

Abstract
Polygenic scores (PGS) combine the effects of common genetic variants1,2 to predict risk or treatment strategies for complex diseases3–7. Adding rare variation to PGS has largely unknown benefit and is methodically challenging. Here we developed a method for constructing rare variant PGS and applied it to calculate genetically modified hemoglobin A1C (HbA1C) thresholds for type 2 diabetes (T2D) diagnosis7–10. The resultant rare variant PGS is highly polygenic (21,293 variants across 154 genes), depends on ultra-rare variants (72.7% observed in <3 people), and identifies significantly more undiagnosed T2D cases than expected by chance (OR = 2.71, P = 1.51 × 10−6). A PGS combining common and rare variants is expected to identify 4.9 million misdiagnosed T2D cases in the USA, nearly 1.5-fold more than the common variant PGS alone. These results provide a method for constructing complex trait PGS from rare variants and suggest that rare variants will augment common variants in precision medicine approaches for common disease.
HbA1C, which measures average blood glucose levels over 2-3 months11, is a widely used T2D diagnostic biomarker. Meeting the diagnostic threshold of 47.53 mmol/mol (6.5% glycated hemoglobin)12 – as opposed to a milder sub-threshold diagnosis of pre-diabetes13 – unlocks a larger therapeutic armamentarium with insurance and treatment implications14,15. HbA1C is influenced by common genetic variants that affect both pathways central to glycemic control7,9,10 and pathways that influence erythrocytic properties such as cell lifespan16. Erythrocytic variants do not affect risk of T2D and can in fact cause diabetes misdiagnoses by altering the expected relationship between measured HbA1C and true blood glucose levels7,9,10,17,18. A PGS of common erythrocytic variants7 identifies a substantial number of undiagnosed T2D cases in the USA – most notably, ~11% of African-Americans carry a G6PD variant (rs1050828)7,19 that shortens erythrocyte lifespan and potentially causes ~0.65 million misdiagnoses7,10.
The identification of common variants that affect HbA1C through erythrocytic pathways advances the accuracy of diabetes diagnosis across people of all genetic backgrounds. Most variants in a population, however, are rare20 and have unknown impacts on HbA1C or the T2D diagnostic threshold. Furthermore, while rare variants have been used to predict21 and diagnose22 Mendelian diseases, it remains unknown whether they could meaningfully contribute to PGS for complex traits such as T2D or HbA1C. Current methodologies for common variant PGS, which rely on effect size estimates for variants from a “discovery” study1–3, do not obviously extend to rare variants, because (a) most individual rare variant effect sizes cannot be accurately estimated23,24 and (b) most rare variants carried by a patient are absent from even large discovery studies20.
To include and evaluate the utility of rare coding variants in a PGS for the HbA1C-based T2D diagnostic threshold, we conducted single-variant and (using burden tests across seven nested variant “masks”; Methods) gene-level association analyses for HbA1C in 87,735 whole exome sequenced (WES) individuals (Supplementary Table 1) from the UK Biobank25,26 (UKB; n ≤ 45,650 Europeans (EU)) and AMP-T2D-GENES study (n ≤ 12,132 EU; 12,369 Hispanics (HS); 6,234 African-Americans (AA); 5,931 East Asians (EA); 5,419 South Asians (SA))27. Thirty-seven predicted high or moderate impact variants and three genes (PIEZO1, GCK, G6PD) produced associations reaching study-specific exome-wide significance thresholds (P ≤ 1.8 × 10−8 and P ≤ 1.0 × 10−7, respectively; Methods), a substantial number relative to expectations from 23 other metabolic phenotypes analyzed (for comparison) in the same samples (Fig. 1a,b, Extended Data Fig. 1, and Supplementary Tables 2–4). Only four of these variants could be considered rare (maximum MAF < 0.01 across all ancestries), and these collectively had a minimal impact on the previous common variant HbA1C PGS (R2 = 0.995 between the augmented and original PGS; Supplementary Note).
Figure 1 |. Rare variant associations for HbA1C are comparatively strong.

a, The number of exome-wide significant predicted high or moderate impact variant associations across 24 quantitative phenotypes. Single variant associations were determined using the efficient mixed-model association expedited (EMMAX) method50, and gene-level associations were determined using burden testing. Yellow: P ≤ 1.8 × 10−8 (as derived from a previously determined threshold49 of P ≤ 4.3 × 10−7 and Bonferroni correction for 24 phenotypes) and exome-wide significant gene-level associations. Blue: P ≤ 1.0 × 10−7 (as derived from the traditional exome-wide significance threshold of P ≤ 2.5 × 10−6 and Bonferroni correction for 24 phenotypes). b, Manhattan plot of all gene-level HbA1C associations determined via burden testing. Those reaching exome-wide significance (P ≤ 1.0 × 10−7; red line) are labeled. c,d, Effect sizes (mmol/mol) for rare variant gene-level associations for G6PD (c) (n = 1,382 for AA; n = 1,930 for EA; n = 41,689 for EU; n = 1,861 for SA; n = 892 for HS) and PIEZO1 (d) (n = 905 for AA; n = 1,340 for EA; n = 42,061 for EU; n = 789 for SA; n = 484 for HS). Previously reported7 nearby common variant associations (n = 7,564 for AA; n = 20,838 for EA; n = 123,665 for EU; n = 8,874 for SA) are shown for comparison. Gene-level effects are displayed from the strongest associated variant mask. AA, African-American; EA, East Asian; EU, European; HS, Hispanic; SA, South Asian; M-A, meta-analysis. Error bars indicate 95% confidence intervals.
Among the three genes with significant associations, PIEZO1 (effect = −0.66 mmol/mol HbA1C; P = 2.8 × 10−23) and G6PD (−2.40 mmol/mol; P = 3.2 × 10−10) encode proteins with known roles in erythrocyte function28–33. Both associations had smaller aggregate effect sizes than the common G6PD rs1050828 variant (−4.15 mmol/mol)7 but larger effect sizes than typical common variant HbA1C associations (~0.26 mmol/mol; Fig. 1c,d and Supplementary Table 5)7. In fact, the PIEZO1 gene-level association explained more HbA1C phenotypic variance than a nearby GWAS association7 (Extended Data Fig. 2, Supplementary Table 6, and Supplementary Note), in contrast to properties of rare variant gene-level associations previously reported for T2D27.
Given their association strength (Fig. 1c,d) and the known erythrocytic roles of their encoded proteins, we hypothesized that PIEZO1 and G6PD might carry rare variants that alter the appropriate HbA1C diagnostic threshold for T2D. To test this hypothesis, we identified individuals in AMP-T2D-GENES (our “test sample”) who (a) had HbA1C levels below 47.53 mmol/mol without antihyperglycemic medication, and (b) carried PIEZO1 or G6PD variants with effect sizes (Methods) sufficient to adjust their HbA1C levels above 47.53 mmol/mol (“reclassify” them). Following previous work7, we then evaluated the proportion of “true” T2D cases (defined by non-HbA1C measurements; Methods) among the reclassified carriers and compared this to the proportion of true cases among individuals matched on cohort and HbA1C level (Methods). To first validate this approach, we applied it to the published common variant HbA1C PGS7 (Extended Data Fig. 3 and Methods) and found that, in our test sample, the common variant PGS reclassified 1.0% of individuals and a greater proportion of true cases than expected by chance (OR = 3.42; P = 3.3 × 10−8). By contrast, the PIEZO1 and G6PD variants reclassified 0.2% of samples and only marginally more true cases than expected by chance (OR = 1.67; P = 0.29).
To explore rare variant associations beyond those reaching exome-wide significance, we conducted two enrichment analyses. First, following previous HbA1C GWAS7, we observed the strongest gene-level HbA1C associations to be moderately enriched for rare variant erythrocyte count (RBC) associations (lowest q-value = 0.04; Methods and Extended Data Fig. 4). Second, following a previous T2D WES study27, we observed P < 0.05 enrichments within 6 of 25 gene sets curated from glycemic effects in mice and 4 of 10 gene sets curated from erythrocytic effects in mice (Fig. 2a and Methods). Moreover, the P < 0.05 gene-level associations within the four enriched erythrocytic gene sets had effect sizes biased toward decreased HbA1C (16/23, binomial P = 0.04; Fig. 2b), consistent with expectations from the gene set annotation and observations in humans16. These results suggest that HbA1C levels are affected by many rare alleles across many genes, in many cases through erythrocytic pathways and thus independently of T2D pathophysiology7,9,10,17,19.
Figure 2 |. Rare variant gene-level HbA1C associations show enrichment for genes involved in glycemic control and erythrocytic pathways in mice.

a, Boxplots displaying the percentile of gene-level P-values (relative to matched genes; grey) for genes thought to impact erythrocyte pathways (blue) and glycemic control (green) in mice. The horizontal dotted line at the 50th percentile indicates the expected median percentile under the null distribution. We used a one-sided Wilcoxon rank sum test to assess significant deviation of percentiles from matched genes; Wilcoxon P-values are displayed for each gene set. The numbers of genes in each gene set are indicated in the Supplementary Note. b, The fraction of genes with a negative effect on HbA1C levels among those (i) with HbA1C gene-level P ≤ 0.05 and (ii) within a significantly enriched (P ≤ 0.05) erythrocytic (n = 4) or glycemic (n = 5) gene set. The bars represent the fractions of genes observed across all gene sets, while the dots represent the fractions of genes observed for each individual gene set. We used a binomial test to assess deviation from the expected fraction of 50%. In a, the box plot indicates minimum, lower quartile, median, upper quartile, and maximum. In b, the error bars indicate standard error.
To evaluate whether this broader collection of rare variants might lead to T2D misdiagnoses, we sought to add them to the existing HbA1C common variant PGS. Existing PGS methods1,3–5,34,35, however, are designed for common variants and do not address how to (a) select genes to include in a rare variant PGS or (b) assign weights to rare variants within the selected genes (including those not seen in the discovery sample). We therefore developed a novel framework for rare coding variant HbA1C PGS that (a) includes genes based on their aggregate P-values and publicly available annotations36–40 (e.g. from knockout mice; Extended Data Fig. 5a) and (b) assigns variant weights based on aggregate effect sizes for the bioinformatically defined23,41,42 masks that contain the variant (Extended Data Fig. 5b and Methods) – for our particular PGS application, we based both gene and weight selection not only on HbA1C association strength but also (to include only variants that might contribute to a T2D misdiagnosis) on evidence of involvement in erythrocytic pathways. We explored nine models with varying criteria for gene and weight selection (Extended Data Figs. 5 and 6 and Supplementary Table 7), selecting weights (or, secondarily, genes and weights; Methods and Supplementary Table 8) using the UKB samples and testing the models in the independent AMP-T2D-GENES samples.
Five (56%) of the PGS models reclassified a significantly greater (OR > 1, P ≤ 5.6 × 10−3 (0.05/9 models)) proportion of true cases than expected by chance (Extended Data Fig. 6 and Supplementary Table 9), with the most significant excess (OR = 2.71, P = 1.5 × 10−6; Fig. 3a) achieved by a model with a “loose” criterion for gene selection and a “nested” method for variant weights. The “loose” criterion requires genes to achieve HbA1C rare variant P < 0.05 and then further filters for evidence of action through erythrocytic pathways according to one of three gene annotations (Methods and Extended Data Fig. 5). The “nested” method assigns each variant a weight equal to the aggregate HbA1C effect size (in the UKB discovery sample) of variants with annotations at least as severe as the variant (i.e. in the most severe nested mask containing the variant; Methods). This “loose, nested” model (Methods) contains 154 genes (Supplementary Table 7) and, across individuals in the AMP-T2D-GENES test sample, 21,293 variants – 21,016 (98.7%) of which have MAF < 0.005 and 15,473 (72.7%) of which are observed in <3 people. The model assigns non-zero weights to 99.7% of individuals in the AMP-T2D-GENES study, 13,573 (64.6%) of whom carry a variant absent from the discovery study (Methods). As a test of model over-fitting, the model reclassified an excess of true cases even when both genes and weights were selected using only the UKB samples (OR = 1.73, P = 0.0056; Extended Data Fig. 7, Supplementary Table 9, and Supplementary Note). As negative controls, models that omitted genes passing the erythrocytic pathway filters (Methods) did not reclassify significantly more true cases than expected by chance, regardless of whether they did (OR = 1.38, P = 0.16; Fig. 3a) or did not (OR = 1.43, P = 0.17) filter for genes annotated as involved in mouse glucose homeostasis.
Figure 3 |. Accuracy and properties of rare and common variant polygenic scores.

We identified “reclassified” individuals with adjusted (but not unadjusted) HbA1C above the T2D diagnostic threshold (47.53 mmol/mol) and compared (via a two-tailed Fisher’s exact test) the fraction of “true” cases among such individuals to the number expected by chance (Methods). a, From top, the forest plot shows Fisher’s exact test odds ratios and 95% confidence intervals for polygenic scores constructed from two exome-wide significant rare variant gene-level associations (PIEZO1/G6PD), the best performing (“loose, nested”) rare variant polygenic score (Erythrocytic Genes), a negative control polygenic score that excludes known erythrocytic genes (Glycemic Genes), a previously published common variant polygenic score7 (Common Erythrocytic Variants), and a polygenic score that combines rare and common variants (Combined Erythrocytic PGS). The area of each diamond is proportional to the number of individuals in our test sample reclassified by the score. b, Fisher’s exact test odds ratios stratified by ancestry. The area of each diamond is now proportional to the number of individuals in the US population that would be reclassified by the score after scaling the ancestral proportions in our test sample to those estimated for the US (Methods). Due to inadequate data regarding East Asian and South Asian percentages of the US population, “Asian” represents a meta-analysis of the South Asian and East Asian results; Supplementary Table 10 shows PGS performance within each ancestry. Europeans are not displayed due to insufficient data in our test sample. c-f, Histograms display the distribution (in mmol/mol HbA1C) of rare variant (gray) and common variant (colored) polygenic scores for each ancestry.
Next, we evaluated whether the “loose, nested” rare variant PGS could augment the previous common variant HbA1C PGS7 (Supplementary Table 10 and Methods). A combined PGS summing the ancestry-appropriate common variant PGS with the rare variant PGS (with additivity justified by the independence of variants in the rare and common variant PGS; Extended Data Fig. 8 and Methods) produced a greater reclassification accuracy in our test sample (OR = 4.4, P = 8.0 × 10−26) than either the rare or common PGS alone. Scaling the ancestry proportions in our test sample to the estimated ancestry proportions of the US population (Methods), the combined PGS is expected to re-classify 4.9 million T2D cases in the US population, nearly 1.5-fold more than the common variant PGS alone (3.4 million) and 2-fold more than the rare variant PGS alone (2.4 million; Fig. 3b). The estimated increase in reclassified cases varies across ancestries and is lower but still substantial for ancestries with high-performing common variant scores (1.2-fold increase for African Americans, 1.25-fold increase for Asians; Supplementary Table 10).
One notable difference between the rare and common variant PGS is that the common variant PGS is undefined for ancestries without GWAS effect size estimates (e.g. Hispanics7), while the rare variant PGS is defined independent of ancestry. While further work is needed to fully justify fixed rare variant weights across ancestries, we observe that (a) our rare variant PGS has comparable accuracy across ancestries in our test sample (Fig. 3b) and (b) within our PGS, rare variant gene-level effect sizes are more homogenous across ancestries than are common variant-level effect sizes (λ = 3.1 for common variant Q statistic vs. λ ≤ 1.6 for gene-level Q statistics; Extended Data Fig. 9 and Methods). This observation does not conflict with the known population-specificity of rare variants20,43: most (68%) variants in the rare variant PGS are ancestry-specific, and the average number of rare variants per individual varies by ancestry (Supplementary Table 11).
These results suggest that rare variants, despite a comparatively modest impact on complex traits27,44, may meaningfully contribute to genetic-based diagnostic strategies for complex disease. They do not suggest, however, that this contribution will be primarily through population-specific large-effect variants20,43,45. The average aggregate effect size for genes in the rare variant PGS is indeed larger than the average effect size of variants in the common variant PGS (0.72 vs. 0.38 mmol/mol; Supplementary Table 12). However, the average rare variant PGS adjustment per individual is smaller than that from the common variant model (0.61 vs. 1.09 mmol/mol; Supplementary Table 13), because individuals tend to carry fewer variants in the rare variant PGS than they do in the common variant PGS (4.7 vs. 8.4) – despite the much larger number of variants in the rare variant PGS (21,293 vs. 22; Supplementary Table 11). These observations are natural consequences of rare variant properties individually (large-effect, ancestry specific) versus in aggregate (polygenic, relatively ancestry non-specific; Fig. 3c–f).
While in a strict sense our study is limited to HbA1C adjustments for diabetes diagnosis, it demonstrates the utility of a rare variant PGS that does not require individual variant effect size estimates but rather assumes that all variants within a mask have the same effect size – this assumption will not hold for all genes but allows the PGS to incorporate variants private to an individual and hold variant weights constant across ancestries. The combined PGS we present could also no doubt be improved, most obviously through methodological advances to include suggestive associations in the common variant PGS (Methods) and optimize the selection of genes and weights in the rare variant PGS – potentially by better using gene annotations (which are more readily available46 than are noncoding variant annotations47) to filter variants included in the model. More broadly, our results suggest that adding rare variants to complex trait PGS will be valuable, albeit through polygenic effects quite distinct from those that might have been expected from early predictions of rare variants with a “high impact” on common disease23,48.
Methods
Additional information about the methods used in this study is available as a Supplementary Note.
AMP-T2D-GENES.
The AMP-T2D-GENES dataset was the focus of a previous T2D exome sequence analysis27. It includes samples from five ancestries (African American, East Asian, European, Hispanic, and South Asian) and six consortia (Supplementary Table 14). We used previously described27 sequence data and SNP array data passing extensive quality control filters (see Supplementary Note), consisting of 6.33 million variants in 45,231 sequenced individuals, 34,974 of which had SNP array data.
In the present study, we analyzed 23 quantitative phenotypes in these samples (Supplementary Table 1). For glycemic traits, we included only T2D control individuals in association analyses (T2D cases were defined as previously described27). All individuals in the AMP-T2D-GENES study provided informed consent, and all samples were approved for use at the respective institution20,27,44,51,52.
UK Biobank.
We obtained data from the UK Biobank26 (UKB) under Project 27892: “Genetic studies of type 2 diabetes, related metabolic traits, and their complications (PI Florez)”. We analyzed 10 quantitative phenotypes (Supplementary Tables 1 and 15), removing T2D cases from glycemic trait analyses with T2D case status determined using a previously published algorithm53. We analyzed UKB exome sequence data from the first release of ~50,000 samples25 – specifically, the PLINK binary file set produced by the functionally equivalent (FE) pipeline. We conducted sample and variant quality control of these data following a similar procedure as for the AMP-T2D-GENES dataset. Full details of this procedure are presented in the Supplementary Note; briefly, it included ancestry and kinship inference using genotype principal components (PCs), exclusion of samples that were outliers on one or more genotype-derived metrics, and exclusion of variants that failed Hardy-Weinberg equilibrium or call rate filters.
Phenotype transformations.
Prior to association analyses, we transformed phenotypes through (a) log-transformations of phenotypes with skewed distributions; (b) adjustment for age, age squared, and sex using residuals from linear regression54; (c) inverse-normal transformations of the calculated residuals for each phenotype; and (d) multiplication of the resulting values by the standard deviation of the original phenotype distribution. UKB phenotypes were additionally (prior to transformations) winsorized to remove extreme values falling outside of five standard deviations from the mean. Transformations for AMP-T2D-GENES were performed separately within each cohort.
We used transformed phenotypes to determine association P-values, but we used raw HbA1C values to determine effect sizes for inclusion in the PGS.
Single variant association analysis.
Within AMP-T2D-GENES, we conducted single variant association analysis following a previously described procedure27 (see Supplementary Note for more details). Within the UKB, we conducted single variant association analysis using linear regression, including covariates for the first nine PCs of ancestry. All coding variants with P ≤ 4.3 × 10−7 and non-coding variants with P ≤ 5 × 10−8 for any phenotype in either dataset are listed in Supplementary Tables 16 and 17.
We combined single variant association results across AMP-T2D-GENES and UKB via a sample size weighted meta-analysis. For coding variants, we used a significance threshold of P ≤ 1.8 × 10−8 (P ≤ 4.3 × 10−7 with Bonferroni correction for 24 phenotypes49); for noncoding variants, we used a significance threshold of P ≤ 2.1 × 10−9 (P ≤ 5 × 10−8 with Bonferroni correction for 24 phenotypes). All variants with significant associations across all phenotypes are listed in Supplementary Table 2.
Variant annotation.
We used the Variant Effect Predictor (VEP)55,56 to annotate variants using the LofTee plugin57, which predicts loss-of-function variants, and the dbNSFP plugin (version 3.2)58, which produces annotations from 15 bioinformatic algorithms. Variant annotations were produced across all ENSEMBL transcripts; for annotating results from the single variant analysis, we used the “--flag-pick-allele” option59 with a previously described ordering criteria for transcripts27. Throughout the text, we use “predicted high or moderate impact variants” to refer to both missense variants and other variants in or near the coding region of a gene that are not synonymous; specifically, these variants are those predicted by VEP to have HIGH or MODERATE impact, spanning the following consequences: transcript_ablation, splice_acceptor_variant, splice_donor_variant, stop_gained, frameshift_variant, stop_lost, start_lost, transcript_amplification, inframe_insertion, inframe_deletion, missense_variant, protein_altering_variant.
Gene-level association analysis.
We used these annotations to, as previously27, group variants into seven nested “variant masks”. These were (in increasing order of variant deleteriousness): (a) “LoFTee”, high confidence variants according to the LoFTee plugin; (b) “16/16”, variants in the LoFTee mask and variants predicted as deleterious by 16 bioinformatic algorithms (see Supplementary Note); (c) “11/11”, variants in the 16/16 mask and variants predicted as deleterious by 11 bioinformatic algorithms; (d) “5/5”, variants in the 11/11 mask and variants predicted as deleterious by 5 bioinformatic algorithms; (e) “5/5 LoFTee LC 1%”, variants in the 5/5 mask and MAF < 1% low-confidence variants according to the LoFTee plugin; (f) “1/5 1%”, variants in the 5/5 LoFTee LC 1% mask and MAF < 1% variants predicted as deleterious by one bioinformatic algorithms; and (g) “0/5 1%” variants in the 1/5 1% mask and all missense variants with MAF < 1%.
For both AMP-T2D-GENES and the UKB, we conducted gene-level association analysis following a previously described procedure27. For each gene, we used the EPACTS software package to perform burden tests across each variant mask, analyzing only unrelated samples. Within AMP-T2D-GENES, we included 10 trans-ancestry PCs, sample subgroup, and sequencing technology as covariates. Within the UKB, we included the first 9 ancestry-specific PCs as model covariates. For genes on the X chromosome, we analyzed males and females separately before combining results via a fixed-effect inverse-variance weighted meta-analysis (implemented via METAL60). All genes with P ≤ 2.5 × 10−6 associations (i.e. threshold not adjusted for multiple hypothesis testing) for any phenotype and mask are listed in Supplementary Table 18 (AMP-T2D-GENES) and Supplementary Table 19 (UKB).
We meta-analyzed the gene-level association results from AMP-T2D-GENES and UKB for each of the seven aforementioned masks by conducting sample-size weighted meta-analyses using the METAL60 software package (Supplementary Table 20). We then used a previously published strategy27 to consolidate P-values for each gene across masks by (a) assigning the gene the lowest P-value across masks; (b) calculating the effective number of gene-level tests for the gene according to the independence of the variants across its masks; and (c) correcting the P-value for the effective number of tests. QQ plots and genomic inflation factors (λ) of the consolidated P-values showed that the tests were, if anything, conservative (all λ < 1). We considered genes with P ≤ 1.0 × 10−7 (P ≤ 2.5 × 10−6 with Bonferroni correction for 24 phenotypes) to be exome-wide significant. All gene-level associations that reach this threshold are listed in Supplementary Table 4.
Proportion of variance explained calculations.
The proportion of variance explained (PVE) by either a SNP or gene-level association was calculated using a previously derived formula61:
where β is the effect size of the genetic association, se(β) is the standard error of β, MAF is minor allele frequency, and N is sample size. For MAF values for gene-level associations, we used the aggregate minor allele frequency across all rare variants included in the associations. For variants and/or genes found on the X chromosome, we treated males as haploid.
Gene set analyses.
We conducted enrichment analyses following a previously described procedure27. For a given set of genes, we (a) calculated the exome-wide percentile for each gene according to its gene-level association P-value; (b) matched each gene to a set of background genes with similar properties (e.g. number of variants, combined allele count); and (c) conducted a one-sided Wilcoxon rank sum test. We analyzed two types of gene sets. First, we searched the mouse genome informatics (MGI) database for genes annotated with terms including ‘erythrocyte’, ‘erythropoiesis’, ‘hematocrit’, ‘glucose’, ‘insulin’, or ‘diabetes’ (see Supplementary Note for the resulting list of 35 gene sets). Second, to test whether genes associated with HbA1C were likewise associated with RBC, or vice versa, we constructed gene sets from the n smallest P-value HbA1C (or RBC) gene-level associations, with n ranging from 1 to 1,000. For each n, we then tested for enrichment either for (a) stronger RBC associations (among the top HbA1C-associated genes) or (b) stronger HbA1C associations (among the top RBC-associated genes). We used the Benjamini and Hochberg method62 to correct for multiple hypothesis testing.
To test whether the direction of effect on HbA1C was consistent across gene-level associations within a gene set, we identified genes in the set with HbA1C association (P ≤ 0.05), assigned a direction of effect to each gene according to the sign of the effect size from the most significant mask, and tested for directional concordance via a binomial test. We used a t-test to evaluate whether the mean proportion of genes with negative effect sizes was greater in erythrocytic vs. glycemic gene sets. We also repeated this analysis for individual variants within each mask, by evaluating (via a t-test) whether there were different proportions of variants with negative effect sizes in the erythrocytic gene sets as compared to the glycemic gene sets (Extended Data Fig. 10). In each case we considered P ≤ 0.05 as significant.
Common variant polygenic adjustment score methodology for HbA1C.
For the common variant PGS, we used a set of variants and effect sizes from a previous GWAS publication (downloaded from www.magicinvestigators.org; Extended Data Fig. 3)7. We calculated a PGS for each AMP-T2D-GENES sample, using available SNP array data, as
where is the common variant adjustment for individual is the estimated effect of the effect allele of a particular variant () included in the PGS, is the dosage of the effect allele for variant for individual , and is the effect allele frequency of . To obtain the adjusted HbA1C value for individual , we subtracted the calculated PGS value from the individual’s reported HbA1C.
Rare variant polygenic adjustment score methodology for HbA1C.
For the rare variant PGS, the key distinction relative to the common variant PGS is the set of variants in the summation, which can vary across individuals and is potentially unbounded – the summation is (in principle) taken over all rare variants that could ever be observed in a population. We resolve this issue by assigning rare variants to a set of pre-defined groups , for each of which the needed parameters are constant and estimable when the model is built. The rare variant PGS then becomes
where is the estimated effect of variant effect alleles within group is the combined effect allelic dosage for all variants in the group, and is the combined effect allele frequency of variants in . As discussed below, we define each variant group within the context of a gene. As for the common variant PGS, to obtain the adjusted HbA1C value for individual , we subtract the calculated PGS value from the individual’s reported HbA1C.
Genes included in the rare variant polygenic score.
To determine genes to be included in the rare variant PGS, we filtered genes according to (a) their likelihood of harboring a true HbA1C association and (b) their likelihood of impacting HbA1C through erythrocytic pathways. We considered genes to have evidence of impacting HbA1C through erythrocytic pathways if they satisfied at least one of three criteria: they either (a) were included in an erythrocytic mouse gene set nominally (P < 0.05) enriched for HbA1C associations; (b) had a nominal (P ≤ 0.05) rare variant gene-level association with RBC; or (c) were located within an RBC GWAS locus.
We explored five strategies for determining genes (Extended Data Fig. 5a):
Strict gene set. These genes demonstrated evidence of impacting HbA1C through erythrocytic pathways (as defined above) and, as evidence of HbA1C association, demonstrated exome-wide significant gene-level rare variant HbA1C associations.
Relaxed gene set. These genes demonstrated evidence of impacting HbA1C through erythrocytic pathways (as defined above) and, as evidence of HbA1C association, demonstrated nominally-significant (P ≤ 0.05) HbA1C gene-level rare variant associations and at least one further line of evidence supporting their association with HbA1C: (a) presence in a glycemic mouse gene set with P < 0.05 HbA1C enrichment or (b) proximity to an HbA1C GWAS association.
Loose gene set. These genes demonstrated evidence of impacting HbA1C through erythrocytic pathways (as defined above) and, as evidence of HbA1C association, demonstrated nominally significant (P ≤ 0.05) HbA1C gene-level rare variant associations.
Negative control gene set. These genes demonstrated nominally significant (P ≤ 0.05) HbA1C rare variant gene-level associations (analogous to the “Loose” gene set). Genes that had evidence of impacting HbA1C through erythrocytic pathways were omitted.
Negative control gene set with glycemic filter (Glycemic gene set). These genes demonstrated nominally significant (P ≤ 0.05) HbA1C gene-level rare variant associations and were (a) included in a glycemic gene set that showed nominally significant (P < 0.05) enrichment for HbA1C associations or (b) were located within a fasting glucose or fasting insulin GWAS locus (analogous to the “Relaxed” gene set). Genes that had evidence of impacting HbA1C through erythrocytic pathways were omitted.
Variant weights in the rare variant polygenic score.
After we define a set of genes for the rare variant PGS, we then define a set of variant groups for each gene , estimated effect sizes for each group , and the estimated combined allele frequency in the population of variants in . We tested three different methods for determining these parameters (Extended Data Fig. 5b). Each method defines groups based on the seven nested masks we analyzed.
The nested variant method assigns an effect to equal to the aggregate effect size for the most stringent mask including . The nested method uses one group for each gene and each mask, each consisting of the variants within that gene and mask but not within any more stringent masks. and are calculated using all variants in the gene and mask – notably, this includes variants also in more stringent masks and therefore not in .
The unique variant method is similar to the nested variant method but attempts to correct for the potential overestimation of effects resulting from variants within more stringent masks. The unique variant method uses the same groups as the nested variant method; however, and are calculated using only variants in .
The weighted variant method attempts to estimate variant weights using data from all masks. Following a previous study27, we assigned each mask a weight ranging from 0 to 1 and then for each gene conducted a weighted burden test in which HbA1C was regressed on all coding variants, each weighted by the value of the most stringent mask that contained it. The weighted variant method then uses the same groups as the nested and unique variant method, with equal to the fraction of individuals that carry variants in The effect size is equal to the weight of the mask multiplied by the weighted effect size estimate for the gene.
Combined polygenic score.
To construct a combined PGS, we first used conditional analysis to evaluate independence between the variants used in the rare and common variant PGS. To do so, we repeated the gene-level analysis for each variant mask used in the rare variant PGS, but conditional upon the variants included in the common variant PGS. This analysis demonstrated that the variants included in the rare variant PGS are (almost entirely) independent of the variants included in the common variant PGS (Extended Data Fig. 8). Therefore, we constructed a combined PGS assuming an additive model where we summed the separate scores for each individual.
Assessing accuracy of the polygenic scores.
To evaluate each PGS, we identified individuals who (a) had reported HbA1C < 47.53 mmol/mol; (b) were not using antihyperglycemic medication; and (c) had PGS-adjusted HbA1C >47.53 mmol/mol. The total number of such “reclassified” individuals was taken as a measure of PGS sensitivity. To measure PGS specificity, we then calculated, of the reclassified individuals, how many were “true” T2D cases that either had (a) a 2-hour glucose measure ≥ 11.1 mmol/L or (b) fasting glucose levels ≥ 7 mmol. We compared the fraction of true T2D cases among those reclassified to the fraction of true cases among individuals matched (10 per reclassified case) on HbA1C level, cohort, and ancestry. We used a Fisher’s exact test to calculate P-values and odds ratios, both within each ancestry and across ancestries; trans-ancestry odds ratios were then calculated via meta-analysis. A P-value ≤ 0.05 was considered significant.
For the rare variant PGS, to avoid over-fitting, we estimated genetic effect sizes using only the UKB samples, and we used AMP-T2D-GENES samples to evaluate the score. As a further exploration of over-fitting, we also evaluated the performance of the models in the more restrictive case in which we used UKB samples to both select the genes and weights in each model. This analysis (Supplementary Tables 8 and 9, and Extended Data Fig. 7) suggested that the results and conclusions from our main models are unlikely to be substantially affected by over-fitting (see Supplementary Note).
Scaling ancestral proportions using NHANES.
To estimate how many true T2D cases in the US population would be reclassified by the various PGS, we estimated US ancestral proportions using the most recent release (2017-2018) of the National Health and Nutrition Examination Survey (NHANES) data, after adjusting for the survey design using the “survey” R package63. We then scaled the ancestry proportions in the AMP-T2D-GENES samples to match the estimated US ancestral proportions, assuming that the total US population size is ~330 million (based on the 2018 US Census Bureau estimates). As NHANES reports an “Asian American” sub-group, but does not further classify individuals as South or East Asian, we combined South and East Asian results from our rare and common variant PGS analysis into a single “Asian” ancestry via an inverse-weighted meta-analysis via METAL60.
Testing for ancestral heterogeneity.
To assess heterogeneity of common or rare variant effect sizes across ancestries, we used a Cochran’s Q test as implemented in the METAL software package60. For the common variant PGS, we downloaded (from www.magicinvestigators.org) previously published ancestry-specific summary statistics7. For the rare variant PGS, we computed ancestry-specific gene-level summary statistics by repeating our analysis procedure specific to each ancestry. We assessed the resulting Q test P-values via QQ-plots and by calculating λ values (Extended Data Fig. 9).
Extended Data
Extended Data Figure 1 |. Single variant HbA1C associations.

Manhattan plot of the single variant associations identified by our meta-analysis. Horizontal lines indicate the threshold used for exome-wide significance for coding variants (red: p≤1.8×10−8 as derived from a previous determined threshold49 p≤4.3×10−7 and Bonferroni correction for 24 phenotypes) and genome-wide significance for non-coding variants (green: p≤2.1×10−9 as derived from the traditional genome-wide significance threshold p≤5×10−8 and Bonferroni correction for 24 phenotypes). Single variant associations were determined via the efficient mixed-model association expedited (EMMAX) method50
Extended Data Figure 2 |. Effect sizes and proportion of variance explained for rare variant HbA1C gene-level associations.
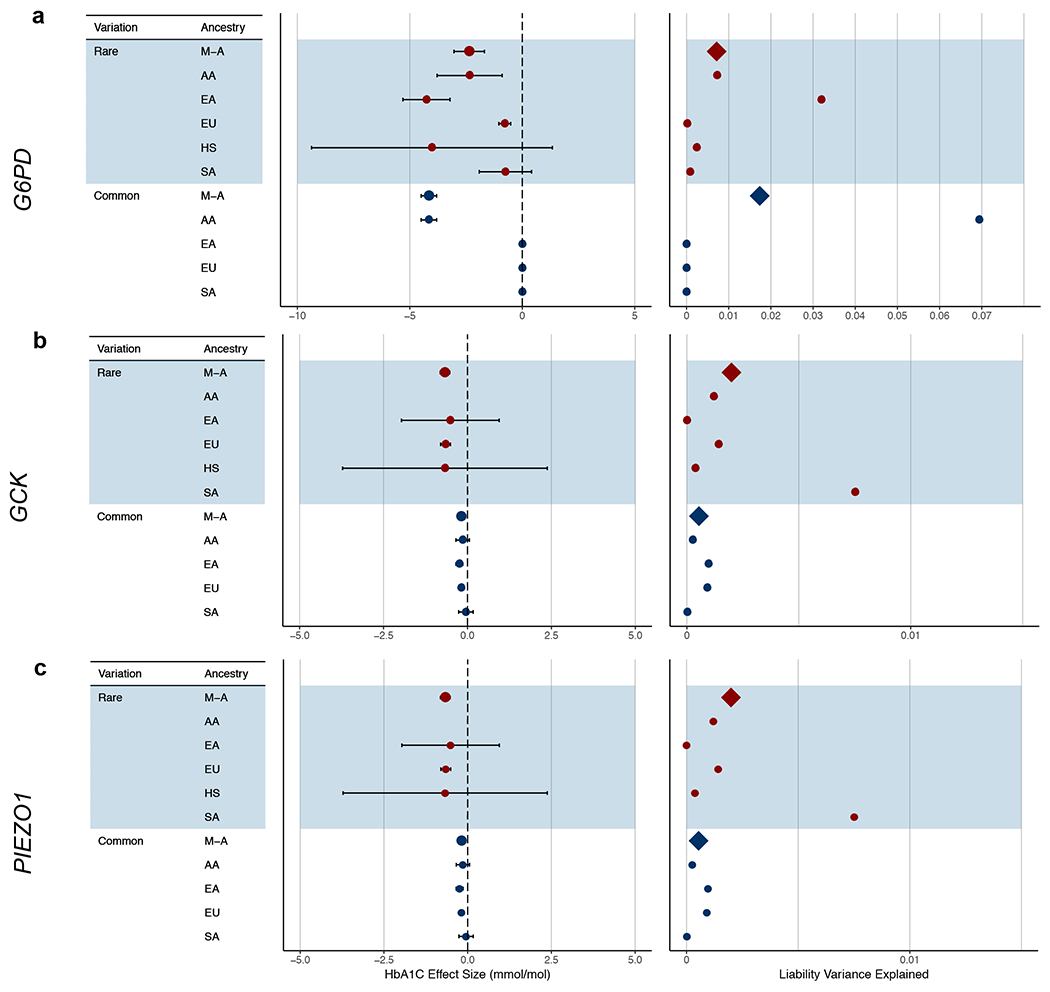
Results are displayed for a, G6PD (N=1,382 for AA; N=1,930 for EA; N=41,689 for EU; N=1,861 for SA; N=892 for HS), b, GCK (N=551 for EA; N=40,241 for EU; N=487 for HS), and (c) PIEZO1 (N=905 for AA; N=1,340 for EA; N=42,061 for EU; N=789 for SA; N=484 for HS). We calculated effect sizes (mmol/mol) and liability variance explained separately for each ancestry and then combined these via a meta-analysis. We performed the calculations for the strongest associated gene-level mask and for the strongest associated common variant within 125kb of the gene as previously reported7 (N=7,564 for AA; N=20,838 for EA; N=123,665 for EU; N=8,874 for SA). Proportion of variance explained is displayed as the proportion of total liability variance. Abbreviations: AA, African-American; EA, East Asian; EU, European; HS, Hispanic; SA, South Asian; M-A, meta-analysis. Error bars indicate 95% confidence intervals.
Extended Data Figure 3 |. Calculating and evaluating common variant polygenic scores.

We calculated common polygenic scores based on effect sizes and results from a previously published multi-ethnic HbA1C GWAS7. We calculated polygenic scores separately for each of the four ancestries in our test sample with available GWAS data, evaluated ancestry-specific odds ratios via a Fisher’s exact tests, and then combined these odds ratios via a fixed-effects meta-analysis to produce a transethnic odds ratio.
Extended Data Figure 4 |. Enrichment analyses of HbA1C and RBC rare variant gene-level associations.
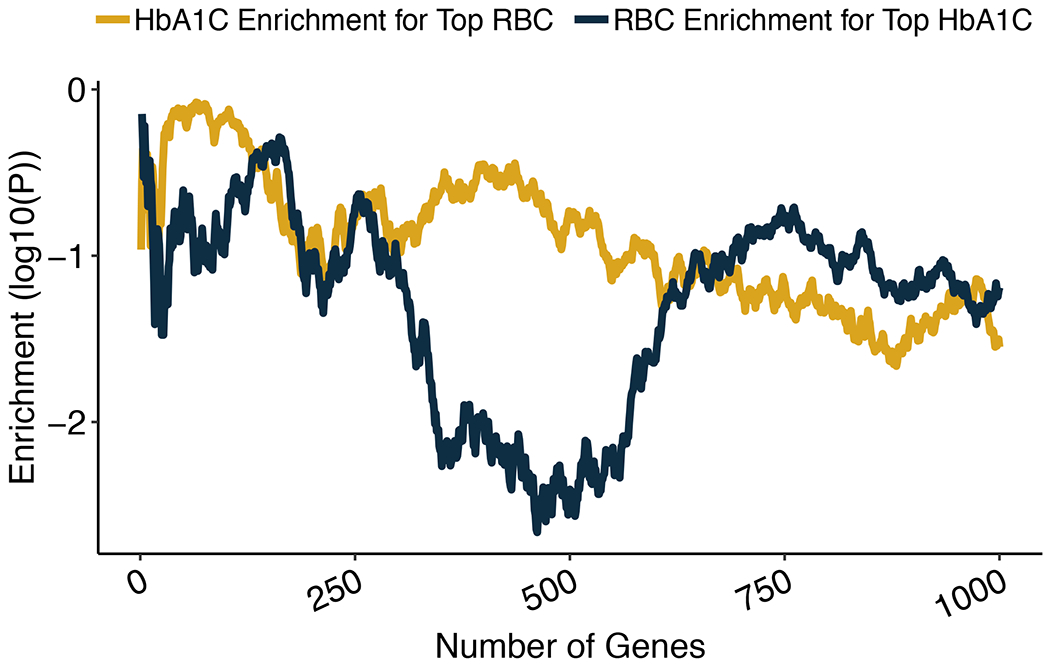
We ranked genes by their HbA1C gene-level p-value and tested the degree to which the top n associations (with n ranging from 1 to 1,000) were enriched for red blood cell count (RBC) gene-level associations. Enrichments were calculated using a one-sided Wilcoxon rank-sum test, comparing the RBC gene-level p-values of the top n HbA1C associations to the RBC gene-level p-values of background genes matched on the number of variants and total allele count; the solid blue line in the plot shows the one-sided Wilcoxon p-values as a function of n. As a negative control, we also conducted the reciprocal analysis in which we tested the top i RBC associations for enrichment for HbA1C associations; the solid yellow shows the one-sided Wilcoxon p-values.
Extended Data Figure 5 |. A framework for constructing polygenic scores that include rare variants.

The framework consists of two steps: a, choosing genes to include in the polygenic score, based on their association p-value and annotation, and b, defining weights for rare variants, based on the masks that include them and the aggregate effect sizes observed for the masks. a, We explored three methods for choosing genes, based on their strength of HbA1C association (blue boxes) and evidence of acting through erythrocytic pathways (red). “GLYCEMIC set” indicates genes located within a glycemic gene set enriched (at p≤0.05) for HbA1C rare variant associations, while “RBC set” indicates genes located within an erythrocytic gene set enriched (at p≤0.05) for HbA1C rare variant associations (the specific gene sets are shown in Figure 2). “HbA1C LOCUS” and “RBC LOCUS” indicates genes located within 125kb of a common variant HbA1C or RBC association, respectively. The two negative controls included only genes that failed the erythrocytic pathway filters (“Excluded”) and applied either the HbA1C association strength filters for the loose gene set (control 1) or the association strength filters for the relaxed gene set (control 2). b, We explored three methods for weighting variants (Methods): the aggregate effect size of the strictest mask that contained the variant (nested), the aggregate effect size of variants unique to the strictest mask that contained the variant (unique), or the aggregate effect size of a weighted burden test for the gene multiplied by the specific weight of the variant (weighted).
Extended Data Figure 6 |. Testing the accuracy of the rare variant polygenic score.

As described in Figure 3 and Methods, for each of the nine rare variant polygenic scores (three variant weighting schemes for each of three gene set definitions; Extended Data Figure 5), we calculated Fisher’s odds ratios and 95% confidence intervals for the fraction of true T2D cases reclassified by the model as compared to the null expectation. The area of the diamond for each odds ratio is proportional to the total number of reclassified individuals in the AMP-T2D-GENES test sample (total N assessed=17,206; see Supplementary Table 9 for model-specific reclassification sample sizes). Error bars indicate 95% confidence intervals of the odds ratios.
Extended Data Figure 7 |. Secondary analysis of rare variant polygenic scores for UKB samples only.

To ensure that the ability of the rare variant polygenic score to reclassify an excess of true cases was not due to over-fitting, we built nine risk scores as in Extended Data Figure 6 but with genes selected from an analysis of only UKB samples (Methods). For each of the nine resulting rare variant polygenic scores, we calculated Fisher’s odds ratios and 95% confidence intervals for the fraction of true T2D cases reclassified by the model as compared to the null expectation. The area of the diamond for each odds ratio is proportional to the total number of reclassified individuals in the AMP-T2D-GENES test sample (total N assessed=17,206; see Supplementary Table 9 for model-specific reclassification sample sizes). Error bars indicate 95% confidence intervals of the odds ratios.
Extended Data Figure 8 |. Impact of adjusting rare variant effects for common variants included in the polygenic score.

Scatterplots indicate HbA1C gene-level effect sizes (mmol/mol) as estimated by burden tests with and without variants from the common variant PGS included as covariates in the test. a-g, Results are shown for each of the seven rare variant masks. We analyzed genes with nominal (p≤0.05) rare variant associations and within 125kb of a variant in the common variant PGS. Results indicate that, on average, rare variant effects remain roughly the same when adjusting for common variants. Spearman’s rank correlation coefficients (i.e. rho) and associated two-sided p-values are indicated on plots, as are the slopes (i.e. beta) and two-sided p-values from linear regression. Blue dotted lines show the linear regression slopes; red lines indicate a slope of 1.
Extended Data Figure 9 |. Testing for heterogeneity across ancestry for variants included in common variant and rare variant polygenic scores.

We used a Cochran’s Q test to evaluate heterogeneity across ancestry-level single-variant and gene-level association results. QQ plots are shown for p-values from a, single-variant Q tests for common variants and b-h, gene-level Q tests for different rare variant masks; included in each analysis were the variants (or genes) included in the corresponding polygenic score. Departures above the diagonal red line suggest heterogeneity beyond the null expectation (blue lines indicate 90% confidence intervals for the null expectation), while lambda values indicate the ratio of the median observed chi square statistic to the median of the expected chi square statistic under the null; larger lambda values indicate larger deviations from the null.
Extended Data Figure 10 |. Fraction of variants found in enriched erythrocytic glycemic gene sets with negative effects on HbA1C levels.

Reported is the fraction of variants with negative HbA1C effect sizes (based on the single variant meta-analysis) within genes (i) with HbA1C gene-level p≤0.05 and (ii) within a significantly enriched (p≤0.05) erythrocytic (N=4) or glycemic (N=5) gene set. a-g, Results are shown for variants within each mask. The bars represent the fractions observed for variants across all gene sets, while the dots represent the fractions observed for variants within each individual gene set. A two-sided t-test was used to assess potentially significant differences; p-values are shown above each plot. Error bars indicate standard error.
Supplementary Material
Acknowledgements
This project was supported by R01DK125490 and UM1DK105554. J.B.C. is supported by a NIDDK Pathway to Independence Award (K99DK127196). A.L. was supported by Grant 2020096 from the Doris Duke Charitable Foundation. J.B.M. was supported by NIH R01 DK078616 and R01HL151855. J.C.F. was supported by NHLBI K24 HL157960. J.I.R. was supported by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756, and is also supported in part by the National Institutes of Health, National Heart, Lung, Long and Blood Institute (NHLBI) contract 1R01HL151855 and the National Institute of Diabetes and Digestive and Kidney Diseases contract UM1DK078616. M.S.U. was supported by K23DK114551.
Competing Interests
As of April 2022, P.D. is an employee and stockholder of Regeneron Pharmaceuticals. J.B.M. is an Academic Associate for Quest Diagnostics Endocrinology R&D. J.C.F. has received consulting honoraria from Novo Nordisk and AstraZeneca, and speaker fees from Merck, Novo Nordisk and AstraZeneca for research lectures over which he had full control of content. All other authors declare no competing interests.
Footnotes
Code availability
All software used in the analysis was open source and is described in the Methods section of the manuscript. Software used: BWA v0.7, Picard v1, GATK v3.4, EPACTS v3.2.4, Plink 1.9, METAL meta-analysis tool, Michigan Imputation Server, Klustakwik v.1.5, FlashPCA2 v.2.0, HAIL v.0.2.61, KING v.2.1.2 , VEP v87, and Tabix v.0.2.6. Previously published scripts (https://www.nature.com/articles/s41586-019-1231-2.pdf) were used to conduct the minimum P-value test and perform the Wilcoxon rank sum test for gene sets. A custom script was written to calculate common and rare variant polygenic scores (available via github: https://github.com/PeterDornbos/dornbos_ps). The type 2 diabetes knowledge portal was used to scan for GWAS loci (https://t2d.hugeamp.org/).
Data availability
Sequence data and phenotypes from the AMP-T2D-GENES study are available via the database of Genotypes and Phenotypes (dbGAP) and/or the European Genome-phenome Archive, as indicated in Supplementary Table 14. Access to data from the UK Biobank can be obtained at https://www.ukbiobank.ac.uk/enable-your-research.
References
- 1.Vilhjálmsson BJ et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi SW, Mak TS & O’Reilly PF Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc 15, 2759–2772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khera AV et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mavaddat N et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet 104, 21–34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler E et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 14, e1002383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leong A & Meigs JB Type 2 diabetes prevention: implications of hemoglobin A1c genetics. Rev. Diabet. Stud 12, 351–362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarnowski C et al. Impact of rare and common genetic variants on diabetes diagnosis by Hemoglobin A1c in multi-ancestry cohorts: the Trans-Omics for Precision Medicine Program. Am. J .Hum. Genet 105, 706–718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soranzo N et al. Common variants at 10 genomic loci influence hemoglobin A₁(C) levels via glycemic and nonglycemic pathways. Diabetes 59, 3229–3239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higgins PJ & Bunn HF Kinetic analysis of the nonenzymatic glycosylation of hemoglobin. J. Biol. Chem 256, 5204–5208 (1981). [PubMed] [Google Scholar]
- 12.American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 44, S15–S33 (2021). [DOI] [PubMed] [Google Scholar]
- 13.American Diabetes Association. 3. Prevention or Delay of Type 2 Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 44, S34–S39 (2021). [DOI] [PubMed] [Google Scholar]
- 14.American Diabetes Association. 6. Glycemic Targets: Standards of Medical Care in Diabetes-2021. Diabetes Care 44, S73–S84 (2021). [DOI] [PubMed] [Google Scholar]
- 15.American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care 44, S111–S124 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Cohen RM et al. Red cell life span heterogeneity in hematologically normal people is sufficient to alter HbA1c. Blood 112, 4284–4291 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chai JF et al. Genome-wide association for HbA1c in Malay identified deletion on SLC4A1 that influences HbA1c independent of glycemia. J. Clin. Endocrinol. Metab 105, dgaa658 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Chen P et al. Multiple nonglycemic genomic loci are newly associated with blood level of glycated hemoglobin in East Asians. Diabetes 63, 2551–2562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet 53, 840–860 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu W et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich JK et al. Determinants of penetrance and variable expressivity in monogenic metabolic conditions across 77,184 exomes. Nat. Commun 12, 3505 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bamshad MJ et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet 12, 745–755 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Cirulli ET & Goldstein DB Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet 11, 415–425 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Abecasis GR, Boehnke M & Lin X Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet 95, 5–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Hout CV et al. Exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flannick J et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luzzatto L, Nannelli C & Notaro R Glucose-6-phosphate dehydrogenase deficiency. Hematol. Oncol. Clin. North Am 30, 373–393 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Pandolfi PP et al. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 14, 5209–5215 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cahalan SM et al. Piezo1 links mechanical forces to red blood cell volume. Elife 4, e07370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faucherre A, Kissa K, Nargeot J, Mangoni ME & Jopling C Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 99, 70–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beutler E G6PD deficiency. Blood 84, 3613–3636 (1994). [PubMed] [Google Scholar]
- 33.Cappellini MD & Fiorelli G Glucose-6-phosphate dehydrogenase deficiency. Lancet 371, 64–74 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Crouch DJM & Bodmer WF Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc. Natl. Acad. Sci. USA 117, 18924–18933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudbridge F Power and predictive accuracy of polygenic risk scores. PLoS Genet. 9, e1003348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CL & Eppig JT The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med 1, 390–399 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mi H, Muruganujan A, Ebert D, Huang X & Thomas PD PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conesa A et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- 39.The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M, Furumichi M, Tanabe M, Sato Y & Morishima K KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altshuler D, Daly MJ & Lander ES Genetic mapping in human disease. Science 322, 881–888 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahajan A et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet 50, 559–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupski JR, Belmont JW, Boerwinkle E & Gibbs RA Clan genomics and the complex architecture of human disease. Cell 147, 32–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuchsberger C et al. The genetic architecture of type 2 diabetes. Nature 536, 41–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Estrada K et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311, 2305–2314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claussnitzer M et al. A brief history of human disease genetics. Nature 577, 179–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bodmer W & Bonilla C Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet 40, 695–701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sveinbjornsson G et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet 48, 314–317 (2016). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 50.Kang HM et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet 42, 348–354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lohmueller KE et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am. J. Hum. Genet 93, 1072–1086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams AL et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eastwood SV et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS One 11, e0162388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hindy G et al. Rare coding variants in 35 genes associate with circulating lipid levels – a multi-ancestry analysis of 170,000 exomes. Am. J. Hum. Genet 109, 81–96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaren W et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X, Wu C, Li C & Boerwinkle E dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat 37, 235–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aken BL et al. Ensembl 2017. Nucleic Acids Res. 45, D635–D642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shim H et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One 10, e0120758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 63.Lumley T survey: analysis of complex survey samples. R package version 4.0 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data and phenotypes from the AMP-T2D-GENES study are available via the database of Genotypes and Phenotypes (dbGAP) and/or the European Genome-phenome Archive, as indicated in Supplementary Table 14. Access to data from the UK Biobank can be obtained at https://www.ukbiobank.ac.uk/enable-your-research.