has recently been reported to influence the subcellular localization of hTERT (56), raising the possibility that cytosolic-nuclear shuttling may be a regulatory mechanism for telomerase function in vivo. Loss of nuclear localization could leave a protein catalytically active but unable to reach its biological substrate. To therefore test whether the DAT domain is required for nuclear localization, an empty vector or one encoding double FLAG-tagged hTERT or representative DAT domain mutants NAAIRS +92 and +122 was transiently transfected into human U2OS cells and the resulting protein detected by indirect immunofluorescence with an anti-FLAG antibody. U2OS cells were chosen because they have a clearly defined nucleus and cytoplasm, which is ideal for monitoring nuclear localization. Wild-type hTERT was found predominantly in the nucleus of U2OS cells (Fig. 6B), although we did observe rare cells in which the signal was dispersed throughout the cell or localized to the cytosol (data not shown). Both DAT domain mutants displayed the same localization as the wild-type protein, being found predominantly in the nucleus, with some cells exhibiting cytosolic signals (Fig. 6B). We thus conclude that the biological dysfunction of the DAT domain mutants cannot be attributed to a failure in nuclear localization.
Finally, based on mounting evidence that hTERT may form homomeric complexes (7, 60), we investigated whether the defects in the DAT or essential domain I-A, I-B, II, and III mutants could be explained by the inability of these mutants to form higher-order complexes. We found that hTERT can indeed form homomeric complexes in vitro when expressed in rabbit reticulocyte lysates. This was determined by the ability to coimmunoprecipitate GST-hTERT and FLAG-hTERT-FLAG by using either an anti-FLAG antibody or an anti-GST antibody. This reaction could occur in the absence or presence of DNA substrate or the hTR subunit, indicating that multimerization is independent of these two parameters (Fig. 7A). The specificity of this interaction was demonstrated by the lack of an association of GST-hTERT with immunoprecipitated HDAC1-FLAG protein, as well as the inability of the FLAG-hTERT-FLAG protein to bind to immunoprecipitated GST (Fig. 7). We next tested whether mutations to any of the essential or DAT domains affected this interaction. GST- and FLAG-tagged hTERT containing representative mutants in the described domains were incubated and immunoprecipitated with an anti-FLAG antibody. In each case, both the GST- and FLAG-tagged protein were coimmunoprecipitated, arguing that the mutations did not affect multimerization, when expressed in rabbit reticulocyte lysates (Fig. 7B). Based on these in vitro experiments, the catalytic and biological defects of these mutants do not appear to be related to an impaired ability to form multimers, although we cannot exclude the possibility that the mutants may not multimerize in vivo.
FIG. 7.

Homomeric complex formation of essential and DAT domain hTERT mutants. (A) Immunoprecipitation of 35S-labeled FLAG-hTERT-FLAG (F-hTERT-F) with either 35S-labeled GST-hTERT or GST in the presence or absence of hTR and Ts oligonucleotide substrate with anti-GST or anti-FLAG antibodies as indicated. (B) 35S-labeled GST-hTERT and F-hTERT-F, wild-type, or NAAIRS substitution in domains I-A, DAT, I-B, II, and III (+50, +92, +152, +386, and +512 mutants, respectively) were incubated together and immunoprecipitated with an anti-FLAG antibody to monitor protein association. As a control, an irrelevant FLAG-tagged protein (HDAC1-F) failed to coimmunoprecipitate GST-hTERT.
DISCUSSION
Functional conservation of N-terminal domains in evolutionarily diverse TERT proteins.
We stably expressed a panel of tandem NAAIRS substitution mutants in telomerase-negative cells and screened for telomerase activity to identify functional domains in the N terminus of hTERT. NAAIRS substitution allows a large region of hTERT to be mutated with a reasonable number of changes, without altering protein length or causing large changes to secondary structure. The use of a cell-based screen allows mutants to be produced in a cellular environment containing factors that may be absent in vitro and allows each mutant to be simultaneously characterized for both in vitro and in vivo activity. The screen revealed four phenotypes for N-terminal mutants: essential (catalytically and biologically inactive), nonessential (catalytically and biologically active), biologically essential (catalytically active, but biologically dead), or slow growth (catalytically active, but biologically impaired). Mutants that give rise to the essential phenotype reside in one of four domains (I-A, I-B, II, and III) which were usually preceded by linker regions in which NAAIRS substitutions had minimal effects on the in vitro and in vivo functions of hTERT. We find that all four domains correspond to the yeast essential domains I, II, and III (19), as well as the similar domains GQ, CP, and QFP (63), defined by alignments of TERT proteins from evolutionarily diverse organisms (Fig. 3).
The domains that we defined as being essential in humans also appear to be conserved at the functional level with those of lower eukaryotes. Domain I in yeast (63) and humans (Fig. 3) is essential for long-term viability. Specific mutations in this region in these organisms, or in the ciliate Tetrahymena (36, 42), have little effect on RNA binding but result in a partial or complete loss of telomerase activity. In humans, the region defined as domain I is divided by the DAT domain and hence was termed domains I-A and I-B. Intriguingly, N-terminal hTERT mutants lacking the first 200 amino acids (including essential domains I-A and I-B and the DAT domain) are nonprocessive in vitro (7), whereas specific NAAIRS substitutions in the same region abrogate catalytic activity (Fig. 1B and Table 1). Perhaps the large deletion removes a portion of hTERT, which functions in an inhibitory manner when mutated by NAAIRS substitution. Nevertheless, both types of analysis define domains I-A and I-B as critical for proper enzyme function, as observed in lower eukaryotes.
Mutations in domains II and III were found to abolish telomerase activity and hTR binding and failed to rescue transformed human cells from crisis. In yeast, these domains are required for enzyme activity, telomere elongation, proliferation and, in the case of two mutants in domain III, RNA binding (19). Similarly, large fragments of TERT minimally encompassing these domains and the conserved T-motif can bind the RNA subunit in humans and Tetrahymena (7, 36). Like domain I, domains II and III appear to be functionally conserved.
We also found that, in addition to the sequence and functional conservation of the N-terminal domains of TERT, the most structured regions of this portion of hTERT mapped to the biologically defined domains of the protein, delineating these regions as important structural domains (Fig. 3). Therefore, with the exception of motifs unique to ciliates (9, 11, 42), the organization and function of the N-terminal domains appears to have remained intact throughout evolution. Thus, despite low sequence homology, the N terminus of TERT proteins do contain evolutionarily conserved functional domains.
The DAT domain in cellular regulation of telomerase.
We identified a region termed the DAT domain defined by 11 contiguous mutants in which in vitro telomerase catalytic activity was dissociated from the ability of telomerase to function efficiently in vivo. Cells expressing DAT domain mutants either entered a period analogous to crisis observed in vector control cells or displayed less dramatic cell death, possibly representing a partial crisis. The latter phenotype, which we termed slow growth, was also found in cells expressing hTERT containing NAAIRS mutations in domain I-A (three-quarters of the slow-growth mutants mapped to either the I-A or the DAT domain). In one case, we found that a mutation in the DAT domain (mutant +86) could give rise to either a slow-growth or a mortal phenotype. Domains I-A, DAT, and I-B also form one continuous structured region that appears to have no obvious role in hTR binding or multimerization. Taken together, we speculate that the function of domains I-A and I-B may therefore be related to that of the DAT domain but that mutations to these two domains are more intrusive to biochemical activity.
Mutations in the DAT domain caused cell death, which was accompanied by a large decrease in telomere length, clearly demonstrating the loss of a novel in vivo telomerase function that is dispensable for biochemical enzyme catalysis in vitro. One aspect of telomere elongation that would not be represented in an in vitro assay for catalytic activity is posttranscriptional regulation of telomerase (15, 29, 39, 56). Recently, mutants have been isolated that affect hTERT entry into the nucleus, suggesting that cellular localization may be involved in coordinating hTERT-mediated telomere elongation (56). Although the DAT domain does not appear to contain any obvious nuclear localization sequence (NLS), nuclear localization could be mediated through a noncanonical NLS or a binding partner. However, we ruled out this possibility, since there were no noticeable defects in cellular localization of hTERT containing mutations in the DAT domain.
Although in vitro assayed telomerase activity purified from Euplotes consists of a single catalytic subunit (38), experiments from both yeast and human systems support the notion that the enzyme may function biologically in a complex containing more than one TERT and RNA subunit (7, 51, 52, 60). Disruption of this interaction could underlie the defect we observed in the DAT domain mutants. However, we find biochemically hTERT forms a homomeric complex in vitro, irrespective of mutations to the DAT domain. Thus, the biologically essential phenotype of the DAT domain cannot be ascribed to a failure of hTERT to multimerize.
Since mutations in the DAT domain neither grossly altered hTERT localization patterns nor homomultimerization, we speculate that this domain could instead be involved in recruitment of telomerase to telomeres. We note that mutations mapping to the corresponding DAT domain region in yeast Est2p had a similar biologically essential phenotype and that the proteins Est1p, Est3p, Cdc13p, and Ku are necessary for biological telomerase function and have been implicated in recruiting telomerase to telomeres in yeast (17, 22, 31, 50, 53). This raises the possibility that the hTERT DAT domain may interact with orthologs of these proteins. Alternatively, the DAT domain may participate in the coordination of 3′G-rich single-strand elongation by telomerase and lagging-strand synthesis of the C-rich strand (1, 15, 53).
Functional domains of hTERT.
The ability of hTERT to elongate telomeres undoubtedly requires complex and precise regulation involving nuclear import, substrate recognition, and coordinated synthesis of the C strand. Since it is not feasible to reconstitute this complex process in vitro, we employed intact human cells to scan hTERT for regions that will further our understanding of these important biologically defined functions. The identification of the biologically essential DAT domain has clearly demonstrated the utility of this approach and represents a definitive step in elucidating the regulation of telomerase function in vivo. Lastly, since the inhibition of telomerase has been shown to prevent cancer cell lines from forming tumors in vivo, all of the essential domains that we identified in hTERT may represent suitable pharmacological targets for the treatment of human cancers.
ACKNOWLEDGMENTS
We thank members of the Counter, Wang, Pendergast, and Yao laboratories for help and advice, Tso-Pang Yao for plasmid pCMV-HDAC1-FLAG, and Sally Kornbluth for critical review of the manuscript. We thank L. A. Cleveland for technical assistance.
This work was supported by grants from the National Institute of Health, administered through the National Cancer Institute (CA82481-01), and the V-Foundation. C.M.C. is a Kimmel Scholar, and B.N.A. and S.S.R.B. are supported by Department of Defense Breast Cancer Research Predoctoral Fellowships.
REFERENCES
- 1.Adams-Martin A, Dionne I, Wellinger R J, Holm C. The function of DNA polymerase alpha at telomeric G tails is important for telomere homeostasis. Mol Cell Biol. 2000;20:786–796. doi: 10.1128/mcb.20.3.786-796.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avilion A A, Piatyszek M A, Gupta J, Shay J W, Bacchetti S, Greider C W. Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res. 1996;56:645–650. [PubMed] [Google Scholar]
- 3.Bacchetti S, Counter C M. Telomeres and telomerase in human cancer. Int J Oncol. 1995;7:423–432. [PubMed] [Google Scholar]
- 4.Bachand F, Autexier C. Functional reconstitution of human telomerase expressed in Saccharomyces cerevisiae. J Biol Chem. 1999;274:38027–38031. doi: 10.1074/jbc.274.53.38027. [DOI] [PubMed] [Google Scholar]
- 5.Bachand F, Autexier C. Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA-protein interactions. Mol Cell Biol. 2001;21:1888–1897. doi: 10.1128/MCB.21.5.1888-1897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachand F, Kukolj G, Autexier C. Expression of hTERT and hTR in cis reconstitutes and active human telomerase ribonucleoprotein. RNA. 2000;6:778–784. doi: 10.1017/s1355838200000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beattie T L, Zhou W, Robinson M O, Harrington L. Polymerization defects within human telomerase are distinct from telomerase RNA and TEP1 binding. Mol Biol Cell. 2000;11:3329–3340. doi: 10.1091/mbc.11.10.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackburn E H, Greider C W. Telomeres. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1995. [Google Scholar]
- 9.Bryan T M, Goodrich K J, Cech T R. Telomerase RNA bound by protein motifs specific to telomerase reverse transcriptase. Mol Cell. 2000;6:493–499. doi: 10.1016/s1097-2765(00)00048-4. [DOI] [PubMed] [Google Scholar]
- 10.Bryan T M, Marusic L, Bacchetti S, Namba M, Reddel R R. The telomere lengthening mechanism in telomerase-negative immortal human cells does not involve the telomerase RNA subunit. Hum Mol Genet. 1997;6:921–926. doi: 10.1093/hmg/6.6.921. [DOI] [PubMed] [Google Scholar]
- 11.Bryan T M, Sperger J M, Chapman K B, Cech T R. Telomerase reverse transcriptase genes identified in Tetrahymena thermophila and Oxytricha trifallax. Proc Natl Acad Sci USA. 1998;95:8479–8484. doi: 10.1073/pnas.95.15.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Counter C M, Avilion A A, Le Feuvre C E, Stewart N G, Greider C W, Harley C B, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Counter C M, Hahn W C, Wei W, Caddle S D, Beijersbergen R L, Lansdorp P M, Sedivy J M, Weinberg R A. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA. 1998;95:14723–14728. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Counter C M, Hirte H W, Bacchetti S, Harley C B. Telomerase activity in human ovarian carcinoma. Proc Natl Acad Sci USA. 1994;91:2900–2904. doi: 10.1073/pnas.91.8.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diede S J, Gottschling D E. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell. 1999;99:723–733. doi: 10.1016/s0092-8674(00)81670-0. [DOI] [PubMed] [Google Scholar]
- 16.Elenbaas B, Spirio L, Koerner F, Fleming M D, Zimonjic D B, Donaher J L, Popescu N C, Hahn W C, Weinberg R A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans S K, Lundblad V. Est1 and Cdc13 as comediators of telomerase access. Science. 1999;286:117–120. doi: 10.1126/science.286.5437.117. [DOI] [PubMed] [Google Scholar]
- 18.Feng J, Funk W D, Wang S S, Weinrich S L, Avilion A A, Chiu C P, Adams R R, Chang E, Allsopp R C, Yu J, Le S, West M D, Harley C B, Andrews W H, Greider C W, Villeponteau B. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 19.Friedman K L, Cech T R. Essential functions of amino-terminal domains in the yeast telomerase catalytic subunit revealed by selection for viable mutants. Genes Dev. 1999;13:2863–2874. doi: 10.1101/gad.13.21.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gou, C., D. Geverd, R. Liao, N. Hamad, C. M. Counter, and D. T. Price. Inhibition of telomerase is related to the lifespan and tumorigenicity of human prostate cancer cells. J. Urol. 166:694–698. [PubMed]
- 21.Graham F L, van der Eb A J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 22.Grandin N, Damon C, Charbonneau M. Cdc13 cooperates with the yeast Ku proteins and stn1 To regulate telomerase recruitment. Mol Cell Biol. 2000;20:8397–8408. doi: 10.1128/mcb.20.22.8397-8408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith J D, Comeau L, Rosenfield S, Stansel R M, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 24.Hahn W C, Counter C M, Lundberg A S, Beijersbergen R L, Brooks M W, Weinberg R A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 25.Hahn W C, Stewart S A, Brooks M W, York S G, Eaton E, Kurachi A, Beijersbergen R L, Knoll J H, Meyerson M, Weinberg R A. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 26.Harley C B, Futcher A B, Greider C W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 27.Harrington L, Zhou W, McPhail T, Oulton R, Yeung D S, Mar V, Bass M B, Robinson M O. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997;11:3109–3115. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hastie N D, Dempster M, Dunlop M G, Thompson A M, Green D K, Allshire R C. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 29.Holt S E, Wright W E, Shay J W. Multiple pathways for the regulation of telomerase activity. Eur J Cancer. 1997;33:761–766. doi: 10.1016/S0959-8049(97)00066-X. [DOI] [PubMed] [Google Scholar]
- 30.Holt S E, Wright W E, Shay J W. Regulation of telomerase activity in immortal cell lines. Mol Cell Biol. 1996;16:2932–2939. doi: 10.1128/mcb.16.6.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes T R, Evans S K, Weilbaecher R G, Lundblad V. The Est3 protein is a subunit of yeast telomerase. Curr Biol. 2000;10:809–812. doi: 10.1016/s0960-9822(00)00562-5. [DOI] [PubMed] [Google Scholar]
- 32.Kilian A, Bowtell D D L, Abud H E, Hime G R, Venter D J, Keese P K, Duncan E L, Reddel R R, Jefferson R A. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997;6:2011–2019. doi: 10.1093/hmg/6.12.2011. [DOI] [PubMed] [Google Scholar]
- 33.Kim N W, Piatyszek M A, Prowse K R, Harley C B, West M D, Ho P L, Coviello G M, Wright W E, Weinrich S L, Shay J W. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 34.Kim N W, Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP) Nucleic Acids Res. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolquist K A, Ellisen L W, Counter C M, Meyerson M, Tan L K, Weinberg R A, Haber D A, Gerald W L. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat Genet. 1998;19:182–186. doi: 10.1038/554. [DOI] [PubMed] [Google Scholar]
- 36.Lai C K, Mitchell J R, Collins K. RNA binding domain of telomerase reverse transcriptase. Mol Cell Biol. 2001;21:990–1000. doi: 10.1128/MCB.21.4.990-1000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lal A, Lash A E, Altschul S F, Velculescu V, Zhang L, McLendon R E, Marra M A, Prange C, Morin P J, Polyak K, Papadopoulos N, Vogelstein B, Kinzler K W, Strausberg R L, Riggins G J. A public database for gene expression in human cancers. Cancer Res. 1999;59:5403–5407. [PubMed] [Google Scholar]
- 38.Lingner J, Hughes T R, Shevchenko A, Mann M, Lundblad V, Cech T R. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- 39.Liu K, Hodes R J, Weng N. Cutting edge: telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphorylation and nuclear translocation. J Immunol. 2001;166:4826–4830. doi: 10.4049/jimmunol.166.8.4826. [DOI] [PubMed] [Google Scholar]
- 40.Masutomi K, Kaneko S, Hayashi N, Yamashita T, Shirota Y, Kobayashi K, Murakami S. Telomerase activity reconstituted in vitro with purified human telomerase reverse transcriptase and human telomerase RNA component. J Biol Chem. 2000;275:22568–22573. doi: 10.1074/jbc.M000622200. [DOI] [PubMed] [Google Scholar]
- 41.Meyerson M, Counter C M, Eaton E N, Ellisen L W, Steiner P, Caddle S D, Ziaugra L, Beijersbergen R L, Davidoff M J, Liu Q, Bacchetti S, Haber D A, Weinberg R A. hEST2, the putative human telomerase catalytic subunit gene, is upregulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 42.Miller M C, Liu J K, Collins K. Template definition by Tetrahymena telomerase reverse transcriptase. EMBO J. 2000;19:4412–4422. doi: 10.1093/emboj/19.16.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell J R, Collins K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol Cell. 2000;6:361–371. doi: 10.1016/s1097-2765(00)00036-8. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell J R, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 45.Morgenstern J P, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990;18:1068. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura T M, Cech T R. Reversing time: origin of telomerase. Cell. 1998;92:587–590. doi: 10.1016/s0092-8674(00)81123-x. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura T M, Morin G B, Chapman K B, Weinrich S L, Andrews W H, Lingner J, Harley C B, Cech T R. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 48.Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H, Nakanishi T, Ide T, Ishikawa F. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet. 1998;18:65–68. doi: 10.1038/ng0198-65. [DOI] [PubMed] [Google Scholar]
- 49.Ouellette M M, Aisner D L, Savre-Train I, Wright W E, Shay J W. Telomerase activity does not always imply telomere maintenance. Biochem Biophys Res Commun. 1999;254:795–803. doi: 10.1006/bbrc.1998.0114. [DOI] [PubMed] [Google Scholar]
- 50.Peterson S E, Stellwagen A E, Diede S J, Singer M S, Haimberger Z W, Johnson C O, Tzoneva M, Gottschling D E. The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat Genet. 2001;27:64–67. doi: 10.1038/83778. [DOI] [PubMed] [Google Scholar]
- 51.Prescott J, Blackburn E H. Telomerase RNA mutations in Saccharomyces cerevisiae alter telomerase action and reveal nonprocessivity in vivo and in vitro. Genes Dev. 1997;11:528–540. doi: 10.1101/gad.11.4.528. [DOI] [PubMed] [Google Scholar]
- 52.Prescott J, Blackburn E H. Functionally interacting telomerase RNAs in the yeast telomerase complex. Genes Dev. 1997;11:2790–2800. doi: 10.1101/gad.11.21.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qi H, Zakian V A. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev. 2000;14:1777–1788. [PMC free article] [PubMed] [Google Scholar]
- 54.Rich J N, Guo C, McLendon R E, Bigner D D, Wang X-F, Counter C M. A genetically tractable model of human glioma formation. Cancer Res. 2001;61:3556–3560. [PubMed] [Google Scholar]
- 55.Sedivy J M. Can ends justify the means?: telomeres and the mechanisms of replicative senescence and immortalization in mammalian cells. Proc Natl Acad Sci USA. 1998;95:9078–9081. doi: 10.1073/pnas.95.16.9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seimiya H, Sawada H, Muramatsu Y, Shimizu M, Ohko K, Yamane K, Tsuruo K. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000;19:2652–2661. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sellers W R, Novitch B G, Miyake S, Heith A, Otterson G A, Kaye F J, Lassar A B, Kaelin W G., Jr Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shay J W, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 59.Stewart N, Bacchetti S. Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology. 1991;180:49–57. doi: 10.1016/0042-6822(91)90008-y. [DOI] [PubMed] [Google Scholar]
- 60.Tesmer V M, Ford L P, Holt S E, Frank B C, Yi X, Aisner D L, Ouellette M, Shay J W, Wright W E. Two inactive fragments of the integral RNA cooperate to assemble active telomerase with the human protein catalytic subunit (hTERT) in vitro. Mol Cell Biol. 1999;19:6207–6216. doi: 10.1128/mcb.19.9.6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinrich S L, Pruzan R, Ma L, Ouellette M, Tesmer V M, Holt S E, Bodnar A G, Lichtsteiner S, Kim N W, Trager J B, Taylor R D, Carlos R, Andrews W H, Wright W E, Shay J W, Harley C B, Morin G B. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet. 1997;17:498–503. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]
- 62.Wilson I A, Haft D H, Getzoff E D, Tainer J A, Lerner R A, Brenner S. Identical short peptide sequences in unrelated proteins can have different conformations: a testing ground for theories of immune recognition. Proc Natl Acad Sci USA. 1985;82:5255–5259. doi: 10.1073/pnas.82.16.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xia J, Peng Y, Mian I S, Lue N F. Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol Cell Biol. 2000;20:5196–5207. doi: 10.1128/mcb.20.14.5196-5207.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Mar V, Zhou W, Harrington L, Robinson M O. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999;13:2388–2399. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou J, Hidaka K, Futcher B. The Est1 subunit of yeast telomerase binds the Tlc1 telomerase RNA. Mol Cell Biol. 2000;20:1947–1955. doi: 10.1128/mcb.20.6.1947-1955.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu J, Wang H, Bishop J M, Blackburn E H. Telomerase extends the lifespan of virus-transformed human cells without net telomere lengthening. Proc Natl Acad Sci USA. 1999;96:3723–3728. doi: 10.1073/pnas.96.7.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]