

Abstract
Specific mRNA degradation mediated by double-stranded RNA (dsRNA) interference (RNAi) is a powerful way of suppressing gene expression in plants, nematodes, and fungal, insect, and protozoan systems. However, only a few cases of RNAi have been reported in mammalian systems. Here, we investigated the feasibility of the RNAi strategy in several mammalian cells by using the enhanced green fluorescent protein gene as a target, either by in situ production of dsRNA from transient transfection of a plasmid harboring a 547-bp inverted repeat or by direct transfection of dsRNA made by in vitro transcription. Several mammalian cells including differentiated embryonic stem (ES) cells did not exhibit specific RNAi in transient transfection. This long dsRNA, however, was capable of inducing a sequence-specific RNAi for the episomal and chromosomal target gene in undifferentiated ES cells. dsRNA at 8.3 nM decreased the cognate gene expression up to 70%. However, RNAi activity was not permanent because it was more pronounced in early time points and diminished 5 days after transfection. Thus, undifferentiated ES cells may lack the interferon response, similar to mouse embryos and oocytes. Regardless of their apparent RNAi activity, however, cytoplasmic extracts from mammalian cells produced a small RNA of 21 to 22 nucleotides from the long dsRNA. Our results suggest that mammalian cells may possess RNAi activity but nonspecific activation of the interferon response by longer dsRNA may mask the specific RNAi. The findings offer an opportunity to use dsRNA for inhibition of gene expression in ES cells to study differentiation.
Frequently, inhibition of gene expression has been caused not only by antisense mRNA but also, in some cases, by expression of sense mRNA which has been used as a control. Moreover, this gene silencing by sense mRNA was shown to be sequence specific for the homologous gene. These initially confusing observations have now been attributed to gene silencing by the production of a minute amount of double-stranded RNA (dsRNA) generated by transcription of the sense mRNA by its promoter and the antisense mRNA by a cryptic promoter within the construct (reviewed in references 3, 6, 11, 26, 27, and 33). The term RNA interference (RNAi) was defined after the discovery that injection of dsRNA into the nematode Caenorhabditis elegans led to specific silencing of the gene homologous to the delivered RNA (12). RNAi was also observed in fruit flies, zebra fish, and other animals, including mice (7, 17, 30, 34, 35). The posttranscriptional gene silencing of C. elegans (18, 19, 28) is closely linked to the mechanism of cosuppression in plants and quelling in fungi (14, 21–23, 29).
Unlike other organisms, accumulation of very small amounts of dsRNA in mammalian cells results in the interferon response. This leads to an overall block of translation by inactivation of an elongation factor by protein kinase. In addition, dsRNA activates a latent 2′,5′-oligoadenylate synthase and increases synthesis of a 2′,5′-oligonucleotide, causing activation of RNase L and nonspecific mRNA degradation. These events result in the onset of apoptosis in mammalian cells (13, 16). The natural function of RNAi and cosuppression appears to be protection of the host genome against invasion by mobile genetic elements such as transposons and viruses, which produce dsRNA in host cells (14, 18, 20, 27). Such considerations have discouraged investigators from using RNAi in mammals. Recently, however, RNAi has been reported in several mammalian systems. Transfection of a plasmid carrying the full-length pro-α1(I) collagen gene into rodent fibroblasts decreased the endogenous pro-α1(I) collagen mRNA up to 90% (1). RNAi activity was also reported in CHO cells, although the amount of dsRNA required for interference was 2,500 times more than in Drosophila S2 cells (32). Sequence-specific RNAi has been demonstrated in the preimplantation mouse embryo and oocytes by direct injection of dsRNA (30, 35). When dsRNA corresponding to an active green fluorescent protein (GFP) gene was injected into mouse zygotes, dsRNAi was effective throughout the blastocyst stage and implantation until embryos reached 6.5 days of development, corresponding to a 40- to 50-fold increase in cell mass (35). With these findings, it becomes critical to determine whether RNAi can be applied in mammalian tissue culture for gene silencing.
The hallmark of RNAi is its specificity. The dsRNA triggers a specific degradation of homologous mRNA only within the region of identity with the dsRNA (37). The ability of a few molecules of dsRNA to eliminate a much larger pool of endogenous mRNA suggests a catalytic or amplification component to the RNAi mechanism. Results from studies of RNAi in plants suggested a mechanism, in which an RNA-primed RNA polymerase can spread gene silencing by dsRNA (28). Another model involves a catalytic RNA degradation generated by the dsRNA molecule and as yet unknown protein components. Recently, dsRNAs were shown to be processed to small 21- to 22-bp sizes in Drosophila embryo extracts (10, 36, 37), cultured S2 cells (2, 15), and C. elegans (24), making it likely that such RNAs serve as the specificity determinants in the RNAi reaction. These results suggest that dsRNA molecules are initially activated by a process that does not require interactions with their cognate mRNA target. Activation would appear to be a limiting step in RNAi, as the reaction is saturated at relatively low levels of dsRNA in vivo (12), potentiated by preincubation with dsRNA (31), and inhibited by excess unrelated dsRNAs (36).
Here, we investigated the feasibility of the RNAi strategy for gene silencing in several mammalian cell lines by using the enhanced GFP (EGFP) gene as a target. Our results show that undifferentiated mouse embryonic stem (ES) cells exhibit a sequence-specific RNAi at a dsRNA concentration similar to that needed in Drosophila S2 cells.We also compared the ability of different mammalian cell types to degrade dsRNA into small pieces of 21 to 22 bp, which is the initial step on RNAi activity.
MATERIALS AND METHODS
Plasmids.
With pEGFP-C1 (Clontech, Palo Alto, Calif.) as the template, a 547-bp fragment encoding a portion of the EGFP gene beginning from the ATG start codon was amplified by PCR with the primers 5′-GCC GTC GAC GGT ACC TCT AGA ACG CGT GCC ATG GTG AGC AAG GGC GAG GAG-3′ and 5′-GCC GCG GCC GCG GCC CTA TTA GCC CTC GAG TAC ATG GTC GGC GAG CTG CAC GCT-3′. A set of restriction sites, SalI, XbaI, and MluI at the 5′ end and EcoRI and NotI at the 3′ end, were incorporated in each primer. After PCR amplification, the 547-bp fragment was digested with NotI and self-ligated. The dimer of the 547-bp fragment was isolated from the agarose gel, purified, ligated to the pGEMT Easy vector (Promega, Madison, Wis.), and used to transform Escherichia coli DH5α (Gibco-BRL, Rockville, Md.) competent cells. The resulting colonies were screened for inverted repeats by restriction enzyme analysis. The sequence of the plasmid harboring an inverted repeat (pGEMT-dsEGFP) was confirmed by DNA sequencing.
To generate control dsRNA, a 629-bp fragment encoding a portion of the lacZ gene (nucleotides 1331 to 1960 from the AUG start codon) was amplified by PCR and ligated to the pGEMT Easy vector. The colonies were screened for plasmids containing the insert in the sense and antisense orientations. The plasmid pSC6-T7-Neo, encoding the T7 RNA polymerase gene under the control of the cytomegalovirus (CMV) promoter was a generous gift from M. Billeter (25). The pActin-lacZ and pIZ/US9-GFP plasmids, encoding the lacZ and EGFP genes under the control of the Drosophila promoters for actin and OpIE2, respectively, were generous gifts from Gregory Hannon. The pCMV-lacZ plasmid was purchased from Clontech.
In vitro transcription of dsRNA.
The pGEMT-dsEGFP construct with an inverted repeat containing a portion of the EGFP gene was linearized with PstI at a unique site located at the 3′ end of the inverted repeat. Using the RiboMax large-scale RNA production system-T7 (Promega, Madison, Wis.), the transcription reaction was performed at 37°C for 3 h, according to the manufacturer's specifications.
Radiolabeled dsRNA was generated by incorporation of [α-32P]UTP during in vitro transcription. After performing the in vitro transcription reaction, RNase-free DNase (Promega, Madison, Wis.) was added to the reaction mixture at 1 U/μg of the template DNA and incubated for 15 min at 37°C. The transcript was further purified by extraction in phenol-chloroform-isoamyl alcohol (25:24:1) and ethanol precipitation. The pellet was washed with 70% ethanol, dried at room temperature, and resuspended in TE buffer (10 mM Tris [pH 7.5] and 1 mM EDTA). To determine the folded structure of the dsRNA, an aliquot of the RNA sample was digested using a mixture of RNase A and T1 (Ambion, Austin, Tex.) at 37°C for 30 min and analyzed on 5% nondenaturing and denaturing polyacrylamide gels containing 40% formamide and 7 M urea. For dsRNA of the lacZ gene, plasmid containing either the sense or antisense lacZ fragment was linearized by restriction enzyme SalI, located downstream of the multiple cloning site. The sense and antisense RNAs were generated separately by in vitro transcription and annealed to generate a 740-bp dsRNA fragment.
Cell culture.
Drosophila S2 cells (generous gift from G. J. Hannon) were maintained at 27°C in 90% Schneider's insect medium (Gibco-BRL, Rockville, Md.) and 10% heat-inactivated fetal bovine serum (FBS). Cells were split every 2 to 3 days to maintain exponential growth. BsrT7/5 (4), a derivative of BHK-21 cells that express the T7 RNA polymerase (generous gift from M. Schnell), were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and penicillin-streptomycin. CHO-K1 cells were maintained in F-12 medium with 10% heat-inactivated FBS. Mouse embryonic stem (ES) cells AB2.2 (Stratagene, La Jolla, Calif.) were maintained in DMEM supplemented with 1,250 U of leukemia inhibitory factor (LIF) (Chemicon, Temecula, Calif.) per ml 15% FBS, 2 mM glutamine, 100 mM β-mercaptoethanol, and 1× nonessential amino acids. Mouse embryonic fibroblast STO cells (American Type Culture Collection, Rockville, Md.) were grown in DMEM with 10% FBS. The STO feeder cells were plated on dishes coated with 0.1% (wt/vol) gelatin, treated with mitomycin C (Sigma, St. Louis, Mo.) at a concentration of 10 μg/ml for 2.5 h at 37°C, and washed three times with phosphate-buffered saline (PBS). ES AB2.2 cells were plated onto mitomycin C-treated STO feeder cells and passaged every 2 days with a daily change of culture medium. For all experiments, ES cells were kept between 17 and 19 passages, counted from the time of isolation of ES cells from the inner cell mass of the blastocysts.
Transfection.
The day before transfection, S2 cells were plated in a 12-well plate (106 cells per well). Various amounts of the PstI-linearized pGEMT-dsEGFP plasmid or dsRNA generated by in vitro transcription were combined with 1 μg of the target plasmid encoding EGFP (pEGFP-C1) and 1 μg of the plasmid encoding the T7 RNA polymerase (pSC6-T7-Neo). In all transfection experiments, a constant amount of total DNA, 5 μg, was maintained by addition of the unrelated pUC19 plasmid. DNA was transfected to S2 cells by the calcium phosphate method. The plasmid encoding β-galactosidase, pCMV-lacZ, was used as a control. CHO-K1 and STO feeder cells were plated in a 12-well plate (105 cells per well) the day before transfection. Various amounts of the PstI-linearized pGEMT-dsEGFP plasmid or in vitro-transcribed dsRNA was combined with 1 μg of the target plasmid encoding EGFP (pEGFP-C1) and 1 μg of the plasmid encoding the T7 RNA polymerase (pSC6-T7-Neo). The DNA mixture was transfected to cells by addition of 7.5 μg of Lipofectamine (Gibco-BRL, Rockville, Md.). The same transfection protocol was used for BsrT7/5 cells except the pSC6-T7-Neo plasmid was not added, as BsrT7/5 already expresses the T7 RNA polymerase (4).
ES cells were grown on feeder STO cells, trypsinized for 5 min, and pipetted extensively to prevent clumping of cells. After addition of 5 volumes of ES medium, cells were put back in the incubator for 45 min. The majority of the STO cells adhere to the plate during this incubation, and ES cells were harvested from the suspension. Various amounts of the PstI-linearized pGEMT-dsEGFP plasmid or dsRNA were combined with 1 μg of the target plasmid encoding EGFP (pEGFP-C1) and 1 μg of the plasmid encoding the T7 RNA polymerase (pSC6-T7-Neo). In all transfection experiments, a constant amount of total DNA, 5 μg, was maintained by addition of the unrelated pUC19 plasmid. A 150-μg M9 peptide (generous gift from Scott Diamond) was then added to the DNA solution in 100 μl of OptiMEM, and the DNA-M9 mixture was further incubated for 15 min at room temperature. Lipofectamine (7.5 μg) was diluted in 100 μl of OptiMEM and added to the DNA-M9 mixture for 45 min. The DNA-M9-Lipofectamine mixture was added to 3 × 105 ES cells in suspension and plated in a 12-well gelatin-coated plate containing 3 × 105 STO feeder cells pretreated with mitomycin C (Sigma, St. Louis, Mo.). The same procedure was used for transfection of ES cells without feeders except that ES cells were plated directly on gelatin plates without STO feeder cells, using medium without LIF .
Quantitation of EGFP and β-galactosidase activities.
All adherent cells were harvested 72 h after transfection by washing with PBS and scraping cells into 100 μl of ice-cold lysate buffer (91.5 mM K2HPO4, 85 mM KH2PO4, and 1 mM dithiothreitol [DTT]). The harvested cells were then subjected to a dry-ice/ethanol freezing and thawing at 37°C for three cycles and centrifuged at 12,000 rpm for 5 min at 4°C. The supernatant was stored at −70°C until use. For ES cells with feeders, 72 h after transfection, cells were trypsinized for 5 min and pipetted extensively. After addition of 5 volumes of ES medium, cells were put back in the incubator for 45 min to allow the STO cells to attach to the plate. After this procedure, ES cells constituted more than 95% of the cells in suspension. ES cells were transferred to a tube, centrifuged, and processed the same way as other cells. The protein concentration of cell lysates was measured with the Pierce reagent (Pierce, Rockford, Ill.) in a 96-well plate. For each lysate, the same amount of protein was used for the fluorescence and chemiluminescence measurements. Fluorescence was measured in relative light units (RLUs) using a 96-well black flat-bottomed plate (Corning Costar, Cambridge, Mass.) and an FL 600 microplate reader (Bio-Tek Instrument, Winooski, Vt.) with KC4 data reduction software on an external personal computer, which controls the reader function and data capture. Excitation was at 485 nm with a 20-nm band-pass filter, and emission was at 530 nm with a 25-nm band-pass filter. To account for the background, each fluorescence reading was subtracted from that of the untransfected cell lysate. The fluorescence reading of each lysate was normalized to that of the lysate prepared from cells transfected without dsRNA, either pGEMT-dsEGFP plasmid or in vitro-transcribed dsRNA.
β-Galactosidase activity was measured by histochemical staining and chemiluminescence. Cells were fixed with 1% glutaraldehyde and stained with X-Gal staining solution [0.1 M sodium phosphate (pH 8.0), 1.3 mM MgCl2, 3 mM K4Fe(CN)6, 3 mM K3Fe(CN)6, and 0.4 mg of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) in N,N-dimethyl formamide] for 4 h. For chemiluminescence measurements, cell lysates were prepared using the Luminescent β-Galactosidase Genetic Reporter System II kit (Clontech, Palo Alto, Calif.). Lysates were analyzed in triplicate by chemiluminescence using the Lumat LB 9507 luminometer (EG&G Berthold, Bad Wildbad, Germany). To account for the background, the chemiluminescence reading was subtracted from that of the untransfected cell lysate. The RLUs of each lysate were normalized to that of the lysate prepared from cells transfected without dsRNA, either pGEMT-dsEGFP plasmid or in vitro-transcribed dsRNA.
Fluorescence microscopy of S2 and ES cells.
For fluorescence microscopy, S2 cells (2 × 106 S2 cells/well of a six-well plate) were plated and transfected with 2.5 μg of pIZ/US9-GFP and an increasing amount, 0, 1.5, or 3.0 μg, of the in vitro-transcribed dsRNA by the calcium phosphate method. ES cells (6 × 105 cells/well of a six-well plate) were mixed with 2.5 μg of pEGFP-C1 plasmid and increasing amounts, 0, 1, and 2 μg, of the in vitro-transcribed dsRNA and plated on the STO feeder cells as described above. Fluorescence micrographs were taken 72 h after transfection.
Analysis of RNA by Northern blotting in ES cells.
ES cells were transfected by three plasmids as described above. Total RNA was purified by the RNAeasy Mini kit (Qiagen, Valencia, Calif.) and quantitated by UV absorbance at 260 nm. A total of 25 μg of RNA was loaded into each lane in a 0.8% formaldehyde denaturing agarose gel, and the Northern blotting was performed using the NorthernMax kit (Ambion, Austin, Tex.). The EGFP probe was a 0.7-kb fragment generated by NheI and BglII restriction enzyme digestions of plasmid pEGFP-C1, and the lacZ probe was a 2.5 kb fragment prepared by PvuII digestion of the pCMV-lacZ plasmid. The probes were labeled by [α-32P]dCTP using the Megaprime DNA labeling system (Amersham, Piscataway, N.J.). A cDNA probe corresponding to the mouse β-actin coding sequence was hybridized as a control.
Generation of ES cells with integrated EGFP gene, transfection with dsRNA, and FACS analysis.
To produce ES cells with an integrated EGFP transgene, 2.5 × 106 AB2.2 cells were transfected with 6 μg of linearized pcDNA3-EGFP by M9 lipofection. G418 (275 μg/ml) selection was started 24 h after transfection. After 10 days of selection with G418, the surviving ES colonies were examined by fluorescence microscopy. Fluorescent colonies were picked and expanded according to established techniques. The number of integrated copies of pcDNA3-EGFP was determined by Southern blot analysis using the EGFP coding region as a probe. Several ES clones with a single copy of the EGFP gene were chosen for use in this study. ES cells were seeded at 105 cells/well of six-well plate in the presence and absence of the feeder layer and transfected with 3 μg of in vitro-transcribed dsRNA-EGFP or dsRNA-lacZ using 1.5 μg of Lipofectamine 2000 (Gibco-BRL, Rockville, Md.). The transfected cells were maintained with daily changes of medium and harvested at various time points to measure GFP fluorescence. Cells were trypsinized, centrifuged, suspended in chilled PBS, and subjected to fluorescence-activated cell sorting (FACS) analysis using a FACScan flow cytometer (Becton Dickinson, San Jose, Calif.). Instrument settings were adjusted to separate live from dead cells, and fluorescence intensity data for 20,000 live cells were collected for each experimental time point. The relative levels of fluorescence for different samples were compared using the geometric means. Instrument settings were kept constant for all samples within each experiment. Data were analyzed using Cell Quest Software (Becton Dickinson).
Generation of small RNA fragments from dsRNA.
Cytoplasmic extracts were isolated as described previously (8). Extracts were prepared from cells in the log phase of growth, and cytoplasmic proteins were extracted in a buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma, Saint Louis, Mo.), and 0.5 mM DTT (Sigma, Saint Louis, Mo.). The final dialysis was performed for 12 h in an excess volume of dialysis buffer (20 mM HEPES [pH 7.9], 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.2 mM PMSF, and 0.5 mM DTT). The protein concentration was measured by the Bradford assay. Cytoplasmic extracts (10 to 50 μg) were incubated with 30 nmol of radiolabeled dsRNA for 1 h at 30°C for S2 cells or 37°C for mammalian cells. The standard reaction was carried out in a 20-μl reaction buffer containing 20 mM HEPES, 2 mM magnesium acetate, 2 mM DTT, 1 mM ATP, 40 mM creatine phosphate, and 100 μg of creatine phosphokinase and 1 U of RNasin (Ambion, Austin, Tex.) per ml. After the reaction, samples were treated with proteinase K (1 mg/ml)–0.5% sodium dodecyl sulfate (SDS) and purified by phenol-chloroform extraction. The size of the dsRNA was examined by a 12% denaturing acrylamide gel. After completion of electrophoresis, the gel was stained with ethidium bromide. The gel was then fixed in a 30% methanol–7% acetic acid solution, dried, and exposed to X-ray film at −80°C.
RESULTS
Generation of dsRNA for EGFP gene.
To generate dsRNA in mammalian cells in situ, we cloned an inverted repeat of a portion of the EGFP gene, extending from the ATG codon to nucleotide 547, under the control of the T7 RNA polymerase (pGEMT-dsEGFP). Using this plasmid, we investigated the feasibility of the RNAi strategy for gene silencing in several mammalian cells by using the EGFP gene as a target. Two strategies were used: (i) in situ production of dsRNA by transient transfection of three plasmids, the target plasmid encoding the EGFP gene (pEGFP-C1), the plasmid harboring a 547-bp inverted repeat of the EGFP gene under control of the T7 promoter (pGEMT-dsEGFP), and the plasmid containing the T7 RNA polymerase cDNA under the control of the CMV promoter (pSC6-T7-Neo) (Fig. 1A), and (ii) direct transfection of in vitro-transcribed dsRNA (547 bp) and the target plasmid pEGFP-C1 (Fig. 1B).
FIG. 1.

Strategy for generation of dsRNA. (A) In situ production of dsRNA of EGFP. An inverted repeat of EGFP, starting from the ATG codon to nucleotide 547 in the coding region, was cloned into the pGEMT vector under control of the T7 promoter (pGEMT-dsEGFP). RNAi activity in mammalian cells was tested by transient transfection of three plasmids, the target plasmid encoding the EGFP gene, the linearized pGEMT-dsEGFP, and plasmid encoding the T7 RNA polymerase cDNA. (B) Direct transfection of dsRNA made by in vitro transcription. Mammalian cells were transfected with dsRNA and the target plasmid encoding the EGFP gene to compare the RNAi activities between in situ production of dsRNA and in vitro-transcribed dsRNA. The dsRNA-EGFP was made by in vitro transcription using the T7 RNA polymerase and the linearized pGEMT-dsEGFP plasmid. (C) Analysis of dsRNA-EGFP transcribed by the T7 RNA polymerase. The pGEMT-dsEGFP plasmid was linearized by PstI, located at the 3′ end of the inverted repeat, to generate a runoff transcript by the T7 RNA polymerase. The transcribed RNA was digested with a mixture of RNases A and T1. The RNA was analyzed in a 5% nondenaturing acrylamide gel. Lane M, 100-bp dsDNA ladder. Lanes 1 and 2 depict in vitro-transcribed RNA with and without RNase treatment, respectively. Lanes 3 and 4 show a control 1.8-kb RNA provided in the RiboMax kit, with and without RNase treatment, respectively. (D) The same RNA samples were electrophoresed on a 5% denaturing acrylamide gel containing 40% formamide and 7 M urea. (E) Analysis of the dsRNA-lacZ transcribed by the T7 RNA polymerase. The sense and antisense RNAs were transcribed from the plasmid linearized by SalI and annealed to make dsRNA. Lanes 1 and 2 depict in vitro-transcribed antisense and sense RNAs, and lane 3 depicts annealed dsRNA in a 1% agarose gel.
The production of dsRNA was confirmed by in vitro transcription of the pGEMT-dsEGFP plasmid by the T7 RNA polymerase. Nondenaturing gel electrophoresis revealed a transcript of approximately 550 bp that did not change in size appreciably upon RNase A and RNase T1 digestion, consistent with a double-stranded structure (Fig. 1C). When the RNA was analyzed in a 5% denaturing acrylamide gel (7 M urea–40% formamide), as expected, the size of the transcript was twice that in the nondenaturing gel (Fig. 1D). Upon RNase digestion, the fragment migrated predominantly to 550 nucleotides in denaturing conditions, indicating cleavage at the connecting loop of the folded dsRNA (Fig. 1D). These results confirm the generation of the 547-bp dsRNA by transcription of the pGEMT-dsEGFP plasmid. As a control, dsRNA for lacZ (740 bp) was generated by annealing the sense and antisense transcripts (Fig. 1E).
Sequence-specific gene silencing by production of dsRNA in situ in S2 cells.
To investigate whether production of a dsRNA in situ by pGEMT-dsEGFP plasmid is sufficient to induce RNAi, we transfected S2 cells with three plasmids: pEGFP-C1, pSC6-T7-Neo, and increasing amounts of pGEMT-dsEGFP. When this plasmid is cotransfected with the gene encoding the T7 RNA polymerase under the control of the CMV promoter, it is expected to make dsRNA of 547 bp in mammalian cells. To test the specificity of RNAi, the plasmid encoding the β-galactosidase, pCMV-lacZ, was added instead of pEGFP-C1 in the control experiment, where all other reagents were kept the same.
Transfection of the pGEMT-dsEGFP plasmid showed a sequence-specific and dose-dependent inhibition of EGFP expression (Fig. 2A). In contrast, β-galactosidase expression was not affected by addition of the pGEMT-dsEGFP plasmid, demonstrating the sequence-specific RNAi. The CMV promoter was not as strong as the Drosophila promoters actin or OpIE2 in S2 cells (data not shown). However, a sufficient amount of protein was generated to measure the EGFP and β-galactosidase activities. S2 cells were also used to compare the efficiency between the in situ production of dsRNA and the direct transfection of dsRNA made by in vitro transcription. Transfection of pIZ/US9-EGFP (the plasmid encoding EGFP under the control of the Drosophila promoter OpIE2) and an increasing amount of dsRNA, ranging from 0 to 3.0 μg, resulted in sequence-specific and dose-dependent inhibition of EGFP expression (Fig. 2B). Again, β-galactosidase expression was not affected by dsRNA (Fig. 2C). These results demonstrate that production of dsRNA in situ by the pGEMT-dsEGFP plasmid is sufficient to produce RNAi in S2 cells.
FIG. 2.
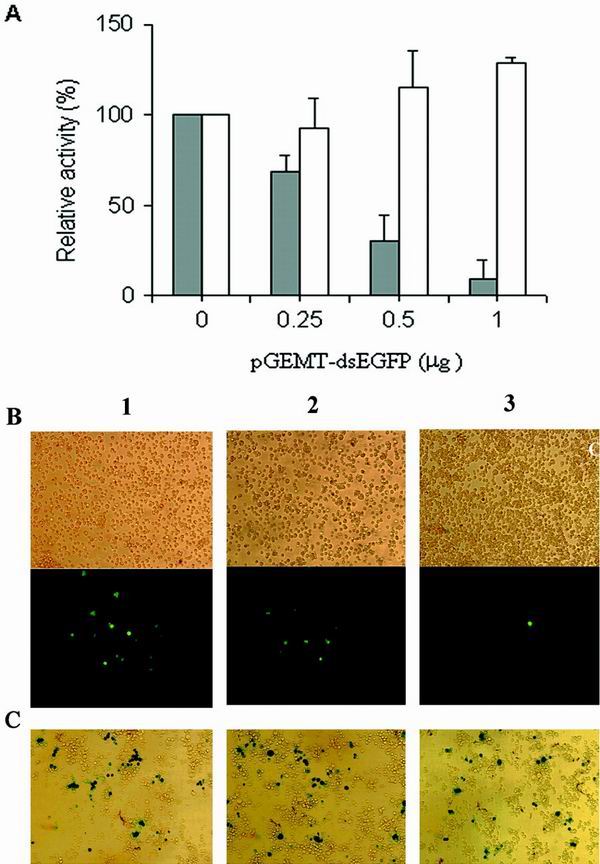
dsRNA produced a sequence-specific and dose-dependent gene silencing in Drosophila S2 cells. (A) Inhibition of EGFP expression by in situ production of dsRNA. S2 cells were transfected with three plasmids, pEGFP-C1 (1 μg), pSC6-T7-Neo (1 μg), and an increasing amount of pGEMT-dsEGFP, ranging from 0.25 to 1 μg. Throughout all transfections, the total amount of DNA was held constant by addition of unrelated pUC19 plasmid. To test the sequence specificity of RNAi, 1 μg of the plasmid encoding lacZ, pCMV-lacZ, was used instead of pEGFP-C1. The RLUs of fluorescence or chemiluminescence were normalized to that of lysate containing no pGEMT-dsEGFP plasmid. The relative activities of cells transfected with pEGFP-C1 plasmid (solid bars) and pCMV-lacZ plasmid (open bars) are shown. Standard deviation indicates the variation among at least three separate transfection experiments performed in duplicate. (B) Sequence-specific and dose-dependent inhibition of EGFP by the in vitro-transcribed dsRNA. S2 cells were transfected with 2.5 μg of pIZ/US9-GFP plasmid and 0, 1.5, or 3.0 μg of the in vitro-transcribed dsRNA-EGFP (lanes 1, 2, and 3, respectively) using a calcium phosphate method. Photographs were taken 72 h later, depicted by a bright field (upper panel) and a fluorescence micrograph (lower panel). (C) β-Galactosidase expression is not inhibited by in-vitro transcribed dsRNA-EGFP. As a control, S2 cells were transfected with 2.5 μg of pActin-lacZ and 0, 1.5, or 3.0 μg of the in vitro-transcribed dsRNA-EGFP by a calcium phosphate method. Histochemical staining was carried out 72 h later.
Several mammalian cells do not exhibit sequence-specific RNAi activity.
We investigated the feasibility of the RNAi strategy for gene silencing in several mammalian cells by using the EGFP gene as a target and transient transfection of three plasmids, pEGFP-C1, pSC6-T7-Neo, and pGEMT-dsEGFP. For most experiments, 105 cells were plated on a 12-well plate and transfected with 1 μg of pEGFP-C1, 1 μg of pSC6-T7-Neo, and an increasing amount of pGEMT-dsEGFP, ranging from 0.25 to 2 μg. Because BsrT7/5 cells already express the T7 RNA polymerase (4), transfection was carried out under identical conditions except that the pSC6-T7-Neo plasmid was omitted. Two cell lines, BsrT7/5 cells and mouse fibroblasts (STO), showed a non-sequence-specific inhibition by dsRNA, indicated by reduction of both EGFP and β-galactosidase activities as increasing amounts of pGEMT-dsEGFP were added (Fig. 3A and 3B). The CHO-K1 cells did not exhibit any inhibition by dsRNA in cognate (EGFP) or noncognate β-galactosidase genes (Fig. 3C). In all experiments, we detected no apparent cytotoxicity, as measured by cell numbers and morphology (data not shown).
FIG. 3.

Several mammalian cells do not show sequence-specific RNAi activity. Three mammalian cell lines, BsrT7/5 (A), STO (B), and CHO-K1 (C), were tested for RNAi activity by transient transfection of three plasmids, pEGFP-C1 (1 μg), pSC6-T7-Neo (1 μg), and increasing amounts of pGEMT-dsEGFP (0.25 to 2 μg). Throughout transfection, the total amount of DNA was held constant by addition of unrelated pUC19 plasmid. To test the sequence specificity of RNAi, 1 μg of the plasmid encoding the β-galactosidase, pCMV-lacZ, was used as a control. The RLUs of fluorescence or chemiluminescence were normalized to that of lysate containing no pGEMT-dsEGFP plasmid. The relative activities of cells transfected with pEGFP-C1 plasmid (solid bars) and pCMV-lacZ plasmid (open bars) are shown. Standard deviation indicates the variation among at least five separate transfections of duplicate samples.
Undifferentiated ES cells exhibit sequence-specific RNAi activity.
Recently, sequence-specific RNAi has been demonstrated in the preimplantation mouse embryo and mouse oocytes by direct injection of dsRNA (30, 35). However, dsRNA in transgenic blastocysts injected as zygotes produced gene silencing for only up to six rounds of cell division (35). These results suggest that undifferentiated cells may have RNAi activity that disappears as the cells differentiate. Here, we investigated whether undifferentiated ES cells respond to dsRNA for gene silencing.
Transfection of pGEMT-dsEGFP plasmid into undifferentiated ES cells maintained on the STO feeder layer showed a sequence-specific and dose-dependent inhibition of EGFP expression, while β-galactosidase expression was not affected (Fig. 4A). In contrast, differentiated ES cells maintained without STO feeder cells showed nonspecific inhibition (Fig. 4B). These cells progressively lost refractive boundaries and flattened to form a patch of giant trophoblastlike cells (data not shown), while undifferentiated ES cells remain as small cells packed tightly in nests (Fig. 5). Direct transfection of the in vitro-transcribed EGFP dsRNA, ranging from 0 to 1.0 μg, also resulted in a dose-dependent and sequence-specific inhibition of EGFP, shown by the fluorescence measurement of cell lysate (Fig. 5A) and by fluorescence microscopy of transfected cells (Fig. 5B). In contrast, β-galactosidase expression was not affected, as measured by either chemiluminescence (Fig. 5A) or histochemical staining (Fig. 5C).
FIG. 4.

Undifferentiated ES cells exhibit RNAi activity. (A) Sequence-specific and dose-dependent inhibition of EGFP by pGEMT-dsEGFP plasmid in ES cells grown on a feeder layer. ES cells were plated on STO feeder cells and transfected with three plasmids, pEGFP-C1 (1 μg), pSC6-T7-Neo (1 μg), and an increasing amount of pGEMT-dsEGFP, ranging from 0.25 to 1 μg. To test the sequence specificity of RNAi, 1 μg of the plasmid encoding β-galactosidase, pCMV-lacZ, was used as a control. The RLUs of fluorescence or chemiluminescence were normalized to that of lysate containing no pGEMT-dsEGFP plasmid. The relative activities of cells transfected with pEGFP-C1 plasmid (solid bars) and pCMV-lacZ plasmid (open bars) are shown. Standard deviation indicates the variation among at least five separate transfection experiments performed in duplicate. (B) Non-sequence-specific inhibition of EGFP by pGEMT-dsEGFP plasmid in differentiated ES cells cultured without the feeder layer. The same experiment was carried out in ES cells plated directly on a gelatin-coated plate with no feeder cells.
FIG. 5.
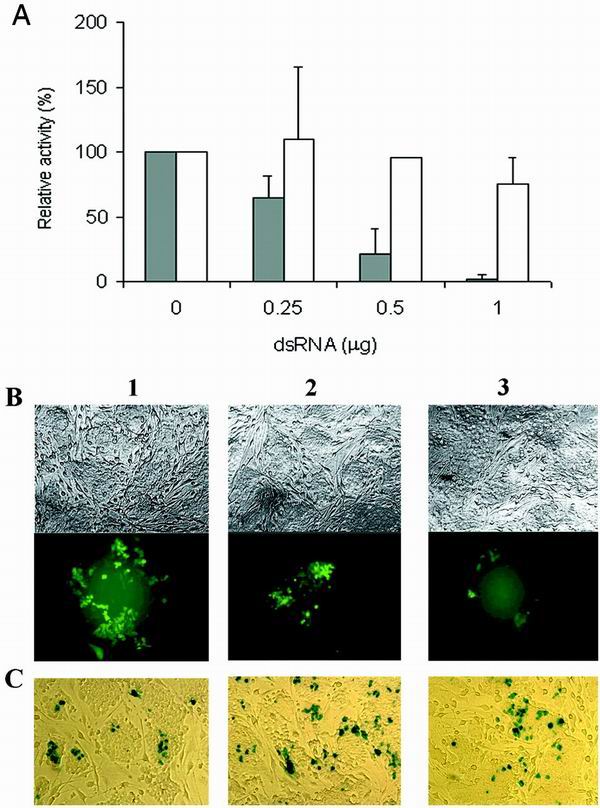
Sequence-specific and dose-dependent inhibition of EGFP expression by in-vitro transcribed dsRNA in undifferentiated ES cells. (A) ES cells were plated on feeder cells and transfected with 1 μg of the pEGFP-C1 plasmid and increasing amounts, 0.25 to 1.0 μg, of the in vitro-transcribed dsRNA. To test the sequence specificity of RNAi, 1 μg of the plasmid encoding the β-galactosidase, pCMV-lacZ, was used as a control. The RLUs of fluorescence or chemiluminescence were normalized to that of lysate containing no dsRNA. The relative activities of cells transfected with pEGFP-C1 plasmid (solid bars) and pCMV-lacZ plasmid (open bars) are shown. Standard deviation indicates the variation among at least three separate transfection performed in duplicate. (B) Fluorescence microscopy of undifferentiated ES cells transfected with 2.5 μg of pEGFP-C1 plasmid and an increasing amount, 0, 1, and 2 μg (lanes, 1, 2, and 3, respectively), of the in vitro-transcribed dsRNA-EGFP. Photographs were taken 72 h later, using a bright field (upper panel) and fluorescence (lower panel). (C) β-Galactosidase expression is not inhibited by in vitro-transcribed dsRNA-EGFP. ES cells were transfected with 2.5 μg of pCMV-lacZ and 0, 1, or 2 μg of in vitro-transcribed dsRNA-EGFP. Histochemical staining was carried out 72 h later.
Thus, only ES cells maintained in an undifferentiated state responded to dsRNA for gene silencing. The sequence-specific inhibition of dsRNA was also shown by a decrease in EGFP mRNA but not β-galactosidase mRNA, as measured by Northern blot analysis of ES cells transfected under identical conditions, in which the protein activity was measured (Fig. 6). These results confirm degradation of cognate EGFP mRNA but not β-galactosidase mRNA.
FIG. 6.

Northern analysis of cognate (EGFP) and the noncognate (β-galactosidase) mRNAs. Undifferentiated ES cells were grown on the feeder layer and transfected by three plasmids, pEGFP-C1 (1 μg), pSC6-T7-Neo (1 μg), and an increasing amount of pGEMT-dsEGFP, 0, 1, or 2 μg (lanes 1, 2, and 3, respectively). As a control, ES cells were transfected with three plasmids, pCMV-lacZ (1 μg), pSC6-T7-Neo (1 μg), and an increasing amount of pGEMT-dsEGFP, 0, 1, or 2 μg (lanes 4, 5, and 6, respectively). Total RNA was isolated from transfected cells, and 25 μg of total RNA was loaded in each lane. The EGFP probe was a 0.7-kb fragment isolated from the pEGFP-C1 plasmid, and the lacZ probe was a 2.5-kb fragment from the pCMV-lacZ plasmid. The probes were labeled by [α-32P]dCTP. A cDNA probe corresponding to the mouse β-actin coding sequence was hybridized as a control.
To investigate RNAi of an integrated gene in ES cells, we generated several ES clones with a single copy of the EGFP gene. We found that dsRNA-EGFP but not dsRNA-lacZ inhibited EGFP gene expression among three different ES clones, as determined by fluorescence microscopy and FACS analysis. Transfection of dsRNA-EGFP but not dsRNA-lacZ resulted in a substantial decrease in fluorescence intensity of the ES cells (Fig. 7A). A representative FACS analysis of one of these clones is shown in Fig. 7B. The EGFP-positive cells were gated, and the relative fluorescence of each peak was measured using the geometric mean fluorescence. EGFP fluorescence decreased over 70% at 48 h after transfection of 8.3 nM dsRNA-EGFP but not by dsRNA-lacZ at the same concentration. Following transfection of dsRNA-EGFP, we observed a new population of cells with reduced fluorescence, indicated as M3 in the middle panel of Fig. 7B. The extent of inhibition was consistent among six independent transfections. Because only 20 to 30% of ES cells were transfected by Lipofectamine 2000 (unpublished observations), the large extent of inhibition by dsRNA suggests that dsRNA can be delivered efficiently to the cytoplasm and inhibits gene expression at a low concentration in mammalian cells (9).
FIG. 7.

Sequence-specific inhibition of EGFP expression by dsRNA in the stable ES-EGFP clone, in which the EGFP gene is integrated as a single copy. (A) Fluorescence microscopy of undifferentiated ES clone untreated or transfected with 3 μg of in vitro-transcribed dsRNA-EGFP or dsRNA-lacZ, respectively. Photographs were taken 72 h later, using a bright field (upper panel) and fluorescence (lower panel). (B) FACS analysis of the ES clone 48 h after transfection. M1 indicates the gating of GFP-negative cells, M2 the gating of GFP-positive cells, and M3 the gating of cells with reduced fluorescence due to RNAi. Untransfected clone (left panel; geometric mean fluorescence: M1 = 6.12, M2= 743.15), cells transfected with dsRNA-EGFP (middle panel; geometric mean fluorescence: M1= 6.16, M2 = 270.93), cells transfected with dsRNA-lacZ (right panel; geometric mean fluorescence: M1 = 6.43, M2= 772.95). (C) Kinetics of RNAi in undifferentiated ES-EGFP clone. The relative geometric mean fluorescence of cells transfected with dsRNA-EGFP (solid bars) or dsRNA-lacZ (open bars) was normalized to the geometric mean fluorescence of untransfected cells. ES cells were split at 72 h after the initial transfection of dsRNA and plated at 2 × 105 cells/well for the later time measurements at 100 and 124 h.
We examined the kinetics of EGFP inhibition after transfection of dsRNA in undifferentiated ES cells. RNAi was more pronounced at early time points and diminished as undifferentiated ES cells replicated, presumably due to dilution of dsRNA per cell (Fig. 7C). Almost no inhibition was observed 5 days after transfection. The stability of EGFP protein may account for the apparent lower inhibition at 24 h than 48 h. The dsRNA-lacZ did not show any inhibition of EGFP expression in all experiments, indicating specific gene silencing activity in undifferentiated ES cells. When the same ES cells were cultured without the feeder layer and LIF, ES cells did not completely differentiate. In this mixed population of ES cells, dsRNA-EGFP produced a reduction in fluorescence similar to that observed in undifferentiated ES cells, but dsRNA-lacZ did not.
The persistence of the RNAi effect in these experiments can be explained by the presence of a mixed population of differentiated and undifferentiated ES cells. However, it was difficult to measure the extent of gene silencing in fully differentiated ES-EGFP cells, since their intrinsic fluorescence decreased substantially upon differentiation. Further analysis of different clones is necessary to draw conclusions for RNAi for endogenous genes in differentiated ES cells. Taken together with the transient-transfection data described above, these results indicate that long dsRNA inhibited episomal and chromosomal target genes in undifferentiated ES cells in a sequence-specific manner.
dsRNA is processed to 21 to 22 nucleotides in mammalian cells.
Small RNAs are associated with a dsRNA-dependent nuclease purified from cultured cells (15), making it likely that such RNAs serve as the specificity determinants in the RNAi reaction. Here, we investigated whether such dsRNA degradation activity may reflect the different RNAi activities among different mammalian cells. The dsRNA degradation activity was detected by the in vitro reaction in which a radiolabeled dsRNA was incubated with cytoplasmic extracts made from various cell types (Fig. 8). Drosophila S2 cells showed the highest activity, which was saturated between 10 and 50 μg of cytoplasmic protein (data not shown). Small RNA fragments were generated by all mammalian cell types tested: cells with a sequence-specific RNAi (undifferentiated ES cells), cells that showed no effect at all (CHO-K1), and cells that showed a nonspecific decrease in gene expression (differentiated ES, STO, and BsrT7/5 cells).
FIG. 8.

Small RNA fragments generated by Drosophila S2 and mammalian cells. Cytoplasmic extracts (50 μg) from various mammalian cells were incubated with 30 nmol of radiolabeled dsRNA for 1 h at 30°C for S2 cells or 37°C for mammalian cells. After the reaction, samples were treated with proteinase K–0.5% SDS. The size of dsRNA was examined on a 12% denaturing acrylamide gel. Probe indicates the radiolabeled dsRNA made by in vitro transcription. Lane M, 10-bp markers.
DISCUSSION
Posttranscriptional gene silencing by dsRNAi is a new tool for studying gene function in many organisms (3, 11, 26, 27, 33). However, only a few cases of RNAi have been reported in mammalian cells (1, 30, 32, 35). Here, we investigated the feasibility of the RNAi strategy for gene silencing in several mammalian cells by using the EGFP gene as a target, either by in situ production of dsRNA from a transient transfection of the plasmid harboring a 547-bp inverted repeat or by direct transfection of dsRNA made by in vitro transcription. In both cases, the dsRNA is generated as a hairpin structure that is resistant to RNase degradation. We reasoned that transient transfection may produce a large amount of stable dsRNA in the cytoplasm because it introduces a high copy number of the plasmid in the cytoplasm, which would then be transcribed by the T7 RNA polymerase. Using transient transfection, we show that undifferentiated ES cells have a sequence-specific RNAi activity that disappears as ES cells differentiate. This long dsRNA also inhibited its cognate gene expression in undifferentiated ES cells with a single integrated copy of the EGFP gene. Thus, both episomal and chromosomal target genes in undifferentiated ES cells were inhibited by the long dsRNA in a sequence-specific manner. Furthermore, the amount of dsRNA effective for RNAi activity in undifferentiated ES cells was similar to the amounts which caused gene silencing in Drosophila S2 cells and showed no apparent toxicity.
Several mammalian cell lines did not exhibit the sequence-specific gene silencing by dsRNA. Two cell lines, BsrT7/5 and mouse embryonic fibroblasts (STO), showed non-sequence-specific inhibition by dsRNA, while CHO-K1 did not exhibit any inhibition by dsRNA on either the cognate or noncognate gene, EGFP or lacZ. When transient cotransfection of plasmid DNA and dsRNA was carried out in several mammalian cells, 293 and NIH 3T3 cells showed no effect at all, while BHK cells showed a nonspecific decrease in gene expression (5). RNAi has been reported in CHO cells, although the amount of dsRNA required for interference was 2,500 times more than in Drosophila S2 cells (32). Because we tested RNAi under identical conditions in S2 and CHO cells, the amount of dsRNA needed to produce RNAi in the S2 cells used in our experiment may not be sufficient to produce RNAi in CHO cells. Recently, a longer dsRNA was shown to induce some sequence-specific silencing in addition to the nonspecific inhibition in mammalian cells (9). It is possible that the reporter system in this study is not sensitive enough to distinguish specific RNAi from the nonspecific inhibition. In all experiments, we detected no apparent cytotoxicity as measured by cell numbers and morphology.
Sequence-specific RNAi has been demonstrated in the preimplantation mouse embryo and mouse oocytes by direct injection of dsRNA (30, 35). The dsRNA in mammalian cells typically activates a protein kinase that phosphorylates and inactivates eIF2a (16). The ensuing inhibition of protein synthesis ultimately results in apoptosis. This sequence-independent response may reflect a form of primitive immune response, since the presence of dsRNA is a common feature of many viral life cycles. Mouse oocytes, however, clearly lack this response, as the oocytes injected with dsRNAs resume meiosis and mature to metaphase II (30). The preimplantation mouse embryo also lacks the response, as embryos injected with dsRNAs develop to the blastocyst stage (35). Specific RNAi activity present in undifferentiated ES cells suggests that undifferentiated ES cells may also lack an interferon response, similar to mouse embryos and oocytes (30, 35). However, RNAi activity was not permanent, since it was more pronounced at early time points and diminished as undifferentiated ES cells replicated, presumably due to dilution of dsRNA per cell.
Posttranscriptional gene silencing by dsRNA requires at least two steps, conversion of the dsRNA into an active species and subsequent targeting of the mRNA for inhibition by these sequence-specific active species. Recent biochemical studies (2, 10, 15, 31, 36, 37) have indicated that RNAi is accomplished by a multicomponent nuclease that targets cognate mRNA for degradation. The specificity of this complex was derived from the incorporation of a small guide sequence that is homologous to the mRNA substrate. These small 21- to 22-nucleotide RNAs, originally identified in plants with active RNAi (14), have also been observed in Drosophila embryos (10, 36, 37) and S2 cells (2, 15). We investigated whether such dsRNA degradation activity may reflect the different RNAi activities among different mammalian cells.
Although Drosophila S2 cells showed the most prominent product of 21 to 22 bp, all mammalian cells tested produced distinct RNA products of the same size. Thus, mammalian cells have the ability to generate 21- to 22-nucleotide fragments from long dsRNA regardless of their apparent RNAi activity. While our manuscript was being reviewed, Tuschl's group reported that 21-nucleotide short interfering RNA (siRNA) was capable of gene silencing in several mammalian cells in which longer dsRNA failed to produce RNAi (9). The apparent lack of RNAi by longer dsRNA in mammalian cells was attributed to nonspecific activation of the interferon response by dsRNA longer than 30 bp, masking the specific RNAi. Therefore, our findings that mammalian cells can generate siRNA regardless of their apparent RNAi activity provide an insight in gene silencing of mammalian cells by siRNA. It would be interesting to compare the extent of gene silencing by siRNA and longer dsRNA in cells that do not show nonspecific inhibition.
ES cells and other stem cells are valuable tools for the study of cell and tissue differentiation and for the creation of animal models of disease. These findings offer an opportunity to use dsRNAi for inhibition of gene expression in ES cells to study differentiation. Stem cells also have the potential for therapeutic use, including the development of replacement tissues if regulation of their differentiation can be understood. The results presented here indicate that RNAi can be used to inhibit gene expression in mouse ES cells and thus may be a useful approach for investigations of stem cell biology in general.
ACKNOWLEDGMENTS
We are grateful to Olga Igoucheva for in vitro RNA degradation study and discussion, Romaica Omaruddin and Haiching Ma for assistance with ES cell culture, Gregory Hannon for Drosophila cells and plasmids, and Scott Diamond for M9 peptides. We thank Tom Tuschl and John Klement for discussion and critical reading of the manuscript.
This work was supported in part by grants from the National Institutes of Health (GM61942, AR38923, and AR44350) to K.Y. and EY12910, the Rosanne H. Silbermann Foundation, and Research to Prevent Blindness to E.A.P.
REFERENCES
- 1.Bahramian M B, Zarbl H. Transcriptional and posttranscriptional silencing of rodent α1(I) collagen by a homologous transcriptionally self-silenced transgene. Mol Cell Biol. 1999;19:274–283. doi: 10.1128/mcb.19.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein E, Caudy A A, Hammond S M, Hannon G J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 3.Bosher J M, Labouesse M. RNA interference: genetic wand and genetic watchdog. Nat Cell Biol. 2000;2:E31–E36. doi: 10.1038/35000102. [DOI] [PubMed] [Google Scholar]
- 4.Buchholz U J, Finke S, Conzelmann K K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol. 1999;73:251–259. doi: 10.1128/jvi.73.1.251-259.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caplen N J, Fleenor J, Fire A, Morgan R A. dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene. 2000;252:95–105. doi: 10.1016/s0378-1119(00)00224-9. [DOI] [PubMed] [Google Scholar]
- 6.Catalanotto C, Azzalin G, Macino G, Cogoni C. Gene silencing in worms and fungi. Nature. 2000;404:245. doi: 10.1038/35005169. [DOI] [PubMed] [Google Scholar]
- 7.Clemens J C, Worby C A, Simonson-Leff N, Muda M, Maehama T, Hemmings B A, Dixon J E. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci USA. 2000;97:6499–6503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dignam J D, Lebovitz R M, Roeder R G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elbashir S M, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 10.Elbashir S M, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fire A. RNA-triggered gene silencing. Trends Genet. 1999;15:358–363. doi: 10.1016/s0168-9525(99)01818-1. [DOI] [PubMed] [Google Scholar]
- 12.Fire A, Xu S, Montgomery M K, Kostas S A, Driver S E, Mello C C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 13.Grant C E, Vasa M Z, Deeley R G. cIRF-3, a new member of the interferon regulatory factor (IRF) family that is rapidly and transiently induced by dsRNA. Nucleic Acids Res. 1995;23:2137–2146. doi: 10.1093/nar/23.12.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton A J, Baulcombe D C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 15.Hammond S M, Bernstein E, Beach D, Hannon G J. An RNA-directed nuclease mediates posttranscriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman R J. Double-stranded RNA-activated protein kinase mediates virus-induced apoptosis: a new role for an old actor. Proc Natl Acad Sci USA. 1999;96:11693–11695. doi: 10.1073/pnas.96.21.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennerdell J R, Carthew R W. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95:1017–1026. doi: 10.1016/s0092-8674(00)81725-0. [DOI] [PubMed] [Google Scholar]
- 18.Ketting R F, Haverkamp T H, van Luenen H G, Plasterk R H. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNase D. Cell. 1999;99:133–141. doi: 10.1016/s0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- 19.Ketting R F, Plasterk R H. A genetic link between cosuppression and RNA interference in C. elegans. Nature. 2000;404:296–298. doi: 10.1038/35005113. [DOI] [PubMed] [Google Scholar]
- 20.Malinsky S, Bucheton A, Busseau I. New insights on homology-dependent silencing of I factor activity by transgenes containing ORF1 in Drosophila melanogaster. Genetics. 2000;156:1147–1155. doi: 10.1093/genetics/156.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meins F., Jr RNA degradation and models for posttranscriptional gene-silencing. Plant Mol Biol. 2000;43:261–273. doi: 10.1023/a:1006443731515. [DOI] [PubMed] [Google Scholar]
- 22.Mette M F, Aufsatz W, van der Winden J, Matzke M A, Matzke A J. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 2000;19:5194–5201. doi: 10.1093/emboj/19.19.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muskens M W, Vissers A P, Mol J N, Kooter J M. Role of inverted DNA repeats in transcriptional and posttranscriptional gene silencing. Plant Mol Biol. 2000;43:243–260. doi: 10.1023/a:1006491613768. [DOI] [PubMed] [Google Scholar]
- 24.Parrish S, Fleenor J, Xu S, Mello C, Fire A. Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol Cell. 2000;6:1077–1087. doi: 10.1016/s1097-2765(00)00106-4. [DOI] [PubMed] [Google Scholar]
- 25.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter M A. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharp P A. RNA interference—2001. Genes Dev. 2001;15:485–490. doi: 10.1101/gad.880001. [DOI] [PubMed] [Google Scholar]
- 27.Sharp P A, Zamore P D. Molecular biology: RNA interference. Science. 2000;287:2431–2433. doi: 10.1126/science.287.5462.2431. [DOI] [PubMed] [Google Scholar]
- 28.Smardon A, Spoerke J M, Stacey S C, Klein M E, Mackin N, Maine E M. EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr Biol. 2000;10:169–178. doi: 10.1016/s0960-9822(00)00323-7. [DOI] [PubMed] [Google Scholar]
- 29.Stam, M., R. de Bruin, R. van R. Blokland. A. van der Hoorn, J. N. Mol, and J. M. Kooter. 2000. Distinct features of posttranscriptional gene silencing by antisense transgenes in single copy and inverted T-DNA repeat loci. Plant J. 21:27–42. [DOI] [PubMed]
- 30.Svoboda P, Stein P, Hayashi H, Schultz R M. Selective reduction of dormant maternal mRNAs in mouse oocytes by RNA interference. Development. 2000;127:4147–4156. doi: 10.1242/dev.127.19.4147. [DOI] [PubMed] [Google Scholar]
- 31.Tuschl T, Zamore P D, Lehmann R, Bartel D P, Sharp P A. Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 1999;13:3191–3197. doi: 10.1101/gad.13.24.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ui-Tei K, Zenno S, Miyata Y, Saigo K. Sensitive assay of RNA interference in Drosophila and Chinese hamster cultured cells using firefly luciferase gene as target. FEBS Lett. 2000;479:79–82. doi: 10.1016/s0014-5793(00)01883-4. [DOI] [PubMed] [Google Scholar]
- 33.Vaucheret H, Fagard M. Transcriptional gene silencing in plants: targets, inducers and regulators. Trends Genet. 2001;17:29–35. doi: 10.1016/s0168-9525(00)02166-1. [DOI] [PubMed] [Google Scholar]
- 34.Wargelius A, Ellingsen S, Fjose A. Double-stranded RNA induces specific developmental defects in zebrafish embryos. Biochem Biophys Res Commun. 1999;263:156–161. doi: 10.1006/bbrc.1999.1343. [DOI] [PubMed] [Google Scholar]
- 35.Wianny F, Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat Cell Biol. 2000;2:70–75. doi: 10.1038/35000016. [DOI] [PubMed] [Google Scholar]
- 36.Yang D, Lu H, Erickson J W. Evidence that processed small dsRNAs may mediate sequence-specific mRNA degradation during RNAi in Drosophila embryos. Curr Biol. 2000;10:1191–1200. doi: 10.1016/s0960-9822(00)00732-6. [DOI] [PubMed] [Google Scholar]
- 37.Zamore P D, Tuschl T, Sharp P A, Bartel D P. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]