

Summary
Background
Macrophages at infection sites are considered as the promising therapeutic targets to prevent sepsis development. The Nrf2/Keap1 system acts as a critical modulator of the antibacterial activity of macrophages. Recently, Keap1–Nrf2 protein–protein interaction (PPI) inhibitors have emerged as safer and stronger Nrf2 activators; however, their therapeutic potential in sepsis remains unclear. Herein, we report a unique heptamethine dye, IR-61, as a Keap1–Nrf2 PPI inhibitor that preferentially accumulates in macrophages at infection sites.
Methods
A mouse model of acute lung bacterial infection was used to investigate the biodistribution of IR-61. SPR study and CESTA were used to detect the Keap1 binding behaviour of IR-61 in vitro and in cells. Established models of sepsis in mice were used to determine the therapeutic effect of IR-61. The relationship between Nrf2 levels and sepsis outcomes was preliminarily investigated using monocytes from human patients.
Findings
Our data showed that IR-61 preferentially accumulated in macrophages at infection sites, enhanced bacterial clearance, and improved outcomes in mice with sepsis. Mechanistic studies indicated that IR-61 potentiated the antibacterial function of macrophages by activating Nrf2 via direct inhibition of the Keap1–Nrf2 interaction. Moreover, we observed that IR-61 enhanced the phagocytic ability of human macrophages, and the expression levels of Nrf2 in monocytes might be associated with the outcomes of sepsis patients.
Interpretations
Our study demonstrates that the specific activation of Nrf2 in macrophages at infection sites is valuable for sepsis management. IR-61 may prove to be a Keap1-Nrf2 PPI inhibitor for the precise treatment of sepsis.
Funding
This work was supported by the National Natural Science Foundation of China (Major program 82192884), the Intramural Research Project (Grants: 2018-JCJQ-ZQ-001 and 20QNPY018), and the Chongqing National Science Foundation (CSTB2022NSCQ-MSX1222).
Keywords: Macrophage-targeting therapy, Small-molecule compound, Keap1–Nrf2 PPI inhibitor, Sepsis
Research in context.
Evidence before this study
Enhancing host antibacterial immunity at infection sites would prevent systemic spread of pathogen, which may be a promising therapeutic strategy for sepsis. Macrophages at infection sites are considered as important therapeutic targets in sepsis owing to their critical role in eradicating local invading pathogens. Activation of Nrf2 by disruption of Keap1 can enhance the antibacterial defence by macrophages and protect against sepsis. Keap1–Nrf2 protein–protein interaction (PPI) inhibitors are safe and effective Nrf2 activators. However, the specific function of Keap1–Nrf2 PPI inhibitors in sepsis remains unknown. More importantly, PPI inhibitors with selectivity for macrophages at infection sites are still lacking.
Added value of this study
In this study, we identified IR-61 as a Keap1–Nrf2 PPI inhibitor with the inherent property of preferential accumulation in macrophages at infection sites. Further analyses revealed that IR-61 potentiated the antibacterial defence by macrophages through Nrf2 and protected mice against sepsis. In addition, we showed that IR-61 augmented bacterial phagocytosis of human macrophages and suggested that the activation of Nrf2 might be a potential strategy for sepsis management.
Implications of all the available evidence
IR-61 is a multifunctional agent that protects against sepsis by selectively activating Nrf2 in macrophages at the infection sites. This strategy holds promise for sepsis management.
Introduction
Sepsis is the main cause of death in critically ill patients, with an overall mortality rate of >25%–30%.1 Failure of the local innate immune system in removing pathogens leads to the systemic dissemination of invading pathogens and hyperactivation of the host immune system, which are considered to be the key factors for sepsis development.2,3 Antibiotics are often used to control bacterial dissemination clinically.4 However, drug resistance and inefficient drug delivery hinder effective antibiotic treatments.5 In addition, only a few randomised clinical trials targeting the hyperactivation of the inflammatory response have been successful.6 An important reason is that targeting a single cytokine cannot prevent all harmful effects of sepsis in the presence of invading pathogens.7 The current view states that an impaired host immune system in sepsis cannot efficiently eliminate the pathogens.8,9 In this context, boosting host antibacterial immunity at infection sites to quickly eradicate pathogens and prevent systemic bacteraemia may be a promising therapeutic strategy for sepsis.
Macrophages constitute the first line of defence against microbial invasion and are mainly responsible for pathogen clearance during sepsis.10 Impairment of macrophage function appears to be the main reason for deficient antibacterial ability in septic hosts.9,11 Several recombinant therapeutic proteins that potentiate the antibacterial defence of macrophages have been tested and demonstrated to improve the outcomes of sepsis in mice.12, 13, 14 Moreover, mesenchymal stem cells (MSCs) also confer protection against sepsis by enhancing the bacterial clearance by macrophages and obtain satisfactory therapeutic effects in clinical trials.15, 16, 17 Unfortunately, problems with the specificity, efficacy, and safety of these agents hinder their clinical applications.18,19 Therefore, new agents targeting macrophages should focus on selectively enhancing the bactericidal ability of macrophages at infection sites, which are expected to greatly improve safety and therapeutic efficacy in sepsis management.
Nuclear factor-erythroid-2-related factor 2 (Nrf2) is the dominant mediator of various cytoprotective responses.20 In addition to its well-described protective effects on tissue damage triggered by oxidative stress and inflammation, Nrf2 activation potentiates the antibacterial ability of macrophages and imparts protection against sepsis.21 Kelch-like ECH-associated protein 1 (Keap1) is the principal negative regulator of Nrf2, and mediates its ubiquitin-dependent proteasomal degradation.22 Recently, Keap1–Nrf2 protein–protein interaction (PPI) inhibitors have been proven to be safer and to confer better cellular protection than electrophilic Nrf2 activators,23 which may have great therapeutic potential in sepsis. However, the currently reported inhibitors lack tissue and cell selectivity in vivo, resulting in limited therapeutic efficacy and uncertain safety risks.24 The development of PPI inhibitors that selectively activate Nrf2 in macrophages at infection sites will facilitate the therapeutic application of PPI inhibitors in sepsis.
In our previous study, we identified a near-infrared (NIR) fluorophore, IR-61, that preferentially accumulated in the mitochondria and suppressed pro-inflammatory cytokines by stimulating the mitochondrial function of macrophages in obesity.25 Interestingly, in this study, we identified IR-61 as a Keap1–Nrf2 PPI inhibitor that enhanced phagocytosis and bactericidal ability of macrophages by activating Nrf2. Importantly, IR-61 preferentially accumulated in macrophages at infection sites following intravenous (i.v.) injection, promoted bacterial clearance, suppressed inflammatory responses, and protected against sepsis. We conclude that IR-61 is a multifunctional agent that enhances antibacterial ability and induces the anti-inflammatory phenotype of macrophages for precise treatment of sepsis.
Methods
Mice
C57BL/6J male mice (RRID: IMSR_JAX:000664) were purchased from the Laboratory Animal Center of the Army Medical University and fed under 12 h light/dark cycle, 21–23 °C temperature range, 40–50% humidity range and free access to food and water. Adult male mice (8–10 weeks) were kept in quarantine for 1 week and then used for producing animal models.
For lung infection model, the mice were anesthetized and then 106 colony-forming units (CFUs) of Escherichia coli (E. coli) (ATCC25922) were intratracheal (i.t.) administered into the mice lungs. The mice were held upright for 2 min after the administration for facilitating the bacteria to enter into the lungs. 4 h later, randomly grouped infected mice were injected with vehicle (PBS) or IR-61 (2 mg kg−1) via i.v. administration.
For LPS-induced sepsis, LPS (10 mg kg−1) was administered by intraperitoneal (i.p.) injection. For E. coli-induced sepsis, 5 × 107 CFUs of live E. coli per mouse were administrated via i.p. injection. Cecal ligation and puncture (CLP) sepsis was induced by a surgical procedure as previously described. Briefly, mice were anesthetized with pentobarbital (80 mg kg−1 i.p.) and a small incision at midline abdominal was made to expose the cecum. Then the cecum was ligated distal to the ileocecal valve and punctured twice with a 22-gauge needle. The abdomen was closed after placing back the cecum in the peritoneal cavity. Mice were randomly grouped and treated with vehicle or IR-61 (2 mg kg−1 i.v.) at 30 min, 3 h, 6 h and 12 h within the first day of sepsis induction and then injected once a day.
Peritoneal macrophages (PMs) preparation
Mice were injected i.p. with 3 mL of 3% thioglycollate to elicit macrophages. Three days later, the mice were sacrificed and injected intraperitoneally with 5 mL PBS. Then macrophages were collected and seeded in culture flasks with DMEM medium containing 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. After 2 h culturing at 37 °C/5% CO2, the cells were rinsed to remove floating cells and remaining attached cells were considered as PMs.
In vivo imaging of IR-61 in mice
E. coli or PBS was i.t. administered into the mice lungs. 4 h later, IR-61 (2 mg kg−1) was injected i.v. into mice and vital tissues (liver, spleen, heart, lung, and kidney) were harvested after 12, 24, and 120 h. NIR fluorescence images were captured using a Kodak In-Vivo FX Professional Imaging System equipped with fluorescence filter sets (excitation/emission 770/830 nm).
Measurement of phagocytosis and bacterial killing
PMs were treated with 10 μM IR-61 for 24 h, and then incubated with fluorescent latex beads (#L3030, Sigma) or pHrodo-labeled E. coli (#P35361, Invitrogen) at a multiplicity of infection (MOI) of 10 for 1 h. For microscopic analysis, the uptake of latex beads or E. coli into cells was captured by a microscope and quantified by estimating the percent of macrophages containing at least one latex bead or measuring fluorescence intensity of internalized E. coli using ImageJ software. For flow cytometry analysis, phagocytosis percentage of latex beads or mean fluorescence intensity (MFI) of internalized E. coli were assessed to determine the effect of IR-61 on macrophages phagocytosis.
PMs (1 × 105 per well) were seeded on each well of a 96-well plate and treated with 10 μM IR-61 for 24 h. Then PMs were infected with live E. coli or live Staphylococcus aureus (S. aureus) (ATCC25923) at 37 °C for 90 min. Next, the cells were washed and incubated with medium containing gentamicin (400 ng mL−1) (#HY-A0276, MCE) for 2 h to remove the remaining extracellular and cell surface-attached bacteria. Thereafter, the cells were lysed in PBS containing 0.1% Triton X-100 (#T9284, Sigma) and lysates were diluted serially in sterile PBS. Each dilution (25 μL of each) was plated and cultured on Luria Bertani (LB) agar plates at 37 °C for 18 h and then the number of CFUs was counted.
Collection of blood, peritoneal lavage fluid (PLF), bronchoalveolar lavage fluid (BALF), and organs
Blood samples were obtained via cardiac puncture, placed into microcentrifuge tubes with heparin and kept on ice for further bacteriological analysis. The remaining blood samples were stood at room temperature for 2 h, centrifuged at 1500 rpm for 20 min and the plasma was stored at −80 °C to further use. To collect PLF, 2 mL of sterile PBS was injected and withdrawn in the peritoneal cavity for 3 times. Then the PLF was recovered and placed on ice for bacteriological examination. The remaining PLF centrifuged at 350 g for 10 min and the supernatant was stored at −80 °C to further use. To obtain BALF, the lungs were aspirated 3 times with 1 mL of sterile PBS. Then BALF was placed on ice for bacteriological examination. The remaining BALF was centrifuged at 350 g for 10 min and the supernatant was stored at −80 °C to further use. The lungs and spleen were excised, then 10 mg tissues were weighed and homogenized in 1 mL sterile PBS for bacteriological examination.
Quantification of bacterial CFUs
Mice were euthanized 24 h after CLP. Blood, PLF, BALF, lung and spleen homogenates were diluted serially in sterile PBS. 25 μL of each dilution was plated and cultured at 37 °C on LB agar. The CFU numbers were counted after 14–18 h incubation.
Enzyme-linked immunosorbent assay
Mice were euthanized 24 h after CLP, and then plasma and PLF were collected. The concentrations of mice inflammatory cytokines: Tnf, IL-6, IL-1β, and IL-10 were directly quantified by Tnf (#E16ME0004, Enogene), IL-6 (#E16ME0006, Enogene), IL-1β (#E16ME0015, Enogene), and IL-10 (#ab46103, Abcam) ELISA kits.
Surface plasmon resonance (SPR) assay
SPR assays were performed on OpenSPR instruments (Nicoya, Canada) to test if IR-61 bound to Keap1. COOH sensor chip was installed on the OpenSPR instrument according to the standard procedure. Then human recombinant Keap1 Protein (#11981-H20B, Sinobiological) was diluted with activation buffer and immobilized on the chip. IR-61 was used at concentrations ranging from 0 to 1 μM and injected separately on the surface of the ligand chip. The parameters of SPR were set as follows: 25 °C; flow rate, 20 μL min−1; contact time, 240 s; disassociation time, 480 s. The kinetic parameters of the binding reactions were calculated and analyzed by Trace Drawer software (Ridgeview Instruments AB, Sweden).
Human samples
Human monocytes from patients meeting sepsis-3 clinical criteria were collected within 24-h Intensive Care Unit (ICU) admission at General Hospital of Western Theater Command. Patients were included who had known or suspected infection with the Sequential Organ Failure Assessment (SOFA) score ≥2 points for organ dysfunction. We excluded patients who were pregnancy or breast-feeding, younger than 18 yrs or older than 80 yrs, immunocompromised or presented immunodeficiency, and receiving chemotherapy within 4 weeks. The clinical data, including age, gender, SOFA score, Acute Physiology and Chronic Health Evaluation II (APACHE II), length of ICU stay, and mortality during the 28-day study period, were recorded in Table S1.
Human monocyte isolation
Human peripheral blood mononuclear cells (hPBMCs) were isolated using a Human peripheral blood monocyte isolation solution kit (#P8680, Sorlabio) according to the manufacturer's protocol. Human monocytes were then isolated from hPBMCs using EasySep™ Human Monocyte Isolation Kit (#19359, Stem cell technologies) following a protocol provided by the manufacturer.
Human monocyte-derived macrophages (hMDMs)
To differentiate monocytes into hMDMs, monocytes were resuspended in RPMI containing 10% FBS, 2 mM l-glutamine, 1% streptomycin/penicillin and 20 ng mL−1 recombinant human M-CSF (#AF-300-25, PeproTech). The medium was changed on day 3, and floating non-adhesive cells were washed off during the medium change. hMDMs were collected from the plate to further use at 6 days after human M-CSF treatment.
Molecular docking assay in silico
To investigate the possible binding mode of IR-61 and Keap1, the crystal structure of human Keap1 (PDB ID: 2Z32) was downloaded from the Protein Data Bank and the structure of IR-61 was reported in previous study. Molecular docking analysis of IR-61 was performed with human Keap1 using AutoDock 4.2.
Cellular thermal shift assay (CETSA)
For the cell lysate CETSA experiments, PMs were harvested and diluted in PBS adding complete protease inhibitor cocktail. The cells were lysed through freeze-thawed three times in liquid nitrogen and centrifuged at 20,000 g for 20 min at 4 °C to separate cell lysates. Then the cell lysates were incubated with PBS or IR-61 for 20 min at room temperature and were divided into smaller aliquots and heated individually at different temperatures (37, 40, 43, 46, 49, 52 °C) for 3 min followed by cooling for 3 min at room temperature. The heated lysates were centrifuged at 20,000 g for 20 min at 4 °C and the supernatants were transferred to further western blot analysis.
Macrophage depletion in vivo
Macrophages in mice were depleted using clodronate liposomes (#CP-005-005, Liposoma). The suspension of control liposomes and clodronate liposomes were delivered i.p. (200 μL) to deplete macrophages 48 h before being infected with E. coli. The macrophage depletion was analyzed by flow cytometry 48 h after treatment based on expression of F4/80.
Adoptive transfer of macrophages
PMs were transfected with siRNA control (siCtrl) or Nrf2 siRNA (siNrf2) for 48 h and then treated with vehicle or IR-61 for another 24 h. At 48 h after macrophage depletion, 107 macrophages were injected i.v. into the mice.
Flow cytometry
IR-61 (2 mg kg−1) was injected i.v. into mice. 24 h later, the lungs were minced and chopped to approximately 10 mm pieces. Then the tissues were digested in DMEM medium containing 2 mg mL−1 of type IV collagenase (#C5138, Sigma), 0.125 mg mL−1 DNase I (#C5025, Sigma) for 30 min at 37 °C and subsequently the resulting suspension passed through a 70 μm cell strainer. After red cell lysis, the cells were blocked with 1% of rat anti mouse CD16/CD32 (#553141, BD Biosciences, RRID: AB_394656) and then stain with anti-mouse CD3 (#100218, Biolegend, RRID: AB_1595492), anti-mouse CD11b (#101205, Biolegend, RRID: AB_312788), anti-mouse Gr1 (#108433, Biolegend, RRID:AB_10900232), anti-mouse CD19 (#115520, Biolegend, RRID: AB_313655), anti-mouse F4/80 (#123116, Biolegend, RRID: AB_893481), and anti-mouse CD11c (#117308, Biolegend, RRID: AB_313777) for 30 min at 4 °C. The acquisition was performed on a FACSCanto flow cytometer with FACSDiva software, and subsequent analysis was performed using FlowJo V10.1.
Western blot analysis
Cells were lysed in IP lysis buffer adding protease inhibitor cocktail. Nuclear proteins were extracted using protein extraction kit (#P0027, Beyotime). Protein concentrations were determined using a BCA kit (Beyotime). Same amounts of proteins were subjected to western blot analysis. Primary antibodies used were: [Anti-β-actin (#AF0003, Beyotime, RRID: AB_2893353), Anti-Keap1 (#10503-2-AP, Proteintech, RRID: AB_2132625), Anti-Nrf2 (#16396-1-AP, Proteintech, RRID: AB_2782956), Anti-β-Trcp (#4394, CST, RRID: AB_10545763), Anti-p62 (#23214, CST, RRID: AB_2798858), Anti-Ubiquitin (#3936, CST, RRID: AB_331292), Anti-Marco (#ab108113, Abcam, RRID: AB_10861943), Anti-Msr1 (#ab151707; #ab123946, Abcam, RRID: AB_10974791), Anti-Lamin A/C (#10298-1-AP, Proteintech, RRID: AB_2296961), Anti–HO–1 (#10701-1-AP, Proteintech, RRID: AB_2118685), Anti-Gclc (#12601-1-AP, Proteintech, RRID: AB_2278734), Anti-Gclm (#14241-1-AP, Proteintech, RRID: AB_2107832), Anti-Gsr (#18257-1-AP, Proteintech, RRID: AB_10598162), Anti-Nqo1 (#67240-1-lg, Proteintech)]. Bound antibodies were detected with HRP-conjugated secondary antibodies (#A0239, Beyotime) by ECL kit (#WBKLS0100, Millipore). All antibodies used in this study were purchased by the company and have been validated.
Quantitative real-time reverse transcript PCR (qRT-PCR)
Total RNA of cells was extracted using Trizol (#15596018, Invitrogen) according to the manufacturer's instructions. cDNA was synthesized by using RevertAid First Strand cDNA Synthesis Kit (#K1622, Invitrogen). Quantitative real-time PCR was carried out in the CFX Connect Real-Time System (Biorad) using a SYBR green qPCR master mix (#RR420B, Takara). The primer sequences for the qRT-PCR are listed in Table S2.
Fluorescence microscopy
For fluorescence analysis of cultured cells, PMs were seed on glass cover slips in 24-well dishes and incubated with PBS or 10 μM IR-61 for 24 h. Then cells were washed three times with PBS and fixed in 4% paraformaldehyde phosphate buffer solution for 15 min at room temperature. Fixed cells were incubated with 0.2% Triton X-100 (#T9284, Sigma) for 10 min for permeabilization and blocked with 10% FBS in PBS for 1 h. The cells were incubated with Nrf2 antibody (#16396-1-AP, Proteintech, RRID: AB_2782956) overnight at 4 °C, followed by another 1 h with the secondary antibody. Cells were imaged using fluorescent microscope (Olympus BX51).
Immunoprecipitation
PMs were treated with PBS or 10 μM IR-61 for 24 h. Pierce Classic IP Kit (#26146, Thermo) was used for immunoprecipitation following to the manufacturer's instructions. In brief, the cells were lysed in ice-cold IP lysis buffer adding protease inhibitor cocktail. The sample was incubated with specific antibody overnight at 4 °C to form immunecomplexes, and then the complexes were added to the Protein A/G Plus Agarose to remove non-bound material. The bound immunecomplexes were dissociated from the Protein A/G by using a low pH elution buffer and subjected to western blot analysis.
Tissue histology
Mice lung and kidney tissues were excised and fixed in 4% paraformaldehyde at room temperature. The fixed tissues were embedded in paraffin and cut into 5 μm thick sections and were stained with hematoxylin–eosin (H&E). These section images were captured by a microscope (Olympus BX51) and blindly assessed by two independent histopathologists.
siRNA treatment
siRNA specifically targeting Nrf2, Marco, β-TrCP or non-specific siRNA was purchased from GenePharma. PMs were cultured in OptiMEM (#31985062, Gibco) and transfected with siRNA using Lipofectamine 3000 (#L3000008, Invitrogen) according to the manufacturer's protocol. Fresh medium was changed for culture at 6 h after transfection, and following experiments were carried out 48 h later.
Statistics
Data are expressed as the mean ± SD. N numbers are attached in the figure legends. All in vivo data represented individual biological replicates. In vitro experiments were repeated at least three times. Sample sizes calculations for in vivo experiments were based on the principle of 3 R's and chosen using PASS 15.0. Pilot data used to calculation of samples sizes were derived from previous experiments in our group. The mean survival rates of control group and IR-61 treatment group were 10% and 70%, respectively calculated from the pilot data. If using α = 0.05 and a power of 0.80, the sample size needed in each group was 8. Sample size calculation for mice bacterial clearance experiments were also based on our pilot data. The mean difference between two groups in CFU (log) of 1.5 and SD = 0.5. If using α = 0.05 and a power of 0.80, the sample size needed in each group was 4. Sample size calculation for cytokine concentrations of mice were based on pilot data too. The mean difference between two groups in cytokines of 150 and SD = 50. If using α = 0.05 and a power of 0.80, the sample size needed in each group was 4. Statistical analyses were conducted with tools of statistics in GraphPad Prism 8.4.3 or SPSS 26.0.
Linear regression adjusting for variable “age” was used to analyzed data in Fig. S9a. Shapiro–Wilk Test was used to assess the assumption of normality (α = 0.05). Two-sided Student's t-test or one-way ANOVA with Bonferroni post-hoc test was applied for normal data. Non-parametric test (Mann–Whitney test) was performed for non-normal data (Fig. 6d (Blood CFU), Fig. 6f (Tnf), and Fig. 6g (BUN)). Statistical significance of survival was determined using a log-rank (Mantel–Cox) test. The Fisher exact test was used for comparison of categorical variables. p < 0.05 was considered statistically significant.
Fig. 6.

IR-61 treatment affords protection against sepsis. (a–c) Mice treated with IR-61 displayed higher survival during LPS sepsis, E. coli sepsis, and CLP sepsis (Vehicle vs IR-61: p = 0.0001 (LPS), p = 0.0230 (E. coli) and p = 0.0029 (CLP); n = 10 mice per group in LPS sepsis and CLP sepsis, n = 12 mice per group in E. coli sepsis). (d) The bacterial clearance in the blood, PLF, spleen, and lung was detected 24 h after IR-61 treatment in the CLP-induced sepsis mice (Vehicle vs IR-61: p = 0.0079 (blood), p = 0.0033 (PLF), p = 0.0387 (spleen), p = 0.0137 (lung); n = 4–5 mice per group). (e, f) Cytokine concentrations in plasma (e) (Vehicle vs IR-61: p = 0.0006 (Tnf), p = 0.0050 (IL-6), p = 0.0001 (IL-1β), p = 0.0012 (IL-10); n = 5 mice per group) and PLF (f) obtained from CLP-induced septic mice treated with IR-61 for 24 h (Vehicle vs IR-61: p = 0.0079 (Tnf), p = 0.0048 (IL-6), p = 0.0005 (IL-1β), p = 0.0217 (IL-10); n = 5 mice per group). (g) Measurements of plasma biochemical parameters for the liver, kidney, heart, and pancreas functions (Vehicle vs IR-61: p = 0.0079 (BUN), p = 0.0003 (ALT), p = 0.0011 (AST), p = 0.0026 (CK), p = 0.0002 (AMYL); n = 5 mice per group). (h) Histopathological analysis of the lung and kidney. Representative images are shown (bars represent 250 μm; n = 5 mice per group). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; Mantel–Cox log-rank test (longitudinal data in panels a–c); two-sided Student's t-test or Mann–Whitney test (graphs in panels d–g)).
Ethics
All animal husbandry and procedures were approved by the Ethics Committee and performed in accordance with the Animal Care and Use Committee Guidelines of the Army Military Medical University (Approve No. AMUWEC2019202). The human studies were approved by the Institutional Review Board of General Hospital of Western Theater Command (Approve No. 2022EC2-ky048). Written informed consent was obtained from each participant.
Role of funders
The funders did not have any role in study design; data collection, analysis, or interpretation; writing of the manuscript; or in the decision to submit the paper for publication.
Results
IR-61 preferentially accumulates in macrophages at infection sites and promotes bacterial clearance
We first investigated the biodistribution of IR-61 in a mouse model of acute lung bacterial infection. The structure of IR-61 was reported previously by our group.25 IR-61 was i.v. injected 4 h after i.t. administration of E. coli or PBS into the lungs of mice, and then the distribution of IR-61 in tissues was detected at the indicated time. We found much more IR-61 in infected lungs than in normal lungs, and the fluorescence signal in infected lungs was the highest among the main tissues of infected mice (Fig. 1a). These results indicated that IR-61 preferentially accumulated at the infection sites. We also determined the cell type preference for IR-61 in the infected lungs by flow cytometry. The results showed that IR-61 was primarily internalised by CD11b+F4/80+ macrophages (p < 0.0001; Fig. 1b and c). These results collectively indicate that IR-61 preferentially accumulates in macrophages at infection sites upon i.v. administration.
Fig. 1.

IR-61 preferentially accumulates in macrophages at infection sites and promotes bacterial clearance. (a)E. coli or PBS was intratracheally administered into the lungs of mice, and then major organs were imaged using a NIR imaging system after i.v. injection of IR-61 at 12, 24, and 120 h (n = 3 mice per group). (b) Staining indicating the IR-61 levels in macrophages (CD11b+F4/80+), neutrophils (CD11b+Gr1+), monocytes (CD11b+Gr1low), dendritic cells (DCs) (CD11c+Gr1low), B cells (CD19+), and T cells (CD3+) isolated from normal lungs and infected lungs (unstained: n = 3 mice; non-infection: n = 5 mice; infection: n = 5 mice). (c) The amount of internalized IR-61 in various immune cells was measured by MFI (infection group: Macrophages vs monocytes p < 0.0001, Macrophages vs DCs p < 0.0001, Macrophages vs neutrophils p < 0.0001, Macrophages vs T cells p < 0.0001, Macrophages vs B cells p < 0.0001; unstained: n = 3 mice; non-infection: n = 5 mice; infection: n = 5 mice). (d) The levels of Tnf, IL-6, IL-1β, and IL-10 in BALF of mice infected with E. coli at 24 h after the treatment with vehicle or IR-61 (Vehicle vs IR-61 p = 0.0017 (Tnf), p = 0.0049 (IL-6), p < 0.0001 (IL-1β), p = 0.0053 (IL-10); n = 5 mice per group). (e) Representative images of H&E-stained lungs of E. coli-infected mice 24 h after the treatment with vehicle or IR-61 (bars represent 250 μm; n = 5 mice per group). (f) CFUs of bacteria in BALF of mice infected with E. coli at 24 h after the treatment with vehicle or IR-61 (Vehicle vs IR-61 p = 0.0009; n = 5 mice per group). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; one-way ANOVA with Bonferroni post-hoc test (graphs in panel c); two-sided Student's t-test (graphs in panels d and f)).
We further explored the therapeutic effects of IR-61 on acute lung infection in mice. Four hours after E. coli administration into the lungs, IR-61 was i.v. injected into mice at 2 mg kg−1. After 24 h, lung tissues and BALF were collected to analyse inflammation or bacterial burden. Our results indicated that IR-61 treatment significantly decreased the levels of Tnf, IL-6, and IL-1β, and increased the levels of IL-10 in BALF (p = 0.0017, p = 0.0049, p < 0.0001, and p = 0.0053 respectively; Fig. 1d). Consistent with the alleviation of the inflammatory response, histological studies of the lung showed that leukocyte infiltration was markedly reduced upon IR-61 treatment (Fig. 1e). As the inflammatory response is often triggered by bacterial infection, we also studied the effect of IR-61 on bacterial clearance in the lungs. Interestingly, we observed that IR-61 remarkably inhibited bacterial proliferation in BALF (p = 0.0009; Fig. 1f). Taken together, we have demonstrated that IR-61 reduces the bacterial burden and inflammatory response at the infection sites.
IR-61 augments bacterial phagocytosis and killing by macrophages
Considering the critical role of macrophages in bacterial clearance, we speculated that IR-61 may regulate the intrinsic antibacterial function of macrophages. To test this hypothesis, the effect of IR-61 on the phagocytic ability of macrophages was studied by monitoring the uptake of fluorescent latex beads and pHrodo-labelled E. coli. Treatment with IR-61 for 24 h was observed to significantly increase the uptake of latex beads and E. coli by PMs (p = 0.0004 and p = 0.0004 respectively; Fig. 2a and b), which was also supported by the result of flow cytometry assay (p < 0.0001 and p < 0.0001 respectively; Fig. 2c and d). Next, we investigated the effect of IR-61 on the bactericidal activity of macrophages. PMs were exposed to S. aureus or E. coli in vitro after IR-61 treatment for 24 h, and the cell lysates were cultured to count bacterial CFUs. We demonstrated that IR-61 potentiated the bactericidal activity of macrophages (p = 0.0003 and p = 0.0203 respectively; Fig. 2e). Taken together, these results indicate that IR-61 directly induces phagocytosis and bactericidal ability of macrophages.
Fig. 2.

IR-61 augments bacterial phagocytosis and killing by macrophages. (a, b) PMs were treated with 10 μM IR-61 for 24 h, and then were incubated with latex beads (a) or pHrodo-labelled E. coli(b) for another 1 h. Uptake was measured using fluorescent microscopy (Control vs IR-61: p = 0.0004 (a) and p = 0.0004 (b); n = 3 plates of PMs per group and random observation of 3 visual fields per plate; bars represent 25 μm). (c, d) Uptake of latex beads (c) or pHrodo-labelled E. coli(d) was measured using flow cytometry (Control vs IR-61: p < 0.0001 (c) and p < 0.0001 (d); n = 3 per group). (e) PMs were treated with 10 μM IR-61 for 24 h, and then were infected with S. aureus or E. coli for 90 min. Cells were lysed after a 2 h incubation with gentamicin and intracellular contents were seeded onto LB agar plates for 18 h to count the CFUs (Control vs IR-61: p = 0.0003 (S. aureus) and p = 0.0203 (E. coli); n = 4 per group). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; two-sided Student's t-test).
Nrf2 mediates the effect of IR-61 on antibacterial activity of macrophages
To elucidate the underlying mechanisms of enhanced phagocytic activity shown by IR-61-treated macrophages, we initially examined the influence of IR-61 on the expression levels of 12 different phagocytic receptors in PMs. The mRNA levels of two class A scavenger receptors, Marco and Msr1, were substantially increased, but there were no differences in the levels of other scavenger receptors (p < 0.0001 and p = 0.0468 respectively; Fig. 3a). Additionally, IR-61 increased the protein levels of Marco and Msr1 (Fig. 3b). Previous studies have revealed that Marco and Msr1 are target genes of Nrf2, and that the promoter of Marco contains an antioxidant response element (ARE).26 We found that the protein levels of Nrf2 were increased in response to IR-61 treatment (Fig. 3b). Moreover, Nrf2 knockdown abolished the effect of IR-61 on Marco and Msr1 expression (Fig. 3c). Next, we investigated whether Nrf2 is responsible for the antibacterial function regulation of macrophages by IR-61. The enhancement in phagocytic activity induced by IR-61 was reversed after Marco knockdown in macrophages (p = 0.6984 and p = 0.2269 respectively; Fig. 3d and e), which was consistent with the observations when macrophages were pre-treated with two class A scavenger receptor antagonists, namely fucoidan and poly-I (p = 0.5587 and p = 0.3457 respectively; Fig. S2a and b). Next, we silenced Nrf2 expression and demonstrated that silencing of Nrf2 abrogated the function of IR-61 to potentiate phagocytic (p = 0.1216 and p = 0.8871 respectively; Fig. 3f and g) and bactericidal activity (p = 0.6576 and p = 0.4892 respectively; Fig. 3h) in macrophages.
Fig. 3.

Nrf2 mediates the effect of IR-61 on antibacterial activity of macrophages. (a) Relative mRNA levels of a panel of phagocytic receptors genes in PMs (Control vs IR-61 p = 0.1514 (Cd11a), p = 0.8793 (Cd11b), p = 0.9663 (Cd16), p = 0.1358 (Cd36), p = 0.4967 (Cd71), p = 0.1188 (Cd86), p = 0.6992 (Cd107), p = 0.2513 (Cd209a), p = 0.2900 (Cd212), p = 0.3309 (F4/80), p < 0.0001 (Marco), p = 0.0468 (Msr1); n = 3 per group). (b) Immunoblots for Marco, Msr1, and Nrf2 in whole cell lysates from the PMs treated with IR-61 for the indicated times. β-actin was used as the loading control. (c) Immunoblots for Marco, Msr1, and Nrf2 in whole cell lysates from PMs treated with IR-61 for 24 h or 48 h after transfection with siCtrl or siNrf2. β-actin was used as the loading control. (d, e) Uptake of latex beads (d) (siCtrl vs siCtrl IR-61 p < 0.0001, Marco siRNA (siMarco) vs siMarco IR-61 p = 0.6984; n = 5 per group) or pHrodo-labelled E. coli(e) (siCtrl vs siCtrl IR-61 p < 0.0001, siMarco vs siMarco IR-61 p = 0.2269; n = 3 per group) was measured using flow cytometry in the PMs treated with IR-61 for 24 h after transfection with siCtrl or siMarco. (f, g) Uptake of latex beads (f) (siCtrl vs siCtrl IR-61 p < 0.0001, siNrf2 vs siNrf2 IR-61 p = 0.1216; n = 3 per group) or pHrodo-labelled E. coli(g) (siCtrl vs siCtrl IR-61 p < 0.0001, siNrf2 vs siNrf2 IR-61 p = 0.8871; n = 5 per group) was measured using flow cytometry in the PMs treated with IR-61 for 24 h after transfection with siCtrl or siNrf2. (h) Intracellular bacterial CFUs were determined in PMs treated with IR-61 for 24 h after transfection with siCtrl or siNrf2 (siCtrl vs siCtrl IR-61 p = 0.0002 (S. aureus), p = 0.0034 (E. coli), siNrf2 vs siNrf2 IR-61 p = 0.6576 (S. aureus), p = 0.4892 (E. coli); n = 4 per group). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; two-sided Student's t-test).
IR-61 activates Nrf2 by preventing its ubiquitin-proteasome degradation
Next, we investigated the mechanism by which IR-61 regulates Nrf2 protein levels. First, we detected the representative antioxidant target genes of Nrf2 to confirm that IR-61 indeed augmented Nrf2 activation. The results showed that IR-61 increased the mRNA levels of Ho1, Nqo1, Gstm1, and Gclc (p < 0.0001 at 24 h; Fig. 4a), as well as the protein levels of Nqo1, Ho1, Gclm, Gsr, and Gclc (Fig. 4b), all of which are downstream target genes of Nrf2. Nuclear translocation of Nrf2 is necessary to activate downstream target genes.27 The immunofluorescence assessment of macrophages indicated that IR-61 increased the amount of Nrf2 in the nucleus (Fig. 4c). The effect of IR-61 on the distribution of Nrf2 in macrophages was further investigated using western blot. As expected, predominant nuclear localisation of Nrf2 was observed in response to IR-61 treatment (Fig. 4d). IR-61 significantly induced the protein expression and nuclear translocation of Nrf2; however, the mRNA levels of Nrf2 remained unchanged (p > 0.9999 at 24 h; Fig. 4e). These data implied that modulation of Nrf2 accumulation by IR-61 might not occur at the transcriptional level. Therefore, we hypothesised that IR-61 might regulate the protein stability of Nrf2. To test this hypothesis, we studied the effect of IR-61 on Nrf2 protein levels when macrophages were treated with the protein synthesis inhibitor, cycloheximide (CHX). Compared to IR-61-treated cells, more rapid degradation of Nrf2 was detected in control cells (Fig. 4f), and the half-life of Nrf2 was shorter in control cells than that in IR-61-treated cells (41 min vs 78 min) (Fig. 4f). Ubiquitination and subsequent proteasome-mediated protein degradation are key mechanisms that control the intracellular levels of Nrf2.28 We demonstrated that the general proteasome inhibitor, MG132, suppressed the effect of IR-61 in terms of increasing the protein levels of Nrf2 (Fig. 4g). In addition, although the levels of total Nrf2 precipitated by the anti-Nrf2 antibody were high, ubiqutin-Nrf2 (Ub-Nrf2) were found to decrease in IR-61-treated macrophages (Fig. 4h). Taken together, these results illustrate that IR-61 stabilises Nrf2 protein by suppressing ubiquitin-proteasome-mediated protein degradation.
Fig. 4.

IR-61 activates Nrf2 by preventing its ubiquitin-proteasome degradation. (a) The mRNA levels of Nrf2 target genes were measured at various time points after treatment with 10 μM IR-61 (Ho1: 0 vs 6 h p = 0.0031, 0 vs 12 h p = 0.0099, 0 vs 24 h p < 0.0001; Nqo1: 0 vs 6 h p < 0.0001, 0 vs 12 h p < 0.0001, 0 vs 24 h p < 0.0001; Gstm1: 0 vs 6 h p = 0.0947, 0 vs 12 h p = 0.0004, 0 vs 24 h p < 0.0001; Gclc: 0 vs 6 h p = 0.0021, 0 vs 12 h p < 0.0001, 0 vs 24 h p < 0.0001; n = 3 per group). (b) The levels of proteins downstream of Nrf2 were measured after treatment with 10 μM IR-61. β-actin was used as the loading control. (c) Immunofluorescence staining of Nrf2 in PMs treated with 10 μM IR-61 for 24 h. Representative images are shown (bars represent 25 μm; n = 3 per group). (d) Protein level of Nrf2 in nucleus was detected at various time points after treatment with 10 μM IR-61. Lamin and β-actin served as markers for nuclear and cytosolic proteins, respectively. (e) The mRNA levels of Nrf2 were measured at various time points after treatment with 10 μM IR-61 (Nrf2: 0 vs 6 h p > 0.9999, 0 vs 12 h p = 0.5833, 0 vs 24 h p > 0.9999; n = 3 per group). (f) Immunoblots for Nrf2 in whole cell lysates from control and IR-61-treated PMs in the presence of 100 mg mL−1 CHX at the various time points (left). Nrf2 band intensity was quantified by ImageJ and Nrf2 levels in the untreated cells were normalized to 1 (right). (g) Immunoblots for Nrf2 in whole cell lysates from control and IR-61-treated PMs in the presence or absence of proteasome inhibitor MG132. (h) The Ub-Nrf2 was measured by co-immunoprecipitation (co-IP) of Nrf2 using a subsequent western blot assay with anti-ubiquitin antibody in control or IR-61-treated macrophages. Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; one-way ANOVA with Bonferroni post-hoc test).
IR-61 promotes Nrf2 stabilization through competitive binding with Keap1
We further explored the molecular mechanisms underlying Nrf2 stabilization by IR-61. Our previous study has indicated that IR-61 temporarily increased ROS levels and activated the ROS/PI3K/AKT pathway in macrophages.25 To confirm the role of ROS in IR-61-mediated accumulation of Nrf2, we pre-treated PMs with N-acetyl-cysteine (NAC) and found that IR-61 promoted Nrf2 accumulation even when ROS were inhibited (Fig. 5a). These results implied that there is an unknown ROS-independent pathway that promotes Nrf2 activation in IR-61-treated macrophages. In addition to Keap1, β-TrCP, a downstream component of the PI3K/AKT/GSK-3 pathway, promoted Nrf2 degradation in a Keap1-independent manner.29 We found that IR-61 induced a significant accumulation of Nrf2 even after β-TrCP knockdown in macrophages (Fig. 5b). Previous studies have indicated that GSK-3 cannot significantly activate Nrf2 in the presence of functional Keap1.30 Hence, we speculated that Keap1 was mainly responsible for IR-61-mediated Nrf2 stabilisation. In addition, IR-61 had no effect on Keap1 expression or the interaction between Keap1 and its negative regulator, p62 (Fig. S3).
Fig. 5.

IR-61 promotes Nrf2 stabilization through competitive binding with keap1. (a) Immunoblots for Nrf2 in whole cell lysates from the PMs incubated in the presence or absence of 5 mM NAC for 4 h and then treated with 10 μM IR-61 for the indicated time. β-actin was used as the loading control. (b) Immunoblots for Nrf2 and β-TrCP in whole cell lysates from PMs treated with IR-61 for 24 h after transfection with siCtrl or β-TrCP siRNA (siβ-TrCP). β-actin was used as the loading control. (c) SPR assay for interaction of IR-61 with Keap1 protein. (d) Immunoblots for Keap1 in whole cell lysates from PMs incubated in the presence (+) or absence (−) of 10 μM IR-61 at different temperatures. (e) Keap1 band intensity was quantified by ImageJ. Blot intensity was normalized to intensity obtained for the 37 °C sample. (f) SPR assay for Nrf2–Keap1 interaction treated with 0, 2.5, 5, and 10 μM IR-61. (g, h) PMs were treated with IR-61 or vehicle for 24 h. Protein levels of Keap1 and Nrf2 as well as the interaction between Keap1 and Nrf2 were assayed using western blot and co-IP. (i) Ligand docking analysis for IR-61 with human Keap1 protein.
Disruption of the Keap1–Nrf2 interaction upon competitive binding with Keap1 is a well-documented mechanism of Nrf2 activation.31 NIR fluorescence signal was detected in separated Keap1 after incubation with IR-61 in vitro (Fig. S4), indicating that IR-61 was bound to Keap1. Consistently, the results from SPR study showed that the extracellular binding affinity of IR-61 with Keap1 was 1.05 μM (Fig. 5c). Target engagement by drugs in cells can be monitored by a CESTA,32 which can be used to detect the cellular Keap1 binding behaviour of IR-61. IR-61 was found to enhance the thermostability of Keap1 and induced an obvious shift in the protein thermal melting curves (Fig. 5d and e). We further investigated whether IR-61 interfered with the interaction between Keap1 and Nrf2. The results using SPR analysis demonstrated that IR-61 disrupted the Keap1–Nrf2 interaction at 2.5, 5, and 10 μM (Fig. 5f). Moreover, immunoprecipitation assay confirmed that IR-61 decreased the interaction between Keap1 and Nrf2 in macrophages (Fig. 5g and h). These results demonstrated that IR-61 exhibited tight Keap1 binding and disrupted the Keap1–Nrf2 interaction both in vitro as well as in cells. In silico molecular ligand docking analysis was performed to predict the underlying binding mode between IR-61 and Keap1. The results showed that IR-61 was fitted into the hydrophobic pocket of Keap1 and formed a hydrophobic interaction with Phe557, Phe335, and Tyr572 (Fig. 5i). In addition, the carboxylate group at the top of IR-61 formed hydrogen bonds with Arg415, Arg483, and Ser508 (Fig. 5i). Altogether, these results verify that IR-61 directly binds to Keap1 to induce Nrf2 release and activation.
IR-61 treatment affords protection against sepsis
Considering the properties of IR-61 in terms of targeting macrophages at infection sites and enhancing their antibacterial activity, we speculated that IR-61 might show therapeutic effects in experimental sepsis. The mortality rate of mice was reduced after treatment with IR-61 compared with vehicle-treated control mice in the models of LPS sepsis, E. coli sepsis, and CLP-induced polymicrobial sepsis (p = 0.0001, p = 0.0230, and p = 0.0029 respectively; Fig. 6a–c). For example, the survival rate of IR-61-treated mice was 70%, and that of vehicle-treated mice was only 10% in CLP sepsis model (Fig. 6c). We further examined the effect of IR-61 treatment on the bacterial load and cytokine production during CLP sepsis. IR-61 treatment significantly reduced the bacterial burden in the blood, PLF, spleen, and lung samples (p = 0.0079, p = 0.0033, p = 0.0387, and p = 0.0137 respectively; Fig. 6d). In addition, markedly decreased levels of Tnf, IL-6, and IL-1β, and increased levels of IL-10, were observed in both plasma (p = 0.0006, p = 0.0050, p = 0.0001, and p = 0.0012 respectively; Fig. 6e) and PLF (p = 0.0079, p = 0.0048, p = 0.0005, and p = 0.0217 respectively; Fig. 6f) after mice were treated with IR-61 as compared to those in vehicle-treated mice. We isolated PMs from CLP-induced septic mice to determine the effect of IR-61 on macrophages in vivo. Macrophages from IR-61-treated mice showed greater phagocytosis of E. coli than that shown by the macrophages from vehicle-treated mice (p < 0.0001; Fig. S5a). In addition, IR-61 treatment enhanced the protein expression of Nrf2, Marco, and Msr1 in PMs isolated from mice with CLP-induced sepsis (Fig. S5b). We also assessed organ injury to confirm the protective function of IR-61 in mice with CLP-induced sepsis. Biochemical examination indicated that IR-61 treatment remarkably attenuated the plasma levels of blood urea nitrogen (BUN; kidney), alanine aminotransferase (ALT; liver), aspartate aminotransferase (AST; liver), creatine kinase (CK; heart), and amylase (AMYL; pancreas) (p = 0.0079, p = 0.0003, p = 0.0011, p = 0.0026, and p = 0.0002 respectively; Fig. 6g). Histological examination revealed reduced injuries in the lungs and kidneys of IR-61-treated mice (Fig. 6h). Furthermore, administration of IR-61 inhibited apoptosis in the septic spleen and intestine (Fig. S6). Taken together, these results demonstrate that IR-61 decreases mortality, bacterial load, inflammatory cytokines, and organ injury during sepsis.
IR-61 improves survival and reduces bacterial burden by activating Nrf2 in macrophages during sepsis
We further evaluated whether IR-61 could manage sepsis by activating Nrf2 in macrophages. Macrophages depletion significantly diminished the survival rate of septic mice treated with IR-61 (p = 0.0990; Fig. 7a and b), implying that macrophages might be related to the protective effect of IR-61 in sepsis. In addition, we found that ML385 (specific Nrf2 inhibitor) effectively inhibited the Nrf2 activity in vivo (Fig. S7) and Nrf2 inhibition abolished the beneficial effects of IR-61 (p = 0.1318; Fig. 7c). To determine whether the improved outcomes of sepsis after IR-61 treatment were mediated mainly by Nrf2 activation in macrophages, we further assessed the survival and bacterial burden of E. coli sepsis after the adoptive transfer of IR-61-treated macrophages. The survival of septic mice who received IR-61-treated macrophages was significantly higher than that of the septic mice who received vehicle-treated macrophages (p = 0.0249; Fig. 7d). However, adoptive transfer of IR-61-treated macrophages with Nrf2 silencing had no effect on the survival of septic mice (p = 0.6436; Fig. 7d). Furthermore, the bacterial burdens in the blood and PLF were reduced in septic mice receiving IR-61-treated macrophages compared with those receiving vehicle-treated macrophages (p < 0.0001 and p = 0.0002 respectively; Fig. 7e). Adoptive transfer of IR-61-treated macrophages with Nrf2 silencing had no effect on the bacterial burden in the blood and PLF of septic mice (p = 0.8098 and p = 0.1467 respectively; Fig. 7e). These results indicate that IR-61 treatment improves the outcomes of sepsis by activating Nrf2 in macrophages.
Fig. 7.

IR-61 improves survival and reduces bacterial burden by activating Nrf2 in macrophages during sepsis. (a) Representative flow cytometry images of F4/80+ macrophages in spleens 2 days after clodronate liposome injection. Quantification of macrophages in control liposome-treated and clodronate liposome-treated mice (Vehicle vs Clodronate: p < 0.0001; n = 4 mice per group). (b) Survival after macrophage depletion and treatment with IR-61 or vehicle in mice with E. coli sepsis (Vehicle + MΦ (+) vs IR-61 + MΦ (+): p = 0.0077, Vehicle + MΦ (−) vs IR-61 + MΦ (−): p = 0.0990; n = 10 mice per group). (c) Mice were given ML385 intraperitoneally 2 h before E. coli injection. Survival of mice treated with vehicle or IR-61 was monitored (Vehicle vs IR-61: p = 0.0272, ML385 + Vehicle vs ML385+IR-61: p = 0.1318; n = 10 mice per group). (d) Survival after transferring vehicle-treated, IR-61-treated, vehicle-treated Nrf2 silenced, or IR-61-treated Nrf2 silenced macrophages in mice with E. coli-induced sepsis (MΦ/Vehicle vs MΦ/IR-61: p = 0.0249, MΦ-Nrf2-/Vehicle vs MΦ-Nrf2-/IR-61: p = 0.6436; n = 10 mice per group). (e) Bacterial burden in the blood or PLF of E. coli-injected mice who received vehicle-treated, IR-61-treated, vehicle-treated Nrf2 silenced, or IR-61-treated Nrf2 silenced macrophages (MΦ/Vehicle vs MΦ/IR-61: p < 0.0001 (blood), p = 0.0002 (PLF), MΦ-Nrf2-/Vehicle vs MΦ-Nrf2-/IR-61: p = 0.8098 (blood), p = 0.1467 (PLF); n = 6 mice per group). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; two-sided Student's t-test (graphs in panels a, e); Mantel–Cox log-rank test (longitudinal data in panels b–d)).
Nrf2 is activated by IR-61 in human macrophages and may associate with sepsis outcomes
We further investigated the effect of IR-61 on human monocyte-derived macrophages. IR-61 treatment increased protein levels of NRF2, MARCO, and MSR1 in human macrophages (Fig. 8a). In addition, IR-61-treated macrophages showed a much greater uptake of pHrodo-labelled E. coli, indicating that IR-61 enhanced the phagocytic ability of human macrophages (p = 0.0022; Fig. 8b). More importantly, ML385 pretreatment abrogated the function of IR-61 to enhance the phagocytosis of human macrophages (p = 0.7328; Fig. S8). Next, we assessed whether NRF2 could be a possible therapeutic target for sepsis in humans. We determined the protein levels of monocytes from patients with sepsis at 24 h after their admission into ICU. Compared with the healthy controls, the expression levels of NRF2, MARCO, and MSR1 were significantly increased in patients with sepsis (Fig. 8c). Furthermore, sepsis non-survivors had decreased expression levels of NRF2, MARCO, and MSR1 compared to that observed in sepsis survivors (Fig. 8c). In addition, analysis of RNA sequencing (RNA-seq) in a well-characterized large cohort of sepsis patients also revealed that both the expression levels of Nrf2 and Marco in patients with sepsis were higher than those in the healthy controls (p = 0.0273 and p < 0.0001 respectively; Fig. 8d), but their expression levels were lower in sepsis non-survivors compared with sepsis survivors (p = 0.0321 and p = 0.0097 respectively; Fig. 8d). These results imply that activation of Nrf2 may be beneficial, and that IR-61 may prove to be a promising candidate agent for sepsis management.
Fig. 8.

Nrf2 is activated by IR-61 in human macrophages and may associate with sepsis outcomes. (a) NRF2, MARCO, and MSR1 protein levels in human macrophages treated with IR-61 for 24 h. β-actin was used as the loading control (n = 4 healthy humans per group). (b) Human macrophages were treated with 10 μM IR-61 for 24 h, and then incubated with pHrodo-labelled E. coli for another 1 h. Uptake was measured by fluorescent microscopy (Control vs IR-61: p = 0.0022; n = 3 plates of human macrophages per group and random observation of 3 visual fields per plate; bars represent 100 μm). (c) NRF2, MARCO, and MSR1 protein levels in circulating CD14+ monocytes isolated from the blood (n = 4 for healthy controls, n = 4 for sepsis survivors, and n = 3 for sepsis non-survivors). (d)Nrf2 and Marco mRNA levels in the peripheral blood (healthy vs sepsis: p = 0.0273 (Nrf2), p < 0.0001 (Marco), survivor vs non-survivor: p = 0.0321 (Nrf2), p = 0.0097 (Marco); n = 22 for healthy controls, n = 51 for sepsis patients, n = 34 for sepsis survivors, and n = 17 for sepsis non-survivors). Data were obtained from GSE95233 dataset. Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; two-sided Student's t-test).
Discussion
Macrophages are important components of the innate immune system and possess a variety of functions, ranging from elimination of invading pathogens to modulation of inflammatory response.33 Macrophages are critical for defence against pathogens and are largely responsible for eradicating bacterial infections in sepsis.9,34 Deficiency of macrophage function seems to be the main reason for insufficient antibacterial defence during sepsis.9,11 The regulation of the antibacterial function of macrophages has been considered as a treatment modality for sepsis. Several recombinant therapeutic proteins that enhance the antibacterial activity of macrophages, such as LETC2, flagellin, and IL-34, have been tested in experimental sepsis models and shown to improve the outcomes of sepsis.12,14,35 However, production capacity, cost, and safety hinder the widespread application of recombinant proteins in sepsis.18 Recently, MSCs administration has emerged as an effective therapeutic strategy for sepsis.36 The promotion of bacterial clearance by macrophages is the key for realizing the therapeutic benefits of MSCs.17 Moreover, MSCs have been shown to exert therapeutic effects in sepsis in clinical trials.15,16 However, some inherent problems associated with MSCs, including genetic instability, heterogeneity, promotion of tumorigenesis, and other side effects, limit their clinical application.19 IR-61 may contribute to the development of macrophage-targeted therapy due to its inherent property of targeting macrophages at infection sites and concurrently modulating their antibacterial defence and pro-inflammatory phenotype. This small molecule can prove to be a safe, cheap, and multifunctional agent with a high delivery efficiency for sepsis management.
Nrf2 is a major regulator of antioxidant responses in many tissues and plays a vital role in cytoprotection against endogenous and exogenous insults.37 By binding to ARE, Nrf2 regulates the expression of a variety of genes, including those encoding proteins associated with cellular redox homeostasis, detoxifying enzymes, macromolecular damage repair, and metabolic alteration enzymes.38,39 These important biological functions of Nrf2 have attracted the interest of researchers in the drug discovery community. Many studies have demonstrated that Nrf2 activation protects against various diseases, particularly chronic age-related and inflammatory diseases, including cancer,40 diabetes,41 neurodegenerative disorders,42 chronic obstructive pulmonary disease,43 and chronic kidney disease.44 Despite its protective role against chronic diseases, some studies have demonstrated a strong relationship between Nrf2 and sepsis.45,46 Nrf2 activation enhances the antibacterial defence capability of macrophages and protects against sepsis.21 However, research pertaining to the pharmaceutical regulation of Nrf2 in the treatment of sepsis is still lacking.
There are two categories of Nrf2 inducers based on the Keap1–Nrf2 interaction, namely covalent Keap1 modifiers and non-covalent Keap1–Nrf2 PPI inhibitors.47 Keap1 contains specific cysteine residues (i.e., Cys151, Cys257, Cys273, Cys288, and Cys297), which act as redox sensors and regulators.48 Most Nrf2 inducers are electrophilic agents that covalently react with the reactive cysteine residues in Keap1.49 This reaction causes conformational changes in Keap1 and blocks Nrf2 ubiquitination.50 Electrophilic Nrf2 activators have been shown to alleviate a variety of diseases, including renal disease, liver disease, and type 2 diabetes mellitus, in clinical trials.51 For example, bardoxolone methyl (CDDO-Me) has shown promising therapeutic effects in chronic kidney disease in early clinical studies.52 However, a phase III study involving CDDO-Me was terminated because of a high rate of cardiovascular events.53 Non-specific binding and uncontrollable reactivity of electrophiles result in side effects and reduced regulation of expected targets.47 Non-covalent Keap1–Nrf2 PPI inhibitors provide a more attractive strategy. Unlike electrophilic agents, Keap1−Nrf2 inhibitors induce Nrf2 activation by competitively and directly disrupting the non-covalent interaction between Keap1 and Nrf2, which is a safer and more effective strategy for cell protection.23,54 PPI inhibitors have shown great therapeutic potential in sepsis. However, targeting specificity and safety remain the main barriers for the clinical use of current PPI inhibitors.24 Nrf2 activation in normal cells disturbs the normal physiological function of ROS, and is associated with an increased risk of tumorigenesis.55,56 Thus, the development of PPI inhibitors that selectively accumulate in macrophages at infection sites may greatly improve the therapeutic effect and reduce side effects in sepsis treatment. However, to the best of our knowledge, PPI inhibitors with inherent tissue- and cell-selective properties have not yet been reported.
IR-61 is designed and synthesised by modifying the N-alkyl side chains around a heptamethine core, representing a small-molecule dye with inherent mitochondrial-targeting and NIR fluorescence properties.57 IR-61 has been demonstrated to target mitochondria and suppress pro-inflammatory cytokines by stimulating the mitochondrial function of macrophages and further inhibit chronic inflammation in obesity.25 In this study, we found that IR-61 preferentially accumulated in macrophages at infection sites following i.v. injection. Unlike traditional strategies of obtaining macrophage-targeting agents by conjugating functional nanocarriers with therapeutic drugs,58 IR-61 possesses inherent macrophage-targeting property, is cheaper and more stable, and has better biocompatibility than most nanomedicines.59,60 In addition to acting in the mitochondria, IR-61 distributed in the cytoplasm can serve as a Keap1–Nrf2 inhibitor, thereby selectively promotes the antibacterial activity of macrophages at infection sites and protects against sepsis. IR-61 is a multifunctional agent that directly inhibits the pro-inflammatory phenotype and enhances bacterial killing of macrophages. These two regulatory effects of IR-61 on macrophages may simultaneously lead to the inflammation alleviation in infected mice. It has good biocompatibility and emerging prospect for clinical applications.
Despite the preliminary observations of an association between Nrf2 expression levels in monocytes and sepsis outcomes in humans, we are aware of several limitations in human study, including single-center source and small sample size. A large sample of sepsis cases from multi-center is needed to confirm relationship between Nrf2 and sepsis outcomes definitively. In addition, although the age of healthy people is lower than that of sepsis patients in this study, the age difference doesn't interfere with our conclusions. A series of studies have confirmed that the Nrf2 protein levels and activity decline with age.61, 62, 63 Moreover, we further analyzed the expression levels of NRF2, MARCO, and MSR1 using linear regression adjusting for variable age. Our results showed that age had no effect on our three outcomes, and the statistical differences among healthy, survivor, and non-survivor groups were still significant based on this regression results (Fig. S9).
As summarized in Fig. 9, we identify the small-molecule dye IR-61 as a Keap1–Nrf2 PPI inhibitor that targets macrophages at infection sites on a mouse infection model, promotes bacterial clearance, inhibits host inflammation response, and improves outcomes of sepsis in mice. Mechanistic studies indicate that IR-61 potentiates the antibacterial activity of macrophages by enhancing Nrf2 activity by inhibiting the Keap1–Nrf2 interaction. Our study has demonstrated that the selective enhancement of Nrf2 in macrophages at infection sites holds promise for sepsis management. More importantly, IR-61 enhances bacterial phagocytosis by human macrophages. To strengthen the results in human setting, we pre-treat the human macrophages with ML385. In addition, we preliminarily investigate the expression of Nrf2 in sepsis using monocytes from human patients and observe an underlying interconnection between the expression levels of Nrf2 in monocytes and sepsis outcomes in humans. In summary, the multifunctional small-molecule agent IR-61 has prospects for being developed as a precise treatment of sepsis. Activation of Nrf2 in macrophages may be a potential strategy for sepsis management.
Fig. 9.

Activation of Nrf2 in macrophages at infection sites imparts protection against sepsis. IR-61 selectively accumulates in macrophages at infection sites upon i.v. administration. Then IR-61 activates Nrf2 mainly by directly inhibiting the Keap1–Nrf2 interaction, which further enhances antibacterial defence of macrophages at infection sites and treat sepsis precisely.
Contributors
Y. Wang, Y. Wang, and C. Shi conceived and designed the project; Y. Wang, Y. Wang, X. Tan, and C. Shi wrote the paper; Y. Wang, B. Tang, H. Li, J. Zheng, C. Zhang, Z. Yang, L. Ma, and J. Zhou acquired the data; Y. Wang, B. Tang, P. Luo, L. Ma, Y. Wang, Z. Chen, Z. Xiao, Y. Wang, L. Long, and C. Shi analyzed and interpreted the data. All authors verified the underlying data and approved the final version of the manuscript.
Data sharing statement
All data are available in the main text or the supplementary materials. All data used in the current study are available from the corresponding author by reasonable request.
Declaration of interests
The authors declare that there is no conflict of interest.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Major program 82192884), Intramural Research Project (grant 2018-JCJQ-ZQ-001 and 20QNPY018), and the Chongqing National Science Foundation (CSTB2022NSCQ-MSX1222).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ebiom.2023.104480.
Contributor Information
Yu Wang, Email: wangyusmmu@163.com.
Chunmeng Shi, Email: shicm@sina.com.
Appendix A. Supplementary data

Figure S1 Flow cytometry gating scheme for determining IR-61 content in the cells.
Figure S2.
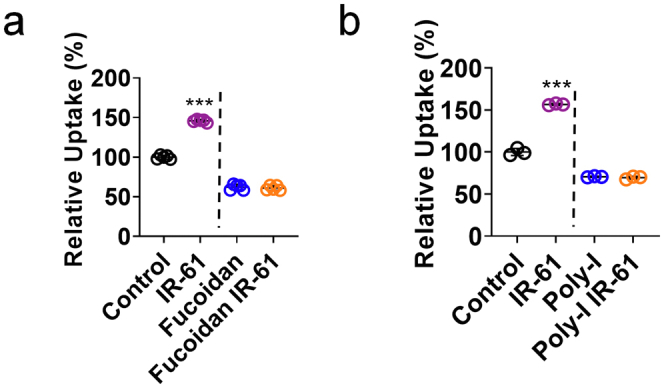
Role of Marco and Msr1 in enhanced phagocytosis of IR-61-treated macrophages. (a, b) PMs were incubated with the class A scavenger receptor antagonists Fucoidan (a) (Control vs IR-61 p < 0.0001, Fucoidan vs Fucoidan IR-61 p = 0.5587;n = 5 per group) and Poly-I (b) (Control vs IR-61 p < 0.0001, Poly-I vs Poly-I IR-61 p = 0.3457;n = 3 per group) and then treated with IR-61. Uptake of pHrodo-labeled E. coli was determined by flow cytometry. Results are presented as the mean ± SD (∗∗∗p < 0.001; two-sided Student’s t-test).
Figure S3.
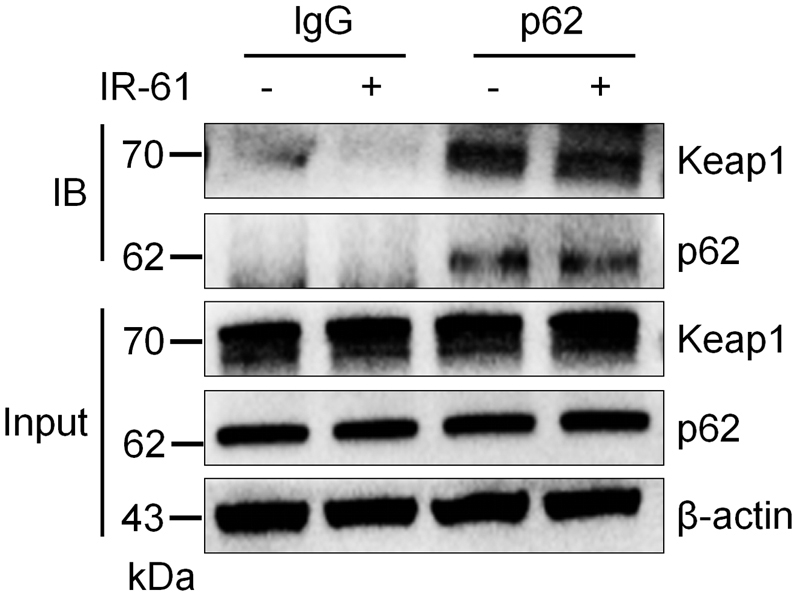
IR-61 has no effect on Keap1 expression or Keap1-p62 interaction. PMs were treated with IR-61 or vehicle for 24 h. The interaction between Keap1/p62 was determined by co-IP assay.
Figure S4.
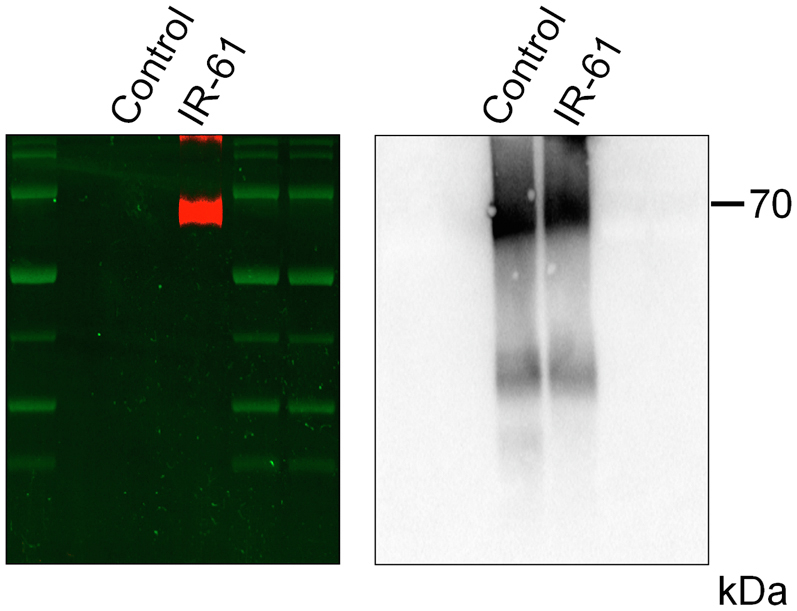
IR-61 binds with Keap1 Protein. Recombinant human Keap1 protein was incubated with IR-61 in 1 M HEPES buffer (pH 7.4) at 37 °C for 30 min, and then the mixture was subjected to western blot. NIR fluorescence band was detected (left panel) and then incubated with Keap1 antibody to scan the protein band (right panel).
Figure S5.

IR-61 promotes phagocytosis and protein levels of Nrf2-Macro/Msr1 in PMs from CLP-induced septic mice. (a) pHrodo-labeled E. coli were injected into the peritoneal cavity 24 h after IR-61 treatment in the CLP-induced septic mice. After 1 h, PMs were extracted and the uptake was determined by flow cytometry (Vehicle vs IR-61 p < 0.0001; n = 6 mice per group). (b) Immunoblots of Nrf2, Marco and Msr1 in whole cell lysates from the PMs in CLP-induced septic mice treated with IR-61 or PBS for 24 h. β-actin was used as the loading control (n = 4 mice per group). Results are presented as the mean ± SD (∗∗∗p < 0.001; two-sided Student’s t-test).
Figure S6.

Apoptosis in spleen and intestine was examined by fluorescent TUNEL labeling. Representative images of TUNEL (green) and DAPI (blue)-stained spleen and intestine samples from vehicle- or IR-61-treated mice 24 h after CLP (bars represent 50 μm; n = 5 mice per group).
Figure S7.
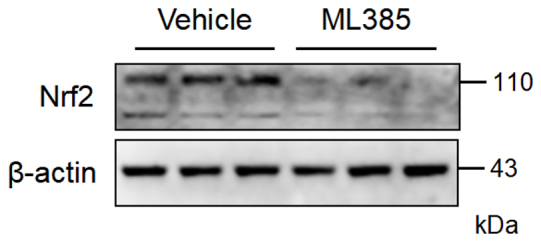
ML385 effectively inhibits the activation of Nrf2 in vivo. Mice were injected intraperitoneally with ML385 or vehicle 2 h before E. coli injection. After 24 h, the PMs were extracted for western blot analysis of Nrf2 levels (n = 3 mice per group).
Figure S8.
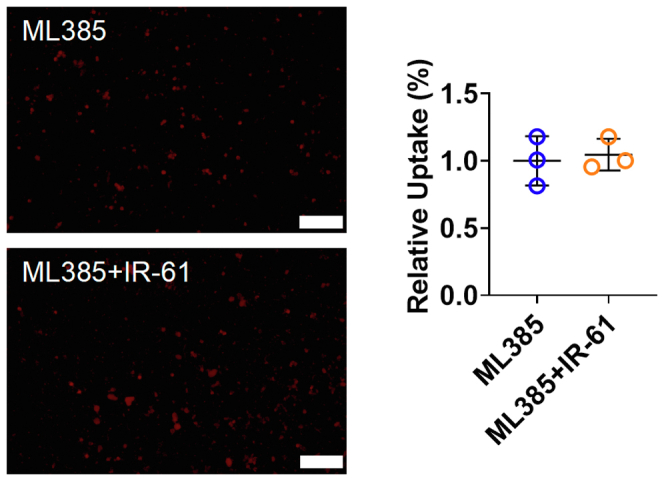
ML385 abrogates the function of IR-61 to enhance the phagocytosis of human macrophages. Human macrophages were treated with ML385 2 h before IR-61 incubation. After 24 h, the cells were incubated with pHrodo-labelled E. coli for another 1 h. Uptake was measured by fluorescent microscopy (ML385 vs ML385+IR-61 p = 0.7328; n = 3 plates of human macrophages per group and random observation of 3 visual fields per plate; bars represent 100 μm). Results are presented as the mean ± SD (two-sided Student’s t-test).
Figure S9.

The statistical analysis of relative protein levels in monocytes from human samples. (a) The relative protein levels of NRF2, MARCO, and MSR1 in Fig. 8c were analyzed using one-way ANOVA with Bonferroni post-hoc test (healthy vs survivor: p < 0.0001 (NRF2), p < 0.0001 (MARCO), p < 0.0001 (MSR1); healthy vs non-survivor: p = 0.0015 (NRF2), p = 0.0047 (MARCO), p = 0.0039 (MSR1); survivor vs non-survivor: p = 0.0029 (NRF2), p = 0.0027 (MARCO), p = 0.0228 (MSR1)). Results are presented as the mean ± SD (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (b) The data in a were further analyzed using linear regression adjusting for variable “age”. The detail parameters are shown.
References
- 1.Cohen J., Vincent J.L., Adhikari N.K., et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614. doi: 10.1016/S1473-3099(15)70112-X. [DOI] [PubMed] [Google Scholar]
- 2.Seeley E.J., Matthay M.A., Wolters P.J. Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol. 2012;303(5):L355–L363. doi: 10.1152/ajplung.00069.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang C.Y., Gao J., Wang Z. Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv Mater. 2018;30(43) doi: 10.1002/adma.201803618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhodes A., Evans L.E., Alhazzani W., et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 5.Papafilippou L., Claxton A., Dark P., et al. Nanotools for sepsis diagnosis and treatment. Adv Healthc Mater. 2021;10(1) doi: 10.1002/adhm.202001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall J.C. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20(4):195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Li Z., Song Y., Yuan P., et al. Antibacterial fusion protein BPI21/LL-37 modification enhances the therapeutic efficacy of hUC-MSCs in sepsis. Mol Ther. 2020;28(8):1806–1817. doi: 10.1016/j.ymthe.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hou J., Chen Q., Wu X., et al. S1PR3 signaling drives bacterial killing and is required for survival in bacterial sepsis. Am J Respir Crit Care Med. 2017;196(12):1559–1570. doi: 10.1164/rccm.201701-0241OC. [DOI] [PubMed] [Google Scholar]
- 9.Csóka B., Németh Z., Szabó I., et al. Macrophage P2X4 receptors augment bacterial killing and protect against sepsis. JCI Insight. 2018;3(11) doi: 10.1172/jci.insight.99431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginhoux F., Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 11.Hotchkiss R.S., Monneret G., Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13(3):260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang X., Yin Y., Yan X., et al. Flagellin attenuates experimental sepsis in a macrophage-dependent manner. Crit Care. 2019;23(1):106. doi: 10.1186/s13054-019-2408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao X., Song Z., Wang C., et al. Interleukin 36α attenuates sepsis by enhancing antibacterial functions of macrophages. J Infect Dis. 2017;215(2):321–332. doi: 10.1093/infdis/jiw535. [DOI] [PubMed] [Google Scholar]
- 14.Lu X.J., Chen J., Yu C.H., et al. LECT2 protects mice against bacterial sepsis by activating macrophages via the CD209a receptor. J Exp Med. 2013;210(1):5–13. doi: 10.1084/jem.20121466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson J.G., Liu K.D., Zhuo H., et al. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med. 2015;3(1):24–32. doi: 10.1016/S2213-2600(14)70291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galstian G.M., Parovichnikova E.N., Makarova P.M., et al. The results of the Russian clinical trial of Mesenchymal Stromal Cells (MSCs) in severe neutropenic patients (pts) with Septic Shock (SS) (RUMCESS trial) Blood. 2015;126(23):2220. [Google Scholar]
- 17.Rabani R., Volchuk A., Jerkic M., et al. Mesenchymal stem cells enhance NOX2-dependent reactive oxygen species production and bacterial killing in macrophages during sepsis. Eur Respir J. 2018;51(4) doi: 10.1183/13993003.02021-2017. [DOI] [PubMed] [Google Scholar]
- 18.O'Flaherty R., Bergin A., Flampouri E., et al. Mammalian cell culture for production of recombinant proteins: a review of the critical steps in their biomanufacturing. Biotechnol Adv. 2020;43 doi: 10.1016/j.biotechadv.2020.107552. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y., Cao X., Qin L. Mesenchymal stem cell-derived extracellular vesicles: a novel cell-free therapy for sepsis. Front Immunol. 2020;11:647. doi: 10.3389/fimmu.2020.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tebay L.E., Robertson H., Durant S.T., et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88(Pt B):108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong X., Thimmulappa R., Craciun F., et al. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011;184(8):928–938. doi: 10.1164/rccm.201102-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itoh K., Mimura J., Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13(11):1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 23.Liu P., Tian W., Tao S., et al. Non-covalent NRF2 activation confers greater cellular protection than covalent activation. Cell Chem Biol. 2019;26(10):1427–1435.e5. doi: 10.1016/j.chembiol.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Z.Y., Lu M.C., You Q.D. Discovery and development of Kelch-like ECH-associated protein 1. Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) Protein-Protein interaction inhibitors: achievements, challenges, and future directions. J Med Chem. 2016;59(24):10837–10858. doi: 10.1021/acs.jmedchem.6b00586. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y., Tang B., Long L., et al. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat Commun. 2021;12(1):102. doi: 10.1038/s41467-020-20315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonilla D.L., Bhattacharya A., Sha Y., et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39(3):537–547. doi: 10.1016/j.immuni.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng S., Essandoh K., Wang X., et al. Tsg101 positively regulates P62-Keap1-Nrf2 pathway to protect hearts against oxidative damage. Redox Biol. 2020;32 doi: 10.1016/j.redox.2020.101453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge W., Zhao K., Wang X., et al. iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for Keap1 binding. Cancer Cell. 2017;32(5):561–573.e6. doi: 10.1016/j.ccell.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Rada P., Rojo A.I., Chowdhry S., et al. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hushpulian D.M., Ammal Kaidery N., Ahuja M., et al. Challenges and limitations of targeting the Keap1-Nrf2 pathway for neurotherapeutics: Bach1 de-repression to the rescue. Front Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.673205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abed D.A., Goldstein M., Albanyan H., et al. Discovery of direct inhibitors of Keap1-Nrf2 Protein–Protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B. 2015;5(4):285–299. doi: 10.1016/j.apsb.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez Molina D., Jafari R., Ignatushchenko M., et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 33.Shapouri-Moghaddam A., Mohammadian S., Vazini H., et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 34.Jin Z., Zhu Z., Liu S., et al. TRIM59 protects mice from sepsis by regulating inflammation and phagocytosis in macrophages. Front Immunol. 2020;11:263. doi: 10.3389/fimmu.2020.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin X., Luo H., Yan X., et al. Interleukin-34 ameliorates survival and bacterial clearance in polymicrobial sepsis. Crit Care Med. 2018;46(6):e584–e590. doi: 10.1097/CCM.0000000000003017. [DOI] [PubMed] [Google Scholar]
- 36.Walter J., Ware L.B., Matthay M.A. Mesenchymal stem cells: mechanisms of potential therapeutic benefit in ARDS and sepsis. Lancet Respir Med. 2014;2(12):1016–1026. doi: 10.1016/S2213-2600(14)70217-6. [DOI] [PubMed] [Google Scholar]
- 37.Baird L., Dinkova-Kostova A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 38.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuadrado A., Rojo A.I., Wells G., et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18(4):295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 40.Sporn M.B., Liby K.T. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12(8):564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhakkiyalakshmi E., Sireesh D., Rajaguru P., et al. The emerging role of redox-sensitive Nrf2-Keap1 pathway in diabetes. Pharmacol Res. 2015;91:104–114. doi: 10.1016/j.phrs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Sivandzade F., Prasad S., Bhalerao A., et al. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019;21:101059. doi: 10.1016/j.redox.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biswal S., Thimmulappa R.K., Harvey C.J. Experimental therapeutics of Nrf2 as a target for prevention of bacterial exacerbations in COPD. Proc Am Thorac Soc. 2012;9(2):47–51. doi: 10.1513/pats.201201-009MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shelton L.M., Lister A., Walsh J., et al. Integrated transcriptomic and proteomic analyses uncover regulatory roles of Nrf2 in the kidney. Kidney Int. 2015;88(6):1261–1273. doi: 10.1038/ki.2015.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thimmulappa R.K., Lee H., Rangasamy T., et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacGarvey N.C., Suliman H.B., Bartz R.R., et al. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2012;185(8):851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pallesen J.S., Tran K.T., Bach A. Non-covalent small-molecule Kelch-like ECH-associated protein 1-nuclear factor erythroid 2-related factor 2 (Keap1-Nrf2) inhibitors and their potential for targeting central nervous system diseases. J Med Chem. 2018;61(18):8088–8103. doi: 10.1021/acs.jmedchem.8b00358. [DOI] [PubMed] [Google Scholar]
- 48.Luo Y., Eggler A.L., Liu D., et al. Sites of alkylation of human Keap1 by natural chemoprevention agents. J Am Soc Mass Spectrom. 2007;18(12):2226–2232. doi: 10.1016/j.jasms.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinkova-Kostova A.T., Kostov R.V., Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys. 2017;617:84–93. doi: 10.1016/j.abb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki T., Yamamoto M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J Biol Chem. 2017;292(41):16817–16824. doi: 10.1074/jbc.R117.800169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuadrado A., Pajares M., Benito C., et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharmacol Sci. 2020;41(9):598–610. doi: 10.1016/j.tips.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y.Y., Yang Y.X., Zhe H., et al. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther. 2014;8:2075–2088. doi: 10.2147/DDDT.S68872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Zeeuw D., Akizawa T., Audhya P., et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369(26):2492–2503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhuang C., Miao Z., Sheng C., et al. Updated research and applications of small molecule inhibitors of Keap1-Nrf2 protein–protein interaction: a review. Curr Med Chem. 2014;21(16):1861–1870. doi: 10.2174/0929867321666140217104648. [DOI] [PubMed] [Google Scholar]
- 55.DeNicola G.M., Karreth F.A., Humpton T.J., et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rojo de la Vega M., Chapman E., Zhang D.D. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X., Chen Z., Luo S., et al. Development of therapeutic small-molecule fluorophore for cell transplantation. Adv Funct Mater. 2016;26(46):8397–8407. [Google Scholar]
- 58.Kim S.S., Ye C., Kumar P., et al. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol Ther. 2010;18(5):993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pei Y., Yeo Y. Drug delivery to macrophages: challenges and opportunities. J Control Release. 2016;240:202–211. doi: 10.1016/j.jconrel.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 60.Jain N.K., Mishra V., Mehra N.K. Targeted drug delivery to macrophages. Expert Opin Drug Deliv. 2013;10(3):353–367. doi: 10.1517/17425247.2013.751370. [DOI] [PubMed] [Google Scholar]
- 61.Kopacz A., Kloska D., Targosz-Korecka M., et al. Keap1 governs ageing-induced protein aggregation in endothelial cells. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bose C., Alves I., Singh P., et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell. 2020;19(11) doi: 10.1111/acel.13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L., Zhang H., Chen B., et al. BaZiBuShen alleviates cognitive deficits and regulates Sirt6/NRF2/HO-1 and Sirt6/P53-PGC-1α-TERT signaling pathways in aging mice. J Ethnopharmacol. 2022;282 doi: 10.1016/j.jep.2021.114653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.