

Abstract
Glutathione S-transferase P1(GSTP1) is known for its transferase and detoxification activity. Based on disease-phenotype genetic associations, we found that GSTP1 might be associated with bone mineral density through Mendelian randomization analysis. Therefore, this study was performed both in vitro cellular and in vivo mouse model to determine how GSTP1 affects bone homeostasis. In our research, GSTP1 was revealed to upregulate the S-glutathionylation level of Pik3r1 through Cys498 and Cys670, thereby decreasing its phosphorylation, further controlling the alteration of autophagic flux via the Pik3r1-AKT-mTOR axis, and lastly altering osteoclast formation in vitro. In addition, knockdown and overexpression of GSTP1 in vivo also altered bone loss outcomes in the OVX mice model. In general, this study identified a new mechanism by which GSTP1 regulates osteoclastogenesis, and it is evident that the cell fate of osteoclasts is controlled by GSTP1-mediated S-glutathionylation via a redox-autophagy cascade.
Keywords: GSTP1, S-glutathionylation, Autophagic flux, Osteoclastogenesis, Pik3r1/AKT/mTOR
Graphical abstract
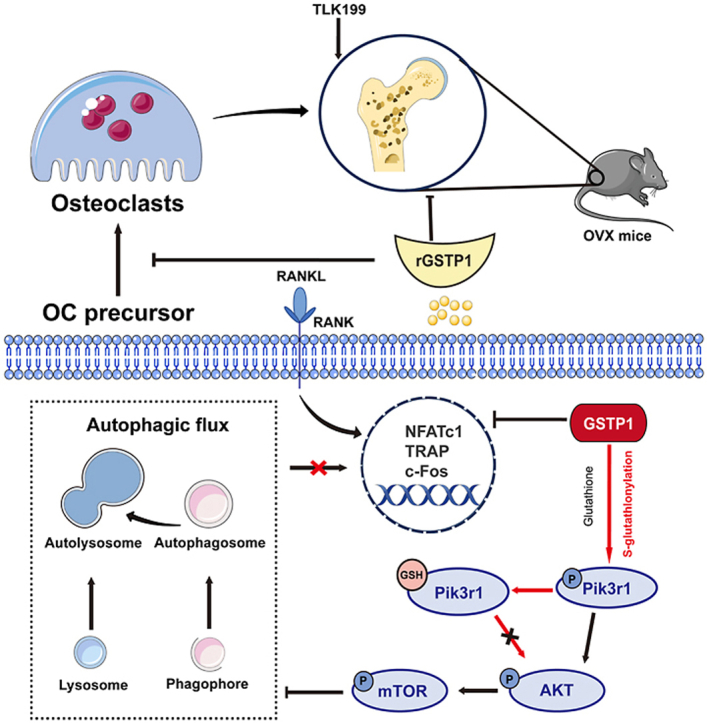
Highlights
-
•
Explore potential disease targets through Mendelian randomization analysis.
-
•
Elaborate the effect of GSTP1 on osteoclastogenesis and bone loss.
-
•
GSTP1 mediates the S-glutathionylation of Cys498 and Cys670 on Pik3r1.
-
•
This redox modification can affect osteoclastogenesis through regulating autophagic flux.
Abbreviations
- BMD
bone mineral density
- MDS
myelodysplastic syndromes
- GSTP1
glutathione S-transferase P1
- rGSTP1
recombinant GSTP1 protein
- GSH
glutathione
- MR
Mendelian Randomization
- GWAS
genome-wide association studies
- IVW
inverse variance weighting
- CLL
Chronic lymphocytic leukemia
- MM
Multiple myeloma
- MPN
Myeloproliferative neoplasms
- WB
Western Blot
- Co-IP
Co-immunoprecipitation
- BMMs
bone marrow monocytes
- PCR
polymerase chain reaction
- SEM
standard error of mean
- siRNA
small interfering RNA
- shRNA
short hairpin RNA
- NFATc1
nuclear factor of activated T cells 1
- Acp5
acid phosphatase 5
- CTSK
cathepsin K
- Pik3r1
phosphoinositide-3-kinase regulatory subunit 1
- AKT
thymoma viral proto-oncogene 1
- mTOR
mechanistic target of rapamycin kinase
- MAR
mineral apposition rate
1. Introduction
Osteoporosis is a metabolic bone disease characterized by decreased bone mass and increased fracture risk, mainly in bone mineral density [1]. Due to the world's population aging, osteoporosis often leads to severe consequences such as fractures, resulting in more expensive healthcare costs, a challenge that should not be underestimated [2]. Therefore, the emphasis on preventing and treating declining bone mineral density and osteoporosis has far-reaching clinical implications. Secondary osteoporosis is defined as osteoporosis that is aggravated by other illnesses or pharmaceutical exposures. It can affect 30% of women, >50% of premenopausal women, and 50%–80% of men [3]. Moreover, the relationship between hematologic disorders and bone health is thought-provoking in many clinical observational studies [4].
The Mendelian randomization method uses a genetic variation to determine whether the observed associations between risk factors and outcomes are consistent with causality [5]. The genetic relationship between dozens of hematologic disorders and bone mineral density using Mendelian randomization studies was explored. It was found that myelodysplastic syndromes (MDS) may influence bone mineral density regarding genetic associations. Next, the known signaling and targeting agents associated with MDS were reviewed [6,7]. By showing a regional map of these targets in the BMD-related GWAS databases, combined with previous literature [8], glutathione S-transferase P1 (GSTP1) was found to be a potential site that may affect bone mineral density.
Osteoclasts play an indispensable role in bone mineral density and associated bone metabolism. Recently, studies on autophagy and osteoclasts have gradually attracted our attention. Previous articles have shown that autophagy has diverse effects on osteoclast differentiation and bone metabolism [9].
As a phase II detoxification enzyme, GSTP1 is well known for its transferase and detoxification activity. In terms of its impact on disease, GSTP1 has multiple roles in the development and progression of several cancers [10]. In addition, several recent studies have reported its great therapeutic potential in lung injury, asthma, and other diseases [11,12]. On the cellular and molecular side, GSTP1 has been associated with autophagy regulation in breast cancer cells [13], pericardial cells [14], and ROS-related pathways [15].
According to recent studies, GSTP1 dramatically accelerates the rate and magnitude of protein S-glutathionylation, which then regulates protein activation or related physiological functions [[16], [17], [18]].
In this study, it was found that GSTP1 can upregulate the level of S-glutathionylation through Cys498 and Cys670 of Pik3r1, thereby inhibiting its phosphorylation, further regulating the change of autophagic flux through the Pik3r1-AKT-mTOR axis, and finally affecting the differentiation of osteoclasts in vitro. Not only that, in vivo, gene-level knockdown of GSTP1 results in a bone-loss phenotype in mice. And it is proposed that recombinant GSTP1 protein (rGSTP1) can rescue bone loss in OVX mice; and TLK199, a targeted inhibitor of GSTP1, accelerates this process. Our study suggests a novel redox-autophagic mechanism and role for GSTP1 in osteoclast differentiation and osteoporosis.
2. Material and methods
2.1. Mendelian randomization analysis
Five hematological illnesses were chosen as the exposure sets [[19], [20], [21], [22], [23]], and heel bone mineral density determined by quantitative ultrasonography(eBMD) was chosen as the outcome set [24,25].
A two-sample MR analysis was performed using the inverse variance weighting (IVW) approach to assess the causal influence of hematological diseases on eBMD. Moreover, we utilized the "MRPRESSO" technique to evaluate possible pleiotropic effects; also, the IVW and MR-Egger regression were used to find heterogeneity, which was quantified using the Cochran Q statistic, with a P value of 0.05 indicating significant heterogeneity. In addition, the "leave-one-out" sensitivity analysis was also used to uncover probable SNPs of significance. All statistical analyses were carried out using the R version 4.0.3 statistical program and the TwoSampleMR package (v0.5.6) and MRPRESSO package (v1.0), both of which were based on the MR-PRESSO vignette and the TwoSampleMR guideline (using RStudio v1.3.1093).
All the specific analysis process is as described in previous study [26].
2.2. Plasmids, antibodies and regents
Si-GSTP1 sequence:
mouse-GSTP1-siRNA-Forward(F): 5′-CCAACUAUGAGAAUGGUAATT-3′
mouse-GSTP1-siRNA-Reverse(R): 5′-UUACCAUUCUCAUAGUUGGTT-3′;
GSTP1-shRNA1: 5′-GCCCAGATGGATATGGTGAAT-3′;
GSTP1-shRNA2: 5′-CCAGATCTCCTTTGCCGATTA-3′;
mCherry-EGFP-LC3 lentiviral virus (Hanbio Biotechnology);
pcDNA3.1-Pik3r1(NM_001077495)-myc-c, pcDNA3.1-Pik3r1(C498A, Cysteine at position 498 is replaced by Alanine)-myc-c, pcDNA3.1-Pik3r1(C656A)-myc-c, pcDNA3.1-Pik3r1(C670A)-myc-c, pcDNA3.1-Pik3r1(C498A,C670A)-myc-c, pcDNA3.1-GSTP1-FLAG (GuanNan. co, Ltd, Hangzhou, China);
TLK199 (MCE, HY-13634A), Rapamycin (MCE, HY-10219); Recombinant GSTP1 protein (rGSTP1) was prepared and purified as previously described [18], and its biological activity has been demonstrated.
Antibodies: GSTP1 (proteintech, 15902-1-AP, 1:1000), β-actin (proteintech, 66009-1-Ig, 1:20000), NFATc1(SantaCruz, sc-7294, 1:300), c-Fos (Abcam, ab190289, 1:1000), Acp5 (proteintech, 11594-1-AP, 1:1000), LC3B (proteintech, 14600-1-AP, 1:1000), P62/SQSTM1 (proteintech, 18420-1-AP, 1:1000), Myc-tag (Beyotime, AF5054, 1:1000), Flag-tag (Beyotime, AF2852, 1:1000), p-Erk1/2 (CST, 4370, 1:1000), Erk1/2(CST, 4695, 1:1000), p-PI3 Kinase p85 (CST, 17366, 1:1000), PI3 Kinase p85 (Abcam, ab191606, 1:1000), PI3 Kinase p110 (proteintech, 20584-1-AP, 1:1000), p-AKT (proteintech, 66444-1-Ig, 1:1000), AKT (proteintech, 10176-2-AP, 1:1000), mTOR (proteintech, 66888-1-Ig, 1:1000), p-mTOR (proteintech, 67778-1-Ig, 1:1000), GSH (Abcam, ab19534, 1:500); Alexa Fluor® 594 (Abcam, ab150116, 1:500), Alexa Fluor® 488 (Abcam, ab150113, 1:500), Goat Anti-rabbit HRP (Fdbio, FDR007, 1:5000), Goat Anti-Mouse HRP (Fdbio, FDM007, 1:5000).
2.3. Primary cell culture and osteoclast differentiation
STR profiles confirmed Raw264.7 (Procell, CL-0190) and HEK-293T (Procell, CL-0005) cell line identity, primary BMMs (bone marrow monocytes) were extracted from the femur and tibia of C57BL/6 mice using the prior experimental method [27], Briefly, 8-week-old C57BL/6 were aseptically dissected, femurs and tibias were harvested, and the medullary cavity was flushed. BMMs were allowed to adhere using medium containing 30 ng/mL of M-CSF. After 3 days of culture, it was used for subsequent differentiation experiments of osteoclasts. The BMMs on the plate were added with RANKL at a concentration of 50 ng/mL on days 0, 1, 3, and 5 (the concentration of RANKL can be adjusted appropriately for different experiments). In the absence of other interventions, differentiated mature osteoclasts could be seen on the 3rd day, and the number of mature osteoclasts reached the threshold on the 5th day.
RAW264.7 and BMMs were cultured and differentiated in 10% FBS (Procell, 164210-50), DMEM (Procell, PM150210), 1% Penicillin-Streptomycin Solution (Procell, PB180120), 30 ng/ml M-CSF (R&D Systems, 416-ML-010) and different concentrations of RANKL (R&D Systems, 462-TEC-010) at 5% CO2, 37 °C.
Without intervention, osteoclast differentiation can be observed under 50 ng/mL RANKL culture for 3–5 days. After fixation with 4% paraformaldehyde for 20min, the cells were stained with TRAP solution (Sigma-Aldrich, CS0740) for 1h. The number of osteoclasts and the number of nuclei were observed and counted under an inverted microscope.
2.4. BMSC cell extraction, differentiation and alkaline phosphatase (ALP) staining
BMSCs were extracted from 8-week-old C57BL/6 mice. Briefly, the tibias and femurs of 8-week-old C57BL/6 mice were aseptically harvested, the bone marrow cavity was flushed and bone marrow cells were collected. Transfer to a Petri dish and retain the adherent cells. After the cells reached 70%–80% confluency, the cells from passage 6 to passage 10 were used for subsequent osteoblast differentiation experiments. BMSCs were cultured in the osteogenic differentiation induction medium (ODM; low-sugar Dulbecco's modified Eagle's medium; 10% fetal bovine serum, 10 mM β-glycerophosphate, 100 nM dexamethasone, 1% penicillin–streptomycin and 0.05 mM l-ascorbic acid-2-phosphate), and the induction medium was replaced every 3 days. When the osteogenic induction reached the 7th day, the cells were stained and analyzed using the ALP Chromogenic Kit (Beyotime, P0321S).
2.5. RT-qPCR
Cells were seeded in 12-well plates (2 × 105 cells/well), and after 3–5 days of culture, total mRNA was extracted using TRIzol reagent (Invitrogen). Then, mRNA reverse transcription and RT-qPCR were performed according to the experimental procedure of One Step RT-PCR Kit (Accurate Biotechnology, AG11606). The sequences of the relevant primers are shown below:
GSTP1: F:5′- ATGCCACCATACACCATTGTC -3′, R:5′- GGGAGCTGCCCATACAGAC -3′;
NFATc1: F:5′-GACCCGGAGTTCGACTTCG -3′, R:5′-TGACACTAGGGGACACATAACTG -3′;
ACP5: F:5′- CACTCCCACCCTGAGATTTGT -3′, R:5′- CATCGTCTGCACGGTTCTG -3′;
c-Fos: F:5′- CGGGTTTCAACGCCGACTA -3′, R:5′- TTGGCACTAGAGACGGACAGA -3′;
CTSK: F:5′-GAAGAAGACTCACCAGAAGCAG -3′, R:5′-TCCAGGTTATGGGCAGAGATT -3′.
2.6. Co-immunoprecipitation and Western Blot
Western blot and co-immunoprecipitation associated with S-glutathionylation were performed under non-reducing, non-denaturing conditions. Initially, cells were harvested, lysed in non-reducing, non-denaturing lysis buffer (Solarbio, R0030), and buffered with Native Gel Sample Loading Buffer (Beyotime, P0016N). The corresponding protein complexes were then immunoprecipitated with the relevant indicated antibodies, incubated overnight with protein A + G Magnetic Beads (Beyotime, P2179M) at 4 °C, and washed with lysis buffer after incubation. Proteins were separated by PAGE gel (Beyotime, P0524M) and native electrophoresis solution (Beyotime, P0556), transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, ISEQ00010), and finally further imaged and quantitatively analyzed.
2.7. Cell transfection and stable cell construction
To knock down or overexpress target proteins, we used RNAiMAX (Thermo Fisher, 13778150) and si-RNA to transfect BMMs, Lipofectamine 3000 (Thermo Fisher, L3000008) and tagging plasmids were used to transfect BMMs or Raw264.7 cells.
Lentiviral infection was used to produce Raw264.7 cell lines stably expressing shRNA or stably expressing mCherry-EGFP-LC3. Lentiviruses were created in HEK 293T cells using the target and packaging vectors pMD2 and psPAX, then infected Raw264.7 cells for two days, and selected with puromycin (10 μg/mL) until stable cell lines were generated. These cells were used for subsequent confocal microscopy imaging or WB experiments.
2.8. Quantification of autophagic flux using flow cytometry
The Raw264.7 cell line stably transfected with mCherry-EGFP-LC3 was used. After different experimental treatments and interventions, cells were trypsinized and resuspended. Flow cytometric analysis was performed using Beckman Coulter CytoFLEX LX and CytExpert v2.4 software, GFP and mCherry fluorescence were excited by 488 and 580 nM lasers. The mCherry/GFP ratio results were generated and analyzed by FlowJo v10 software.
2.9. Transmission electron microscope (TEM)
The samples were fixed overnight at 4 °C with 2.5% glutaraldehyde, then embedded and sliced (LEICA EM UC7), and finally the microscopic cell morphology was observed under a transmission electron microscope (Hitachi H-7650).
2.10. Protein molecular docking and conservation analysis
The 3D structure of Pik3r1 was obtained from the Uniprot website (https://www.uniprot.org/) and obtained using Alphafold. Since the protein homology is greater than 30%, the predicted structure can be used for docking. At the same time, the structural formula of the ligand molecule glutathione was downloaded from Pubchem data (https://pubchem.ncbi.nlm.nih.gov/), and the structure in pdb format was obtained by PyMOL software conversion. In this study, the analytical docking was performed through the Molecular Operating Environment (MOE v2019.1001, Chemical Computing Group Inc., Montreal, QC, Canada). Before docking, select the implicit solvation model of the force field and reaction field (R-field) of AMBER10: EHT. The protein's protonation state and hydrogen orientation were optimized using LigX at 300 K and pH 7.0, and the energy was minimized by MOE using default parameters. Covalent docking analysis was performed using MOE software. Use "GBVI/WSA dG" for scoring. GBVI/WSA dG is a force field-based scoring function that determines the binding free energy (kcal/mol) of a ligand at a given pose.
Conservation analysis of the target protein was performed using the online tool The Consurf Server (https://consurf.tau.ac.il/quick_help.php).
The sequences of the target proteins were aligned between different species by Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
2.11. Mass spectrometry
Raw264.7 cells were plated in 10 cm culture dishes, and the plasmid was transfected to overexpress GSTP1. At the same time, M-CSF (30 ng/mL) and RANKL (100 ng/mL) were added on days 0, 1, and 3 to promote osteoclast formation. On the third day, cell lysates were collected and incubated overnight at 4 °C with GSH antibody to pull down S-glutathionylated proteins. Electrophoresis experiments were performed under non-reducing and non-denaturing conditions, and proteins were visualized on the gel by silver staining (Biosharp, BL620A). Take the gel with a molecular weight of 70-100kD for subsequent enzymatic hydrolysis and mass spectrometry detection. Thermo Fisher Q Exactive was interfaced to a Thermo Fisher Easy-nLC 1000 system for nano LC/MS/MS analysis of the digest. The detection method is positive ions, the precursor ion scanning range is 300–1800 m/z, the resolution of the primary mass spectrometer is 70,000 at 200 m/z, the AGC (Automatic gain control) target is 1e6, the Maximum IT is 50 ms, and the dynamic exclusion time (Dynamic exclusion) is 30.0s. The mass-to-charge ratio of Peptides and peptide fragments are collected according to the following method: 20 fragment spectra (MS2 scan) are collected after each full scan (full scan), MS2 Activation Type is HCD, Isolation window is 2 m/z, and MS2 resolution The rate is 17,500 at 200 m/z, the Normalized Collision Energy is 27eV, and the Underfill is 0.1%.
2.12. Generation of Pik3r1-KO Raw264.7 cell line
For Pik3r1 KO, the single guide RNAs (sgRNAs) were designed using the online CRISPR design tool (Red CottonTM, Guangzhou, China, https://en.rc-crispr.com/). The exon region of Pik3r1 was selected to be targeted by CRISPR/Cas9 genome editing. A ranked list of sgRNAs was generated with specificity and efficiency scores. The pair of oligos for two targeting sites was annealed and ligated to the YKO-RP006 vector (Ubigene Biosciences Co., Ltd., Guangzhou, China). The plasmids containing each target sgRNA sequences were transfected into cells with Lipofectamine 3000 (Thermo Fisher Scientific). 24–48 h after the transfection, puromycin were added to screen the cells. After antibiotic selection a certain number of cells were diluted by limited dilution method and inoculated into 96-well plate. Selection of single clones were performed after 2–4 weeks and selected Pik3r1 KO clones were validated by PCR and Sanger sequencing. The sgRNAs and primers for CRISPR design are shown in Supplementary Fig. 11.
2.13. Mice
All the animal experiments are complied with ethical standards of the Animal Care and Welfare Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine. All mice were kept in the specific pathogen-free animal care room with a temperature of 23 ± 3 °C, humidity of 55 ± 10% and a 12-h light/dark cycle (lights on from 8:00 a.m. to 8:00 p.m.). All mice were given ad libitum access to water and normal rodent chow.
2.14. Ovariectomy (OVX)-induced osteoporosis model
The OVX-induced osteoporosis model was established according to the previous method [27]. 13-week-old female C57BL/6J mice were selected for SHAM surgery or OVX surgery.
We conducted four batches of animal experiments. The first two batches of mice were used to explore the effect of GSTP1 alterations on bone in physiological state for 2 months, while the latter two groups were used to explore the effects of GSTP1 on bone loss under OVX conditions. The first batch of mice was divided into three groups of 5 mice each, the SHAM group (LV-NC lentivirus), the GSTP1 knockdown group (LV-GSTP1 lentivirus) and the LV-GSTP1+LY3023414 (10 mg/kg, ip) group. The lentiviral suspension (1.5 × 108 TU/ml, 50 μl) was injected into the bone marrow cavity every 10 days.
The second batch of mice was divided into three groups of 5 mice each, the SHAM-operated group, the TLK199 group, and the rGSTP1 group. Among them, TLK199 (50 mg/kg) was injected intraperitoneally every other day, and rGSTP1 (100 mg/kg) was injected by tail vein every other day.
The third batch lasted for 1 month, the mice were randomly divided into SHAM group, OVX group and TLK199 group, 5 mice in each group, and intraperitoneal injection of normal saline or TLK199 mice (50 mg/kg) every other day.
The last batch was available for two months. The mice were placed into three groups, each with 5 mice: SHAM, OVX, and rGSTP1. The mice were given a tail vein injection of either normal saline or rGSTP1 (100 mg/kg) every other day.
Mice were sacrificed at the expected time, and femurs were collected and fixed with 4% paraformaldehyde for subsequent analysis.
2.15. Calcein labeling
Calcein solution (20 mg/kg) was injected intraperitoneally on the 10th and 3rd days before the mice were sacrificed, and after the samples were collected, the femur tissues were separated. 4%PFA was used to fix bone tissue for at least 48 h. The tissue is then dehydrated in 60%–70% - 80%–90% - 95% alcohol. Each step takes anywhere for 24–72 h. Next, absolute ethyl alcohol is used twice to dehydrate and 12 h–24 h each time. The same is true for xylene.
After that, prepare three reagents: immersion solution I: methyl methacrylate 400 ml and dibutyl phthalate 100 ml mixing; immersion solution II: methyl methacrylate 400 ml and dibutyl phthalate 100 ml mix, add 15g benzoyl peroxide stirring mixture; Immersion solution III: Methyl methacrylate 400 ml mixed with dibutyl phthalate 100 ml, adding 25g benzoyl peroxide stirring mixture. Put tissue into three cylinders of permeates penetrating fluid at 4 °C for 24–72 h in turn. The tissue was put into the embedding bottle, add appropriate amount of immersion solution III, then vacuumed for 5 h and polymerized in a 37 °C water bath. Routine sections of approximately 10 μm were performed by a hard tissue slicer (Leica, HistoCore AUTOCUT). Finally, observe relevant parameters under a fluorescence microscope, such as mineral apposition rate (MAR: interlabel width/labeling period).
2.16. Microcomputed tomography (Micro-CT) and analysis
Micro-CT (Bruker SKYSCAN1276) was used to scan different groups of distal femur samples. According to the instrument instructions, the X-ray energy was set to 180μA/60 kV, and the equidistant resolution was set to 10 μm. Using CTAn and CTVol software, 3D modeling analysis was performed on the scan data, and the same area under the femoral growth plate was selected for calculation of trabecular bone parameters, including bone volume to total volume ratio (BV/TV), trabecular bone number (Tb. N), trabecular bone thickness (Tb. Th), and trabecular separation (Tb. Sp).
BV/TV refers to the ratio of mineralized bone volume (BV) to a given target total volume (TV). Tb.N and Tb.Th represent the average number of trabecular bone and the average thickness of trabecular bone in a given bone segment, respectively, and their values tend to be proportional to bone strength. Tb. Sp represents the average degree of separation of trabecular bone within a given medullary canal region, generally the higher the degree of separation, the greater the bone loss.
2.17. Immunohistochemistry and histology staining
The fixed mouse femurs were decalcified with 10% EDTA solution for 3 weeks before paraffin embedding and sectioning. Afterwards, H&E (hematoxylin and eosin) staining, TRAP staining (with fast green alcohol solution for background staining) and immunohistochemistry were performed. The number of osteoclasts on each bone surface (N.Oc/BS) and the ratio of osteoclasts to the area of the bone surface (Oc.S/BS) were then analyzed by microscopy, and bone tissue-related morphometric measurements were performed as before.
2.18. Statistical analysis
GraphPad Prism 8.0 was used to examine all statistics, which were represented as mean ± SEM. Statistical differences were analyzed by one/two-way ANOVA or two-tailed Student's t-test according to the actual situation. Experimental data for each quantitative analysis were replicated at least three times. Statistical significance was defined as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
3. Results
3.1. The effect of GSTP1 on bone mineral density is more likely to be achieved by osteoclasts than osteoblasts
This study performed a Mendelian randomization analysis (Supplementary Figs. S1A–H) to demonstrate the potential association between MDS and bone mineral density (Fig. 1A). Then, the known signal transduction and action sites of MDS were summarized. Through the region plots of these sites in the bone mineral density-related GWAS databases, it was found that GSTP1 may affect BMD (Fig. 1B). It is known that BMD is mainly affected by osteoblasts and osteoclasts.
Fig. 1.

The effect of GSTP1 on bone mineral density is more likely to be achieved by osteoclasts than osteoblasts. (A) Mendelian randomization analysis revealed the potential causal effect of MDS on BMD. (B) Region plots of known MDS therapeutic targets in the BMD-related GWAS databases. (C) scRNA-seq biological analysis of bone marrow cells from younger female (49 years old) and older female (60 years old, postmenopausal). (D) Violin plots of GSTP1 expression in different cell populations in bone marrow of two samples. (E) Violin plot comparing GSTP1 expression in bone marrow monocytes of young and old female after data normalization. (F) Protein expression and quantitative results of GSTP1 during the differentiation of BMMs. (G) Protein expression and quantitative results of GSTP1 during the differentiation of Raw264.7 cells. (H) Protein expression and quantification of GSTP1 in BMMs of SHAM-operated and OVX groups(C57BL/6). (I) H&E/TRAP staining and immunohistochemistry of the distal femur showed differences in the expression of GSTP1 between the SHAM-operated and OVX groups(C57BL/6). Green arrows indicate the degree of OVX-induced osteoporosis exhibited by H&E staining, and yellow arrows indicate the immunohistochemistry results of GSTP1 expression on the bone surface.
All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Based on sequencing results from previous studies (GSE37676) and our experimental validation,It was proved that there was no significant difference in the expression level of GSTP1 during the osteogenic differentiation of BMSCs and Mc3T3 cells (Supplementary Figs. S2A–C). Moreover, Knockdown of GSTP1 did not significantly affect Alkaline Phosphatase (ALP) activity (Supplementary Figs. S2D–F), an indicator revealing early phase of osteogenesis. At the same time, the results of qPCR showed that the expressions of related osteo-specific genes RUNX2 were not significantly changed (Supplementary Fig. S2G). These preliminary conclusions indicated that GSTP1 had little relationship with osteoblasts.
To further explore GSTP1 expression in bone marrow cellularity, we used previous bone marrow single-cell RNA-sequencing (scRNA-seq) datasets [28] for biological analysis (Fig. 1C). It showed that GSTP1 is highly expressed in bone marrow monocytes (Fig. 1D), ranking second in both young and old single-cell clusters (Supplementary Figs. S3A and B). And the expression of GSTP1 was lower in older female (with relatively lower bone density) than in young female (Fig. 1E). Osteoclasts are mainly derived from bone marrow monocytes, so these findings guide us to investigate the crucial role of GSTP1 in osteoclastogenesis.
During the differentiation of osteoclasts, whether in BMMs or Raw264.7 cells, the results of qPCR (Supplementary Figs. S4A and B) and Western Blot (Fig. 1F and G) showed that the expression of GSTP1 decreased with the days of differentiation, which is consistent with the sequencing data (GSE176265) from previous study (Supplementary Fig. S4C).
In addition, about BMMs extracted from murine medullary cavity, the results of WB (Fig. 1H) and immunohistochemistry (Fig. 1I) demonstrated that GSTP1 in ovariectomized (OVX) group was significantly lower than SHAM group, which is consistent with the findings of qPCR (Supplementary Fig. S4D) and previous sequencing data of GSE68303 (Supplementary Fig. S4E).
Overall, the potential effects of GSTP1 on bone mineral density are more likely to occur through the action of osteoclasts than osteoblasts.
3.2. GSTP1 plays an inhibitory role in osteoclastogenesis
We intend to explore the specific role of GSTP1 in osteoclastogenesis. The results showed that the pharmacological inhibitor of GSTP1 (TLK199) promoted osteoclast formation in a dose-dependent manner (Fig. 2A and B) at non-toxic concentrations (Supplementary Figs. S5A–C). Meanwhile, the findings of qPCR showed that TLK199 also had a dose-dependent up-regulation effect on the expression of osteoclast-related genes including NFATc1, Acp5, c-Fos, and CTSK when the murine bone marrow monocytes (BMMs) were stimulated with M-CSF and RANKL for 3 days (Fig. 2C). To clarify the effect of GSTP1 on osteoclasts, we transfected BMMs with NC-siRNA, GSTP1-siRNA, and also knocked down GSTP1 levels by transfecting GSTP1-shRNA in Raw264.7 cells. TRAP staining revealed that the number of osteoclasts and nuclei formed in the GSTP1-siRNA group were more than those in the NC-siRNA group (Fig. 2D and E), and the expression of osteoclast-specific proteins including NFATc1, Acp5, and c-Fos also had higher levels in the GSTP1-siRNA group (Fig. 2F). In Raw264.7 cells, the same WB results were also validated in the GSTP1-shRNA group (Fig. 2G).
Fig. 2.

GSTP1 plays an inhibitory role in osteoclastogenesis. (A) TRAP staining of different concentrations of TLK199 on BMMs under M-CSF and RANKL treatment. (B) Quantitative analysis of cell size and number of nuclei of TRAP-positive cells. (C) The qPCR results of osteoclast-specific genes at day 3 with different concentrations of TLK199. (D) TRAP staining of GSTP1-siRNA or NC-siRNA on BMMs under M-CSF and RANKL treatment. (E) Quantitative analysis of cell size and number of nuclei of TRAP-positive cells. (F) WB results and quantification of osteoclast-specific proteins during osteoclastogenesis when GSTP1 was knocked down by siRNA in BMMs. (G) WB results and quantification of osteoclast-specific proteins during osteoclastogenesis when GSTP1 was stably knocked down by shRNA in Raw264.7 cells. (H) TRAP staining of rGSTP1 on BMMs under M-CSF and RANKL treatment. (I) Quantitative analysis of cell size and number of nuclei of TRAP-positive cells. (J) WB results and quantification of osteoclast-specific proteins during osteoclastogenesis when BMMs were stimulated with rGSTP1. (K) WB results and quantification of osteoclast-specific proteins during osteoclastogenesis when Flag-GSTP1 plasmid was used to overexpress GSTP1 in Raw264.7 cells.
The concentration of RANKL used in knockdown of GSTP1 was 30 ng/mL, while the concentration used in overexpression experiments was 100 ng/mL. All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Conversely, we used exogenous recombinant GSTP1 protein (rGSTP1) to intervene in osteoclast differentiation of BMMs, and transfected Raw264.7 cells with pcDNA3.1, Flag-GSTP1 to overexpress GSTP1. The results of TRAP staining suggested that rGSTP1 could inhibit the osteoclast differentiation of BMMs (Fig. 2H and I), and down-regulated the expression of osteoclast-specific proteins including NFATc1 and Acp5 (Fig. 2J). The identical WB outcomes in the Flag-GSTP1 group in Raw264.7 cells were likewise confirmed (Fig. 2K).
Taken together, these findings collectively imply that GSTP1 might play an inhibitory role in osteoclastogenesis.
3.3. GSTP1 inhibits osteoclastogenesis through regulating autophagic flux
To investigate how GSTP1 affects osteoclasts, we initially looked at the morphological microstructure of Raw264.7 cells following rGSTP1 stimulation. Under the condition of M-CSF and RANKL stimulation, the images of transmission electron microscope (TEM) showed that rGSTP1 group presented more autophagic vesicles and phagocytic vesicles (Fig. 3A). Previous research has revealed that autophagy has a wide range of impacts on osteoclast differentiation [9], so we intend to investigate whether GSTP1 regulates autophagy levels during osteoclast differentiation. The mammalian autophagy protein LC3 is a marker of autophagosomes, and the amount of LC3-II is generally strongly connected to the number of autophagosomes. As an autophagy substrate, p62 is effectively destroyed by autophagy after being preferentially incorporated into autophagosomes by direct binding to LC3. As a result, the overall cellular expression of p62 was found to be inversely linked with autophagic activity [29,30].
Fig. 3.

GSTP1 inhibits osteoclastogenesis through regulating autophagic flux. (A) TEM images of Raw264.7 cells differentiated with RANKL (100 ng/mL) stimulation in rGSTP1 group and SHAM group, green arrows represent autophagosomes and phagosomes. (B) WB analysis of the effects of rGSTP1 on LC-I, LC3-II and p62 protein expression during BMMs differentiation. (C) WB analysis of the effects of siRNA knockdown of GSTP1 on LC-I, LC3-II and p62 protein expression during BMMs differentiation. (D) WB analysis of the effects of overexpression of GSTP1 with Flag-GSTP1 plasmid on LC-I, LC3-II and p62 proteins expression during Raw264.7 differentiation. (E) Raw264.7 cells stably knocked down GSTP1 were treated with M-CSF and RANKL, WB analysis of LC-I, LC3-II and p62 proteins expression during Raw264.7 differentiation. (F) Raw264.7 cells stably transfected with mCherry-GFP-LC3 were constructed. Experiments were performed with different concentrations of rGSTP1, and cells were observed using a confocal microscope after stimulation with M-CSF and RANKL for 3 days. (G) Under the same conditions as (F), the ratio of mCherry/GFP in different groups was quantified by flow cytometry. (H) Raw264.7 cells stably transfected with mCherry-GFP-LC3 were constructed. After transfecting cells with Si-NC and Si-GSTP1, these cells were observed by confocal microscopy with M-CSF and RANKL for 3 days. (I) Under the same conditions as (H), the ratio of mCherry/GFP in different groups was quantified by flow cytometry. (J) The autophagy activator rapamycin reversed the staining profile and quantification of TRAP-positive cells promoted by low levels of GSTP1. (K) Quantitative analysis of cell size of nuclei of TRAP-positive cells.
All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
First, we validated the regulation of autophagic flux by GSTP1 in BMMs. On the third day of differentiation of BMMs, exogenously elevated rGSTP1 dramatically enhanced the LC3-II/I ratio and decreased the protein level of SQSTM1/p62 (Fig. 3B, Supplementary Fig. S6A). Furthermore, when GSTP1 was knocked down in BMMs using GSTP1-siRNA, decreased levels of GSTP1 downregulated the autophagic flux of osteoclast formation in the first three days (Fig. 3C, Supplementary Fig. S6B).
Then, we further validated this conclusion with Raw264.7 cells. The level of GSTP1 was up-regulated by transfecting the plasmid with Flag tag, WB results showed that cells transfected with Flag-GSTP1 showed higher LC3-II/I ratio and lower SQSTM1/p62 protein level during osteoclast differentiation (Fig. 3D, Supplementary Fig. S6C). On the contrary, in cells stably transfected with the GSTP1-shRNA plasmid, WB findings showed that the low-level GSTP1 group had a lower LC3-II/I ratio and a greater SQSTM1/p62 protein level with M-CSF and RANKL stimulation (Fig. 3E, Supplementary Fig. S6D).
In addition to this, a more reliable autophagic flux reporter system based on tandem fusions of LC3 with acid-insensitive mCherry (or other red fluorescent proteins such as RFP) and acid-sensitive GFP was used to study autophagosome maturation and disintegration. At first, both GFP and mCherry fluorescence are detectable in the phagophore or autophagosome as yellow dots; Due to the destruction of GFP by acidic lysosomal proteases during the fusion of autophagosomes with lysosomes, green fluorescence is eliminated, leaving LC3 to only emit red fluorescence [29]. Then we created Raw264.7 cells that stably expressed mCherry-GFP-LC3. On the third day of differentiation, compared with the sham group, fluorescence following exogenous rGSTP1 stimulation displayed more yellow and red dots, which respectively represent autophagosomes and autophagolysosomes (Fig. 3F, Supplementary Fig. S6E). Additionally, the autophagic flux can be described more intuitively using the mCherry:GFP ratio [31,32]. According to the examination of flow cytometry, cells stimulated by rGSTP1 displayed a greater ratio of mCherry: GFP (Fig. 3G). In contrast, the fluorescence showed that when Raw264.7 cells were transfected with GSTP1-siRNA, there were less yellow and red spots than the NC-siRNA group, which was more visible on the third day of M-CSF and RANKL stimulation (Fig. 3H, Supplementary Fig. S6F). Meanwhile, the results of flow cytometry analysis showed that when GSTP1 was knocked down, the cells had a lower ratio of mCherry: GFP (Fig. 3I).
Finally, we intend to evaluate the role of GSTP1-regulated autophagic flux in osteoclast differentiation. Since GSTP1 is down-regulated during osteoclast differentiation and correspondingly down-regulates the level of autophagy, we chose a common autophagy activator, rapamycin, to rescue the low-level autophagy mediated by GSTP1-siRNA. And the results showed that the osteoclastogenesis promoted by lower levels of GSTP1 was also reversed. (Fig. 3J and K).
So, the above findings imply that GSTP1 can regulate osteoclastogenesis by regulating autophagic flux during differentiation.
3.4. GSTP1 regulates autophagic flux through the Pik3r1/AKT/mTOR signaling axis
The previous results showed that the autophagy activator rapamycin suppressed low-level GSTP1-promoted osteoclastogenesis. Since it is known that rapamycin targets mTOR and earlier study has indicated that mTOR plays an essential role in mammalian autophagy [33], we wondered whether the regulation of autophagy by GSTP1 during osteoclast differentiation is accomplished through mTOR. Exogenous rGSTP1 was shown to reduce the amount of phosphorylated mTOR (Ser2448) during the differentiation of BMMs (Fig. 4A, Supplementary Fig. S7A). The involvement of mTOR in autophagy is mostly activated via the ERK or PI3K-AKT pathways [34,35].
Fig. 4.

GSTP1 regulates autophagic flux through the Pik3r1/AKT/mTOR signaling axis. (A) BMMs were differentiated with M-CSF and RANKL (100 ng/mL) for 5 days, the WB results showed that rGSTP1(50 ng/mL) affects the expression of p-mTOR. (B) WB results showed that rGSTP1(50 ng/mL) affects the expression of p-Pik3r1, but not p-ERK1/2. (C) WB results showed that rGSTP1(50 ng/mL) affects the expression of p-AKT. (D) Raw264.7 cells were transfected with pcDNA3.1 or Flag-GSTP1 plasmid, and stimulated with M-CSF and RANKL (100 ng/mL) for 5 days. The WB results showed that the overexpression of GSTP1 regulated the level of p-Pik3r1, p-AKT, p-mTOR. (E) Raw264.7 cells transfected with shRNA-GSTP1 or shRNA-Scramble plasmids, and stimulated with M-CSF and RANKL (30 ng/mL) for 5 days. WB results show that low level of GSTP1 regulate the expression of p-Pik3r1. (F) WB results show that low level of GSTP1 regulate the expression of p-AKT. (G) WB results show that low level of GSTP1 regulate the expression of p-mTOR. (H) BMMs were differentiated with M-CSF and RANKL (50 ng/mL) for 5 days, One group used Si-GSTP1 and the other group used Si-GSTP1 + LY3023414 (10 nM, a non-toxic concentration, which alone did not inhibit osteoclast differentiation). WB results showed the differences in the expression of p-Pik3r1, p-AKT, AKT, ACP5 proteins between the two groups. (I) TRAP staining of Si-NC,Si-GSTP1 and Si-GSTP1+ LY3023414(10 nM) on the differentiation ability of BMMs under MCF and RANKL treatment. (J) Quantitative analysis of cell size and number of nuclei of TRAP-positive osteoclasts.
All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Following that, we hope to elucidate how this procedure is achieved. We differentiated BMMs under the stimulation of M-CSF and RANKL for 5 days, according to the findings, there was no difference between the rGSTP1 group and the control group in terms of the level of phosphorylated ERK1/2 (Thr202/Tyr204). Conversely, rGSTP1 suppressed the phosphorylated PI3K (Tyr458) protein level (Fig. 4B, Supplementary Fig. S7B). As a result, we preliminarily concluded that the regulation of autophagy by GSTP1 through mTOR dose not via the ERK1/2 signaling cascade during osteoclast formation. PI3Ks are heterodimers composed of the regulatory subunit p85 (Pik3r1) and the catalytic subunit p110 [36], and the WB results demonstrated that rGSTP1 did not modify the phosphorylation level of p110 during the differentiation of BMMs (Supplementary Figs. S8A and B), but did down-regulate the phosphorylated Pik3r1 level (Fig. 4B, Supplementary Fig. S7B). Correspondingly, phosphorylated AKT (Ser473), the downstream target of Pik3r1, was subsequently downregulated (Fig. 4C, Supplementary Fig. S7C). Similarly, when we transfected Raw264.7 with the Flag-GSTP1 plasmid, overexpression of GSTP1 inhibited the phosphorylated Pik3r1-AKT-mTOR signaling cascade (Fig. 4D), and this inhibition was most pronounced on days 3 and 5 under the stimulation of M-CSF and RANKL (Supplementary Fig. S7D).
For additional validation, we used Raw264.7 cells that had been stably transfected with GSTP1-shRNA. WB results demonstrated that reduced levels of GSTP1 increased the Pik3r1-AKT-mTOR signaling pathway when stimulated with M-CSF and RANKL for 5 days (Fig. 4E–G, Supplementary Figs. S9A–C).
Knockdown of GSTP1 promotes osteoclastogenesis. To clarify that this effect is indeed achieved through the Pik3r1-AKT-mTOR signaling cascade, we selected PI3K inhibitors (LY3023414) to try to reverse the osteoclast differentiation promoted by low levels of GSTP1. The WB results confirmed that LY3023414 could inhibit the phosphorylation of Pik3r1 and the phosphorylation level of AKT (Fig. 4H, Supplementary Fig. S9D), which was consistent with the previously reported pharmacological effects. Next, the WB results showed that the expression level of Acp5 was reversed (Fig. 4H, Supplementary Fig. S9D), the results of TRAP staining also showed that the osteoclast differentiation promoted by GSTP1 knockdown was reversed by LY3023414 (Fig. 4I and J).
All of the preceding findings indicate that the autophagic flux mediated by GSTP1 during osteoclastogenesis is more likely to be completed via the Pik3r1-AKT-mTOR signaling cascade.
3.5. GSTP1 upregulates S-glutathionylation of Pik3r1 to inhibit its phosphorylation
Given the previous results suggesting that GSTP1 regulates cellular activities by downregulating phosphorylation of downstream kinases, we further explored how it occurs. Previous research has shown that GSTP1-mediated S-glutathionylation of downstream kinases is critical in a variety of cellular metabolisms [12]. S-glutathionylation is a key posttranslational regulatory alteration of protein cysteine (Cys) thiols, which can act as global inhibitors of cellular metabolism in ROS-related pathogenesis [37]. Recent studies have also found that Raw264.7 cells have a significant S-glutathionylation occupancy in several organelles [38,39], so we speculated whether the regulation of downstream kinase activity by GSTP1 during osteoclast differentiation is related to S-glutathionylation.
WB findings revealed that the S-glutathionylation level was continually changing during osteoclast formation, and GSTP1 overexpression increased the total S-glutathionylation level (Fig. 5A). Then we examined the S-glutathionylation levels of GSTP1-mediated signaling cascade-related proteins, and preliminary results showed that GSTP1 might upregulate the S-glutathionylation levels of Pik3r1 (Fig. 5B), but not p110 (Fig. 5C), AKT (Fig. 5D) or mTOR (Fig. 5E).
Fig. 5.

GSTP1 upregulates S-glutathionylation of Pik3r1 to inhibit its phosphorylation. The differentiation of BMMs was induced with M-CSF and RANKL, while rGSTP1 and Si-GSTP1 were used to up-regulate or down-regulate GSTP1 levels in cells, respectively. Then the related experiments are further completed. (A) S-glutathionylation levels of overall proteins on days 0, 1, 3 of osteoclast formation, and changes in S-glutathionylation levels when GSTP1 was upregulated. (B) Co-immunoprecipitation of Pik3r1, and WB analysis of S-glutathionylation levels. (C) Co-immunoprecipitation of p110, and WB analysis of S-glutathionylation levels. (D) Co-immunoprecipitation of AKT, and WB analysis of S-glutathionylation levels. (E) Co-immunoprecipitation of mTOR, and WB analysis of S-glutathionylation levels. (F) BMMs were treated with rGSTP1. Co-immunoprecipitation of Pik3r1, and WB analysis of Pik3r1 and GSH were used to detect the S-glutathionylation level of Pik3r1. (G) BMMs were treated with rGSTP1. Co-immunoprecipitation of GSH, and WB analysis of Pik3r1 was used to detect the S-glutathionylation level of Pik3r1. (H) BMMs were transfected with Si-GSTP1 and Si-NC. Co-immunoprecipitation of Pik3r1, and WB analysis of Pik3r1 and GSH were used to detect the S-glutathionylation level of Pik3r1. (I) BMMs were transfected with Si-GSTP1 and Si-NC. Co-immunoprecipitation of GSH, and WB analysis of Pik3r1 was used to detect the S-glutathionylation level of Pik3r1.
Co-IP findings are obtained from different blots using the same samples. All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, to better understand the regulation function of this impact in osteoclastogenesis, we stimulated Raw264.7 cells with M-CSF and RANKL, the results of immunofluorescence also showed that the exogenous rGSTP1 group increased the co-localization of Pik3r1 and GSH (Supplementary Fig. S10A). When we decreased GSTP1 levels with GSTP1-siRNA, their co-localization levels decreased accordingly (Supplementary Fig. S10B).
Additionally, co-immunoprecipitation results showed that exogenous rGSTP1 enhanced the S-glutathionylation level of Pik3r1 during osteoclastogenesis on the third day of differentiation, and down-regulated the phosphorylation level of Pik3r1 (Fig. 5F–G, Supplementary Figs. S10C–D). In contrast, when GSTP1-siRNA was used to downregulate the level of GSTP1, co-immunoprecipitation findings showed that S-glutathionylation of Pik3r1 was further downregulated, and correspondingly, the phosphorylation of the kinase was improved (Fig. 5H–I, Supplementary Figs. S10E–F).
Taken together, GSTP1 can upregulate the S-glutathionylation of Pik3r1 during osteoclastogenesis and further suppress the kinase activity.
3.6. Cys498 and Cys670 are the sites of S-glutathionylation of Pik3r1
Since protein Cys thiols are modified post-translationally by the process of S-glutathionylation, we explored the more microscopic protein molecular structure of Pik3r1. Firstly, preliminary sequence alignment in different species (Fig. 6A) and the Consurf software [40] were used to show that 4 cysteines (Cys498, Cys656, Cys659, Cys670) in Pik3r1 were evolutionarily conserved (Fig. 6B), indicating that cysteines play an important role in the regulation of Pik3r1 function.
Fig. 6.

Cys498 and Cys670 are the sites of S-glutathionylation of Pik3r1. (A) Alignment of full-length amino acid sequences of Pik3r1 proteins from different species, blue highlights indicate cysteine positions in murine Pik3r1, and orange underlines indicate conserved cysteine residues shown in the sequences. (B) Further analysis of Pik3r1 using Consurf software revealed evolutionarily conserved cysteine residues. (C) S-glutathionylation of Cys498 and Cys670 from Pik3r1 were detected by mass spectrometry. The 493IFEEQCQTQER503 and 669HCVINK674 peptides including glutathionylated cysteine 498 and cysteine670; S-glutathionylation caused a 305 Da rise in the MW. (D) The three-dimensional structure of the complexes of the receptor proteins Pik3r1-CYS498, Pik3r1-CYS670 and the ligand glutathione, corresponding to the middle and right side of the figure respectively for the ligand-receptor interaction 3D pocket and 2D display diagram. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
After that, Raw264.7 cells were cultured with M-CSF and RANKL under the condition of overexpressing GSTP1, and cell lysates were extracted on the third day of osteoclast formation. S-glutathionylated proteins were then pulled down using GSH antibody. Electrophoresis was carried out under non-reducing, non-denaturing conditions and mass spectrometry was performed on gels in the specified molecular weight range. The assay results showed a 305 Da increase in the MW (molecular weight) of the 493IFEEQCQTQER503 and 669HCVINK674 peptides, indicating that S-glutathionylation occurred at the Cys498 and Cys670 (Fig. 6C).
In addition, the results of molecular protein docking also showed that Cys498 and Cys670 were more likely to interact with GSH. The 2D image clearly shows that after glutathione is covalently docked with the protein Pik3r1-CYS498, glutathione forms hydrogen bonds with GLN499 and GLU502, and has electrostatic interactions with LYS532 and GLU502; Similarly, the 2D pattern of the docking of Pik3r1-CYS670 and the ligand GSH shows that the small molecule GSH forms hydrogen bonds with HIS669, HIS668, ARG649, and VAL671, and there is an electrostatic interaction with ARG649 (Fig. 6D).
To sum up, Cys498 and Cys670 are the sites of S-glutathionylation of Pik3r1.
3.7. GSTP1-mediated glutathionylation of Pik3r1 regulates osteoclastogenesis
To clarify the role of GSTP1-mediated S-glutathionylation of Pik3r1 in osteoclastogenesis, we first knocked out Pik3r1 in Raw264.7 cells using CRISPR/Cas9 gene editing technology to exclude the influence of endogenous Pik3r1, then alanine was used to replace the cysteine site where S-glutathionylation may occur, and three single-site mutation plasmids and one double-site mutation plasmid were constructed. Finally, these mutant plasmids and the wild-type myc-Pik3r1 plasmid were transfected into Pik3r1-KO Raw264.7 cells to complete subsequent experiments (Fig. 7A).
Fig. 7.

GSTP1-mediated glutathionylation of Pik3r1 regulates osteoclastogenesis (A) Flowchart of cell biology experiments to verify GSTP1-mediated S-glutathionylation of Pik3r1. (B) WB results of Pik3r1 knockout and GSTP1 expression levels in Raw264.7 cells. (C) WB results of myc-tag protein and GSTP1 expression levels after transfection of myc-Pik3r1 and myc-Pik3r1(C498A, C670A) plasmids. (D) Co-immunoprecipitation of myc-tag, and WB analysis of S-glutathionylation levels. (E) Raw264.7 cells (Pik3r1-KO) were transfected with myc-Pik3r1, myc-Pik3r1(C498A), myc-Pik3r1(C670A) plasmids respectively. Co-immunoprecipitation of myc-tag, and WB analysis of GSH was used to detect the S-glutathionylation level under Rank1(50 ng/mL) stimulation for 3 days. (F) Raw264.7 cells (Pik3r1-KO) were transfected with myc-Pik3r1, myc-Pik3r1(C498A, C670A) plasmids respectively. Co-immunoprecipitation of myc-tag, and WB analysis of GSH was used to detect the S-glutathionylation level under the stimulation of RANKL (50 ng/mL) for 3 days. (G) Raw264.7 cells (Pik3r1-KO) were transfected with myc-Pik3r1, myc-Pik3r1(C498A, C670A) plasmids respectively, while transfected with pcDNA3.1 or Flag-GSTP1. WB results showed myc-tag, Flag-GSTP1, p-AKT, p-mTOR expression level under Rank1(50 ng/mL) stimulation for 3 days. (H) Under the same conditions as (G), WB results showed myc-tag, Flag-GSTP1, LC-I, LC3-II and p62 expression level. (I) Under the same conditions as (G), WB results showed myc-tag, Flag-GSTP1, NFATc1 and c-Fos expression level. (J) Under the same conditions as (G), the ratio of mCherry/GFP in different groups was quantified by flow cytometry. (K) Raw264.7 cells (Pik3r1-KO) were transfected with myc-Pik3r1, myc-Pik3r1(C498A, C670A) plasmids respectively, while transfected with pcDNA3.1 or Flag-GSTP1. TRAP staining of RAW264.7 cells with different conditions under Rank1(150 ng/mL) stimulation for 5 days.
Co-IP findings are obtained from different blots using the same samples. All WB quantifications in this study were based on the grayscale values exhibited by the bands, all data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
According to the flowchart, we first knocked out Pik3r1 in Raw264.7 cells according to the CRISPR/Cas9 gene editing strategy (Supplementary Fig. S11A). PCR and sequencing results verified the knockout of Pik3r1 (Supplementary Fig. S11B). In addition, WB results further confirmed that the knockout of Pik3r1 did not affect the level of GSTP1 (Fig. 7B, Supplementary Fig. S12A), and transfection with equal amounts of pcDNA3.1-Myc-Pik3r1 (WT) and pcDNA3.1-Myc-Pik3r1 (C498A, C670A) did not affect the level of GSTP1 (Fig. 7C, Supplementary Fig. S12B). Cytotoxicity experiments demonstrated that there was no significant difference in the proliferation level of Pik3r1-KO cells after complementing the mutant plasmid (Supplementary Fig. S12C). Moreover, under the conditions of M-CSF (30 ng/mL) and RANKL (100 ng/mL) stimulation for 3 days, the results of co-immunoprecipitation showed that Raw264.7 cells overexpressing GSTP1 exhibited higher levels of S-glutathionylation (Fig. 7D), indicating that transfection plasmids can perform similar biological activities in cells. All of the above laid the premise foundation for the follow-up experiments.
Next, we verified the differences in S-glutathionylation levels of cells that mutated plasmids in transfection units. During osteoclast formation, the results of co-immunoprecipitation showed that the S-glutathionylation of Pik3r1 upregulated by GSTP1 in cells transfected with these C498A and C670A mutant plasmids was lower than that of wild type (Fig. 7E, Supplementary Fig. S12D), but not significantly different in cells transfected with C656A mutant plasmids (Supplementary Fig. S12E). Furthermore, we mutated Cys498 and Cys670 at the same time, and transformed the double-mutated plasmid into Pik3r1-KO cells. Under the same conditions, co-immunoprecipitation results showed that the up-regulated S-glutathionylation level by GSTP1 was completely abolished (Fig. 7F), which strongly demonstrated that GSTP1-mediated S-glutathionylation of Pik3r1 during osteoclastogenesis is achieved through Cys498 and Cys670.
At last, the cascade regulation of previously discovered pathways and phenotypes by GSTP1-mediated S-glutathionylation of Pik3r1 was validated. WB results showed that the phosphorylation cascade level of AKT-mTOR inhibited by overexpressed GSTP1 could be reversed by de-S-glutathionylation of Pik3r1 (Fig. 7G, Supplementary Fig. S12F), levels of associated proteins indicative of increased autophagic flux were substantially reversed (Fig. 7H, Supplementary Fig. S12G), and the characteristic proteins related to osteoclast formation inhibited by GSTP1 were also partially rescued (Fig. 7I, Supplementary Fig. S12H). Likewise, the reversal of autophagic flux demonstrated by flow cytometry had a similar outcome (Fig. 7J), and TRAP staining also revealed that the differentiation level of GSTP1-inhibited Raw264.7 cells was eventually partially rescued (Fig. 7K).
So far, we have proved that Cys498 and Cys670 are the two sites of S-glutathionylation of Pik3r1 mediated by GSTP1 through exhaustive experiments in vitro, and this redox-related modification can regulate autophagic flux through the Pik3r1-AKT-mTOR cascade and ultimately inhibit osteoclastogenesis.
3.8. TLK199 aggravated bone loss in OVX mice in vivo, whereas rGSTP1 dose the opposite
To explore the role of GSTP1 in vivo, we conducted a series of animal experiments (Fig. 8A).
Fig. 8.

TLK199 aggravated bone loss in OVX mice in vivo, whereas rGSTP1 dose the opposite. (A) Flowchart of the animal experiments. (B) Representative Micro-CT images of mice in the sham, OVX and OVX + TLK199 groups after one month. (C) Quantitative analysis of bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp) and trabecular number (Tb.N). (D) TRAP staining of the distal femur slices in the sham-operated group, the OVX group and the OVX + TLK199 group after one month (fast green as the background color). (E) Representative Micro-CT images of mice in the SHAM, OVX and OVX + rGSTP1 groups after two month. (F) Quantitative analysis of bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp) and trabecular number (Tb.N). (G) TRAP staining of the distal femur slices in the SHAM-operated, the OVX and the OVX + rGSTP1 groups after two month. All data are presented as mean ± SEM. Experimental data for each quantitative analysis were replicated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Firstly, we investigated the role of GSTP1 in physiological state. LV-GSTP1 lentivirus was used to specifically knock down GSTP1 expression levels in bone marrow, and the results of WB and immunohistochemistry both confirmed the decreased expression level of GSTP1 in bone marrow (Supplementary Figs. S13A and B). Micro-CT results showed that specifically knockdown of GSTP1 aggregated bone loss of physiological mice compared with the sham (LV-NC) group (Supplementary Figs. S13C and D). Previous conclusions suggested that Pik3r1 may be a key target of GSTP1 knockdown to promote osteoclast differentiation, so we used Pik3r1 inhibitor (LY3023414) in vivo, and as expected, the bone loss induced by GSTP1 knockdown was reversed by LY3023414 (Supplementary Figs. S13C and D).
Meanwhile, the pharmacological inhibition induced by TLK199 in vivo had a similar effect on bone loss (Supplementary Figs. S13E and F), and the degree of inhibition of GSTP1 expression in bone marrow was better than that of LV-GSTP1 (Supplementary Figs. S13G and H). However, injection of rGSTP1 under physiological conditions did not significantly change bone homeostasis in the short term (Supplementary Figs. S13G and H).
Furthermore, considering the action of GSTP1 on osteoclasts, we are more interested in the pharmacological suppression of GSTP1 as well as the effect of recombinant GSTP1 protein (rGSTP1) in osteoporosis ovariectomized osteoporosis mouse model.
TLK199, an FDA-approved orphan medication, has completed the Phase II clinical study for MDS and is the most selective GSTP1 inhibitor to date [41]. We treated OVX mice with TLK199 (50 mg/kg) and saline for one month respectively, and the treatment method was intraperitoneal injection every other day, then established three groups: sham operation group (SHAM), OVX group and OVX + TLK199 group. Micro-CT results showed that OVX mice treated with TLK199 exhibited less bone mass (Fig. 8B), lower BV/TV, Tb.N and higher Tb.Sp (Fig. 8C); Additionally, TRAP staining revealed that the TLK199 group had more osteoclasts than the OVX group did in terms of both number and area on the femoral surface trabecular bone (Fig. 8D, Supplementary Fig. S14A).
The biological activity of rGSTP1 protein in vivo has been demonstrated and reported in previous studies [42,43],So we treated OVX mice with rGSTP1 protein (100 mg/kg) and saline by tail vein injection every other day for two months. Three groups were established, SHAM group, OVX group, OVX + rGSTP1 group. Micro-CT findings demonstrate that OVX-induced bone loss can be reversed by intravenously injecting rGSTP1 protein (Fig. 8E), OVX mice treated with rGSTP1 protein had increased BV/TV, Tb.N, Tb.Th, and reduced Tb.Sp (Fig. 8F). Moreover, TRAP staining revealed that in the intervention group using GSTP1 recombinant protein, N.Oc/BS and Oc.S/BS were lower on the trabecular bone surface of the femur (Fig. 8G, Supplementary Fig. S14B). Finally, we supplemented calcein labeling experiments and found that rGSTP1 did not affect bone formation rate in vivo (Supplementary Fig. S14C), and TLK199 or rGSTP1 did not affect mouse body weight and growth in vivo (Supplementary Fig. S14D).
In conclusion, TLK199, a specific pharmacological inhibitor of GSTP1, exacerbated bone loss in OVX mice, whereas exogenous GSTP1 recombinant protein reversed osteoporosis in OVX mice.
4. Discussion
Osteoporosis is still an age-related metabolic disorder of current concern, and bone mineral density is a reliable reference indicator. Our team concentrated on the genetic link between illnesses. In exploring the causal relationship between blood diseases and bone mineral density, it was found that MDS may have a certain relationship with bone mineral density through Mendelian randomization, which is aligns with previous reports.
We focused on TLK199, an FDA-approved orphan medication for MDS that is a pharmacological inhibitor of GSTP1 [41,51]. The region plots of GSTP1 in the BMD-related GWAS databases revealed that GSTP1 was probably correlated with BMD, which is consistent with earlier research [8].
Firstly, we looked at the expression of GSTP1 during the osteoclastic and osteoblastic differentiation processes as well as its influence on osteoclastogenesis in vitro. The findings indicated that the effect of GSTP1 on bone mineral density was more likely to be achieved through influencing osteoclasts. The role of GSTP1 in cancer cells and endothelial cells has been widely documented, but the role in osteoclasts is still unknown. Thus, this work investigated the probable mechanism of GSTP1 in osteoclasts in depth.
We determined the inhibitory effect of GSTP1 on osteoclastogenesis by knockdown or overexpression of GSTP1. At the same time, the microscopic cell morphology under transmission electron microscope showed there were more and more obvious autophagosomes and phagosomes during the osteoclast differentiation process stimulated by recombinant GSTP1 recombinant protein. In recent years, studies on autophagy and bone metabolism have emerged in an endless stream, and the regulatory role of autophagy in bone metabolism is also diverse [44]. The results showed that knockdown of GSTP1 further inhibited autophagic flux during osteoclastogenesis, whereas osteoclast differentiation promoted by low levels of GSTP1 was reversed when autophagy levels were upregulated using rapamycin. This effect is also similar with the results of some previous studies [[45], [46], [47]].
As a result, we hypothesized that GSTP1 influenced osteoclast formation by controlling autophagy levels. It was discovered that autophagy levels are regulated by the Pik3r1/AKT/mTOR signaling pathway. The role of PI3K-AKT-mTOR pathway-mediated autophagy in osteoclasts is also consistent with previous findings [48,49].
However, it is worth noting that many previous articles in the study of bone metabolism believed that the increase in the level of autophagy often promoted the differentiation of osteoclasts [50]. In our study, autophagic flux upregulated by GSTP1 precisely inhibited osteoclast differentiation. This result perfects the relationship between autophagy and bone metabolism. Different proteins regulate different molecules of autophagy, resulting in diverse outcomes of osteoclast formation fate, and the contrary insights of altered autophagic flux on osteoclast formation still require deeper mechanistic elucidation.
GSTP1 is a glutathione transferase that, in addition to adding GSH to various electrophiles, can also transfer GSH to cysteine residues of proteins and upregulate S-glutathionylation of these proteins. This modification mediated by GSTP1 changes the activities and functions of downstream target proteins, and plays a vital role in various cellular metabolism and homeostasis maintenance [12,18,42,51].
The level of cellular oxidative stress can regulate a variety of cellular metabolisms, thereby determining cell fate, and reversible cysteine residue modification in target proteins can also modify protein activity and function, hence influencing signaling cascades [52].
As one of the least common amino acids encoded by the human genome, these cysteine-related pathways are generally evolutionarily well conserved. Also, cysteine residues can be used to organize the cellular activity, and the accompanying redox regulation organizes the metabolism of various cellular functions [53].
The management of autophagy by redox-related changes can follow cells throughout their lifespan. As one type of bone metabolism directly related to the oxidative stress environment, the differentiation fate of osteoclasts has attracted a lot of research [54,55].
It is the first study to discover that GSTP1-mediated cysteine -related S-glutathionylation regulates the formation fate of osteoclasts. Not only provides strong new evidence for this connection, but also perfects a novel regulatory modification mechanism for the differentiation of osteoclasts.
However, our study still has some limitations. Firstly, in the fluorescence colocalization results of GSH and Pik3r1, the changes of GSH showed the level of global GSH expression, but not specific to Pik3r1. So, this colocalization result can only prove the change of their colocalization degree non-specifically. Secondly, our research is aimed at how GSTP1 regulates the process of osteoclast differentiation, and the soluble factors released after osteoclast formation are also very important for osteoporosis. Whether GSTP1 has an effect on these factors needs further study. Finally, our in vivo experiments are specific to this model of OVX-induced osteoporosis, and other strains and disease models need more studies to confirm.
Collectively, the above data represent a novel mechanism by which GSTP1 regulates osteoclastogenesis. GSTP1 first upregulates S-glutathionylation of Pik3r1, a modification that inhibits kinase phosphorylation activity and regulates autophagic flux through the Pik3r1-AKT-mTOR signaling cascade, thereby inhibiting osteoclastogenesis and altering its formation destiny. Targeting GSTP1 in osteoclastogenesis may provide a potential therapeutic direction for osteoporosis. Given the positive therapeutic effect of TLK199 in patients with MDS in clinical trials and the relatively clear evidence for lung-related diseases, our results suggest that in the future clinical use of TLK199 should be paid attention to prevent the occurrence of osteoporosis in patients. Moreover, the clinical application of targeted agonists and recombinant proteins of GSTP1 for osteoporosis and some osteoclast-related immune and metabolic diseases is also worthy of further exploration.
CRediT authorship contributions
Xiaoxiao Ji: Data curation, Investigation, Visualization, Writing - original draft. Jianqiao Hong: Conceptualization. Weinan Yang: Investigation. Minjun Yao: Investigation. Jie Wang: Formal analysis. Guangyao Jiang: Resources. Yibo Wang: Investigation. Congsun Li: Investigation. Jiyan Lin: Investigation. Haochen Mou: Investigation. Chaozhong Li: Software. Sihao Li: Resources. Yazhou Chen: Data curation. Minming Shi: Funding acquisition. Wei Wang: Funding acquisition. Fei Lu: Data curation. Haobo Wu: Writing – review. Xiang Zhao: Funding acquisition. Yiying Qi: Writing – review. Shigui Yan: Writing – review.
Funding
This research was supported by National Natural Science Foundation of China under Grant (No. 81902279), Zhejiang Province Medical and Health project (NO.2020391395) and Natural Science Foundation of Zhejiang Province (LY20H170002).
Ethics statement
All the animal experiments are complied with ethical standards of the Animal Care and Welfare Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine.
Data availability statement
Data will be provided upon justifiable request.
Declaration of competing interest
The authors claim to have no conflicts of interests.
Acknowledgements
We thank all participants for their financial and technical support for this study, and we are particularly grateful for the technical support by JingYao. Chen, Q. Huang of the Core Facilities, Zhejiang University School of Medicine.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102635.
Contributor Information
Haobo Wu, Email: 2505014@zju.edu.cn.
Xiang Zhao, Email: flyingzhao@zju.edu.cn.
Yiying Qi, Email: qiyiying@zju.edu.cn.
Shigui Yan, Email: zrjwsj@zju.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Lorentzon M., Cummings S.R. Osteoporosis: the evolution of a diagnosis. J. Intern. Med. 2015;277(6):650–661. doi: 10.1111/joim.12369. [DOI] [PubMed] [Google Scholar]
- 2.Cummings S.R., Melton L.J. Epidemiology and outcomes of osteoporotic fractures. Lancet. 2002;359(9319):1761–1767. doi: 10.1016/S0140-6736(02)08657-9. [DOI] [PubMed] [Google Scholar]
- 3.Ebeling P.R., Nguyen H.H., Aleksova J., Vincent A.J., Wong P., Milat F. Secondary osteoporosis. Endocr. Rev. 2022;43(2):240–313. doi: 10.1210/endrev/bnab028. [DOI] [PubMed] [Google Scholar]
- 4.Steer K., Stavnichuk M., Morris M., Komarova S.V. Bone health in patients with hematopoietic disorders of bone marrow origin: systematic review and meta-analysis. J. Bone Miner. Res. 2017;32(4):731–742. doi: 10.1002/jbmr.3026. [DOI] [PubMed] [Google Scholar]
- 5.Emdin C.A., Khera A.V., Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–1926. doi: 10.1001/jama.2017.17219. [DOI] [PubMed] [Google Scholar]
- 6.Naing A., Sokol L., List A.F. Developmental therapeutics for myelodysplastic syndromes. J. Natl. Compr. Cancer Netw. 2006;4(1):78–82. doi: 10.6004/jnccn.2006.0008. [DOI] [PubMed] [Google Scholar]
- 7.Platzbecker U., Kubasch A.S., Homer-Bouthiette C., Prebet T. Current challenges and unmet medical needs in myelodysplastic syndromes. Leukemia. 2021;35(8):2182–2198. doi: 10.1038/s41375-021-01265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mlakar S.J., Prezelj J., Marc J. Testing GSTP1 genotypes and haplotypes interactions in Slovenian post-/pre-menopausal women: novel involvement of glutathione S-transferases in bone remodeling process. Maturitas. 2012;71(2):180–187. doi: 10.1016/j.maturitas.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 9.Wang S., Deng Z., Ma Y., et al. The role of autophagy and mitophagy in bone metabolic disorders. Int. J. Biol. Sci. 2020;16(14):2675–2691. doi: 10.7150/ijbs.46627. Published 2020 Jul 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee A., Gupta S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018;433:33–42. doi: 10.1016/j.canlet.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Lei X., Du L., Yu W., Wang Y., Ma N., Qu B. GSTP1 as a novel target in radiation induced lung injury. J. Transl. Med. 2021;19(1):297. doi: 10.1186/s12967-021-02978-0. Published 2021 Jul 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van de Wetering C., Manuel A.M., Sharafi M., et al. Glutathione-S-transferase P promotes glycolysis in asthma in association with oxidation of pyruvate kinase M2. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong X., Yang Y., Zhou Y., et al. Glutathione S-transferases P1 protects breast cancer cell from adriamycin-induced cell death through promoting autophagy. Cell Death Differ. 2019;26(10):2086–2099. doi: 10.1038/s41418-019-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y., Li N., Chen T., et al. Sirt3 promotes sensitivity to sunitinib-induced cardiotoxicity via inhibition of GTSP1/JNK/autophagy pathway in vivo and in vitro. Arch. Toxicol. 2019;93(11):3249–3260. doi: 10.1007/s00204-019-02573-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J., Li X.Y., Liu Y.J., et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy. 2022;18(6):1240–1255. doi: 10.1080/15548627.2021.1984656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manevich Y., Feinstein S.I., Fisher A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. U. S. A. 2004;101(11):3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye Z.W., Zhang J., Ancrum T., Manevich Y., Townsend D.M., Tew K.D. Glutathione S-transferase P-mediated protein S-glutathionylation of resident endoplasmic reticulum proteins influences sensitivity to drug-induced unfolded protein response. Antioxidants Redox Signal. 2017;26(6):247–261. doi: 10.1089/ars.2015.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Dong X., Zheng S., et al. GSTpi regulates VE-cadherin stabilization through promoting S-glutathionylation of Src. Redox Biol. 2020;30 doi: 10.1016/j.redox.2019.101416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedoszytko B., Sobalska-Kwapis M., Strapagiel D., et al. Results from a genome-wide association study (GWAS) in mastocytosis reveal new gene polymorphisms associated with WHO subgroups. Int. J. Mol. Sci. 2020;21(15):5506. doi: 10.3390/ijms21155506. Published 2020 Jul 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berndt S.I., Camp N.J., Skibola C.F., et al. Meta-analysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Nat. Commun. 2016;7 doi: 10.1038/ncomms10933. Published 2016 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao E.L., Nandakumar S.K., Liao X., et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature. 2020;586(7831):769–775. doi: 10.1038/s41586-020-2786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGraw K.L., Cheng C.H., Chen Y.A., et al. Non-del(5q) myelodysplastic syndromes-associated loci detected by SNP-array genome-wide association meta-analysis. Blood Adv. 2019;3(22):3579–3589. doi: 10.1182/bloodadvances.2019000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Went M., Sud A., Försti A., et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma [published correction appears in Nat Commun. 2019 Jan 10;10(1):213] Nat. Commun. 2018;9(1):3707. doi: 10.1038/s41467-018-04989-w. Published 2018 Sep. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemp J.P., Morris J.A., Medina-Gomez C., et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet. 2017;49(10):1468–1475. doi: 10.1038/ng.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S.K. Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture [published correction appears in PLoS One. PLoS One. 2019 Mar 13;14(3) doi: 10.1371/journal.pone.0200785. 2018;13(7): e0200785. Published 2018 Jul 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji X., Hong J., Qu Z., et al. HemoglobinA1c is a risk factor for changes of bone mineral density: a mendelian randomization study. Front. Endocrinol. 2022;13 doi: 10.3389/fendo.2022.942878. Published 2022 Jul 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng J., Zhang W., Wang C., et al. Catalpol suppresses osteoclastogenesis and attenuates osteoclast-derived bone resorption by modulating PTEN activity. Biochem. Pharmacol. 2020;171 doi: 10.1016/j.bcp.2019.113715. [DOI] [PubMed] [Google Scholar]
- 28.Han X., Zhou Z., Fei L., et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581(7808):303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X.J., Chen S., Huang K.X., Le W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013;34(5):595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gump J.M., Thorburn A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy. 2014;10(7):1327–1334. doi: 10.4161/auto.29394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinsey C.G., Camolotto S.A., Boespflug A.M., et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019;25(4):620–627. doi: 10.1038/s41591-019-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y.C., Guan K.L. mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 2015;125(1):25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S.Y. mTOR-targeted cancer therapy: great target but disappointing clinical outcomes, why? Front. Med. 2021;15(2):221–231. doi: 10.1007/s11684-020-0812-7. [DOI] [PubMed] [Google Scholar]
- 35.Mendoza M.C., Er E.E., Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 37.Mailloux R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020;32 doi: 10.1016/j.redox.2020.101472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan J., Zhang T., Gaffrey M.J., et al. Stochiometric quantification of the thiol redox proteome of macrophages reveals subcellular compartmentalization and susceptibility to oxidative perturbations. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su D., Gaffrey M.J., Guo J., et al. Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic. Biol. Med. 2014;67:460–470. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashkenazy H., Abadi S., Martz E., et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahadevan D., Sutton G.R. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expet Opin. Invest. Drugs. 2015;24(5):725–733. doi: 10.1517/13543784.2015.1021003. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y., Xue W., Zhang W., et al. Histone methyltransferase G9a protects against acute liver injury through GSTP1. Cell Death Differ. 2020;27(4):1243–1258. doi: 10.1038/s41418-019-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whalen R., Rockey D.C., Friedman S.L., Boyer T.D. Activation of rat hepatic stellate cells leads to loss of glutathione S-transferases and their enzymatic activity against products of oxidative stress. Hepatology. 1999;30(4):927–933. doi: 10.1002/hep.510300404. [DOI] [PubMed] [Google Scholar]
- 44.Starling S. A role for autophagy in bone biology. Nat. Rev. Endocrinol. 2019;15(8):438–439. doi: 10.1038/s41574-019-0223-5. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez-García O., García-López E., Loredo V., et al. Rapamycin induces growth retardation by disrupting angiogenesis in the growth plate. Kidney Int. 2010;78(6):561–568. doi: 10.1038/ki.2010.173. [DOI] [PubMed] [Google Scholar]
- 46.Smink J.J., Bégay V., Schoenmaker T., Sterneck E., de Vries T.J., Leutz A. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 2009;28(12):1769–1781. doi: 10.1038/emboj.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hussein O., Tiedemann K., Murshed M., Komarova S.V. Rapamycin inhibits osteolysis and improves survival in a model of experimental bone metastases. Cancer Lett. 2012;314(2):176–184. doi: 10.1016/j.canlet.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 48.Ma J., Du D., Liu J., et al. Hydrogen sulphide promotes osteoclastogenesis by inhibiting autophagy through the PI3K/AKT/mTOR pathway. J. Drug Target. 2020;28(2):176–185. doi: 10.1080/1061186X.2019.1624969. [DOI] [PubMed] [Google Scholar]
- 49.Zhao H., Sun Z., Ma Y., et al. Antiosteoclastic bone resorption activity of osteoprotegerin via enhanced AKT/mTOR/ULK1-mediated autophagic pathway. J. Cell. Physiol. 2020;235(3):3002–3012. doi: 10.1002/jcp.29205. [DOI] [PubMed] [Google Scholar]
- 50.Yin X., Zhou C., Li J., et al. Autophagy in bone homeostasis and the onset of osteoporosis [published correction appears in Bone Res. 2020 Oct 6;8:36] Bone Res. 2019;7(28) doi: 10.1038/s41413-019-0058-7. Published 2019 Oct 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMillan D.H., van der Velden J.L., Lahue K.G., et al. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase π. JCI Insight. 2016;1(8) doi: 10.1172/jci.insight.85717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Go Y.M., Jones D.P. Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 2013;48(2):173–181. doi: 10.3109/10409238.2013.764840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008;295(4):C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weng Y., Wang H., Li L., Feng Y., Xu S., Wang Z. Trem2 mediated Syk-dependent ROS amplification is essential for osteoclastogenesis in periodontitis microenvironment. Redox Biol. 2021;40 doi: 10.1016/j.redox.2020.101849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun W., Qiao W., Zhou B., et al. Overexpression of Sirt1 in mesenchymal stem cells protects against bone loss in mice by FOXO3a deacetylation and oxidative stress inhibition. Metabolism. 2018;88:61–71. doi: 10.1016/j.metabol.2018.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be provided upon justifiable request.